

Abstract
The vast majority of people with cystic fibrosis (CF) are now eligible for CF transmembrane regulator (CFTR) modulator therapy. The remaining individuals with CF harbor premature termination codons (PTCs) or rare CFTR variants with limited treatment options. Although the clinical modulator response can be reliably predicted using primary airway epithelial cells, primary cells carrying rare CFTR variants are scarce. To overcome this obstacle, cell lines can be created by overexpression of mouse Bmi-1 and human TERT (hTERT). Using this approach, we developed 2 non-CF and 6 CF airway epithelial cell lines, 3 of which were homozygous for the W1282X PTC variant. The Bmi-1/hTERT cell lines recapitulated primary cell morphology and ion transport function. The 2 F508del-CFTR cell lines responded robustly to CFTR modulators, which was mirrored in the parent primary cells and in the cell donors’ clinical response. Cereblon E3 ligase modulators targeting eukaryotic release factor 3a (eRF3a) rescued W1282X-CFTR function to approximately 20% of WT levels and, when paired with G418, rescued G542X-CFTR function to approximately 50% of WT levels. Intriguingly, eRF3a degraders also diminished epithelial sodium channel (ENaC) function. These studies demonstrate that Bmi-1/hTERT cell lines faithfully mirrored primary cell responses to CFTR modulators and illustrate a therapeutic approach to rescue CFTR nonsense mutations.
Keywords: Pulmonology, Therapeutics
Keywords: Genetic diseases, Ion channels, Translation

Introduction
Cystic fibrosis (CF) is a life-limiting genetic disease affecting approximately 70,000 people worldwide (1). Severe pathology develops in the lungs, where absent or dysfunctional CF transmembrane regulator (CFTR) protein leads to the accumulation of thick airway mucus, impaired mucus transport, chronic infection and inflammation, and, eventually, bronchiectasis (2). Historically, treatments have been limited to symptom management. However, the 2012 US FDA approval of the first small-molecule CFTR modulator ivacaftor ushered in a new era of CF precision medicine (3). In contrast to previous treatment approaches, CFTR modulators treat the underlying cause of disease by directly acting on the CFTR protein to correct folding, trafficking, function, or stability. With additional CFTR modulator approvals (4–6) culminating in the 2019 approval of a triple-combination therapy, elexacaftor, tezacaftor, and ivacaftor (trade name Trikafta) (7), as many as 90% of individuals with CF are now eligible for an FDA-approved modulator therapy.
However, developing therapies for the remaining individuals with CF has proven challenging. This is in part because this patient group harbors a wide range of rare CFTR variants. Indeed, more than 1200 CFTR variants are carried by 5 or fewer individuals worldwide (8, 9). For these individuals, a well-powered clinical trial is not possible. Thus, to extend life-changing treatment to all people with CF, the approach for evaluating candidate therapies must evolve.
The FDA set the precedent for such a change in 2017, when they expanded the use of ivacaftor to patient populations harboring 1 of 23 relatively rare CFTR variants (10). Although drug label expansions are common, this instance was particularly groundbreaking because the FDA based their decision purely on in vitro data (11) rather than a clinical trial. This new paradigm relies on the fidelity of in vitro systems to accurately predict clinical responses.
Primary CF human bronchial epithelial cells (HBECs) obtained at the time of lung transplantation have served as the gold standard to assess CFTR rescue in vitro (12–14). However, the supply of CF explant lungs is limited, particularly for rare CFTR variants. HBECs can also be obtained by bronchial brushing, but the procedure is invasive and yields low cell numbers (15). A readily available alternative to HBECs is human nasal epithelial cells (HNECs), which are increasingly being used as a model for the lower airways. HNECs can be obtained via nasal curettage, a nonsurgical and well-tolerated method (16). A direct comparison of paired HBEC and HNEC samples demonstrated that mature airway cell markers and CFTR activity with and without modulator treatment are preserved (15). However, like bronchial brushing, nasal curettage yields a limited supply of cells.
Previous work by our laboratory and others has shown that expression of mouse B cell–specific Moloney murine leukemia virus integration site 1 (Bmi-1) and human telomerase reverse transcriptase (hTERT) enables robust expansion of bronchial epithelial cells that can be differentiated and assayed for CFTR function for up to 15 passages (17, 18). Other groups have applied this method to nasopharyngeal biopsies (19), but, to our knowledge, this technique has not been used for nasal curettage samples. We hypothesized that Bmi-1 and hTERT expression would enhance HNEC growth properties, while maintaining their ability to differentiate into a polarized, pseudostratified epithelium. We propose that this method could be applied to rare-genotype HBECs and HNECs as they become available to create the cellular resources required for personalized medicine and drug discovery.
Here, we created 5 nasal and 3 bronchial Bmi-1/hTERT cell lines that recapitulated primary cell morphology and ion transport function for at least 15 passages. By examining cell lines with F508del/F508del and F508del/S492F genotypes and comparing them with the parent primary cells, we assessed the fidelity of CFTR modulator responses and the percentage of WT CFTR activity restored. We also compared the in vitro cell culture response to VX-445/VX-661 to the in vivo cell donors’ clinical responses to Trikafta.
We then used this approach to evaluate a potential modulator therapy for patients homozygous for W1282X, a nonsense mutation that generates a premature termination codon (PTC) in the CFTR transcript, predisposing CFTR mRNA to nonsense-mediated decay (NMD) and absent or truncated protein. Without a targetable protein, modulator therapies are ineffective, leaving these patients without treatment options. Recent studies illustrate that the cereblon (CRBN) E3 ubiquitin ligase modulator CC-90009 promotes PTC readthrough by knockdown of a key player in translation termination, the eukaryotic release factor 3a (eRF3a) (20). Using 1 nasal and 2 bronchial W1282X/W1282X cell lines, we tested the effects of CC-90009, again showing fidelity between cell lines and primary cells. Not only did we see robust CFTR rescue, but we also observed a dramatic reduction in epithelial sodium channel (ENaC) activity. We then assessed a second eRF3a degrader, SJ6986 (21), and obtained similar results, suggesting that eRF3a degradation represents a generalizable mechanism for the rescue of CFTR PTC variants. Finally, we tested CC-90009 and SJ6986 in a previously published but modified G542X/G542X bronchial cell line and observed dramatic synergy with the aminoglycoside G418 and robust CFTR functional rescue. We believe the cell lines and data generated here will facilitate the development of treatments for people with CF who currently lack an approved CFTR modulator therapy.
Results
Generation of Bmi-1/hTERT nasal and bronchial cell lines and culture optimization.
Primary cell samples were transduced with a lentivirus containing mouse Bmi-1 and hTERT separated by the T2A self-cleaving peptide sequence (Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI154571DS1). By these methods, we generated 2 non-CF nasal, 3 CF nasal, and 2 CF bronchial cell lines (Table 1). We confirmed successful vector integration at passage 6 (P6) and P15 by hTERT activity and Bmi-1 protein expression (Supplemental Figure 1, B–F). Conditionally reprogrammed cell (CRC) culture improves HNEC growth capacity (22–24). However, the CRC technique requires coculturing with irradiated or mitomycin-treated NIH3T3 feeder cells. We found that nasal cells could also be effectively expanded in EpiX media (Propagenix) as a feeder-free alternative. A comparison of a representative nasal cell line (UNCNN2T) and its parent cells in CRC and EpiX culture conditions (Supplemental Figure 1G) along with the growth properties of the other cell lines we generated can be found in the Supplemental Figure 2. Cell line differentiation was optimized by comparing UNC air-liquid interface (ALI) media (25), Vertex ALI media (26), and Pneumacult ALI media (STEMCELL Technologies). Pneumacult ALI reproducibly generated a mucociliary epithelial morphology (Supplemental Figure 3) and was used for the remainder of the study except where indicated.
Table 1. Demographics of the primary cell donors.

Bmi-1/hTERT nasal cell lines model primary cell morphology and function.
H&E and Alcian blue–periodic acid–Schiff (AB-PAS) staining of a representative non-CF nasal cell line (UNCNN2T) revealed a well-differentiated epithelium at P6 and P15 that was morphologically similar to the parent primary cells at P2 (Figure 1A). These results were confirmed by whole-mount immunostaining, which illustrated the presence of MUC5AC-producing goblet cells and α-tubulin+ ciliated cells (Figure 1, B–D). Measurements with a 24-channel transepithelial current clamp amplifier (TECC-24) device demonstrated that parent HNEC electrophysiology was also recapitulated by the UNCNN2T cell line at mid- and late-passage stages but with increased CFTR activity at P5 compared with that of parent cells (Figure 1, E–G). From this, we concluded that Bmi-1/hTERT nasal cell lines model primary HNEC morphology and function for at least 15 passages. Representative histology and whole-mount immunostaining of all other nasal cell lines are shown in Supplemental Figure 4.
Figure 1. Nasal cell lines model primary cell morphology and ion transport function.

