

Abstract
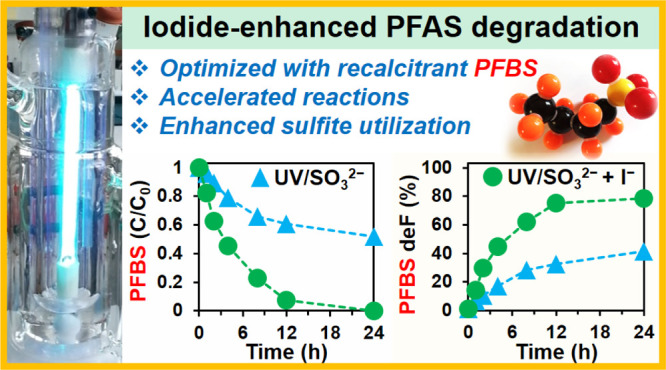
The addition of iodide (I–) in the UV/sulfite system (UV/S) significantly accelerated the reductive degradation of perfluorosulfonates (PFSAs, CnF2n+1SO3–) and perfluorocarboxylates (PFCAs, CnF2n+1COO–). Using the highly recalcitrant perfluorobutane sulfonate (C4F9SO3–) as a probe, we optimized the UV/sulfite + iodide system (UV/S + I) to degrade n = 1–7 PFCAs and n = 4, 6, 8 PFSAs. In general, the kinetics of per- and polyfluoroalkyl substance (PFAS) decay, defluorination, and transformation product formations in UV/S + I were up to three times faster than those in UV/S. Both systems achieve a similar maximum defluorination. The enhanced reaction rates and optimized photoreactor settings lowered the EE/O for PFCA degradation below 1.5 kW h m–3. The relatively high quantum yield of eaq– from I– made the availability of hydrated electrons (eaq–) in UV/S + I and UV/I two times greater than that in UV/S. Meanwhile, the rapid scavenging of reactive iodine species by SO32– made the lifetime of eaq– in UV/S + I eight times longer than that in UV/I. The addition of I– also substantially enhanced SO32– utilization in treating concentrated PFAS. The optimized UV/S + I system achieved >99.7% removal of most PFSAs and PFCAs and >90% overall defluorination in a synthetic solution of concentrated PFAS mixtures and NaCl. We extended the discussion over molecular transformation mechanisms, development of PFAS degradation technologies, and the fate of iodine species.
Keywords: PFAS, PFBS, defluorination, laser flash photolysis, hydrated electron, reactive iodine species, brine treatment, energy consumption
Short abstract
Adding iodide in the UV/sulfite system significantly enhanced the reaction rate, energy efficiency, and chemical utilization for reductive defluorination of various per- and polyfluoroalkyl substance pollutants.
Introduction
Per- and polyfluoroalkyl substances (PFASs) have become global pollutants1 due to their (i) wide applications since the 1940s, (ii) high recalcitrance in natural environments, and (iii) diverse toxicities to animals2,3 and health concerns for humans.4−7 While carbon sorption,8,9 ion exchange,9−11 and membrane filtration12,13 can capture PFAS from polluted water, the subsequent treatment of the concentrated PFAS remains challenging.14−16
Among the rapidly developing technologies for PFAS degradation,17−23 photochemical treatment with UV-generated electron (eaq–) has demonstrated great promise for environmental remediation.24−34 The C–F bond can be reductively converted into C–H by eaq– (eq 1),25,29 which can be generated from UV irradiation of SO32– (eq 2) with a quantum yield (QY) at 0.12–0.14 mol/einstein35,36
| 1 |
| 2 |
Although the 254 nm irradiation used in common water treatment applications is not the maximum absorption wavelength of SO32–,36 the cost-effectiveness of PFAS degradation by UV/sulfite (UV/S) has been greatly enhanced by tuning reaction conditions, such as pH adjustment.37 At pH 12.0, the defluorination has reached 73–93% for n = 2–8 perfluorocarboxylates (CnF2n+1COO–, PFCAs) and 79–84% for n = 4, 6, 8 perfluorosulfonates (CnF2n+1SO3–, PFSAs).38 The C–H bonds in the UV/S treatment residue allow for further defluorination by oxidation with HO•, resulting in near-complete defluorination.38 However, PFSAs are intrinsically more recalcitrant than PFCAs for UV/S treatment. The short-chain n = 4 perfluorobutane sulfonate (PFBS, C4F9SO3–) is even more challenging than longer-chain n = 6, 8 PFSAs.25,29 Multiple spikes of sulfite and >24 h are required to degrade all parent PFBS,38 resulting in high chemical dosage and energy consumption for an effective treatment.
Before the application of UV/S for pollutant degradation,26,36 UV/iodide (UV/I) had been developed earlier for PFAS degradation.24,25,39 The generation of eaq– from iodide (eq 3) has a QY at 0.22–0.29 mol/einstein40,41
| 3 |
A previous study reported >95% defluorination of perfluorooctanoate (C7F15COO–, PFOA, an extensively studied “representative” PFAS) by UV/I at pH 9.2 and ambient temperature.42 Although a higher concentration of eaq– could be expected from UV/I than UV/S, our use of UV/I only achieved PFOA defluorination of up to 60% (see Table A2 of the Supporting Information of ref (29)). The low efficiency could be attributed to eaq– scavenging by reactive iodine species (RIS, e.g., I2, I2•–, and I3–) and dissolved oxygen (DO).25,39 While DO removal by N2 was necessary for UV/I, N2 purge is not necessary for UV/S29 because the initial DO can be rapidly scavenged by sulfite.43 Therefore, we added sulfite to simplify the DO removal for UV/I. To our surprise, the degradation and defluorination of the highly recalcitrant PFBS were significantly accelerated even at a 10-fold higher concentration than our previous studies (i.e., 250 vs 25 μM). To date, the UV/sulfite + iodide (UV/S + I) system has only been examined for reductive degradation of monochloroacetate44 and bromate45 in 2018. The first report on dechlorination by UV/S + I44 has revealed key mechanistic insights to understand the enhanced degradation of highly recalcitrant PFAS. Herein, we systematically optimized the UV/S + I system using the PFBS as the challenging probe, elucidated the reaction mechanisms using various model fluorinated compounds, and showcased the significantly enhanced system efficiencies in both chemical dosing and energy consumption for the degradation of legacy PFSAs and PFCAs.
Materials and Methods
Chemicals
Sodium sulfite (Na2SO3), potassium iodide (KI), sodium hydroxide (NaOH), and sodium bicarbonate (NaHCO3) were purchased from Fisher Chemical. PFSAs (n = 4, 6, 8 CnF2n+1SO3–, PFSAs) and PFCAs (n = 1–8 CnF2n+1COO–) were purchased in bulk quantities (i.e., 0.1–5 g) and used as received. The information on CAS numbers, purities, and vendors are listed in Supporting Information.
Photochemical Reaction
The optimization of UV/S + I system parameters used the previously reported photochemical reactor configuration (Ace Glass parts #7864-10, #7874-38, and #7506-14, wrapped with aluminum foil).29,46 A 600 mL solution containing 25 μM PFBS, 5 mM NaHCO3, and predetermined concentrations of KI and Na2SO3 and pH (adjusted with NaOH) was irradiated by an 18 W low-pressure mercury lamp (GPH212T5L/HO) in the closed photoreactor with jacketed water cooling at 20 °C. For the reduction of energy consumption, the optimized solution condition was further applied in a new reactor configuration, where a 2000 mL solution was irradiated by a 10 W low-pressure mercury lamp (GPH212T5L, with the same geometry as the 18 W lamp) in a quartz sleeve placed in the center of a 2 L tall-form beaker (wrapped with aluminum foil and covered by a plastic cap with parafilm sealing). Because the 2 L beaker did not have a water cooling jacket, the solution was gradually heated from 20 to 36 °C in the first 4 h by the UV lamp and then maintained around 36 °C due to the balance with heat dissipation.
Water Sample Analysis
The parent PFAS and transformation products (TPs) were analyzed by liquid chromatography–high-resolution quadrupole Orbitrap mass spectrometer (LC–HRMS/MS). Both suspect and non-target screening were conducted to identify TPs. Short-chain (n = 1 and 2) PFCAs and their TPs were analyzed by ion chromatography (IC). The degradation kinetics were fit with C/C0 > 0.2 data using the pseudo-first-order kinetic model. Rationales for this fitting method are described in Text S1. The released fluoride ion (F–) was measured by an ion-selective electrode (ISE) and validated by IC.29,38 Detailed operation parameters of LC–HRMS/MS, IC, and ISE have been reported previously29,32,37,38 and also included in the Supporting Information. The defluorination percentage (deF%) is defined as the molar ratio between the released F– in solution and the total F in the parent PFAS compounds.
