

Abstract
Basic science breakthroughs in T cell biology and immune-tumor cell interactions ushered in a new era of cancer immunotherapy. Twenty years ago, cancer immunoediting was proposed as a framework to understand the dynamic process by which the immune system can both control and shape cancer and in its most complex form occurs through three phases termed elimination, equilibrium, and escape. During cancer progression through these phases, tumors undergo immunoediting, rendering them less immunogenic and more capable of establishing an immunosuppressive microenvironment. Therefore, cancer immunoediting integrates the complex immune-tumor cell interactions occurring in the tumor microenvironment and sculpts immunogenicity beyond shaping antigenicity. However, with the success of cancer immunotherapy resulting in durable clinical responses in the last decade and subsequent emergence of immuno-oncology as a clinical subspecialty, the phrase “cancer immunoediting” has recently, at times, been inappropriately restricted to describing neoantigen loss by immunoselection. This focus has obscured other mechanisms by which cancer immunoediting modifies tumor immunogenicity. While establishment of the concept of cancer immunoediting and definitive experimental evidence supporting its existence was initially obtained from pre-clinal models in the absence of immunotherapy, cancer immunoediting is a continual process that also occurs during immunotherapy in human cancer patients. Herein, we discuss the known mechanisms of cancer immunoediting obtained from preclinical and clinical data with an emphasis on how a greater understanding of cancer immunoediting may provide insights into immunotherapy resistance and how this resistance can be overcome.
Introduction
Cancer immunotherapy has emerged as a pillar of cancer therapy. Immune checkpoint therapy (ICT) with monoclonal antibodies targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1)/PD-L1 are standard of care treatments for certain patients with a range of different cancer types (1–5). Adoptive cell therapy (ACT) regimens using chimeric antigen receptor (CAR) T cells are FDA-approved treatments for subsets of leukemia, lymphoma, and advanced multiple myeloma patients (6). However, twenty years ago there was less interest in immuno-oncology and only relatively recently has there been a shift in focus from cancer cells to the tumor microenvironment (TME) and the critical interactions between immune cells and tumor cells.
The immune system can both restrain tumor progression and paradoxically promote tumor evolution and progression. Cancer immunoediting was proposed in the early 2000’s as a framework to understand the complex interactions between immune cells and tumors (7, 8). Cancer immunoediting initiates after transformation has occurred and may lead to elimination of transformed cells. If, however, the immune system does not completely eliminate the growing tumor cells, the cancer may enter a state of immune-mediated equilibrium. Immunological shaping of the tumor and establishment of a suppressive TME may lead to tumor escape and subsequent outgrowth. Cancer immunoediting is a continual process that occurs not only during the development and progression of tumors, but also occurs in patients treated with cancer immunotherapies, where the therapy affects the immunoediting process. This review will provide a brief history of cancer immunoediting and will place the concept in current context with a focus on emerging evidence in humans and implications for immunotherapy treatments moving forward.
Cancer Immunosurveillance
For a century, the concept that the immune system could detect and kill cancer cells, called cancer immunosurveillance, was wrought with controversy. Icons of 20th-century immunology, including Paul Ehrlich, F. Macfarlane Burnet and Lewis Thomas hypothesized that cancer immunosurveillance occurs, but definitive evidence was lacking until the turn of the 21st-century (8). With robust mouse tumor models, Robert Schreiber and colleagues provided initial evidence that cancer immunosurveillance, indeed, occurs in immunocompetent hosts (9). Moreover, Schreiber and colleagues demonstrated that the immune system both destroys cancer cells and shapes its outgrowth. This dual function of immunity to both control cancer (elimination, equilibrium) and promote cancer (escape) became the foundation for the cancer immunoediting hypothesis (7, 8). In this context, the original idea of cancer immunosurveillance becomes part of the elimination phase of cancer immunoediting. It has mistakenly become synonymous with immunoediting as detection and destruction of cancer cells by antigen-specific T cells. Rather, the cancer immunoediting hypothesis integrates the complex tumor-immune cell interactions occurring within the TME to sculpt immunogenicity beyond shaping antigenicity.
Cancer Immunoediting: an integrative hypothesis
Cancer immunoediting is a concept that emphasizes the dual host-protective and tumor-sculpting actions of the immune system. It is postulated that cancer immunoediting consists of three phases: Elimination, Equilibrium, and Escape (Figure 1). Cancer immunoediting engages after transformation has occurred and non-immunological intrinsic tumor suppressive mechanisms have failed. Elimination is the first phase of cancer immunoediting, whereby the innate and adaptive immune system act in concert to destroy the nascent tumor, thus leading to tumor “elimination”, the main tenet of the cancer immunosurveillance hypothesis. If, however, the immune system fails to eradicate the growing tumor cells, the cancer may enter the equilibrium phase where its outgrowth is immunologically restrained, but the cancer is not eliminated (10). The equilibrium phase can be a prolonged event, but further immunological sculpting of the tumor and establishment of a suppressive TME may lead to the escape phase of cancer immunoediting, where the clinically apparent disease of cancer manifests. It is the escape phase of cancer immunoediting that is often overlooked. During this phase, tumor cells may recruit immune cells to create an immunosuppressive TME and immune cells may induce tumor cells to express immune checkpoint molecules. Much of the initial experimental evidence supporting the cancer immunoediting hypothesis came from studies in the mouse MCA sarcoma model (8, 11). These studies uncovered the importance of several immune cell populations (e.g., T cells, NK cells) and molecules (e.g., type I and type II IFNs, FASL, TRAIL, Perforin) during cancer immunoediting. These findings have largely been consistent with subsequent data from a variety of animal tumor models and human cancer patients with additional cell types and molecules having also been uncovered as being integral during cancer immunoediting. The specific cells and effector molecules known to be involved in cancer immunoediting have been reviewed extensively elsewhere (11, 12).
Figure 1: The cancer immunoediting process.
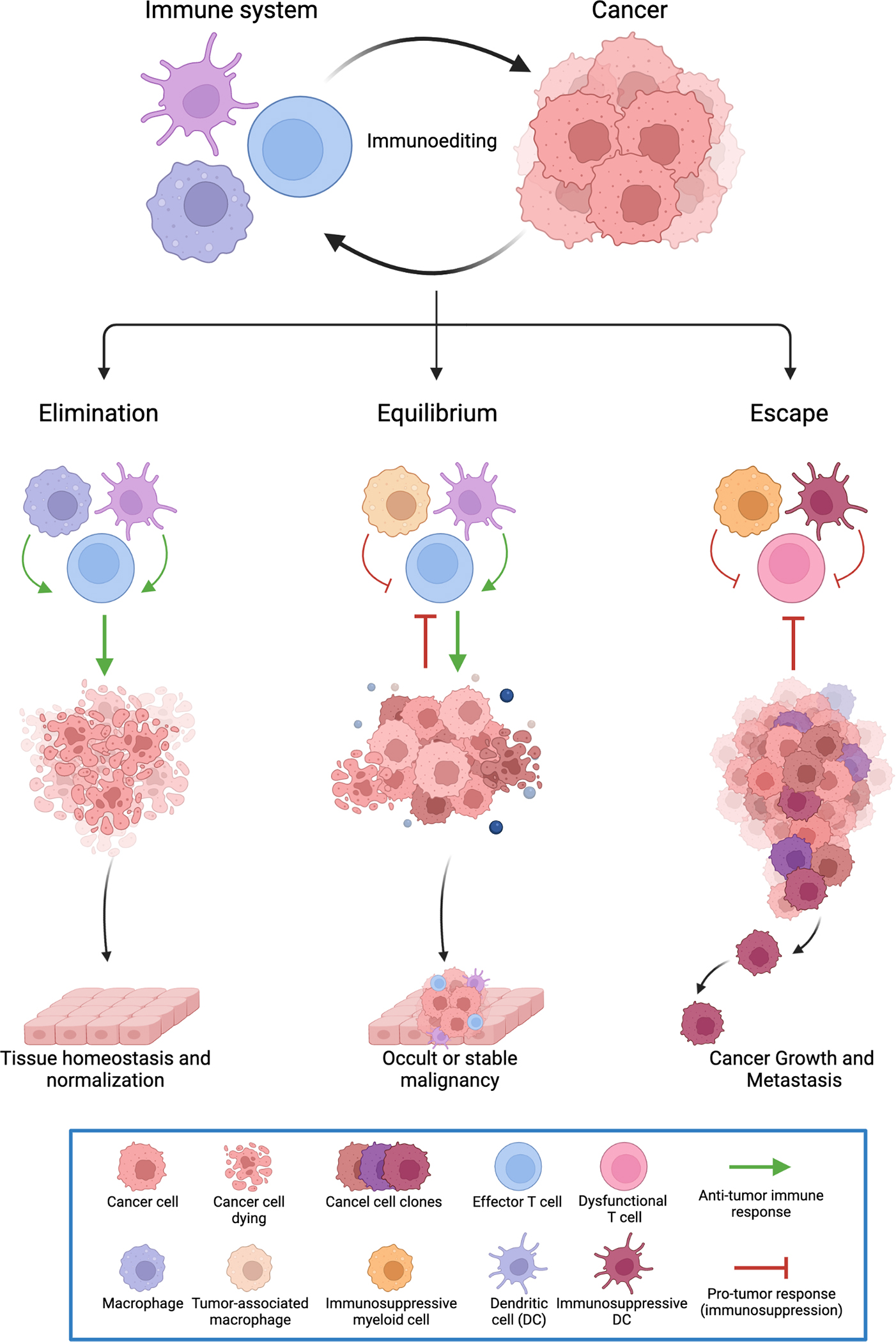
The cancer immunoediting hypothesis provides a framework to decipher the dynamic and complex interactions between immune cells and cancer cells within the tumor microenvironment. These interactions may be anti-tumor or pro-tumor; thereby highlighting the dual role of immune cells in both preventing tumors from growing and/or enhancing tumor growth. Cancer immunoediting consists of three phases: Elimination, Equilibrium, and Escape. Elimination is the first phase of cancer immunoediting, whereby the innate and adaptive immune system act in concert to destroy the nascent tumor, thus leading to tumor destruction and tissue homeostasis or normalization. When the immune system fails to eradicate the tumor cells, the cancer may enter the equilibrium phase where its outgrowth is immunologically restrained, but the cancer is not eliminated. Further immunological sculpting of the tumor and establishment of a suppressive tumor microenvironment may lead to the escape phase of cancer immunoediting, where the cancer becomes clinically apparent disease and spreads to other organs.