(A) H&E and AB-PAS staining of UNCNN2T (non-CF) P2 parent cells and cell line at P6 and P15. Scale bar: 50 μm. (B–D) Whole-mount immunostaining of UNCNN2T P2 parent cells (B) and cell line at P6 (C) and P15 (D). α-Tubulin (white), MUC5AC (green), phalloidin (F-actin, red), and Hoechst (nuclei, blue). Scale bar: 25 μm. (E–G) TECC-24 measurements of UNCNN2T P2 parent cells and cell line at P5 and P15. (E) TECC-24 tracing representing 3–4 replicates. Acute addition of 6 μM benzamil (Benz), 10 μM FSK, and an inhibitor mixture (Inh mix) consisting of CFTRinh-172, GlyH-101, and bumetanide (each at 20 μM), is indicated by arrows. (F) Basal Ieq and change in Ieq (ΔIeq) in response to benzamil, FSK, and the inhibitor mixture. n = 3–4. (G) Baseline conductance values. n = 3–4. All data were analyzed using an ordinary linear model and are presented as the mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Bmi-1/hTERT nasal cell lines predict the primary cell response to CFTR modulators.
The goal of developing patient-derived cell lines is to generate a model in which the primary cell and ultimately the clinical response to CFTR-targeted therapies can be predicted. To test the ability of Bmi-1/hTERT nasal cell lines to predict CFTR modulator responses, we treated our F508del/F508del nasal cell line (UNCX4T) with CFTR corrector combinations or a vehicle control (DMSO). Here, and in all subsequent TECC-24 assays, CF cells were pretreated with test compounds and treated acutely with a CFTR potentiator (either genistein or VX-770 as indicated) following CFTR activation with forskolin (FSK). Thus, UNCX4T cells were treated with VX-445 and VX-661, the 2 correctors in Trikafta, or a recently described triple-corrector (3C) combination that combined VX-809 with 2 additional CFTR correctors, 3151 and 4172 (27). UNCX4T cells were then assayed for CFTR function using a TECC-24 device (Figure 2, A, C, and D). VX-445/VX-661 and 3C rescued 23.0% ± 1.4% and 19.5% ± 4.8% of WT CFTR function, respectively (mean ± SD), as determined by dividing the FSK response by the average non-CF nasal cell line response (i.e., 46.5 ± 5.0 μA/cm2) (Figure 1F and Supplemental Figure 2H). These data align with preclinical studies of VX-445/VX-661 and clinical observations of Trikafta in F508del homozygous populations (28). VX-445/VX-661 and 3C treatment also significantly rescued CFTR ion transport in the parent nasal cells compared with the DMSO control, rescuing 27.3% ± 5.1% and 26.7% ± 3.9% of WT CFTR function, respectively (mean ± SD) (Figure 2, B–D). These data suggest that the 3C combination might be as effective as Trikafta in rescuing F508del-CFTR and could represent a therapeutic candidate for those who cannot tolerate or do not respond to Trikafta treatment.
Figure 2. Nasal cell lines predict primary cell and clinical response to CFTR modulators.

(A and B) TECC-24 tracings of the UNCX4T nasal cell line (F508del/F508del) (A) and parent primary cells (B) treated with 0.1% DMSO and VX-445 and VX-661 (each at 5 μM), or a triple corrector combination (3C) containing VX-809/3151/4172 (each at 5 μM). Tracings are representative of 3–4 replicates. Acute addition of the potentiator 10 μM genistein is indicated by arrows. (C and D) ΔIeq of UNCX4T and parent cells in response to FSK (C) and the inhibitor mixture (D). n = 3–4. (E and F) TECC-24 tracings of the UNCX3T nasal cell line (F508del/S492F) (E) and parent primary cells (F) pretreated with DMSO, VX-445 and VX-661, or 3C. Tracings are representative of 3–4 replicates. (G and H) ΔIeq of UNCX3T and parent cells in response to FSK (G) and the inhibitor mixture (H). n = 3–4. (I) Change in the percentage of predicted FEV1 before and after Trikafta initiation in the UNCX4T donor (I) and the UNCX3T donor (J). Blue data points indicate FEV1 measured during SYMDEKO therapy, orange data points indicate FEV1 measured during Trikafta therapy, and red data points indicate FEV1 measured during a CF exacerbation. The treatment course for the UNCX4T and UNCX3T donors is indicated above the respective FEV1 plots and includes the timeline of i.v. antibiotics (green), XOLAIR for ABPA (dark blue), prednisone for ABPA (purple), antibiotics for treatment of Mycobacterium avium complex (MAC) (pink). (K) Change in weight in kilograms of the UNCX3T cell donor after Trikafta initiation. Gastrostomy tube use and subsequent removal are indicated by an orange bar. All data were analyzed using an ordinary linear model and are presented as the mean ± SD. Post hoc comparisons were performed using the general linear hypothesis test. **P < 0.01 and ***P < 0.001.
Next, we assessed a F508del/S492F compound heterozygous cell line (UNCX3T) for its response to VX-445/VX-661 and 3C (Figure 2, E, G, and H). UNCX3T responded well to VX-445/VX-661 treatment, recapitulating 22.3% ± 0.7% of WT CFTR function (mean ± SD). However, 3C treatment was less effective, producing 14.6% ± 1.5% of WT function (mean ± SD). These findings were mirrored in the UNCX3T parent cells (Figure 2, F–H). Because 3C treatment produced a lower CFTR functional response in UNCX3T, which carries only 1 copy of F508del, we posit that this modulator combination does not rescue S492F-CFTR as effectively as F508del-CFTR. Even so, the response to 3C fell well within the therapeutic window (i.e., 10% of WT CFTR function over baseline) (29) and could serve as an alternative therapy. From these studies, we concluded that Bmi-1/hTERT nasal cell lines generated from CF donors can be used to accurately predict the primary cell response to FDA-approved CFTR modulators and those in preclinical development.
Nasal cell line functional rescue correlates with the clinical response to Trikafta in 2 patients with CF.
All nasal curettage samples were obtained prior to the 2019 FDA approval of Trikafta. At the time of cell collection, the UNCX4T cell donor (F508del/F508del) was prescribed tezacaftor/ivacaftor (trade name SYMDEKO), whereas the UNCX3T cell donor (F508del/S492F) was not eligible for CFTR modulators. After nasal cell harvest and cell line generation, both donors became eligible for Trikafta and initiated treatment. Trikafta therapy was highly effective in these individuals, with an 11% and 22% increase in the percentage of predicted forced expired volume in 1 second (FEV1) over a 6-month baseline in the UNCX4T and UNCX3T donors, respectively (Figure 2, I and J). For the UNCX3T donor, Trikafta therapy promoted other significant changes in health, including a reduced frequency of pulmonary exacerbations and need for i.v. antibiotics, cessation of XOLAIR treatment for allergic bronchopulmonary aspergillosis (ABPA), and gastrostomy tube removal following improved weight gain and retention (Figure 2, J and K). Overall, the robust functional response to Trikafta that was observed in the UNCX4T and UNCX3T nasal cell lines in vitro correlated with the cell donors’ positive clinical response to therapy.
W1282X-CFTR is rescued by the CRBN modulator CC-90009.
Current therapies are not effective at rescuing the truncated protein generated by the W1282X-CFTR variant. One proposed treatment strategy is to promote ribosomal readthrough of the PTC to generate full-length protein (30, 31). Low levels of readthrough can be accomplished with high concentrations of aminoglycosides in overexpression cell lines (32). However, clinical readthrough agents are largely ineffective (33, 34), probably because of NMD, a surveillance pathway by which the cell detects and eliminates PTC-containing mRNA transcripts (35). Indeed, a recent study found substantial degradation of the CFTR transcript in an individual homozygous for the W1282X variant, with the mutated CFTR mRNA expressed at only 2.1% of WT levels (36). Thus, many groups have hypothesized that effective treatment of nonsense mutations will also require NMD inhibition (31, 36, 37). Studies in which NMD is bypassed by expressing intron-less cDNA copies of W1282X-CFTR have demonstrated that the truncated protein exhibits defective cellular trafficking and gating that can be augmented by CFTR modulators (38, 39). Yet in primary cells, CFTR modulators alone do not rescue function (30). We therefore hypothesized that effective rescue of W1282X-CFTR would require a combination of therapeutic approaches to (a) promote PTC readthrough, (b) inhibit NMD, and (c) modulate the resulting CFTR protein.
Recently, a class of CRBN E3 ligase modulators have been shown to significantly improve PTC readthrough by aminoglycoside compounds (20). One of these agents, CC-90009, is currently under investigation for the treatment of relapsed and refractory acute myeloid leukemia (AML) in a phase I clinical trial (NCT02848001). CC-90009 mediates the interaction between CRBN and eRF3a, also known as the G1 to S phase transition 1 (GSPT1) protein, which functions as a key player in stop codon recognition and translation termination. Upon interaction with CRBN, eRF3a is ubiquitinated and targeted for proteasomal degradation. Thus, we hypothesized that CC-90009 would further enhance therapeutic readthrough and rescue of W1282X-CFTR.
We optimized the CC-90009 dose by treating primary HBECs with escalating concentrations from 0.01 to 10 μM and probing for eRF3a protein knockdown (Supplemental Figure 5A). Knockdown of 85% was achieved with CC-90009 concentrations of 0.1 μM or higher. We then assessed cytotoxicity by measuring lactate dehydrogenase (LDH) release and loss of transepithelial resistance in ALI cultures treated with CC-90009 doses ranging from 0.1 to 1 μM (Supplemental Figure 5, B–D). Cytotoxicity was not observed with 0.1 μM CC-90009, although it was seen at higher doses. Cell morphology was also not altered by 0.1 μM CC-90009 treatment (Supplemental Figure 5E). From this, we concluded that 0.1 μM CC-90009 effectively reduced eRF3a protein expression without causing cytotoxicity or aberrant changes in cell morphology.
