

ABSTRACT
With the rapid clinical development of immune checkpoint inhibitors (ICIs), the standard of care in cancer management has evolved rapidly. However, immunotherapy is not currently beneficial for all patients. In addition to intrinsic tumor factors, other etiologies of resistance to ICIs arise from the complex interplay between cancer and its microenvironment. Recognition of the essential role of the tumor microenvironment (TME) in cancer progression has led to a shift from a tumor-cell-centered view of cancer development, to the concept of a complex tumor ecosystem that supports tumor growth and metastatic dissemination. The expansion of immunosuppressive cells represents a cardinal strategy deployed by tumor cells to escape detection and elimination by the immune system. Regulatory T lymphocytes (Treg), myeloid-derived suppressor cells (MDSCs), and type-2 tumor-associated macrophages (TAM2) are major components of these inhibitory cellular networks, with the ability to suppress innate and adaptive anticancer immunity. They therefore represent major impediments to anticancer therapies, particularly immune-based interventions. Recent work has provided evidence that, beyond their direct cytotoxic effects on cancer cells, several conventional chemotherapeutic (CT) drugs and agents used in targeted therapies (TT) can promote the elimination or inactivation of suppressive immune cells, resulting in enhanced antitumor immunity. In this review, we will analyze findings pertaining to this concept, discuss the possible molecular bases underlying the selective targeting of these immunosuppressive cells by antineoplastic agents (CT and/or TT), and consider current challenges and future prospects related to the integration of these molecules into more efficient anticancer strategies, in the era of immunotherapy.
KEYWORDS: Cancer, chemotherapy, immunotherapy, targeted therapy, immunosuppressive cells, tumor microenvironment
Graphical abstract (Made with Biorender)
1. Background
During the twentieth century, cancer treatment focused on the discovery of new cytotoxic drugs or new synergic chemotherapy (CT) combinations. The molecular mechanisms of CT-induced cell death were also described in greater detail, leading to an enhanced understanding of altered signaling pathways in tumors. Consequently, for more than two decades now, the development of targeted therapies (TT) has made it possible to advance the management of cancer patients toward personalized medicine. This era started with the leading molecule, imatinib, which inhibits the tyrosine kinase BCR-ABL expressed by chronic myeloid leukemia cells.1 Following this success, other molecules focalized attention on the different steps of tumoral pathways, i.e. molecules targeting extracellular receptors (monoclonal antibodies, mAb), tyrosine kinase inhibitors (TKI) or molecules targeting intracellular proteins such as serine-threonine kinase inhibitors (BRAF or MEK inhibitors). Nevertheless, the efficacy of targeted therapies sometimes remains low over the long term, because of adaptative clonal resistance and limited to a certain proportion of patients, despite often very significant clinical responses. More recently, immune response stimulation with monoclonal antibody targeting immune checkpoints (known as immune checkpoint inhibitors, ICIs) has yielded an increase in overall survival in many tumor settings. These therapeutic options consider cancer management not only from the viewpoint of the cancer cell characteristics, but also with regard to its immune contexture.
With the growing therapeutic arsenal in oncology, a major challenge is to use these treatments in the most rational way possible, using the combinations that seem most likely to benefit to the growing number of patients. This is particularly true for ICI resistant tumors, in which immune response stimulation with conventional CT or TT is considered a relevant strategy. This concept emerged via the capacities of CT and TT to positively modulate antitumor immune response. Consequently, identifying combinations integrating CT and/or TT that are able to positively modulate the tumor microenvironment (TME) with regard to immunotherapy efficacy has become a major issue.
Currently, it is possible to distinguish three main ways in which CT and TT could exert a positive effect on anti-tumor immunity. First, changes in intrinsic host characteristics caused by CT and TT may play a role in immune response by inducing modifications in the vascular (angiogenesis) or neuroendocrine systems, or by modulating the composition of the mucosal-associated microbiota.2,3 Second, the “on-target” or “cancer-cell dependent” effect (direct cytotoxic effect on cancer cells) enhances adjuvant potency and antigenicity of malignant dying cells. In this domain, immunogenic cell death (ICD), a regulated form of cell death, is mechanistically linked to release of damage-associated molecular patterns (DAMPs), and pro-inflammatory factors from dying tumor cells. Among these factors, calreticulin, extracellular adenosine triphosphate (ATP), high-mobility group box 1 (HMGB1), cancer cell-derived nucleic acid, and annexin A1 could stimulate innate immune cells and increase recruitment of Th1 CD4 + T cells/cytotoxic CD8 + T cells in the tumor.4 Finally, the “off target” or “immune-cell dependent” effects acting on immune cells of tumor microenvironment either by the activation of immune effector cells or the depletion of immune cells with immunosuppressive properties or switching them to a pro-inflammatory and antitumor phenotype. Taken together, these three axes of immunomodulation can have complementary effects, making it possible to create the best conditions for tumor response to immunotherapy. The growing literature led Galluzzi et al. and Petroni et al. to, respectively, review the “cancer-cell” and “immune-cell dependent” effects of CT and TT on the immune system.5,6 However, to the best of our knowledge, there is no review that describes the “immune-cell dependent” effects CT and TT in the context of the standard of clinical care, and the genetic specificity of tumors, to suggest immunologically synergistic CT/TT/ICI combinations.
The TME includes many cells interconnected by numerous signaling pathways that are responsible for initiating an anti- or a protumoral landscape. For example, CD8+ T cells,7 TH1 CD4+ T cells,8 memory T cells, natural killer (NK) cells9 or dendritic cells (DCs)10 are involved in anti-tumor immunity, while, on the other hand, immunosuppressive cells including regulatory T cells (Treg),11 myeloid-derived suppressor cells (MDSCs), M2 phenotype tumor associated macrophages (TAM),12 cancer associated fibroblasts (CAF)13 and tumor cells themselves are involved in pro-tumoral effects. The relative quantity of these different populations and the interaction between them can help to qualify a tumor as “hot”, “cold”, “excluded” or “immunosuppressed”, and more or less likely to respond to ICIs. One of the possible actions of CT and TT is therefore to modulate immunosupressive populations thereby making the tumor more sensitive to immunotherapy. We will focus mainly on this point in this review. To achieve this aim, CT and TT are likely to modulate the immune signaling pathways between these populations, particularly the immunosuppressive pathways, often up-regulated in tumors. These immunosuppressive pathways can be defined according to their underlying mechanisms: enzyme-dependent, cytokine-dependent and immune checkpoint-dependent (Figure 1).
Figure 1.
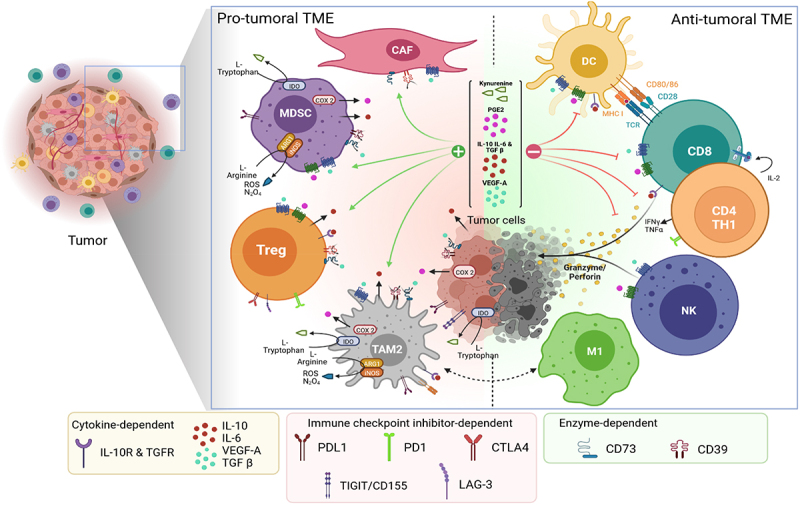
Modulation of the tumor microenvironment: enzyme-dependent, immune checkpoint inhibitor-dependent and cytokine-dependent pathways (Made with Biorender). Treg: regulatory-T cell, MDSC: myeloid-derived suppressor cell, TAM2: tumor associated macrophages of phenotype 2, CAF: cancer associated fibroblast, M1: macrophage of phenotype 1, NK: Natural killer cells, CD8: CD8 + T cell, CD4: CD4 + T cell, DC: Dendritic cell, IDO: Indoleamine 2, 3-dioxygenase, ROS: Reactive oxygen species, COX2: Cyclo-oxygenase type 2, PGE2: Prostaglandin E2, VEGF-A: Vascular endothelial growth factor A., TGFβ: Transforming growth factor beta, TNFa: Tumor necrosis factor alfa, IL-(r): Interleukin (receptor).
Among the enzymatic-dependent immunosuppressive pathways, CD39 and CD73, two ectonucleotidases differently expressed on tumor cells (but also on Treg, MDSCs or TAM), are involved in the degradation of pro-inflammatory ATP, and consequently, the release of adenosine, which in turn inhibits immune-effective cells and maintains immunosuppressive cells in an active state.14 In a similar way, the enzyme indoleamine 2, 3-dioxygenase (IDO) metabolizes tryptophan into kynurenine in the presence of IFNγ. Tryptophan is an amino acid essential for the survival of immune cells, and the lack of tryptophan in the TME results in inhibition of T cell proliferation. Moreover, the production of kynurenine leads to the differentiation of Foxp3+ Treg lymphocytes. High expression of IDO by cancer cell is associated with poor prognosis and reduced overall survival in patients with solid tumors.15
Focusing on the macrophages, M1 phenotype macrophages produce inducible nitric oxide synthase (iNOS), which uses L-arginine as a substrate to produce nitric oxide.16 Macrophages of the M1 phenotype, also called “killer” macrophages, are associated with anti-tumor immunity. On the other hand, M2 macrophages constitutively produce the enzyme arginase 1 (Arg1), involved in altering the proliferative capacities of T lymphocytes by blocking them in the G0/G1 cell cycle phase. TT and CT can be used to influence the M1/M2 ratio, in order to modulate the anti-tumor immune response. We could also briefly mention cyclo-oxygenase (COX) enzymes, which are responsible for the production of prostanoids, including the prostaglandin E2 (PGE2). PGE2 has been shown to play an immunosuppressive role by inhibiting CD8+ T cells, NK and DCs and by promoting differentiation and activation of MDSC and Treg.17 S.Zelenay et al. is investigating the underlying mechanisms by which pharmacological inhibition of PGE2 synthesis or signaling improved the efficacy of ICI therapy.18,19
Regarding the cytokine-dependent pathway, Kitamura et al. suggested that interleukin-6 (IL-6)/ signal transducer and activator of transcription 3 (STAT3) activation in the TME is able to inhibit DC maturation and the activation of effector T cells in cancers. Indeed, previous studies have indicated that IL-6 suppresses the antigen presentation ability of DC through activation of STAT3.20,21 Moreover, angiogenesis plays a central role in both local tumor growth and distant metastasis in breast cancer.22 There is evidence that vascular endothelial growth factor A (VEGF-A) can act as an immunosuppressive factor via several mechanisms, such as inhibiting DC function and maturation, enhancing expression of programmed cell death-ligand 1 (PD-L1) by DCs and PD-1 and other checkpoints involved in CD8 + T cells exhaustion,23 promoting infiltration into the tumor of immunosuppressive T regulatory cells, TAMs and MDSCs, as well as inhibiting cytotoxic CD8+ T cell infiltration into tumors.24,25 In addition, vascular normalization in tumor by VEGF-targeted therapy can improve oxygen levels, drug delivery, and immune cell infiltration.26,27 For all these reasons, it is clear that VEGF-targeted therapies could modulate the tumor-induced immunosuppressive microenvironment, thereby enhancing TH1 T-cell response and increasing tumor infiltration by T cells. Other cytokines, such as the IL-10 and TGFβ pathways, also promote immunosuppressive conditions through Treg activation.28
Finally, the immune checkpoint inhibitor-dependent pathway notably includes programmed cell death protein 1 (PD-1), an immune-inhibitory receptor expressed in activated T cells and involved in the regulation of T-cell functions.29 PD-1 is able to interact with its ligand, programmed cell death programmed death-ligand 1 (PD-L1, CD274), expressed on various cell types, including tumor cells. Following PD-1/PD-L1 binding, T lymphocyte inactivation occurs. Thus, multiple tumor types are able to generate an immunosuppressive TME and avoid T cell cytolysis with PD-L1 expression. The overexpression of PD-L1 has also been associated with poor prognosis and evasion of T cell recognition in several cancers.30 Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), known as CD152, is another immune checkpoint that can be expressed by tumor cells, and which downregulates T cells and inhibits anti-tumor response.31 There are many other inhibitory checkpoints, such as LAG3 or TIM-3. Immunotherapies act by overcoming these immune brakes.32 The T-cell immunoglobulin and immunoreceptor tyrosine-based inhibitory domain (TIGIT) is also an immune-inhibitory molecule on T cells. Cancer cells may evade cancer immunity by expressing TIGIT ligands, such as CD155.33 CD155-positivity is associated with aggressive tumor behavior, and could be a significant predictor of a poor prognosis.34 The TIGIT/CD155 axis contributes to resistance to ICIs, including both primary and acquired resistance, and may be a therapeutic target for combination with immunotherapy.35 However, there is currently no evidence of modulation of this axis by chemotherapy or targeted therapies.
In summary, we propose to review mainly the “immune-cell dependent” effects on immunosuppressive TME of current CT and TT, alone or in combination, in different subsets of solid tumors, including non-squamous-non-small cell lung cancer (NS-NSCLC), breast cancer (BC), hepatocellular carcinoma (HCC), pancreatic cancer (PANC), colorectal cancer (CRC) and melanoma (MEL). We have chosen these cancer sites because of their high incidence and/or the interesting prospects offered by these treatment combinations in these settings. Pre-clinical and clinical perspectives of therapeutic associations with ICIs will be proposed. It should be noted that, in addition to chemotherapies and targeted therapies, radiotherapy is also able to positively modulate the anti-tumor immune response, with interesting results when combined with immunotherapy.36 Boustani et al. discussed the main preclinical and clinical evidence on strategies that can lead to an enhanced response to PD-1/PD-L1 blockade in combination with radiation therapy by studying the optimal dose and fractionation.37 Nevertheless, in this review, we have chosen to focus on systemic treatments.
2. Non squamous-non-small cell lung cancer
Non-Small Cell Lung Cancer (NSCLC) is the most frequent thoracic cancer, and non-squamous NSCLC (NS-NSCLC) is the most common histological subtype.38 NS-NSCLC are characterized by various molecular alterations. The most common of these genetic alterations are epidermal growth factor receptor (EGFR) and KRAS activating mutations: EGFR insertions/deletions are found in approximately 15% of NS-NSCLC, while KRAS mutation incidence reaches 30% in Western countries.39 Additional driver mutations in lung adenocarcinoma occur with a lower frequency, including ALK gene rearrangements, ROS1 and RET translocations or HER2 and BRAF mutations.40 For patients with activating genomic alterations, the development of targeted approaches led to targeted therapies that could be used as standard treatment.41,42 These patients do not currently benefit from immunotherapy. Indeed, EGFR mutation correlates, for example, with uninflamed phenotype and weak immunogenicity.43 Conversely, patients without targetable oncogenic addiction are classically treated with cytotoxic chemotherapies and ICIs, used either concomitantly or sequentially. Anti-PD-1/PD-L1 ICIs are now the standard of care in the first-line treatment of advanced NS-NSLC, as monotherapy44 for patients with high expression of PD-L1 (>50%) or in association with chemotherapy (pemetrexed and platinum-based drugs) (PD-L1 ≥ 1%).45 In second or further lines of treatment, other cytotoxic drugs can be used, such as taxanes, gemcitabine or vinorelbine. All of these treatments can modulate the anti-tumor immune response, in different ways, especially by acting on immunosuppressive TME.
2.1. Conventional chemotherapies in NS-NSCLC
Beyond the induction of immunogenic cell death, chemotherapies used in NS-NSCLC are able to modulate the tumor microenvironment. MDSCs, a cell population accumulating at the tumor site and in peripheral lymphoid organs, can mediate immunosuppression through multiple mechanisms.46 The most well-known mechanisms are inhibition of T-cell proliferation,47 induction of an immunosuppressive environment (induction of Treg, anergy of NK cells through membrane-bound TGFβ)48 and induction of M2 phenotype macrophages.49 Collectively, drugs that are capable of acting on these cells are likely to improve anti-tumor immunity. Anti-metabolite drugs, such as gemcitabine, are chemotherapeutic agents that deplete MDSCs in both animal models and in patients.46,50–52 A retrospective study raised the question of the impact of neoadjuvant chemotherapy with cisplatin and docetaxel on TAMs.53 Indeed, TAMs have been implicated in tumor invasion, immune suppression, and metastasis.54 They are also known to be a prognostic factor, with a negative association between the tumor infiltrating macrophage density and survival of patients with early-stage NSCLC after surgical resection.55,56 Feng et al. confirmed this prognostic role and the impact of the association of neoadjuvant cisplatin-docetaxel chemotherapy on these key cells.53 It is important to note that these studies did not differentiate phenotype 1 or 2 TAM. M2-phenotype TAM express high level of hemoglobin scavenger receptor (CD163) to favor tumor cell progression. It would seem more relevant to look at the roles of these chemotherapies on the CD163+/CD68+ tumor ratio (representing type 1 and type 2 TAM markers).57 However, there is limited data on this subject.
Moreover, several chemotherapeutic drugs have been reported to deplete Treg cells, which is associated with better prognosis for patients. CD4+ CD25+ Foxp3+ Treg cells are immunosuppressive, and their accumulation could inhibit effective immune response in cancer patients, leading to tumor development and progression. Li et al. highlighted the effect of docetaxel on the depletion of Treg populations in peripheral blood mononuclear cells (PBMCs) of patients suffering from NSCLC.58 Moreover, Treg percentages were higher in patients with NSCLC than control subjects with nonmalignant lung disease in the study conducted by Cheng Chen et al.59 This team found that the percentage of CD4+ CD25+ Foxp3+ cells increased in line with tumor progression, and was significantly reduced following chemotherapy with gemcitabine plus cisplatin. Furthermore, the association of cisplatin and vinorelbine has shown an “immune-cell dependent” effect on Treg lymphocytes, by modulating both their function and their number in a sharp and lasting way.60 Roselli et al. showed that this same association prompts modifications of the cytotoxic T lymphocyte (CTL)/Treg ratio, toward a ratio favorable to antitumor immunity.61 Another frequently used chemotherapy for the management of NS-NSCLC, i.e. paclitaxel, a mitotic inhibitor, also displays depletive effects on Treg populations. Indeed, paclitaxel is capable of boosting antitumoral immunity by inducing Treg apoptosis via upregulation of the death receptor CD95 and by downregulating the inhibitory function of Treg cells in NSCLC.62,63 Thus, conventional cytotoxic chemotherapies stimulate the immune response in different ways of immunosuppressive TME modulation, providing a rationale for associating them with immunotherapies, but also with targeted therapies, which are also able to stimulate the anti-tumor immune response.
2.2. Targeted therapies in NS-NSCLC
2.2.1. Egfr inhibitors
As mentioned above, EFGR and KRAS mutations are the most frequently encountered somatic mutations in NS-NSCLC. Consequently, we focus here on therapies targeting these abnormalities, which may play a role in the composition of immunosuppressive tumor microenvironment. The era of targeted therapies in lung cancer began in 2003 with the approval in Japan of gefitinib, an EGFR tyrosine kinase inhibitor (TKI) that inhibits EGFR in a reversible manner.64 This was followed by the development and marketing of new generation EGFR TKIs such as afatinib, or more recently, osimertinib, tested in the FLAURA trial. In this study, osimertinib, an irreversible EGFR-TKI inhibitor, showed a major increase in median progression-free survival of 18.9 months compared to standard EGFR TKIs (gefitinib or erlotinib).65 EGFR-targeted therapies are able to modulate the TME in EGFR-driven lung tumors and to enhance the anti-tumor immune response. Indeed, previous studies have demonstrated that inhibition of EGFR by EGFR-TKIs modulates the TME through several mechanisms, including attenuation of the suppressive function of Tregs and enhancement of the antitumor activity of cytotoxic T cells.66 Jia et al. showed that EGFR-TKIs had a rapid effect on the immune microenvironment, by increasing cytotoxic CD8+ T cell levels, raising DCs, eradicating Foxp3+ Tregs, and by promoting M1 macrophage polarization.67 These results were in accordance with other studies.68,69 Another interesting study demonstrated the effect of the flavonoid drug melafolone on the TME. Melafolone is a molecule capable of inhibiting both EGFR and COX-2, resulting in the promotion of effector CD8+ T cell infiltration. It constitutes a good candidate against resistance to checkpoint blockade therapy for human lung cancer.70 Indeed, COX-2 upregulates PD-L1 on MDSCs and TAMs in murine MBT-2 tumors71 and is also associated with PD-L1 expression in resected tissue specimens of human lung adenocarcinoma.72 In the same way, immunoglobulin-like transcript 4 (ILT4) appears to be a crucial immunosuppressive molecule, and is induced by activation of EGFR-AKT and ERK1/2 signaling in NSCLC cells. ILT4 overexpression suppresses tumor immunity by recruiting M2-like TAMs and impairing T cell response, while ILT4 inhibition prevented immunosuppression and tumor promotion. Thus, in a murine model, Chen et al. identified novel mechanisms for EGFR-mediated tumor immune escape, and provided promising immunotherapeutic strategies for patients with EGFR-activated NSCLC (ILT4 antagonism and immunotherapy combination).73
2.2.2. KRAS Inhibitors
Another common genetic alteration in lung adenocarcinoma is KRAS activating mutation.The majority (95%) of KRAS mutations in NS-NSCLC occur in codons 12 (>80%) and 13. Nevertheless, KRAS having been for a long time non-targetable directly, some therapies targeting the downstream molecules of the KRAS signaling pathway have emerged. These include MEK and mTOR inhibitors, as well as ligands of the retinoid X receptor (RXR). Targeting MEK using trametinib in KRAS G12D mutated murine lung cancer in association with an anti PD-1 showed an increase in CD4+ and CD8+ T cells and a significant reduction in MDSCs.74 These findings suggest a potential therapeutic approach for lung cancers, showing synergy between targeted therapy using MEKi and immunotherapies. These effects on MDSCs could be synergic with the mechanism we previously described. Indeed, our team demonstrated that it was possible to restore CXCL10 secretion and sensitivity to chemo-immunotherapy by inhibiting the MEK pathway in KRAS-mutated tumors.75 Allegrezza et al. also showed the role of the MEK inhibitor trametinib on MDSCs by modulating the chemoattractant factors of these cells in the TME.76 Concerning the oncogenic MAPkinases pathway, SHP2 is an upstream oncoprotein overexpressed in a variety of cancer cell types and which regulates cell survival, differentiation and proliferation through this signaling pathway. SHP2 inhibitors (SHP2i) could influence the TME with depletion of type 2 macrophages, CXCL10 secretion, and also by promoting B and T cell infiltration in KRAS- and EGFR-mutant NSCLC. Nevertheless, SHP2i are also responsible for an increase in MDSCs in the tumor through the production of CXCR2 ligands. Accordingly, Tang et al. suggested combining SHP2 and CXCR2 inhibitors in these tumors to promote TH1 polarization of CD4+ T cells and to increase tumor CD8+ T cell infiltration.77 Through this intervention on the TME, the authors were able to show an improvement in survival in several NSCLC models.