Laser Flash Photolysis
A transient absorption spectrometer (LP980, Edinburgh Instruments) equipped with 266 nm laser pulse (Nd:YAG)34 was used to measure the yield and lifetime of eaq– produced from UV/S, UV/I, and UV/S + I. The sample solution (2.5 mL) containing KI and Na2SO3 at predetermined dosage was filled in a 1 cm × 1 cm × 3 cm quartz cell and then purged with N2 for 30 min. The solution was then excited by the laser beam along with the 1 cm optical path. The absorption by eaq– and I3– was measured at 70034 and 350 nm,47 respectively. The lifetime of eaq– and I3– was calculated by eq 4
| 4 |
where Rt is the signal response at time t, B is the amplitude of the absorption curves at time zero, τ represents the time required for the response to decay to 37% of the original signal strength, and the fitted τ is defined as the lifetime.34
Results and Discussion
Significantly Accelerated PFBS Degradation by UV/S + I
Previously, we have used PFOA as the probe to optimize the UV/S system (pH 12.0 and 10 mM Na2SO3).37 However, when the target pollutant was switched to PFBS, the optimized UV/S only achieved 48% decay of the parent PFBS (Figure 1a) and 42% overall defluorination after 24 h (Figure 1b).38 To enhance the degradation, two more spikes of 10 mM Na2SO3 at 8 and 16 h (i.e., 30 mM Na2SO3 in total) boosted the parent PFBS decay and overall defluorination to 86 and 69%, respectively.38 In sharp contrast, the addition of 2 mM KI to 10 mM Na2SO3 in the UV/S system substantially accelerated the reaction by reaching >99% decay of the parent PFBS and 78% overall defluorination after 24 h, without additional Na2SO3 spikes during the reaction. Thus, I– not only accelerated the reaction but also significantly enhanced the utilization efficiency of SO32–. Notably, for most samples from different reaction settings and sampling times, the deF% from the reacted portion of the PFBS was consistent (i.e., 83 ± 2%). Therefore, the decay of the parent PFBS is the rate-limiting step and the following defluorination from TPs is fast.29
Figure 1.

(a) Parent compound decay and (b) defluorination of PFBS by UV/S (10 mM), UV/I (2 mM), and UV/S (10 mM) + I (2 mM); PFBS degradation by UV/S + I at varied (c,d) iodide concentrations, (e) sulfite concentrations, and (f) solution pH. Default reaction conditions: PFBS (0.025 mM), carbonate (5 mM), pH 12.0, 254 nm irradiation (an 18 W low-pressure Hg lamp in 600 mL solution), and 20 °C.
We continued using the highly recalcitrant PFBS to further optimize the UV/S + I system. The examination on varying concentrations of I–, SO32–, and pH (Figure 1c–f) confirmed that the initially used 2 mM I–, 10 mM SO32–, and pH 12.0 constitute the optimal reaction conditions for 0.025 mM PFBS. The use of ≤5 mM SO32– led to early termination of defluorination (Figure 1e) probably due to the early depletion of sulfite. Further elevated concentrations of either SO32– (e.g., 20 mM) or I– (e.g., 5 mM) did not result in a better performance. In our previous study on the UV/S system, 10 mM SO32– was found to be necessary to achieve the maximum defluorination of 0.025 mM PFOA.37
Energy Efficiency of the UV/S + I System
We further confirmed the high energy efficiency of UV/S + I from the rapid degradation of n = 6, 8 PFSAs and n = 1–7 PFCAs (Table 1). The first-order rate constants for the parent compound decay were 2.4–4.1-fold of those by UV/S, where all conditions were the same except for the absence of iodide. The term EE/O, which is defined as the electrical energy consumed for lowering the pollutant concentration by one order-of-magnitude (i.e., C/C0 = 0.1 or 90% removal), has been frequently used for water technology evaluation48
where P is the power of the UV lamp (kW), k is the first-order rate constant (h–1), and V is the volume of water (m3). To probe the possibility of further lowering the EE/O by UV/S + I, we decreased the power of the UV lamp from 18 to 10 W and increased the volume of water from 600 to 2000 mL. The decay of the parent PFAS in the new configuration of 10 W: 2000 mL was 20–50% slower than in the previous configuration of 18 W: 600 mL. Hence, the EE/O values were reduced by 3–5-fold from modifying the reactor configuration (see Text S2 for an extended discussion). Together with the acceleration by iodide, the EE/O for n = 2–7 PFCAs by UV/S + I have become <1.5 kW h/m3 (Table 1). Interestingly, the maximum deF% of PFCAs by UV/S in the 10 W: 2000 mL configuration was significantly lower than that in the 18 W: 600 mL configuration (Figure S1). In stark comparison, defluorination of PFCAs by UV/S + I in the 10 W: 2000 mL configuration only showed a lower initial rate, but the maximum deF% after 4 h was the same as in the 18 W: 600 mL configuration.
Table 1. DeF%, Rate Constant, EE/O, and EE/Max.deF of PFCAs and PFSAs by UV/S and UV/S + I Treatmenta.
| defluorination
after 1 h (%) |
parent compound decay k (h–1)b |
EE/O (kW h m–3)b |
EE/Max.deF (kW h m–3)b |
|||||
|---|---|---|---|---|---|---|---|---|
| PFCA(CnF2n+1COO–) | UV/S | UV/S + I | UV/S | UV/S + I | UV/S | UV/S + I | UV/S | UV/S + I |
| n = 1 TFA | 84 ± 1.2 | 100 ± 1.3 | 3.7 (2.4) | 8.6 (6.8) | 18.6 (4.8) | 7.8 (1.7) | <60 (10) | <22.8 (3.8) |
| n = 2 PFPrA | 47 ± 0.4 | 75 ± 0.9 | 6.7 (4.9) | 17.2 (8.9) | 10.2 (2.3) | 4.2 (1.3) | <240 (40) | <60 (20) |
| n = 3 PFBA | 52 ± 2.1 | 77 ± 0.8 | 5.8 (3.5) | 19.2 (9.9) | 12.0 (3.3) | 3.6 (1.2) | <240 (40) | <120 (20) |
| n = 4 PFPeA | 42 ± 3.9 | 73 ± 1.5 | 5.5 (3.4) | 16.9 (8.7) | 12.6 (3.4) | 4.2 (1.3) | <240 (40) | <120 (20) |
| n = 5 PFHxA | 51 ± 3.1 | 79 ± 1.3 | 6.1 (4.1) | 19.7 (10.1) | 11.4 (2.8) | 3.6 (1.1) | <240 (40) | <60 (20) |
| n = 6 PFHpA | 41 ± 2.7 | 77 ± 3.6 | 5.4 (3.7) | 17.5 (9.4) | 12.6 (3.1) | 4.2 (1.2) | <240 (60) | <60 (20) |
| n = 7 PFOA | 39 ± 1.5 | 88 ± 0.3 | 5.2 (3.1) | 15.4 (8.1) | 13.2 (3.7) | 4.2 (1.4) | <240 (60) | <60 (20) |
| defluorination
after 24 h (%) |
parent compound decay k (h–1) |
EE/O (kW h m–3) |
EE/Max.deF (kW h m–3) |
|||||
|---|---|---|---|---|---|---|---|---|
| PFSA (CnF2n+1SO3–) | UV/S | UV/S + I | UV/S | UV/S + I | UV/S | UV/S + I | UV/S | UV/S + I |
| n = 4 PFBS | 43 ± 1.8 | 78 ± 1.2 | 0.05 (0.02) | 0.19 (0.05) | 1320 (576) | 366 (230) | ∼1320 (576) | ∼366 (230) |
| n = 6 PFHxS | 60 ± 3.1 | 84 ± 1.3 | 0.38 (0.29) | 1.56 (0.61) | 181 (39.7) | 44.4 (18.9) | <360 (60) | <120 (40) |
| n = 8 PFOS | 84 ± 0.9 | 92 ± 0.7 | 0.78 (0.65) | 1.90 (1.01) | 88.8 (17.7) | 36.6 (11.5) | <240 (40) | <60 (40) |
Reaction conditions were the following: individual PFCA/PFSA (0.025 mM), Na2SO3 (10 mM), KI (2 mM), NaHCO3 (5 mM), pH 12.0, 254 nm irradiation (an 18 W low-pressure Hg lamp in 600 mL solution), and 20 °C.
Values in parentheses are from an “energy-saving” setting (a 10 W lamp in 2 L solution, solution constituents not changed) at 20–36 °C due to the gradual heating by the UV lamp without a cooling water bath.
For the much more recalcitrant PFSAs, the EE/O were also lowered to 11.5–230 kW h/m3. As elucidated in our previous study,29 one primary mechanism for parent PFCA decay is hydrodefluorination of the α −CF2– moiety, and one primary mechanism for parent PFSA decay is the reductive cleavage of the C–S bond. Because both mechanisms require the reaction with eaq–, the significantly accelerated degradation of both PFCAs and PFSAs by UV/S + I can be attributed to the enhanced availability of eaq–.