Having established a safe dose, we then treated a W1282X/W1282X nasal cell line (UNCX2T) with combinations of CC-90009, an inhibitor of NMD (Smg1i), an aminoglycoside (G418), and a CFTR corrector (VX-809) and compared the results with a vehicle control (DMSO) and single drug controls (Figure 3, A–C). CFTR function (i.e., FSK and CFTRinh-172 response) was undetectable in vehicle-treated cells or cells treated with Smg1i or G418 alone. However, all combinations that included CC-90009 resulted in substantial CFTR rescue. Unexpectedly, the CC-90009 single drug control rescued 19.1% ± 3.5% of WT CFTR function, indicating that CC-90009 was primarily responsible for the rescue seen in the tested combinations. Unlike previous reports (20), we did not observe synergy between G418 and CC-90009 to rescue W1282X-CFTR. The sufficiency of CC-90009 to rescue CFTR was confirmed in a panel of 3 W1282X/W1282X cell lines (UNCX2T, UNCCF9T, and UNCCF10T) and in the parent primary cells (Figure 3, D–G), where CFTR was rescued to 21.7% ± 5.0% and 19.5% ± 6.0% of WT CFTR function, respectively. From this, we concluded that CC-90009 could function as a single agent to rescue W1282X-CFTR to approximately 20% of WT function.
Figure 3. The eRF3a degrader CC-90009 rescues W1282X-CFTR in a panel of cell lines and parent primary cells.

(A–C) TECC-24 measurements of the UNCX2T cell line (W1282X/W1282X) treated with 0.1% DMSO, 200 μM G418, 0.3 μM SMG1i, 0.1 μM CC-90009, and 3 μM VX-809, alone or in combination as indicated. Acute addition of the potentiator 10 μM VX-770 is indicated by an arrow. (A) TECC-24 tracing representing 3–4 replicates. (B and C) ΔIeq in response to FSK (B) and CFTRinh-172 (C). Data were analyzed using ordinary linear models. n = 3–4. (D–I) TECC-24 measurements of a panel of W1282X/W1282X cell lines and parent cells treated with 0.1% DMSO or 0.1 μM CC-90009. (D and E) TECC-24 tracing of the UNCX2T cell line (D) and parent cells (E). Tracings are representative of the W1282X/W1282X panel containing 3 cell donors with 6 replicates per donor. (F–I) ΔIeq in response to FSK (F) and CFTRinh-172 (G), benazmil (H), and basal Ieq (I). Data were analyzed using a linear mixed-effects model with the donor as a random effect factor. n = 6 per donor. Post hoc comparisons were performed using the general linear hypothesis test. All data are presented as the mean ± SD. ***P < 0.001.
CC-90009 diminishes ENaC function and expression.
ENaC plays a vital role in the lungs, balancing CFTR-mediated fluid secretion by regulating sodium absorption across the airway epithelium. Upon treatment with CC-90009, we noted a striking and unexpected decrease in ENaC function, indicated by the response to benzamil, a potent ENaC inhibitor (Figure 3H). Indeed, CC-90009 diminished the benzamil response by 93.4% ± 3.8% and 84.3% ± 6.1% in our panel of W1282X/W1282X cell lines and parent cells, respectively. The basal equivalent current (Ieq) was also reduced in the treated cells (Figure 3I), indicating reduced sodium absorption at baseline. To understand this decrease in function, we assessed the mRNA expression of the 3 ENaC subunits a, b, and g (gene names SCNN1A, SCNN1B, and SCNN1G, respectively) (40) (Figure 4A). Treatment with CC-90009 decreased SCNN1B and SCNN1G mRNA expression in all cell lines and primary cells, while SCNN1A mRNA expression was unchanged in the cell lines and increased in the parent cells. By Western blotting, we observed a dose-dependent decrease in ENaCa protein levels to 26% of baseline expression with the 0.1 μM CC-90009 dose (Figure 4B). From this, we concluded that CC-90009 modulates ENaC expression at both the mRNA and protein levels. Even though eRF3a is the only reported target of CC-90009 (41), our data suggest that ENaC or an ENaC regulator may be a previously undescribed target.
Figure 4. CC-90009 reduces ENaC expression and rescues CFTR by promoting readthrough and full-length CFTR production.

(A) Relative SCNN1A, SCNN1B, SCNN1G, and CFTR mRNA levels by RT-qPCR in a panel of W1282X/W1282X cell lines and parent cells. Data were analyzed using a linear mixed-effects model with the donor as a random effect factor and are presented as the mean ± SD. n = 2–6 for each cell line. Post hoc comparisons were performed using the general linear hypothesis test. *P < 0.05 and ***P < 0.001. (B) Western blot for ENaCa in UNCCF9T parent cells treated with escalating doses of CC-90009. ENaCa expression normalized to GAPDH and relative to the 0.1% DMSO control is quantified below. Cells were grown in Vertex ALI media to increase the level of ENaC expression for detectability by Western blotting. (C) Western blot for eRF3a in a panel of W1282X/W1282X cell lines and parent cells treated with 0.1% DMSO or 0.1 μM CC-90009. eRF3a expression normalized to GAPDH and relative to the paired DMSO control is quantified below. (D) CFTR IP–Western blot (top) of the UNCX2T cell line (W1282X/W1282X) treated with 0.1% DMSO or 0.1 μM CC-90009 or the UNCNN2T non-CF cell line. A nonspecific band was observed in all samples and is indicated by a pound sign. CFTR expression normalized to actin and relative to the UNCNN2T non-CF control is quantified below. The same blot was reprobed for eRF3a (bottom). eRF3a expression normalized to actin and relative to the DMSO control is quantified below. All samples were run on the same blot and imaged at the same intensity level.
CC-90009 rescues W1282X-CFTR by promoting PTC readthrough.
As expected, CC-90009 promoted eRF3a knockdown in all of the cell lines and primary cells in our W1282X/W1282X panel (Figure 4C). eRF3 plays a known role in SURF (SMG1-UPF1-eRF1-eRF3) complex formation, the first step of NMD (42). Previous work by Baradaran-Heravi et al. demonstrated that degradation of eRF3a by CC-90009 leads to partial NMD suppression and an increase in PTC-bearing transcripts (20). We assessed CFTR mRNA expression by real-time quantitative PCR (RT-qPCR) and observed a similar increase in expression, indicating that CC-90009 suppressed NMD (Figure 4A). This partial suppression of NMD may explain why we observed no added benefit from treatment with SMG1i (Figure 3, A–C).
eRF3a plays a vital role in PTC recognition. Thus, we hypothesized that the CFTR functional rescue seen in TECC-24 assays would be primarily attributed to PTC readthrough and generation of a full-length CFTR protein. To test this, we immunoprecipitated CFTR followed by Western blotting (i.e., IP-Western blotting) using an antibody targeting a CFTR C-terminal epitope. With this approach, only full-length CFTR would produce a positive signal. We treated a W1282X/W1282X nasal cell line (UNCX2T) with and without CC-90009 and compared the IP-Western results with those from a non-CF nasal cell line control (UNCNN2T). Concurrent with eRF3a knockdown, we observed a positive CFTR signal in UNCX2T cells treated with CC-90009 but not in the DMSO control (Figure 4D). This band appeared to have the same molecular weight as CFTR in the non-CF UNCNN2T cell line, further confirming full-length CFTR production. Full-length CFTR abundance was low in UNCX2T cells treated with CC-90009, at approximately 12% of UNCNN2T expression levels. However, work from Boucher and colleagues has shown that even low levels of CFTR restoration can lead to significant functional rescue (43). From this, we concluded that CC-90009 rescued W1282X-CFTR through partial suppression of NMD, readthrough of the PTC, and generation of full-length protein.
An alternate eRF3a degrader, SJ6986, rescues W1282X-CFTR function and inhibits ENaC function.
Nishiguchi et al. recently generated a library of CRBN-modulating chemicals and identified several selective eRF3a degraders (21). One of these degraders, SJ6986, demonstrated higher potency than CC-90009 in their screening assays. Thus, we hypothesized that SJ6986 would also rescue W1282X-CFTR by promoting eRF3a knockdown. To establish an optimal dose, we treated a W1282X/W1282X nasal cell line (UNCX2T) with increasing concentrations of SJ6986 from 0.2 to 0.8 μM (Supplemental Figure 6). We observed low toxicity and significant CFTR rescue at the 0.2 μM dose. Next, we assessed CFTR function in UNCX2T cells treated with SJ6986 and G418, either alone or in combination, and compared it with a vehicle control (DMSO; Figure 5, A–C). SJ6986 significantly rescued CFTR function but to a lesser extent than was seen with CC-90009, rescuing 4.6% ± 3.2% of WT function. We also observed synergy between SJ6986 and G418, with the combination rescuing 13.7% ± 4.0% of WT CFTR function (Figure 5B). As with CC-90009, we observed a decrease in ENaC function, with an 82.9% ± 4.4% reduction in the benzamil response from baseline (Figure 5D). The basal Ieq was also reduced in SJ6986-treated cells, indicating lower baseline sodium absorption (Figure 5E). At the mRNA level, SCNN1A and SCNN1B expression remained unchanged. However, SCNN1G expression levels were significantly reduced by SJ6986 treatment (Figure 5F). We found that ENaCa protein expression was also substantially decreased by SJ6986 treatment (Figure 5G). From this, we concluded that, like CC-90009, SJ6986 rescued W1282X-CFTR function and reduced ENaC expression and function.
