

Abstract
Regulatory elements activate promoters by recruiting transcription factors (TFs) to specific motifs. Notably, TF-DNA interactions often depend on cooperativity with colocalized partners, suggesting an underlying cis-regulatory syntax. To explore TF cooperativity in mammals, we here analyze ~500 mouse and human primary cells by combining an atlas of TF motifs, footprints, ChIP-Seq, transcriptomes, and accessibility. We uncover two TF groups that colocalize with most expressed factors, forming stripes in hierarchical clustering maps. The first group includes lineage-determining factors that occupy DNA elements broadly, consistent with their key role in tissue-specific transcription. The second one, dubbed universal stripe factors (USFs), comprise ~30 SP, KLF, EGR, and ZBTB family members that recognize overlapping GC-rich sequences in all tissues analyzed. Knockouts and single molecule tracking reveal that USFs impart accessibility to colocalized partners and increase their residence time. Mammalian cells have thus evolved a TF superfamily with overlapping DNA binding that facilitate chromatin accessibility.
Graphical Abstract
eTOC
Transcription factors are known to cooperate with each other in their interaction with cognate DNA binding motifs. Zhao et al. report a superfamily of ~30 factors that recognizes overlapping GC-rich sequences in mammalian genomes and that renders chromatin accessible to binding partners.
DNA regulatory elements in higher organisms control the spatiotemporal expression of genes during development and homeostasis (Bulger and Groudine, 2010). Of an average size of ~400 bps (Kim et al., 2019), regulatory elements contain binding motifs for multiple transcription factors (TFs), which play distinct roles in PolII activation. At promoters, TFs help recruit PolII and determine the burst size of mRNA synthesis; at enhancers they control tissue-specific transcription and mRNA burst frequency (Larsson et al., 2019; Stavreva et al., 2019). The importance of these activities is highlighted by the fact that more than 80% of GWAS-identified polymorphisms associated with disease fall within regulatory elements (Giral et al., 2018; Maurano et al., 2012; Visel et al., 2009). To understand how such non-coding variants cause disease (a primary goal of translational research), several strategies are currently being implemented, including SNP enrichment methods (Cano-Gamez and Trynka, 2020), the statistical colocalization of variants to eQTLs (Giambartolomei et al., 2014), and genome-wide CRISPR screens (Bourges et al., 2020; Kampmann, 2020). While these techniques are powerful, a full understanding of the variant-to-function problem will require the deciphering of the cis-regulatory code (Jindal and Farley, 2021).
Also known as enhancer grammar or syntax, the cis-regulatory code posits that the affinity, arrangement and spacing of DNA motifs determine the recruitment of regulatory proteins (Rickels and Shilatifard, 2018; Zeitlinger, 2020). An extreme example of this is the INF-β enhanceosome, in which the precise position of motifs in the DNA helix promotes cooperative binding of TFs (Maniatis et al., 1998). At the β enhancer, TF cooperativity is thought to occur through binding-induced structural changes in DNA conformation (Panne et al., 2007). In other settings however, protein-protein interactions play a more direct role. A well-studied example is AP-1, which in hematopoietic enhancers forms strong ternary complexes with adjacent ETS, IRF or NFAT proteins binding at fixed distances (Bassuk and Leiden, 1995; Chen et al., 1998; Macian et al., 2001; Murphy et al., 2013).
Whereas rules of enhancer syntax are apparent for overlapping or adjacently bound TFs, binding cooperativity between factors separated by longer distances has also been observed. Studies in macrophages for instance have shown that, in addition to the aforementioned partners, AP-1 is stabilized by dozens of TFs recognizing motifs at variable distances from it (Link et al., 2018). Most regulatory elements are thus believed to rely on some form of TF cooperativity that help recruit cofactors, outcompete nucleosomes, and ultimately activate PolII. The difficulty of accurately measuring and integrating these parameters into a single model has rendered the deciphering of the cis-regulatory code a particularly challenging task. However, basic rules of TF cooperativity have often been inferred from motif distributions, a feature that has provided valuable insights into the function of a fraction of cis-regulatory elements (Morgunova and Taipale, 2017; Spitz and Furlong, 2012). In this report we have sought to identify TF cooperativity at a genome-wide scale. Our strategy combined analyses of chromatin accessibility (ATAC- or DHS-Seq), RNA-Seq, TF ChIP-Seq, footprinting and DNA motifs maps from a large panel of mouse and human cells and tissues. The results identify a cluster of TFs that bind overlapping GC-rich sequences and provides accessibility to most DNA regulatory elements and longer residence times to nuclear chromatin.
Results
A comprehensive map of TF binding sites in mice and humans
To dissect TF cooperativity genome-wide we first defined TF binding motifs in mice and humans by combining JASPAR (Khan et al., 2018) TRANSFAC (Wingender, 2008), and CIS-BP-defined TF position weight matrices (PWMs, (Weirauch et al., 2014)) into a database that was manually curated to remove redundancies. We identified 3,756 unique PWMs for a total of 897 mouse TFs, and 5,937 PWMs, for 1,309 human TFs (Figure 1A). Based on this database we next generated a comprehensive map of predicted TF binding motifs, with 254 and 535 million sites in the mouse and human genomes respectively (Figure 1A). To determine which motifs are accessible, we compiled all available ATAC-Seq and DHS-Seq experiments from GEO and ENCODE databases. Of 5,553 samples analyzed, 3,517 (63%) passed ENCODE quality standards (TSS enrichment metric, Supplementary Table 1A). Next, we combined all replicates and complemented the samples with 96 ATAC-Seq experiments we processed from selected immune and somatic cells, including early stages of hematopoiesis, innate lymphoid cells (ILCs), most stages of B and T cell development, and various cells of the nervous system (Supplementary Table 1B). In total, 239 mouse and 251 human unique samples were available for subsequent analysis (Figure 1A, Supplementary Tables 1A-C).
Figure 1. A comprehensive map of TF motifs in the mouse and human genomes.

(A) JASPAR, TRANSFAC, and CIS-BP databases were merged to define 3,756 and 5,937 PWMs for mouse and human TFs respectively. Analysis of the two genomes resulted in 254 and 535 million TF binding motifs. Based on DHS and ATAC-Seq data, 76 and 182 million motifs were identified in accessible DNA in the two genomes. (B) Bar graph showing the number of accessible DNA elements found in a single cell type (unique), shared between multiple cell types, or present in all cell types (common) in mice and humans. Throughout the text, DNA elements refer to 201bp ATAC-Seq (or DHS-Seq) or summits, detected by MACS2. (C) Pie charts show the percentage of cell-specific or common DNA elements associated with or distal to promoters in the mouse genome.
We identified 1.3M and 1.7M unique accessible elements in the mouse and human genomes respectively, the vast majority of which were either cell type-specific or shared between a subset of cells and tissues (Figure 1B). Only a relatively small number of accessible sites (8,638 in mouse, 15,660 in human) were common to all samples examined (Figure 1B). As expected, nearly all (99%) cell-specific elements were promoter-distal, implying that tissue-specific enhancers are enriched in this population (Figure 1C). Conversely, nearly 50% of common elements overlapped with promoters (Figure 1C). Mapping of TF motifs over the accessible genome revealed that about one third of mouse (76M of 254M) and human (182M of 535M) motifs are available for TF binding (Figure 1A).