From mice to humans: a shift in nomenclature
With the success of immune checkpoint therapy (ICT) translated to human cancer patients, a new era in oncology began (Figure 2) (13). The advances in cancer immunotherapy in the late 20th century were primarily driven by immunologists studying fundamental mechanisms of T cell activation and suppression (14–16). In some pre-clinical mouse models of cancer, antibodies targeting CTLA-4 and PD-1/PD-L1 were shown to be effective in treating cancers (1, 17, 18). Successful clinical trials for treatment of melanoma with ipilimumab (anti-CTLA-4) (3) resulted in FDA approval in 2011, followed by anti-PD-1 therapies pembrolizumab (4) and nivolumab (5) for melanoma resulted in FDA approval in 2014. On the heels of these therapy approvals, a sub-specialty within the clinical field of oncology emerged, called immuno-oncology, that is dedicated to the study and development of cancer immunotherapies, the immunologic monitoring of responses and adverse events of patients treated with cancer immunotherapies, and integration of tailored clinical treatment paradigms that vary widely from previously used chemotherapy methodologies (19).
Figure 2: The development of immuno-oncology.
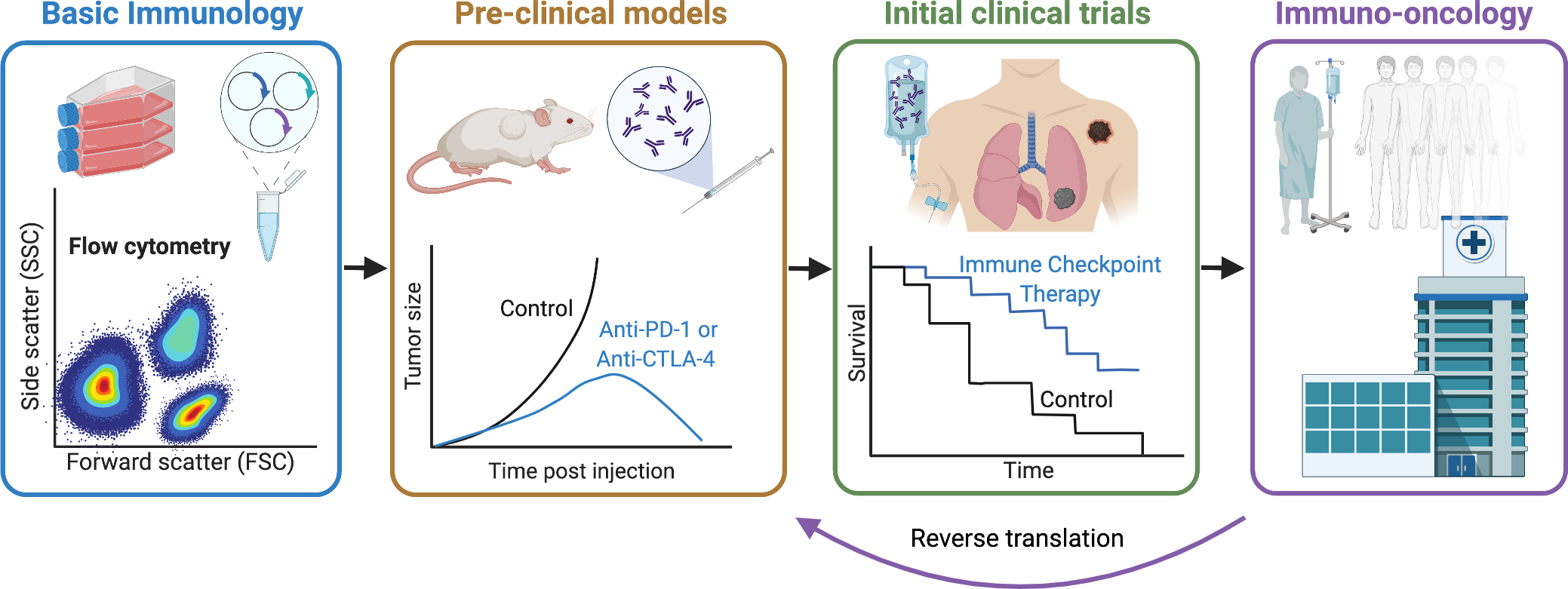
Fundamental insights into T cell activation and inhibition in the late 20th century provided the rationale to targeting immune inhibitory receptors such as CTLA-4 and PD-1 or PD-L1 to treat cancer in pre-clinical cancer models. At the beginning of the 21st century, successful targeting of “immune checkpoints” ushered in the first clinical trials in human cancer patients. Cancer immunotherapy provided greater response rates with longer duration than traditional chemotherapy for some cancers. Subsequently, the subspecialty immuno-oncology was borne dedicated to the study and development of cancer immunotherapies, the immunologic monitoring of responses and adverse events of patients treated with cancer immunotherapies and tailored clinical treatment paradigms. Insights gained from immuno-oncology are often then brought back to the research laboratory in ‘reverse translation’ to overcome challenges such as resistance to immunotherapy.
The impact of immunotherapy on oncology cannot be overstated. In 2010, the International Society for Biological Therapy of Cancer (iSBTc) changed its name to Society for Immunotherapy of Cancer (SITC) (20). In the same year the National Institutes of Health announced the formation of the Cancer Immunotherapy Trials Network. International collaborations between Cancer Research Institute and the Association for Cancer Immunotherapy among others helped to build the emerging framework of immuno-oncology that has accelerated clinical trial development for cancer patients receiving cancer immunotherapies (19). Since 2014, anti-PD-1/PD-L1 therapy that has now been approved for over 20 cancer indications and there were 4,400 clinical trials involving anti-PD-1/PD-L1 therapy as of September 2020 (21). Beyond anti-PD-1/PD-L1 therapy, there are thousands of cancer immunotherapy clinical trials currently ongoing globally with over 500 unique targets (22). This furious pace is a testament to the success of previous immunotherapies (anti-PD-1/PD-L1 and anti-CTLA-4) but has become a daunting landscape to navigate.
It is no surprise, then, that the nomenclature of immune responses against cancer has shifted towards those used in clinical settings. How we discuss the interactions between immune cells and cancers is now more focused on clinical outcomes of patients in response to therapy rather than the cellular dynamics of tumor cells and immune cells occurring in the TME. Therefore, in the scenario when the immune system completely eradicates cancer (elimination) in response to immunotherapy, the patient is designated a complete responder (Figure 3). Patients whose cancers enter an equilibrium state with the immune system in response to immunotherapy are often referred to as a partial responder, durable responder or have durable benefit. Finally, patients whose tumors eventually escape immune attack are referred to as non-durable responders or non-responders. This last scenario may be due to primary or secondary resistance to immunotherapy, where primary resistance occurs when tumors fail to respond to immunotherapy (non-responders) and secondary (acquired) resistance occurs when a tumor that initially responds to immunotherapy progressively grows (non-durable responders) (23). More work is needed to dissect the complex and dynamic process of immunoediting that is occurring within cancer patients treated with immunotherapy to improve clinical outcomes.
