

Abstract
Arterial hypertension (HTA) is associated with liver disease, but causality remains unclear. We investigated whether genetic predisposition to HTA is associated with liver disease in the population, and if antihypertensive medication modifies this association. Participants of the Finnish health-examination surveys, FINRISK 1992–2012 and Health 2000 (n = 33,770), were linked with national electronic healthcare registers for liver-related outcomes (K70-K77, C22.0) and with the drug reimbursement registry for new initiation of antihypertensive medication during follow-up. Genetic predisposition to HTA was defined by polygenic risk scores (PRSs). During a median 12.9-year follow-up (409,268.9 person-years), 441 liver-related outcomes occurred. In the fully-adjusted Cox-regression models, both measured systolic blood pressure and clinically defined HTA were associated with liver-related outcomes. PRSs for systolic and diastolic blood pressure were significantly associated with liver-related outcomes (HR/SD 1.19, 95% CI 1.01–1.24, and 1.12, 95% CI 1.01–1.25, respectively). In the highest quintile of the systolic blood pressure PRS, new initiation of antihypertensive medication was associated with reduced rates of liver-related outcomes (HR 0.55, 95% CI 0.31–0.97). HTA and a genetic predisposition for HTA are associated with liver-related outcomes in the population. New initiation of antihypertensive medication attenuates this association in persons with high genetic risk for HTA.
Subject terms: Hepatology, Epidemiology
Introduction
Arterial hypertension (HTA) is one of the most important risk factors for cardiovascular morbidity and mortality1. Observational studies, dating back to the 1970s, also link HTA with chronic liver disease, especially non-alcoholic fatty liver disease (NAFLD)2–4. NAFLD is currently the most common chronic liver disease affecting around 25% of the adult population5.
Although NAFLD and HTA share many metabolic features, the relationship between NAFLD and HTA seems to be independent of these other metabolic components4. Specifically, HTA has been associated with elevated liver enzymes, liver steatosis and advanced fibrosis, independent of body mass index (BMI), metabolic factors (e.g. abdominal obesity, diabetes, dyslipidemia), alcohol use and other relevant confounders in numerous studies3,6–18. For example, a cross-sectional study based on data from 16,082 individuals in 26 cohorts found systolic blood pressure (SBP) to be independently associated with advanced liver fibrosis17. Furthermore, a systematic review of studies on NAFLD with paired liver biopsies identified HTA as a predictor of more rapid liver fibrosis progression over time, whereas metabolic syndrome, diabetes or BMI failed to predict such progression19. Recently, large population studies found HTA to be associated with severe liver-related outcomes including hepatocellular carcinoma independently of relevant confounders20–24. HTA was even reported to have a stronger risk effect for future liver cirrhosis than other metabolic traits such as diabetes, dyslipidemia, and obesity21.
Despite these previous observations, it still remains unclear whether the association between HTA and liver disease is causal or due to unmeasured or inadequately measured confounding secondary to, for example, alcohol use, other lifestyle factors, or associated metabolic conditions. Assuming that a causal relationship between HTA and liver disease exists, it remains unclear whether this relationship is modifiable by antihypertensive medication.
Many lifestyle factors may affect blood pressure over time, and therefore genetic predisposition to HTA may serve as a better marker than measured blood pressure to study associations with other conditions, such as liver disease. Polygenic risk scores (PRS) provide an overall measure of an individual’s genetic predisposition to develop a condition such as HTA25,26.
The aim of this study was to analyze whether HTA is associated with liver-related outcomes. For this purpose, we analyzed both clinically defined HTA and a recently published hypertension-PRS in a large general population cohort with linked electronic healthcare registry data for liver-related outcomes. By linkage with national drug imbursement registries, we further investigated whether use of antihypertensive medication influences the association between the genetic predisposition for HTA and liver disease.
Methods
Data were from the Finnish health-examination surveys, FINRISK 1992, 1997, 2002, 2007, and 2012, as well as the Health 2000 Survey. The FINRISK studies are cross-sectional health-examination surveys conducted in Finland every five years in a systematic and standardized fashion since 1972 by the Finnish Institute for Health and Welfare (previously National Public Health Institute). The surveys provide data on representative samples of adults (25–74 years) from 4 to 6 regions in Finland. The samples were randomly drawn from the Finnish National Population Register and were stratified by region, gender, and 10-year age groups27.
The Health 2000 Survey, conducted in 2000–2001, was also coordinated by the Finnish Institute for Health and Welfare (previously National Public Health Institute), and the cohort is considered representative of the adult Finnish population through a regional two-stage stratified cluster sampling procedure28.
Data were collected at baseline via interviews, questionnaire, health examination, and blood sampling as previously described27,28. All participants provided signed informed consent, and the studies were approved by the Coordinating Ethical Committee of the Helsinki and Uusimaa Hospital District (previously studies also approved by the institutional review board of the National Public Health Institute, both in Helsinki, Finland). The FINRISK and Health 2000 sample collections were transferred to THL Biobank in 2015 after approval of the Coordinating Ethical Committee of the Helsinki and Uusimaa Hospital District. All methods were performed in accordance with the relevant guidelines and regulations and with the Declaration of Helsinki.
SBP and diastolic blood pressure (DBP) were measured two or three times from the right arm in the sitting position using the mercury sphygmomanometer following at least 5 min of rest; we used the mean value of these measurements in this study27,28. Respondents were asked to report how often they consumed alcoholic beverages during the previous year and the average amount they consumed per week during the previous month. Average alcohol intake (100% ethanol in grams per day) was calculated based on these data. Smoking status was categorized as never smokers, previous smokers and current smokers. Exercise was assessed by asking how often the subject performs leisure-time physical exercise for at least 20–30 min so that he/she is at least slightly out of breath and sweaty. Diabetes was defined either by a fasting serum glucose ≥ 7.0 mmol/L (126 mg/dL), taking diabetic medication, or by having a prior known diabetes diagnosis. BMI (kg/m2) and waist circumference (cm) were measured at baseline27,28.