Figure 5. Another eRF3a degrader, SJ6986, rescues CFTR and decreases ENaC expression.

(A–E) TECC-24 measurements of the UNCX2T cell line (W1282X/W1282X) treated with 0.1% DMSO, 200 μM G418, and 0.2 μM SJ6986, alone or in combination. (A) TECC-24 tracing representing 4 replicates. (B–D) ΔIeq in response to FSK (B), CFTRinh-172 (C), and benzamil (D). (E) Basal Ieq. n = 4. (F) Relative SCNN1A, SCNN1B, SCNN1G, and CFTR mRNA levels by RT-qPCR in the UNCX2T cell line treated with 0.1% DMSO or 0.2 μM SJ6986. n = 4–6. (G) Western blot for ENaCa in the UNCX2T cell line treated with 0.1% DMSO, 0.1 μM CC-90009, or 0.2 μM SJ6986. ENaCa expression normalized to GAPDH and relative to the DMSO control is quantified below. Cells were grown in Vertex ALI media to increase the level of ENaC expression for detectability by Western blotting. (H) CFTR IP–Western blot (top) of the UNCX2T cell line treated with 0.1% DMSO, 0.1 μM CC-90009, or 0.2 μM SJ6986 or the untreated UNCNN2T non-CF cell line. The final lane (UNCNN2T) also appears in Figure 4D. A nonspecific band observed in all samples is indicated by a pound sign. CFTR expression normalized to actin and relative to the UNCNN2T non-CF control is quantified below. The same blot was reprobed for eRF3a (bottom). eRF3a expression normalized to actin and relative to the DMSO control is quantified below. All data were analyzed using ordinary linear models and are presented as the mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
We hypothesized that W1282X-CFTR functional rescue with SJ6986 was due to PTC readthrough and generation of full-length CFTR protein. To test this, we performed CFTR IP-Western blotting using an antibody targeting a CFTR C-terminal epitope. In the UNCX2T cell line (W1282X/W1282X), we observed a positive CFTR signal accompanied by eRF3a knockdown in cells treated with CC-90009 or SJ6986 but not in vehicle-treated cells (Figure 5H). By RT-qPCR, we found that CFTR mRNA expression was also increased by SJ6986, indicating partial NMD suppression (Figure 5F). From this, we concluded that SJ6986 rescued W1282X-CFTR by the same mechanism as CC-90009 through partial suppression of NMD, readthrough of the PTC, and generation of full-length protein.
eRF3a degraders synergize with G418 to rescue the G542X-CFTR variant.
Previously, our laboratory published a report on a Bmi-1/hTERT bronchial cell line homozygous for the G542X-CFTR variant (UNCCF7T) (44). This cell line was created with separate lentiviruses carrying mouse Bmi-1 and hTERT. In the present study, to boost Bmi-1/hTERT expression, UNCCF7T was transduced with the optimized lentivirus containing both genes (Supplemental Figure 1A). This supplemented cell line was named UNCCF13T to distinguish it from its parent cell line (Table 1).
To test whether G542X-CFTR could be rescued by either CC-90009 or SJ6986, we treated our G542X/G542X cell line (UNCCF13T) with CC-90009, SJ6986, G418, Smg1i, and VX-809, either alone or in combination and compared the results with a vehicle control (DMSO) (Figure 6). The CC-90009 and SJ6986 single-drug controls were less effective at rescuing G542X-CFTR than rescuing W1282X-CFTR, with only marginal functional rescue seen with CC-90009 and no rescue seen with SJ6986. We observed low but statistically significant levels of functional rescue with G418 alone. However, both eRF3a degraders synergized dramatically with G418. The CC-90009/G418 and SJ6986/G418 combinations rescued 48.3% ± 3.6% and 43.9% ± 4.7% of WT CFTR function, respectively. No further improvement was seen with added NMD inhibition (i.e., Smg1i) or CFTR modulation (i.e., VX-809). The discrepancy between our findings with W1282X-CFTR and G542X-CFTR suggests that sensitivity to and synergy between eRF3a degraders and G418 were highly dependent on mutation context, a phenomenon that has been described previously (45, 46).
Figure 6. eRF3a degraders synergize with G418 to rescue G542X-CFTR function.

(A–D) TECC-24 measurements of the UNCCF13T cell line (G542X/G542X) treated with 0.1% DMSO or combinations of 200 μM G418, 0.1 μM CC-90009, 0.2 μM SJ6986, 0.3 μM Smg1i, and 3 μM VX-809. (A and B) Change in Ieq in response to FSK (A) and CFTRinh-172 (B). (C and D) TECC-24 tracings representing 4 replicates. All data were analyzed using ordinary linear models and are presented as the mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001.
Discussion
The extension of CFTR modulators to 90% of people with CF represents a tremendous victory. However, developing targeted therapies for the remaining 10% will be challenging. This challenge is heightened by the limited availability of rare genotype primary cells. For this reason, the field has turned to cell lines for drug discovery efforts. Preclinical studies rely heavily on Fischer rat thyroid (FRT) cells engineered to express high levels of mutant CFTR (11, 47, 48). While informative, FRT cells do not capture the spectrum of response to CFTR modulators that is routinely observed in clinical trials, even in patient cohorts with the same genotype (6, 7, 49, 50). The inability to mimic patient-specific differences in CFTR regulation and function is an inherent shortcoming of the model (1, 26). Further, because of its robust CFTR overexpression, the FRT model has a greater chance of producing false-positive “hits” (51). This was documented in a study of the W1282X variant, in which the modulators VX-809 and VX-770 rescued CFTR function in the FRT model but not in patient-derived nasal epithelial cells (38). Thus, FRT and other overexpression cell lines may not accurately recapitulate mutations that pathologically decrease CFTR transcript and protein levels.
Although primary HBECs are the gold standard for CF disease modeling, they can only be expanded for approximately 10 population doublings using conventional growth methods (12). In addition, extensive population doubling often results in squamous differentiation and reduced ion transport (14). Here, we describe the development of Bmi-1/hTERT cell lines derived from primary airway epithelial cells that could be expanded for at least 30 population doublings (15 passages) while recapitulating primary cell morphology and CFTR function. We believe the ability to amply expand and assay CFTR function in patient-derived cells represents a major advantage of this model.
In this study, we directly compared the functional response of patient-derived Bmi-1/hTERT cell lines and the primary cells from which they were developed. Across 5 CF donors (3 nasal and 2 bronchial cell lines carrying a range of CFTR mutations and using both FDA-approved CFTR modulators and those in preclinical development, we found that patient-derived cell lines accurately predicted CFTR rescue in the parent primary cells. In this study, we created 2 nasal cell lines from donors carrying the F508del variant to serve as a positive control for an effective modulator response. By testing a Trikafta-like triple combination of VX-445, VX-661, and genistein, we provide proof of concept that CFTR functional rescue in patient-derived cell lines correlated with a positive clinical response to therapy. Although the patient cohort was small, to our knowledge, we are the first group to demonstrate a consensus between patient-derived cell lines, parent primary cells, and patient clinical outcomes. These data highlight the promise of personalized medicine and the predictive power of patient-derived cell lines.
Nonsense mutations in the CFTR transcript produce little to no CFTR protein. Thus, therapeutic efforts have largely focused on promoting ribosomal readthrough of the causative PTC. The best-characterized therapeutic agents for inducing readthrough are aminoglycosides (like G418) and ataluren (PTC124). Aminoglycosides have not been approved for clinical use, largely because of high toxicity (52–55). However, a small study demonstrated that short-term intranasal administration of an aminoglycoside partially rescued the CF phenotype of elevated nasal potential difference (56, 57). Ataluren was recently approved by the European Medicines Agency (but not yet by the US FDA) for treatment of Duchenne muscular dystrophy. However, a phase III clinical trial of ataluren in CF was unsuccessful, with no change in the primary or secondary endpoints of FEV1 improvement or the rate of pulmonary exacerbations (34). A recent study demonstrated that aminoglycosides and ataluren promote ribosomal readthrough by distinct mechanisms (58). Aminoglycosides bind directly to the ribosome and increase the probability of near-cognate transfer RNA (tRNA) insertion. By contrast, early evidence indicates that ataluren functions by inhibiting release factor activity (58). Here, we explored an approach to directly inhibit the release factor eRF3a.
During translation, PTCs cause the ribosome to stall and recruit eRF1 and eRF3a. Upon PTC recognition, eRF1 binds the ribosomal A site and, through its interaction with eRF3a, hydrolyzes the peptidyl-tRNA to prompt the release of nascent polypeptides (59, 60). In vitro reporter assays have demonstrated that depletion of either eRF1 or eRF3a by small interfering RNAs (siRNAs) or antisense oligonucleotides (ASOs) promotes PTC readthrough (61, 62). This work was confirmed by Huang et al., who assessed PTC readthrough by knocking down eRF1 and eRF3a with ASOs in a murine hemophilia model (63). Only modest PTC readthrough was observed. However, the group reported striking levels of synergy with aminoglycoside treatment. Another study by Sharma et al. demonstrated PTC readthrough in CFTR following small-molecule eRF1 knockdown (64). This group reported aminoglycoside synergy in FRT and 16HBE gene–edited cell models. In this study, we explored a class of compounds that degrades eRF3a.