TF combinatorial information
To comprehensively define TF combinations, we calculated the colocalization frequency of all possible TF motif pairs in ATAC-Seq or DHS-Seq summits (201 bp). Only expressed TFs were included. To visualize the results, we generated heat maps based on agglomerative hierarchical clustering (Figure 2A, Supplementary Figure 1A and Methods). Our analysis classified motif pairs into 4 main groups: overlapped, colocalized, non-significant and excluded. At one extreme we found factors that recognize the same or highly overlapping DNA motifs (≥ 50% colocalization, Figure 2B). In heat maps from mouse or human cells, such pairs are clustered into well-defined TF families, such as the ATF-FOS-JUN, FOX, IRF-STAT, and NFKB among others (Figure 2A, Supplementary Figure 1A-B). Interestingly however, we also found motifs that are significantly excluded from each other (FDR ≤ 0.01), showing little or no colocalization (≤ 1%, Supplementary Table 1D). Most of these motifs map to mutually exclusive AT and GC rich sequences, which are rarely found in the same DNA element (e.g. MBD2 and FOXN3, Figure 2B). A third group comprised TF motifs that do not show statistically significant colocalization or exclusion and thus were classified as “non-significant” (Figure 2B). These were by far the most abundant group, comprising nearly 90% of all pairs (Supplementary Figure 1C).
Figure 2. TF combinatorial information.
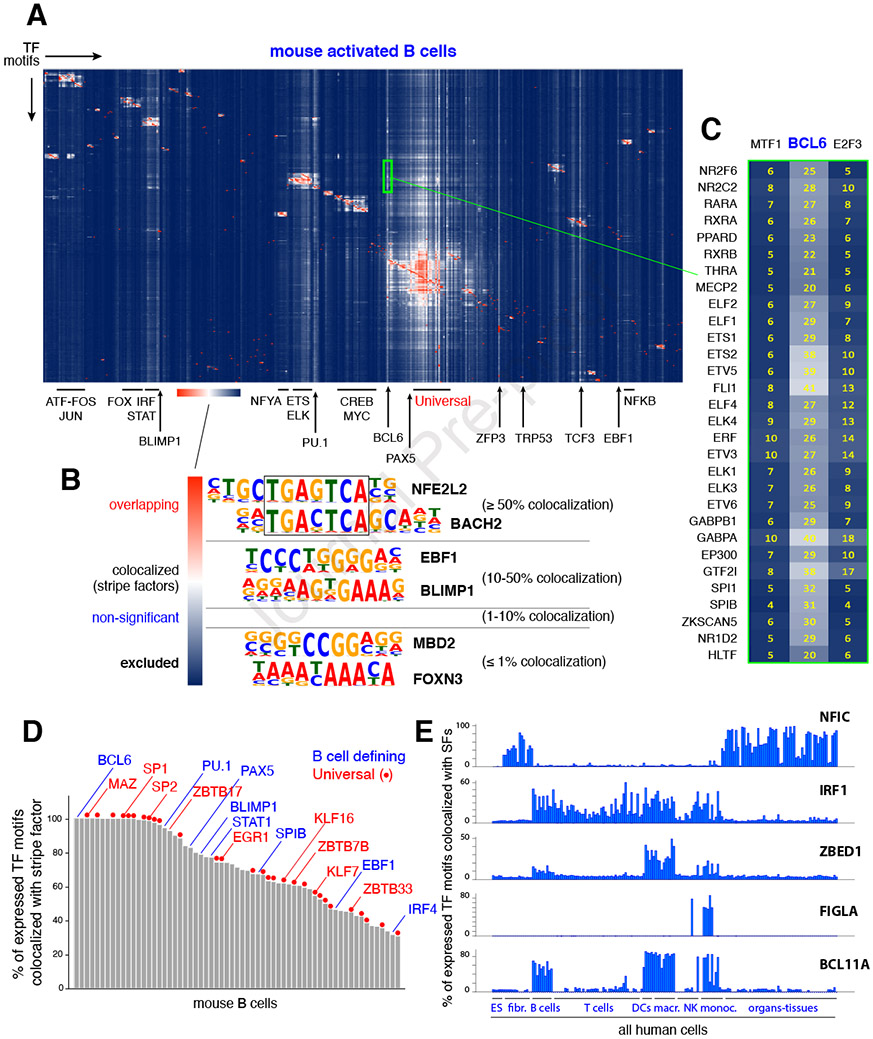
(A) Agglomerative hierarchical clustering showing the frequency of colocalization for all known TF pairs in mouse B cells. Family clusters are highlighted with bars while stripe factors are denoted with arrows. The color scale shown at the bottom illustrates the relative colocalization between functional motif pairs: red represents ≥ 50%; white represents 10-50%; blue represents non-significant to excluded (≤ 1%). (B) Hierarchical clustering identifies 4 main TF motif pair groups: overlapping (≥ 50% colocalization), colocalized (10-50%, stripe factors being a special case), non-significant, and excluded (≤ 1% colocalization). (C) Close up view of the BCL6 stripe (green rectangle in panel A). As an example, 25% of all elements containing the NR2F6 TF motif (top in the list) also contain the BCL6 motif. (D) Bar graph showing all 63 mouse B cell stripe factors classified based on the percentage of expressed TFs that are colocalized with them. Examples of B cell-defining factors (blue) and universal stripe factors (red) are shown. (E) Examples of TFs that display stripe profiles in defined cell types.
The final category, and from a syntax standpoint perhaps the most interesting one, included motifs that are colocalized at high frequency (10-50%) but that do not necessarily overlap (FDR ≤ 0.01, Figure 2B). Factors recognizing such motifs are predicted to colocalize with most TFs expressed in each cell type. Consequently, in heat maps they create prominent vertical white stripes (Figures 2A, 2C, and Supplementary Figures 1A and 1D). Notably, “stripe” factors in B lymphocytes comprised known drivers of B cell ontogeny, including BCL6, PU.1, PAX5, BLIMP1, STAT1, SPIB, EBF1 and IRF4 (Figure 2A and 2D). Their colocalization with all expressed TFs ranged from 34% for IRF4 to as high as 100% for BCL6 (Figure 2D). Hierarchical clustering maps of human ES cells also showed vertical stripes associated with pluripotency factors (KLF4, NANOG), as well as TFs known to play key roles in stemness: AR, TBX1, TFAP2C, E2F8 and SALL4 (Supplementary Figure Figures 1A, 2A and (Chen et al., 2009; Kregel et al., 2013; Li et al., 2008; Pastor et al., 2018; Takahashi and Yamanaka, 2006)).