The FINRISK and Health 2000 samples were genotyped using Illumina CoreExome, OMNIExpress, and 610 K arrays. Individuals with non-Finnish genetic ancestry, as defined using principal component analysis, or obscure sex were excluded. Quality control before phasing and imputation excluded variants with missingness > 5%, call rate < 95%, minor allele count (MAC) < 3 (if Zcalled) or MAC < 10 (if called using Illumina GenCal), INFO < 0.8, minor allele frequency < 0.001%, Hardy–Weinberg equilibrium p-value < 1 * 10–10, and heterozygosity exceeding ± 4 standard deviations. The quality control was performed simultaneously on all data. Prior to imputation, the haplotypes were estimated using Eagle 2.3.529. Imputation was performed with Beagle 4.130 using high-coverage, population-specific SISu v3 reference panel of 3775 whole-genome sequences at the Institute for Molecular Medicine Finland (FIMM).
PRSs for SBP and DBP were derived from GWAS where over 1 million people were studied26. To generate PRSs we used GWAS summary statistics from discovery meta-analysis comprising 757,601 individuals from the UK Biobank and International Cancer Benchmarking Partnership (ICBP) samples that were publicly available31. PRS was constructed by calculating the weighted sum of risk alleles which an individual carry. PRS weights were calculated with PRS-CS32,33 using European 1000 Genomes Project samples as an external LD reference panel. Polygenic score for each individual was produced using PLINK’s –score command34. The mean of the PRS was scaled to zero with R 3.6.0 to reflect SD scaling in addition to mean centering. The final variant counts of the PRSs were 1,072,098 for SBP and 1,073,588 for DBP.
Follow-up data were obtained from several national electronic healthcare registers through linkage using the unique personal identity code assigned to all Finnish residents. The Finnish Drug Reimbursement Register covers all reimbursed pharmacy prescription drug purchases since 1964, purchase timestamps, and respective Anatomical Therapeutic Chemical Classification System codes (ATC). In the present study, exposure to antihypertensive medication required at least three purchase events for drugs with ATC codes C02, C03, C07, C08, and C09, and start of exposure was the time-point of the first purchase event. All of these drugs are reimbursed prescription drugs in Finland and therefore registry coverage is complete. Data for hospitalizations were obtained from the Care Register for Health Care (HILMO), which covers all hospitalizations in Finland since 1969. One or several ICD-diagnoses are assigned to each hospitalization at discharge; these diagnosis codes are systematically recorded in the HILMO register. Data for malignancies were obtained from the Finnish Cancer Registry, with nationwide cancer records since 1953. Vital status and cause of death data were obtained from Statistics Finland. In Finland, each person who dies is by law assigned a cause of death (in accordance with the ICD) to the official death certificate, issued by the treating physician based on medical or autopsy evidence, or forensic evidence when necessary; the death codes are then verified by medical experts at the register and recorded according to systematic coding principle. Data collection to all these registries is mandatory by law and general quality is consistent and virtually 100% complete35,36.
Study endpoints were fatal and non-fatal liver disease requiring hospital admission or causing liver cancer or liver-related death, and defined by the ICD-10 codes K70-K77 or C22.0, or ICD-8/9 codes 570–573 or 155.0 in any of the follow-up registries. Regarding hospitalizations, we only considered liver events recorded as either the main cause of hospitalization or as the first or second side cause.
From the combined FINRISK and Health 2000 dataset (n = 43,105), we excluded subjects with missing registry-follow-up (n = 1506), missing genetic data (n = 5221), chronic viral hepatitis (n = 100), one individual from each related pair with a kinship coefficient > 0.2 or duplicates (n = 2270), or a record of liver disease at or before baseline (n = 238). The remaining cohort comprised 33,770 subjects.
Statistical analyses
For comparing groups, we used Chi Square, Student’s t-test or Mann Whitney tests as appropriate. Associations between baseline variables and SBP PRS and DBP PRS was analyzed by linear regression adjusted for age and sex. Cox regression analyses were performed with time to the first liver event as the outcome. The following HTA-related exposure variables were analyzed in separate Cox models: measured SBP, measured DBP, SBP PRS, DBP PRS, elevated blood pressure (systolic blood pressure ≥ 130 or diastolic ≥ 85 mmHg or antihypertensive medication use as defined by Alberti et al.37), and HTA status. HTA status was defined in line with recent guidelines38 by a blood pressure cutoff of ≥ 140 (systolic) or ≥ 90 (diastolic) mmHg and baseline antihypertensive medication use in the following categories: (a) normal blood pressure, no medication; (b) high blood pressure, previously unknown; (c) high blood pressure, previously known, no medication; (d) high blood pressure despite medication; and (e) normal blood pressure during medication. Two sets of adjustments were considered: adjustment for (a) age and sex, or adjustment for (b) age, sex, BMI, waist circumference, weekly alcohol use, fraction of alcohol use as wine, low-density lipoprotein (LDL) cholesterol, high-density lipoprotein (HDL) cholesterol, triglycerides, diabetes, exercise habits, smoking status and baseline cardiovascular disease. In addition to this, Cox regression analyses with genetic risk scores as covariates were also adjusted for genotyping chip, and the first three principal components of genetic structure. Two-way interactions for the liver outcome were tested between the HTA PRSs and alcohol use, BMI, waist circumference, diabetes, and prevalent cardiovascular disease. A subgroup analysis was performed by the presence of the metabolic syndrome at baseline. Metabolic syndrome was defined according to the Joint Interim Statement criteria37, using the following cutoffs: waist circumference ≥ 94 cm for men and ≥ 80 cm for women; serum triglycerides ≥ 1.7 mmol/L; serum HDL-cholesterol < 1.0 mmol/L for men and < 1.3 mmol/L for women; and fasting serum glucose ≥ 5.6 mmol/L or diabetes medication.
To investigate whether antihypertensive medication modifies the association between HTA and liver-related outcomes, we analyzed interaction effects between antihypertensive medication use and SBP PRS and DBP PRS by Cox regression with confounder adjustments as above. To avoid prevalent-user bias, we excluded subjects with antihypertensive medication use at or before baseline from these analyses. To mitigate immortal-time bias, medication exposure was modelled as a time-varying variable using the tmerge function of the R survival package. We also performed a sensitivity analysis excluding subjects with antihypertensive medication initiated > 5 years after baseline. The functional form of the SBP PRS and DBP PRS in interaction with antihypertensive medication was plotted using restricted cubic splines. For each Cox regression model and for each covariate used, we evaluated the proportional hazards assumption both visually by plotting Schoenfeld residuals against time and by testing with the score test whether the slope of the corresponding time-dependent coefficient deviates from zero (R’s cox.zph-function). Data were analyzed with R software version 3.6.139.