At the cutting edge of research, it has also been possible to develop direct inhibitors of KRAS mutations. MRTX849 (adagrasib) has also shown promising results in combination with anti-PD1 agents in KRAS G12C mutated tumors.78 This KRAS inhibitor increases major histocompatibility complex (MHC) class I expression, decreases tumor infiltration of MDSCs, increases M1 polarization of macrophages, and increases the number of DCs, CD4+ and CD8+ T cells. Thus, by modifying the immunosuppressive TME, MRTX849 has been shown to induce encouraging responses in combination with immunotherapy in mouse models. AMG510 (sotorasib), another KRAS inhibitor, has demonstrated an impact on the TME, alone and in association with anti-PD-1, by increasing infiltration of CD8+ T cells, DCs, including CD103+ cross-presenting DCs, and macrophages with M1 polarization.79 This phenomenom is linked with a cancer-cell dependent effect due to the specificity of inhibitors against the mutated form of KRAS. It actualy not clear how KRAS inhibitors can modulate the recruitment of cytotoxic and suppressive cells but it’s likely that these effects are associated with changes in the cytokine/chemokine expression profile of tumor cells. This point should be explored. In the same way, KRAS-mutated lung cells secrete pro-inflammatory cytokines able to activate the Janus kinase 1/2 (JAK 1/2) signaling pathway, promoting tumor cell survival.80 The use of ruxolitinib, a JAK 1/2 inhibitor, in a KRAS G12D mutated model of human lung cancer, showed a depletion of TAM and MDSCs, changing the TME toward an anti-tumorigenic state.81
2.2.3. mTOR Inhibitors
Targeting mTOR has also shown interesting results by inhibiting tumor growth in brain metastasis of lung cancer, acting on tumor-associated microglia/macrophages and alleviating primary T cell apoptosis in co-culture with H1975.82 mTOR inhibitors could also suppress PD-L1 in EGFR and ALK-driven lung cancer, mediated in part by mTORC2/AKT/GSK3β-dependent proteasomal degradation. Moerland et al. showed that targeting downstream molecular pathways seemed to have a similar impact with the use of MSU-42011, a selective ligand of RXR.83 In combination with cisplatin and pemetrexed, MSU-42011 demonstrated the ability to reduce tumor-promoting macrophages, decreased the number of immunosuppressive MDSCs, and increased infiltration and activation of CD8 + T cells into the lung. RXR is also important in the regulation of Th1/Th2 CD4 + T cell responses by DCs.84
2.2.4. IDO Inhbitors
In addition to targeting these mutations, there are a multitude of targets in lung cancer that show substantial therapeutic promise. One example, among others, is the possibility of targeting the IDO1 considered as an enzyme-dependent immunosuppressive pathway. Among its other functions, this molecule is involved in Treg differentiation, but is also able to induce MDSCs and suppress intra-tumoral CD8+ T and NK cells.85 Moreover, resistance to anti PD-1 drugs in lung cancer seems to be associated with over-expression of IDO1. Taking into account these mechanisms, INCB023843, an IDO1 inhibitor, decreased IDO1 expression in MDSCs in a murine model of lung cancer resistant to anti PD-1 immunotherapy, leading to MDSC depletion and overcoming this resistance.86
Several ongoing clinical studies are assessing the efficacy and safety of different combination therapies, including classical chemotherapies, innovative targeted therapy, or immunotherapy, with the main objective of influencing the TME, with a view to prolonging patient survival. Examples of these trials (from ClinicalTrials.gov) are listed in Table 1. We also present some studies concerning small cell lung cancer (SCLC) for which associations around immunotherapy are also developing.
Table 1.
Summary of therapeutic trials in NSCLC and SCLC combining targeted therapy and/or chemotherapy with immunotherapy. This is a non-exhaustive list taken from ClinicalTrials.gov (with NCT identifier). Clinical trials concerning NSCLC (non small cell lung carcinoma) are indicated in the black box, and SCLC (Small cell lung carcinoma) in the red box. NSCLC: Non small cell lung carcinoma, aPD-L1: anti programmed death ligand 1, MTORi: mammalian target of rapamaycin inhibitors aPD-1: anti programmed cell death protein 1, VEGF(R) i: vascular endothelial growth factor (receptor) inhibitor, aCTLA-4: anti Cytotoxic T-lymphocyte antigen 4, TKi: tyrosine kinase inhibitor, MEKi: MAPK-ERK inhibitor, IDOi: indoleamine 2,3-DiOxygenase inhibitor, EGFRi: epidermal growth factor receptor inhibitor, BRAFi: B-raf inhibitor, NA: not applicable, SCLC: Small cell lung carcinoma, PARPi: Poly (ADP-ribose) polymerase inhibitor.
| NCT identifier | Phase | Tumor type | Immunotherapies | Targeted therapies | Chemotherapies |
|---|---|---|---|---|---|
| NCT04348292 | I | Resectable NSCLC | Durvalumab (a PD-L1) |
Sirolimus (MTORi) |
None |
| NCT03991819 | I | Advanced NSCLC | Pembrolizumab (a PD-1) |
Binimetinib (MEKi) |
None |
| NCT04507906 | I/II | Advanced NSCLC | Nivolumab (a PD-1) |
Anlotinib (VEGFRi) |
None |
| NCT03377023 | I/II | Advanced NSCLC | Nivolumab (a PD-1) Ipilimumab (a CTLA-4) |
Nintedanib (multiple TKi) |
None |
| NCT02658890 | I/II | Advanced NSCLC | Nivolumab (a PD-1) Ipilimumab (a CTLA-4) |
BMS-986205 (IDOi) | None |
| NCT03562871 | I/II | Advanced NSCLC | Pembrolizumab (a PD-1) |
IO102 (IDOi) | Carboplatin Pemetrexed |
| NCT03581487 | I/II | Advanced NSCLC | Durvalumab (a PD-L1) Tremelimumab (a CTLA-4) |
Selumetinib (MEKi) |
None |
| NCT03600701 | II | Advanced NSCLC | Atezolizumab (a PD-L1) |
Cobimetinib (MEKi) | None |
| NCT03689855 | II | Advanced NSCLC | Atezolizumab (a PD-L1) |
Ramucirumab (VEGFRi) | None |
| NCT03786692 | II | Advanced NSCLC | Atezolizumab (a PD-L1) |
Bevacizumab (VEGFi) | Carboplatin Pemetrexed |
| NCT04670913 | II | Advanced NSCLC | Camrelizumab (a PD-1) |
Apatinib (VEGFRi) | None |
| NCT03527108 | II | Advanced NSCLC | Nivolumab (a PD-1) |
Ramucirumab (VEGFRi) | None |
| NCT03971474 | II | Advanced NSCLC | Pembrolizumab (a PD-1) |
Ramucirumab (VEGFRi) |
None |
| NCT04340882 | II | Advanced NSCLC | Pembrolizumab (a PD-1) |
Ramucirumab (VEGFRi) |
Docetaxel |
| NCT04512430 | II | Resectable NSCLC EGFR mutated |
Atezolizumab (a PD-L1) |
Bevacizumab (VEGFi) |
Pemetrexed Carboplatin |
| NCT04517526 | II | Advanced NSCLC EGFR mutated |
Durvalumab (a PD-L1) |
Bevacizumab (VEGFi) |
Pemetrexed Cisplatin Carboplatin |
| NCT04120454 | II | Advanced NSCLC EGFR mutated |
Pembrolizumab (a PD-1) |
Ramucirumab (VEGFRi) |
None |
| NCT04989322 | II | Advanced NSCLC EGFR, ALK and ROS aberrations |
Pembrolizumab (a PD-1) |
Lenvatinib (multiple TKi) | Carboplatin Pemetrexed |
| NCT03178552 | II/III | Advanced NSCLC | Atezolizumab (a PD-L1) |
Bevacizumab (VEGFi) Vemurafenib (BRAFi) Cobimetinib (MEKi) |
Carboplatin Pemetrexed |
| NCT03976375 | III | Advanced NSCLC | Pembrolizumab (a PD-1) |
Lenvatinib (multiple TKi) |
None |
| NCT04973293 | NA | Resectable NSCLC | Sintilimab (a PD-1) |
Bevacizumab (VEGFi) |
Carboplatin Pemetrexed |
| NCT03830918 | I/II | Advanced SCLC | Atezolizumab (a PD-L1) |
Niraparib (PARPi) |
Temozolomide |
| NCT04728230 | I/II | Advanced SCLC | Durvalumab (a PD-L1) |
Olaparib (PARPi) |
Carboplatin Etoposide |
3. Breast cancer
Despite a few cases of long-responder patients, it is important to develop therapeutic combination in order to sensitize BC to immunotherapy. Indeed, some commonly used chemotherapies or targeted therapy have shown that they may impact on the TME by targeting the immunosuppressive microenvironment or couteract the immune-exclusion profile. These possibilities of turning a “cold” breast tumor into a “warm” one are summarized in Ledys et al.‘s review.87
3.1. Conventional chemotherapies in breast cancer
Chemotherapy is part of standard of care in management of breast cancer (BC). Chemotherapies for BC include antimetabolites, alkylating and intercalating agents and mitotic spindle poisons. All of these therapeutic classes are able to modulate the TME, potentially making it more sensitive to the addition of immunotherapy, whether in the early or late stage. Among the antimetabolite drugs, 5-fluorouracil (5-FU) and gemcitabine have been proven to deplete MDSCs.88 5-FU also facilitates antigen uptake by DCs and subsequent cross-presentation of tumor antigens, making tumor cells more sensitive to lysis by CD8+ T cells.89 Gemcitabine, in addition to its effect on MDSCs, promotes cross-presentation of tumor antigens to T cells by DCs, promotes CD8+, CD4+ T cells, NK cells, and reduces Treg proliferation, resulting in anti-tumor immunity.90 Concerning intercalating agents, the effect of anthracyclines on the induction of immunogenic cell death is now well known. There is also the possibility that these drugs modulate the TME by sensitizing tumors to the action of DCs and CD8+ T lymphocytes. This therapeutic class, widely used in breast cancer, is also able to promote elimination of MDSCs, explaining its usefulness for restoring anti-tumor immunity.91 Ladoire et al. previously reported that an increased ratio of CD8+ TILs to Treg T cells after anthracycline-based neoadjuvant chemotherapy was predictive of pathologic complete response and survival in BC.92 Mitotic spindle poisons such as taxanes (paclitaxel and docetaxel) are widely used in BC and may have discordant effects on TME. On the one hand, paclitaxel enhances the TIL subset, notably CD8 T cells, in a neoadjuvant therapeutic strategy,93 especially in triple-negative breast cancer (TNBC).94 On the other hand, paclitaxel was also shown to increases TAMs in a PyMT/MMTV mouse model of BC, and effect that is overcome by addition of PLX3397, an inhibitors of both CSF1R and c-kit.95 Taxanes can also increase serum levels of IFNγ, IL-6 and the cytotoxic function of NK cells.96 Carson et al. studied the immune response of BC patients treated in an adjuvant setting with or without a taxane. The use of taxanes was associated with an increase in the production of T lymphocytes and an increase in the cytotoxicity of NK cells.97 Furthermore, docetaxel, another antimicrotubule agent, has a depletive effect on Tregs.61 Alkylating agents also have interesting properties for association with immunotherapy. Cyclophosphamide and cisplatin are, respectively, able to increase tumor infiltration by CD4+ and CD8+ T cells, and to deplete TAMs in a 4T1 triple negative BC model.98 Indeed, although cyclophosphamide has a lymphoablative effect at high doses, its use at lower doses promotes immunostimulation by inducing inhibition of regulatory T cells and by restoring NK effector functions.99 Therefore, there is a genuine rationale for combining this treatment with immunotherapy.
To summarize, Park et al. recently provided evidence in support of the majority of these observations by showing changes in the TME when cytotoxic chemotherapies were used in the neoadjuvant setting with anthracycline, cyclophosphamide, and taxane in patients with BC.100 An increase in TILs and CD8+ T cells was demonstrated, as well as up-regulation of inflammatory signatures predictive of response to immunotherapy. TNBC patients seemed to be the most likely to show anti-tumor immune stimulation. The benefit of adding immunotherapy to neoadjuvant chemotherapy was clinically confirmed in a prospective trial evaluating the combination of pembrolizumab with chemotherapy in localized TNBC.101
Nevertheless, and particularly since the discordant results of the IMPASSION 130102 and 131103 trials in TNBC, it also seems relevant to look at treatments associated with chemotherapy, which may modulate the TME, in particular by favoring the TH2 or Treg polarization of CD4 + T cells or by favoring the CD8 + T cells exhaustion.104 Thus, the use of glucocorticoids for anti-allergenic purposes with paclitaxel could be partly responsible for the negative results of the IMPASSION 131 study because of its negative impact on the TME. Conversely, nab-paclitaxel, another taxane which does not require anti-allergic corticosteroids, has been shown to improve survival in combination with immunotherapy in TNBC. Beyond chemotherapies, this highlights the need to keep in mind the potentially harmful role of certain premedication’s on chemo-immunotherapy combination.
3.2. Targeted therapies in breast cancer
3.2.1. ANTI VEGF/VEGFR THERAPIES
Since the implication of VEGF and its receptor in tumor growth was first described and regarding this immunosuppressive cytokine-dependent pathway, a number of targeted therapies have been developed. An anti-VEGF used in the management of TNBC, bevacizumab, has suggested a depletive effect on the Treg population in PBMCs of non-progressor patients, in association with durvalumab.105 Anti-VEGFR2 therapies were shown to modulate immunosuppression induced in hyperangiogenic tumor. Apatinib, a VEGFR2 inhibitor, converted an immunosuppressive TME into a pro-inflammatory TME.106 DC101, an anti-VEGFR2 mAb, has shown that when administered in low doses, it has the ability to reprogram the immunosuppressive TME by depleting MDSCs, and promoting M1 polarization of TAMs, ultimately leading to recruitment of CD8+ T cells.107 Interestingly, Reguera-Nuñez et al. showed that nintedanib, a VEGFR tyrosine kinase inhibitor, in combination with paclitaxel and PD-L1 blockade, increased mouse survival in an advanced metastatic EMT-6 BC model.108 Studies evaluating the combination of bevacizumab + atezolizumab + endocrine therapy in hormone-receptor positive breast cancer (HR+BC) (the MORPHEUS HR+ BC trial, NCT03280563) or bevacizumab + atezolizumab + selicrelumab (a CD40 agonist antibody) in TNBC BC (the MORPHEUS TNBC trial, NCT03424005) are currently recruiting. A phase II clinical trial evaluating the efficacy and safety of neoadjuvant therapy with sintilimab and apatinib combined chemotherapy in TNBC is also currently ongoing (NCT04722718).
3.2.2. ANTI-HER2 ANTIBODIES
Human Epidermal Growth Factor Receptor-2 (HER-2) is overexpressed in about 20 to 30% of BCs. Targeting this receptor has changed the therapeutic strategy in early stage or metastatic HER2+ breast cancer. Trastuzumab, a humanized monoclonal antibody targeting HER2, has shown interesting effects on TME cells, with a complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity (ADCC) by NK cells, monocytes and granulocytes. Indeed, trastuzumab was associated with increased tumor infiltration of NK cells.109 Gennari et al. also reported that patients with objective response to trastuzumab had higher numbers of infiltrating leukocytes and higher ADCC activity.110 Moreover, a clinical study analyzing PBMCs from patients receiving trastuzumab showed depletion of CD4+CD25+Foxp3+ Treg in PBMCs.111 Several pre-clinical studies have highlighted the positive effect of trastuzumab in increasing CD8+ T cell tumor infiltration by acting on the M1 polarization of macrophages, with the involvement of IL-21 secreted by tumor cells.112,113 These observations were supported by the study of Girguolo et al. that showed an increase in TIL levels following HER2 blockade in early HER2-enriched BC subtypes.114 A randomized phase III trial is currently evaluating taxanes/trastuzumab/pertuzumab with or without atezolizumab in first-line HER2-positive metastatic BC (NCT03199885) A randomized phase III trial is currently evaluating taxanes/trastuzumab/pertuzumab with or without atezolizumab (NCT03199885) in first-line HER2-positive metastatic BC. A similar study is investigating a similar combination as a neoadjuvant treatment (NCT03747120). Nevertheless, a randomized phase III trial evaluated atezolizumab with neoadjuvant dose-dense doxorubicin/cyclophosphamide–paclitaxel and pertuzumab-trastuzumab for high-risk, HER2-positive early breast cancer did not increase the percentage of pathologic complete response.113 Nonetheless, it seems to be interesting to note better results on low PD-L1 level population highlighting the potential off-target role of anti-HER2 antibodies. Moreover, it is difficult to discuss therapies targeting HER2 without mentioning the antibody-drug-conjugate (ADC) revolution. Trastuzumab deruxtecan (DS-8201) is an ADC combining an anti-HER2 antibody, a cleavable linker, and a cytotoxic topoisomerase I inhibitor. This treatment showed durable antitumor activity in a pretreated patient population with HER2-positive metastatic BC.115 Following these results, many other ADCs are in development. Their properties on the immune system and TME are also being studied. d’Amico et al. elucidated the immune-mediated mechanisms of a novel HER2-targeting ADC bearing a potent anthracycline derivate as payload (T-PNU) in a human HER2-expressing syngeneic BC model resistant to trastuzumab.116 The immunostimulatory properties of T-PNU profoundly reshape the transcriptional and immune profiles within the TME. Following these observations, the authors hypothesized that there would be improved anti-tumor efficacy of immunotherapy by associating a treatment with ADCs carrying an anthracycline payload. It is highly possible that such combinations will be studied clinically in the near future. A phase I dose escalation study of SBT6050 (an ADC including a TLR8 agonist) alone and in combination with PD-1 inhibitors in subjects with advanced solid tumors expressing HER2 is currently recruiting (NCT04460456). Indeed, a candidate drug that acts as aTLR7/8 TLR8 agonist showed promising preclinical results in overcoming tumor resistance to checkpoint blockade: combination treatment increased T cell, tumor-infiltrating DCs, and M1 TAMs, ultimately enhancing the recruitment of CD8+ T cells to tumors.117 In an abstract presenting results of the phase I interim analyses, SBT6050 induced myeloid (MCP-1, IP-10 and Il-6) and NK/T cell activation at all dose levels.118
3.2.3. ENDOCRINE THERAPIES AND CDKs 4/6 INHIBITORS
Endocrine therapies are frequently used in the management of HR+ BC, in the localized or metastatic settings. These include different therapeutic classes, namely: selective estrogen receptor modulators (SERMs, tamoxifen), estrogen receptor down-regulators (fulvestrant) or aromatase inhibitors (letrozole, anastrozole, exemestane). These treatments are known to have an effect on anti-tumor immunity and the TME. SERMs can decrease intratumoral levels of CCL2 and CCL5, thus promoting TAM polarization toward the M1 phenotype.119 Conversely, tamoxifen can also promote an immunosuppressive landscape, inducing CD4+ T-cell polarization toward a Th2 phenotype through the inhibition of DC differentiation, maturation and function, suppressing the cytotoxic immune activity through the inhibition of CD8 + T cells.120,121 Aromatase inhibitors may also decrease naïve T-cell differentiation into T-regulatory cells (Foxp3+ T cells), resulting in a more favorable CD8+/Foxp3+ ratio.122,123 Several combinations are being evaluated in the MORPHEUS HR+ BC clinical trial (NCT03280563), especially associations of atezolizumab and fulvestrant and/or tamoxifen/exemestane. For the treatment of HR+ HER2-negative advanced BC, cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors (especially abemaciclib, palbociclib or ribociclib) represented a major step forward. CDK4/6 inhibitors, in conjunction with their protein regulator cyclin D1, are involved in the regulation of cell cycle progression. These treatments have been shown to play a role in the regulation of the TME, with effects on depletion of circulating and tumor burden of Foxp3+ Treg.124–126 In addition, abemaciclib is able to significantly decrease the Treg/CD8+ T cell ratio, suggesting that CDK4/6 inhibitors might enhance the susceptibility of such tumors to immune checkpoint blockade. A clinical trial evaluating pembrolizumab, endocrine therapy, and palbociclib for the treatment of postmenopausal patients with newly diagnosed metastatic stage IV ER-positive BC is currently recruiting (NCT02778685). There is also an arm evaluating the association of atezolizumab + abemaciclib + fulvestrant in the MORPHEUS HR+ BC trial (NCT03280563). Finally, ribociclib significantly reduces the frequency of immunosuppressive myeloid subsets, notably MDSCs.124,127 Results of the clinical trial evaluating ribociclib in combination with an immunotherapy drug called PDR001 plus fulvestrant are pending (NCT03294694).
3.2.4. POLY (ADP-RIBOSE) POLYMERASE (PARP) INHIBITORS
About 20% of TNBCs are BRCA-mutated, with homologous recombination deficiency. The inhibition of poly (ADP-ribose) polymerase (PARP) has emerged as a new therapeutic strategy in early or advanced BRCA-mutated BC.128,129 It is now accepted that PARP inhibitors (e.g. olaparib, niraparib, talazoparib) can play a role in anti-tumor immunity, in particular through the cGAS/STING signaling pathway.130 These observations could explain why STING agonism enhances anti-tumor immune response and therapeutic efficacy of PARP inhibition in BRCA-associated BC.131 Interestingly, Ding et al. showed that olaparib increased recruitment of DCs, decreased MDSCs in the tumor and in the blood, and increased intratumoral CD4+ and CD8+ T cells and the production of IFNγ by these cells in vivo in BRCA1 deficient ovarian models.132 These results were confirmed with talazoparib (in mammary tumors of BRCA-deficient mice), also able to deplete MDSCs and to enhance tumor infiltration of CD45+CD3+ subset cells,133 and with niraparib (in MDA-MB-436, a BRCA1 mutant triple-negative breast cancer cell line), shown to increase tumor infiltration of CD8+ T cells.134 There is therefore a real biological rationale for combining PARP inhibitors and immunotherapy. In tumor models, niraparib increased antitumor activity and synergistic response in both BRCA-deficient and BRCA-proficient tumor cells.134 In terms of clinical trials, the TOPACIO study evaluated the combination of niraparib and pembrolizumab, which provided promising antitumor activity in patients with advanced TNBC, with numerically higher response rates in those with tumor BRCA mutations.135 The MEDIOLA trial, which tested the combinatinon of olaparib and durvalumab in germline BRCA-mutated BC showed also promising antitumour activity.136 Nevertheless, apart from interesting response rates, it is still difficult to know the benefit of adding immunotherapy to these PARP inhibitors in patients with a BRCA-mutated BC. Due to these findings and in view of the rationale for combining immunotherapy/PARPi or immunotherapy/chemotherapy, a clinical trial is currently evaluating the best strategy between chemotherapy + anti PD-1 or PARPi + anti PD-1 in metastatic TNBC (NCT04191135).