Figure 3: Cancer immunoediting occurs naturally and during cancer immunotherapy.
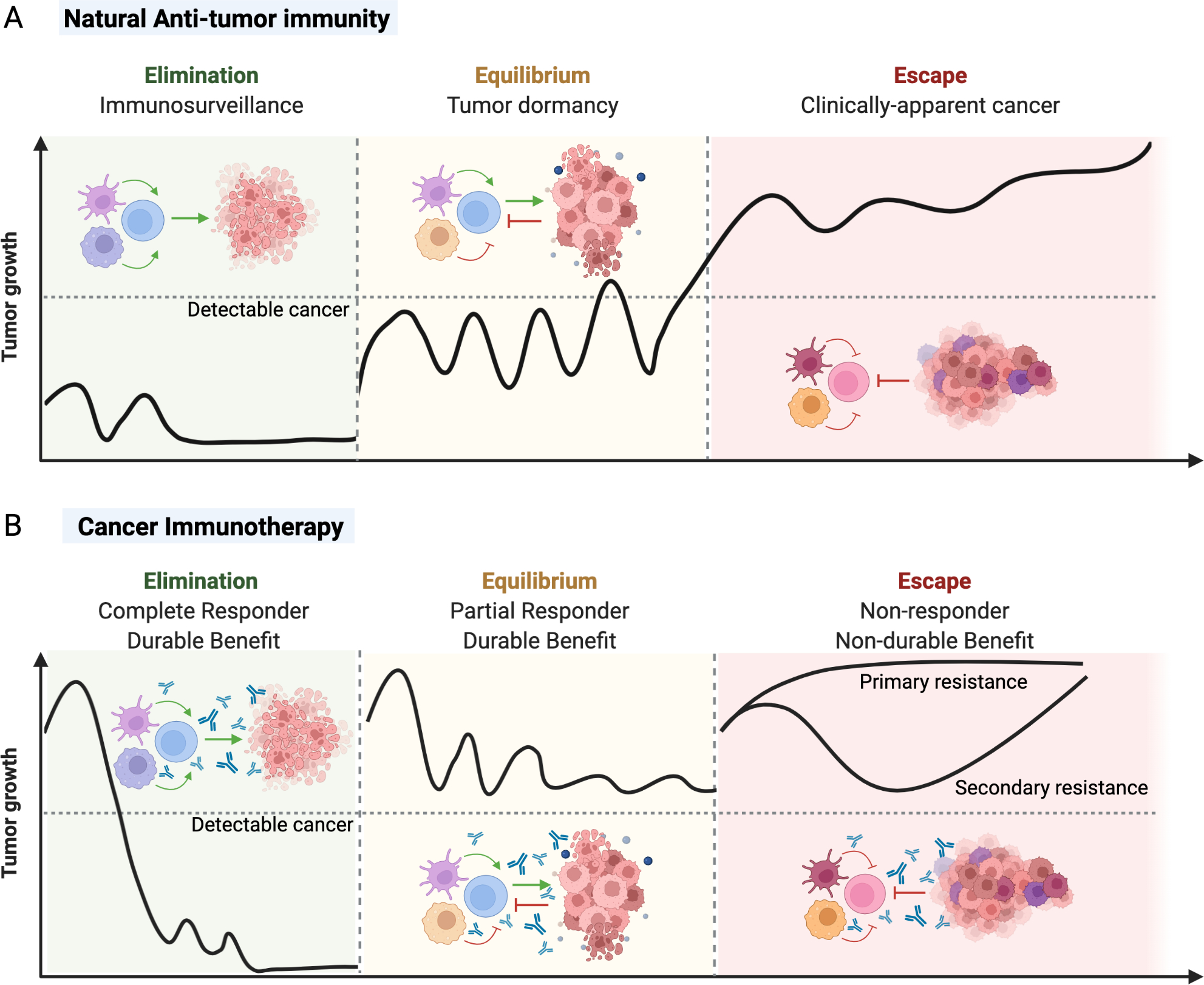
A) Cancer immunoediting was initially described during natural immune-tumor cell interactions in the absence of immunotherapy. During natural anti-tumor immunity, the immune system may detect and destroy a nascent tumor during the elimination phase, resulting in immunosurveillance and prevention of clinically detectable cancer. Tumors may enter a dynamic equilibrium between pro-tumor and anti-tumor effects, resulting in tumor dormancy. However, subsequent immunoediting of tumor immunogenicity may result in the development of an immunosuppressive tumor microenvironment and dysfunctional anti-tumor immune response that outweighs anti-tumor effects, resulting tumor escape and subsequent cancer. B) Cancer immunoediting is a continually process that also occurs during cancer immunotherapy of established advanced cancers in human patients. If successful, cancer immunotherapy can correct or normalize a dysfunctional immune response in the tumor microenvironment and drive the elimination of the tumor with resultant durable benefit. If complete elimination of the tumor is not achieved with immunotherapy, there could be an establishment of the equilibrium phase where the patient’s cancer partially responds to therapy and still has durable benefit. However, many cancers fail to respond to specific immunotherapies and continue to escape immune control. These non-responders either never respond to immunotherapy (primary resistance) or initially respond to treatment, but then undergo further immunoediting and subsequent escape (secondary or acquired resistance).
Cancer immunoediting: more than neoantigens
A fundamental principle of cancer immunoediting is that recognition of tumor antigens by T cells can drive the immunological elimination or sculpting of an emergent cancer. Indeed, preclinical work has demonstrated that epigenetic silencing of highly immunogenic tumor antigens (24) and T cell immunoselection against tumor clones expressing strong rejection antigens (25) represent mechanisms of cancer immunoediting. In human cancer patients, there is evidence of immunoediting via neoantigen loss in certain solid tumor types (26). By comparing the rate of predicted neoantigens formed from nonsilent mutations (per total nonsilent mutations) to the observed mutational rate of silent mutations observed in TCGA datasets, fewer predicted neoantigens than would be expected in colorectal and kidney clear cell cancers were found, suggesting that tumor neoantigens were subject to immune selection pressure. A separate study reported that untreated non-small-cell lung cancer (NSCLC) patients displaying high intratumoral immune infiltration exhibited enhanced hypermethylation of promoter regions of genes encoding predicted mutant neoantigens and thus, lack of neoantigen expression (27). These hypermethylation events occurred less frequently in the corresponding non-mutant form of the same genes in other tumors. Analysis of metastatic tumors from pancreatic ductal adenocarcinoma patients revealed selective loss of “high-quality” neoantigens (with characteristics associated with long-term survivors) upon metastatic progression (28). In metastatic colorectal cancer patients, longitudinal analysis of tumor evolution at distinct metastatic sites revealed a lower frequency of immunogenic mutations than expected in tumors with a high immune infiltrate containing proliferating T cells located in proximity to tumor cells, suggesting that cancer immunoediting favored the outgrowth of escape metastatic clones with fewer immunogenic neoantigens (29).
In addition to shaping the tumor antigenome, tumor-intrinsic loss of antigen presentation likely represents an additional mechanism of cancer immunoediting that, in some circumstances, may lead to primary or secondary resistance to immunotherapy (30–33). Whole-exome cancer sequencing studies have indicated a relatively high frequency of somatic changes, including putative loss-of-function mutations in genes encoding human leukocyte antigen (HLA)/major histocompatibility complex (MHC) and B2M (encoding an essential component of HLA/MHC class I (HLA-I/MHC-I)) (26, 34–39). Although downregulation of HLA-I/MHC-I molecules can prompt natural killer (NK) cell-mediated tumor cell killing, tumor cell shedding of NK co-stimulatory molecules, tumor upregulation of anti-apoptotic molecules, and cytokines frequently observed within the tumor microenvironment can facilitate avoidance of immune-mediated tumor killing (40–42). Nevertheless, NK cells (and other innate lymphoid cells (ILCs)), as well as NKT cells do, indeed participate in the cancer immunoediting process (43, 44).