Ethics approval
The studies were approved by the Coordinating Ethical Committee of the Helsinki and Uusimaa Hospital District (previously studies also approved by the institutional review board of the National Public Health Institute, both in Helsinki, Finland). The FINRISK and Health 2000 sample collections were transferred to THL Biobank in 2015 after approval of the Coordinating Ethical Committee of the Helsinki and Uusimaa Hospital District.
Patient consent
All participants provided signed informed consent.
Results
The study comprised 33,770 individuals with a mean age of 49.6 years and 54% women (Table 1). Mean SBP and DBP were 135 (SD 20) and 81 (SD 11) mmHg, respectively. Seventy-two percent had elevated blood pressure (blood pressure ≥ 130/ ≥ 85 mmHg or antihypertensive medication). Measured SBP and DBP correlated significantly with the respective SBP and DBP PRSs (Supplementary Fig. 1). Of baseline variables, age, sex, measured blood pressures, diabetes, metabolic syndrome, cardiovascular disease, alcohol intake, fraction of alcohol intake as wine, exercise, and triglycerides were associated with both SBP and DBP PRSs when assessed using age- and sex-adjusted linear regression analysis (Table 1 and Supplementary Table 1).
Table 1.
Baseline demographics.
| All subjects | Medication-naïve non-initiator | Medication naïve new-initiator | P | P for association with SBP PRSb | P for association with DBP PRSb | |
|---|---|---|---|---|---|---|
| Persons | 33,770 | 16,150 | 9576 | |||
| Age | 49.6 (13.9) | 43.8 (12.5) | 50.4 (12.2) | < 0.001 | 0.008 | 0.011 |
| Women | 18,075 (53.5) | 8643 (53.5) | 5094 (53.2) | 0.626 | 0.022 | 0.025 |
| SBP | 134.6 (19.8) | 126.7 (15.8) | 140.6 (19.8) | < 0.001 | < 0.001 | < 0.001 |
| DBP | 80.7 (11.4) | 77.3 (10.3) | 84.5 (11.3) | < 0.001 | < 0.001 | < 0.001 |
| Pulse pressure | 54.0 (15.8) | 49.5 (12.8) | 56.2 (16.2) | < 0.001 | < 0.001 | < 0.001 |
| Elevated blood pressurea | 24,140 (71.5) | 7029 (43.6) | 9289 (97.0) | < 0.001 | < 0.001 | < 0.001 |
| Body mass index | 26.8 (4.7) | 25.5 (4.1) | 27.3 (4.7) | < 0.001 | 0.152 | 0.023 |
| Waist circumference | 90.3 (13.8) | 86.5 (12.4) | 91.0 (13.7) | < 0.001 | 0.492 | 0.339 |
| Diabetes | 2589 (7.7) | 470 (2.9) | 586 (6.1) | < 0.001 | < 0.001 | < 0.001 |
| Metabolic syndrome | 11,135 (33.0) | 2758 (17.1) | 3910 (40.8) | < 0.001 | < 0.001 | < 0.001 |
| Cardiovascular disease | 1229 (3.6) | 77 (0.5) | 148 (1.5) | < 0.001 | < 0.001 | < 0.001 |
| Weekly alcohol use (grams/week) | 75.0 (137.4) | 77.9 (134.6) | 75.8 (136.5) | 0.237 | 0.003 | 0.024 |
| Fraction of alcohol use as wine | 0.23 (0.34) | 0.24 (0.34) | 0.22 (0.34) | 0.010 | < 0.001 | 0.002 |
| Smoking status | < 0.001 | |||||
| Current | 8013 (23.9) | 4241 (26.4) | 2447 (25.7) | 0.395 | 0.262 | |
| Former | 7590 (22.6) | 3147 (19.6) | 2171 (22.8) | 0.216 | 0.062 | |
| Never | 17,920 (53.5) | 8691 (54.1) | 4900 (51.5) | Ref | Ref | |
| Exercise | < 0.001 | |||||
| At least 2 times a week | 16,137 (57.3) | 7561 (58.4) | 5010 (54.4) | Ref | Ref | |
| 2–4 times a month | 7526 (26.7) | 3642 (28.1) | 2703 (29.3) | < 0.001 | 0.002 | |
| Less often | 4481 (15.9) | 1753 (13.5) | 1505 (16.3) | 0.078 | 0.647 | |
| Low-density lipoprotein cholesterol | 3.41 (0.97) | 3.34 (0.95) | 3.59 (0.97) | < 0.001 | 0.497 | 0.659 |
| High-density lipoprotein cholesterol | 1.43 (0.38) | 1.46 (0.38) | 1.41 (0.38) | < 0.001 | 0.001 | 0.108 |
| Triglycerides | 1.47 (1.00) | 1.33 (0.89) | 1.56 (1.06) | < 0.001 | < 0.001 | < 0.001 |
Data are as mean (SD) or n (%).
HTA, arterial hypertension; SBP, systolic blood pressure; DBP, diastolic blood pressure; PRS, polygenic risk score; Ref, reference group.
aEither measured blood pressure ≥ 130 (systolic) or ≥ 85 (diastolic) mmHg or antihypertensive medication use at baseline.
bBy age- and sex-adjusted linear regression analysis.
During a median follow-up of 12.9 years (IQR 7.8–17.8, range 0.1–23.0, 409,268.9 person-years), we observed 441 liver-related outcomes (262 among men and 179 among women) and 3642 deaths (2147 among men and 1495 among women).
In separate Cox regression models adjusted for age and sex, measured SBP, DBP and pulse pressure were associated with incident liver-related outcomes (Table 2). In the fully adjusted models, SBP and pulse pressure remained significantly associated with liver-related outcomes. Elevated blood pressure as a categorical variable (blood pressure ≥ 130/85 mHg or antihypertensive medication) exhibited a hazards ratio (HR) of 1.8 in the fully adjusted model (Table 2). When considering the multicategorical HTA status (categories = (a) normal blood pressure, no medication; (b) high blood pressure, previously unknown; (c) high blood pressure, previously known, no medication; (d) high blood pressure despite medication; and (e) normal blood pressure during medication, with high blood pressure defined as ≥ 140 (systolic) or ≥ 90 (diastolic) mmHg) as the independent variable in the fully adjusted model, all categories depicting HTA showed increased rates of liver-related outcomes compared to those with normal blood pressure without medication (Table 2 and Fig. 1). The highest rates of liver outcomes were observed among those with high blood pressure despite medication and those with previously unknown HTA, but the confidence intervals were overlapping between categories.