Of note, some studies have revealed that olaparib could also induce a pro-tumor phenotype of macrophages in the TME.137 However, interestingly, these macrophages seemed to be dependent on the colony-stimulating factor 1 receptor axis (CSF1/CSF1R). Mehta et al. reported that the combination of olaparib with an anti-CSF1R had the potential to decrease PD-L1 expression by myeloid cells, to reduce pro-tumor TAMs and to overcome PARP inhibitor resistance in BRCA1-deficient TNBC.137 With this understanding and considering that TAMs are strongly involved in BC tumorigenesis, targeting the CSF1R axis appears to be an attractive therapeutic avenue. Of particular note is its ability to deplete type 2-phenotype TAM and to have a synergistic effect with cisplatin, promoting the release of interferon (IFN) and leading to a more immunogenic TME.138
3.2.5. OTHER TARGETED THERAPIES ACTING ON THE BC TME
EGFR expression is common in BC, especially in triple negative and basal-type breast carcinomas. Cetuximab is a monoclonal antibody that acts on the extracellular EGF-receptor by recruiting NK cells, leading to tumor cell death via antibody-dependent cell-mediated cytotoxicity (ADCC). Juliá et al. highlighted cetuximab’s effect promoting DC maturation in TNBC cell lines.139 In addition, when associated with photo-immunotherapy, cetuximab promoted phenotype-M2 macrophage toward phenotype-M1.140 However, there is currently no clinical trial evaluating a combination of anti-EGFR and immunotherapy in BC, and this remains a therapeutic possibility to be explored.
Another interesting point to highlight concerns Bruton’s tyrosine kinase (BTK) inhibitors. BTK plays a crucial role in oncogenic pathways and is notably known for its involvement in B cell malignancies. Therefore, BTK inhibitors have emerged including ibrutinib, which can inhibit phosphorylation of BTK and efficiently reduce the phosphorylation of the receptor tyrosine kinases ErbB1, ErbB2 and ErbB3, thereby suppressing AKT and MAPK signaling in ErbB2-positive BC cell lines.141 Dubovsky et al. suggested that ibrutinib may also enhance the antitumor immune response by modulating the Th1/Th2 CD4+ T cell ratio.142 Treatment of mice bearing EMT-6 mammary tumors with ibrutinib resulted in a reduced frequency of MDSCs in both the spleen and tumor.143 Varikuti et al. demonstrated that ibrutinib was able to deplete and reprogram MDSCs to mature DCs, which boosts antitumor Th1 immune response and improves infiltration of cytotoxic T lymphocytes due to enhanced tumor-derived antigen presentation to CD8+ T cells.144 A combination of immunotherapy and ibrutinib was shown to suppress tumor growth in preclinical models of TNBC.145 However, a phase 1/2 clinical trial reported limited antitumor activity with a combination of ibrutinib-immunotherapy (durvalumab).146
Alpelisib, another treatment used for advanced HR+ PIK3CA-mutated BC, seems able to modulate anti-tumor immunity. Indeed, this PI3K inhibitor could significantly deplete MDSCs and Treg populations, either alone147 or in association with ribociclib,124 thereby improving responsiveness of BC including TNBC to immunotherapy by increasing PD-L1 expression in preclinical models.148 A clinical trial (NCT04317105) evaluating copanlisib, another PI3K inhibitor, with immunotherapy (nivolumab ± ipilimumab) is currently recruiting patients with advanced solid tumor and changes in the PIK3CA gene. Regarding other therapeutics that act on the PI3K/AKT/mTOR pathway, we can also mention ipatasertib, which was initially evaluated in a phase III trial in combination with paclitaxel in patients with metastatic TNBC.149 This study was negative in terms of its primary endpoint, with no improvement in progression-free survival. However, ipatasertib efficiently depletes Foxp3+ regulatory T cells in the TME, resulting in increased infiltration of effector T cells.150 For these reasons, a trial evaluating ipatasertib in combination with atezolizumab in patients with advanced solid tumors with PI3K pathway hyperactivation is currently ongoing (IceCAP, NCT03673787).
Yin et al. demonstrated the effect of alisertib, an Aurora A kinase inhibitor, on TME in a murine model of BC.151 Indeed, they illustrated that alisertib had the potential to sensitize the malignant BC to anti-PD-L1 therapy by promoting infiltration and activation of effector T cells. This phenomenon entails selective depletion of tumor-promoting myeloid cells, including MDSCs and TAMs.
It is also relevant to mention the place of CD73 inhibitors among the enzymatic-dependent immunosuppressive pathways. CD73 is overexpressed in several cancers and reduced anti-tumor immunity in BC. Monoclonal antibodies directed against CD73 could help to reprogram the TME by decreasing the adenosine mediated immunosuppression particularly as a synergistic immunotherapeutic combination with immunotherapy. The SYNERGY trial investigates the efficacy and safety of the combination of chemotherapy (paclitaxel + carboplatin) with immunotherapy (durvalumab [anti-PD-L1] ± MEDI9447 [anti-CD73]) in previously untreated locally recurrent inoperable or metastatic TNBC (NCT03616886).
Finally, another way of targeting MDSCs is to target Bcl-xL (proteins with cell death repressor activities) with a sublethal dose of ABT-737 (a Bcl-2 and Bcl-xL inhibitor). This molecule has proven to be an effective strategy to overcome MDSCs apoptotic-resistance in a mouse model, leading to suppression of MDSC accumulation in the tumor-bearing host.152 Moreover, Venetoclax, another Bcl-2 inhibitor, has been evaluated in association with palbociclib, and was shown to reduce Foxp3+ Treg populations by decreasing their proliferation capacity.126 There is currently no reports of any ongoing therapeutic trial combining these modalities with immunotherapy. A summary of main clinical trials in BC (from ClinicalTrials.gov) is given in Table 2.
Table 2.
Summary of therapeutic trials combining targeted therapy and/or chemotherapy with immunotherapy. This is a non-exhaustive list from the ClinicalTrials.gov website (NCT identifier). HR: Hormone Receptor, HER2: Human Epidermal Growth Factor Receptor-2, aPD-1: anti programmed cell death protein 1, CDKi: Cycline-Dependent kinase inhibitor, AKTi: Protein kinase B/ akt inhibitor, VEGF(R)i: Vascular Endothelial Growth Factor (receptor) inhibitor, aPD-L1: anti programmed death ligand 1, IL-6i: Interleukin-6 inhibitor, TNBC: Triple Negative Breast Cancer, ADC: Antibody Drug Conjugate, aCTLA-4: anti Cytotoxic T-lymphocyte antigen 4, PI3K(i): Phopshatidylinositol 3 kinase (inhibitor), MEKi: MAPK-ERK inhibitor, PARPi: Poly (ADP-ribose) polymerase inhibitor.
| NCT identifier | Phase | Tumor type | Immunotherapies | Targeted therapies | Chemotherapies | ||
|---|---|---|---|---|---|---|---|
| NCT03294694 | I | Advanced HR+ HER2- | PDR001 (aPD-1) |
Ribociclib (CDKi) Fulvestrant |
None | ||
| NCT03280563 | I/II | Advanced HR+ HER2- | Atezolizumab (aPD-L1) |
Abemaciclib (CDKi) Ipatasertib (AKTi) Exemestane/Fulvestrant/Tamoxifen Bevacizumab (VEGFi) |
None | ||
| NCT03424005 | I/II | Advanced TNBC | Atezolizumab (aPD-L1) |
Bevacizumab (VEGFi) Ipatasertib (AKTi) Selicrelumab (CD40 agonist) Tocilizumab (IL-6i) Sacituzumab-govitecan (ADC) |
Capecitabine Gemcitabine Carboplatin Eribulin Nab-Paclitaxel |
||
| NCT03853707 | I/II | Advanced TNBC | Atezolizumab (aPD-L1) |
Ipatasertib (AKTi) |
Capecitabine Carboplatin Paclitaxel |
||
| NCT03616886 | I/II | Advanced TNBC | Durvalumab (aPD-L1) |
Oleclumab (CD73i) | Carboplatin Paclitaxel |
||
| NCT04317105 | I/II | Advanced PIK3CA mutation/PTEN loss |
Nivolumab (aPD-1) Ipilimumab (aCTLA-4) |
Copanlisib (PI3Ki) |
None | ||
| NCT03673787 | I/II | Advanced PI3K Pathway Hyperactivation |
Atezolizumab (aPD-L1) |
Ipatasertib (AKTi) |
None | ||
| NCT03395899 | II | Resectable HR+ HER2- |
Atezolizumab (aPD-L1) |
Bevacizumab (VEGFi) Cobimetinib (MEKi) Ipatasertib (AKTi) |
None | ||
| NCT04722718 | II | Resectable TNBC | Sintilimab (aPD-1) |
Apatinib (VEGFRi) |
Nab-paclitaxel Carboplatin |
||
| NCT03747120 | II | Resectable HER2+ | Pembrolizumab (aPD-1) |
Trastuzumab Pertuzumab (HER2i) |
Paclitaxel | ||
| NCT03202316 | II | Advanced | Atezolizumab (aPD-L1) |
Cobimetinib (MEKi) |
Eribulin | ||
| NCT02778685 | II | Advanced HR+ HER2- | Pembrolizumab (aPD-1) |
Bevacizumab (VEGFi) Letrozole/Fulvestrant Palbociclib |
None | ||
| NCT04739670 | II | Advanced TNBC | Atezolizumab (aPD-L1) |
Bevacizumab (VEGFi) |
Carboplatin Gemcitabine |
||
| NCT04460456 | II | Advanced HER+ | Pembrolizumab Cemiplimab (aPD-1) |
SBT6050 (ADC) |
None | ||
| NCT04191135 | II/III | Advanced TNBC | Pembrolizumab (aPD-1) |
Olaparib (PARPi) |
Carboplatin Gemcitabine |
||
| NCT03199885 | III | Advanced HER+ | Atezolizumab (aPD-L1) |
Trastuzumab Pertuzumab (HER2i) |
Paclitaxel | ||
4. Hepatocellular carcinoma and pancreatic cancer
In patients with unresectable hepatocellular carcinoma (HCC), the combination of atezolizumab and bevacizumab (an anti VEGF agent) was found to be associated with better progression-free and overall survival, response rate, and preservation of quality of life compared to sorafenib.153 Several active intrinsic immune-evasion pathways, including overexpression of VEGF, have been linked to the development and progression of liver cancer.154,155 Considering this immunosuppressive cytokine-dependent pathway, anti-VEGF therapies reduce VEGF-mediated immunosuppression within the tumor and its microenvironment, may enhance anti PD-1 and anti-PD-L1 efficacy by reversing VEGF-mediated immunosuppression and promoting T-cell infiltration in the tumor.156,157 Hegde et al. reported that anti-VEGF reprograms the TME from an immunosuppressive to an immune permissive microenvironment in human cancers.158 The combination of bevacizumab and atezolizumab has since become the standard of first-line treatment.
Motz et al. demonstrated that tumor-derived VEGF-A and PGE2 cooperatively induced Fas ligand (FasL) expression on human endothelial cells, which acquired the ability to kill effector CD8+ T cells.157 They showed that this was not the case for Treg cells, due to higher levels of cellular FLICE (FADD-like-1β-converting enzyme) inhibitory protein (c-FLIP) expression in Tregs. Regarding these enzyme and cytokine-dependent immunosuppressive pathways and using preclinical models, they further showed that pharmacologic inhibition of VEGF and PGE2 attenuated tumor endothelial FasL expression, produced a significant increase in the influx of tumor-rejecting CD8+ over Foxp3+ T cells, which was FasL-dependent, and led to CD8-dependent tumor growth suppression. In particular, the role of VEGF-A in promoting tumor growth is not solely mediated by its classical role in angiogenesis, but rather extends to include vascular mechanisms controlling mobilization of antitumor immunity. Because of the intimate relationship between angiogenesis and immunosuppression and the likely overwhelming redundancy of pathways controlling both mechanisms, combinatorial strategies inhibiting both arms will be required for effective tumor control. Moreover, the number of CD83+ tumor-infiltrating DCs has been shown to inversely correlate with lymph node metastasis and tissue expression of VEGF and transforming growth factor β (TGFβ) in human breast cancer specimens.159 In addition to these observations, Roland et al. showed that the reduction of macrophage infiltration is an important aspect of anti VEGF therapy.156
In parallel to anti-VEGF therapy, multitarget TKI have shown marked effects on the TME through their action on many oncogenic pathways. For example, tivozanib reduces Tregs and MDSCs in PBMC subsets through downregulation of the c-Kit/ERK2 pathway. Furthermore, tivozanib seems to decrease exhausted TILs, including CD4+ and CD8+ expressing PD-1, and was shown to be associated with survival of HCC patients.160 Sunitinib or sorafenib are other TKIs frequently used in the second line of treatment for HCCs. Sunitinib reduces Treg frequency and decreases their immunosuppressive activity notably IL-10 and TGFβ release. By the same token, it enhances the cytotoxic activity of CD8+ T cells.161 Regarding sorafenib, the effects seem paradoxical. On the one hand, it decreases exhausted CTL subsets and decreases expression of PD-1 in CD8+, stops proliferation and induces Treg apoptosis, slowing their immunosuppressive effects.162,163 On the other hand, it counterintuitively increased Ly6G+ MDSCs in a mouse model of HCC.164 Nevertheless, Cao et al. demonstrated the depletive effect of sorafenib on MDSCs.165 Regarding these observations, a clinical trial evaluating the combination of sorafenib + atezolizumab versus sorafenib alone is ongoing (NCT04770896). Other TKIs; such as lenvatinib, cabozantinib or apatinib are also being used as potential partners to immunotherapy in other clinical trials (NCT04770896, NCT04044651, NCT03755791, NCT04639180).
There are other possible axes to stimulate anti-tumor immunity and act on the TME of HCC. For example, acting on the PI3K/AKT/mTOR axis seems to have effects on the Treg subset. Yuan et al. showed that HCC cells produce and express amphiregulin, which is able to activate the immunosuppressive functions of intratumoral CD4+Foxp3+ regulatory T cells.166 This molecule can trigger the activation of mechanistic target of rapamycin complex 1 (mTORC1) in Treg cells. Using an amphiregulin neutralizing antibody could block this phenomenon, restoring anti-tumor immunity. Moreover, rapamycin, which inhibits mTORC1 and mTORC2, is a well-known inhibitor that is especially useful in preventing the rejection of kidney transplants by inhibiting T cell and B cell activation. However, it seems also to have a paradoxical effect on the tumor by reducing the immunosuppressive functions of Tregs and promoting CD8+ T cell anti-tumor immunity.147
It is also possible to try to act on MDSCs. Tadalafil, a phosphodiesterase type 5 inhibitor (PDE5i) seems to be capable of abolishing the suppressive functions of MDSCs by acting on both NOS2 and Arg1. Furthermore, it blocks chemotaxis by inhibiting CX3CL1 and IL-13 release, preventing accumulation of MDSCs in the TME of murine HCC models.167 The treatment of MDSCs with a PDE5i reversed MDSCs suppressor function and enhanced cytokine-induced killer activity against human HCC cell lines in vitro, as reported by Yu et al.168 A clinical trial is currently evaluating tadalafil in combination with immunotherapy in metastatic HCC or pancreatic cancer (NCT03785210).
Cytokines and their receptors may have a role in anti-tumor immunity, and developing targeted therapies acting on these molecules is a further possibility. For example, Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) is known to induce the extrinsic apoptosis pathway.169 The development of molecules targeting TRAIL and its receptor, TRAIL-R, represent a new therapeutic axis that has garnered interest in the last few years in anti-tumor treatment, especially in HCC.170,171 DS-8273a, an agonistic TRAIL-R2 antibody, in a phase I trial including various digestive cancers such as HCC, showed the potential to maintain selective depletion of MDSCs for at least 28 days, which may provide a sufficient window of therapeutic activity in combination with immunotherapies.172 We could also cite the chemokine receptor type 4 (CXCR4), a G protein-coupled receptor, involved in homing and chemotaxis of the immune system.173 In a model of HCC (i.e. HCA-1), the addition of AMD3100, a CXCR4 inhibitor, overcame the sorafenib-induced increase in F4/80+ TAMs, CD11b+Gr-1+ MDSCs and Foxp3+ Tregs. Moreover, the addition of PD-1 treatment to this association promoted infiltration and switched on CTLs in the tumor.174 For these reasons, the combination of cemiplimab (an anti PD-1) and motixafortide (a CXCR4 inhibitor) is being studied in advanced pancreatic cancer (NCT04543071).
To continue on the subject of pancreatic cancer, CSF-1, also known as M-CSF, is involved in chemoattraction of myeloid subsets including TAM2, promoting tumor progression and propensity to metastasize.175 By targeting the CSF1/CSF1R axis, Mitchem et al. showed that anti-CSF1Rs are able to limit gemcitabine-induced pro-tumoral TAM infiltration in pancreatic ductal adenocarcinoma. This combotherapy also increased tumor infiltration of CTLs and depleted Treg subsets.176 C-X-C motif Chemokine receptor type 2 (CCR2) is a CCL2 receptor also expressed by myeloid cells. Again with the goal of modulating the TME, adding a CCR2 inhibitor to standard FOLFIRINOX chemotherapy has an antitumor immune tendency by decreasing TAM and Treg subsets and promoting CD8+, CD4+ T cells in patients with borderline resectable or locally advanced pancreatic cancer.177 Interestingly, with a mathematical model, Shafiekhani et al. combined 5-FU chemotherapy and anti-CD25 immunotherapy to improve clinical outcome and therapeutic efficacy.178 Indeed, anti-CD25 could decrease the abundance of tumor infiltrating regulatory T cells.
Another possibility concerns the inhibition of CD73, whose role among the enzymatic-dependent immunosuppressive pathways has been developed previously in this review. Regarding immunohistochemistry staining, all HCC and all pancreatic ductal adenocarcinoma (PDAC) expressed CD73 ectonucleotidase in Amedeo Sciarra et al. study.179 Moreover, CD73 expression correlated with morphological tumor grade. Oleclumab (a CD73 inhibitor) is thus being evaluated in combination with durvalumab (an anti PD-L1) and chemotherapy in patients with resectable/borderline resectable pancreatic cancer (NCT04940286).
Interestingly, combinations of treatments similar to BC are under development, sharing the same rationale for association. For example, there is the study of PARPi in combination with immunotherapy in BRCA-mutated pancreatic cancer (NCT04548752), or the development of ADCs such as anetumab-ravtansine in combination with chemo-immunotherapy (NCT03816358). Main clinical trials evaluating these combinations are listed in Table 3.
Table 3.
Summary of therapeutic trials combining targeted therapy and/or chemotherapy with immunotherapy in HCC and pancreatic cancer. This is a non-exhaustive list from the ClinicalTrial.gov website (NCT identifier). Clinical trials concerning HCC are indicated in the black box, and pancreatic cancers in the red box. HCC: Hepatocellular carcinoma, aPD-1: anti programmed cell death protein 1, TKi: Tyrosine kinase inhibitor, aPD-L1: anti programmed death ligand 1, VEGF(R)i: Vascular Endothelial Growth Factor (receptor) inhibitor, aCTLA-4: anti Cytotoxic T-lymphocyte antigen 4, FGFRi: Fibroblast growth factor receptor inhibitorsTLR: Toll-Like-Receptor, ADC: Antibody Drug Conjugate, PDE5i: Phosphodiesterase-5 inhibitor, CXCR4i: C-X-C chemokine receptor 4 inhibitor, PARPi: Poly (ADP-ribose) polymerase inhibitor.
| NCT identifier | Phase | Tumor type | Immunotherapies | Targeted therapies | Chemotherapies |
|---|---|---|---|---|---|
| NCT05286320 | I/II | Advanced HCC | Pembrolizumab (aPD-1) |
Lenvatinib (multiple TKi) |
None |
| NCT04721132 | II | Resectable HCC | Atezolizumab (aPD-L1) |
Bevacizumab (VEGFi) |
None |
| NCT05194293 | II | Resectable HCC | Durvalumab (aPD-L1) |
Regorafenib (multiple TKi) |
None |
| NCT05168163 | II | Advanced HCC | Atezolizumab (aPD-L1) |
Cabozantinib Lenvatinib (multiple TKi) |
None |
| NCT03937830 | II | Advanced HCC | Durvalumab (aPD-L1) Tremelimumab (aCTLA-4) |
Bevacizumab (VEGFi) |
Doxorubicin (chemoembolization) |
| NCT04828486 | II | Advanced HCC | Pembrolizumab (aPD-1) |
Futibatinib (FGFRi) |
None |
| NCT04442581 | II | Advanced HCC | Pembrolizumab (aPD-1) |
Cabozantinib (multiple TKi) |
None |
| NCT04044651 | II/III | Advanced HCC | Nivolumab (aPD-1) |
Lenvatinib (multiple TKi) |
None |
| NCT04639180 | III | Resectable HCC | Camrelizumab (aPD-1) |
Apatinib (VEGFRi) |
None |
| NCT05198609 | III | Advanced HCC | Camreluzimab (aPD-1) |
Apatinib (VEGFRi) |
Fluorouracil Oxaliplatin |
| NCT04770896 | III | Advanced HCC | Atezolizumab (aPD-L1) |
Lenvatinib Sorafenib (multiple TKi) |
None |
| NCT03755791 | III | Advanced HCC | Atezolizumab (aPD-L1) |
Cabozantinib (multiple TKi) |
None |
| NCT04787991 | I | Advanced Pancreatic Cancer | Nivolumab (aPD-1) Ipilimumab (aCTLA-4) |
Hydroxychloroquine | Gemcitabine Nab-paclitaxel |
| NCT04050085 | I | Advanced Pancreatic Cancer | Nivolumab (aPD-1) |
SD-101 (TLR-9 agonist) |
None |
| NCT03816358 | I/II | Advanced Pancreatic Cancer | Nivolumab (aPD-1) Ipilimumab (aCTLA-4) |
Anetumab Ravtansine (ADC) |
Gemcitabine |
| NCT03214250 | I/II | Advanced Pancreatic Cancer | Nivolumab (aPD-1) |
APX005M (CD40 agonist) |
Gemcitabine Nab-paclitaxel |
| NCT02451982 | II | Resectable Pancreatic Cancer | Nivolumab (aPD-1) |
Urelumab (CD137 agonist) |
Cyclophosphamide |
| NCT04940286 | II | Resectable Pancreatic Cancer | Durvalumab (aPD-L1) |
Oleclumab (CD73i) |
Gemcitabine Nab-paclitaxel |
| NCT03727880 | II | Resectable Pancreatic Cancer | Pembrolizumab (aPD-1) |
Defactinib (Focal Adhesion Kinase inhibitor) |
None |
| NCT03785210 | II | Advanced HCC or Pancreatic cancer | Nivolumab (aPD-1) |
Tadalafil (PDE5i) |
None |
| NCT04543071 | II | Advanced Pancreatic Cancer | Cemiplimab (aPD-1) |
Motixafortide (CXCR4i) |
Gemcitabine Nab-paclitaxel |
| NCT04548752 | II | Advanced Pancreatic Cancer BRCA 1/2 mutated |
Pembrolizumab (aPD-1) |
Olaparib (PARPi) |
None |
5. Colorectal cancer
5.1. Conventional chemotherapies
There are currently different treatment strategies for colorectal cancer (CRC), but the cornerstone of management in metastatic stages is the use of chemotherapy combinations. The main findings relating to “immune-cell dependent” effects of chemotherapies have been described in models of digestive cancers.180 The most frequently used drugs are 5-fluorouracil (5FU), irinotecan and platinum salts (oxaliplatin). These cytotoxic chemotherapies can modulate the immunosuppressive effect of the TME.