Alterations of tumor-intrinsic IFN-γ signaling can also lead to altered antigen processing and defects in tumor expression of HLA-I/MHC-I and, although not as commonly expressed in solid tumors, HLA-II/MHC-II (45). Human melanoma and lung adenocarcinoma cell lines that were found to be unresponsive to IFN-γ displayed defects in antigen presentation (9, 46) and in preclinical models, tumors rendered insensitive to IFN-γ resist immune rejection via defects in HLA-I/MHC-I antigen presentation (30). More recent work involving CRISPR screens in human tumor cell lines and mouse tumor models revealed that knocking out genes encoding components of IFN-γ signaling was associated with diminished T cell recognition of tumor cells and insensitivity to ICT (47, 48). Consistent with these findings, loss-of-function mutations in genes essential for IFN-γ signaling have been observed in tumors from some patients with primary resistance to immunotherapy (33, 49, 50). It should also be noted that IFN-γ can suppress tumor growth through mechanisms not directly related to antigen presentation, including by directly suppressing tumor cell proliferation (46, 49).
While cancer immunoediting shapes the tumor antigenome and subsequent antigenicity, the phrase “cancer immunoediting” has recently, at times, been inappropriately restricted to describing neoantigen loss by immunoselection. This focus has obscured other mechanisms by which cancer immunoediting modifies tumor immunogenicity. Avoidance of immune elimination and progression to either the equilibrium or escape phases of cancer immunoediting can occur through additional mechanisms including adaptive immune resistance, whereby cancer cells hijack pathways designed to limit inflammatory responses (Figure 4) (51, 52). Upregulation of immune checkpoint molecules (e.g., PD-L1) production of immunosuppressive cytokines; recruitment of T regulatory cells (Tregs) and suppressive myeloid cells (i.e., alternatively activated macrophages and myeloid derived suppressor cells (MDSC)); and activation of pathways including WNT-β-catenin to suppress dendritic cell (DC) recruitment and DC-mediated priming of antigen-specific CD8+ T cells and infiltration of T cells (T cell exclusion) are further mechanism by which cancer can escape immune control (2, 53–56). Additionally, PTEN loss can increase PD-L1 expression on tumor cells and has been linked to a pro-tumor microenvironment characterized by immunosuppressive cytokines and other molecules such as IDO1, as well as Treg and suppressive myeloid cell infiltration (57). It is noteworthy that B cells also likely participate in the process of cancer immunoediting and have recently been implicated in responses to cancer immunotherapy (58–60). This is in contrast to certain pre-clinical models of cancer immunoediting, where B cells did not play a major role (11).
Figure 4: Adaptive immune resistance.
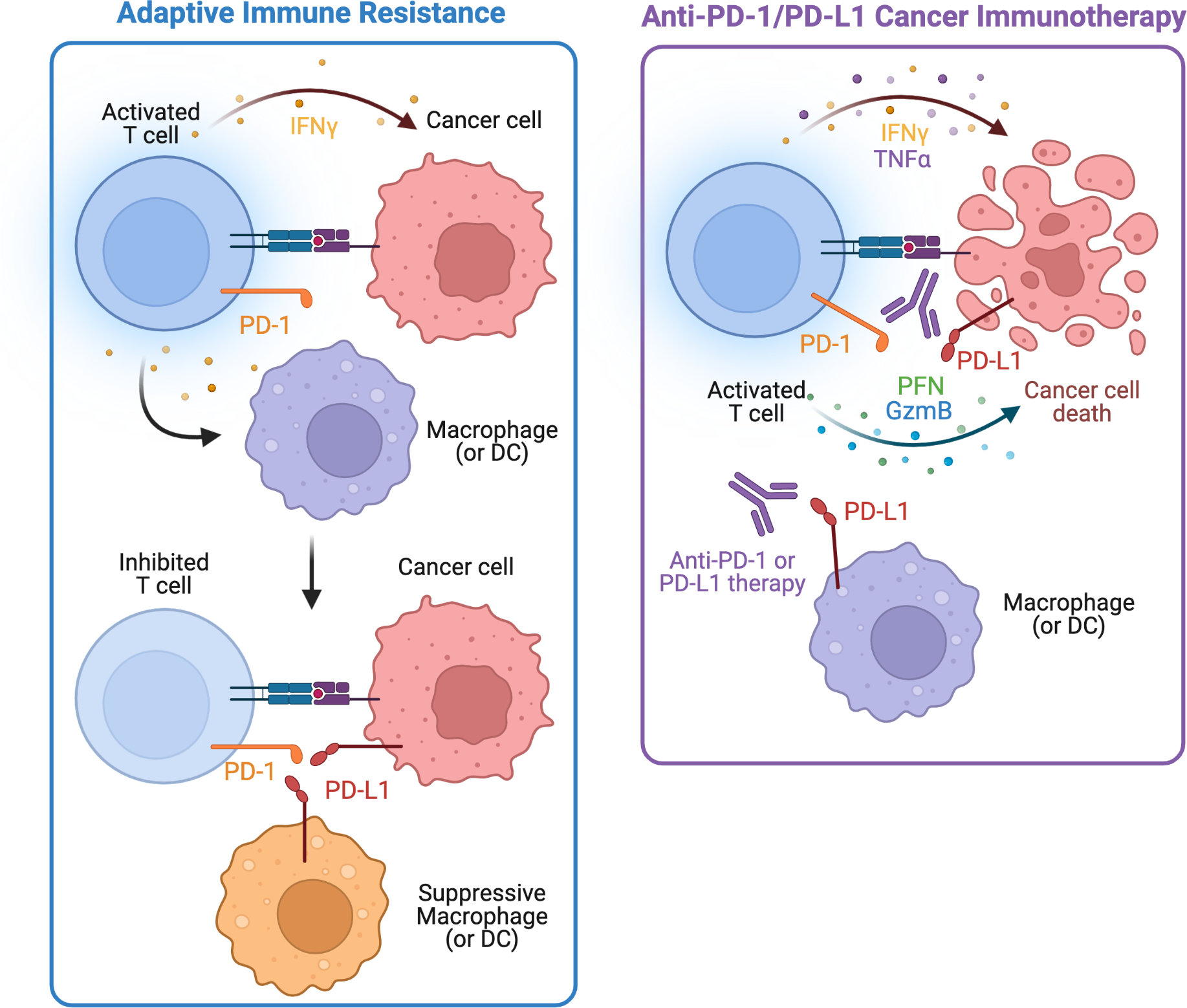
Upon T cell detection of cancer and activation, antigen-specific T cells produce IFN-γ to eliminate cancer. However, some cancers and myeloid cells such as macrophages or dendritic cells (DCs) upregulate PD-L1 (also known as B7-H1) in response to immune attack and IFN-γ signaling. Subsequent upregulation of PD-L1 binds to activated T cells expressing PD-1 and inhibits T cells in a process termed adaptive immune resistance. Targeting this local, dysfunctional immune response within the tumor microenvironment with anti-PD-1 or anti-PD-L1 immunotherapy, reduces the T cell inhibition, allowing the T cells to become re-activated and eliminate cancer cells through cytokines (IFN-γ, TNFα) and cytolytic programs with perforin (PFN) and granzyme B (GzmB). Targeting adaptive immune resistance with anti-PD-1/PD-L1 therapy is an example of normalization cancer immunotherapy.
Cancer Immunoediting: How immunity shapes the tumor microenvironment
During the early successes of immune-checkpoint blockade therapy and the emergence of immuno-oncology, Hanahan and Weinberg proposed the next generation of cancer hallmarks (61). Cancer immunoediting integrates the emerging hallmark of ‘avoiding immune destruction’ and enabling characteristic of ‘tumor-promoting inflammation’ (61). In the decade since, there is plethora of human data to indicate that the immune system, indeed, is a firmly established hallmark of cancer. Moreover, immune-directed treatment is likely the most successfully targeted hallmark for therapy. Immune cells are key contributors to the TME, a dysfunctional tissue that may exceed the complexity of normal tissues. Immunity shapes cancer formation and sustains tumor progression during the escape phase. Following the cancer immunoediting hypothesis and the success of ICT, there is intense research focusing on how immune cells and tumor cells interact to shape the TME. Untangling TME heterogeneity is a key goal in developing next generation cancer immunotherapies through rational clinical trial design.