Table 2.
Associations between hypertension and liver-related outcomes using both measured blood pressure, use of antihypertensive medication and polygenic risk scores (PRSs).
| n/N | HRa (95% CI) | P | HRb (95% CI) | P | |
|---|---|---|---|---|---|
| Measured SBP (per 1 SD) | 441/33,770 | 1.22 (1.10–1.34) | < 0.001 | 1.16 (1.03–1.30) | 0.01 |
| Measured DBP (per 1 SD) | 441/33,770 | 1.17 (1.07–1.29) | 0.001 | 1.06 (0.95–1.20) | 0.29 |
| Measured pulse pressure (per 1 SD) | 441/33,770 | 1.12 (1.02–1.23) | 0.019 | 1.12 (1.00–1.24) | 0.04 |
| Elevated blood pressurec | |||||
| No | 61/9602 | 1.00 | 1.00 | ||
| Yes | 380/24,140 | 1.92 (1.44–2.54) | < 0.001 | 1.79 (1.27–2.51) | 0.001 |
| HTA statusd | |||||
| Normal blood pressure, no medication | 104/11,361 | 1.00 | 1.00 | ||
| High blood pressure, previously unknown | 72/4444 | 1.47 (1.07–2.01) | 0.02 | 1.73 (1.20–2.49) | 0.003 |
| High blood pressure, previously known, no medication | 70/3304 | 2.07 (1.51–2.83) | < 0.001 | 1.67 (1.12–2.48) | 0.01 |
| High blood pressure despite medication | 40/2478 | 1.86 (1.25–2.76) | 0.002 | 1.75 (1.08–2.86) | 0.02 |
| Normal blood pressure during medication | 9/870 | 1.57 (0.78–3.14) | 0.21 | 1.48 (0.63–3.51) | 0.37 |
| SBP PRS (per 1 SD) | 441/33,770 | 1.12 (1.02–1.23) | 0.02 | 1.19 (1.01–1.24) | 0.04 |
| DBP PRS (per 1 SD) | 441/33,770 | 1.13 (1.03–1.24) | 0.01 | 1.12 (1.01–1.25) | 0.03 |
Analyses are by Cox regression analyses.
CI, confidence interval; HR, hazards ratio; HTA, arterial hypertension; SBP, systolic blood pressure; DBP, diastolic blood pressure; PRS, polygenic risk score.
aAdjusted for age and sex. Analyses including polygenic risk scores (PRS) are additionally adjusted for genotyping chip and the first three principal components of genetic structure.
bAdjusted for age, sex, body mass index, waist circumference, weekly alcohol use, fraction of alcohol use as wine, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, triglycerides, diabetes, exercise habits, smoking status (current, former, never smoker) and baseline cardiovascular disease. Analyses including polygenic risk scores (PRS) are additionally adjusted for genotyping chip and the first three principal components of genetic structure.
cEither measured blood pressure ≥ 130 (systolic) or ≥ 85 (diastolic) mmHg or antihypertensive medication use at baseline.
dHigh blood pressure defined as ≥ 140 (systolic) or ≥ 90 (diastolic) mmHg.
Figure 1.

Associations by Cox regression between different categories of baseline arterial hypertension and liver-related outcomes. High blood pressure is defined as measured blood pressure ≥ 140 (systolic) or ≥ 90 (diastolic) mmHg. Error bars reflect 95% confidence intervals.
In separate Cox regression models considering the PRSs as independent variables, both the SBP and DBP PRSs were significantly associated with liver-related outcomes in both age- and sex-adjusted models and in the fully adjusted models (Table 2). Interaction terms between SBP PRS or DBP PRS and alcohol use, BMI, waist circumference, diabetes, and prevalent cardiovascular disease were all non-significant (Supplementary Table 2).
We next studied possible effect modification from the metabolic syndrome. In Cox regression analysis with age, sex, alcohol use, SBP or DBP PRS, metabolic syndrome, and the interaction terms between the PRSs and metabolic syndrome as covariates, the risk for liver-related outcomes increased along with the SBP PRS and DBP PRS both among those with and without the metabolic syndrome (Supplementary Fig. 2). Both SBP PRS and DBP PRS were significantly associated with liver-related outcomes among subjects without baseline metabolic syndrome (SBP PRS: HR 1.19, 95% CI 1.00–1.42, P = 0.049; DBP PRS: HR 1.28, 95% CI 1.07–1.53, P = 0.006, adjusted for age, sex, and alcohol use).
Antihypertensive medication modifies the association between genetic predisposition for HTA and liver-related outcomes
After excluding 8044 individuals with antihypertensive medication use at or before baseline, the cohort comprised 16,150 medication-naïve non-initiators and 9576 medication-naïve new initiators (Table 1). Among the new initiators, mean time from study baseline to initiation of medication was 7.4 years (median 6.3 years, IQR 3.1–10.6). During follow-up, there were 180 liver-related outcomes among non-initiators and 141 among new initiators.
SBP PRS and DBP PRS were significantly associated with liver-related outcomes independently of antihypertensive medication exposure in the fully adjusted models (Table 3). Initiation of medication after baseline was non-significant. However, when accounting for non-linearity of the PRSs, there was a tendency towards reduced rates of liver outcomes among medication new initiators at the highest end of the SBP and DBP PRSs (Figs. 2 and 3). In fact, in the highest quintile of the SBP PRS (n = 5058 with 80 liver-related outcomes), new initiation of antihypertensive medication was associated with a reduced rate of liver-related outcomes (HR 0.55, 95% CI 0.31–0.97, P = 0.039, fully adjusted Cox model).
Table 3.