In a FOLFIRI-like therapeutic model combining CPT11 (irinotecan) and 5-FU, Kanterman et al. showed that 5-FU overcomes CPT11-induced apoptosis resistance of MDSCs and depletes this myeloid subpopulation by acting on their maturation and activity.181 Identical resultats were found by our team. In a context of FOLFOX-bevacizumab regimen, decrease of MDSCs after the first administration of chemotherapy is associated with a better progression-free survival.182 In addition, when associated with radiotherapy, 5-FU showed its ability to increase infiltration of CTLs, notably CD8+, CD4+ and NK T cells.183,184 Recently, we also showed that the use of tipiracile/trifluridine, an anti-metabolite administered after 5-FU escape, could synergize with oxaliplatin to sensitize the CT26 preclinical model to anti-PD-1 immunotherapy. This synergy is dependent on both an effect on the tumor cell and an effect on the suppressor cells. Indeed, these two drugs synergize to induce immunogenic tumor cell death but also promote depletion of type 2 macrophages.185
Oxaliplatin is also frequently used and could enhance the immune response against tumors by decreasing regulatory/suppressor cells (Tregs and MDSCs) and by increasing the ratio of cytotoxic CD8 + T cells to immunosuppressive cell populations in the TME.186 In the same manner, Zhu et al. demonstrated that in addition to inducing immunogenic cell death as well described by other teams,187 oxaliplatin is able to modulate this tumor microenvironment.188 Indeed, they found that the number of mature DCs was increased after immature DCs were cocultured with oxaliplatin-treated H22 cells (corresponding to a hepatocarcinoma model). Numbers of CD8+ T cells and mature DCs were found to be increased in vivo whereas, the number of Treg cells was decreased.
These beneficial effects in vitro were confirmed by Pfirschke et al. in vivo. Indeed, oxaliplatin and immunotherapy (anti-CTLA-4) combination was able to reject CT26 tumors in approximately 40% of mice analyzed.189 In another interesting preclinical study using two mouse colorectal cancer models (CT26 and MC38), Dosset et al. found that a combination of a 5-FU/oxaliplatin and PD-1 blockade therapy induced complete and long-lasting tumor response.190 As described above, this association was able to induce recruitment of an effective T cell population in the TME, creating a favorable environment for the action of immunotherapy. This immune recruitment has already been shown to be associated with improved survival in colorectal cancer patients, especially since the advent of immunotherapy.191 In parallel, Dosset et al. also demonstrate that this FOLFOX regimen induce the expression of checkpoints inhibitors (PD-1 and PD-L1) on the activated CD8 T cells and on tumor cells themselves, responsible of tumor immune resistance. Therefore, the addition of checkpoint inhibitors allowed FOLFOX-recruited CD8 T cells to induce an effective anti-tumor immune response. Interestingly, the authors found a decreased tumor T cell infiltration after chemotherapy administration. These results suggest that the immune checkpoint inhibitors should be given concomitantly or early after FOLFOX therapy. In humans, the effectiveness of this combination is currently being evaluated. Promising data have been obtained for patients with MSS colon tumors treated with FOLFOX and dual immunotherapy with anti-PD-L1/anti-CTLA4.192 To finish on the impact of chemotherapies on the TME in digestive tumors, we can also briefly mention gemcitabine, wich has also shown its capacity to deplete MDSCs193 and Tregs.194 It can also induce an increase in pro-tumor TAMs.176 Cyclophosphamide has similar effects by decreasing Tregs and restoring the function of T and NK cells in a colon cancer model.99,195
5.2. Conventional targeted therapies
The place of immunotherapy in the management of locally advanced or metastatic CRC remains limited. Currently in France, the only marketing authorization is for pembrolizumab in colorectal tumors with microsatellite instability (MSI+). In this situation, immunotherapy is used alone, with good results, but for a limited number of patients (<10%). There is thus a strong rationale for developing therapeutic combinations to increase anti-tumor immunity. A distinction is classically made between mutated and non-mutated RAS/RAF colorectal tumors.
Cetuximab and panitumumab are two mAbs directed against EGFR and frequently included in treatment strategies for RAS/RAF wild type (WT) CRC. Zhao et al. showed that cetuximab is able to restore an antitumoral TME by modulating and reprogramming functions of TAMs from the M2-like phenotype toward an M1-like phenotype, including the suppression of IL-6 expression in TAMs.196 Indeed, the EGFR axis is involved in M2 polarization through the PI3K/AKT/mTOR pathway.197 The inhibition of PI3K or EGFR with a monoclonal antibody is able to push this polarization toward anti-tumoral M1 polarization. Furthermore, Abu-Eid et al. reported that PI3K-Akt inhibitors reduced tumor growth in CT26 mouse models, due to the selective inhibition of Tregs. However, they showed that the antitumor effect achieved via inhibition of PI3K-Akt can be exhibited through other mechanisms, such as enhanced survival of CD8 + T cells (Akt signaling drives CD8 + T cell differentiation).198 VEGF and VEGFR inhibitors are broadly prescribed in the standard treatment of CRC, particularly in patients with RAS/RAF mutated colon cancer. The increase in CD4+Foxp3+ Tregs, M2-like macrophages and MDSCs, and the decrease in CD8 + T cells are well-known immune features in tumors overexpressing VEGFA. As previously described and regarding this cytokine-dependent immunosuppressive pathway, anti-VEGF therapies may enhance immunotherapy efficacy by reversing VEGF-mediated immunosuppression and promoting T-cell infiltration in tumors.24,199,200 As a reminder, TAMs may induce the production of IL-10 or TGF-β, able to suppress CD8+ T cell and DC functions, and able to stimulate Treg cells.201 TGF-β can be produced through the VEGF/VEGFR2 signaling pathway, and can promote metastasis through its action on epithelial–mesenchymal transition. Min et al. showed that anti-VEGFR therapies may help to control the immune inhibitory functions of M2-TAMs in CRC.202 Manzoni et al. first reported in 2010 that bevacizumab, an anti-VEGF frequently used for the management of CRC, is able to increase B and T cells.203 The expansion of T lymphocytes could imply an improvement in DC-presenting capacity. These effects were associated with improved clinical outcome. A study evaluating the efficacy and safety of multiple immunotherapy-based treatment combinations, including bevacizumab, in patients with metastatic CRC is currently ongoing (NCT03555149).
Multi-target TKIs have other effects on the TME. For example, regorafenib was shown to inhibit angiogenic receptor tyrosine kinases (VEGFR1-3) and also, through its action on the PDGFR axis, was able to deplete CAF in KM12SM human colon cancer cells by acting on PDGFR expressed by CAFs.204,205 This molecule is currently used in the management of CRC, after all standard therapies, and is being evaluated in combination with immunotherapy in the aforementioned Morpheus study (NCT03555149). Similarly, other dedicated clinical trials are ongoing (NCT04126733 combining regorafenib with nivolumab; NCT04110093: regorafenib with carelizumab, sintilimab and toripalimab, three PD-1 inhibitors). NCT04126733 trial also reported reasonable adverse events with Regorafenib-Nivolumab combination.206
For patients with BRAF-mutated CRC, the standard of treatment in second-line therapy is based on the use of anti-EGFR, anti-BRAF and anti-MEK. Selumetinib, a MEK inhibitor, was shown to deplete myeloid subsets including TAMs, enhancing the response to associated immunotherapy with an anti-CTLA-4 in a KRAS-mutant CT26 mouse colorectal cancer model.207 In addition, it reduces expression of Arg1 associated with the down-regulation of COX-2 within tumor cells, two enzymes whose involvement in immunosuppression has previously been described.207 These observations are supported by the findings of Eruslanov et al. who reported that the drug LM1685, by inhibiting COX-2, seems to prevent tumor-induced arginase activation in myeloid cells, including TAMs and MDSCs.208 Cobimetinib, another MEK inhibitor, had a similar effect on M2-like macrophages and MDSCs. In addition, it also increases the CD8+/Treg ratio.209 Taken together, these data suggest the growing interest of anti-MEK in improving the responsiveness to immunotherapy as highlighted in the study NCT04044430 (combining encorafenib, binimetinib, and nivolumab in BRAF-mutated CRC). Moreover, anti PD-L1 immunotherapy yielded a response in microsatellite-stable (MSS) metastatic CRC in combination with MEK inhibitors.210
SHP2 is an oncogenic protein involved in many signaling pathways that contribute to tumor growth and survival, including receptor tyrosine kinase pathways. Inhibitors of the protein tyrosine phosphatase-2 (SHP2) can inhibit receptor tyrosine kinase (RTK) signaling. Liu et al. showed that in BRAF V600E CRC models, TNO155, an SHP2 inhibitor, synergized with BRAF and MEK inhibitors by blocking ERK feedback activation by different RTKs.211 In addition, the association of TNO155 showed immunomodulatory effects. This molecule inhibited RAS activation by the CSF1R pathway, and inhibited TAM 2 and MDSCs in the tumor. This treatment may also enhance PD-1 blockade efficacy83 and is being evaluating in conjunction with an anti PD-1, spartalizumab in selected malignancies, including CRC (NCT04000529). In a same way, by inhibiting CSF-1 R in a MC38 murine model of colon adenocarcinoma, RG7155 has shown its capacities to strongly reduce F4/80+ tumor-associated macrophages accompanied by an increase of the CD8+/CD4+ T cell ratio.212
Another possibility concerns the development of an anti-CCR5. Chemokine ligand 5 (CCL5) is a chemokine released by CD4+ and CD8+ T cells, and this phenomenon could be used by tumor cells to metastasize to the liver. C-C chemokine receptor 5 (CCR5) is the natural G-coupled protein receptor of CCL5, and is involved in M2 polarization of TAMs in CRC. Halama et al. showed that maraviroc, a CCR5 inhibitor, exerted anti-tumor effects by reprogramming macrophages toward the M1 phenotype via STAT3 regulation.213 A phase I clinical trial (PICASSO study, NCT03274804) evaluated the combination of an anti-PD-1 (pembrolizumab) with CCR5 inhibition for the treatment of refractory microsatellite-stable metastatic CRC. Unfortunately, the observed clinical efficacy remained limited, apart for some situations of prolonged stabilization.214
In studies of Bruton’s tyrosine kinase (BTK) inhibitors, the effect on colorectal tumor cell lines was studied. In the same way as for breast cell lines, ibrutinib had an effect on MDSCs, leading to a decrease in this population, thus promoting CD8+ infiltration.143 The NCT03332498 trial, recently closed to enrollment, is evaluating the combination of Pembrolizumab and Ibrutinib in advanced refractory colon cancer.
In order to potentiate anti-tumor immunity, it is also possible to target the cell cycle of tumors cells and apoptosis, with interesting effects on the TME. For example, the protein p53 is controlled by mouse double minute 2 homolog (MDM2), an ubiquitin E3 ligase, which regulates p53 levels and proteasomal degradation. The MDM2 protein is overexpressed in many cancers, causing disruption of the p53-MDM2 axis, and linked to tumorigenesis. It represents a novel target for cancer therapy. Several MDM2 antagonists have been developed, such as APG 115. This molecule can change the polarization of macrophages, in favor of an anti-tumor M1 phenotype state, and could activate CD4+ T cells. Furthermore, inhibiting MDM2 upregulates PD-L1 expression on tumor cells and may lead to high immunogenicity, confirmed by an improvement in the cytotoxic activity of CD8+ T cells when an anti PD-1 is added to APG 115.215
Still concerning cell cycle modulation, CDK4/6 inhibitors (abemaciclib or palbociclib) in preclinical CRC models showed similar results to the breast cell-line studies. These molecules were shown to induce depletion of circulating and tumor burden Foxp3+ Tregs by slowing their proliferation.59–61 This observation suggests that CDK4/6 inhibitors might enhance the susceptibility of such tumors to immune checkpoint blockade. Other treatment options are also under investigation, such as venetoclax, which induces apoptosis by inhibiting BCL-2 and showed an increase in intra-tumoral CD8 + T cells in a MC38 preclinical model.216
Finally, Yes‐associated protein 1 (YAP1) is activated in CRC and is associated with tumor growth. By inhibiting YAP1, Yang et al. showed that the accumulation of tumor-associated MDSCs could be modulated, by decreasing granulocyte–macrophage colony‐stimulating factor tumoral release.217 There is thus an opening for the development of treatments combining these molecules with immunotherapy in the future. Table 4 summarizes examples of clinical trials combining CT and TT with immunotherapy in CRC.
Table 4.
Summary of therapeutic trials combining targeted therapy – chemotherapy – immunotherapy in colorectal cancers. This is a non-exhaustive list from the ClinicalTrial.gov website (NCT identifier). CRC: Colorectal Cancer, aPD-1: anti programmed cell death protein 1, VEGF(R)i: Vascular Endothelial Growth Factor (receptor) inhibitor, MSI: Microsatellite Instability, COXi: Cyclo-Oxygenase-2 Inhibitor, aPD-L1: anti programmed death ligand 1, HDACi: Histone De-Acetylase Inhibitor, TKi: Tyrosine Kinase inhibitor, BRAFi: B-raf inhibitor, EGFRi: Epidermal Growth Factor Receptor inhibitor, PI3Ki: Phosphatidylinositol 3-kinase inhibitor, BTKi: Bruton’s Tyrosine Kinase (BTK) inhibitors, aCTLA-4: anti Cytotoxic T-lymphocyte antigen 4, MEKi: MAPK-ERK inhibitor.
| NCT identifier | Phase | Tumor type | Immunotherapies | Targeted therapies | Chemotherapies |
|---|---|---|---|---|---|
| NCT02298959 | I | Advanced CRC | Pembrolizumab (aPD-1) |
Ziv-aflibercept (VEGF-trap) |
None |
| NCT03926338 | I/II | Resectable CRC MSI |
Toripalimab (aPD-1) |
Celecoxib (COXi) | None |
| NCT03555149 | I/II | Advanced CRC | Atezolizumab (aPD-L1) |
Bevacizumab (VEGFi) Regorafenib (multiple TKi) Isatuximab (HDACi) AB928 (Dual Adenosine Receptor Antagonist |
None |
| NCT04110093 | I/II | Advanced CRC | Camrelizumab Nivolumab Sintilimab Toripalimab (aPD-1) |
Regorafenib (multiple TKi) |
None |
| NCT04017650 | I/II | Advanced CRC | Nivolumab (aPD-1) |
Encorafenib (BRAFi) Cetuximab (EGFRi) |
None |
| NCT03711058 | I/II | Advanced CRC | Nivolumab (aPD-1) |
Copanlisib (PI3Ki) |
None |
| NCT03332498 | I/II | Advanced CRC | Pembrolizumab (aPD-1) |
Ibrutinib (BTKi) |
None |
| NCT03377361 | I/II | Advanced CRC | Nivolumab (aPD-1) Ipilimumab (aCTLA-4) |
Trametinib (MEKi) |
None |
| NCT04715633 | II | Resectable CRC MSI |
Camrelizumab (aPD-1) |
Apatinib (VEGFRi) |
None |
| NCT04745130 | II | Advanced CRC | Sintilimab (aPD-1) |
Regorafenib (multiple TKi) Cetuximab (EGFRi) |
None |
| NCT04866862 | II | Advanced CRC | Camrelizumab (aPD-1) |
Fruquintinib (VEGFRi) |
None |
| NCT03608046 | II | Advanced CRC | Avelumab (aPD-L1) |
Cetuximab (EGFRi) |
Irinotecan |
| NCT04271813 | II | Advanced CRC | Sintilimab (aPD-L1) |
Anlotinib (VEGFRi) |
None |
| NCT04527068 | II | Advanced CRC | Tripleitriumab (aPD-1) |
Bevacizumab (VEGFi) |
None |
| NCT04262687 | II | Advanced CRC | Pembrolizumab (aPD-1) |
Bevacizumab (VEGFi) |
Oxaliplatin Capecitabine |
| NCT02997228 | III | Advanced CRC | Atezolizumab (aPD-L1) |
Bevacizumab (VEGFi) |
Oxaliplatin Fluorouracil |
6. Melanoma
Melanoma is a highly chemoresistant tumor and the use of cytotoxic chemotherapy is no longer part of the standard treatment in first or second-line therapy. For example, dacarbazine, an alkylating agent, was previously used, and was shown to stimulate NKG2D expression on murine tumor cells, leading to the recruitment of CD8+ and NK T cells into the tumor.218,219 However, the breakthrough of immunotherapy as a therapeutic standard has resulted in very low rates of use of conventional cytotoxic chemotherapies in the setting of melanoma. Indeed, melanoma is considered as one of the most immunogenic solid tumors, and many immunotherapeutic approaches have been developed to circumvent disease progression. For patients without BRAF mutation, the combination of two immunotherapies, namely ipilimumab (a CTLA-4 inhibitor) and nivolumab (a PD-1 inhibitor) has become the standard first-line treatment for patients with unresectable or metastatic melanoma.220 In a complementary manner, BRAF mutation is a major oncogenic mutation found in approximately 50% of human melanomas, and confers constitutive activation of the MAPK pathway, responsible for cell growth, survival, and differentiation. Significant progress has been made in understanding the molecular bases of melanoma oncogenesis. These advances have led to the development of highly effective targeted therapies, which have also been shown to increase survival in advanced melanoma. For this reason, the combination of a MEK inhibitor with a BRAF inhibitor has become a new therapeutic standard in advanced BRAF-mutated melanoma.221,222 This combination seems to have an effect on anti-tumor immunity and can modulate the TME by enhancing the presence of tumor-specific antigens and the level of intratumor CD8+ T cells.223–225 Similarly, Ferrari de Andrade et al. reported that BRAF inhibitors were able to enhance proliferation and activation of NK cells.226 Conversely, some teams underline the increase of immunosuppressive populations in the TME under the effects of these treatments, with notably an increase of Tregs or MDSCs.221,224 Despite this, the combination of anti-BRAF, anti-MEK, and immunotherapy remained associated with a good antitumoral activity in a mouse model of BRAF-mutated melanoma. These therapies therefore seem to be of interest in terms of the immune system, underpinning attempts to associate BRAF and MEK inhibitors with immunotherapy in clinical trials (NCT03235245).
Beyond BRAF and MEK inhibitors, other targeted therapies (new or old) are being associated with immunotherapies, thanks to the identification of interesting mechanisms from an immune point of view. Zhang et al. showed that axitinib, a VEGFR TKi, increased infiltration of CD8+ T cells and downregulated the amount of MDSCs and type 2 macrophages, by inhibiting ARG1 and NOS2.227 For these reasons, and due to the often intense vascularization of melanomas, it is hypothesized that axitinib can metabolically remodel the TME to render it more sensitive to immunotherapy, specifically by increasing T cell infiltration through HIF1alpha activity resulting from hypoxia (NCT04493203). This mechanism is also reported with anti-VEGF therapy.228 In the same way, anlotinib, a novel multi-target TKI (VEGFR2, PDGFR, FGFR) affects infiltration of CD8+ T cells and depletion of the Foxp3+ T cell population, prompting its association with immunotherapy.229,230 Cabozantinib inhibits multiple-receptor tyrosine kinases, including c-MET or vascular endothelial growth factor receptor 2 (VEGFR2), and is approved for use in solid malignancies such as renal cell carcinoma, hepatocellular carcinoma or medullary thyroid cancer. This treatment has also been shown to have immunomodulatory effects in vitro and in murine models. In addition, c-Met has been found to induce overexpression of PD-L1 and to induce tolerogenic DCs in mouse models as describe by Balan et al. and Ilangumaran et al.231,232 For all these reasons, Jain et al. hypothesized that combining cabozantinib with pembrolizumab would have the potential to improve response rate in metastatic or recurrent melanoma (NCT03957551).233 Other combinations are under clinical evaluation, such as a combination of cabozantinib with nivolumab and ipilimumab (NCT05200143).
Still considering the cytokine-dependent immunosuppressive pathway, interesting results from a preclinical study underline that the association of vactosertib, a TGFβ receptor inhibitor, with checkpoint inhibitor immunotherapy, can slow the expansion of melanoma-associated fibroblasts (MAF), and increase CD8 + T cell infiltration.234 In parallel to these observations, several teams have shown that adenosine signaling reduces effector functions of cytotoxic lymphocytes, while also promoting recruitment and polarization of immunosuppressive cell types, including MDSCs and T reg cells.235,236 A2AR inhibition enhances infiltration of cytotoxic NK cells and CD8+ T lymphocytes. For these reasons, it has been suggested that A2AR inhibition may be a useful therapeutic addition to both BRAF and MEK inhibition, in patients with melanoma.237
Among the enzymatic-dependent immunosuppressive pathways, we could also mention Epacadostat, an IDO1 selective inhibitor that showed promising antitumour activity in the ECHO-202/KEYNOTE-037 study in advanced melanoma. Indeed, IDO1 is correlated with poor prognosis in patients with melanoma, making the use of IDO1 inhibitor interesting.238 However, the corresponding phase III study was negative, with no clinical benefit from the addition of this IDO1 inhibitor to immunotherapy in unresectable or metastatic melanoma.239
Moreover as in other tumors, inhibition of the BTK and Pi3K/AKT pathway has shown its effect on Treg and MDSC depletion.143,147 Accordingly, a currently ongoing trial (NCT03021460) is evaluating the combination of ibrutinib plus pembrolizumab in advanced melanoma.