As the goal of PFAS degradation is not merely removing 90% of the parent compound, we propose a new metric that calculates the electrical energy consumed by the time (t) upon reaching 90% of the maximum defluorination (EE/Max.deF, Table 1)
Because defluorination involves multiple pathways and cannot be quantified with a simple kinetic model, the EE/Max.deF values are estimated as a range based on the time when the first sample showing >90% of the maximum defluorination was collected. The maximum defluorination was determined in the sample taken at 24 h when sulfite had been fully depleted (see Figure 6c).
Figure 6.
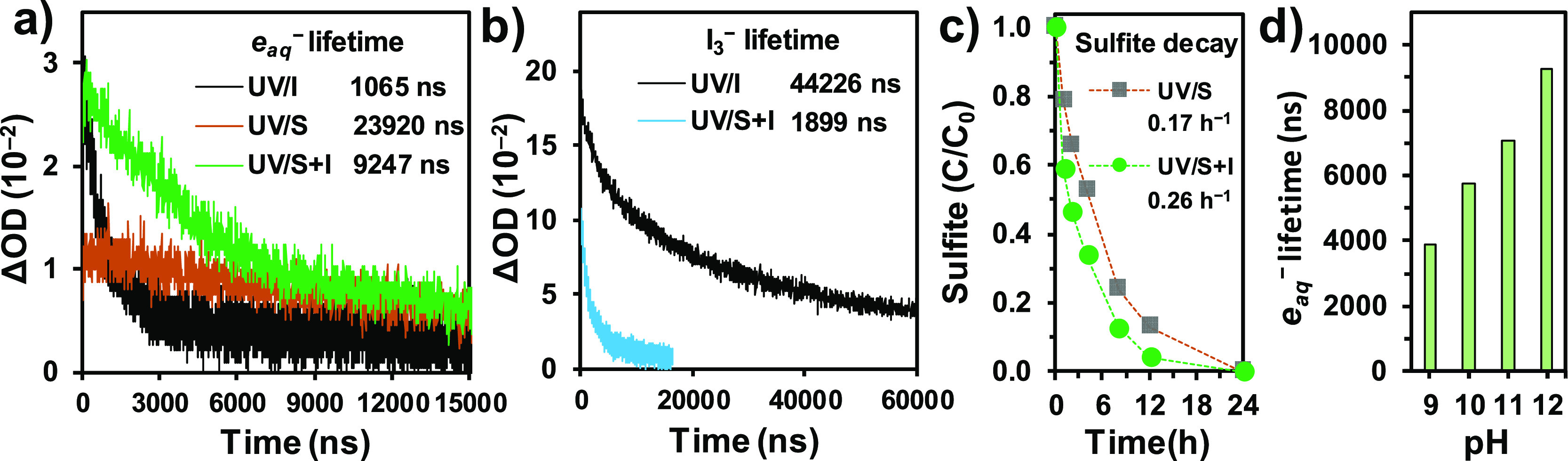
(a) Kinetic profiles of transient absorption at 700 nm after the 266 nm laser pulse excitation of I, S, and S + I solutions; (b) kinetic profiles of transient absorption at 350 nm after 266 nm laser pulse excitation of I and S + I solutions; (c) decay of sulfite in UV/S and UV/S + I in the absence of PFAS; and (d) lifetime of eaq– in UV/S + I at varied solution pH. Conditions: Na2SO3 (S, 10 mM), KI (I, 2 mM), and pH 12.0.
For PFCAs, the parent compound decay was much faster than the stepwise defluorination (Figure 2a vs b,c). The decay and defluorination of n ≥ 2 PFCAs followed similar time profiles, so the average values are shown in those figures. Still, their EE/Max.deF values by UV/S + I were <20 kW h m–3. For PFSA degradation, the parent compound decay and defluorination were much more synchronous (Figure 2d–f) than PFCA degradation. Because >90% parent compound decay of PFSAs is the pre-requisite for reaching 90% of the maximum defluorination, the EE/Max.deF values for PFSAs were also higher than their EE/O. The current reactor configuration and solution composition of UV/S + I have resulted in the low-end EE/O values for PFOA/PFOS degradation among existing reports on various technologies.16,19,23,49 Currently, the comparison of EE/Max.deF with other treatment methods is not feasible due to the lack of reported defluorination-time profiles, especially for PFBS degradation.
Figure 2.

(a) Parent compound decay and (b) defluorination of n = 1–7 PFCAs by UV/S (10 mM), and UV/S (10 mM) + I (2 mM). Panel (c) shows the magnified defluorination profiles for n = 2–7 PFCAs (averaged) within the first 1 h. (d–f) Parent compound decay and defluorination of n = 4, 6, 8 PFSAs. Reaction conditions: individual PFAS (0.025 mM), NaHCO3 (5 mM), pH 12.0, 254 nm irradiation (an 18 W low-pressure Hg lamp in 600 mL solution), and 20 °C.
Mechanistic Insights from Reaction Kinetics and TPs
To understand how the new UV/S + I system accelerated both parent compound decay and defluorination, we used n = 1 and 2 PFCAs (CnF2n+1COO–) as mechanistic probes. Short-chain PFCAs have a limited number of fluorinated carbons, and many possible TPs have commercial standards for quantitation. The decay and defluorination of the three n = 1 structures, CF3–COO– (trifluoroacetate, TFA), CHF2–COO– (difluoroacetate, DFA), and CH2F–COO– (monofluoroacetate, MFA) by UV/S + I were significantly faster than those by UV/S (Figure 3a–c vs d–f). While the complete defluorination by UV/S took 2–4 h, only 1 h was needed for UV/S + I. The formation of acetate (CH3–COO–) indicates the hydrodefluorination pathway, where F atoms were stepwise replaced by H atoms via reduction with eaq– (Figure 3g). The generation (and degradation) of the stepwise hydrodefluorination TPs (e.g., DFA and MFA from TFA) was also faster in the UV/S + I system. Meanwhile, the various gaps of the acetate yield from 100% indicate the competing decarboxylation pathway (Figure 3g). At the end of reactions where 100% defluorination was achieved, the acetate yields from DFA and MFA by UV/S + I (72 and 93%, respectively) were higher than those by UV/S (45 and 72%, respectively). The preferred hydrodefluorination indicates enhanced availability of eaq– in the UV/S + I system.
Figure 3.
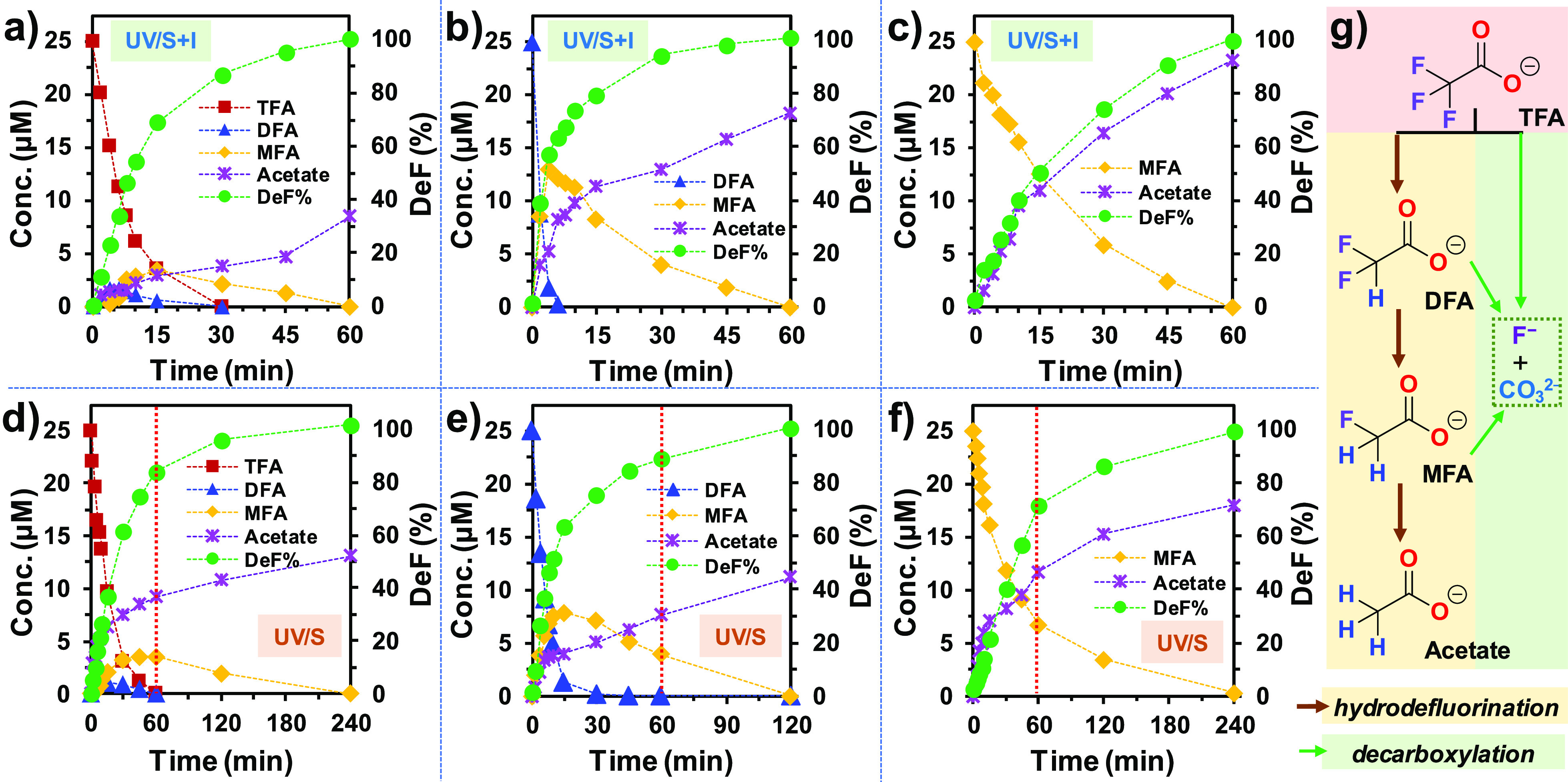
Parent compound decay, TP formation, and defluorination of tri-, di-, and monofluoro acetates by (a–c) UV/S + I and (d–f) UV/S and (g) reaction schemes. Reaction conditions: individual PFAS (0.025 mM), Na2SO3 (10 mM), KI (2 mM), NaHCO3 (5 mM), pH 12.0, 254 nm irradiation (an 18 W low-pressure Hg lamp in 600 mL solution), and 20 °C. The vertical red dotted lines in panels (d–f) indicate the 1 h time window where 100% defluorination by UV/S + I was achieved.