Joint associations and interaction between hypertension polygenic risk scores (PRSs) and antihypertensive medication for liver-related outcomes among 25,726 subjects that were medication-naïve at baseline. Analyses are by Cox regression analyses.
| Overall (n = 25,726) | Excluding subjects with medication started > 5 years after baseline (n = 23,855) | |||
|---|---|---|---|---|
| HRa (95% CI) | P | HRa (95% CI) | P | |
| Model with SBP PRS and medication | ||||
| SBP PRS (per 1 SD) | 1.28 (1.13–1.46) | < 0.001 | 1.33 (1.14–1.55) | < 0.001 |
| Antihypertensive medication | 0.93 (0.72–1.21) | 0.598 | 0.76 (0.53–1.08) | 0.127 |
| Interaction term SBP PRS * antihypertensive medication | 0.79 (0.63–0.98) | 0.038 | 0.73 (0.53–1.02) | 0.064 |
| Model with DBP PRS and medication | ||||
| DBP PRS (per 1 SD) | 1.25 (1.10–1.42) | < 0.001 | 1.37 (1.18–1.59) | < 0.001 |
| Antihypertensive medication | 0.94 (0.72–1.21) | 0.615 | 0.71 (0.49–1.03) | 0.074 |
| Interaction term DBP PRS * antihypertensive medication | 0.81 (0.65–1.02) | 0.072 | 0.93 (0.67–1.29) | 0.661 |
CI, confidence interval; HR, hazards ratio; SBP, systolic blood pressure; DBP, diastolic blood pressure; PRS, polygenic risk score.
aAdjusted for age, sex, body mass index, waist circumference, weekly alcohol use, fraction of alcohol use as wine, low-density lipoprotein cholesterol, high-density lipoprotein cholesterol, triglycerides, diabetes, exercise habits, smoking status (current, former, never smoker) and baseline cardiovascular disease, genotyping chip and the first three principal components of genetic structure.
Figure 2.
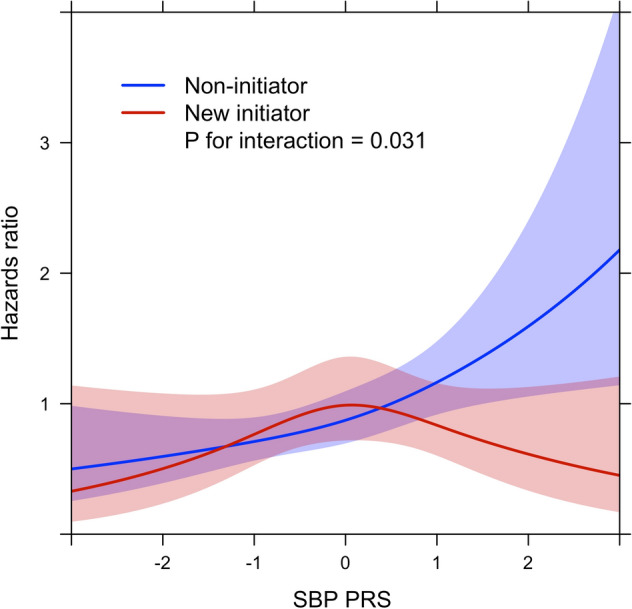
Interaction between systolic blood pressure polygenic risk score (SBP PRS) and new initiation of antihypertensive medication for liver-related outcomes by time-dependent Cox regression analysis using restricted cubic splines. SBP PRS is standardized as per 1 SD. The light red and light blue areas reflect 95% confidence intervals.
Figure 3.
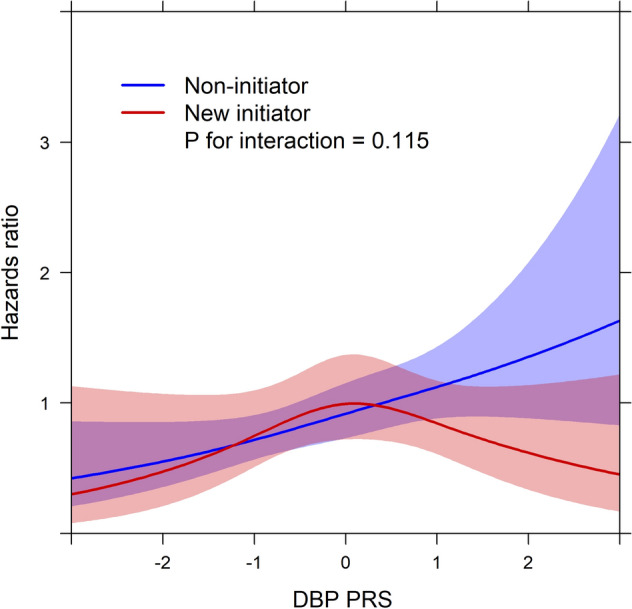
Interaction between diastolic blood pressure polygenic risk score (DBP PRS) and new initiation of antihypertensive medication for liver-related outcomes by time-dependent Cox regression analysis using restricted cubic splines. DBP PRS is standardized as per 1 SD. The light red and light blue areas reflect 95% confidence intervals.
In a sensitivity analysis excluding 1871 subjects with antihypertensive medication initiated > 5 years after baseline, the findings were fairly similar to those in the overall population (Table 3).
Discussion
We found that genetic predisposition to HTA is independently associated with incident liver-related outcomes in the general population. A similar association was found for HTA defined by blood pressure measurements and antihypertensive medication use at baseline. Furthermore, initiation of antihypertensive medication after study baseline was associated with a reduction in the rate of liver-related outcomes in individuals with a high genetic risk of HTA compared to a lack of antihypertensive medication exposure in such individuals.
Strengths of our study include the large and well-characterized health-examination survey cohort considered representative of the general population, with linkage to comprehensive and reliable national healthcare registers27,28. Data regarding antihypertensive medication use from the drug reimbursement register are also virtually complete. We were able to account for medications started any time during the follow-up, which allowed modelling drug exposure in a time-dependent fashion. Genetic risk of hypertension, as assessed by the previously developed PRSs26, overcomes issues with the time-varying nature of measured blood pressures and hypertension as exposures, and is subject to less confounding than single blood pressure measurements. Furthermore, measured blood pressure may be a confounder on the causal pathway between alcohol and liver disease; specifically, alcohol intake may be underestimated and/or high blood pressure might reflect increased sensitivity to alcoholic toxicity. The PRSs should not be affected by such issues.
Study limitations include the inability to account for time-varying confounding since phenotype variables were recorded only at study baseline. Still, the PRSs are unaffected by changes in lifestyle and environmental factors over time. Regarding antihypertensive medication, initiation of medication late after study baseline probably means that the person has over time acquired additional risk factors of HTA, many of which are also risk factors for liver disease. In this situation, adjustment for time-varying confounding would expectedly amplify the beneficial impact on liver-related outcomes of antihypertensive medication in the high-risk PRS groups, but this remains to be shown.