In total, due to chemoresistance and the decline in the use of chemotherapy in the management of melanomas, chemo-immunotherapy does not currently seem to be preferred, despite its possible effects on anti-tumor immune response. Nevertheless, as described in this review, there is a real immune rationale around the combination of targeted therapies and immunotherapies, which has led to several clinical trials. Beyond the immune interest, combining immunotherapy with targeted therapies could also limit the risks of early relapse after stopping targeted therapies and favor prolonged responses.
7. Conclusions perspectives
In the era of immunotherapy, understanding the cellular and molecular mechanisms involved in suppressing the anti-tumor immune response in TME is essential. Since the demonstration of the efficacy of immunotherapy as a single agent in several types of cancer, the current objective is to be able to combine it with other therapeutic strategies that will make it possible to overcome resistance and/or amplify its effects. If TT and CT are originally intended to impact cancer cell proliferation by altering the signaling of oncogenic pathways, these therapies are also able to directly or indirectly influence the actors of the anti-tumor immune response. In most cases, the combination of these drugs is based on the upward modulation of the anti-tumor immune response. Concerning more specifically immunosuppression, TTs and CTs can have a favorable influence by depletion of immunosuppressive cells (Tregs, MDSCs, CAFs, TAM2), by reprogramming of these cells (MDSCs to DC, TAM2 to TAM1), by inhibition of soluble immunosuppressive factors or by decreasing the expression of inhibitory immune checkpoints. It is in this context that many combinations involving targeted therapy or chemotherapy with immunotherapy are becoming the new standard of treatment. In lung cancer, for example, the combination of the platinum salt/pemetrexed doublet with anti-PD-1 antibody immunotherapy amplifies the anti-tumor immune response by increasing immunogenic cell death. In the case of hepatocellular cancer, the combination of an anti-angiogenic agent with immunotherapy amplifies the immune response by decreasing the immunosuppressive signals related in particular to VEGF-A signaling. This review of the litterature has allowed us to expose the current landscape of CT± TT ± ICIs combinations under evaluation in some solid tumors by specifying the biological mechanisms impacted (Figure 2). We were able to show that pre-clinical and clinical research on this topic is very active and that a large number of clinical trials are underway with encouraging preliminary results (Figure 3). Nevertheless, we could notice that chemotherapies and targeted therapies are likely to be beneficial but sometimes harmful on the tumor microenvironment, making it difficult to predict in advance the final effect of such combinations on the immune system and sensitization to immunotherapy. We were also able to note that the doses were also likely to affect positively or negatively the anti-tumor immune response, as well as the sequence of administration of these different treatments or the use of premedication’s to chemotherapy. This observation leads us to briefly discuss the timing of drugs administrations in such combinations. In a clinical trial involving patients with advanced melanoma, Vilain et al. stated that changes in immune profiling in the TME during the early phase of immunotherapy treatment could better predict the treatment success rate, in an actively suppressed immune system against cancer.240 This team underlines the potential interest of privileging immunotherapy before chemotherapy, notably by creating a more permissive tumor microenvironment and by increasing tumor antigenicity. Other and our teams emphasize the importance of chemotherapy prior to the addition of immunotherapy particularly in a context of colorectal cancer. Moreover, Parra and colleagues compared the TME changes on surgically resectable NSCLC patients, according to whether they had received neoadjuvant chemotherapy.241 It was confirmed that the overall immune cell infiltration increased and the fraction of T cells were significantly elevated in the tissues of patients who underwent neoadjuvant chemotherapy. Therefore, it may be necessary to consider the sequential administration of immunotherapy targeting the altered immune contexture after neoadjuvant chemotherapy. Liu et al. demonstrated increased T cell infiltration and PD-1/PD-L1 expression in the TME through the combination of cisplatin and crizotinib in orthotopic murine NSCLC models and reported a sensitizing effect of anti–PD-1 treatment.242 Thus, these results suggest that sequential ICI administration after chemotherapy with or without TT may become a promising strategy to optimized anti-tumor strategy. Altogether, the sequence of administration of immunotherapies should be thoroughly deliberated in view of the cancer-immunity cycle and therapy-induced dynamic changes in the immune contexture of the TME.243 It is therefore essential to continue to explore this research axis and to study the “cancer-cell” and “immune-cell dependent” effects of TT and CT in order to find new therapeutic associations to amplify the effectiveness of immunotherapy in cancer.
Figure 2.
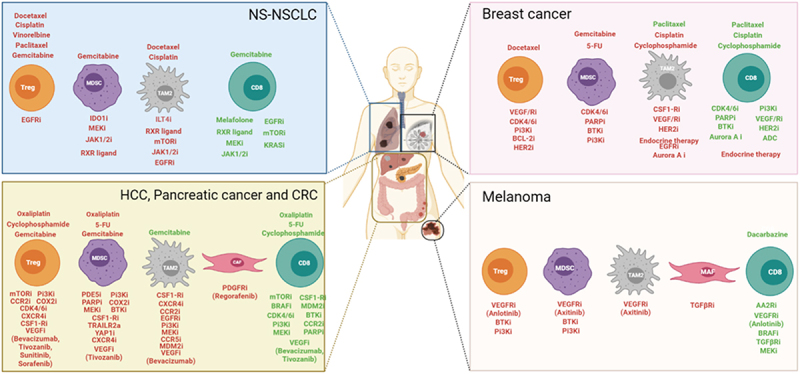
Summary of the effects of chemotherapy and targeted therapy on the tumor microenvironment in NS-NSCLC, breast cancer, pancreatic cancer, colorectal cancer, hepatocellular carcinoma and melanoma Chemotherapies and targeted therapies are separated in each situation. Treatments that modulate upwards are represented in green, and those that modulate downwards are represented in red. NS-NSCLC: Non squamous non-small cell carcinoma, HCC: Hepatocellular carcinoma, CRC: Colorectal cancer, Treg: regulatory T celsl, MDSC: myeloid-derived suppressor cell, TAM2: tumor associated macrophages of phenotype 2, CAF: cancer associated fibroblast, MAF: Melanoma associated fibroblast, CD8: CD8 T cell, i: inhibitor, a: agonist, 5-FU: 5-Fluorouracil. (Made with Biorender).
Figure 3.

Combination of targeted therapy and immunotherapy with or without chemotherapy in different solids tumors. Heatmap represents recent or ongoing clinical trials combining targeted therapy (classified in function of target pathway), immunotherapy (anti-PD-(l)1 or anti-CTLA-4 or both) and chemotherapy. This non-exhaustive list was done in July 2022 (from ClinicalTrials.gov) within the main cancers presented in this review. NSCLC: non small cell lung carcinoma, SCLC: small cell lung carcinoma, BRCA: breast carcinoma, CCR: colorectal cancer, HCC: hepatocellular carcinoma, PANC: pancreatic cancer, MEL: melanoma. Color code is specified bellow the heat map.
Acknowledgments
We wish to thank Fiona Ecarnot, PhD (EA3920, University of Franche-Comté and Besançon, France) for English correction and helpful comments.
Funding Statement
R.B., L.G., L.K., F.G, S.L. and E.L. are supported by the “Ligue Nationale contre le Cancer (Labelled teams)”, the “Ligue Nationale contre le Cancer Grand-Est”, the “Institut National du Cancer (INCa)”, the “Association pour la Recherche sur le Cancer (ARC)”, the “Fondation AMGEN”, the “LabEx LipSTIC”, the “Region Bourgogne-Franche-Comté (BFC)”, and the “I-Site-BFC program”. This study was also supported by the Georges-François Leclerc Cancer Center.
Abbreviations
| Immune checkpoint inhibitors (ICIs) |
| Tumor microenvironment (TME) |
| Regulatory T lymphocytes (Treg) |
| Myeloid-derived suppressor cells (MDSCs) |
| Tumor-associated macrophages (TAMs) |
| Chemotherapies (CT) |
| Targeted Therapies (TT) |
| Tyrosine Kinase Inhibitor (TKI) |
| Monoclonal antibodies (mAb) |
| Immunogenic cell death (ICD) |
| High-Mobility Group Box 1 (HMGB1) |
| Natural Killer (NK) |
| Dendritic cells (DCs) |
| Cancer Associated Fibroblast (CAF) |
| Indoleamine 2,3-DiOxygenase (IDO) |
| Inducible Nitric Oxide Synthase (iNOS) |
| Arginase 1 (Arg1) |
| Cyclo-oxygenase (COX) |
| Prostaglandin E2 (PGE2) |
| Signal Transducer and Activator of Transcription 3 (STAT3) |
| Vascular Endothelial Growth Factor A (VEGF-A) |
| Programmed cell Death-Ligand 1 (PD-L1) |
| Programmed cell Death protein 1 (PD-1) |
| Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) |
| T-cell immunoglobulin and immunoreceptor Tyrosine-based inhibitory domain (TIGIT) |
| Non-Squamous-Non-Small Cell Lung Cancer (NS-NSCLC) |
| Breast Cancer (BC) |
| HepatoCarcinoma Cancer (HCC) |
| ColoRectal Cancer (CCR) |
| Peripheral Blood Mononuclear Cells (PBMCs) |
| Cytotoxic T lymphocyte (CTL) |
| Immunoglobulin-like transcript 4 (ILT4) |
| Retinoid X receptor (RXR) |
| Cisplatin-Pemetrexed (CP) |
| Optineurin (OPTN) |
| Janus kinase 1/2 (JAK 1/2) |
| Tumor-infiltrating lymphocytes (TILs) |
| Breast Cancer (BC) |
| Triple-negative breast cancer (TNBC) |
| Hormone-receptor breast cancer (HR+BC) |
| Human Epidermal Growth Factor Receptor-2 (HER-2) |
| Antibody-drug-conjugates (ADC) |
| Cyclin-dependent kinases 4 and 6 inhibitors (CDK4/6i) |
| Poly (ADP-ribose) polymerase (PARP) |
| Colony-stimulating factor 1 receptor axis (CSF1/CSF1R) |
| Interferon (IFN) |
| Antibody-dependent cell-mediated cytotoxicity (ADCC) |
| Temporary Use Authorization (ATU) |
| Phosphodiesterase type 5 (PDE5) |
| Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) |
| Chemokine Receptor type 4 (CXCR4) |
| C-Chemokine receptor type 2 (CCR2) |
| Microsatellite-instable (MSI+) |
| Protein tyrosine phosphatase-2 (SHP2) |
| C-C chemokine receptor type 5 (CCR5) |
| Mouse Double Minute 2 (MDM2) |
| Yes‐associated protein 1 (YAP1) |
Disclosure statement
F.G, and S.L received fees for oral communication and travel grants from Lilly, Pfizer, Novartis, Bristol-Myers Squibb, Roche, Ipsen, Janssen Oncology, and Sanofi. Other authors declare no conflict of interest.
Author Contributions
Writing review and editing, R.B, L.G., L.K., F.G, S.L and E.L.; supervision E.L; L.G and R.B prepared all figures and tables. All authors have read and agreed to the published version of the manuscript.
References
- 1.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S.. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–29. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 2.Viaud S, Daillère R, Boneca IG, Lepage P, Langella P, Chamaillard M, Pittet MJ, Ghiringhelli F, Trinchieri G, Goldszmid R. Gut microbiome and anticancer immune response: Really hot Shot! Cell Death Differ. 2015;22(2):199–214. doi: 10.1038/cdd.2014.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillère R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ. The intestinal microbiota modulates the anticancer immune effects of Cyclophosphamide. Science. 2013;342(6161):971–976. doi: 10.1126/science.1240537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW. Molecular mechanisms of cell death: Recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Galluzzi L, Humeau J, Buqué A, Zitvogel L, Kroemer G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat Rev Clin Oncol. 2020;17(12):725–741. doi: 10.1038/s41571-020-0413-z. [DOI] [PubMed] [Google Scholar]
- 6.Petroni G, Buqué A, Zitvogel L, Kroemer G, Galluzzi L. Immunomodulation by targeted anticancer agents. Cancer Cell. 2021;39(3):310–345. doi: 10.1016/j.ccell.2020.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer. 2021;124(2):359–367. doi: 10.1038/s41416-020-01048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li T, Wu B, Yang T, Zhang L, Jin K. the outstanding antitumor capacity of CD4+ T helper Lymphocytes. Biochim Biophys Acta Rev Cancer. 2020;1874(2):188439. doi: 10.1016/j.bbcan.2020.188439. [DOI] [PubMed] [Google Scholar]
- 9.Ben-Shmuel A, Biber G, Barda-Saad M. Unleashing Natural Killer Cells in the Tumor microenvironment-the next generation of immunotherapy? Front Immunol. 2020;11:275. doi: 10.3389/fimmu.2020.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marciscano AE, Anandasabapathy N. The role of dendritic cells in Cancer and Anti-Tumor Immunity. Semin Immunol. 2021;52:101481. doi: 10.1016/j.smim.2021.101481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Togashi Y, Shitara K, Nishikawa H. Regulatory T Cells in Cancer Immunosuppression - Implications for Anticancer Therapy. Nat Rev Clin Oncol. 2019;16(6):356–371. doi: 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- 12.Szebeni GJ, Vizler C, Nagy LI, Kitajka K, Puskas LG. Pro-Tumoral Inflammatory Myeloid Cells as Emerging Therapeutic Targets. Int J Mol Sci. 2016;17(11):E1958. doi: 10.3390/ijms17111958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben Baruch B, Mantsur E, Franco-Barraza J, Blacher E, Cukierman E, Stein R. CD38 in cancer-associated fibroblasts promotes pro-Tumoral Activity. Lab Investig J Tech Methods Pathol. 2020;100(12):1517–1531. doi: 10.1038/s41374-020-0458-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allard B, Longhi MS, Robson SC, Stagg J. The ectonucleotidases CD39 and CD73: novel Checkpoint Inhibitor Targets. Immunol Rev. 2017;276(1):121–144. doi: 10.1111/imr.12528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moon YW, Hajjar J, Hwu P, Naing A. Targeting the Indoleamine 2,3-dioxygenase Pathway in Cancer. J Immunother Cancer. 2015;3(1):51. doi: 10.1186/s40425-015-0094-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weisser SB, McLarren KW, Kuroda E, Sly LM. Generation and characterization of murine alternatively activated Macrophages. Methods Mol Biol Clifton NJ. 2013;946:225–239. doi: 10.1007/978-1-62703-128-8_14. [DOI] [PubMed] [Google Scholar]
- 17.Wang D, DuBois RN. The role of prostaglandin E(2) in Tumor-Associated immunosuppression. Trends Mol Med. 2016;22(1):1–3. doi: 10.1016/j.molmed.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonavita E, Bromley CP, Jonsson G, Pelly VS, Sahoo S, Walwyn-Brown K, Mensurado S, Moeini A, Flanagan E, Bell CR. Antagonistic inflammatory phenotypes dictate tumor fate and response to immune checkpoint blockade. Immunity. 2020;53(6):1215–1229.e8. doi: 10.1016/j.immuni.2020.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, Chakravarty P, Girotti MR, Marais R, Quezada SA. Cyclooxygenase-dependent Tumor Growth through evasion of immunity. Cell. 2015;162(6):1257–1270. doi: 10.1016/j.cell.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kitamura H, Ohno Y, Toyoshima Y, Ohtake J, Homma S, Kawamura H, Takahashi N, Taketomi A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017;108(10):1947–1952. doi: 10.1111/cas.13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Park S-J, Nakagawa T, Kitamura H, Atsumi T, Kamon H, Sawa S-I, Kamimura D, Ueda N, Iwakura Y, Ishihara K. IL-6 Regulates in Vivo Dendritic Cell Differentiation through STAT3 Activation. J Immunol Baltim Md. 2004;1950(173):3844–3854. doi: 10.4049/jimmunol.173.6.3844. [DOI] [PubMed] [Google Scholar]
- 22.Schneider BP, Miller KD. Angiogenesis of breast cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23(8):1782–1790. doi: 10.1200/JCO.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 23.Voron T, Colussi O, Marcheteau E, Pernot S, Nizard M, Pointet A-L, Latreche S, Bergaya S, Benhamouda N, Tanchot C. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T Cells in Tumors. J Exp Med. 2015;212(2):139–148. doi: 10.1084/jem.20140559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ohm JE, Carbone DP. VEGF as a mediator of Tumor-associated immunodeficiency. Immunol Res. 2001;23(2–3):263–272. doi: 10.1385/IR:23:2-3:263. [DOI] [PubMed] [Google Scholar]
- 25.Huang H, Langenkamp E, Georganaki M, Loskog A, Fuchs PF, Dieterich LC, Kreuger J, Dimberg A. VEGF suppresses T-Lymphocyte infiltration in the Tumor microenvironment through Inhibition of NF-ΚB-Induced Endothelial Activation. FASEB J Off Publ Fed Am Soc Exp Biol. 2015;29:227–238. doi: 10.1096/fj.14-250985. [DOI] [PubMed] [Google Scholar]
- 26.Ramjiawan RR, Griffioen AW, Duda DG. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis. 2017;20(2):185–204. doi: 10.1007/s10456-017-9552-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang J, Yan J, Liu B. Targeting VEGF/VEGFR to Modulate Antitumor Immunity. Front Immunol. 2018;9:978. doi: 10.3389/fimmu.2018.00978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can treg cells be a New Therapeutic Target? Cancer Sci. 2019;110(7):2080–2089. doi: 10.1111/cas.14069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 Antigen on the surface of stimulated mouse T and B Lymphocytes. Int Immunol. 1996;8(5):765–772. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 30.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 31.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. Pillars Article: CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405–413. doi: 10.1016/1074-7613(94)90071-X. [DOI] [PubMed] [Google Scholar]
- 32.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawashima S, Inozume T, Kawazu M, Ueno T, Nagasaki J, Tanji E, Honobe A, Ohnuma T, Kawamura T, Umeda Y. TIGIT/CD155 axis mediates resistance to immunotherapy in patients with melanoma with the inflamed Tumor Microenvironment. J Immunother Cancer. 2021;9(11):e003134. doi: 10.1136/jitc-2021-003134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oyama R, Kanayama M, Mori M, Matsumiya H, Taira A, Shinohara S, Takenaka M, Yoneda K, Kuroda K, Tanaka F. CD155 expression and its clinical significance in non-small cell Lung Cancer. Oncol Lett. 2022;23(5):166. doi: 10.3892/ol.2022.13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kučan Brlić P, Lenac Roviš T, Cinamon G, Tsukerman P, Mandelboim O, Jonjić S. Targeting PVR (CD155) and its receptors in Anti-Tumor Therapy. Cell Mol Immunol. 2019;16(1):40–52. doi: 10.1038/s41423-018-0168-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Patel KR, Martinez A, Stahl JM, Logan SJ, Perricone AJ, Ferris MJ, Buchwald ZS, Chowdhary M, Delman KA, Monson DK. Increase in PD-L1 expression after pre-operative radiotherapy for soft Tissue Sarcoma. Oncoimmunology. 2018;7(7):e1442168. doi: 10.1080/2162402X.2018.1442168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boustani J, Lecoester B, Baude J, Latour C, Adotevi O, Mirjolet C, Truc G. Anti-PD-1/Anti-PD-l1 drugs and radiation therapy: Combinations and optimization strategies. Cancers. 2021;13(19):4893. doi: 10.3390/cancers13194893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y, Beer DG, Powell CA, Riely GJ, Van Schil PE. International association for the study of Lung Cancer/American Thoracic Society/European Respiratory Society International Multidisciplinary Classification of Lung Adenocarcinoma. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. 2011;6:244–285. doi: 10.1097/JTO.0b013e318206a221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dogan S, Shen R, Ang DC, Johnson ML, D’Angelo SP, Paik PK, Brzostowski EB, Riely GJ, Kris MG, Zakowski MF. Molecular epidemiology of EGFR and KRAS mutations in 3,026 Lung Adenocarcinomas: Higher susceptibility of women to smoking-related KRAS-Mutant Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2012;18(22):6169–6177. doi: 10.1158/1078-0432.CCR-11-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Barta JA, Powell CA, Wisnivesky JP. Global Epidemiology of Lung Cancer. Ann Glob Health. 2019;85(8). doi: 10.5334/aogh.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, Zhou C, Reungwetwattana T, Cheng Y, Chewaskulyong B. Overall survival with osimertinib in untreated, EGFR-Mutated advanced NSCLC. N Engl J Med. 2020;382(1):41–50. doi: 10.1056/NEJMoa1913662. [DOI] [PubMed] [Google Scholar]