However, for the degradation of TFA, the acetate yield by UV/S + I (34%) was lower than that by UV/S (52%). The calculated C–F bond dissociation energy (BDE) in TFA (116.8 kcal mol–1) is significantly higher than those in DFA (109.7 kcal mol–1) and MFA (108.6 kcal mol–1).29 We have also found that the C–F bonds in CF3CH2–COO– (121.5 kcal mol–1) and HCF2CH2–COO– (118.8 kcal mol–1) were both sluggish in the reaction with eaq–.37,50 Therefore, the dominating pathway for TFA degradation by UV/S + I should be decarboxylation.
The degradation of CF3CF2–COO– (PFPrA, see Table 1 for acronyms) using UV/S + I was also significantly faster than that by UV/S (Figure 4a,b vs c,d). The recalcitrant hydrodefluorination TP, CF3CH2–COO–, has very strong sp3 C–F bonds (Figure 4e) and thus is accumulated in the solution. Both UV/S + I and UV/S achieved a similar deF% of PFPrA (79% in Figure 4a and 76% in 4c), suggesting that UV/S + I cannot cleave additional C–F bonds that are recalcitrant under UV/S treatment. However, upon reaching the maximum deF%, the yield of CF3CH2–COO– from UV/S + I treatment was higher than that from UV/S treatment (62 vs 39 from 250 μM of PFPrA), indicating enhanced availability of eaq– for hydrodefluorination of the relatively weak C–F bonds. Notably, the limit of deF% via hydrodefluorination is 40% (i.e., two α C–F bonds out of the five C–F bonds in PFPrA are cleaved), whereas the overall deF% reached 79%. The deeper defluorination is thus contributed by the decarboxylation pathway, which mineralized the α CF2 moiety and yielded TFA (Figure 4e). TFA can be 100% defluorinated by either pathway (Figure 3).
Figure 4.

Parent compound decay, TP formation, and defluorination of PFPrA by (a,b) UV/S + I and (c,d) UV/S. Panels (b,d) show magnified time profiles within the first hour of reaction. Panel (e) shows the proposed reaction schemes. Reaction conditions: PFPrA (0.025 mM), Na2SO3 (10 mM), KI (2 mM), NaHCO3 (5 mM), pH 12.0, 254 nm irradiation (an 18 W low-pressure Hg lamp in 600 mL solution), and 20 °C.
It is worth noting that other degradation pathways also occurred. As shown by the F balance counting F– and known TPs from hydrodefluorination (CF3CFH–COO– and CF3CH2–COO–) and from decarboxylation (TFA, DFA, and MFA were negligible), the maximum gap reached >50% at the beginning (15 min for UV/S + I and 1 h for UV/S). After the maximum deF% was achieved (8 h for UV/S + I and 24 h for UV/S), the missed F balance was decreased to 6% for UV/S + I and 15% for UV/S. While we are investigating novel TPs with other mechanistic hypotheses, we noted that most F-containing TPs did not escape the system as gaseous products because the following oxidation with HO• (generated from heat-activated persulfate at pH > 12) achieved ∼100% defluorination.38
The abovementioned mechanistic insights were further corroborated by the results from C7F15–COO– (PFOA). The generation and the following degradation of the decarboxylation product, C6F13–COO– (PFHpA), and the hydrodefluorination product, C7F14H–COO–, were much faster by UV/S + I than by UV/S (Figure 5a vs b). We note that the detected concentration of a TP is the result of the balance between generation and degradation at the same time. Although the maximum concentrations of those two TPs under UV/S + I and UV/S were similar, their total generated amounts under UV/S + I should be more than those under UV/S.
Figure 5.

Parent compound decay and TP formation for the degradation of (a,b) PFOA and (c,d) PFBS by UV/S + I and UV/S. Panel (e) shows the proposed reaction schemes. Reaction conditions were the following: individual PFAS (0.025 mM), Na2SO3 (10 mM), KI (2 mM), NaHCO3 (5 mM), pH 12.0, 254 nm irradiation (an 18 W low-pressure Hg lamp in 600 mL solution), and 20 °C.
TP analysis for the degradation of C4F9–SO3– (PFBS) also confirmed that the formation of the hydrodefluorination product, C4F7H2–SO3–, was significantly enhanced by UV/S + I (Figure 5c vs d). According to the calculated C–F BDEs for PFSAs,29 the most probable location for the initial hydrodefluorination is on the β carbon (Figure 5e). Further hydrodefluorination only yielded C4F6H3–SO3–, which accumulated under UV/S + I treatment. Hence, the high overall deF% of 79% should also be attributed to C–S cleavage upon the reaction between C4F9–SO3– and eaq– to yield n ≤ 3 PFCAs (Figure 5e). However, PFCA TPs (e.g., PFBA) were not detected, probably due to the rate-limiting decay of the PFBS and the rapid degradation of daughter PFCAs.
Mechanistic Insights from Reactive Species Analyses
The significantly accelerated parent compound decay and the increased yield of hydrodefluorination products by UV/S + I prompted us to explain the enhanced availability of eaq–. We compared the abundance and lifetime of eaq– produced from UV/S + I, UV/S, and UV/I by measuring the absorption at 700 nm in laser flash photolysis (LFP) experiments (Figure 6a). As expected from the reported QY, the abundance of eaq– generated by the 266 nm laser from 2 mM I– (absorbance 0.012, QY = 0.22–0.29 mol/einstein)40,41 was approximately two times higher than that from 10 mM SO32– (absorbance 0.016, QY = 0.12–0.14 mol/einstein)35,36 at pH 12. However, the eaq– from UV/I has a 22-fold shorter lifetime than that from UV/S. Besides the quenching by H+ (eq 5), a series of RIS can rapidly form (eqs 6–8) and become strong scavengers of eaq– (eqs 9–11).25,39 This may be one reason for the limited performance of UV/I for PFAS degradation.39,44,51,52
| 5 |
| 6 |
| 7 |
| 8 |
| 9 |
| 10 |
| 11 |
For comparison, UV/S + I generated eaq– in a similar abundance to UV/I. Meanwhile, the lifetime of eaq– in UV/S + I was eightfold longer than that in UV/I (Figure 6a). Besides serving as the source of eaq– (eq 2), SO32– has also been found to rapidly reduce RIS back to I– (eqs 12–15)53,54
| 12 |
| 13 |
| 14 |
| 15 |
As I3– has a characteristic absorption at 352 nm,39,44 the rapid quenching of I3– by SO32– was also confirmed by the LFP experiment (Figure 6b). The use of 10 mM SO32– shortened the lifetime of I3– by 23-fold. Although a radical reaction network remains challenging to build, a simplified interpretation is that SO32– donates up to two electrons to convert RIS back to I– via eqs 12–15. This interpretation is consistent with (i) the faster consumption of SO32– in UV/S + I than in UV/S (Figure 6c), (ii) the generation of eaq– in high abundance and long lifetime in UV/S + I, and (iii) the enhanced reductive PFAS degradation. These experimental findings complement the previous study on monochloroacetate degradation, which conducted meticulous kinetic measurements for a comprehensive mechanistic understanding of the UV/S + I system.44 Moreover, SO32– is an instant scavenger of DO without UV irradiation.43 Upon adding Na2SO3 in water, the measured DO immediately dropped from 7.3 to <0.1 mg/L. Thus, the use of SO32– effectively prevented eaq– quenching by DO,25,26 and the UV/S + I system does not need N2 purge.