In total, our analysis predicts 183 mouse and 288 human stripe factors (Supplementary Table 1E). Published ChIP-Seq experiments supported this classification by showing that pluripotency (KLF4, NANOG) and hematopoietic (PU.1, PAX5, BCL6) factors occupy a sizable fraction of regulatory elements across the genome (36-57%, Supplementary Figure 2B and (Chronis et al., 2017; Hu et al., 2016; Huang et al., 2013)). Such broad occupancy explains their colocalization with most TFs expressed in the given cell type. A survey of the human samples provided additional examples of known and novel cell-defining TFs as stripe factors, including NFIC in non-hematopoietic lineages, IRF1 in hematopoietic linages, ZBED1 in DCs and macrophages, FIGLA in monocytes, and BCL11A in B cells, DCs, macrophages and monocytes (Figure 2E). The data is thus consistent with the model where cell identity factors colocalize with large numbers of expressed TFs, a feature that supports their key role in development and homeostasis.
The co-occurrence of stripe and other TF motifs within regulatory elements was tested against 150,000 random 201bp regions from the genome. The analysis showed that 78.9% of TF colocalizations were significantly enriched in DHS summits compared to random regions (FDR <= 0.01, see Methods and Supplementary Figure 2C). To further validate the high colocalization of stripe factors, we analyzed binding profiles of 108 TFs by ChIP-Seq in HEK293 cells (Consortium et al., 2012). Notably, while motif analysis predicted 24 stripe factors in this group, ChIP-Seq identified 64 (Figure 3A). This discrepancy may be due to the observation that ChIP-Seq experiments detect both direct (motif+) and indirect (motif−) binding events (Liang et al., 2014; Yao et al., 2017), a feature that should increase colocalization between TFs. To directly test this idea, we repeated the analysis but only considered ChIP-Seq peaks that included cognate TF motifs. This time the analysis identified 28 stripe factors, including all 24 defined by motif analysis only (Figure 3A and Supplementary Table 1F). A survey of MCF7, GM12878 and K562 cells showed similar findings (Supplementary Figure 2D and Supplementary Table 1F). The data thus corroborate the presence of stripe factors in mammalian cells and reveal that some TFs can display high colocalization possibly through indirect binding. Because the latter group cannot be linked to specific DNA motifs, we have not considered them further in this study.
Figure 3. Universal Stripe Factors.

(A) Agglomerative hierarchical clustering of 108 TFs expressed in HEK293 cells based on TF motifs (left), ChIP-Seq peaks (middle), or ChIP-Seq peaks carrying cognate motifs (right). The number of stripe factors are included below each graph. (B) Heat map sections showing the USF cluster in human CD19+ B cells based on motifs (left) or footprinting (right) analysis. (C) Closeup view of the clustering. Numbers in each square (e.g. 72%) denote the percentage of elements that are positive for a “row” TF motif (E2F4) that also contain a “column” TF motif (KLF12). (D) The most common PWM motif assigned to human USFs. (E) Location of C2H2 ZFs (red squares) in human USFs.
Characterization of Universal Stripe Factors
We noticed that ~30 stripe factors form a very large cluster in heat maps from all mouse and human samples, either by motif, footprinting or ChIP-Seq analyses (Figure 3A-B, Supplementary Figures 1A, 2E and Supplementary Table 1F). Because of such broad distribution, we dubbed the subset universal stripe factors (USFs). Among them we found members of the SP and KLF families, which are known to bind DNA with three conserved C2H2 ZFs (Kaczynski et al., 2003). In addition, we found TFs not previously associated with the SP-KLF group, including members of the EGR and ZBTB families, as well as a subset of zinc finger proteins (ZFP-ZNF), MAZ, PATZ1, and RREB1 among others (Figure 3C and Supplementary Table 1G). Their clustering with SP and KLF factors is explained by their recognition of very similar, often overlapping G-rich DNA sequences (Figure 3D), likely through multiple C2H2 ZFs, which ranged in number from 3 in EGRs to as many as 24 in ZNF658 (Figure 3E and Supplementary Figure 3A).
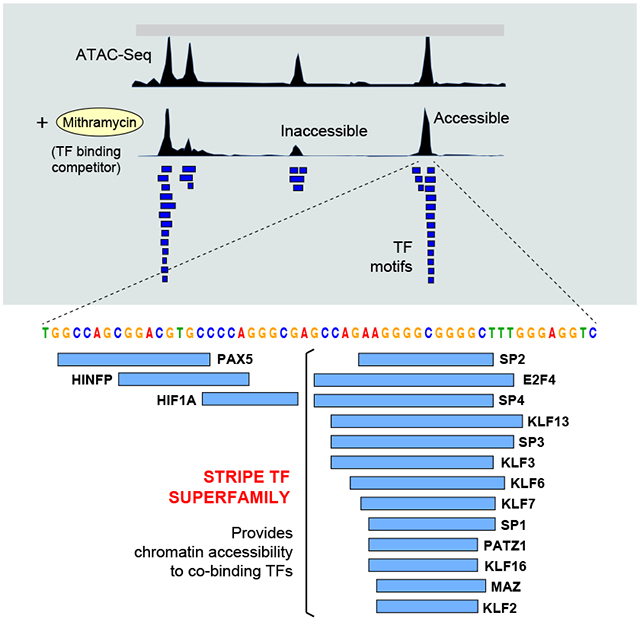
Previous studies with glucocorticoid receptors showed that the recognition of DNA motifs by multiple TFs increases the accessibility for co-binding factors, a process dubbed assisted loading (Voss et al., 2011). We thus wondered whether USFs as a group impart accessibility to regulatory DNA. To test this idea, we cultured activated B cells with the mithramycin (MTM) analogue EC-8042 (Nunez et al., 2012). MTM binds broadly to NC/GC/GN motifs in the minor groove of DNA and has been shown to interfere with TF occupancy, even for factors binding to the major groove (Hou et al., 2016; Vizcaino et al., 2014). At high concentration, MTM is cytotoxic to human tumors (Federico et al., 2020). However, at lower concentrations (50nM) it slows down proliferation of primary B cells without affecting viability (Supplementary Figure 3B). ATAC-Seq analysis showed that under such conditions, ~40% of DNA elements become at least 2-fold less accessible, while the remaining 60% are more resistant to treatment (Figure 4A). Notably, USF motifs were enriched in the resistant group (p < 2.2e-16, Figure 4B). Conversely, the number of MTM motifs was similar between resistant and sensitive elements (Figure 4C), demonstrating that resistance to MTM competition correlates with the presence of USF motifs. To extend this result to the factors themselves, we performed ChIP-Seq for SP1, RREB1, and EGR1 in CH12 B cells. As non-USF controls we probed PU.1, IRF4 and RELA. We found a strong correlation between USF occupancy and resistance to MTM treatment. On average, USFs were enriched 1.3-1.5-fold at elements that were resistant, whereas non-USFs showed no such enrichment (Figure 4D). Figure 4E and Supplementary Figure 3C provide individual examples of this global trend at the Hilpda and Snx2 loci.
Figure 4. USF recruitment correlates with accessibility and resistance to MTM.