- 42.Tomasini P, Egea J, Souquet-Bressand M, Greillier L, Barlesi F. Alectinib in the treatment of ALK-positive metastatic non-small cell lung cancer: Clinical trial evidence and experience with a focus on brain metastases. Ther Adv Respir Dis. 2019;13:1753466619831906. doi: 10.1177/1753466619831906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong Z-Y, Zhang J-T, Liu S-Y, Su J, Zhang C, Xie Z, Zhou Q, Tu H-Y, Xu C-R, Yan L-X. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Oncoimmunology. 2017;6(11):e1356145. doi: 10.1080/2162402X.2017.1356145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S. Pembrolizumab versus chemotherapy for PD-L1-positive non-small-cell lung cancer. N Engl J Med. 2016;375(19):1823–1833. doi: 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 45.Gadgeel S, Rodríguez-Abreu D, Speranza G, Esteban E, Felip E, Dómine M, Hui R, Hochmair MJ, Clingan P, Powell SF. Updated analysis from KEYNOTE-189: Pembrolizumab or placebo plus pemetrexed and platinum for previously untreated metastatic nonsquamous non-small-cell lung cancer. J Clin Oncol Off J Am Soc Clin Oncol. 2020;38(14):1505–1517. doi: 10.1200/JCO.19.03136. [DOI] [PubMed] [Google Scholar]
- 46.Wang Z, Till B, Gao Q. Chemotherapeutic agent-mediated elimination of myeloid-derived suppressor cells. Oncoimmunology. 2017;6:e1331807. doi: 10.1080/2162402X.2017.1331807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Han Y, Guo Q, Zhang M, Cao X. Cancer-expanded myeloid-derived suppressor cells induce anergy of nk cells through membrane-bound TGF-beta 1. J Immunol Baltim Md. 2009;1950(182):240–249. doi: 10.4049/jimmunol.182.1.240. [DOI] [PubMed] [Google Scholar]
- 49.Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol Baltim Md. 2007;1950(179):977–983. doi: 10.4049/jimmunol.179.2.977. [DOI] [PubMed] [Google Scholar]
- 50.Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9(7–8):900–909. doi: 10.1016/j.intimp.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 51.Sawant A, Schafer CC, Jin TH, Zmijewski J, Tse HM, Roth J, Sun Z, Siegal GP, Thannickal VJ, Grant SC. Enhancement of antitumor immunity in lung cancer by targeting myeloid-derived suppressor cell pathways. Cancer Res. 2013;73(22):6609–6620. doi: 10.1158/0008-5472.CAN-13-0987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qu X, Felder MAR, Perez Horta Z, Sondel PM, Rakhmilevich AL. Antitumor effects of anti-CD40/CpG immunotherapy combined with gemcitabine or 5-fluorouracil chemotherapy in the B16 melanoma model. Int Immunopharmacol. 2013;17(4):1141–1147. doi: 10.1016/j.intimp.2013.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feng P-H, Yu C-T, Wu C-Y, Lee M-J, Lee W-H, Wang L-S, Lin S-M, Fu J-F, Lee K-Y, Yen T-H. Tumor-associated macrophages in stage IIIA PN2 non-small cell lung cancer after neoadjuvant chemotherapy and surgery. Am J Transl Res. 2014;6:593–603. [PMC free article] [PubMed] [Google Scholar]
- 54.Chiu K-C, Lee C-H, Liu S-Y, Chou Y-T, Huang R-Y, Huang S-M, Shieh Y-S. Polarization of tumor-associated macrophages and gas6/Axl signaling in oral squamous cell carcinoma. Oral Oncol. 2015;51(7):683–689. doi: 10.1016/j.oraloncology.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 55.Chen JJW, Lin Y-C, Yao P-L, Yuan A, Chen H-Y, Shun C-T, Tsai M-F, Chen C-H, Yang P-C. Tumor-associated macrophages: The double-edged sword in cancer progression. J Clin Oncol Off J Am Soc Clin Oncol. 2005;23(5):953–964. doi: 10.1200/JCO.2005.12.172. [DOI] [PubMed] [Google Scholar]
- 56.Buehler M, Tse B, Leboucq A, Jacob F, Caduff R, Fink D, Goldstein DR, Heinzelmann-Schwarz V. Meta-analysis of microarray data identifies GAS6 expression as an independent predictor of poor survival in Ovarian Cancer. BioMed Res Int. 2013;2013:238284. doi: 10.1155/2013/238284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mills CD. M1 and M2 macrophages: oracles of health and disease. Crit Rev Immunol. 2012;32(6):463–488. doi: 10.1615/critrevimmunol.v32.i6.10. [DOI] [PubMed] [Google Scholar]
- 58.Li J-Y, Duan X-F, Wang L-P, Xu Y-J, Huang L, Zhang T-F, Liu J-Y, Li F, Zhang Z, Yue D-L. Selective depletion of regulatory T cell subsets by docetaxel treatment in patients with nonsmall cell lung cancer. J Immunol Res. 2014;2014:286170. doi: 10.1155/2014/286170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen C, Chen Z, Chen D, Zhang B, Wang Z, Le H. Suppressive effects of gemcitabine plus cisplatin chemotherapy on regulatory T cells in nonsmall-cell lung cancer. J Int Med Res. 2015;43(2):180–187. doi: 10.1177/0300060514561504. [DOI] [PubMed] [Google Scholar]
- 60.Gameiro SR, Caballero JA, Higgins JP, Apelian D, Hodge JW. Exploitation of differential homeostatic proliferation of T-Cell subsets following Chemotherapy to enhance the efficacy of vaccine-mediated antitumor responses. Cancer Immunol Immunother CII. 2011;60(9):1227–1242. doi: 10.1007/s00262-011-1020-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roselli M, Cereda V, Di Bari MG, Formica V, Spila A, Jochems C, Farsaci B, Donahue R, Gulley JL, Schlom J. Effects of conventional therapeutic interventions on the number and function of regulatory t cells. Oncoimmunology. 2013;2(10):e27025. doi: 10.4161/onci.27025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weiss E-M, Schmidt A, Vobis D, Garbi N, Lahl K, Mayer CT, Sparwasser T, Ludwig A, Suri-Payer E, Oberle N. Foxp3-mediated suppression of CD95L expression confers resistance to activation-induced cell death in regulatory T cells. J Immunol Baltim Md. 2011;1950(187):1684–1691. doi: 10.4049/jimmunol.1002321. [DOI] [PubMed] [Google Scholar]
- 63.Zhang L, Dermawan K, Jin M, Liu R, Zheng H, Xu L, Zhang Y, Cai Y, Chu Y, Xiong S. Differential impairment of regulatory t cells rather than effector t cells by paclitaxel-based chemotherapy. Clin Immunol Orlando Fla. 2008;129(2):219–229. doi: 10.1016/j.clim.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 64.Takahashi K, Saito H, Hasegawa Y, Ando M, Yamamoto M, Kojima E, Sugino Y, Kimura T, Nomura F, Ogasawara T. First-line gefitinib therapy for elderly patients with non-small cell lung cancer harboring EGFR mutation: Central Japan Lung Study Group 0901. Cancer Chemother Pharmacol. 2014;74(4):721–727. doi: 10.1007/s00280-014-2548-z. [DOI] [PubMed] [Google Scholar]
- 65.Soria J-C, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, Dechaphunkul A, Imamura F, Nogami N, Kurata T. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378(2):113–125. doi: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 66.Wang S, Zhang Y, Wang Y, Ye P, Li J, Li H, Ding Q, Xia J. Amphiregulin confers regulatory t cell suppressive function and tumor invasion via the EGFR/GSK-3β/Foxp3 Axis. J Biol Chem. 2016;291(40):21085–21095. doi: 10.1074/jbc.M116.717892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jia Y, Li X, Jiang T, Zhao S, Zhao C, Zhang L, Liu X, Shi J, Qiao M, Luo J. EGFR-targeted therapy alters the tumor microenvironment in EGFR-driven lung tumors: Implications for combination therapies. Int J Cancer. 2019;145(5):1432–1444. doi: 10.1002/ijc.32191. [DOI] [PubMed] [Google Scholar]
- 68.Peng H, Chen B, Huang W, Tang Y, Jiang Y, Zhang W, Huang Y. Reprogramming tumor-associated macrophages to reverse EGFRT790M resistance by Dual-targeting codelivery of gefitinib/vorinosta. Nano Lett. 2017;17(12):7684–7690. doi: 10.1021/acs.nanolett.7b03756. [DOI] [PubMed] [Google Scholar]
- 69.Mascia F, Schloemann DT, Cataisson C, McKinnon KM, Krymskaya L, Wolcott KM, Yuspa SH. Cell autonomous or systemic EGFR blockade alters the immune-environment in squamous cell carcinomas. Int J Cancer. 2016;139(11):2593–2597. doi: 10.1002/ijc.30376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tang H, Liu Y, Wang C, Zheng H, Chen Y, Liu W, Chen X, Zhang J, Chen H, Yang Y. Inhibition of COX-2 and EGFR by melafolone improves anti-PD-1 therapy through vascular normalization and PD-L1 downregulation in lung cancer. J Pharmacol Exp Ther. 2019;368(3):401–413. doi: 10.1124/jpet.118.254359. [DOI] [PubMed] [Google Scholar]
- 71.Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/MPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cell. Proc Natl Acad Sci U S A. 2017;114(5):1117–1122. doi: 10.1073/pnas.1612920114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shimizu K, Okita R, Saisho S, Maeda A, Nojima Y, Nakata M. Prognostic value of cox-2 and PD-l1 expression and its relationship with tumor-infiltrating lymphocytes in resected lung adenocarcinoma. Cancer Manag Res. 2017;9:741–750. doi: 10.2147/CMAR.S146897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen X, Gao A, Zhang F, Yang Z, Wang S, Fang Y, Li J, Wang J, Shi W, Wang L. ILT4 inhibition prevents TAM- and dysfunctional t cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in NSCLC with EGFR activation. Theranostics. 2021;11(7):3392–3416. doi: 10.7150/thno.52435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee JW, Zhang Y, Eoh KJ, Sharma R, Sanmamed MF, Wu J, Choi J, Park HS, Iwasaki A, Kaftan E. The Combination of MEK inhibitor with immunomodulatory antibodies targeting programmed death 1 and programmed death ligand 1 results in prolonged survival in kras/P53-driven lung cancer. J Thorac Oncol Off Publ Int Assoc Study Lung Cancer. 2019;14:1046–1060. doi: 10.1016/j.jtho.2019.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Limagne E, Nuttin L, Thibaudin M, Jacquin E, Aucagne R, Bon M, Revy S, Barnestein R, Ballot E, Truntzer C. MEK inhibition overcomes chemoimmunotherapy resistance by inducing CXCL10 in cancer cells. Cancer Cell. 2022;40(2):136–152.e12. doi: 10.1016/j.ccell.2021.12.009. [DOI] [PubMed] [Google Scholar]
- 76.Allegrezza MJ, Rutkowski MR, Stephen TL, Svoronos N, Perales-Puchalt A, Nguyen JM, Payne KK, Singhal S, Eruslanov EB, Tchou J. Trametinib drives t-cell-dependent control of KRAS-mutated tumors by inhibiting pathological myelopoiesis. Cancer Res. 2016;76(21):6253–6265. doi: 10.1158/0008-5472.CAN-16-1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tang KH, Li S, Khodadadi-Jamayran A, Jen J, Han H, Guidry K, Chen T, Hao Y, Fedele C, Zebala JA. Combined Inhibition of SHP2 and CXCR1/2 promotes antitumor t-cell response in NSCLC. Cancer Discov. 2022;12(1):47–61. doi: 10.1158/2159-8290.CD-21-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Briere DM, Li S, Calinisan A, Sudhakar N, Aranda R, Hargis L, Peng DH, Deng J, Engstrom LD, Hallin J. The KRASG12C Inhibitor MRTX849 reconditions the tumor immune microenvironment and sensitizes tumors to checkpoint inhibitor therapy. Mol Cancer Ther. 2021;20(6):975–985. doi: 10.1158/1535-7163.MCT-20-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N. The Clinical KRAS(G12C) Inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575(7781):217–223. doi: 10.1038/s41586-019-1694-1. [DOI] [PubMed] [Google Scholar]
- 80.Zhu Z, Aref AR, Cohoon TJ, Barbie TU, Imamura Y, Yang S, Moody SE, Shen RR, Schinzel AC, Thai TC. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014;4(4):452–465. doi: 10.1158/2159-8290.CD-13-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mohrherr J, Haber M, Breitenecker K, Aigner P, Moritsch S, Voronin V, Eferl R, Moriggl R, Stoiber D, Győrffy B. JAK-stat inhibition impairs k-ras-driven lung adenocarcinoma progression. Int J Cancer. 2019;145(12):3376–3388. doi: 10.1002/ijc.32624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang Q, Zhang Y, Chen Y, Qian J, Zhang X, Yu K. A novel MTORC1/2 inhibitor (MTI-31) inhibits tumor growth, epithelial-mesenchymal transition, metastases, and improves antitumor immunity in preclinical models of lung cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2019;25(12):3630–3642. doi: 10.1158/1078-0432.CCR-18-2548. [DOI] [PubMed] [Google Scholar]
- 83.Moerland JA, Zhang D, Reich LA, Carapellucci S, Lockwood B, Leal AS, Krieger-Burke T, Aleiwi B, Ellsworth E, Liby KT. The novel rexinoid Msu-42011 is effective for the treatment of preclinical kras-driven lung cancer. Sci Rep. 2020;10(1):22244. doi: 10.1038/s41598-020-79260-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Du X, Tabeta K, Mann N, Crozat K, Mudd S, Beutler B. an essential role for rxr alpha in the development of Th2 responses. Eur J Immunol. 2005;35(12):3414–3423. doi: 10.1002/eji.200535366. [DOI] [PubMed] [Google Scholar]
- 85.Prendergast GC, Malachowski WJ, Mondal A, Scherle P, Muller AJ. Indoleamine 2,3-dioxygenase and its therapeutic inhibition in cancer. Int Rev Cell Mol Biol. 2018;336:175–203. doi: 10.1016/bs.ircmb.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li A, Barsoumian HB, Schoenhals JE, Cushman TR, Caetano MS, Wang X, Valdecanas DR, Niknam S, Younes AI, Li G. Indoleamine 2,3-dioxygenase 1 inhibition targets anti-PD1-resistant lung tumors by blocking myeloid-derived suppressor cells. Cancer Lett. 2018;431:54–63. doi: 10.1016/j.canlet.2018.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ledys F, Kalfeist L, Galland L, Limagne E, Ladoire S. Therapeutic associations comprising anti-PD-1/PD-L1 in breast cancer: Clinical challenges and perspectives. Cancers. 2021;13(23):5999. doi: 10.3390/cancers13235999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vincent J, Mignot G, Chalmin F, Ladoire S, Bruchard M, Chevriaux A, Martin F, Apetoh L, Rébé C, Ghiringhelli F. 5-fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced t cell-dependent antitumor immunity. Cancer Res. 2010;70(8):3052–3061. doi: 10.1158/0008-5472.CAN-09-3690. [DOI] [PubMed] [Google Scholar]
- 89.Galetto A, Buttiglieri S, Forno S, Moro F, Mussa A, Matera L. Drug- and cell-mediated antitumor cytotoxicities modulate cross-presentation of tumor antigens by myeloid dendritic cells. Anticancer Drugs. 2003;14:833–843. doi: 10.1097/00001813-200311000-00010. [DOI] [PubMed] [Google Scholar]
- 90.Nowak AK, Lake RA, Marzo AL, Scott B, Heath WR, Collins EJ, Frelinger JA, Robinson BWS. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T Cells. J Immunol Baltim Md. 2003;1950(170):4905–4913. doi: 10.4049/jimmunol.170.10.4905. [DOI] [PubMed] [Google Scholar]
- 91.Zhang Z, Yu X, Yu X, Wang Z, Wu P, Huang J. Anthracyclines potentiate anti-tumor immunity: A new opportunity for chemoimmunotherapy. Cancer Lett. 2015;369(2):331–335. doi: 10.1016/j.canlet.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 92.Ladoire S, Arnould L, Apetoh L, Coudert B, Martin F, Chauffert B, Fumoleau P, Ghiringhelli F. pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of Tumor-Infiltrating Foxp3+ regulatory T cells. Clinical Cancer Research. 2008;14(8):2413–2420. doi: 10.1158/1078-0432.CCR-07-4491. [DOI] [PubMed] [Google Scholar]
- 93.Demaria S, Volm MD, Shapiro RL, Yee HT, Oratz R, Formenti SC, Muggia F, Symmans WF. Development of tumor-infiltrating lymphocytes in breast cancer after neoadjuvant paclitaxel chemotherapy. Clin Cancer Res Off J Am Assoc Cancer Res. 2001;7:3025–3030. [PubMed] [Google Scholar]
- 94.Ono M, Tsuda H, Shimizu C, Yamamoto S, Shibata T, Yamamoto H, Hirata T, Yonemori K, Ando M, Tamura K. Tumor-infiltrating lymphocytes are correlated with response to neoadjuvant chemotherapy in triple-negative breast cancer. Breast Cancer Res Treat. 2012;132(3):793–805. doi: 10.1007/s10549-011-1554-7. [DOI] [PubMed] [Google Scholar]
- 95.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67. doi: 10.1158/2159-8274.CD-10-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tsavaris N, Kosmas C, Vadiaka M, Kanelopoulos P, Boulamatsis D. Immune changes in patients with advanced breast cancer undergoing chemotherapy with Taxanes. Br J Cancer. 2002;87(1):21–27. doi: 10.1038/sj.bjc.6600347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Carson WE, Shapiro CL, Crespin TR, Thornton LM, Andersen BL. cellular immunity in breast cancer patients completing taxane treatment. Clin Cancer Res Off J Am Assoc Cancer Res. 2004;10(10):3401–3409. doi: 10.1158/1078-0432.CCR-1016-03. [DOI] [PubMed] [Google Scholar]
- 98.Orecchioni S, Talarico G, Labanca V, Calleri A, Mancuso P, Bertolini F. Vinorelbine, cyclophosphamide and 5-fu effects on the circulating and intratumoural landscape of immune cells improve anti-PD-L1 efficacy in preclinical models of breast cancer and Lymphoma. Br J Cancer. 2018;118(10):1329–1336. doi: 10.1038/s41416-018-0076-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B. Metronomic cyclophosphamide regimepleten selectively Des CD4+CD25+ Regulatory T Cells and Restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother CII. 2007;56(5):641–648. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Park YH, Lal S, Lee JE, Choi Y-L, Wen J, Ram S, Ding Y, Lee S-H, Powell E, Lee SK. Chemotherapy Induces Dynamic Immune Responses in Breast Cancers That Impact Treatment Outcome. Nat Commun. 2020;11(1):6175. doi: 10.1038/s41467-020-19933-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, Denkert C, Park YH, Hui R, Harbeck N. Pembrolizumab for Early Triple-Negative Breast Cancer. New England Journal of Medicine. 2020;382(9):810–821. doi: 10.1056/NEJMoa1910549. [DOI] [PubMed] [Google Scholar]
- 102.Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, Diéras V, Henschel V, Molinero L, Chui SY. Atezolizumab plus Nab-Paclitaxel as First-Line Treatment for Unresectable, Locally Advanced or Metastatic Triple-Negative Breast Cancer (IMpassion130): updated Efficacy Results from a Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2020;21(1):44–59. doi: 10.1016/S1470-2045(19)30689-8. [DOI] [PubMed] [Google Scholar]
- 103.Miles D, Gligorov J, André F, Cameron D, Schneeweiss A, Barrios C, Xu B, Wardley A, Kaen D, Andrade L. Primary Results from IMpassion131, a Double-Blind, Placebo-Controlled, Randomised Phase III Trial of First-Line Paclitaxel with or without Atezolizumab for Unresectable Locally Advanced/Metastatic Triple-Negative Breast Cancer. Ann Oncol Off J Eur Soc Med Oncol. 2021;32(8):994–1004. doi: 10.1016/j.annonc.2021.05.801. [DOI] [PubMed] [Google Scholar]
- 104.Kalfeist L, Galland L, Ledys F, Ghiringhelli F, Limagne E, Ladoire S. Impact of Glucocorticoid Use in Oncology in the Immunotherapy Era. Cells. 2022;11(770):770. doi: 10.3390/cells11050770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Quintela-Fandino M, Holgado E, Manso L, Morales S, Bermejo B, Colomer R, Apala JV, Blanco R, Muñoz M, Caleiras E. Immuno-Priming Durvalumab with Bevacizumab in HER2-Negative Advanced Breast Cancer: a Pilot Clinical Trial. Breast Cancer Res BCR. 2020;22(1):124. doi: 10.1186/s13058-020-01362-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang Q, Gao J, Di W, Wu X. Anti-Angiogenesis Therapy Overcomes the Innate Resistance to PD-1/PD-L1 Blockade in VEGFA-Overexpressed Mouse Tumor Models. Cancer Immunol Immunother CII. 2020;69(9):1781–1799. doi: 10.1007/s00262-020-02576-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang Y, Yuan J, Righi E, Kamoun WS, Ancukiewicz M, Nezivar J, Santosuosso M, Martin JD, Martin MR, Vianello F. Vascular Normalizing Doses of Antiangiogenic Treatment Reprogram the Immunosuppressive Tumor Microenvironment and Enhance Immunotherapy. Proc Natl Acad Sci U S A. 2012;109(43):17561–17566. doi: 10.1073/pnas.1215397109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Reguera-Nuñez E, Xu P, Chow A, Man S, Hilberg F, Kerbel RS. Therapeutic Impact of Nintedanib with Paclitaxel and/or a PD-L1 Antibody in Preclinical Models of Orthotopic Primary or Metastatic Triple Negative Breast Cancer. J Exp Clin Cancer Res. 2019;38(1):16. doi: 10.1186/s13046-018-0999-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, Cabaret V, Fermeaux V, Bertheau P, Garnier J. Trastuzumab-Based Treatment of HER2-Positive Breast Cancer: an Antibody-Dependent Cellular Cytotoxicity Mechanism? Br J Cancer. 2006;94(2):259–267. doi: 10.1038/sj.bjc.6602930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gennari R, Menard S, Fagnoni F, Ponchio L, Scelsi M, Tagliabue E, Castiglioni F, Villani L, Magalotti C, Gibelli N. Pilot Study of the Mechanism of Action of Preoperative Trastuzumab in Patients with Primary Operable Breast Tumors Overexpressing HER2. Clin Cancer Res Off J Am Assoc Cancer Res. 2004;10(17):5650–5655. doi: 10.1158/1078-0432.CCR-04-0225. [DOI] [PubMed] [Google Scholar]