Cancer Immunoediting and the Cancer-Immunity Cycle
The cancer-immunity cycle (62) seeks to highlight the critical steps involved in establishing effective anti-tumor immunity. These steps are based on our fundamental understanding of general immune activation including antigen processing, antigen presentation, T cell priming, T cell infiltration, T cell recognition and T cell effector responses to generate anti-tumor immunity. Cancer immunoediting is involved throughout the proposed cancer-immunity cycle steps and is particularly relevant at the final steps of T cell and tumor cell interactions that ultimately result in elimination, equilibrium, and escape. From a clinical care perspective, it is these final steps of T cell recognition and killing that occur within the TME that are the most critical and has been a major focus of immuno-oncology (Figure 5). Upon T cell recognition and attempted killing with release of IFN-γ, tumor cells upregulate expression of PD-L1 as an escape mechanism (63). This process of tumors adapting to immune attack to enter the escape phase of cancer immunoediting, is often referred to as adaptive resistance (Figure 4) (64). Unequivocal data exists that targeting the localized dysfunctional immune responses within the TME (i.e., anti-PD-1/PD-L1 therapy), referred to as normalization cancer immunotherapy (65, 66), results in the most effective immunotherapy. Normalization cancer immunotherapy seeks to identify a specific and local tumor escape mechanism that can be selectively targeted within the TME to limit systemic toxicity, re-program a dysfunctional immune response, and restore a normal anti-tumor immune response that results in cancer cell elimination and tissue homeostasis (65, 66). The iterative, complex, and dynamic interactions of immune cells and tumor cells undergoing cancer immunoediting results in significant TME heterogeneity. Stratification of the diverse immune compositions within the TME to select for the best immunotherapy for an individual patient is needed to optimize clinical outcomes.
Figure 5: Cancer immunoediting shapes tumor immunogenicity with impacts for patient-centered cancer immunotherapy.
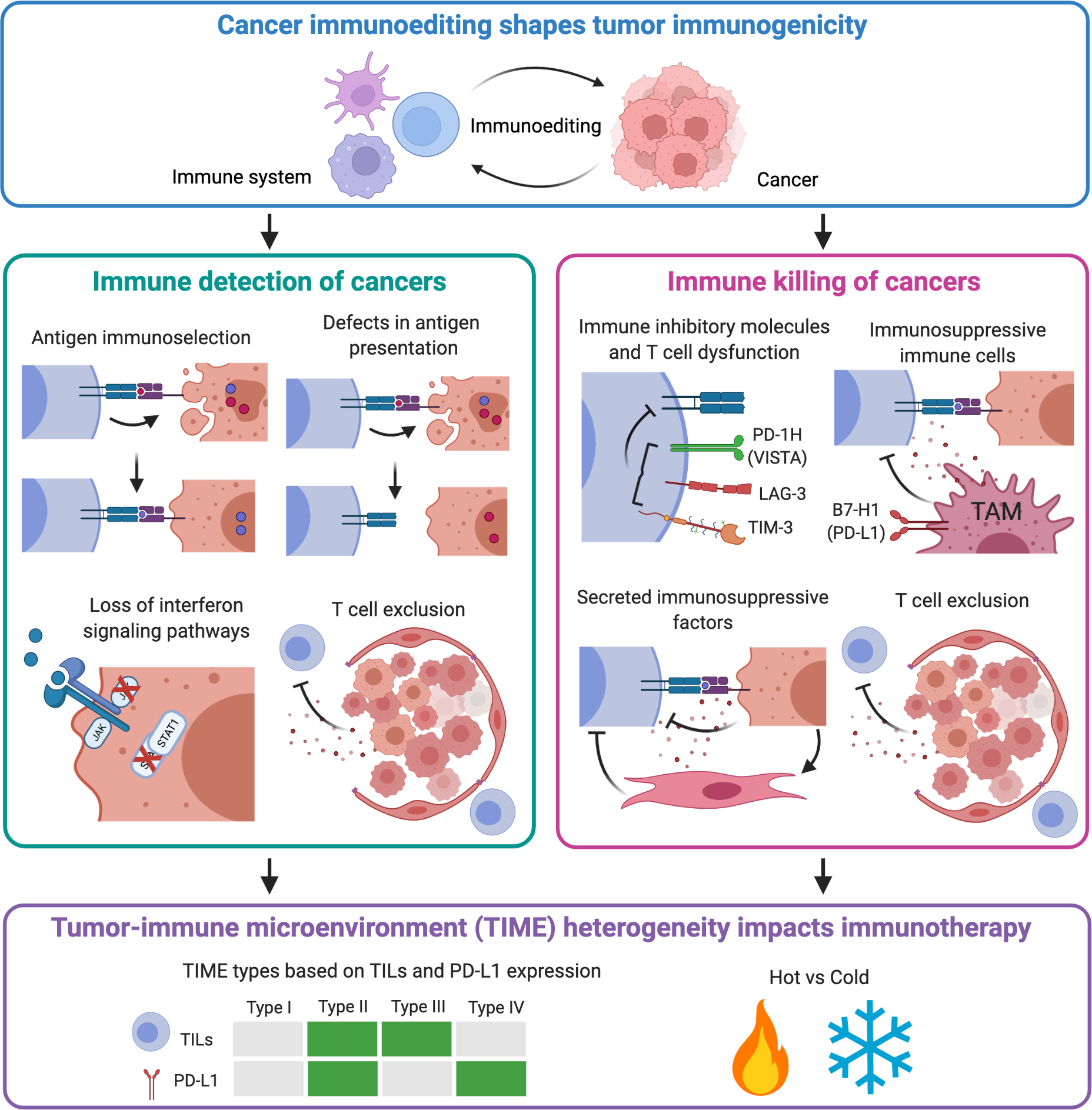
The dynamic immune-tumor cell interactions generate significant tumor microenvironment heterogeneity. Two key features for anti-tumor immunity and elimination of cancer include immune detection of cancers and immune killing of cancer. The cancer immunoediting process sculpts tumor immunogenicity through a variety of mechanisms that affect either immune detection or killing of cancers. Escape from immune detection may include antigen immunoselection, defects in antigen presentation, loss of interferon signaling pathways and T cell exclusion. Escape from immune killing may include expression of immune inhibitory molecules (“immune checkpoints”) that inhibit T cell function, secreted immunosuppressive factors, recruitment of immunosuppressive immune cells, and exclusion of T cells. T cell exclusion by either secreted factors or stromal barriers inhibits both immune detection and killing of caners. Together, immunoediting of the tumor microenvironment creates unique tumor-immune microenvironments (TIME) that stratifies responses to immunotherapies. For example, classifying tumors by the presence of tumor infiltrating T cells (TILs) and expression of immunotherapy target PD-L1 creates four distinct TIME subclassifications that affects response to anti-PD immunotherapy (63, 67): type I (neither PD-L1 nor TILs present); type II (both PD-L1 and TILs present); type III (no PD-L1, but TILs present); and type IV (PD-L1 present, but no TILs). More broadly, TIME can be considered as either ‘hot’ tumors with significant immune infiltration or “cold” tumors with little infiltration or anti-tumor immunity. Unique immunotherapy strategies are needed for each unique TIME for a patient-centered caner immunotherapy.
Cancer Immunoediting and the Tumor-Immune Microenvironment (TIME)
The ongoing interplay between immune cells and cancer cells during the cancer immunoediting process within the TME creates unique immune environments, called tumor-immune microenvironment (TIME) that has significant implications for cancer therapy (63, 67). Classification and the clinical predictive relevance of such heterogeneity of TIME was first revealed by Galon et al., where they demonstrated that the density and location of intratumoral CD4+ Th1, cytotoxic, and memory T cells correlated with clinical outcome in colorectal cancer patients (68). Unique patterns of T cell infiltration, T-cell inflammatory gene signatures, presence of B cells and tertiary lymphoid structures, PD-L1 expression, and tumor mutational burden have all been used to classify TIME (58, 59, 63, 67, 69–71). These T-cell inflammatory gene signatures are often used to designate whether a tumor is “hot” or “cold” (Figure 5). One of the first strategies to define TIME subtypes used two parameters: PD-L1 expression and presence of infiltrating T cells (TILs) in the TME. In this case, four distinct TIME subtypes are identified: type I (neither PD-L1 nor TILs present); type II (both PD-L1 and TILs present); type III (no PD-L1, but TILS present); and type IV (PD-L1 present, but no TILs) (63, 67) (Figure 5). A recent iteration that seeks to classify the TIME indirectly uses the concepts of cancer immunoediting to develop the concept of the ‘cancer-immune set point’ (72). The cancer-immune set point is the inherent immunological status of a tumor that results from an equilibrium of factors that promote or suppress anti-tumor immunity. Using the cancer-immune set point, the TIME may be segregated into three phenotypes: “immune-inflamed”, “immune-excluded”, or “immune-desert” (72). A critical aspect of the cancer-immune set point is the integration of the frequency of peptide-MHC-TCR interactions by tumor antigen-specific CD8+ T cells and the strength of TCR signaling. Two recent studies in human cancer patients demonstrate that the quality and quantity of tumor antigens drive T cell responses within the TME. In the first, single-cell RNA sequencing and single TCR sequencing were coupled with CITE-seq from four melanoma patients to interrogate tumor specific CD8+ T cells vs non-tumor reactive T cells (73). Melanoma-specific CD8+ T cells displayed an exhausted phenotype that was proportional to the abundance of melanoma antigens, suggesting that chronic T cell-tumor cell interactions “edits” T cells to become more dysfunctional (73). In the second study, combined single-cell RNA sequencing and single TCR sequencing was performed from 15 non-small cell lung cancer patients treated with neoadjuvant anti-PD-1 therapy (74). CD8+ T cells that were specific for mutation-associated neoantigens expressed an incomplete cytolytic program when compared to viral antigen-specific CD8+ T cells. Additionally, tumor-antigen specific CD8+ T cells from non-responders showed less TCR signaling and upregulated immune checkpoints and other inhibitors of T cell activation (74). Taken together, these studies and others, suggest that cancer immunoediting of tumor antigen-specific T cells and tumors bearing those antigens not only shapes antigenicity, but can reprogram T cell functional states, resulting in impaired immunotherapy responses. Bystander T cells that infiltrate the TME that are not cancer-specific are the most abundant infiltrating T cells in many cancers (75) and may play key roles in immunoediting and subsequent immunotherapy of cancer. Less is known about the contribution of bystander T cells to immunoediting, and immunotherapy as compared to cancer-specific T cells, but bystander T cells are activated with effector functions (76) and may be targeted in the future (77). Immunoediting of the TIME through genomic instability and subsequent neoantigen generation, T cell recognition, and resultant tumor cell immunosuppressive programs stratifies tumors with different TIMEs that has implications for cancer immunotherapy strategies (Figure 5) (78, 79).