Baseline alcohol use was defined by questionnaires with its inherent limitations. Also, data on medication type and doses used were unavailable, and the statistical power to address this was considered inadequate. The drug exposure analysis represents intention to treat, and duration of treatment was not explicitly defined. Antihypertensive medications can also be used for other indications than blood pressure treatment; nonetheless, this should not affect the interaction effects between antihypertensive medication and genetic HTA risk for liver outcomes.
Numerous previous studies have reported associations between HTA and liver enzymes, steatosis, fibrosis, and liver-related outcomes3,6–24. One study even found that the association with liver-related outcomes was stronger for HTA than for other metabolic traits21. None of these previous studies, however, used polygenic risk scores to define hypertension.
Although our findings together with previous ones support the existence of a link between HTA and liver disease, possible pleiotropy and residual confounding may still prevent definitive conclusions. For the same reasons, the assumptions of Mendelian randomization analysis do not hold40, and we did therefore not engage in Mendelian randomization analyses in this study. Future studies with larger sample sizes should investigate specific genetic variants to elucidate possible mechanisms between HTA and liver disease.
Proposed mechanisms linking HTA with liver disease include activation of the renin–angiotensin–aldosterone system (RAAS), insulin resistance, and endothelial dysfunction-related arterial stiffness4. Experimental studies suggest a role of the RAAS in the progression of liver fibrosis41–45, and RAAS inhibitors have been associated with beneficial effects on the liver in a multitude of animal studies46–48. It has also been shown that activated human hepatic stellate cells, which are responsible for fibrogenesis in the liver, express components of the RAAS and synthesize angiotensin II, suggesting that locally generated angiotensin II could participate in tissue remodeling in the human liver43. A trend towards improvements in liver enzyme levels with the use of RAAS inhibitors has been found in clinical trials of NAFLD patients, but results are inconsistent and studies have generally been small with short duration of follow-up49. One observational study with a median follow-up of 36 months reported that the use of RAAS inhibitors in patients with NAFLD and type 2 diabetes was associated with a slower liver fibrosis progression rate50. Prospective trials with long follow-up are needed to confirm whether a good blood pressure control can contribute to slowing down the progression of liver fibrosis and decreasing the risk for liver-related clinical outcomes, and whether such potential effects are related to specific medication types (e.g. RAAS inhibitors) or antihypertensive treatment in general.
In conclusion, HTA was independently associated with liver disease. Such an association was found both for HTA defined clinically and by polygenic risk scores. New initiation of antihypertensive medication was associated with reduced rates of liver-related outcomes in persons with high genetic HTA risk.
Supplementary Information
Acknowledgements
FINRISK and Health 2000 data used for the research were obtained from THL Biobank (study number: BB2019_31). We thank all study participants for their generous participation at THL Biobank and the FINRISK 1992-2012 studies and Health 2000 Survey. FinnGen is acknowledged for imputation of the genotyped data. The data used for the research was imputed with the THL Biobank's SISu v3 imputation reference panel. We thank the Sequencing Informatics Team, FIMM Human Genomics, University of Helsinki, for the work done in preparation of the reference panel data. We thank Pietro Della Briotta Parolo for developing CS-PRS pipeline (https://github.com/FINNGEN/CS-PRS-pipeline).
Abbreviations
- HTA
Arterial hypertension
- PRS
Polygenic risk score
- NAFLD
Non-alcoholic fatty liver disease
- BMI
Body mass index
- SBP
Systolic blood pressure
- DBP
Diastolic blood pressure
- MAC
Minor allele count
- ATC
Anatomical Therapeutic Chemical Classification System codes
- HILMO
Care Register for Health Care
- LDL
Low-density lipoprotein
- HDL
High-density lipoprotein
- HR
Hazards ratio
- CI
Confidence interval
Author contributions
F.Å. designed the study, performed analyses and wrote the first draft; K.K. calculated polygenic risk scores and provided critical revision; V.M. participated in study design, critical revision; AB, statistical consultancy, critical revision; V.S., T.N., S.M., A.L. and M.P. participated in design and data collection of the cohort data, critical revision of manuscript; P.L. and M.F., participated in study design, critical revision; A.J. participated in design and data collection of the cohorts, participated in study design, critical revision of manuscript, senior author.
Funding
Dr. Åberg was supported by the Mary and Georg Ehrnrooth Foundation, Medicinska Understödsföreningen Liv och Hälsa, Finska Läkaresällskapet, Academy of Finland (#338544), and Sigrid Jusélius Foundation. Dr. Männistö was supported by The Finnish Medical Foundation and Mary and Georg Ehrnrooth Foundation. Dr. Luukkonen was supported by the Novo Nordisk and Sigrid Jusélius Foundations. Dr. Salomaa was supported by the Finnish Foundation for Cardiovascular Research. The researchers are all independent of the funders. The funding sources had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Data availability
FINRISK and Health 2000 data are available from the THL biobank based on a research application, as explained on the website of the THL biobank (https://thl.fi/en/web/thl-biobank/for-researchers).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-022-20084-z.