- 111.Horlock C, Stott B, Dyson PJ, Morishita M, Coombes RC, Savage P, Stebbing J. The Effects of Trastuzumab on the CD4+CD25+FoxP3+ and CD4+IL17A+ T-Cell Axis in Patients with Breast Cancer. Br J Cancer. 2009;100(7):1061–1067. doi: 10.1038/sj.bjc.6604963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bloom MJ, Jarrett AM, Triplett TA, Syed AK, Davis T, Yankeelov TE, Sorace AG. Anti-HER2 Induced Myeloid Cell Alterations Correspond with Increasing Vascular Maturation in a Murine Model of HER2+ Breast Cancer. BMC Cancer. 2020;20(1):359. doi: 10.1186/s12885-020-06868-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xu M, Liu M, Du X, Li S, Li H, Li X, Li Y, Wang Y, Qin Z, Fu Y-X. Intratumoral Delivery of IL-21 Overcomes Anti-Her2/Neu Resistance through Shifting Tumor-Associated Macrophages from M2 to M1 Phenotype. J Immunol Baltim Md. 2015;1950(194):4997–5006. doi: 10.4049/jimmunol.1402603. [DOI] [PubMed] [Google Scholar]
- 114.Griguolo G, Serna G, Pascual T, Fasani R, Guardia X, Chic N, Paré L, Pernas S, Muñoz M, Oliveira M. Immune Microenvironment Characterisation and Dynamics during Anti-HER2-Based Neoadjuvant Treatment in HER2-Positive Breast Cancer. NPJ Precis Oncol. 2021;5(1):23. doi: 10.1038/s41698-021-00163-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Modi S. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Metastatic Breast Cancer: plain Language Summary of the DESTINY-Breast01 Study. Future Oncol Lond Engl. 2021;17(26):3415–3423. doi: 10.2217/fon-2021-0427. [DOI] [PubMed] [Google Scholar]
- 116.D’Amico L, Menzel U, Prummer M, Müller P, Buchi M, Kashyap A, Haessler U, Yermanos A, Gébleux R, Briendl M. A Novel Anti-HER2 Anthracycline-Based Antibody-Drug Conjugate Induces Adaptive Anti-Tumor Immunity and Potentiates PD-1 Blockade in Breast Cancer. J Immunother Cancer. 2019;7(1):16. doi: 10.1186/s40425-018-0464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wang L, Gao Y, Zhang G, Li D, Wang Z, Zhang J, Hermida LC, He L, Wang Z, Si J. Enhancing KDM5A and TLR Activity Improves the Response to Immune Checkpoint Blockade. Sci Transl Med. 2020;12(560):eaax2282. doi: 10.1126/scitranslmed.aax2282. [DOI] [PubMed] [Google Scholar]
- 118.Emens L, Hunder N, Klempner S, Hamilton E, Beeram M, Odegard V, Hamke S. 317 A Phase 1/1b Study of SBT6050, a HER2-Directed Monoclonal Antibody Conjugated to a Toll-like Receptor 8 Agonist, in Subjects with Advanced HER2-Expressing Solid Tumors. J Immunother Cancer. 2020;8(1). doi: 10.1136/jitc-2020-SITC2020.0317. [DOI] [Google Scholar]
- 119.Svensson S, Abrahamsson A, Rodriguez GV, Olsson A-K, Jensen L, Cao Y, Dabrosin C. CCL2 and CCL5 Are Novel Therapeutic Targets for Estrogen-Dependent Breast Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2015;21(16):3794–3805. doi: 10.1158/1078-0432.CCR-15-0204. [DOI] [PubMed] [Google Scholar]
- 120.Nalbandian G, Paharkova-Vatchkova V, Mao A, Nale S, Kovats S. The Selective Estrogen Receptor Modulators, Tamoxifen and Raloxifene, Impair Dendritic Cell Differentiation and Activation. J Immunol Baltim Md. 2005;1950(175):2666–2675. doi: 10.4049/jimmunol.175.4.2666. [DOI] [PubMed] [Google Scholar]
- 121.Behjati S, Frank MH. The Effects of Tamoxifen on Immunity. Curr Med Chem. 2009;16(24):3076–3080. doi: 10.2174/092986709788803042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chan MSM, Wang L, Felizola SJA, Ueno T, Toi M, Loo W, Chow LWC, Suzuki T, Sasano H. Changes of Tumor Infiltrating Lymphocyte Subtypes before and after Neoadjuvant Endocrine Therapy in Estrogen Receptor-Positive Breast Cancer Patients–an Immunohistochemical Study of Cd8+ and Foxp3+ Using Double Immunostaining with Correlation to the Pathobiological Response of the Patients. Int J Biol Markers. 2012;27(4):e295–304. doi: 10.5301/JBM.2012.10439. [DOI] [PubMed] [Google Scholar]
- 123.Generali D, Bates G, Berruti A, Brizzi MP, Campo L, Bonardi S, Bersiga A, Allevi G, Milani M, Aguggini S. Immunomodulation of FOXP3+ Regulatory T Cells by the Aromatase Inhibitor Letrozole in Breast Cancer Patients. Clin Cancer Res Off J Am Assoc Cancer Res. 2009;15(3):1046–1051. doi: 10.1158/1078-0432.CCR-08-1507. [DOI] [PubMed] [Google Scholar]
- 124.Teo ZL, Versaci S, Dushyanthen S, Caramia F, Savas P, Mintoff CP, Zethoven M, Virassamy B, Luen SJ, McArthur GA. Combined CDK4/6 and PI3Kα Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res. 2017;77(22):6340–6352. doi: 10.1158/0008-5472.CAN-17-2210. [DOI] [PubMed] [Google Scholar]
- 125.Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, Khan N, Ubellacker JM, Xie S, Metzger-Filho O. CDK4/6 Inhibition Triggers Anti-Tumour Immunity. Nature. 2017;548(7668):471–475. doi: 10.1038/nature23465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Whittle JR, Vaillant F, Surgenor E, Policheni AN, Giner G, Capaldo BD, Chen H-R, Liu HK, Dekkers JF, Sachs N. Dual Targeting of CDK4/6 and BCL2 Pathways Augments Tumor Response in Estrogen Receptor-Positive Breast Cancer. Clin Cancer Res Off J Am Assoc Cancer Res. 2020;26(15):4120–4134. doi: 10.1158/1078-0432.CCR-19-1872. [DOI] [PubMed] [Google Scholar]
- 127.Huang H, Zhou J, Chen H, Li J, Zhang C, Jiang X, Ni C. The Immunomodulatory Effects of Endocrine Therapy in Breast Cancer. J Exp Clin Cancer Res CR. 2021;40(1):19. doi: 10.1186/s13046-020-01788-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tutt ANJ, Garber JE, Kaufman B, Viale G, Fumagalli D, Rastogi P, Gelber RD, de Azambuja E, Fielding A, Balmaña J. Adjuvant Olaparib for Patients with BRCA1 – or BRCA -Mutated Breast Cancer. N Engl J Med. 2021;384(25):2394–2405. doi: 10.1056/NEJMoa2105215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, Delaloge S, Li W, Tung N, Armstrong A. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N Engl J Med. 2017;377(6):523–533. doi: 10.1056/NEJMoa1706450. [DOI] [PubMed] [Google Scholar]
- 130.Parkes EE, Savage KI, Lioe T, Boyd C, Halliday S, Walker SM, Lowry K, Knight L, Buckley NE, Grogan A. Activation of a CGAS-STING-Mediated Immune Response Predicts Response to Neoadjuvant Chemotherapy in Early Breast Cancer. Br J Cancer. 2022;126(2):247–258. doi: 10.1038/s41416-021-01599-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Flood BA, Higgs EF, Li S, Luke JJ, Gajewski TF. STING Pathway Agonism as a Cancer Therapeutic. Immunol Rev. 2019;290(1):24–38. doi: 10.1111/imr.12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ding L, Kim H-J, Wang Q, Kearns M, Jiang T, Ohlson CE, Li BB, Xie S, Liu JF, Stover EH. PARP Inhibition Elicits STING-Dependent Antitumor Immunity in Brca1-Deficient Ovarian Cancer. Cell Rep. 2018;25(11):2972–2980.e5. doi: 10.1016/j.celrep.2018.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang D, Baldwin P, Leal AS, Carapellucci S, Sridhar S, Liby KT. A Nano-Liposome Formulation of the PARP Inhibitor Talazoparib Enhances Treatment Efficacy and Modulates Immune Cell Populations in Mammary Tumors of BRCA-Deficient Mice. Theranostics. 2019;9(21):6224–6238. doi: 10.7150/thno.36281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang Z, Sun K, Xiao Y, Feng B, Mikule K, Ma X, Feng N, Vellano CP, Federico L, Marszalek JR. Niraparib Activates Interferon Signaling and Potentiates Anti-PD-1 Antibody Efficacy in Tumor Models. Sci Rep. 2019;9(1):1853. doi: 10.1038/s41598-019-38534-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Vinayak S, Tolaney SM, Schwartzberg L, Mita M, McCann G, Tan AR, Wahner-Hendrickson AE, Forero A, Anders C, Wulf GM. Open-Label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019;5(8):1132–1140. doi: 10.1001/jamaoncol.2019.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Domchek SM, Postel-Vinay S, Im S-A, Park YH, Delord J-P, Italiano A, Alexandre J, You B, Bastian S, Krebs MG. Olaparib and Durvalumab in Patients with Germline BRCA-Mutated Metastatic Breast Cancer (MEDIOLA): an Open-Label, Multicentre, Phase 1/2, Basket Study. Lancet Oncol. 2020;21(9):1155–1164. doi: 10.1016/S1470-2045(20)30324-7. [DOI] [PubMed] [Google Scholar]
- 137.Mehta AK, Cheney EM, Hartl CA, Pantelidou C, Oliwa M, Castrillon JA, Lin J-R, Hurst KE, de Oliveira Taveira M, Johnson NT. Targeting Immunosuppressive Macrophages Overcomes PARP Inhibitor Resistance in BRCA1-Associated Triple-Negative Breast Cancer. Nat Cancer. 2021;2(1):66–82. doi: 10.1038/s43018-020-00148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Salvagno C, Ciampricotti M, Tuit S, Hau C-S, van Weverwijk A, Coffelt SB, Kersten K, Vrijland K, Kos K, Ulas T. Therapeutic Targeting of Macrophages Enhances Chemotherapy Efficacy by Unleashing Type I Interferon Response. Nat Cell Biol. 2019;21(4):511–521. doi: 10.1038/s41556-019-0298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Juliá EP, Mordoh J, Levy EM. Cetuximab and IL-15 Promote NK and Dendritic Cell Activation In Vitro in Triple Negative Breast Cancer. Cells. 2020;9(7):E1573. doi: 10.3390/cells9071573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Emami F, Pathak S, Nguyen TT, Shrestha P, Maharjan S, Kim JO, Jeong J-H, Yook S. Photoimmunotherapy with Cetuximab-Conjugated Gold Nanorods Reduces Drug Resistance in Triple Negative Breast Cancer Spheroids with Enhanced Infiltration of Tumor-Associated Macrophages. J Control Release Off J Control Release Soc. 2021;329:645–664. doi: 10.1016/j.jconrel.2020.10.001. [DOI] [PubMed] [Google Scholar]
- 141.Grabinski N, Ewald F. Ibrutinib (ImbruvicaTM) Potently Inhibits ErbB Receptor Phosphorylation and Cell Viability of ErbB2-Positive Breast Cancer Cells. Invest New Drugs. 2014;32(6):1096–1104. doi: 10.1007/s10637-014-0141-2. [DOI] [PubMed] [Google Scholar]
- 142.Dubovsky JA, Beckwith KA, Natarajan G, Woyach JA, Jaglowski S, Zhong Y, Hessler JD, Liu T-M, Chang BY, Larkin KM. Ibrutinib Is an Irreversible Molecular Inhibitor of ITK Driving a Th1-Selective Pressure in T Lymphocytes. Blood. 2013;122(15):2539–2549. doi: 10.1182/blood-2013-06-507947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, McMichael E, Duggan M, Campbell A, Keller K. Myeloid-Derived Suppressor Cells Express Bruton’s Tyrosine Kinase and Can Be Depleted in Tumor-Bearing Hosts by Ibrutinib Treatment. Cancer Res. 2016;76(8):2125–2136. doi: 10.1158/0008-5472.CAN-15-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Varikuti S, Singh B, Volpedo G, Ahirwar DK, Jha BK, Saljoughian N, Viana AG, Verma C, Hamza O, Halsey G. Ibrutinib Treatment Inhibits Breast Cancer Progression and Metastasis by Inducing Conversion of Myeloid-Derived Suppressor Cells to Dendritic Cells. Br J Cancer. 2020;122(7):1005–1013. doi: 10.1038/s41416-020-0743-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sagiv-Barfi I, Kohrt HEK, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic Antitumor Immunity by Checkpoint Blockade Is Enhanced by Ibrutinib, an Inhibitor of Both BTK and ITK. Proc Natl Acad Sci U S A. 2015;112(9):E966–972. doi: 10.1073/pnas.1500712112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Hong D, Rasco D, Veeder M, Luke JJ, Chandler J, Balmanoukian A, George TJ, Munster P, Berlin JD, Gutierrez M. A Phase 1b/2 Study of the Bruton Tyrosine Kinase Inhibitor Ibrutinib and the PD-L1 Inhibitor Durvalumab in Patients with Pretreated Solid Tumors. Oncology. 2019;97(2):102–111. doi: 10.1159/000500571. [DOI] [PubMed] [Google Scholar]
- 147.Ali K, Soond DR, Pineiro R, Hagemann T, Pearce W, Lim EL, Bouabe H, Scudamore CL, Hancox T, Maecker H. Inactivation of PI(3)K P110δ Breaks Regulatory T-Cell-Mediated Immune Tolerance to Cancer. Nature. 2014;510(7505):407–411. doi: 10.1038/nature13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, Su X, Wang Y, Gonzalez-Angulo AM, Akcakanat A. PD-L1 Expression in Triple-Negative Breast Cancer. Cancer Immunol Res. 2014;2:361–370. doi: 10.1158/2326-6066.CIR-13-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dent R, Kim S-B, Oliveira M, Isakoff SJ, Barrios CH, O’Shaughnessy J, Lu X, Wongchenko M, Bradley D, Mani A. IPATunity130: a Pivotal Randomized Phase III Trial Evaluating Ipatasertib (IPAT) + Paclitaxel (PAC) for PIK3CA/AKT1/PTEN-Altered Advanced Triple-Negative (TN) or Hormone Receptor-Positive HER2-Negative (HR+/HER2–) Breast Cancer (BC). J Clin Oncol. 2018;36(15_suppl):TPS1117. doi: 10.1200/JCO.2018.36.15_suppl.TPS1117. [DOI] [Google Scholar]
- 150.Tiu C, Biondo A, Welsh LC, Jones TL, Zachariou A, Prout T, Turner AJ, Daly R, Vivanco I, Yap C. Abstract CT120: results of the Glioblastoma Multiforme (GBM) Cohort of Phase 1 Trial Ice-CAP (NCT03673787): preliminary Evidence of Antitumour Activity of Ipatasertib (Ipa) and Atezolizumab (A) in Patients (Pts) with PTEN Loss. Cancer Res. 2021;81(13_Supplement):CT120–CT120. CT120. doi: 10.1158/1538-7445.AM2021-CT120. [DOI] [Google Scholar]
- 151.Yin T, Zhao Z-B, Guo J, Wang T, Yang J-B, Wang C, Long J, Ma S, Huang Q, Zhang K. Aurora A Inhibition Eliminates Myeloid Cell-Mediated Immunosuppression and Enhances the Efficacy of Anti-PD-L1 Therapy in Breast Cancer. Cancer Res. 2019;79(13):3431–3444. doi: 10.1158/0008-5472.CAN-18-3397. [DOI] [PubMed] [Google Scholar]
- 152.Hu X, Bardhan K, Paschall AV, Yang D, Waller JL, Park MA, Nayak-Kapoor A, Samuel TA, Abrams SI, Liu K. Deregulation of Apoptotic Factors Bcl-XL and Bax Confers Apoptotic Resistance to Myeloid-Derived Suppressor Cells and Contributes to Their Persistence in Cancer. J Biol Chem. 2013;288(26):19103–19115. doi: 10.1074/jbc.M112.434530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim T-Y, Kudo M, Breder V, Merle P, Kaseb AO. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi: 10.1056/NEJMoa1915745. [DOI] [PubMed] [Google Scholar]
- 154.Zhu AX, Duda DG, Sahani DV, Jain RK. HCC and Angiogenesis: possible Targets and Future Directions. Nat Rev Clin Oncol. 2011;8(5):292–301. doi: 10.1038/nrclinonc.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Morse MA, Sun W, Kim R, He AR, Abada PB, Mynderse M, Finn RS. The Role of Angiogenesis in Hepatocellular Carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2019;25(3):912–920. doi: 10.1158/1078-0432.CCR-18-1254. [DOI] [PubMed] [Google Scholar]
- 156.Roland CL, Dineen SP, Lynn KD, Sullivan LA, Dellinger MT, Sadegh L, Sullivan JP, Shames DS, Brekken RA. Inhibition of Vascular Endothelial Growth Factor Reduces Angiogenesis and Modulates Immune Cell Infiltration of Orthotopic Breast Cancer Xenografts. Mol Cancer Ther. 2009;8(7):1761–1771. doi: 10.1158/1535-7163.MCT-09-0280. [DOI] [PubMed] [Google Scholar]
- 157.Motz GT, Santoro SP, Wang L-P, Garrabrant T, Lastra RR, Hagemann IS, Lal P, Feldman MD, Benencia F, Coukos G. Tumor Endothelium FasL Establishes a Selective Immune Barrier Promoting Tolerance in Tumors. Nat Med. 2014;20(6):607–615. doi: 10.1038/nm.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hegde PS, Wallin JJ, Mancao C. Predictive Markers of Anti-VEGF and Emerging Role of Angiogenesis Inhibitors as Immunotherapeutics. Semin Cancer Biol. 2018;52:117–124. doi: 10.1016/j.semcancer.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 159.Iwamoto M, Shinohara H, Miyamoto A, Okuzawa M, Mabuchi H, Nohara T, Gon G, Toyoda M, Tanigawa N. Prognostic Value of Tumor-Infiltrating Dendritic Cells Expressing CD83 in Human Breast Carcinomas. Int J Cancer. 2003;104(1):92–97. doi: 10.1002/ijc.10915. [DOI] [PubMed] [Google Scholar]
- 160.Kalathil SG, Wang K, Hutson A, Iyer R, Thanavala Y. Tivozanib Mediated Inhibition of C-Kit/SCF Signaling on Tregs and MDSCs and Reversal of Tumor Induced Immune Suppression Correlates with Survival of HCC Patients. Oncoimmunology. 2020;9(1):1824863. doi: 10.1080/2162402X.2020.1824863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liu D, Li G, Avella DM, Kimchi ET, Kaifi JT, Rubinstein MP, Camp ER, Rockey DC, Schell TD, Staveley-O’Carroll KF. Sunitinib Represses Regulatory T Cells to Overcome Immunotolerance in a Murine Model of Hepatocellular Cancer. Oncoimmunology. 2017;7(1):e1372079. doi: 10.1080/2162402X.2017.1372079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kalathil SG, Lugade AA, Miller A, Iyer R, Thanavala Y. PD-1+ and Foxp3+ T Cell Reduction Correlates with Survival of HCC Patients after Sorafenib Therapy. JCI Insight. 2016;1(11):e86182. doi: 10.1172/jci.insight.86182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Chen M-L, Yan B-S, Lu W-C, Chen M-H, Yu S-L, Yang P-C, Cheng A-L. Sorafenib Relieves Cell-Intrinsic and Cell-Extrinsic Inhibitions of Effector T Cells in Tumor Microenvironment to Augment Antitumor Immunity. Int J Cancer. 2014;134(2):319–331. doi: 10.1002/ijc.28362. [DOI] [PubMed] [Google Scholar]
- 164.Chang C-J, Yang Y-H, Chiu C-J, Lu L-C, Liao -C-C, Liang C-W, Hsu C-H, Cheng A-L. Targeting Tumor-Infiltrating Ly6G+ Myeloid Cells Improves Sorafenib Efficacy in Mouse Orthotopic Hepatocellular Carcinoma. Int J Cancer. 2018;142(9):1878–1889. doi: 10.1002/ijc.31216. [DOI] [PubMed] [Google Scholar]
- 165.Cao M, Xu Y, Youn J, Cabrera R, Zhang X, Gabrilovich D, Nelson DR, Liu C. Kinase Inhibitor Sorafenib Modulates Immunosuppressive Cell Populations in a Murine Liver Cancer Model. Lab Investig J Tech Methods Pathol. 2011;91(4):598–608. doi: 10.1038/labinvest.2010.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yuan C-H, Sun X-M, Zhu C-L, Liu S-P, Wu L, Chen H, Feng M-H, Wu K, Wang F-B. Amphiregulin Activates Regulatory T Lymphocytes and Suppresses CD8+ T Cell-Mediated Anti-Tumor Response in Hepatocellular Carcinoma Cells. Oncotarget. 2015;6(31):32138–32153. doi: 10.18632/oncotarget.5171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, Dolcetti L, Bronte V, Borrello I. Phosphodiesterase-5 Inhibition Augments Endogenous Antitumor Immunity by Reducing Myeloid-Derived Suppressor Cell Function. J Exp Med. 2006;203:2691–2702. doi: 10.1084/jem.20061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Yu SJ, Ma C, Heinrich B, Brown ZJ, Sandhu M, Zhang Q, Fu Q, Agdashian D, Rosato U, Korangy F. Targeting the Crosstalk between Cytokine-Induced Killer Cells and Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma. J Hepatol. 2019;70(3):449–457. doi: 10.1016/j.jhep.2018.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liu S, Qiu J, He G, He W, Liu C, Cai D, Pan H. TRAIL Promotes Hepatocellular Carcinoma Apoptosis and Inhibits Proliferation and Migration via Interacting with IER3. Cancer Cell Int. 2021;21(1):63. doi: 10.1186/s12935-020-01724-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Yuan X, Gajan A, Chu Q, Xiong H, Wu K, Wu GS. Developing TRAIL/TRAIL Death Receptor-Based Cancer Therapies. Cancer Metastasis Rev. 2018;37(4):733–748. doi: 10.1007/s10555-018-9728-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ganten TM, Koschny R, Sykora J, Schulze-Bergkamen H, Büchler P, Haas TL, Schader MB, Untergasser A, Stremmel W, Walczak H. Preclinical Differentiation between Apparently Safe and Potentially Hepatotoxic Applications of TRAIL Either Alone or in Combination with Chemotherapeutic Drugs. Clin Cancer Res Off J Am Assoc Cancer Res. 2006;12(8):2640–2646. doi: 10.1158/1078-0432.CCR-05-2635. [DOI] [PubMed] [Google Scholar]