Cancer Immunoediting during immunotherapy
As clinically apparent tumors have entered the escape phase of cancer immunoediting, the goal of therapy is to drive elimination or, at the very least, hold cancer in equilibrium. The process of cancer immunoediting not only transpires during natural tumor progression but also likely occurs in response to immunotherapy (Figure 3) (80). This can result in secondary (acquired) resistance to immunotherapy, whereby a clinical response is observed (incomplete elimination or equilibrium), followed by subsequent progression (secondary escape). For example, cancer patients achieving objective responses to ICT frequently experience durable responses, but delayed relapses are sometimes observed often even despite continuous therapy (81). Due to the sheer number of patients treated with ICT, secondary resistance in this context has been the most frequently examined (82). Although secondary resistance is not well-understood, potential mechanisms that have been described and may be grouped into those that affect immune detection of cancers and those that affect immune killing of cancers (Figure 5).
First, cancer immunoediting can alter the antigenome during tumor progression in the absence of immunotherapy and this mechanism of cancer immunoediting also likely occurs during immunotherapy. In preclinical models, anti-PD-L1 therapy altered the mutational landscape and decreased the number of subclonal mutations predicted to function as neoantigens (83), suggesting potentiated immunoediting. In humans, analysis of 42 patient-matched pretreatment and resistant tumors from NSCLC patients treated with anti-PD-1 monotherapy or combined anti-PD-1 and anti-CTLA-4 revealed altered neoantigen landscape in multiple cases of secondary or acquired resistance (84). Most mutations that were eliminated were predicted neoantigens with loss transpiring from loss of heterozygosity (LOH), deletion of chromosomal regions containing truncal mutations, or elimination of neoantigen-expressing tumor subclones. A separate study suggested that ACT may also reshape the tumor neoantigen repertoire. Sequential tumor samples and intratumoral T cells from two melanoma patients that received ACT that included both CD8+ and CD4+ tumor-specific T cells showed that tumor cells displayed loss of multiple T cell-recognized neoantigens, either by LOH of the mutant allele or reduced expression of the mutant genes encoding the neoantigens (85). This indicated immunoediting by loss of expression of immunogenic antigens and suggested that therapeutic induction of broad neoantigen-specific T cell responses should pursued to avoid immunotherapy resistance. While the focus of the aforementioned studies was on neoantigens, alterations in expression of non-mutant shared antigens have been observed in response to immune selection pressure either exerted naturally or by vaccination with melanoma-associated peptides derived from Melan A/MART-1, tyrosinase, and pmel/gp100 (86). In addition, while CAR T cell therapies have demonstrated noteworthy efficacy against certain hematopoietic malignancies, relapses do occur in a fraction of patients and these escape malignancies often exhibit down-modulation or complete loss of CAR T cell-specific antigens (6, 87, 88).
Second, cancer immunoediting via tumor intrinsic alterations in antigen presentation has been observed in many tumor types and may lead to immunotherapy resistance (82). Some of the first clinical data demonstrating loss of β2M (and thus surface HLA-I/MHC-I expression) upon immunotherapy came from the Rosenberg group, whereby B2M mutations were detected in progressing tumors from melanoma patients who initially experienced a clinical response after receiving multiple forms of immunotherapy, including IL-2, IFNα, and/or ACT (89). Similar observations were made by other groups in case reports that noted acquired loss of HLA-I/MHC-I expression in relapsing metastatic melanoma lesions, including in a melanoma patient who experienced disease relapse after treatment with multiple forms of immunotherapy (36, 38, 90). Secondary resistance to ICT has been associated with acquired defects in β2M, including homozygous truncating and frameshift mutations and LOH (32, 91, 92). In a cancer vaccine setting, patients treated with a personalized mRNA vaccine displayed remarkable vaccine-induced tumor-specific T cell responses and evidence of reduced metastasis after vaccination (93). Of those experiencing disease recurrence, one patient showed complete response and regression of metastatic tumors following anti-PD1 therapy, while another patient who relapsed failed anti-PD1 therapy, but was found to have a B2M mutation and loss of HLA-I/MHC-I expression.
Third, acquired defects in IFN-γ signaling pathways have been shown to mediate tumor escape in preclinical models and have also been observed in patients at the time of secondary resistance (46). In preclinical orthotopic pancreatic ductal adenocarcinoma models, objective responses were observed upon anti-PD-1/PD-L1 blockade. However, tumor escape variants emerged and were found to have defects in IFN-γ-induced TAP1 expression, which is required for peptide transport into the ER and subsequent loading onto MHC-I (94). In humans, acquired loss-of-function mutations and LOH in genes encoding JAK1 or JAK2 were identified in two patients who responded to ICT but subsequently progressed (31). In a separate report, genomic analysis of melanoma tumors before and after nivolumab (anti-PD-1) in patients who first progressed on ipilimumab (anti-CTLA-4) or were ipilimumab-naive suggested immunotherapy can drive differential clonal evolution within tumors and select against potential neoantigenic mutations, in particular if tumors are not completely eliminated by treatment (95). Consistent with immunotherapy-induced cancer immunoediting, the authors observed a reduction in overall tumor mutational burden during anti-PD-1 therapy, with progressive disease correlating with subsequent selection of tumor clones containing mutations within the CDKN2A gene and the IFN-γ signaling pathway, events that are known to promote tumor escape. Subsequent studies have linked CDKN2A gene expression with response to immunotherapy (96) and CDKN2A loss-of-function predicts immunotherapy resistance (97). This may be due to allelic overlap where loss of CDKN2A results in JAK2 loss, rendering tumor cells insensitive to IFN-γ signaling (98).
In addition, tumor escape and/or secondary resistance may be facilitated by induction of additional immunosuppressive pathways that inhibit destruction of cancers following cancer cell recognition (Figure 5). During ICT, upregulation of distinct immune checkpoints has been observed (95, 99). Since many immune checkpoints are induced by T cell activation and inflammation in the TME, it is not entirely surprising that these would be expressed in response to anti-CTLA-4 and/or anti-PD-1/PD-L1. Although the checkpoint upregulated may depend on the tumor type, their upregulation nevertheless may facilitate primary or acquired resistance. Several studies have found increased expression of LAG3, TIM3, and VISTA in the TME at the time of relapse after initially responding to ICT (95, 100, 101). While preclinical models and clinical data suggest upregulation of these additional immune checkpoints can enable tumor outgrowth and mediate primary resistance, it is still unclear whether this is a common mechanism of acquired resistance.
In cases of acquired resistance, PTEN loss and enhanced WNT-β-catenin signaling has been observed. George et al. described a patient with metastatic uterine leiomyosarcoma who experienced a clinical response to anti-PD-1 (pembrolizumab) for more than 2 years. At the time of acquired resistance, biallelic PTEN loss was detected (102). A separate study described two melanoma patients who developed acquired immunotherapy resistance, with one patient acquiring biallelic PTEN loss after initially responding to anti-CTLA-4 and anti-PD-1 and the other patient developing subsequent metastases displaying enhanced expression of β-catenin after demonstrating a durable partial response to a melanoma-associated peptide and IL-12 vaccine (103).