(A) Dot plot showing ATAC-Seq signals at DNA elements from untreated vs. MTM-treated (50nM) activated B cells. Boxes identify resistant and sensitive populations. (B) Box plot showing the fraction of USFs per ATAC-Seq summit that are resistant (blue) or sensitive (red) to MTM treatment. The peaks were classified into 4 populations based on RPKM values as shown in panel A. (C) Number of MTM binding motifs per summit of ATAC-Seq peaks as shown in panel B. (D) ChIP-Seq signal intensity for 3 USF and 3 control TFs at elements that are resistant (blue) or sensitive (red) to MTM treatment. (E) Example of MTM-resistant and sensitive elements (based on ATAC-Seq) at the Hilpda locus in mouse B cells. Binding of SP1 (USF) and IRF4 (based on ChIP-Seq) and predicted TF motifs are included. Red asterisk below denotes overlapping USF motifs.
To corroborate the results with an orthogonal method we measured the impact of single nucleotide polymorphisms (SNPs) on USF recruitment and regulatory DNA accessibility. ATAC-Seq and ChIP-Seq for USF and non-SF controls were performed in activated B cells from inbred mouse strains C57BL/6J, BALB/c AnPt, CAST/EiJ, and their F1 progeny (Figure 5A). To accurately map SNPs, the three genomes were sequenced to depths of 75x-100x, revealing 0.1 (C57BL/6J), 5.1 (BALB/c AnPt), and 22.6 (CAST/EiJ) million SNPs relative to the mm10 reference genome (Supplementary Figure 4A). We next identified all ATAC-Seq peaks carrying single SNPs overlapping with USF (SP1, MAZ, RREB1) or non-USF (YY1, NRF1, IKAROS) binding motifs and measured accessibility. The results clearly showed that SNPs affecting recruitment of USFs (> 4-fold) have a significantly greater impact on accessibility than those targeting non-USF binding (p = 5.4e−9, Figure 5B). For example, a G-A change at the SP1 motif within the Gm10505 gene impairs binding of SP1 and colocalized factors, and it reduces overall accessibility. Conversely, an SNP at the NRF1 motif upstream of the Uqcc3 gene only impacted NRF1 binding (Figure 5C). Additional examples are provided in Supplementary Figure 4B. These and the MTM results are thus consistent with a model where recruitment of USFs provides greater accessibility to mammalian cis-elements.
Figure 5. USFs provide accessibility to regulatory DNA.

(A) Experimental strategy to assess accessibility at ATAC-Seq peaks where a single SNPs impacts recruitment of USFs or non-SFs. Mice images were obtained from the Jackson lab with permission. (B) Bar graph shows fold change in ATAC-Seq signals at elements where SNPs reduce occupancy of USFs (pink) or non-SF (grey) controls in F1 B cells. (C) Examples of ATAC-Seq peaks carrying single SNPs targeting SP1 (left) or NRF1 (right) binding motifs. BALB/c and Castaneous alleles are depicted in black and purple respectively.
USFs regulate the recruitment and dynamics of colocalized proteins
On average, USF motifs are present in 68% and 74% of ATAC- or DHS-Seq peaks in mouse and human cell types respectively, a feature that suggests these factors might regulate DNA accessibility across the mammalian genome. To directly test this idea, we deleted USF genes from different TF families in CH12 B cells: Sp1, Klf16, Zbtb7a, and Maz (Supplementary Figure 5A). We found that the number of affected regulatory elements increased with the number of deleted factors, with decreased accessibility being the predominant trend (Figure 6A). Using a > 2-fold cut off, Zbtb7a−/− B cells showed 881 changes, while in Sp1−/−Klf16−/−Zbtb7a−/−Maz−/− quadruple knockout (4KO) cells as many as 9,409 elements were affected (Figure 6A). In addition, a scatter plot showed many elements being affected below the 2-fold cut off, indicating that loss in accessibility in 4KO cells was broad (Figure 6B).
Figure 6. Deletion of USFs affects recruitment of TF partners.

(A) Bar graph depicting the number of elements affected in accessibility (ATAC-Seq) >2 fold upon loss of individual or different combinations of USFs. (B) Scatter plot shows ATAC-Seq signals (rpkm values) in WT and 4KO CH12 B cells, lacking SP1, MAX, KLF16 and ZBTB7A. (C) Box plot shows ChIP-Seq signals of 5 TFs in WT and 4KO cells. Signal reduction (100% - median fold change value on each peak) were 30% for NRF1, 17% for YB1, 22% for YY1, 52% for SMAD3 and 29% for SMAD7. (D) Comparison of transcriptomes in WT and 4KO cells normalized using spike in controls in TFGβ activated cells (red dots and line).
To assess the impact of USF deletion on colocalized TFs we performed ChIP-Seq for NRF1, YB1, YY1, SMAD3 and SMAD7, the last two measured in TGFβ-treated cells (see Methods). We chose these factors because their expression was mostly unaltered in 4KO cells (Supplementary Figure 5B). In all cases, we found a reduction in recruitment, which ranged from 17% for NFYB to 52% for SMAD3 (Figure 6C). Importantly, these global changes were also reflected in the transcriptome, which showed a 7-26% reduction relative to control (Figure 6D and Methods), including at gene targets for the chosen TFs (Supplementary Figure 6). These data are thus consistent with the notion that USFs provide broad accessibility to DNA elements and facilitate recruitment of colocalized factors.
Loss of accessibility in the absence of USFs predicts an impact in the binding dynamics of colocalized factors. To directly test this idea, we fused the HaloTag peptide (Los et al., 2008) to the N-terminus of SMAD3 and SMAD7 in WT and 4KO cells. We selected SMAD proteins for this experiment because we can readily control their nuclear import by TGFβ treatment, a feature that allows measuring background nuclear fluorescence in untreated cells. By means of highly inclined and laminated optical sheet (HiLO) microscopy and the JF549 fluorophore (Grimm et al., 2016) we tracked single molecules in real time in the presence of TGF-β at 6h for SMAD3 (Figure 7A) and at 24h for SMAD7 (Supplementary Figure 7A), following their peak of nuclear expression as previously reported (Giroux et al., 2010; Luwor et al., 2013; Qiu et al., 2005). The mean square displacement (MSD, i.e. particle movement over time) analysis revealed three main SMAD diffusive populations (P1 to P3, Supplementary Figure 7B). P1 displayed long exploration areas and a mean square displacement (MSD, i.e. particle movement over time) consistent with 2D Brownian motion, with an effective diffusion coefficient of 2.7 +/− 0.24 μm2/s for SMAD3 and 2.1 +/− 0.02 μm2/s for SMAD7 (Supplementary Figure 7B). Conversely, P2 and P3 represented sub-diffusive low-mobility states which have been linked to chromatin binding and a confined state respectively (Garcia et al., 2021a).
Figure 7. Lack of USF recruitment impacts residence time of colocalized factors.

(A) Micrograph showing particle trajectories of HALO-SMAD3 in WT CH12 B cells. Colors represent different tracks. Bar = 1 μm. (B) Survival probability (%) of HALO-SMAD3 molecules in WT (blue) or 4KO (red) CH12 B cells. The data was fitted to power-law or biexponential curves respectively. (C) Box plot showing binding time distribution (in seconds) of HALO-SMAD3 molecules expressed in WT or 4KO cells. (D) Pie charts showing percentage of the different diffusive states of SMAD3 (left) or SMAD7 (right) displaying residence times of 5-10” (white), 10-20” (grey) or > 20” (yellow). Upper pie charts represent data from WT cells, lower from 4KO.