In addition, the lifetime of eaq– at pH from 9 to 12 showed an increasing trend (Figure 6d). Because the speciation of both I– and SO32– is not impacted at pH > 10, the longer lifetime at higher pH is primarily attributed to the reduced eaq– quenching by H+ (eq 5). This result is consistent with the excellent PFBS degradation kinetics at pH 12 (Figure 1f).
Iodide Significantly Enhanced the Utilization of Sulfite
In our previous studies on the UV/S system, the typical concentration of PFOA was 25 μM because higher concentrations resulted in inferior performance. For example, using 10 mM SO32– at pH 12, the defluorination of 25 μM of individual n = 2–7 PFCAs reached 73–93% at 8 h. The kinetics did not show a significant dependence on the fluoroalkyl chain length (n). However, the deF% from 250 μM (or 0.25 mM) of individual PFCAs were significantly lowered and exhibited an obvious decrease with the increased n (Figure 7a). For PFOA, 0.25 mM of C7F15–COO– contained 3.75 mM of the C–F bond. If 100% defluorination could be achieved solely by the hydrodefluorination mechanism (eq 1), up to 7.5 mM of eaq– would be consumed. However, the use of 10 mM SO32– in UV/S only achieved 34% defluorination at 24 h, where most SO32– had been depleted (Figure 6c). Under UV irradiation, the major portion of SO32– that was not utilized for PFAS degradation probably generated H2 gas.35 However, this aspect warrants further investigation. Additional spikes of 10 mM SO32– enabled further defluorination, but a total of 40 mM SO32– only defluorinated PFOA up to 73% (Figure 7b). In stark contrast, the initially added 10 mM SO32– and 2 mM I– directly achieved 84% defluorination from the 0.25 mM PFOA within 12 h. Thus, the utilization of sulfite was significantly enhanced by the addition of iodide. Further increasing the PFOA concentration to 0.5 mM led to significantly lower defluorination (Figure 7c). Hence, we increased the concentration of all PFCAs and PFSAs from 25 to 250 μM. The UV/S + I treatment of PFCAs achieved a similar deF% for both concentrations (Figure 7d). The UV/S + I treatment of n = 4, 6, 8 PFSAs (0.25 mM) without spiking additional SO32– also achieved 68, 81, and 87% of defluorination, respectively (Figure 7e). These values are consistent with the maximum deF% of n = 4, 6, 8 PFSAs (25 μM) by UV/S with additional SO32– spikes (69, 72, and 84%, respectively).38
Figure 7.
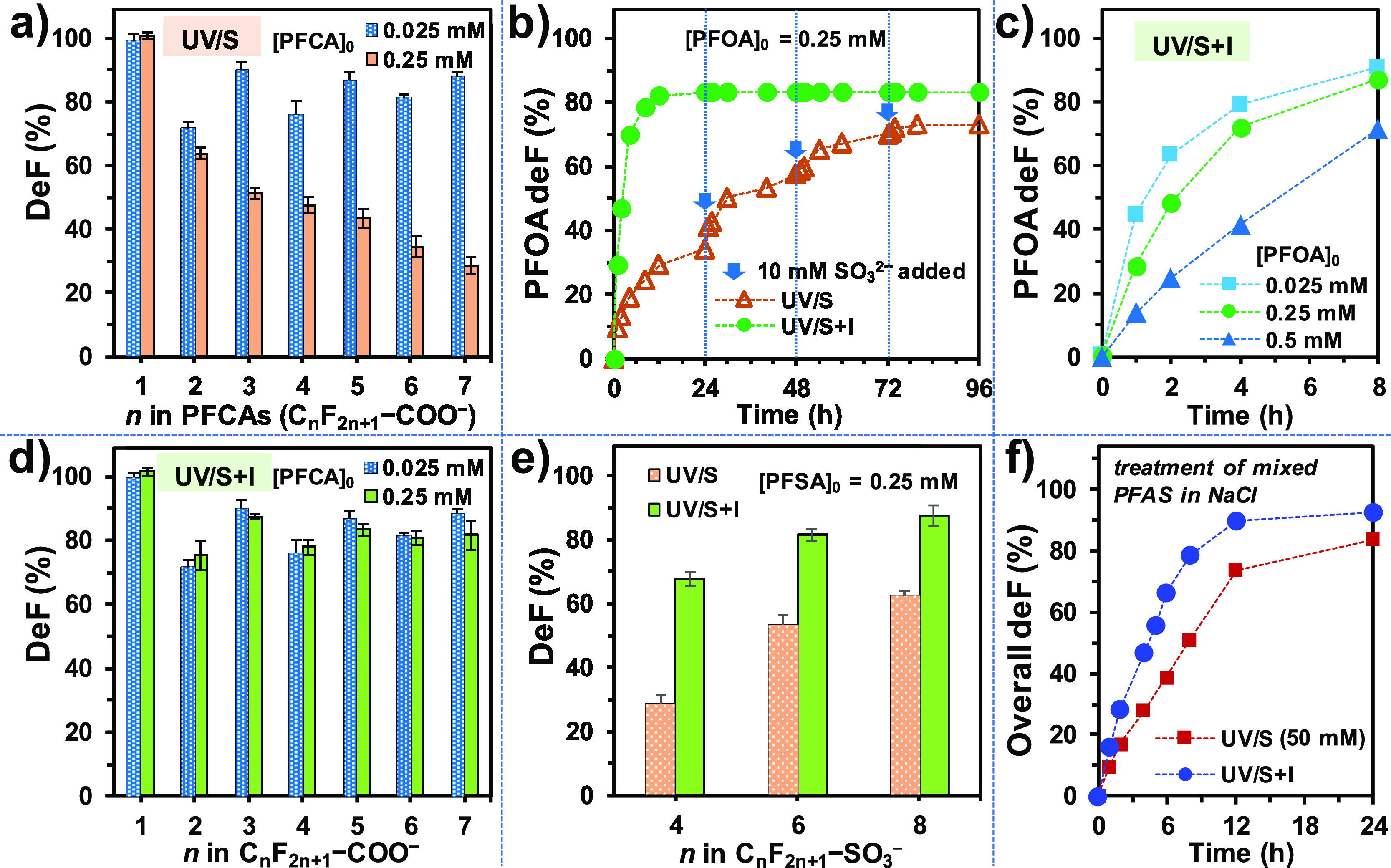
(a) DeF% of PFCAs by UV/S after 8 h; (b) time profiles for the defluorination from 0.25 mM PFOA by UV/S (with SO32– spikes every 24 h) and UV/S + I (no SO32– spike); (c) time profiles for the defluorination from various [PFOA]0 by UV/S + I; (d) deF% of PFCAs after 24 h by UV/S + I; (e) deF% of PFSAs by UV/I and UV/S + I after 24 h; (f) time profiles for the defluorination from the mixed PFAS (see Table 2) in NaCl brine after a 1:1 dilution with deionized (DI) water. Default reaction conditions: 10 mM Na2SO3 (10 mM), KI (2 mM for UV/S + I), NaHCO3 (5 mM), pH 12.0, and 254 nm irradiation (an 18 W low-pressure Hg lamp for 600 mL solution), and 20 °C.