It is important to acknowledge that while there is solid evidence for cancer immunoediting potentiating acquired resistance to immunotherapy, the specific means by which this occurs is often, in part, implied based on circumstantial information. Furthermore, the mechanisms behind tumor editing likely differs depending on the immunotherapy employed as well as the cancer type/location. In regards to the latter, distinct mechanisms of cancer immunoediting were observed when comparing preclinical genetically-engineered mouse models (GEMM) of sarcoma and lung cancer, whereby despite both tumor models expressing the same Kras mutation and harboring deletion of p53, the means by which cancer immunoediting shaped the immunogenicity and outgrowth of tumors differed between the two tumor models (24, 104). It is therefore of upmost importance to further define cancer immunoediting across a spectrum of tumor types and immunotherapies to better anticipate and respond to acquired resistance. In human cancer patients, metastatic lesions demonstrate unique cancer mutations, likely due to genetic and epigenetic factors (105). Different TME within the same patient may help to explain dissociated responses of individual metastatic lesions within the same patient to immunotherapy (106). Thus, identifying unique mechanism of cancer immunoediting within distinct metastatic lesions will also be critical in developing novel immunotherapies.
Conclusions
Cancer Immunoediting remains a useful framework to understand the complex and dynamic relationships between cancer cells, immune cells, and stromal cells from initial tumor cell transformation to cancer development and ultimately cancer immunotherapy. As many patients do not experience complete tumor elimination, some patients may entire equilibrium and transform cancer to a chronic disease, requiring new and emerging immunotherapies to keep cancer at bay. The early and ongoing dynamic immune cell and tumor cell interactions occurring in the TME underscore the case for earlier use of immunotherapies rather than late-stage, advanced cancers. This approach is becoming more common with adjuvant therapy or neoadjuvant therapy with surgical, chemotherapy or radiation treatment. Immuno-oncology has transformed cancer care, but significant challenges remain. There needs to be a continued focus on the dynamic immune cell and tumor cell interactions within the TME, the underlying principle of cancer immunoediting, to identify mechanisms of immunotherapy resistance and employ strategies to overcome resistance. Understanding the natural immunity to cancer may provide insights to “normalize” the dysfunctional immune response to cancers occurring in the TME (65, 66). Cancer immunoediting during immunotherapy may help identify new targets or optimal approaches to achieve normalization cancer immunotherapy.
Acknowledgements:
We apologize to all the investigators whose research could not be included due to space limitations. M.M.G is a Cancer Prevention and Research Institute of Texas (CPRIT) Scholar in Cancer Research and a Parker Institute for Cancer Immunotherapy (PICI) Bridge Scholar and is supported by the University of Texas MD Anderson Cancer Center Support Grant (CCSG) New Faculty Award supported by the National Cancer Institute (NCI) (P30CA016672) and a CPRIT Scholar in Cancer Research Grant (RR190017).
M.D.V. is supported by a Physician-Scientist Career Development Award from the Dermatology Foundation and Dermatology Fellow Awards from the Melanoma Research Alliance (https://doi.org/10.48050/pc.gr.89581 and https://doi.org/10.48050/pc.gr.141701). Figures created with BioRender.com.
Footnotes
Competing Interests/Disclosures: Spouse of M.D.V. is an employee of Regeneron Pharmaceuticals. M.M.G. receives a personal honorarium of $1000.00 USD per year from Springer Nature Ltd for his role as an Associate Editor for the journal Nature Precision Oncology.
References:
- 1.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–6. [DOI] [PubMed] [Google Scholar]
- 2.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372(26):2521–32. [DOI] [PubMed] [Google Scholar]
- 5.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21(3):145–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107–11. [DOI] [PubMed] [Google Scholar]
- 8.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991–8. [DOI] [PubMed] [Google Scholar]
- 9.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95(13):7556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, et al. Adaptive immunity maintains occult cancer in an equilibrium state. Nature. 2007;450(7171):903–7. [DOI] [PubMed] [Google Scholar]
- 11.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–71. [DOI] [PubMed] [Google Scholar]
- 12.Teng MW, Galon J, Fridman WH, Smyth MJ. From mice to humans: developments in cancer immunoediting. J Clin Invest. 2015;125(9):3338–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5(12):1365–9. [DOI] [PubMed] [Google Scholar]
- 16.Chen L, Ashe S, Brady WA, Hellstrom I, Hellstrom KE, Ledbetter JA, et al. Costimulation of antitumor immunity by the B7 counterreceptor for the T lymphocyte molecules CD28 and CTLA-4. Cell. 1992;71(7):1093–102. [DOI] [PubMed] [Google Scholar]
- 17.Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8(8):793–800. [DOI] [PubMed] [Google Scholar]
- 18.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoos A, Britten C. The immuno-oncology framework: Enabling a new era of cancer therapy. Oncoimmunology. 2012;1(3):334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balwit JM, Kalinski P, Sondak VK, Coulie PG, Jaffee EM, Gajewski TF, et al. Review of the 25th annual scientific meeting of the International Society for Biological Therapy of Cancer. J Transl Med. 2011;9:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xin Yu J, Hodge JP, Oliva C, Neftelinov ST, Hubbard-Lucey VM, Tang J. Trends in clinical development for PD-1/PD-L1 inhibitors. Nat Rev Drug Discov. 2020;19(3):163–4. [DOI] [PubMed] [Google Scholar]
- 22.Upadhaya S, Neftelino ST, Hodge JP, Oliva C, Campbell JR, Yu JX. Combinations take centre stage in PD1/PDL1 inhibitor clinical trials. Nat Rev Drug Discov. 2020. [DOI] [PubMed] [Google Scholar]
- 23.Kluger HM, Tawbi HA, Ascierto ML, Bowden M, Callahan MK, Cha E, et al. Defining tumor resistance to PD-1 pathway blockade: recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J Immunother Cancer. 2020;8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature. 2012;482(7385):405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482(7385):400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution. Nature. 2019;567(7749):479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Balachandran VP, Luksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551(7681):512–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angelova M, Mlecnik B, Vasaturo A, Bindea G, Fredriksen T, Lafontaine L, et al. Evolution of Metastases in Space and Time under Immune Selection. Cell. 2018;175(3):751–65 e16. [DOI] [PubMed] [Google Scholar]
- 30.Dighe AS, Richards E, Old LJ, Schreiber RD. Enhanced in vivo growth and resistance to rejection of tumor cells expressing dominant negative IFN gamma receptors. Immunity. 1994;1(6):447–56. [DOI] [PubMed] [Google Scholar]
- 31.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med. 2016;375(9):819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8(1):1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017;7(2):188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505(7484):495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shukla SA, Rooney MS, Rajasagi M, Tiao G, Dixon PM, Lawrence MS, et al. Comprehensive analysis of cancer-associated somatic mutations in class I HLA genes. Nat Biotechnol. 2015;33(11):1152–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao F, Sucker A, Horn S, Heeke C, Bielefeld N, Schrors B, et al. Melanoma Lesions Independently Acquire T-cell Resistance during Metastatic Latency. Cancer Res. 2016;76(15):4347–58. [DOI] [PubMed] [Google Scholar]
- 37.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell. 2017;171(6):1259–71 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrors B, Lubcke S, Lennerz V, Fatho M, Bicker A, Wolfel C, et al. HLA class I loss in metachronous metastases prevents continuous T cell recognition of mutated neoantigens in a human melanoma model. Oncotarget. 2017;8(17):28312–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Datar IJ, Hauc SC, Desai S, Gianino N, Henick B, Liu Y, et al. Spatial Analysis and Clinical Significance of HLA Class-I and Class-II Subunit Expression in Non-Small Cell Lung Cancer. Clin Cancer Res. 2021;27(10):2837–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groh V, Wu J, Yee C, Spies T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature. 2002;419(6908):734–8. [DOI] [PubMed] [Google Scholar]
- 41.Doubrovina ES, Doubrovin MM, Vider E, Sisson RB, O’Reilly RJ, Dupont B, et al. Evasion from NK cell immunity by MHC class I chain-related molecules expressing colon adenocarcinoma. J Immunol. 2003;171(12):6891–9. [DOI] [PubMed] [Google Scholar]
- 42.Ghiringhelli F, Menard C, Terme M, Flament C, Taieb J, Chaput N, et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-beta-dependent manner. J Exp Med. 2005;202(8):1075–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gross E, Sunwoo JB, Bui JD. Cancer immunosurveillance and immunoediting by natural killer cells. Cancer J. 2013;19(6):483–9. [DOI] [PubMed] [Google Scholar]