References
- 1.Brouwers S, Sudano I, Kokubo Y, Sulaica EM. Arterial hypertension. Lancet. 2021;398:249–261. doi: 10.1016/S0140-6736(21)00221-X. [DOI] [PubMed] [Google Scholar]
- 2.Ramsay LE. Liver dysfunction in hypertension. Lancet. 1977;2:111–114. doi: 10.1016/S0140-6736(77)90121-0. [DOI] [PubMed] [Google Scholar]
- 3.Brookes MJ, Cooper BT. Hypertension and fatty liver: Guilty by association? J. Hum. Hypertens. 2007;21:264–270. doi: 10.1038/sj.jhh.1002148. [DOI] [PubMed] [Google Scholar]
- 4.Oikonomou D, Georgiopoulos G, Katsi V, Kourek C, Tsioufis C, Alexopoulou A, et al. Non-alcoholic fatty liver disease and hypertension: Coprevalent or correlated? Eur. J. Gastroenterol. Hepatol. 2018;30:979–985. doi: 10.1097/MEG.0000000000001191. [DOI] [PubMed] [Google Scholar]
- 5.Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, et al. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018;15:11–20. doi: 10.1038/nrgastro.2017.109. [DOI] [PubMed] [Google Scholar]
- 6.Long MT, Zhang X, Xu H, Liu C-T, Corey KE, Chung RT, et al. Hepatic fibrosis associates with multiple cardiometabolic disease risk factors: The Framingham Heart Study. Hepatology. 2021;73:548–559. doi: 10.1002/hep.31608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bedogni G, Miglioli L, Masutti F, Tiribelli C, Marchesini G, Bellentani S. Prevalence of and risk factors for nonalcoholic fatty liver disease: The Dionysos nutrition and liver study. Hepatology. 2005;42:44–52. doi: 10.1002/hep.20734. [DOI] [PubMed] [Google Scholar]
- 8.Wong VW-S, Chu WC-W, Wong GL-H, Chan RS-M, Chim AM-L, Ong A, et al. Prevalence of non-alcoholic fatty liver disease and advanced fibrosis in Hong Kong Chinese: A population study using proton-magnetic resonance spectroscopy and transient elastography. Gut. 2012;61:409–415. doi: 10.1136/gutjnl-2011-300342. [DOI] [PubMed] [Google Scholar]
- 9.Suomela E, Oikonen M, Virtanen J, Parkkola R, Jokinen E, Laitinen T, et al. Prevalence and determinants of fatty liver in normal-weight and overweight young adults. The Cardiovascular Risk in Young Finns Study. Ann Med. 2015;47:40–46. doi: 10.3109/07853890.2014.966752. [DOI] [PubMed] [Google Scholar]
- 10.Dixon JB, Bhathal PS, O’Brien PE. Nonalcoholic fatty liver disease: Predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology. 2001;121:91–100. doi: 10.1053/gast.2001.25540. [DOI] [PubMed] [Google Scholar]
- 11.Donati G, Stagni B, Piscaglia F, Venturoli N, Morselli-Labate AM, Rasciti L, et al. Increased prevalence of fatty liver in arterial hypertensive patients with normal liver enzymes: Role of insulin resistance. Gut. 2004;53:1020–1023. doi: 10.1136/gut.2003.027086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aneni EC, Oni ET, Martin SS, Blaha MJ, Agatston AS, Feldman T, et al. Blood pressure is associated with the presence and severity of nonalcoholic fatty liver disease across the spectrum of cardiometabolic risk. J. Hypertens. 2015;33:1207–1214. doi: 10.1097/HJH.0000000000000532. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Zhang T, Zhang C, Tang F, Zhong N, Li H, et al. Identification of reciprocal causality between non-alcoholic fatty liver disease and metabolic syndrome by a simplified Bayesian network in a Chinese population. BMJ Open. 2015;5:e008204. doi: 10.1136/bmjopen-2015-008204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang T, Zhang C, Zhang Y, Tang F, Li H, Zhang Q, et al. Metabolic syndrome and its components as predictors of nonalcoholic fatty liver disease in a northern urban Han Chinese population: A prospective cohort study. Atherosclerosis. 2015;240:144–148. doi: 10.1016/j.atherosclerosis.2015.02.049. [DOI] [PubMed] [Google Scholar]
- 15.Gawrieh S, Wilson LA, Cummings OW, Clark JM, Loomba R, Hameed B, et al. Histologic findings of advanced fibrosis and cirrhosis in patients with nonalcoholic fatty liver disease who have normal aminotransferase levels. Am. J. Gastroenterol. 2019;114:1626–1635. doi: 10.14309/ajg.0000000000000388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Labenz C, Huber Y, Kalliga E, Nagel M, Ruckes C, Straub BK, et al. Predictors of advanced fibrosis in non-cirrhotic non-alcoholic fatty liver disease in Germany. Aliment. Pharmacol. Ther. 2018;48:1109–1116. doi: 10.1111/apt.14976. [DOI] [PubMed] [Google Scholar]
- 17.Bazerbachi F, Haffar S, Wang Z, Cabezas J, Arias-Loste MT, Crespo J, et al. Range of normal liver stiffness and factors associated with increased stiffness measurements in apparently healthy individuals. Clin. Gastroenterol. Hepatol. 2019;17:54–64.e1. doi: 10.1016/j.cgh.2018.08.069. [DOI] [PubMed] [Google Scholar]
- 18.Nabi O, Lacombe K, Boursier J, Mathurin P, Zins M, Serfaty L. Prevalence and risk factors of nonalcoholic fatty liver disease and advanced fibrosis in general population: The French Nationwide NASH-CO Study. Gastroenterology. 2020;159:791–793.e2. doi: 10.1053/j.gastro.2020.04.048. [DOI] [PubMed] [Google Scholar]
- 19.Singh, S., Allen, A. M., Wang, Z., Prokop, L. J., Murad, M. H., & Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015;13:643–654.e1–9; quiz e39–40. [DOI] [PMC free article] [PubMed]
- 20.Björkström K, Franzén S, Eliasson B, Miftaraj M, Gudbjörnsdottir S, Trolle-Lagerros Y, et al. Risk factors for severe liver disease in patients with type 2 diabetes. Clin. Gastroenterol. Hepatol. 2019;17:2769–2775.e4. doi: 10.1016/j.cgh.2019.04.038. [DOI] [PubMed] [Google Scholar]
- 21.Kanwal F, Kramer JR, Li L, Dai J, Natarajan Y, Yu X, et al. Effect of metabolic traits on the risk of cirrhosis and hepatocellular cancer in nonalcoholic fatty liver disease. Hepatology. 2020;71:808–819. doi: 10.1002/hep.31014. [DOI] [PubMed] [Google Scholar]
- 22.Borena W, Strohmaier S, Lukanova A, Bjørge T, Lindkvist B, Hallmans G, et al. Metabolic risk factors and primary liver cancer in a prospective study of 578,700 adults. Int. J. Cancer. 2012;131:193–200. doi: 10.1002/ijc.26338. [DOI] [PubMed] [Google Scholar]
- 23.Stocks T, Van Hemelrijck M, Manjer J, Bjørge T, Ulmer H, Hallmans G, et al. Blood pressure and risk of cancer incidence and mortality in the metabolic syndrome and cancer project. Hypertension. 2012;59:802–810. doi: 10.1161/HYPERTENSIONAHA.111.189258. [DOI] [PubMed] [Google Scholar]
- 24.Azuma S, Asahina Y, Kakinuma S, Azuma K, Miyoshi M, Inoue E, et al. Diabetic retinopathy as a risk factor associated with the development of hepatocellular carcinoma in nonalcoholic fatty liver disease. Dig. Dis. 2019;37:247–254. doi: 10.1159/000493580. [DOI] [PubMed] [Google Scholar]
- 25.Torkamani A, Wineinger NE, Topol EJ. The personal and clinical utility of polygenic risk scores. Nat. Rev. Genet. 2018;19:581–590. doi: 10.1038/s41576-018-0018-x. [DOI] [PubMed] [Google Scholar]
- 26.Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat. Genet. 2018;50:1412–1425. doi: 10.1038/s41588-018-0205-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Borodulin K, Tolonen H, Jousilahti P, Jula A, Juolevi A, Koskinen S, et al. Cohort profile: The National FINRISK Study. Int. J. Epidemiol. 2018;47:696–696i. doi: 10.1093/ije/dyx239. [DOI] [PubMed] [Google Scholar]
- 28.Aromaa, A., & Koskinen, S. Health and Functional Capacity in Finland: Baseline Results of the Health 2000 health Examination Survey. Publications of National Public Health Institute, Series B 12/2004 Helsinki, Finland (2004).