Small-molecule eRF3a degraders have been described as potent tumoricidal agents, with activity against AML (41, 65–70). Baradaran-Heravi et al. were the first to assess eRF3a degraders for ribosomal readthrough activity (20). In a panel of human disease models, the authors found that CC-90009 and its analog CC-885 promoted PTC readthrough via eRF3a degradation and that readthrough was further enhanced with the inclusion of aminoglycosides (20). This group also knocked down eRF3a by siRNA and confirmed that the PTC readthrough they observed with CC-90009 was a direct consequence of eRF3a degradation rather than an off-target effect of the compound.
We found that treatment with the eRF3a degraders CC-90009 and SJ6986 substantially rescued CFTR function in patient-derived cell lines carrying the W1282X- and G542X-CFTR variants. By demonstrating consistent findings between 2 distinct eRF3a degraders, we propose that eRF3a degradation is a generalizable mechanism for CFTR PTC rescue. The robust functional rescue seen in our G542X/G542X cell line provided evidence for successful ribosomal readthrough, since CFTR proteins truncated before the 1172 residue are known to have no residual channel function (71). Further, we observed full-length CFTR protein expression by CFTR IP-Western blotting. Assessing the amino acid substitutions at the PTC site in CFTR would be highly informative but is limited by low CFTR protein levels in primary airway epithelial cells. Future studies to understand the amino acid substitutions induced by readthrough agents will require extensive protein purification and/or different model systems.
Genotype-dependent sensitivity to readthrough agents has been described previously (45, 46). Although we did not observe synergy between G418 and CC-90009 to rescue W1282X-CFTR, we did observe synergy between G418 and CC-90009 in the G542X-CFTR context and between G418 and SJ6986 in both genotypes. We speculate that synergy between eRF3a degraders and aminoglycosides depends on the CFTR genotype, the baseline level of the eRF3a degrader and aminoglycoside responsiveness, and the degree of eRF3a protein degradation by different compounds and doses. Mutation-dependent sensitivity to readthrough compounds further underscores the importance of personalized medicine approaches to assess drug efficacy.
CC-90009 reportedly has little effect on the proteome apart from eRF3a (67, 70). However, Aliouat et al. documented over 2000 genes with significant changes in mRNA expression and protein translation after eRF3a knockdown (72). In the present study, we report an unexpected decrease in ENaC function as a result of CC-90009 or SJ6986 treatment. These findings could have implications for the ongoing clinical evaluation of CC-90009 for the treatment of AML (NCT02848001). Early clinical trial results indicate hypotension as a dose-limiting toxicity and a treatment-emergent adverse event (68). Sodium reabsorption via ENaC is known to determine extracellular fluid volume and regulate blood pressure (73, 74). Thus, we posit that the adverse hypotension observed in CC-90009 clinical trials could be explained by off-target ENaC inhibition. We speculate that ENaC is affected by eRF3a degraders by 1 of 2 possible mechanisms: (a) eRF3a loss directly affects ENaC or an ENaC regulator, or (b) ENaC or an ENaC regulator could share structural similarity with the eRF3a site that binds CC-90009 and SJ6986. Future work will be required to understand the connection between eRF3a degraders and ENaC functional knockdown. Although the effects on ENaC were unexpected, these too could prove therapeutic in the lungs. Increasing airway hydration through ENaC inhibition has been proposed previously as a CF treatment strategy (75).
One concern of therapeutically degrading eRF3a is the possibility of normal termination codon (NTC) readthrough. However, NTCs are more highly regulated than PTCs (63). Further, studies of ataluren-induced readthrough via release factor inhibition demonstrated no adverse effect on translation termination at NTCs (53, 76). While these studies set the precedent that readthrough therapies are safe, future work will be important to determine whether eRF3a degradation affects translation termination at NTCs.
In summary, here we describe a pipeline for developing patient-derived airway epithelial cell lines to model rare CFTR variants. Bmi-1/hTERT nasal and bronchial cell lines are highly representative of primary cell morphology, function, and response to CFTR modulators and correlated with the cell donors’ clinical response to Trikafta therapy. We present the finding that eRF3a degradation by CC-90009 or SJ6986 promoted robust functional rescue of the CFTR PTC variants W1282X and G542X. We observed an off-target but potentially beneficial reduction of ENaC activity following treatment with both eRF3a degraders, and follow-up studies are required to understand the mechanism. Overall, eRF3a degradation may be a promising therapeutic strategy to develop treatments for people with CF carrying PTC variants.
Methods
Primary cell isolation and tissue culture.
Primary HNECs were obtained by nasal curettage from 2 non-CF and 3 CF donors, yielding 2.1 × 106 cells on average. Demographics and CFTR genotypes are summarized in Table 1. Freshly obtained samples were transferred into lactated Ringer’s solution. Cell dissociation was performed by treatment with DTT (0.5 mg/mL; MilliporeSigma, D0632) and DNase (10 μg/mL; MilliporeSigma, DN25) for 15 minutes. Cells were disaggregated by incubation with Accutase (MilliporeSigma, A6964) for 10 minutes. HBECs from explanted CF transplant lungs were obtained and isolated as previously described (25). HNECs and HBECs were cultured by CRC culturing methods (14, 77) unless otherwise indicated. Briefly, cells were cocultured with irradiated NIH3T3 fibroblasts (provided by Richard Schlegel and Xuefeng Liu at Georgetown University, Washington, DC, who received an original stock from Howard Green at Harvard University, Cambridge, Massachusetts, USA) in the presence of the rho-kinase inhibitor Y-27632. Primary cells were cultured with an antibiotic-antifungal cocktail for the first 2 days.
Production and titering of the Bmi-1/hTERT lentivirus.
HEK293T cells (ATCC) were thawed and passaged a minimum of 2 times in 100 mm tissue culture dishes (Corning, 430167) with DMEM with 10% FBS and 1% penicillin-streptomycin before transfecting the cells at 85%–95% confluence with 8 μg pCDH-hTERT-T2A-Bmi-1, 8 μg psPAX, and 4 μg VSVG plasmids, using Lipofectamine 2000 (Invitrogen, Thermo Fisher Scientific, 11668-019) according to the manufacturer’s protocol. Cells were maintained at 37°C in 5% CO2. Media were changed and collected every 24 hours for up to 72 hours and stored at 4°C. A portion of virus-containing medium was aliquoted and frozen at –80°C, and the remainder was concentrated with the Lenti-X Concentrator kit (Clontech, 631231). Viral titering was performed using 15 μL unconcentrated virus or 1.5 μL concentrated virus with the Lenti-X RT-qPCR Titration Kit (Clontech, 632165). Results were read on an ABI QuantStudio 6 RT-PCR machine.
Creation of Bmi-1/hTERT growth-enhanced cell lines.
Primary cells were cultured in CRC conditions to P1. At approximately 30% confluence, irradiated NIH3T3 cells were removed with trypsin, and the media were replaced with irradiated NIH3T3-conditioned media (78) containing polybrene (10 μg/mL) and hTERT-T2A-Bmi-1 lentivirus (Supplemental Figure 1A) at an MOI of 2. After 24 hours, cells were washed with 1× PBS and transduced again with fresh virus to increase transduction efficiency. After 48 hours of lentivirus exposure, CRC culture conditions were restored by adding irradiated NIH3T3 cells. Cells were passaged using Accutase at 70%–90% confluence. Growth curves were created by calculating population doublings as the log base 2 of the ending cell number divided by the starting cell number.
Validation of hTERT expression and activity.
hTERT expression was determined using quantitative analyses of telomerase activity by the combination of the conventional telomeric repeat amplification protocol (TRAP) with RT-qPCR (79, 80). Cells were lysed with a buffer containing 0.5% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate), 10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.1 mM benzamidine, 10 U/mL RNasin, and 10% glycerol. The protein concentration was measured by detergent-compatible (DC) protein assay (Bio-Rad, 500-0112), and 5 mM β-mercaptoethanol was added to the samples. Each 40 μL reaction contained 2 μg cell lysate diluted in 0.1 mg/mL BSA, 1× TRAP reaction buffer (20 mM Tris-HCl [pH 8.3], 1.5 mM MgCl2, 10 mM EGTA, and 50 mM KCl, 50 μM of each deoxynucleotide triphosphate, 910 nM telomerase substrate (TS) forward primer [see sequence below], and 0.4 μg T4 gene protein [New England BioLabs (NEB), M0300S]). The reaction mixture was incubated at 33°C for 5 hours followed by 95°C for 10 minutes to inactivate telomerase. RT-qPCR with iQ SYBR Green Supermix (Bio-Rad, 170-8882) was then performed to quantitate the substrate molecules to which telomeric repeats were added. Each 25 μL reaction contained 300 nM TS forward and reverse primer and 1 μL of the product from the first step. A standard curve was produced by 10-fold serial dilution of HEK293 cell extracts, from 4 μg to 4 ng. All samples were run in triplicate. The TS primer sequences were as follows: 5′-AATCCGTCGAGCAGAGTT-3′ (forward) and 5′-CCCTTACCCTTACCCTTACCCTAA-3′ (reverse).
Western blot analysis.