Motivated by these positive results, we further challenged the UV/S + I system to treat a synthetic brine, which was prepared based on the NaCl content and quantified PFAS constituents in a recently reported “still bottom” waste brine from ion-exchange resin regeneration (Table 2).16 Natural organic matters and other potential inhibiting species that require pretreatment efforts were not included in the present study. Individual PFAS component concentrations were chosen as the averaged values of the six reported brines.16 Commercially unavailable structures (e.g., n = 5, 7, 9 PFSAs), minor structures (e.g., n = 9 PFCA and n = 8 sulfonamide), and unknown PFAS precursors were not included. Hence, the synthetic brine contained 3 wt% NaCl and a total of 10.9 mM of C–F bonds in the mixture of three fluorotelomer sulfonates (FTSAs, n = 4, 6, 8 CnF2n+1–CH2CH2–SO3–), six PFCAs (n = 3–8), and three PFSAs (n = 4, 6, 8). We diluted the brine for twofold, so that the eaq– generated from 10 mM SO32– and 2 mM I– would suffice the need for cleaving all C–F bonds (5.5 mM after dilution) via the reductive pathway (eq 1). The UV/S + I treatment achieved 90 and 93% defluorination from this brine sample after 12 and 24 h, respectively (Figure 7f). The UV/S treatment using 50 mM SO32– exhibited an inferior rate and extent of defluorination. Quantitation of the individual PFAS in the treated brine found that a majority of parent structures have been removed for >99.5%. The outstanding substrates exhibiting high recalcitrance included PFBS (94% removal) and 4:2 FTS (52% removal). Hence, short-chain fluorotelomers are not ideal for direct UV/S + I treatment. The isolation of the very short fluoroalkyl chain by −CH2– moieties voids the favorable mechanistic factors for PFAS degradation: (1) vulnerable C–F bonds in the middle of long fluoroalkyl chains, (2) weak α C–F bonds in PFCAs, and (3) the dissociation of C–S bonds in PFSAs.29
Table 2. Treatment of a Concentrated PFAS Mixture in NaCl Brine by UV/S + Ia.
| PFAS category | compound | PFAS concentration (μg L–1) | total fluorine in C–F bonds (μg L–1) | residual PFAS after 24 h (μg L–1)b | parent PFAS removal ratio (%) |
|---|---|---|---|---|---|
| FTSAs | 4:2 FTS | 131 | 68 | 63 | 51.9 |
| 6:2 FTS | 27,950 | 16,124 | 296 | 98.9 | |
| 8:2 FTS | 875 | 535 | <53 | >93.9 | |
| PFCAs | PFBA | 1950 | 1212 | <5 | >99.7 |
| PFPeA | 5872 | 3803 | <13 | >99.7 | |
| PFHxA | 16,433 | 10,936 | <8 | >99.9 | |
| PFHpA | 7492 | 5083 | <9 | >99.8 | |
| PFOA | 101,550 | 69,896 | <10 | >99.9 | |
| PFNA | 228 | 159 | <12 | >94.7 | |
| PFSAs | PFBS | 4988 | 2522 | 279 | 94.4 |
| PFHxS | 72,550 | 40,893 | <88 | >99.8 | |
| PFOS | 86,950 | 56,155 | <100 | >99.8 | |
| sum | 327.0 mg L–1 | 207.4 mg L–1 (10.9 mM) | <0.9 mg L–1 | >99.7 |
Reaction conditions: the 3% NaCl brine with the tabulated components was diluted with 1:1 DI water, Na2SO3 (10 mM), KI (2 mM), NaHCO3 (5 mM), pH 12.0, 254 nm irradiation (an 18 W low-pressure Hg lamp in 600 mL solution), and 20 °C.
Concentrations without a definitive value indicate the limit of quantitation by the HRMS instrument in our study. These values are the HRMS-detected concentration multiplied by 2 (due to the 1:1 dilution before treatment). Solid-phase extraction for accurate quantitation in low concentration ranges (e.g., the prevalent 70 ng L–1 reference for drinking water) was not used in this proof-of-concept study.
Implications to PFAS Degradation Research and Engineering
Our experimental results collectively suggest that the use of iodide in UV/S + I significantly benefits PFAS degradation by (i) increasing the concentration of eaq– (Figure 6a) and (ii) increasing the utilization of sulfite (Figure 7b). Mechanistic probing experiments (Figures 3–5) identified similar transformation pathways at much higher rates by UV/S + I than by UV/S. While all reaction steps were significantly accelerated upon the addition of I–, the maximum deF% remained the same. The consumptions of both electricity (Figure 2 and Table 1) and chemical (Figure 7) by UV/S + I are significantly lower than those by UV/S. Although the results have established a highly efficient photochemical system and provided mechanistic insights to the best of our current research capability, this single report cannot cover all details from fundamental understanding to practical application. For example, the interactions between various sulfur and iodine species are much more complex than the proposed, as shown in eqs 5–15.39,44,55 However, a comprehensive and accurate reaction network modeling can be established upon deeper understandings of reactions for individual reactive species with PFASs and their TPs. Further elucidation of PFAS degradation pathways and mechanisms should be prioritized for future research efforts. After all, we have provided detailed data sets that confirm UV/S + I as a potential engineering solution for PFAS destruction. Although iodinated gaseous products were reported in earlier studies using UV/I,25 the accumulation of polar iodinated organics from UV/S + I treatment is less likely because (i) C–I bonds are highly vulnerable to eaq– reduction and UV photolysis44 and (ii) RIS are rapidly scavenged by SO32–.
For PFAS degradation technology development, we highlight the importance of investigating a broad range of structures rather than the legacy PFOA/PFOS. We have shown that the reactivity of PFAS highly depends on the chain length and functional groups.29,32,50 This study provides an example of using the highly challenging C4 PFBS as the probe for technology development. As expected, the optimized UV/S + I system exhibits significantly enhanced performance for all PFCAs and C6 and C8 PFSAs. Still, like UV/S, the UV/S + I system is mechanistically limited for treating short-chain fluorotelomers.38 Thus, oxidation is required both before and after the UV/S + I treatment to ensure efficient and complete defluorination of most PFAS pollutants.38 NOM and other potential inhibiting species in real “still bottom” brines will also be addressed in our near-future study on sequential treatment strategies.
Lastly, iodide is a relatively expensive chemical enriched from seawater, but we do not consider the cost of iodide as a major barrier for practical applications of UV/S + I. The volume of waste brine from ion-exchange resin regeneration is minimal. The real cost of both the electricity and chemicals for PFAS destruction in the waste brine is to be divided by the large volumes of drinking water or groundwater treated by ion exchange. However, when iodide recycling is necessary, iodine species in the treated brine can be rapidly transformed into either I– (e.g., eqs 12–15) or I2 (e.g., by H2O2)56 for facile separation. I– has a much higher affinity to ordinary anion exchange resins than common anions (e.g., F–, Cl–, SO42–, and HCO3–/CO32–),57 and I2 can be extracted by common organic solvents from water. For example, at 25 °C the solubility of I2 in diethyl ether (25 wt%) is substantially higher than that in water (∼300 mg/L).58
Acknowledgments
Financial support was provided by the Strategic Environmental Research and Development Program Project ER18-1289 (for Z.L., J.G., and J.L.) and Project ER20-1541 (for Y.Y. and Y.M.).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.1c07608.