- 29.Loh P-R, Danecek P, Palamara PF, Fuchsberger C, Reshef Y, Finucane H, et al. Reference-based phasing using the haplotype reference consortium panel. Nat. Genet. 2016;48:1443–1448. doi: 10.1038/ng.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Browning BL, Browning SR. Genotype imputation with millions of reference samples. Am. J. Hum. Genet. 2016;98:116–126. doi: 10.1016/j.ajhg.2015.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.GRASP Updates. https://grasp.nhlbi.nih.gov/FullResults.aspx. Accessed 16 Oct 2021.
- 32.Ge T. PRS-CS. Python (2021).
- 33.Ge T, Chen C-Y, Ni Y, Feng Y-CA, Smoller JW. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 2019;10:1776. doi: 10.1038/s41467-019-09718-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sund R. Quality of the Finnish Hospital Discharge Register: A systematic review. Scand. J. Public Health. 2012;40:505–515. doi: 10.1177/1403494812456637. [DOI] [PubMed] [Google Scholar]
- 36.Pukkala E. Biobanks and registers in epidemiologic research on cancer. Methods Mol. Biol. 2011;675:127–164. doi: 10.1007/978-1-59745-423-0_5. [DOI] [PubMed] [Google Scholar]
- 37.Alberti KGMM, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, et al. Harmonizing the metabolic syndrome: A joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 38.Unger T, Borghi C, Charchar F, Khan NA, Poulter NR, Prabhakaran D, et al. International society of hypertension global hypertension practice guidelines. Hypertension. 2020;75:1334–1357. doi: 10.1161/HYPERTENSIONAHA.120.15026. [DOI] [PubMed] [Google Scholar]
- 39.R Core Team. R: Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (2022). https://www.R-project.org/
- 40.Latvala A, Ollikainen M. Mendelian randomization in (epi)genetic epidemiology: An effective tool to be handled with care. Genome Biol. 2016;17:156. doi: 10.1186/s13059-016-1018-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pereira RM, dos Santos RAS, da Costa Dias FL, Teixeira MM, Simões e Silva AC. Renin-angiotensin system in the pathogenesis of liver fibrosis. World J. Gastroenterol. 2009;15:2579–2586. doi: 10.3748/wjg.15.2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshiji H, Kuriyama S, Yoshii J, Ikenaka Y, Noguchi R, Nakatani T, et al. Angiotensin-II type 1 receptor interaction is a major regulator for liver fibrosis development in rats. Hepatology. 2001;34(4 Pt 1):745–750. doi: 10.1053/jhep.2001.28231. [DOI] [PubMed] [Google Scholar]
- 43.Bataller R, Sancho-Bru P, Ginès P, Lora JM, Al-Garawi A, Solé M, et al. Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology. 2003;125:117–125. doi: 10.1016/S0016-5085(03)00695-4. [DOI] [PubMed] [Google Scholar]
- 44.Park JG, Mok JS, Han YI, Park TS, Kang KW, Choi CS, et al. Connectivity mapping of angiotensin-PPAR interactions involved in the amelioration of non-alcoholic steatohepatitis by Telmisartan. Sci. Rep. 2019;9:4003. doi: 10.1038/s41598-019-40322-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu Y, Ma KL, Zhang Y, Wen Y, Wang GH, Hu ZB, et al. Lipid disorder and intrahepatic renin-angiotensin system activation synergistically contribute to non-alcoholic fatty liver disease. Liver Int. 2016;36:1525–1534. doi: 10.1111/liv.13131. [DOI] [PubMed] [Google Scholar]
- 46.Yokohama S, Yoneda M, Haneda M, Okamoto S, Okada M, Aso K, et al. Therapeutic efficacy of an angiotensin II receptor antagonist in patients with nonalcoholic steatohepatitis. Hepatology. 2004;40:1222–1225. doi: 10.1002/hep.20420. [DOI] [PubMed] [Google Scholar]
- 47.Moreno M, Gonzalo T, Kok RJ, Sancho-Bru P, van Beuge M, Swart J, et al. Reduction of advanced liver fibrosis by short-term targeted delivery of an angiotensin receptor blocker to hepatic stellate cells in rats. Hepatology. 2010;51:942–952. doi: 10.1002/hep.23421. [DOI] [PubMed] [Google Scholar]
- 48.Namisaki T, Noguchi R, Moriya K, Kitade M, Aihara Y, Douhara A, et al. Beneficial effects of combined ursodeoxycholic acid and angiotensin-II type 1 receptor blocker on hepatic fibrogenesis in a rat model of nonalcoholic steatohepatitis. J. Gastroenterol. 2016;51:162–172. doi: 10.1007/s00535-015-1104-x. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Xu H, Wu W, Ye J, Fang D, Shi D, et al. Clinical application of angiotensin receptor blockers in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis. Oncotarget. 2018;9:24155–24167. doi: 10.18632/oncotarget.23816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pelusi S, Petta S, Rosso C, Borroni V, Fracanzani AL, Dongiovanni P, et al. Renin-angiotensin system inhibitors, type 2 diabetes and fibrosis progression: An observational study in patients with nonalcoholic fatty liver disease. PLoS ONE. 2016;11:e0163069. doi: 10.1371/journal.pone.0163069. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
FINRISK and Health 2000 data are available from the THL biobank based on a research application, as explained on the website of the THL biobank (https://thl.fi/en/web/thl-biobank/for-researchers).