Cells were rinsed with cold PBS and lysed in RIPA buffer (Boston Bioproducts, BP-115D) with a Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific, 1861280) for 30 minutes on ice and spun at 16,000g for 15 minutes to remove the insoluble fraction. The protein concentration was determined using the DC protein assay (Bio-Rad, 5000112). For each sample, 30 μg protein was loaded onto an SDS-PAGE gel and transferred onto a nitrocellulose membrane. To validate Bmi-1 expression, blots were incubated with anti–Bmi-1 antibody (Cell Signaling Technology, 5856S), followed by goat anti–rabbit IgG (H+L) DyLight800 (Invitrogen, Thermo Fisher Scientific, SA5-10036). Signals were detected and quantified using an Odyssey Image scanner (LI-COR). To determine the effect of CC-90009 on ENaCa and eRF3a expression, blots were incubated with anti-ENaCa (mAb UNC1 19.2.1) (81) and anti-eRF3a antibody (Abcam, ab49878), followed by peroxidase-linked donkey anti–mouse IgG (Thermo Fisher Scientific, 45000679) and donkey anti–rabbit IgG (Thermo Fisher Scientific, 45000682), respectively. Signals were detected using Clarity Western ECL substrate (Bio-Rad, 170-5061), imaged with the Sapphire Biomolecular Imager (Azure Biosystems), and quantified with ImageQuant TL. To normalize protein loading, the blots were reprobed with anti-GAPDH (Proteintech, 60004-1-Ig). Western blot images were cropped around the known molecular weight of the respective proteins. The corresponding complete, unedited blots are included in the supplemental material.
CFTR IP-Western blotting.
CFTR was immunoprecipitated as described previously (82) in Nonidet P-40 buffer (1% NP-40 Alternative [Calbiochem], 150 mM NaCl, 50 mM Tris, 1 mM EGTA, 4 μg/mL leupeptin, 8 μg/mL aprotinin, 200 μg/mL Pefabloc, 484 μg/mL benzamidine, and 14 μg/mL E64, pH 7.4) using an affinity-purified rabbit polyclonal antibody 155 against amino acids 1465–1480 of human CFTR (83), followed by pulldown with protein A/G Plus-Agarose (Santa Cruz Biotechnology). Samples were separated on 4%–20% gradient SDS-PAGE gels (Bio-Rad) and then transferred onto nitrocellulose. Blots were probed with mouse monoclonal anti-CFTR antibodies 596 and 217 (CFTR Antibody Distribution Program; 1:2000 each) and then with IR Dye 680 goat anti–mouse IgG (Invitrogen, Thermo Fisher Scientific). Anti-actin (Cell Signaling Technology) was used as a loading control. Protein bands were visualized using the Sapphire Biomolecular Imager (Azure Biosystems).
RNA purification and RT-qPCR.
Total RNA was purified using the Direct-zol RNA Miniprep kit (Zymo Research, R2052) and quantified using a NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific). TaqMan assays for CFTR (Integrated DNA Technologies, Hs.PT.58.28207352), SCNN1A (Hs01013028_m1), SCNN1B (Hs00165722_m1), and SCNN1G (Hs00168918_m1) were performed with the Luna Universal Probe One-Step RT-qPCR Kit (NEB, E3006L). Expression of each gene was normalized to 18S (Hs99999901_s1). All TaqMan probes were purchased from Applied Biosystems except the CFTR probe. RT-qPCR was run on Applied Biosystem’s QuantStudio 6 (Thermo Fisher Scientific).
Mycoplasma testing, DNA fingerprinting, and CFTR genotyping.
Mycoplasma contamination was evaluated by the MycoAlert Plus Mycoplasma Detection Kit (Lonza, LT07-710) with the MycoAlert Assay Control Set (Lonza, LT07-518) according to the manufacturer’s instructions. Mycoplasma-specific enzymes were undetectable in Bmi-1/hTERT cells between P6 and P7 or in the parent primary cells at P2. Luminometry results were read on a FLUOstar Omega microplate reader (BMG LABTECH). Cell line authentication was performed by Labcorp’s Cell Line Testing Division on July 13, 2021, using the PowerPlex 16HS (Promega) for short tandem repeat (STR) DNA profiling. Each cell line was unique among human cell lines and samples present in Cellosaurus (version 1.4.4). Further, the STR profile of each cell line matched that of the parent cells. CFTR genotyping was performed by clinically approved laboratories and retrieved from the clinical record.
Culture at an ALI.
Cell culture inserts were coated with human placenta type IV collagen (MilliporeSigma, C7521). Cells expanded by the CRC culture method were seeded at 2.5 × 105 cells on 12 mm Snapwells (Corning Costar, 3407) or at 3.3 × 104 cells on 6.5 mm Transwells (Corning Costar, 3470) or HTS Transwell 24-well Permeable Support (Corning Costar, 3378) and transitioned to ALI when cells reached confluence. Cells were maintained in Pneumacult ALI medium (STEMCELL Technologies, no. 05001) for 21–32 days after seeding. For drug treatment, compounds were added to basolateral media, and 10 μL or 50 μL was added apically for 6.5 mm and 12 mm Transwells, respectively.
Whole-mount immunostaining and imaging.
ALI cultures were fixed for 15 minutes on days 21–32 in 4% paraformaldehyde and stained for α-tubulin (MilliporeSigma, MAB1864; 3 μg/mL), MUC5AC (Thermo Fisher Scientific, 45M1; 4 μg/mL), F-actin (phalloidin; Invitrogen, Thermo Fisher Scientific, A22287), and DNA (Hoechst 33342; Invitrogen, Thermo Fisher Scientific, H3570) using species-specific secondary antibodies as previously described (84). Cultures were imaged using a Leica TCS SP8 with a ×63 oil objective.
Electrophysiology measurements.
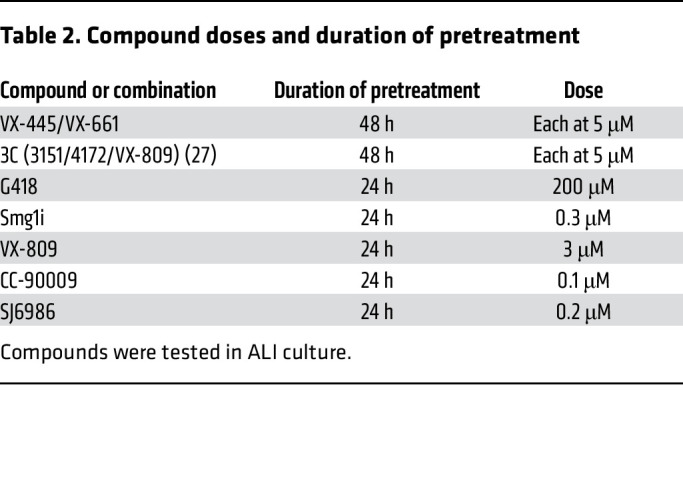
The transepithelial electrical resistance and the Ieq were recorded using a TECC-24 as previously described (85). To determine the baseline resistance, 8 initial measurements were collected at approximately 2 minute intervals. Changes in Ieq were determined after the addition of benzamil, FSK, a CFTR potentiator (either genistein or VX-770 as indicated), and a CFTR inhibitor 172 (CFTRinh-172) or an inhibitor mixture containing CFTRinh-172, GlyH-101, and bumetanide as indicated. TECC-24 assays were performed on days 21–32 after seeding. Cell cultures were treated with test compounds prior to the TECC-24 assay as indicated in Table 2.
Table 2. Compound doses and duration of pretreatment.
Statistics.
All data were analyzed using ordinary linear models or linear mixed-effects models when multiple donors were studied, with the donor as a random effect factor. We performed log transformation before statistical testing when the variance differed greatly between groups, as indicated in the figure legends. Analyses were performed with either the lm function in base R or the lmer function in the R packages lme4 and lmerTest. Post hoc comparisons were performed using the general linear hypothesis test from the multcomp R package. A P value of less than 0.05 was considered significant.
Study approval.
Protocols for informed consent to obtain cells via nasal curettage and from explanted CF lungs were approved under the University of North Carolina’s Office of Human Research Ethics/Institutional Review Board studies 98-1015 and 03-1396, respectively.
Author contributions
REL, CAL, LH, and SHR conceived the project and designed the experiments with the assistance of JWT and NAC. The Bmi-1/hTERT lentiviral vector was prepared by JTM and LCM. Nasal curettage and review of clinical records were performed by MRK and AJK. Cell line generation and growth curve analyses were performed by REL, CAL, and LH. TMM, CAL, SCG, and REL performed whole-mount immunostaining and confocal imaging. Western blots and RT-qPCR was performed by LH and ECBS. CFTR IP-Western blotting were performed by DMC and MG. Electrophysiology experiments were performed by REL, ECBS, and SCG. REL, LH, JWT, and NAC assessed CC-90009’s effects on PTC readthrough. All of the statistical analysis was performed by HD. REL and CAL composed the manuscript and figures. All authors read, edited, and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
The authors thank the cell donors; Dr. Laurie Snyder and the BioRepository and Precision Pathology Center Staff at Duke University; Drs. Hannah Blau, Huda Mussaffi, and Dario Prais at Schneider Children’s Hospital in Israel; and Dr. Mordechai Kramer at Rabin Medical Center in Israel for facilitating the procurement of CF lungs. We thank William Trey Brown for providing the medical records and Eric Roe for his help with reference formatting. We thank Robert Bridges at Chicago Medical School, and Ashvani Singh, Corina Balut, and the engineering team at Abbvie, North Chicago, for their generous assistance in establishing the TECC-24 system at the University of North Carolina. We are grateful to the Marsico Lung Institute Tissue Procurement and Cell Culture Core for providing cells and media and to Richard Boucher for discussions about electrophysiology. We thank Richard Schlegel and Xuefeng Liu at Georgetown University for providing NIH3T3J2 cells. CFTR modulators were provided by Dr. Gergely Lukacs at McGill University (Montreal, Quebec, Canada). This work was supported by Emily’s Entourage grant 17-5600; Cystic Fibrosis Foundation grants RANDEL17XX0, RANDEL20XX2, and BOUCHE19R0; and NIH grants 1T32GM133364, 1F31HL158197, 5KL2TR002490, 5R01HL071798, and 2P30DK065988.