- 44.Wagner M, Koyasu S. Cancer Immunoediting by Innate Lymphoid Cells. Trends Immunol. 2019;40(5):415–30. [DOI] [PubMed] [Google Scholar]
- 45.Chen M, Chen R, Jin Y, Li J, Hu X, Zhang J, et al. Cold and heterogeneous T cell repertoire is associated with copy number aberrations and loss of immune genes in small-cell lung cancer. Nat Commun. 2021;12(1):6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sucker A, Zhao F, Pieper N, Heeke C, Maltaner R, Stadtler N, et al. Acquired IFNgamma resistance impairs anti-tumor immunity and gives rise to T-cell-resistant melanoma lesions. Nat Commun. 2017;8:15440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547(7664):413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patel SJ, Sanjana NE, Kishton RJ, Eidizadeh A, Vodnala SK, Cam M, et al. Identification of essential genes for cancer immunotherapy. Nature. 2017;548(7669):537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167(2):397–404 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017;168(4):707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ribas A Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015;5(9):915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70. [DOI] [PubMed] [Google Scholar]
- 55.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523(7559):231–5. [DOI] [PubMed] [Google Scholar]
- 56.Gangoso E, Southgate B, Bradley L, Rus S, Galvez-Cancino F, McGivern N, et al. Glioblastomas acquire myeloid-affiliated transcriptional programs via epigenetic immunoediting to elicit immune evasion. Cell. 2021;184(9):2454–70 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016;6(2):202–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577(7791):561–5. [DOI] [PubMed] [Google Scholar]
- 59.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577(7791):549–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cui C, Wang J, Fagerberg E, Chen PM, Connolly KA, Damo M, et al. Neoantigen-driven B cell and CD4 T follicular helper cell collaboration promotes anti-tumor CD8 T cell responses. Cell. 2021;184(25):6101–18 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. [DOI] [PubMed] [Google Scholar]
- 62.Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. [DOI] [PubMed] [Google Scholar]
- 63.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4(127):127ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim TK, Herbst RS, Chen L. Defining and Understanding Adaptive Resistance in Cancer Immunotherapy. Trends Immunol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sanmamed MF, Chen L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell. 2018;175(2):313–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vesely MD, Chen L. Normalization Cancer Immunotherapy for Melanoma. J Invest Dermatol. 2020;140(6):1134–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer--response. Clin Cancer Res. 2013;19(19):5542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313(5795):1960–4. [DOI] [PubMed] [Google Scholar]
- 69.Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75(11):2139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol. 2019;16(3):151–67. [DOI] [PubMed] [Google Scholar]
- 71.Van den Eynde M, Mlecnik B, Bindea G, Fredriksen T, Church SE, Lafontaine L, et al. The Link between the Multiverse of Immune Microenvironments in Metastases and the Survival of Colorectal Cancer Patients. Cancer Cell. 2018;34(6):1012–26 e3. [DOI] [PubMed] [Google Scholar]
- 72.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541(7637):321–30. [DOI] [PubMed] [Google Scholar]
- 73.Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature. 2021;596(7870):119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EH, et al. Transcriptional programs of neoantigen-specific TIL in anti-PD-1-treated lung cancers. Nature. 2021;596(7870):126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simoni Y, Becht E, Fehlings M, Loh CY, Koo SL, Teng KWW, et al. Bystander CD8(+) T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575–9. [DOI] [PubMed] [Google Scholar]
- 76.Maurice NJ, Taber AK, Prlic M. The Ugly Duckling Turned to Swan: A Change in Perception of Bystander-Activated Memory CD8 T Cells. J Immunol. 2021;206(3):455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Meier SL, Satpathy AT, Wells DK. Bystander T cells in cancer immunology and therapy. Nat Cancer. 2022;3(2):143–55. [DOI] [PubMed] [Google Scholar]
- 78.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smyth MJ, Ngiow SF, Ribas A, Teng MW. Combination cancer immunotherapies tailored to the tumour microenvironment. Nat Rev Clin Oncol. 2016;13(3):143–58. [DOI] [PubMed] [Google Scholar]
- 80.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515(7528):577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang DY, Eroglu Z, Ozgun A, Leger PD, Zhao S, Ye F, et al. Clinical Features of Acquired Resistance to Anti-PD-1 Therapy in Advanced Melanoma. Cancer Immunol Res. 2017;5(5):357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schoenfeld AJ, Hellmann MD. Acquired Resistance to Immune Checkpoint Inhibitors. Cancer Cell. 2020;37(4):443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Efremova M, Rieder D, Klepsch V, Charoentong P, Finotello F, Hackl H, et al. Targeting immune checkpoints potentiates immunoediting and changes the dynamics of tumor evolution. Nat Commun. 2018;9(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of Neoantigen Landscape during Immune Checkpoint Blockade in Non-Small Cell Lung Cancer. Cancer Discov. 2017;7(3):264–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature. 2016;536(7614):91–5. [DOI] [PubMed] [Google Scholar]
- 86.Jager E, Ringhoffer M, Karbach J, Arand M, Oesch F, Knuth A. Inverse relationship of melanocyte differentiation antigen expression in melanoma tissues and CD8+ cytotoxic-T-cell responses: evidence for immunoselection of antigen-loss variants in vivo. Int J Cancer. 1996;66(4):470–6. [DOI] [PubMed] [Google Scholar]
- 87.Majzner RG, Mackall CL. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018;8(10):1219–26. [DOI] [PubMed] [Google Scholar]
- 88.Hamieh M, Dobrin A, Cabriolu A, van der Stegen SJC, Giavridis T, Mansilla-Soto J, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Restifo NP, Marincola FM, Kawakami Y, Taubenberger J, Yannelli JR, Rosenberg SA. Loss of functional beta 2-microglobulin in metastatic melanomas from five patients receiving immunotherapy. J Natl Cancer Inst. 1996;88(2):100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yamshchikov GV, Mullins DW, Chang CC, Ogino T, Thompson L, Presley J, et al. Sequential immune escape and shifting of T cell responses in a long-term survivor of melanoma. J Immunol. 2005;174(11):6863–71. [DOI] [PubMed] [Google Scholar]
- 91.Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017;7(12):1420–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Lower M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 2017;547(7662):222–6. [DOI] [PubMed] [Google Scholar]
- 94.Burrack AL, Spartz EJ, Raynor JF, Wang I, Olson M, Stromnes IM. Combination PD-1 and PD-L1 Blockade Promotes Durable Neoantigen-Specific T Cell-Mediated Immunity in Pancreatic Ductal Adenocarcinoma. Cell Rep. 2019;28(8):2140–55 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171(4):934–49 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Banchereau R, Leng N, Zill O, Sokol E, Liu G, Pavlick D, et al. Molecular determinants of response to PD-L1 blockade across tumor types. Nat Commun. 2021;12(1):3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gutiontov SI, Turchan WT, Spurr LF, Rouhani SJ, Chervin CS, Steinhardt G, et al. CDKN2A loss-of-function predicts immunotherapy resistance in non-small cell lung cancer. Sci Rep. 2021;11(1):20059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Horn S, Leonardelli S, Sucker A, Schadendorf D, Griewank KG, Paschen A. Tumor CDKN2A-Associated JAK2 Loss and Susceptibility to Immunotherapy Resistance. J Natl Cancer Inst. 2018;110(6):677–81. [DOI] [PubMed] [Google Scholar]
- 99.Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, et al. VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat Med. 2017;23(5):551–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw RPM, et al. Negative immune checkpoint regulation by VISTA: a mechanism of acquired resistance to anti-PD-1 therapy in metastatic melanoma patients. Mod Pathol. 2017;30(12):1666–76. [DOI] [PubMed] [Google Scholar]
- 102.George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity. 2017;46(2):197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Trujillo JA, Luke JJ, Zha Y, Segal JP, Ritterhouse LL, Spranger S, et al. Secondary resistance to immunotherapy associated with beta-catenin pathway activation or PTEN loss in metastatic melanoma. J Immunother Cancer. 2019;7(1):295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.DuPage M, Cheung AF, Mazumdar C, Winslow MM, Bronson R, Schmidt LM, et al. Endogenous T cell responses to antigens expressed in lung adenocarcinomas delay malignant tumor progression. Cancer Cell. 2011;19(1):72–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–45. [DOI] [PubMed] [Google Scholar]
- 106.Humbert O, Chardin D. Dissociated Response in Metastatic Cancer: An Atypical Pattern Brought Into the Spotlight With Immunotherapy. Front Oncol. 2020;10:566297. [DOI] [PMC free article] [PubMed] [Google Scholar]