To define how deletion of USFs impacts the dynamics of SMAD binding, we measured their residence time by long-exposure single molecule tracking (SMT (Paakinaho et al., 2017; Presman et al., 2017)). In WT cells, SMAD3 and SMAD7 residence times were best fit by a power-law distribution, implying that both factors exhibit a broad distribution of effective binding affinities (Figure 7B and Supplementary Figure 7C). Notably, in 4KO cells this power-law behavior transitioned to a biexponential one (Figure 7B and Supplementary Figure 7C), implying that chromatin contacts become more homogenous for SMADs in the absence of USFs. Consistent with this idea, the overall dwelling time of SMADs dropped from a broad range of 10-100 s in WT to a narrower range of 10-50 s in 4KO cells (Figure 7C and Supplementary Figure 7D). Accordingly, SMAD3 and SMAD7 trajectory population fractions that had residence times >20 s decreased from ~15% in WT to ~6% in 4KO cells (Figure 7D). Conversely, the free diffusion of either SMAD3 or SMAD7 was unchanged with the loss of USFs (Supplementary Figure 7B). Taken together these findings demonstrate that USFs impact not only the accessibility but also the residence time of colocalized proteins at chromatin.
Discussion
In this study we have explored TF combinatorial binding in the mouse and human genomes with comprehensive motif maps and ChIP-Seq datasets. TF recruitment in mammalian cells has been shown to depend on several parameters. First, the affinity of the TF DNA binding domain for cognate sequences, which vary considerably from site to site due to motif degeneracy, suboptimization and allelic variants (Farley et al., 2015; Rowan et al., 2010; Zandvakili et al., 2018). Second, nucleosome competition, a feature that is influenced by steric hindrance and the local concentration of TFs and unbound histones (Joseph et al., 2017; Zhu et al., 2018). Third, the presence of cooperative TF partners that stabilize DNA contacts through protein-protein interactions or the recruitment of specific cofactors that form multiprotein complexes (Murphy et al., 2013; Panne et al., 2007; Reiter et al., 2017). TF cooperation is a well-described phenomenon among proteins that form heterodimers and bind closely spaced or overlapping motifs (Kerppola and Curran, 1991). Our analysis identified multiple clusters of such factors, including the well-characterized AP-1 group, composed of ATF, FOS, JUN and MAF (Shaulian and Karin, 2002). Another example in hematopoietic cells was the NF-κB family, consisting of RELA, RELB, c-REL, NF-κB1 and NF-κB2, which form up to 15 different dimers through combinatorial associations (Oeckinghaus and Ghosh, 2009).
Unexpectedly, the analysis also uncovered a distinct group of factors that frequently pairs with most TFs expressed in the cell, creating vertical stripes in motif or ChIP-Seq hierarchical heatmaps. The resulting TF combinations differ from those formed by heterodimers in that the pairs bind non-overlapping motifs. Based on their expression across cells and tissues, stripe factors fall into two main subsets: restricted or cell-specific and broadly or ubiquitously expressed ones. The idea that cell-specific factors colocalize and might collaborate with most TFs expressed in the cell is consistent with their fundamental transcriptional role during ontogeny (Barozzi et al., 2014; Nutt et al., 2007; Schaefer and Lengerke, 2020). This group includes well known cell-defining proteins PU.1, BCL6, BLIMP1, SOX2, NANOG, and KLF4 among others.
The second group, which we named universal stripe factors, is composed of about 30 members clustered in heat maps from all cells and tissues examined. It comprises the SP, KLF, EGR and ZBTB families, as well as a subset of zinc finger proteins. Remarkably, as a unit these factors recognize the same or highly overlapping DNA motifs in ~70% of DNA elements, a feature that may help explain their function. Three sets of experiments shed light on this issue. First, in the presence of the general binding competitor MTM, DNA elements enriched for USFs were more resistant to the loss of accessibility caused by the drug. Second, SNP-induced loss of USF binding in F1 mouse cells compromised accessibility more frequently than for non-stripe factors. Finally, chromatin accessibility and binding dynamics of colocalized factors were broadly changed when selected USFs were deleted. The strong inference is that USFs enable proteins within the same regulatory element to engage chromatin.
Precisely how USFs facilitate accessibility is unclear. One possibility is that by binding overlapping sequences they may help evict nucleosomes by mass action. A similar mechanism was previously proposed for TF collaborative binding (Deplancke et al., 2016; Mirny, 2010) or assisted loading (Voss et al., 2011), where TFs assist one another by outcompeting nucleosomes for DNA contacts. This binding interdependency can be explained by the fact that the affinity of nucleosomes for DNA is substantially greater than for any one TF alone (Polach and Widom, 1996). By binding en masse to overlapping GC sequences, USFs are well poised for this function. In support of this view, there is strong evidence that higher GC-content correlates with increased DNA accessibility (Comings et al., 1975; Hammelman et al., 2020). These considerations, together with the ubiquitous expression and broad binding of USFs in mouse and human cells indicate that mammalian cells might have evolved USFs at least in part to help establish regulatory elements and the recruitment of TF partners across the genome.
Limitations of the study
The hierarchical clustering maps uncovered TFs that display stripe profiles only with ChIP-Seq data. A subtraction analysis (Figure 3A and Supplementary Figure 2D) suggests that this phenomenon is likely due to indirect TF binding because the “extra” ChIP-Seq peaks that generate the stripes lack cognate DNA motifs. Previous reports have proposed that such peaks result from “tethering” or protein-protein contacts (Biddie et al., 2011; Neph et al., 2012). Our study does not address whether that type of binding impacts regulatory DNA accessibility. However, our unpublished data indicate that motif− ChIP-Seq peaks tend to have fewer reads than motif+ ones, suggestive of weaker binding. In addition, Figure 5B shows that YY1 and IKAROS (which are classified as indirect stripe factors in human cells) exert little or no impact in accessibility when their binding is compromised. This contrasts with USFs (SP1, MAZ or RREB1), which have a significant impact. Future studies will be necessary to determine whether indirect TF binding plays a physiological role in mammalian cells, or they are simply the result of TFs scanning of chromatin.
Another limitation of our study is that it does not specify whether USFs create accessibility, as pioneer factors do, or they simply help to maintain it. We favor the latter possibility because of 15 mammalian pioneer factors described in the literature only 4 were classified as stripe factors (EBF1, KLF4, P53 and PU.1).
Finally, the analysis of USF knockouts was limited to those that did not compromise viability when deleted in CH12 B cells. Several combinations which could have shed additional light on USF function were never obtained.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to the lead contact: Rafael Casellas (rafael.casellas@nih.gov).
Materials availability
Cell lines, including CH12 Zbtb7a−/− cells, CH12 Maz−/− cells, CH12 Klf16−/− cells, CH12 Sp1−/− cells, CH12 Sp1−/− Zbtb7a−/− double KO cells, CH12 Klf16−/− Zbtb7a−/− double KO cells, CH12 Sp1−/− Klf16−/− Zbtb7a−/− triple KO cells, CH12 Sp1−/− Klf16−/− Zbtb7a−/− Maz−/− quadruple KO cells, CH12 Halotag-Smad3 cells and CH12 Halotag-Smad7 cells can be obtained by contacting Supriya Vartak (supriya.vartak@nih.gov).