Details of PFAS chemicals; quantitation of PFAS parent compounds, TPs, and anions; extended discussions on kinetic data fitting and photoreactor configurations; and defluorination profiles in the two photoreactor configurations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ng C.; Cousins I. T.; DeWitt J. C.; Glüge J.; Goldenman G.; Herzke D.; Lohmann R.; Miller M.; Patton S.; Scheringer M.; Trier X.; Wang Z. Addressing urgent questions for PFAS in the 21st Century. Environ. Sci. Technol. 2021, 55, 12755–12765. 10.1021/acs.est.1c03386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rericha Y.; Cao D.; Truong L.; Simonich M.; Field J. A.; Tanguay R. L. Behavior effects of structurally diverse per- and polyfluoroalkyl substances in zebrafish. Chem. Res. Toxicol. 2021, 34, 1409–1416. 10.1021/acs.chemrestox.1c00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta S.; Reddam A.; Liu Z.; Liu J.; Volz D. C. High-content screening in zebrafish identifies perfluorooctanesulfonamide as a potent developmental toxicant. Environ. Pollut. 2020, 256, 113550. 10.1016/j.envpol.2019.113550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen G. T. H.; Nocentini A.; Angeli A.; Gratteri P.; Supuran C. T.; Donald W. A. Perfluoroalkyl substances of significant environmental concern can strongly inhibit human carbonic anhydrase isozymes. Anal. Chem. 2020, 92, 4614–4622. 10.1021/acs.analchem.0c00163. [DOI] [PubMed] [Google Scholar]
- Harris M. H.; Rifas-Shiman S. L.; Calafat A. M.; Ye X.; Mora A. M.; Webster T. F.; Oken E.; Sagiv S. K. Predictors of per- and polyfluoroalkyl substance (PFAS) plasma concentrations in 6–10 year old American children. Environ. Sci. Technol. 2017, 51, 5193–5204. 10.1021/acs.est.6b05811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nian M.; Luo K.; Luo F.; Aimuzi R.; Huo X.; Chen Q.; Tian Y.; Zhang J. Association between prenatal exposure to PFAS and fetal sex hormones: Are the short-chain PFAS safer?. Environ. Sci. Technol. 2020, 54, 8291–8299. 10.1021/acs.est.0c02444. [DOI] [PubMed] [Google Scholar]
- Zheng G.; Schreder E.; Dempsey J. C.; Uding N.; Chu V.; Andres G.; Sathyanarayana S.; Salamova A. Per- and polyfluoroalkyl substances (PFAS) in breast milk: Concerning trends for current-use PFAS. Environ. Sci. Technol. 2021, 55, 7510–7520. 10.1021/acs.est.0c06978. [DOI] [PubMed] [Google Scholar]
- Xiao X.; Ulrich B. A.; Chen B.; Higgins C. P. Sorption of poly- and perfluoroalkyl substances (PFASs) relevant to aqueous film-forming foam (AFFF)-impacted groundwater by biochars and activated carbon. Environ. Sci. Technol. 2017, 51, 6342–6351. 10.1021/acs.est.7b00970. [DOI] [PubMed] [Google Scholar]
- McCleaf P.; Englund S.; Östlund A.; Lindegren K.; Wiberg K.; Ahrens L. Removal efficiency of multiple poly- and perfluoroalkyl substances (PFASs) in drinking water using granular activated carbon (GAC) and anion exchange (AE) column tests. Water Res. 2017, 120, 77–87. 10.1016/j.watres.2017.04.057. [DOI] [PubMed] [Google Scholar]
- Fang Y.; Ellis A.; Choi Y. J.; Boyer T. H.; Higgins C. P.; Schaefer C. E.; Strathmann T. J. Removal of per-and polyfluoroalkyl substances (PFASs) in aqueous film-forming foam (AFFF) using ion-exchange and nonionic resins. Environ. Sci. Technol. 2021, 55, 5001–5011. 10.1021/acs.est.1c00769. [DOI] [PubMed] [Google Scholar]
- Liu Y.-L.; Sun M. Ion exchange removal and resin regeneration to treat per- and polyfluoroalkyl ether acids and other emerging PFAS in drinking water. Water Res. 2021, 207, 117781. 10.1016/j.watres.2021.117781. [DOI] [PubMed] [Google Scholar]
- Steinle-Darling E.; Reinhard M. Nanofiltration for trace organic contaminant removal: Structure, solution, and membrane fouling effects on the rejection of perfluorochemicals. Environ. Sci. Technol. 2008, 42, 5292–5297. 10.1021/es703207s. [DOI] [PubMed] [Google Scholar]
- Appleman T. D.; Higgins C. P.; Quiñones O.; Vanderford B. J.; Kolstad C.; Zeigler-Holady J. C.; Dickenson E. R. V. Treatment of poly- and perfluoroalkyl substances in US full-scale water treatment systems. Water Res. 2014, 51, 246–255. 10.1016/j.watres.2013.10.067. [DOI] [PubMed] [Google Scholar]
- Sonmez Baghirzade B.; Zhang Y.; Reuther J. F.; Saleh N. B.; Venkatesan A. K.; Apul O. G. Thermal regeneration of spent granular activated carbon presents an opportunity to break the forever PFAS cycle. Environ. Sci. Technol. 2021, 55, 5608–5619. 10.1021/acs.est.0c08224. [DOI] [PubMed] [Google Scholar]
- Liu S.; Zhao S.; Liang Z.; Wang F.; Sun F.; Chen D. Perfluoroalkyl substances (PFASs) in leachate, fly ash, and bottom ash from waste incineration plants: Implications for the environmental release of PFAS. Sci. Total Environ. 2021, 795, 148468. 10.1016/j.scitotenv.2021.148468. [DOI] [PubMed] [Google Scholar]
- Singh R. K.; Multari N.; Nau-Hix C.; Woodard S.; Nickelsen M.; Mededovic Thagard S.; Holsen T. M. Removal of poly- and per-fluorinated compounds from ion exchange regenerant still bottom samples in a plasma reactor. Environ. Sci. Technol. 2020, 54, 13973–13980. 10.1021/acs.est.0c02158. [DOI] [PubMed] [Google Scholar]
- Qanbarzadeh M.; Wang D.; Ateia M.; Sahu S. P.; Cates E. L. Impacts of reactor configuration, degradation mechanisms, and water matrices on perfluorocarboxylic acid treatment efficiency by the UV/Bi3O(OH)(PO4)2 photocatalytic process. ACS ES&T Engg 2021, 1, 239–248. 10.1021/acsestengg.0c00086. [DOI] [Google Scholar]
- Zhu Y.; Xu T.; Zhao D.; Li F.; Liu W.; Wang B.; An B. Adsorption and solid-phase photocatalytic degradation of perfluorooctane sulfonate in water using gallium-doped carbon-modified titanate nanotubes. Chem. Eng. J. 2021, 421, 129676. 10.1016/j.cej.2021.129676. [DOI] [Google Scholar]
- Yang S.; Fernando S.; Holsen T. M.; Yang Y. Inhibition of perchlorate formation during the electrochemical oxidation of perfluoroalkyl acid in groundwater. Environ. Sci. Technol. Lett. 2019, 6, 775–780. 10.1021/acs.estlett.9b00653. [DOI] [Google Scholar]
- Huang D.; Wang K.; Niu J.; Chu C.; Weon S.; Zhu Q.; Lu J.; Stavitski E.; Kim J.-H. Amorphous Pd-loaded Ti4O7 electrode for direct anodic destruction of perfluorooctanoic acid. Environ. Sci. Technol. 2020, 54, 10954–10963. 10.1021/acs.est.0c03800. [DOI] [PubMed] [Google Scholar]
- Lewis A. J.; Joyce T.; Hadaya M.; Ebrahimi F.; Dragiev I.; Giardetti N.; Yang J.; Fridman G.; Rabinovich A.; Fridman A. A.; McKenzie E. R.; Sales C. M. Rapid degradation of PFAS in aqueous solutions by reverse vortex flow gliding arc plasma. Environ. Sci.: Water Res. Technol. 2020, 6, 1044–1057. 10.1039/c9ew01050e. [DOI] [Google Scholar]
- Saleem M.; Biondo O.; Sretenović G.; Tomei G.; Magarotto M.; Pavarin D.; Marotta E.; Paradisi C. Comparative performance assessment of plasma reactors for the treatment of PFOA; reactor design, kinetics, mineralization and energy yield. Chem. Eng. J. 2020, 382, 123031. 10.1016/j.cej.2019.123031. [DOI] [Google Scholar]
- Singh R. K.; Brown E.; Thagard S. M.; Holsen T. M. Treatment of PFAS-containing landfill leachate using an enhanced contact plasma reactor. J. Hazard. Mater. 2021, 408, 124452. 10.1016/j.jhazmat.2020.124452. [DOI] [PubMed] [Google Scholar]
- Huang L.; Dong W.; Hou H. Investigation of the reactivity of hydrated electron toward perfluorinated carboxylates by laser flash photolysis. Chem. Phys. Lett. 2007, 436, 124–128. 10.1016/j.cplett.2007.01.037. [DOI] [Google Scholar]
- Park H.; Vecitis C. D.; Cheng J.; Choi W.; Mader B. T.; Hoffmann M. R. Reductive defluorination of aqueous perfluorinated alkyl surfactants: Effects of ionic headgroup and chain length. J. Phys. Chem. A 2009, 113, 690–696. 10.1021/jp807116q. [DOI] [PubMed] [Google Scholar]
- Song Z.; Tang H.; Wang N.; Zhu L. Reductive defluorination of perfluorooctanoic acid by hydrated electrons in a sulfite-mediated UV photochemical system. J. Hazard. Mater. 2013, 262, 332–338. 10.1016/j.jhazmat.2013.08.059. [DOI] [PubMed] [Google Scholar]