Version 1. 07/28/2022
In-Press Preview
Version 2. 09/15/2022
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2022, Lee et al. This is an open access article published under the terms of the Creative Commons Attribution 4.0 International License.
Reference information: J Clin Invest. 2022;132(18):e154571. https://doi.org/10.1172/JCI154571.
Contributor Information
Rhianna E. Lee, Email: rhianna_lee@med.unc.edu.
Catherine A. Lewis, Email: catlewis@email.unc.edu.
Lihua He, Email: lihua_he@med.unc.edu.
Emily C. Bulik-Sullivan, Email: emily_bulik-sullivan@med.unc.edu.
Samuel C. Gallant, Email: gallants@ad.unc.edu.
Teresa M. Mascenik, Email: mascenikteresa@gmail.com.
Hong Dang, Email: dangh@email.unc.edu.
Deborah M. Cholon, Email: deborah_cholon@med.unc.edu.
Martina Gentzsch, Email: martina_gentzsch@med.unc.edu.
Lisa C. Morton, Email: lisa_morton@med.unc.edu.
John T. Minges, Email: minges@unc.edu.
Jonathan W. Theile, Email: jtheile@ligand.com.
Neil A. Castle, Email: ncastle@icagen.com.
Michael R. Knowles, Email: michael_knowles@med.unc.edu.
Adam J. Kimple, Email: adam_kimple@med.unc.edu.
Scott H. Randell, Email: randell@med.unc.edu.
References
- 1.Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nat Rev Genet. 2015;16(1):45–56. doi: 10.1038/nrg3849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ratjen F, et al. Cystic fibrosis. Nat Rev Dis Primers. 2015;1:15010. doi: 10.1038/nrdp.2015.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramsey BW, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med. 2011;365(18):1663–1672. doi: 10.1056/NEJMoa1105185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rowe SM, et al. Tezacaftor-ivacaftor in residual-function heterozygotes with cystic fibrosis. N Engl J Med. 2017;377(21):2024–2035. doi: 10.1056/NEJMoa1709847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taylor-Cousar JL, et al. Tezacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del. N Engl J Med. 2017;377(21):2013–2023. doi: 10.1056/NEJMoa1709846. [DOI] [PubMed] [Google Scholar]
- 6.Wainwright CE, et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N Engl J Med. 2015;373(3):220–231. doi: 10.1056/NEJMoa1409547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Middleton PG, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381(19):1809–1819. doi: 10.1056/NEJMoa1908639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Durmowicz AG, et al. The U.S. Food and Drug Administration’s experience with ivacaftor in cystic fibrosis. Establishing efficacy using in vitro data in lieu of a clinical trial. Ann Am Thorac Soc. 2018;15(1):1–2. doi: 10.1513/AnnalsATS.201708-668PS. [DOI] [PubMed] [Google Scholar]
- 9. Clinical and Functional Translation fo CFTR. CFTR2. https://cftr2.org/ Updated April 29, 2022. Accessed July 27, 2022.
- 10. US Food & Drug Administration (FDA). Drugs@FDA: FDA-Approved Drugs. https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=203188 Updated December 21, 2020. Accessed July 27, 2022.
- 11.Van Goor F, et al. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros. 2014;13(1):29–36. doi: 10.1016/j.jcf.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 12.Randell SH, et al. Primary epithelial cell models for cystic fibrosis research. Methods Mol Biol. 2011;742:285–310. doi: 10.1007/978-1-61779-120-8_18. [DOI] [PubMed] [Google Scholar]
- 13.Van Goor F, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol. 2006;290(6):L1117–L1130. doi: 10.1152/ajplung.00169.2005. [DOI] [PubMed] [Google Scholar]
- 14.Gentzsch M, et al. Pharmacological rescue of conditionally reprogrammed cystic fibrosis bronchial epithelial cells. Am J Respir Cell Mol Biol. 2017;56(5):568–574. doi: 10.1165/rcmb.2016-0276MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brewington JJ, et al. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight. 2018;3(13):99385. doi: 10.1172/jci.insight.99385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mosler K, et al. Feasibility of nasal epithelial brushing for the study of airway epithelial functions in CF infants. J Cyst Fibros. 2008;7(1):44–53. doi: 10.1016/j.jcf.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Fulcher ML, et al. Novel human bronchial epithelial cell lines for cystic fibrosis research. Am J Physiol Lung Cell Mol Physiol. 2009;296(1):L82–L91. doi: 10.1152/ajplung.90314.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munye MM, et al. BMI-1 extends proliferative potential of human bronchial epithelial cells while retaining their mucociliary differentiation capacity. Am J Physiol Lung Cell Mol Physiol. 2017;312(2):L258–L267. doi: 10.1152/ajplung.00471.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yip YL, et al. Efficient immortalization of primary nasopharyngeal epithelial cells for EBV infection study. PLoS One. 2013;8(10):e78395. doi: 10.1371/journal.pone.0078395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baradaran-Heravi A, et al. Effect of small molecule eRF3 degraders on premature termination codon readthrough. Nucleic Acids Res. 2021;49(7):3692–3708. doi: 10.1093/nar/gkab194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishiguchi G, et al. Identification of potent, selective, and orally bioavailable small-molecule GSPT1/2 degraders from a focused library of cereblon modulators. J Med Chem. 2021;64(11):7296–7311. doi: 10.1021/acs.jmedchem.0c01313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kmit A, et al. Extent of rescue of F508del-CFTR function by VX-809 and VX-770 in human nasal epithelial cells correlates with SNP rs7512462 in SLC26A9 gene in F508del/F508del cystic fibrosis patients. Biochim Biophys Acta Mol Basis Dis. 2019;1865(6):1323–1331. doi: 10.1016/j.bbadis.2019.01.029. [DOI] [PubMed] [Google Scholar]
- 23.Reynolds SD, et al. Airway progenitor clone formation is enhanced by Y-27632-dependent changes in the transcriptome. Am J Respir Cell Mol Biol. 2016;55(3):323–336. doi: 10.1165/rcmb.2015-0274MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolf S, et al. Conditional reprogramming of pediatric airway epithelial cells: a new human model to investigate early-life respiratory disorders. Pediatr Allergy Immunol. 2017;28(8):810–817. doi: 10.1111/pai.12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fulcher ML, et al. Well-differentiated human airway epithelial cell cultures. Methods Mol Med. 2005;107:183–206. doi: 10.1385/1-59259-861-7:183. [DOI] [PubMed] [Google Scholar]
- 26.Neuberger T, et al. Use of primary cultures of human bronchial epithelial cells isolated from cystic fibrosis patients for the pre-clinical testing of CFTR modulators. Methods Mol Biol. 2011;741:39–54. doi: 10.1007/978-1-61779-117-8_4. [DOI] [PubMed] [Google Scholar]
- 27.Veit G, et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat Med. 2018;24(11):1732–1742. doi: 10.1038/s41591-018-0200-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keating D, et al. VX-445-tezacaftor-ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N Engl J Med. 2018;379(17):1612–1620. doi: 10.1056/NEJMoa1807120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dave K, et al. Entering the era of highly effective modulator therapies. Pediatr Pulmonol. 2021;56(suppl 1):S79–S89. doi: 10.1002/ppul.24968. [DOI] [PubMed] [Google Scholar]
- 30.Mutyam V, et al. Therapeutic benefit observed with the CFTR potentiator, ivacaftor, in a CF patient homozygous for the W1282X CFTR nonsense mutation. J Cyst Fibros. 2017;16(1):24–29. doi: 10.1016/j.jcf.2016.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valley HC, et al. Isogenic cell models of cystic fibrosis-causing variants in natively expressing pulmonary epithelial cells. J Cyst Fibros. 2019;18(4):476–483. doi: 10.1016/j.jcf.2018.12.001. [DOI] [PubMed] [Google Scholar]
- 32.Shalev M, Baasov T. When proteins start to make sense: fine-tuning aminoglycosides for PTC suppression therapy. Medchemcomm. 2014;5(8):1092–1105. doi: 10.1039/C4MD00081A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kerem E, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2(7):539–547. doi: 10.1016/S2213-2600(14)70100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Konstan MW, et al. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: the international, randomized, double-blind, placebo-controlled ataluren confirmatory trial in cystic fibrosis (ACT CF) J Cyst Fibros. 2020;19(4):595–601. doi: 10.1016/j.jcf.2020.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Linde L, et al. Nonsense-mediated mRNA decay affects nonsense transcript levels and governs response of cystic fibrosis patients to gentamicin. J Clin Invest. 2007;117(3):683–692. doi: 10.1172/JCI28523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aksit MA, et al. Decreased mRNA and protein stability of W1282X limits response to modulator therapy. J Cyst Fibros. 2019;18(5):606–613. doi: 10.1016/j.jcf.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laselva O, et al. Functional rescue of c.3846G>A (W1282X) in patient-derived nasal cultures achieved by inhibition of nonsense mediated decay and protein modulators with complementary mechanisms of action. J Cyst Fibros. 2020;19(5):717–727. doi: 10.1016/j.jcf.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Haggie PM, et al. Correctors and potentiators rescue function of the truncated W1282X-cystic fibrosis transmembrane regulator (CFTR) translation product. J Biol Chem. 2017;292(3):771–785. doi: 10.1074/jbc.M116.764720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang W, et al. Robust stimulation of W1282X-CFTR channel activity by a combination of allosteric modulators. PLoS One. 2016;11(3):e0152232. doi: 10.1371/journal.pone.0152232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Canessa CM, et al. Amiloride-sensitive epithelial Na+ channel is made of three homologous subunits. Nature. 1994;367(6462):463–467. doi: 10.1038/367463a0. [DOI] [PubMed] [Google Scholar]