Data and code availability
RNA-seq, ChIP-seq, ATAC-seq, whole genome sequencing and other deep-sequencing data reported in this paper have been deposited at GEO and are publicly available as of the date of publication. Accession numbers can be found at GSE164906. This paper also analyzes existing, publicly available data. These accession numbers for the datasets are listed in the supplementary table 1H.
Original code to determine TF colocalization has been archived at Zenodo (DOI: 10.5281/zenodo.6642964) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from Yongbing Zhao upon request (im@ybzhao.com).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All strains of mice used in the study were purchased from Jackson Laboratory. All animal experiments were conducted according to institutional animal care and safety guidelines at National Institutes of Health. To study the impact of SNPs on accessibility, F1 progeny of (i) CAST/EiJ and BALB/cJ and (ii) C57BL/6J and BALB/cJ were generated.
Primary cells and cell lines
Primary B cells were isolated from the spleens of 6–8-week-old male mice using the EasySep™ Mouse B Cell isolation kit from StemCell Technologies Inc., according to manufacturer’s instructions. Cells were cultured in RPMI-1640 media supplemented with 10% FBS (Gemini), 1% Penicillin-Streptomycin, 1% HEPES, 1% Sodium Pyruvate, 1% Glutamax, 1% NEAA, 50 μM β-mercaptoethanol (Thermo Fisher Scientific), 50μg/ml LPS (Sigma), 2.5 μg/ml IL-4 (Sigma), and 0.5 μg/ml anti-CD180 (BD Bioscience). CH12 B lymphoma cells were cultured in RPMI-1640 media supplemented with 10% FBS (Gemini), 1% Penicillin- Streptomycin and 50 μM of β-mercaptoethanol (Thermo Fisher Scientific). Platinum-A cells were cultured in high glucose DMEM containing 1% Penicillin-Streptomycin, 1% Sodium Pyruvate and 1% Glutamax. All cells were cultured at 37°C and 5% CO2 in a humidified incubator, and were routinely tested for Mycoplasma contamination.
METHOD DETAILS
CRISPR Cas9 engineering of CH12 B cells
Suitable sgRNA targets were identified using the sgRNA online tool https://crispr.zhaopage.com, and sgRNAs were cloned into the pSPCas9(BB)-2A-GFP (pX458, Addgene #48138) vector. To delete a target gene, CH12 B cells were nucleofected using the SF Cell Line 4D-Nucleofector™ X Kit with the 4D- Nucleofector™ device (Pulse code: CM150). For each target gene, we used a plasmid containing an sgRNA targeting a sequence upstream the first exon, and a plasmid containing an sgRNA targeting a sequence downstream the last exon (1 μg of each pX458 plasmid per 1–2 million cells). 24 h following transfection, cells were single cell GFP sorted into 96-well plates, and DNA was extracted after about 10 days, using mouse direct PCR kit (Bimake). Genotyping was performed using specific screening primers. Using this protocol, CH12 Zbtb7a−/− cells, CH12 Maz−/− cells, CH12 Klf16−/− cells and CH12 Sp1−/− cells were generated. This nucleofection-sorting-screening cycle was repeated consecutively to obtain CH12 Sp1−/− Zbtb7a−/− double KO cells, CH12 Klf16−/− Zbtb7a−/− double KO cells, CH12 Sp1−/− Klf16−/− Zbtb7a−/− triple KO cells and CH12 Sp1−/− Klf16−/− Zbtb7a−/− Maz−/− quadruple KO cells. Each genotype was confirmed by performing at least two biological replicates.
Chromatin immunoprecipitation sequencing (ChIP-seq)
Cultured cells were fixed with 1% formaldehyde (Sigma) for 10 min at room temperature and the reaction was quenched with 125 mM glycine (Sigma). Ten million fixed cells per sample were washed with PBS, snap-frozen and stored at −80°C until further processing. Before use, the cells were resuspended in 850 μl of RIPA buffer (10 mM Tris pH 7.6, 1 mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100) freshly supplemented with Complete Mini EDTA free proteinase inhibitor (Roche). Sonication was performed using Bioruptor sonicator (Diagenode) at high amplitude for 20 cycles of 30 sec sonication followed by 30 sec of pause. Chromatin was incubated overnight at 4°C under slow rotation with 5 μg of the antibody of interest pre bound to 40 μl of Dynabeads Protein A (or G) for 40 min at room temperature under agitation. After immunoprecipitation, the beads were washed twice with RIPA buffer, twice with RIPA buffer containing 0.3M NaCl, twice with LiCl buffer (0.25 M LiCl, 0.5% Igepal-630, 0.5% sodium deoxycholate), once with LiCl buffer (0.25M LiCl, 0.5% NP-40, 0.5% NaDOC), once with TE pH 8.0 containing 0.2% Triton X-100, and once with TE pH 8.0. Crosslinks were reversed by incubating the beads at 65°C for 4 hours in the presence of 0.3% SDS and 1 mg/ml Proteinase K (Thermo Fisher Scientific). ChIP DNA was purified by ChIP DNA clean and concentrator column (Zymo Research). Libraries were prepared using the Ovation Ultralow Library System V2 kit and paired-end sequencing was performed on NovaSeq6000 (Illumina). For each sample we sequenced at least two biological replicates.
ATAC-seq
To perform ATAC-seq analysis, primary activated B cells or CH12 B cells were seeded at the concentration of 150,000 cells/ml and harvested after 72 h of culture. For each sample, 10,000 cells were centrifuged at 500g for 5 min at 4°C, washed once with 50 μl of cold PBS buffer, then resuspended in 50 μl of cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630). Cells were then immediately centrifuged at 500g for 10 min at 4°C to permeabilize and isolate the nuclei. Permeabilized nuclei were resuspended in 50 μl of transposition mix (25 μl 2x TD Buffer (Illumina Cat #FC-121-1030), 2.5 μl Tn5 Transposes (Illumina Cat #FC-121-1030) and 22.5 μl nuclease free water), and incubated for 30 minutes at 37°C. At the end of the transposase reaction, DNA was isolated using the DNA Clean & Concentrator kit (Zymo Research), and PCR amplified using primers that include Illumina adaptors. Each PCR reaction contained 10 μl Transposed DNA, 9.7 μl nuclease free water, 2.5 μl 25μM Customized Nextera PCR Primer 1 (Ad1_NoMX), 2.5 μl 25μM Customized Nextera PCR Primer 2 with a Barcode (Ad2.x),0.3 μl 100x SYBR Green I (Invitrogen Cat#S-7563), and 25 μl NEBNext High-Fidelity 2x PCR Master Mix (New England Labs Cat #M0541); and was performed according to the following cycles program: (1) 72°C, 5 min; (2) 98°C, 30 sec; (3) 98°C, 10 sec; (4) 63°C, 30 sec; (5) 72°C, 1 min; (6) Repeat steps 3-5, 4x. Since the tagged DNA fragments must be further amplified to provide sufficient material for sequencing, to reduce GC and size bias due to PCR, an appropriate, minimal number of additional PCR cycles (N) was determined using qPCR, preventing saturation of the amplification reaction. Each pilot qPCR reaction included the following: 5 μl of previously amplified DNA, 4.41 μl nuclease-free H2O , 0.25 μl 25 μM Custom Nextera PCR Primer 1, 0.25 μl 25 μM Custom Nextera PCR Primer 2, 0.09 μl 100 × SYBR Green I, 5 μl NEBNext High-Fidelity 2 × PCR Master Mix. PCR was performed according to the following cycles program: (1) 98°C, 30 sec; (2) 98°C, 10 sec; (3) 63°C, 30 sec; (4) 72°C, 1 min; (5) Repeat steps 2-4, 19x. To calculate the additional numbers of cycles (N) required to amplify the library, we plotted the Rn value (fluorescent signal from SYBR Green I, corrected for background signal) versus cycle number, and N was determined as the cycle number that corresponds to approximately one-fourth of the maximum fluorescent intensity. Once N was established, the remaining 45 μl of previously amplified DNA were further amplified using the above-mentioned PCR conditions. At the end of the second PCR amplification, each sample was purified using the DNA Clean & Concentrator kit (Zymo Research) and paired-end sequencing was performed on NovaSeq6000 (Illumina). For each sample we sequenced at least two biological replicates.