- 172.Dominguez GA, Condamine T, Mony S, Hashimoto A, Wang F, Liu Q, Forero A, Bendell J, Witt R, Hockstein N. Selective Targeting of Myeloid-Derived Suppressor Cells in Cancer Patients Using DS-8273a, an Agonistic TRAIL-R2 Antibody. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23(12):2942–2950. doi: 10.1158/1078-0432.CCR-16-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kawaguchi N, Zhang -T-T, Nakanishi T. Involvement of CXCR4 in Normal and Abnormal Development. Cells. 2019;8(2):E185. doi: 10.3390/cells8020185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y, Ochiai H, Kitahara S, Unan EC, Reddy TP. CXCR4 Inhibition in Tumor Microenvironment Facilitates Anti-Programmed Death Receptor-1 Immunotherapy in Sorafenib-Treated Hepatocellular Carcinoma in Mice. Hepatol Baltim Md. 2015;61(5):1591–1602. doi: 10.1002/hep.27665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Stanley ER, Chitu V. CSF-1 Receptor Signaling in Myeloid Cells. Cold Spring Harb Perspect Biol. 2014;6(6):a021857. doi: 10.1101/cshperspect.a021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D. Targeting Tumor-Infiltrating Macrophages Decreases Tumor-Initiating Cells, Relieves Immunosuppression, and Improves Chemotherapeutic Responses. Cancer Res. 2013;73(3):1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nywening TM, Wang-Gillam A, Sanford DE, Belt BA, Panni RZ, Cusworth BM, Toriola AT, Nieman RK, Worley LA, Yano M. Targeting Tumour-Associated Macrophages with CCR2 Inhibition in Combination with FOLFIRINOX in Patients with Borderline Resectable and Locally Advanced Pancreatic Cancer: a Single-Centre, Open-Label, Dose-Finding, Non-Randomised, Phase 1b Trial. Lancet Oncol. 2016;17:651–662. doi: 10.1016/S1470-2045(16)00078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Shafiekhani S, Dehghanbanadaki H, Fatemi AS, Rahbar S, Hadjati J, Jafari AH. Prediction of Anti-CD25 and 5-FU Treatments Efficacy for Pancreatic Cancer Using a Mathematical Model. BMC Cancer. 2021;21(1):1226. doi: 10.1186/s12885-021-08770-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Sciarra A, Monteiro I, Ménétrier-Caux C, Caux C, Gilbert B, Halkic N, La Rosa S, Romero P, Sempoux C, de Leval L. CD73 Expression in Normal and Pathological Human Hepatobiliopancreatic Tissues. Cancer Immunol Immunother CII. 2019;68(3):467–478. doi: 10.1007/s00262-018-2290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Duffy AG, Greten TF. Immunological Off-Target Effects of Standard Treatments in Gastrointestinal Cancers. Ann Oncol. 2014;25(1):24–32. doi: 10.1093/annonc/mdt349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Kanterman J, Sade-Feldman M, Biton M, Ish-Shalom E, Lasry A, Goldshtein A, Hubert A, Baniyash M. Adverse immunoregulatory effects of 5FU and CPT11 chemotherapy on myeloid-derived suppressor cells and colorectal cancer outcomes. Cancer Res. 2014;74:6022–6035. doi: 10.1158/0008-5472.CAN-14-0657. [DOI] [PubMed] [Google Scholar]
- 182.Limagne E, Euvrard R, Thibaudin M, Rébé C, Derangère V, Chevriaux A, Boidot R, Végran F, Bonnefoy N, Vincent J. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predicts the efficacy of a FOLFOX-Bevacizumab drug treatment regimen. Cancer Res. 2016;76(18):5241–5252. doi: 10.1158/0008-5472.CAN-15-3164. [DOI] [PubMed] [Google Scholar]
- 183.Lim SH, Chua W, Cheng C, Descallar J, Ng W, Solomon M, Bokey L, Wong K, Lee MT, de Souza P. Effect of neoadjuvant chemoradiation on tumor-infiltrating/associated lymphocytes in locally advanced rectal cancers. Anticancer Res. 2014;34(11):6505–6513. [PubMed] [Google Scholar]
- 184.Khallouf H, Märten A, Serba S, Teichgräber V, Büchler MW, Jäger D, Schmidt J. 5-fluorouracil and interferon-α immunochemotherapy enhances immunogenicity of murine pancreatic cancer through upregulation of NKG2D ligands and MHC class I. J Immunother Hagerstown Md. 2012;1997(35):245–253. doi: 10.1097/CJI.0b013e31824b3a76. [DOI] [PubMed] [Google Scholar]
- 185.Limagne E, Thibaudin M, Nuttin L, Spill A, Derangère V, Fumet J-D, Amellal N, Peranzoni E, Cattan V, Ghiringhelli F. Trifluridine/tipiracil plus oxaliplatin improves PD-1 blockade in colorectal cancer by inducing immunogenic cell death and depleting macrophages. Cancer Immunol Res. 2019;7(12):1958–1969. doi: 10.1158/2326-6066.CIR-19-0228. [DOI] [PubMed] [Google Scholar]
- 186.Gonzalez-Aparicio M, Alzuguren P, Mauleon I, Medina-Echeverz J, Hervas-Stubbs S, Mancheno U, Berraondo P, Crettaz J, Gonzalez-Aseguinolaza G, Prieto J. Oxaliplatin in Combination with liver-specific expression of interleukin 12 reduces the immunosuppressive microenvironment of tumours and eradicates metastatic colorectal cancer in mice. Gut. 2011;60(3):341–349. doi: 10.1136/gut.2010.211722. [DOI] [PubMed] [Google Scholar]
- 187.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 188.Zhu H, Shan Y, Ge K, Lu J, Kong W, Jia C. Oxaliplatin induces immunogenic cell death in hepatocellular carcinoma cells and synergizes with immune checkpoint blockade therapy. Cell Oncol Dordr. 2020;43(6):1203–1214. doi: 10.1007/s13402-020-00552-2. [DOI] [PubMed] [Google Scholar]
- 189.Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y. Immunogenic chemotherapy sensitizes tumors to checkpoint blockade therapy. Immunity. 2016;44(2):343–354. doi: 10.1016/j.immuni.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Dosset M, Vargas TR, Lagrange A, Boidot R, Végran F, Roussey A, Chalmin F, Dondaine L, Paul C, Lauret Marie-Joseph E. PD-1/PD-L1 pathway: An adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology. 2018;7(6):e1433981. doi: 10.1080/2162402X.2018.1433981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 192.Ghiringhelli F, Chibaudel B, Taieb J, Bennouna J, Martin-Babau J, Fonck M, Borg C, Cohen R, Thibaudin M, Limagne E. Durvalumab and tremelimumab in combination with FOLFOX in patients with RAS-mutated, microsatellite-stable, previously untreated metastatic colorectal cancer (MCRC): Results of the first intermediate analysis of the phase Ib/II MEDETREME trial. J Clin Oncol. 2020;38:3006. doi: 10.1200/JCO.2020.38.15_suppl.3006. [DOI] [Google Scholar]
- 193.Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, Guenterberg K, Kondadasula SV, Chaudhury AR, La Perle KM. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res. 2011;71(15):5101–5110. doi: 10.1158/0008-5472.CAN-10-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Homma Y, Taniguchi K, Nakazawa M, Matsuyama R, Mori R, Takeda K, Ichikawa Y, Tanaka K, Endo I. Changes in the immune cell population and cell proliferation in peripheral blood after gemcitabine-based chemotherapy for pancreatic cancer. Clin Transl Oncol Off Publ Fed Span Oncol Soc Natl Cancer Inst Mex. 2014;16:330–335. doi: 10.1007/s12094-013-1079-0. [DOI] [PubMed] [Google Scholar]
- 195.Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol. 2004;34(2):336–344. doi: 10.1002/eji.200324181. [DOI] [PubMed] [Google Scholar]
- 196.Zhao Y, Liu X, Huo M, Wang Y, Li Y, Xu N, Zhu H. Cetuximab enhances the anti-tumor function of macrophages in an IL-6 dependent manner. Life Sci. 2021;267:118953. doi: 10.1016/j.lfs.2020.118953. [DOI] [PubMed] [Google Scholar]
- 197.Lian G, Chen S, Ouyang M, Li F, Chen L, Yang J. Colon Cancer Cell Secretes EGF to Promote M2 polarization of TAM through EGFR/PI3K/AKT/MTOR Pathway. Technol. in Cancer. Res. Treat. 2019;18:1533033819849068. doi: 10.1177/1533033819849068.10.7150/thno.36281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Abu-Eid R, Samara RN, Ozbun L, Abdalla MY, Berzofsky JA, Friedman KM, Mkrtichyan M, Khleif SN. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol Res. 2014;2(11):1080–1089. doi: 10.1158/2326-6066.CIR-14-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Johnson B, Osada T, Clay T, Lyerly H, Morse M. Physiology and therapeutics of vascular endothelial growth factor in tumor immunosuppression. Curr Mol Med. 2009;9(6):702–707. doi: 10.2174/156652409788970634. [DOI] [PubMed] [Google Scholar]
- 200.Lapeyre-Prost A, Terme M, Pernot S, Pointet A-L, Voron T, Tartour E, Taieb J. Immunomodulatory activity of VEGF in cancer. Int Rev Cell Mol Biol. 2017;330:295–342. doi: 10.1016/bs.ircmb.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 201.Wrzesinski SH, Wan YY, Flavell RA. Transforming growth factor-beta and the immune response: Implications for anticancer therapy. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13(18):5262–5270. doi: 10.1158/1078-0432.CCR-07-1157. [DOI] [PubMed] [Google Scholar]
- 202.Min AKT, Mimura K, Nakajima S, Okayama H, Saito K, Sakamoto W, Fujita S, Endo H, Saito M, Saze Z. Therapeutic potential of anti-VEGF receptor 2 therapy targeting for M2-tumor-associated macrophages in colorectal cancer. Cancer Immunol Immunother CII. 2021;70(2):289–298. doi: 10.1007/s00262-020-02676-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Manzoni M, Rovati B, Ronzoni M, Loupakis F, Mariucci S, Ricci V, Gattoni E, Salvatore L, Tinelli C, Villa E. Immunological effects of bevacizumab-based treatment in metastatic colorectal cancer. Oncology. 2010;79(3–4):187–196. doi: 10.1159/000320609. [DOI] [PubMed] [Google Scholar]
- 204.Takigawa H, Kitadai Y, Shinagawa K, Yuge R, Higashi Y, Tanaka S, Yasui W, Chayama K. Multikinase inhibitor regorafenib inhibits the growth and metastasis of colon cancer with abundant stroma. Cancer Sci. 2016;107(5):601–608. doi: 10.1111/cas.12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Schirripa M, Borelli B, D’Aurizio R, Lubrano S, Cremolini C, Zucchelli G, Antoniotti C, Marmorino F, Prete AA, Murgioni S. Early modifications of circulating micrornas levels in metastatic colorectal cancer patients treated with regorafenib. Pharmacogenomics J. 2019;19(5):455–464. doi: 10.1038/s41397-019-0075-3. [DOI] [PubMed] [Google Scholar]
- 206.Fakih M, Raghav KPS, Chang DZ, Bendell JC, Larson T, Cohn AL, Huyck TK, Cosgrove D, Fiorillo JA, Garbo LE. Single-Arm, phase 2 study of regorafenib plus nivolumab in patients with mismatch repair-proficient (PMMR)/microsatellite stable (MSS) colorectal cancer (CRC). J Clin Oncol. 2021;39(15_suppl):3560. doi: 10.1200/JCO.2021.39.15_suppl.3560. [DOI] [Google Scholar]
- 207.Poon E, Mullins S, Watkins A, Williams GS, Koopmann J-O, Di Genova G, Cumberbatch M, Veldman-Jones M, Grosskurth SE, Sah V. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J Immunother Cancer. 2017;5(1):63. doi: 10.1186/s40425-017-0268-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal advance: Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE₂ catabolism in myeloid cells. J Leukoc Biol. 2010;88(5):839–848. doi: 10.1189/jlb.1209821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Baumann D, Hägele T, Mochayedi J, Drebant J, Vent C, Blobner S, Noll JH, Nickel I, Schumacher C, Boos SL. Proimmunogenic Impact of MEK inhibition synergizes with agonist anti-CD40 Immunostimulatory antibodies in tumor therapy. Nat Commun. 2020;11:2176. doi: 10.1038/s41467-020-15979-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Desai J, Hong YS, Kim JE, Kim TW, Han SW, Bang Y-J, Chee CE, Heong VYM, McRee A, Chow LQ. Efficacy and safety of cobimetinib (Cobi) and atezolizumab (Atezo) in an expanded phase 1b study of microsatellite-Stable (MSS) metastatic colorectal cancer (MCRC). Ann Oncol. 2016;27:vi155. doi: 10.1093/annonc/mdw370.19. [DOI] [Google Scholar]
- 211.Liu C, Lu H, Wang H, Loo A, Zhang X, Yang G, Kowal C, Delach S, Wang Y, Goldoni S. Combinations with allosteric SHP2 inhibitor TNO155 to block receptor tyrosine kinase signaling. Clin Cancer Res Off J Am Assoc Cancer Res. 2021;27(1):342–354. doi: 10.1158/1078-0432.CCR-20-2718. [DOI] [PubMed] [Google Scholar]
- 212.Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel LP, Feuerhake F, Klaman I. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25(6):846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 213.Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, Suetterlin T, Brand K, Krauss J, Lasitschka F. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by anti-CCR5 therapy in cancer patients. Cancer Cell. 2016;29(4):587–601. doi: 10.1016/j.ccell.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 214.Haag GM, Springfeld C, Grün B, Apostolidis L, Zschäbitz S, Dietrich M, Berger A-K, Weber TF, Zoernig I, Schaaf M. Pembrolizumab and maraviroc in refractory mismatch repair proficient/microsatellite-stable metastatic colorectal cancer – the PICCASSO phase I trial. Eur J Cancer. 2022;167:112–122. doi: 10.1016/j.ejca.2022.03.017. [DOI] [PubMed] [Google Scholar]
- 215.Fang DD, Tang Q, Kong Y, Wang Q, Gu J, Fang X, Zou P, Rong T, Wang J, Yang D. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J Immunother Cancer. 2019;7(1):327. doi: 10.1186/s40425-019-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Kohlhapp FJ, Haribhai D, Mathew R, Duggan R, Ellis PA, Wang R, Lasater EA, Shi Y, Dave N, Riehm JJ. Venetoclax increases intratumoral effector t cells and antitumor efficacy in combination with immune checkpoint blockade. Cancer Discov. 2021;11(1):68–79. doi: 10.1158/2159-8290.CD-19-0759. [DOI] [PubMed] [Google Scholar]
- 217.Yang R, Cai -T-T, Wu X-J, Liu Y-N, He J, Zhang X-S, Ma G, Li J. Tumour YAP1 and PTEN expression correlates with tumour-associated myeloid suppressor cell expansion and reduced survival in colorectal cancer. Immunology. 2018;155(2):263–272. doi: 10.1111/imm.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hervieu A, Mignot G, Ghiringhelli F. Dacarbazine mediate antimelanoma effects via NK cells. Oncoimmunology. 2013;2(4):e23714. doi: 10.4161/onci.23714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hervieu A, Rébé C, Végran F, Chalmin F, Bruchard M, Vabres P, Apetoh L, Ghiringhelli F, Mignot G. Dacarbazine-mediated Upregulation of NKG2D ligands on tumor cells activates nk and cd8 t cells and restrains melanoma growth. J Invest Dermatol. 2013;133(2):499–508. doi: 10.1038/jid.2012.273. [DOI] [PubMed] [Google Scholar]
- 220.Lebbé C, Meyer N, Mortier L, Marquez-Rodas I, Robert C, Rutkowski P, Menzies AM, Eigentler T, Ascierto PA, Smylie M. Evaluation of two dosing regimens for nivolumab in combination with ipilimumab in patients with advanced melanoma: Results from the phase IIIb/IV checkmate 511 trial. J Clin Oncol Off J Am Soc Clin Oncol. 2019;37(11):867–875. doi: 10.1200/JCO.18.01998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367(18):1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, Demidov LV, Hassel JC, Rutkowski P, Mohr P. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367(2):107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- 223.Kakavand H, Wilmott JS, Menzies AM, Vilain R, Haydu LE, Yearley JH, Thompson JF, Kefford RF, Hersey P, Long GV. PD-L1 expression and tumor-infiltrating lymphocytes define different subsets of MAPK inhibitor-treated melanoma patients. Clin Cancer Res Off J Am Assoc Cancer Res. 2015;21(14):3140–3148. doi: 10.1158/1078-0432.CCR-14-2023. [DOI] [PubMed] [Google Scholar]
- 224.Hu-Lieskovan S, Mok S, Homet Moreno B, Tsoi J, Robert L, Goedert L, Pinheiro EM, Koya RC, Graeber TG, Comin-Anduix B. Improved antitumor activity of immunotherapy with BRAF and MEK Inhibitors in BRAF(V600E) melanoma. Sci Transl Med. 2015;7(279):279ra41. doi: 10.1126/scitranslmed.aaa4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Liu L, Mayes PA, Eastman S, Shi H, Yadavilli S, Zhang T, Yang J, Seestaller-Wehr L, Zhang S-Y, Hopson C. The BRAF and MEK Inhibitors dabrafenib and trametinib: Effects on immune function and in combination with immunomodulatory antibodies targeting PD-1, PD-L1, and CTLA-4. Clin Cancer Res Off J Am Assoc Cancer Res. 2015;21(7):1639–1651. doi: 10.1158/1078-0432.CCR-14-2339. [DOI] [PubMed] [Google Scholar]
- 226.Ferrari de Andrade L, Ngiow SF, Stannard K, Rusakiewicz S, Kalimutho M, Khanna KK, Tey S-K, Takeda K, Zitvogel L, Martinet L. Natural killer cells are essential for the ability of BRAF inhibitors to control BRAFV600E-mutant metastatic melanoma. Cancer Res. 2014;74(24):7298–7308. doi: 10.1158/0008-5472.CAN-14-1339. [DOI] [PubMed] [Google Scholar]
- 227.Zhang X, Fang X, Gao Z, Chen W, Tao F, Cai P, Yuan H, Shu Y, Xu Q, Sun Y. Axitinib, a selective inhibitor of vascular endothelial growth factor receptor, exerts an anticancer effect in melanoma through promoting antitumor immunity. Anticancer Drugs. 2014;25(2):204–211. doi: 10.1097/CAD.0000000000000033. [DOI] [PubMed] [Google Scholar]
- 228.de Almeida PE, Mak J, Hernandez G, Jesudason R, Herault A, Javinal V, Borneo J, Kim JM, Walsh KB. Anti-VEGF treatment enhances CD8+ T-cell antitumor activity by amplifying hypoxia. Cancer Immunol Res. 2020;8(6):806–818. doi: 10.1158/2326-6066.CIR-19-0360. [DOI] [PubMed] [Google Scholar]
- 229.Yuan M, Zhu Z, Mao W, Wang H, Qian H, Wu J, Guo X, Xu Q. Anlotinib combined with anti-PD-1 antibodies therapy in patients with advanced refractory solid Tumors: A single-center, observational, prospective study. Front Oncol. 2021;11:683502. doi: 10.3389/fonc.2021.683502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Liu S, Qin T, Liu Z, Wang J, Jia Y, Feng Y, Gao Y, Li K. Anlotinib alters tumor immune microenvironment by downregulating PD-L1 expression on vascular endothelial cells. Cell Death Dis. 2020;11(5):309. doi: 10.1038/s41419-020-2511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Balan M, Mier Y, Teran E, Waaga-Gasser AM, Gasser M, Choueiri TK, Freeman G, Pal S. Novel roles of c-met in the survival of renal cancer cells through the regulation of HO-1 and PD-L1 expression. J Biol Chem. 2015;290(13):8110–8120. doi: 10.1074/jbc.M114.612689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ilangumaran S, Villalobos-Hernandez A, Bobbala D, Ramanathan S. The hepatocyte growth factor (hgf)-met receptor tyrosine kinase signaling pathway: Diverse roles in modulating immune cell functions. Cytokine. 2016;82:125–139. doi: 10.1016/j.cyto.2015.12.013. [DOI] [PubMed] [Google Scholar]
- 233.Jain J, Frees M, Yasin H, Bonner J, Freesmeier M, Garje R, Venur VA, Milhem M, Zakharia Y. 427 A phase 1b/2 study of cabozantinib in combination with pembrolizumab in advanced melanoma. J Immunother Cancer. 2020:8. doi: 10.1136/jitc-2020-SITC2020.0427. [DOI] [Google Scholar]
- 234.Zhao F, Evans K, Xiao C, DeVito N, Theivanthiran B, Holtzhausen A, Siska PJ, Blobe GC, Hanks BA. Stromal fibroblasts mediate anti-PD-1 resistance via MMP-9 and dictate TGFβ inhibitor sequencing in melanoma. Cancer Immunol Res. 2018;6(12):1459–1471. doi: 10.1158/2326-6066.CIR-18-0086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Young A, Mittal D, Stagg J, Smyth MJ. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov. 2014;4(8):879–888. doi: 10.1158/2159-8290.CD-14-0341. [DOI] [PubMed] [Google Scholar]
- 236.Beavis PA, Divisekera U, Paget C, Chow MT, John LB, Devaud C, Dwyer K, Stagg J, Smyth MJ, Darcy PK. Blockade of A2A receptors potently suppresses the metastasis of CD73+ Tumors. Proc Natl Acad Sci U S A. 2013;110(36):14711–14716. doi: 10.1073/pnas.1308209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Young A, Ngiow SF, Madore J, Reinhardt J, Landsberg J, Chitsazan A, Rautela J, Bald T, Barkauskas DS, Ahern E. Targeting adenosine in braf-mutant melanoma reduces Tumor growth and metastasis. Cancer Res. 2017;77(17):4684–4696. doi: 10.1158/0008-5472.CAN-17-0393. [DOI] [PubMed] [Google Scholar]
- 238.Weinlich G, Murr C, Richardsen L, Winkler C, Fuchs D. Decreased serum tryptophan concentration predicts poor prognosis in malignant melanoma patients. Dermatol Basel Switz. 2007;214(1):8–14. doi: 10.1159/000096906. [DOI] [PubMed] [Google Scholar]
- 239.Long GV, Dummer R, Hamid O, Gajewski TF, Caglevic C, Dalle S, Arance A, Carlino MS, Grob -J-J, Kim TM. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind Study. Lancet Oncol. 2019;20(8):1083–1097. doi: 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- 240.Vilain RE, Menzies AM, Wilmott JS, Kakavand H, Madore J, Guminski A, Liniker E, Kong BY, Cooper AJ, Howle JR. Dynamic changes in PD-L1 expression and immune infiltrates early during treatment predict response to PD-1 blockade in Melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2017;23(17):5024–5033. doi: 10.1158/1078-0432.CCR-16-0698. [DOI] [PubMed] [Google Scholar]
- 241.Parra ER, Villalobos P, Behrens C, Jiang M, Pataer A, Swisher SG, William WN, Zhang J, Lee J, Cascone T. Effect of neoadjuvant chemotherapy on the immune microenvironment in non-small cell Lung Carcinomas as determined by multiplex immunofluorescence and image analysis approaches. J Immunother Cancer. 2018;6(1):48. doi: 10.1186/s40425-018-0368-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Liu P, Zhao L, Pol J, Levesque S, Petrazzuolo A, Pfirschke C, Engblom C, Rickelt S, Yamazaki T, Iribarren K. Crizotinib-induced immunogenic cell death in non-small cell Lung Cancer. Nat Commun. 2019;10(1):1486. doi: 10.1038/s41467-019-09415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kwon M, Jung H, Nam G-H, Kim I-S. The right timing, right combination, right sequence, and right delivery for cancer immunotherapy. J Control Release Off J Control Release Soc. 2021;331:321–334. doi: 10.1016/j.jconrel.2021.01.009. [DOI] [PubMed] [Google Scholar]