- 41.Surka C, et al. CC-90009, a novel cereblon E3 ligase modulator, targets acute myeloid leukemia blasts and leukemia stem cells. Blood. 2021;137(5):661–677. doi: 10.1182/blood.2020008676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Causier B, et al. Conservation of nonsense-mediated mRNA decay complex components throughout eukaryotic evolution. Sci Rep. 2017;7(1):16692. doi: 10.1038/s41598-017-16942-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johnson LG, et al. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet. 1992;2(1):21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 44.Gentzsch M, et al. Restoration of R117H CFTR folding and function in human airway cells through combination treatment with VX-809 and VX-770. Am J Physiol Lung Cell Mol Physiol. 2016;311(3):L550–L559. doi: 10.1152/ajplung.00186.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clarke LA, et al. Integrity and stability of PTC bearing CFTR mRNA and relevance to future modulator therapies in cystic fibrosis. Genes (Basel) 2021;12(11):1810. doi: 10.3390/genes12111810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Venturini A, et al. Comprehensive analysis of combinatorial pharmacological treatments to correct nonsense mutations in the CFTR gene. Int J Mol Sci. 2021;22(21):11972. doi: 10.3390/ijms222111972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Goor F, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106(44):18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van Goor F, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A. 2011;108(46):18843–18848. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Boeck K, et al. Efficacy and safety of ivacaftor in patients with cystic fibrosis and a non-G551D gating mutation. J Cyst Fibros. 2014;13(6):674–680. doi: 10.1016/j.jcf.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 50.Donaldson SH, et al. Tezacaftor/ivacaftor in subjects with cystic fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am J Respir Crit Care Med. 2018;197(2):214–224. doi: 10.1164/rccm.201704-0717OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Awatade NT, et al. Human primary epithelial cell models: promising tools in the era of cystic fibrosis personalized medicine. Front Pharmacol. 2018;9:1429. doi: 10.3389/fphar.2018.01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dabrowski M, et al. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol Med. 2018;24(1):25. doi: 10.1186/s10020-018-0024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagel-Wolfrum K, et al. Targeting nonsense mutations in diseases with translational read-through-inducing drugs (TRIDs) BioDrugs. 2016;30(2):49–74. doi: 10.1007/s40259-016-0157-6. [DOI] [PubMed] [Google Scholar]
- 54.Keeling KM, et al. Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum Genet. 2014;15:371–394. doi: 10.1146/annurev-genom-091212-153527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xie J, et al. New developments in aminoglycoside therapy and ototoxicity. Hear Res. 2011;281(1–2):28–37. doi: 10.1016/j.heares.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilschanski M, et al. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Respir Crit Care Med. 2000;161(3 pt 1):860–865. doi: 10.1164/ajrccm.161.3.9904116. [DOI] [PubMed] [Google Scholar]
- 57.Wilschanski M, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349(15):1433–1441. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 58.Ng MY, et al. Ataluren and aminoglycosides stimulate read-through of nonsense codons by orthogonal mechanisms. Proc Natl Acad Sci U S A. 2021;118(2):e2020599118. doi: 10.1073/pnas.2020599118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frolova L, et al. Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA. 1996;2(4):334–341. [PMC free article] [PubMed] [Google Scholar]
- 60.Zhouravleva G, et al. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J. 1995;14(16):4065–4072. doi: 10.1002/j.1460-2075.1995.tb00078.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Carnes J, et al. Stop codon suppression via inhibition of eRF1 expression. RNA. 2003;9(6):648–653. doi: 10.1261/rna.5280103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chauvin C, et al. Involvement of human release factors eRF3a and eRF3b in translation termination and regulation of the termination complex formation. Mol Cell Biol. 2005;25(14):5801–5811. doi: 10.1128/MCB.25.14.5801-5811.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang L, et al. Targeting translation termination machinery with antisense oligonucleotides for diseases caused by nonsense mutations. Nucleic Acid Ther. 2019;29(4):175–186. doi: 10.1089/nat.2019.0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sharma J, et al. A small molecule that induces translational readthrough of CFTR nonsense mutations by eRF1 depletion. Nat Commun. 2021;12(1):4358. doi: 10.1038/s41467-021-24575-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matyskiela ME, et al. A novel cereblon modulator recruits GSPT1 to the CRL4 (CRBN) ubiquitin ligase. Nature. 2016;535(7611):252–257. doi: 10.1038/nature18611. [DOI] [PubMed] [Google Scholar]
- 66.Lu G, et al. Elucidating the mechanism of action of CC-90009, a novel cereblon E3 ligase modulator, in AML via genome-wide CRISPR screen. Blood. 2019;134(suppl_1):405. doi: 10.1182/blood-2019-125492. [DOI] [Google Scholar]
- 67.Lopez-Girona A, et al. CC-90009, a novel cereblon E3 ligase modulator, targets GSPT1 for degradation to induce potent tumoricidal activity against acute myeloid leukemia (AML) Blood. 2019;134(supplement_1):2703. doi: 10.1182/blood-2019-127892. [DOI] [Google Scholar]
- 68.Uy GL, et al. Clinical activity of CC-90009, a cereblon E3 ligase modulator and first-in-class GSPT1 degrader, as a single agent in patients with relapsed or refractory acute myeloid leukemia (R/R AML): first results from a phase I dose-finding study. Blood. 2019;134(suppl_1):232. doi: 10.1182/blood-2019-123966. [DOI] [Google Scholar]
- 69.Fan J, et al. Pharmacodynamic responses to CC-90009, a novel cereblon E3 ligase modulator, in a phase I dose-escalation study in relapsed or refractory acute myeloid leukemia (R/R AML) Blood. 2019;134(suppl_1):2547. doi: 10.1182/blood-2019-124291. [DOI] [Google Scholar]
- 70.Jin L, et al. A novel cereblon E3 ligase modulator eradicates acute myeloid leukemia stem cells through degradation of translation termination factor GSPT1. Blood. 2019;134(supplement_1):3940. doi: 10.1182/blood-2019-128450. [DOI] [Google Scholar]
- 71.Cui L, et al. Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol. 2007;365(4):981–994. doi: 10.1016/j.jmb.2006.10.086. [DOI] [PubMed] [Google Scholar]
- 72.Aliouat A, et al. Divergent effects of translation termination factor eRF3A and nonsense-mediated mRNA decay factor UPF1 on the expression of uORF carrying mRNAs and ribosome protein genes. RNA Biol. 2020;17(2):227–239. doi: 10.1080/15476286.2019.1674595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pratt JH. Central role for ENaC in development of hypertension. J Am Soc Nephrol. 2005;16(11):3154–3159. doi: 10.1681/ASN.2005050460. [DOI] [PubMed] [Google Scholar]
- 74.Song J, et al. Regulation of blood pressure, the epithelial sodium channel (ENaC), and other key renal sodium transporters by chronic insulin infusion in rats. Am J Physiol Renal Physiol. 2006;290(5):F1055–F1064. doi: 10.1152/ajprenal.00108.2005. [DOI] [PubMed] [Google Scholar]
- 75.Shei RJ, et al. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr Opin Pharmacol. 2018;43:152–165. doi: 10.1016/j.coph.2018.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Welch EM, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- 77.Suprynowicz FA, et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc Natl Acad Sci U S A. 2012;109(49):20035–20040. doi: 10.1073/pnas.1213241109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu X, et al. Conditional reprogramming and long-term expansion of normal and tumor cells from human biospecimens. Nat Protoc. 2017;12(2):439–451. doi: 10.1038/nprot.2016.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fu B, et al. Keratinocyte growth conditions modulate telomerase expression, senescence, and immortalization by human papillomavirus type 16 E6 and E7 oncogenes. Cancer Res. 2003;63(22):7815–7824. [PubMed] [Google Scholar]
- 80.Liu X, et al. HPV E7 contributes to the telomerase activity of immortalized and tumorigenic cells and augments E6-induced hTERT promoter function. Virology. 2008;375(2):611–623. doi: 10.1016/j.virol.2008.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gentzsch M, et al. The cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelila Na+ channel. J Biol Chem. 2010;285(42):32227–32232. doi: 10.1074/jbc.M110.155259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cholon DM, et al. Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci Transl Med. 2014;6(246):246ra296. doi: 10.1126/scitranslmed.3008680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kartner N, et al. Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell. 1991;64(4):681–691. doi: 10.1016/0092-8674(91)90498-N. [DOI] [PubMed] [Google Scholar]
- 84.Ghosh A, et al. Chronic e-cigarette exposure alters the human bronchial epithelial proteome. Am J Respir Crit Care Med. 2018;198(1):67–76. doi: 10.1164/rccm.201710-2033OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liang F, et al. High-throughput screening for readthrough modulators of CFTR PTC mutations. SLAS Technol. 2017;22(3):315–324. doi: 10.1177/2472630317692561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.