Biotag vector cloning and transduction
Smad3, Smad7, Zbtb7a, Klf13, and Pax5 were PCR amplified with gene specific primers and Q5 High Fidelity polymerase (NEB) from cDNA of CH12 or primary activated B cells using Superscript III reverse transcription (Invitrogen). Biotag was cloned at the N-terminus of Smad3, Smad7, Zbtb7a, or Klf13 and combined with P2A-mOrange or P2A-GFP by stitch PCR. Primer information listed below.
| Primer | Sequence |
|---|---|
| Smad3-for NBiotag-F | GAAAATCGAATGGCACGAAGGCGCGCCGAGCTCGAGGGTTATGTCGTCCATCCTGCCCTTC |
| Smad3-for Nbiotag-R | ACTTCCTCTGCCCTCAGACACACTGGAACAGCGGATG |
| T2AmOr-S3 NBiotag-F | TGTTCCAGTGTGTCTGAGGGCAGAGGAAGTCTTCTAACATG |
| T2AmOr-for NBiotag-R | TCGTTGTGGGAGGTGATGTCCAACTTGATGTCGACGATGTAGGCGCCGGG |
| Smad7 for NBiotag-F | GAAAATCGAATGGCACGAAGGCGCGCCGAGCTCGAGGGTTATGTTCAGGACCAAACGATCTGC |
| Smad7 for Nbiotag-R | ACTTCCTCTGCCCTCCCGGCTGTTGAAGATGACCTC |
| T2AmOr-S7 nBiotag-F | ATCTTCAACAGCCGGGAGGGCAGAGGAAGTCTTCTAACATG |
| Zbtb7a-for NBiotag-F | GCACGAAGGCGCGCCGAGCTCGAGGGCTGGCGGCGTGGACGGCCC |
| Zbtb7a-for NBiotag-R | GGCTGAAGTTAGTAGCTCCGCTTCCGGTTGCGAAGTTACCCTCGGCGGTG |
| Klf13-for NBiotag-F | GCACGAAGGCGCGCCGAGCTCGAGGGCAGCCGCCGCCTATGTGGAC |
| Klf13-for NBiotag-R | GGCTGAAGTTAGTAGCTCCGCTTCCGGGCGAGCTGGCCGGGCTGATG |
| P2A-GFP-F | GGAAGCGGAGCTACTAACTTCAGCCTGCTGAAGCAGGCTGGAGACGTGGAGGAGAACCCTGGACCTGTGAGCAAGGGCGAGGAGCTGTTC |
| P2A-GFP-R | CACTGTGCTGGCGGCCCGGTCG |
Stitch PCR products were cloned into the retroviral vector pMy using restriction enzyme digestion and ligation. Retroviral particles were produced in Platinum-A retroviral packaging cell line by transfecting the cloned pMy vectors in presence of Lipofectamine LTX and Plus reagent (Life technologies). Infectious retrovirus was harvested 48 h post transfection. For Smad3 and Smad7 transduction, CH12 B cells (wild-type and QKO) were transduced with Vector1 (pMy-biotagSMAD3-T2A-mOrange or pMy-biotagSMAD7-T2A-mOrange) and Vector2 (pMy-BirA-T2A-EGFP) by centrifugation for 90 min at 2,500 rpm, at 32°C in the presence of 6 mg/ml polybrene. After 6 h, cells were diluted to 0.2 million cells per ml using reconstituted RPMI medium. A second infection was performed in a similar manner. 48 hours after the second infection, B cells were harvested and GFP + mOrange double positive cells were sorted using a BD FACSAria III cell sorter (Becton Dickinson). For Zbtb7a and Klf13 transduction, wild-type CH12 B cells were transduced with Vector1 (pMy-biotag-ZBTB7A-P2A-GFP or pMy-biotag-KLF13-P2A-GFP) and Vector2 (pMy-BirA-T2A-mOrange) following the same protocol used for Smad3 and Smad7 transduction.
Biotag ChiP-seq
Sorted cells (10-20 million) were crosslinked for 10 min at 37°C with 1% (v/v) formaldehyde and quenched with 0.125 M glycine. For SMAD3 and SMAD7 ChIP-seq, cells were treated with TGFβ (5 ng/ml; 240-B R&D systems) for 6 or 24 h before crosslinking. Crosslinked cell samples were then sonicated using a Covaris sonicator to obtain DNA fragments of 200–500 bp in length. Samples were incubated with 50 μl of Dynabeads M-280 Streptavidin Beads (Invitrogen) overnight at 4°C in RIPA buffer (10 mM Tris [pH 7.6], 1 mM EDTA, 0.1% [w/v] SDS, 0.1% [w/v] sodium deoxycholate and 1% [v/v] Triton X-100). Beads were washed twice with Wash buffer 1 (2% [v/v] SDS), once with Wash buffer 2 (0.1% [v/v] deoxycholate, 1% [v/v], once with Wash buffer 3 (250 mM LiCl, 0.5% [v/v] NP-40, 0.5% [v/v] deoxycholate, 1 mM EDTA, and 10 mM Tris-HCl [pH 8.1]), and then twice with TE buffer (10 mM Tris-HCl [pH 7.5] and 1 mM EDTA). ChIP DNA was then extracted for 4 h at 65°C in Tris-EDTA buffer with 0.3% (w/v) SDS and 1 mg/ml Proteinase K (Thermo Fisher Scientific). DNA was purified by ChIP DNA clean and a concentrator kit (Zymo research). Libraries were prepared using the Ovation Ultralow Library System V2 kit and 50 bp paired-end sequencing was performed on NovaSeq 6000 (Illumina). For each sample we sequenced at least two biological replicates.