- Tian H.; Gao J.; Li H.; Boyd S. A.; Gu C. Complete defluorination of perfluorinated compounds by hydrated electrons generated from 3-indole-acetic-acid in organomodified montmorillonite. Sci. Rep. 2016, 6, 32949. 10.1038/srep32949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.; Zhang C.; Xing L.; Zhou Q.; Dong W.; Hoffmann M. R. UV/nitrilotriacetic acid process as a novel strategy for efficient photoreductive degradation of perfluorooctanesulfonate. Environ. Sci. Technol. 2018, 52, 2953–2962. 10.1021/acs.est.7b05912. [DOI] [PubMed] [Google Scholar]
- Bentel M. J.; Yu Y.; Xu L.; Li Z.; Wong B. M.; Men Y.; Liu J. Defluorination of per- and polyfluoroalkyl substances (PFASs) with hydrated electrons: Structural dependence and implications to PFAS remediation and management. Environ. Sci. Technol. 2019, 53, 3718–3728. 10.1021/acs.est.8b06648. [DOI] [PubMed] [Google Scholar]
- Cui J.; Gao P.; Deng Y. Destruction of per- and polyfluoroalkyl substances (PFAS) with advanced reduction processes (ARPs): A critical review. Environ. Sci. Technol. 2020, 54, 3752–3766. 10.1021/acs.est.9b05565. [DOI] [PubMed] [Google Scholar]
- Bao Y.; Huang J.; Cagnetta G.; Yu G. Removal of F–53B as PFOS alternative in chrome plating wastewater by UV/sulfite reduction. Water Res. 2019, 163, 114907. 10.1016/j.watres.2019.114907. [DOI] [PubMed] [Google Scholar]
- Bentel M. J.; Yu Y.; Xu L.; Kwon H.; Li Z.; Wong B. M.; Men Y.; Liu J. Degradation of perfluoroalkyl ether carboxylic acids with hydrated electrons: Structure-reactivity relationships and environmental implications. Environ. Sci. Technol. 2020, 54, 2489–2499. 10.1021/acs.est.9b05869. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Li C.; Gao J.; Dong H.; Chen Y.; Wu B.; Gu C. Efficient reductive destruction of perfluoroalkyl substances under self-assembled micelle confinement. Environ. Sci. Technol. 2020, 54, 5178–5185. 10.1021/acs.est.9b06599. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Teng Y.; Mi N.; Jin X.; Yang D.; Wang C.; Wu B.; Ren H.; Zeng G.; Gu C. Highly efficient hydrated electron utilization and reductive destruction of perfluoroalkyl substances induced by intermolecular interaction. Environ. Sci. Technol. 2021, 55, 3996–4006. 10.1021/acs.est.0c07927. [DOI] [PubMed] [Google Scholar]
- Huang C.; Linkous C. A.; Adebiyi O.; T-Raissi A. Hydrogen production via photolytic oxidation of aqueous sodium sulfite solutions. Environ. Sci. Technol. 2010, 44, 5283–5288. 10.1021/es903766w. [DOI] [PubMed] [Google Scholar]
- Li X.; Ma J.; Liu G.; Fang J.; Yue S.; Guan Y.; Chen L.; Liu X. Efficient reductive dechlorination of monochloroacetic acid by sulfite/UV process. Environ. Sci. Technol. 2012, 46, 7342–7349. 10.1021/es3008535. [DOI] [PubMed] [Google Scholar]
- Bentel M. J.; Liu Z.; Yu Y.; Gao J.; Men Y.; Liu J. Enhanced degradation of perfluorocarboxylic acids (PFCAs) by UV/sulfite treatment: Reaction mechanisms and system efficiencies at pH 12. Environ. Sci. Technol. Lett. 2020, 7, 351–357. 10.1021/acs.estlett.0c00236. [DOI] [Google Scholar]
- Liu Z.; Bentel M. J.; Yu Y.; Ren C.; Gao J.; Pulikkal V. F.; Sun M.; Men Y.; Liu J. Near-quantitative defluorination of perfluorinated and fluorotelomer carboxylates and sulfonates with integrated oxidation and reduction. Environ. Sci. Technol. 2021, 55, 7052–7062. 10.1021/acs.est.1c00353. [DOI] [PubMed] [Google Scholar]
- Park H.; Vecitis C. D.; Cheng J.; Dalleska N. F.; Mader B. T.; Hoffmann M. R. Reductive degradation of perfluoroalkyl compounds with aquated electrons generated from iodide photolysis at 254 nm. Photochem. Photobiol. Sci. 2011, 10, 1945–1953. 10.1039/c1pp05270e. [DOI] [PubMed] [Google Scholar]
- Sauer M. C.; Crowell R. A.; Shkrob I. A. Electron photodetachment from aqueous anions. 1. Quantum yields for generation of hydrated electron by 193 and 248 nm laser photoexcitation of miscellaneous inorganic anions. J. Phys. Chem. A 2004, 108, 5490–5502. 10.1021/jp049722t. [DOI] [Google Scholar]
- Iwata A.; Nakashima N.; Kusaba M.; Izawa Y.; Yamanaka C. Quantum yields of hydrated electrons by UV laser irradiation. Chem. Phys. Lett. 1993, 207, 137–142. 10.1016/0009-2614(93)87004-m. [DOI] [Google Scholar]
- Qu Y.; Zhang C.; Li F.; Chen J.; Zhou Q. Photo-reductive defluorination of perfluorooctanoic acid in water. Water Res. 2010, 44, 2939–2947. 10.1016/j.watres.2010.02.019. [DOI] [PubMed] [Google Scholar]
- Wong G. T. F.; Zhang L.-S. Chemical removal of oxygen with sulfite for the polarographic or voltammetric determination of iodate or iodide in seawater. Mar. Chem. 1992, 38, 109–116. 10.1016/0304-4203(92)90070-q. [DOI] [Google Scholar]
- Yu K.; Li X.; Chen L.; Fang J.; Chen H.; Li Q.; Chi N.; Ma J. Mechanism and efficiency of contaminant reduction by hydrated electron in the sulfite/iodide/UV process. Water Res. 2018, 129, 357–364. 10.1016/j.watres.2017.11.030. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Wang J.; Yan D.; Wang L.; Liu X. Efficient reduction of bromate by iodide-assisted UV/sulfite process. Catalysts 2018, 8, 652. 10.3390/catal8120652. [DOI] [Google Scholar]
- Tenorio R.; Liu J.; Xiao X.; Maizel A.; Higgins C. P.; Schaefer C. E.; Strathmann T. J. Destruction of per- and polyfluoroalkyl substances (PFASs) in aqueous film-forming foam (AFFF) with UV-sulfite photoreductive treatment. Environ. Sci. Technol. 2020, 54, 6957–6967. 10.1021/acs.est.0c00961. [DOI] [PubMed] [Google Scholar]
- Kireev S. V.; Shnyrev S. L. Study of molecular iodine, iodate ions, iodide ions, and triiodide ions solutions absorption in the UV and visible light spectral bands. Laser Phys. 2015, 25, 075602. 10.1088/1054-660x/25/7/075602. [DOI] [Google Scholar]
- Bolton J. R.; Bircher K. G.; Tumas W.; Tolman C. A. Figures-of-merit for the technical development and application of advanced oxidation processes. J. Adv. Oxid. Technol. 1996, 1, 13–17. 10.1515/jaots-1996-0104. [DOI] [Google Scholar]
- Nzeribe B. N.; Crimi M.; Mededovic Thagard S.; Holsen T. M. Physico-chemical processes for the treatment of per- and polyfluoroalkyl substances (PFAS): A review. Crit. Rev. Environ. Sci. Technol. 2019, 49, 866–915. 10.1080/10643389.2018.1542916. [DOI] [Google Scholar]
- Gao J.; Liu Z.; Bentel M. J.; Yu Y.; Men Y.; Liu J. Defluorination of omega-hydroperfluorocarboxylates (ω-HPFCAs): Distinct reactivities from perfluoro and fluorotelomeric carboxylates. Environ. Sci. Technol. 2021, 55, 14146–14155. 10.1021/acs.est.1c04429. [DOI] [PubMed] [Google Scholar]
- Buxton G. V.; Greenstock C. L.; Helman W. P.; Ross A. B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O–) in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. 10.1063/1.555805. [DOI] [Google Scholar]
- Elliot A. J. A pulse radiolysis study of the reaction of OH– with I2 and the decay of I2–. Can. J. Chem. 1992, 70, 1658–1661. 10.1139/v92-207. [DOI] [Google Scholar]
- Yiin B. S.; Margerum D. W. Nonmetal redox kinetics: reactions of iodine and triiodide with sulfite and hydrogen sulfite and the hydrolysis of iodosulfate. Inorg. Chem. 1990, 29, 1559–1564. 10.1021/ic00333a023. [DOI] [Google Scholar]
- Neta P.; Huie R. E.; Ross A. B. Rate constants for reactions of inorganic radicals in aqueous solution. J. Phys. Chem. Ref. Data 1988, 17, 1027–1284. 10.1063/1.555808. [DOI] [Google Scholar]
- Li X.; Fang J.; Liu G.; Zhang S.; Pan B.; Ma J. Kinetics and efficiency of the hydrated electron-induced dehalogenation by the sulfite/UV process. Water Res. 2014, 62, 220–228. 10.1016/j.watres.2014.05.051. [DOI] [PubMed] [Google Scholar]
- Bray W. C.; Liebhafsky H. A. Reactions involving hydrogen peroxide, iodine and iodate ion. I. Introduction. J. Am. Chem. Soc. 1931, 53, 38–44. 10.1021/ja01352a006. [DOI] [Google Scholar]
- Gregor H. P.; Belle J.; Marcus R. A. Studies on ion-exchange resins. XIII. Selectivity coefficients of quaternary base anion-exchange resins toward univalent anions. J. Am. Chem. Soc. 1955, 77, 2713–2719. 10.1021/ja01615a011. [DOI] [Google Scholar]
- Hildebrand J. H.; Benesi H. A.; Mower L. M. Solubility of iodine in ethyl alcohol, ethyl ether, mesitylene, p-xylene, 2,2-dimethylbutane, cyclohexane and perfluoro-n-heptane. J. Am. Chem. Soc. 1950, 72, 1017–1020. 10.1021/ja01158a096. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.