EC-8042 treatment
For displacing DNA-bound transcription factors, 24 h activated mouse B cells were treated with the Mithramycin derivative EC-8042 (10, 50 and 100 nM) and cells were cultured for another 24, 48 or 72 h for optimizing treatment conditions. 0.01% DMSO treated cells served as vehicle control. Cells were harvested and assessed for proliferation (cell number) and viability (trypan blue exclusion) at each denoted timepoint. At least three biological replicates were performed for EC-8042 dose titration and time course experiments (three technical replicates per biological replicate).
TGFβ treatment
CH12 B cells were treated with TGFβ (5 ng/ml; 240-B R&D systems) for 6 or 24 h before harvesting.
Generation of Halo-Smad3/Halo-Smad7 knock-in cell lines
sgRNAs recognizing N-terminus of Smad3 or Smad7 were designed with the Optimized CRISPR Design tool (http://zlab.bio/guide-design-resources) and cloned into pX458-GFP vector (Addgene). Smad3 and Smad7 were amplified using gene specific primers and Q5 High Fidelity polymerase (NEB) from genomic DNA of CH12 cells obtained by DNeasy Blood and Tissue kit (Qiagen). Halo-tag was introduced at the N-terminus of Smad3 and Smad7 by overlapping (stitch) PCR. 500-600 bp homologous arms were used to construct donor DNA for Halo-tag knock-in, with silent mutations introduced on 5′ arms to avoid Cas9/sgRNA cutting of the donor DNA. Donor DNA was inserted into pCR-Blunt II-Topo vector (Thermo Fisher). The donor DNA and targeting sgRNA were nucleofected into CH12 B cells using the SF Cell Line 4D-Nucleofector™ X Kit. 48 hours post transfection, B cells were harvested and single-cell GFP positives were sorted into 96-well plates using a BD FACSAria III cell sorter (Becton Dickinson). 10-12 days post sorting, colonies were picked and genotyped to identify homozygous knock-in clones. sgRNA sequences and primers used for cloning and genotyping are listed below.
| Primer | Sequence |
|---|---|
| sgRNA_Smad3_N-terminus | GTGACCCTTCGGTGCCAGCC |
| sgRNA_Smad7_N-terminus | TAGCCGGCAAACGACTTTTC |
| Smad3-5HA-F1 | GAGGGATCCTTAAGGGCGAATTCTGCAGATGCGGCGACTGCGCTGGGAAGGAGGCTG |
| Smad3-5HA-R1 | GTACCGATTTCTGCCATAGCTGACAACGCAGAGTTACCGGCGCCCCC |
| Halo-for Smad3-F1 | GTAACTCTGCGTTGTCAGCTATGGCAGAAATCGGTACTGGC |
| Halo-for Smad3-R1 | GTGAAGGGCAGGATGGACGACATGATCGCGTTATCGCTCTGAAAGTACAG |
| Smad3-3HA-F1 | CGATAACGCGATCATGTCGTCCATCCTGCCCTTCACCCCCCCGATC |
| Smad3-3HA-R1 | CATGCTCGAGCGGCCGCCAGTGTGATGGATCCTCCACGAGCGCGGGGCGGGAGGCGGCGGAG |
| Smad7-5HA-F1 | GAGGGATCCTTAAGGGCGAATTCTGCAGATTGCTTAGCAAGGGGGAAAGAGGCTTTTTCCTC |
| Smad7-5HA-R1 | CCGATTTCTGCCATGCGGGGCGAGGAGGCGAGGATAAAATTCATTCGCAGGTTAAGGAG |
| halo-for Smad7-F1 | CCTCGCCTCCTCGCCCCGCATGGCAGAAATCGGTACTGGC |
| halo-for smad7-R1 | CGTTTGGTCCTGAACATGATCGCGTTATCGCTCTGAAAGTACAG |
| Smad7-3HA-F1 | CTTTCAGAGCGATAACGCGATCATGTTCAGGACCAAACGATCTGCGCTCGTC |
| Smad7-3HA-R1 | CATGCTCGAGCGGCCGCCAGTGTGATGGATCTCTTGACTTCCGAGGAATGCCTGAGATCC |
Single Molecule Imaging
CH12 B cells were incubated with Janelia Fluor Dye-conjugated Halo ligand for 30 minutes at 37°C. JF549 (F.C. 2 nM, Janelia Research Campus) was used in MSD and residence time experiments. Post staining, cells were washed with PBS three times, resuspended in phenol red-free medium, plated onto poly-L-Lysine coated coverslip chamber (ibidi, 80824) and incubated at 37°C for 30 minutes for fast attachment.
Single-molecule imaging experiments were performed using a 100x, 1.35 NA silicone oil objective (Olympus) on a previously described home-built widefield microscope (Kieffer-Kwon et al., 2017) with a multi-band dichroic (405/488/561/633) BrightLine quad-band bandpass filter (Semrock, USA). We removed the cylindrical lenses from the emission path when conducting imaging, using 561 nm laser line for illumination of JF549. The lasers were focused into the back pupil plane of the objective to generate collimated illumination. An xy translation stage with a mirror was placed in a plane conjugated to the back pupil plane to change the angle of the laser beam at the sample plane for generating inclined illumination. A DAQ (BNC-2110, National Instruments) and AOTF (Acousto-optics) were used to modulate the intensity and wavelength of multiple laser lines. The microscope, AOTF, lasers and the camera were controlled through Micro-manager. Imaging was performed with an electron multiplying charge coupled device (EM-CCD) camera (Photometrics, Evolve 512 Delta). Cells were maintained at 37°C with 5% CO2 and proper humidity control and the objective was similarly heated to 37°C, using a microscopy incubation system (Tokai Hit, INU) for live-cell experiments. The experiment was performed in three biological replicates. For each sample, at least 50 cells were analyzed for single molecule tracking analysis.
Residence time analysis
To measure the residence time (RT) of endogenous Halo-Smad3 and Halo-Smad7, we labeled knock-in CH12 cells with 2nM JF549. Photobleaching correction was performed by tracking transiently expressed Halo-H2B. Images were continuously acquired for 800 frames in the JF549 channel at 561 nm with long image acquisition time (500 ms). An improved method accounting for photobleaching effects was applied to the residence times analysis (Garcia et al., 2021a). Briefly, the dwell time distribution of histone H2B was measured at the focal plane under precise SMT acquisition conditions and then fitted to a triple exponential and double exponential model to calculate photobleaching parameters; model selection was used to determine the best predictive model. The dwell time distribution was obtained by calculating the ensemble distribution of bound times for Smad3 and Smad7 in different cells from each biological replicate and corrected by dividing the exponential component estimated in the H2B dwell time distribution analysis (S(t) = eγtSE(t), where S(t) corresponds to the survival distribution after photobleaching correction, SE(t) the empirical survival distribution and γ the photobleaching rate). After photobleaching correction, the dwell time distribution was fitted to different models. The best predicting model was selected using Bayesian Inference Criterion (BIC) and the evidence in decibels of a power-law model with respect to a double exponential model. Evidence of 30 Db corresponds to a probability higher than 0.999 that the power-law model better describes the data in comparison with the double exponential model. For Halo-Smad3 and Halo-Smad7, the evidence was 33.6 Db (ΔBIC of 90.8) and 30.1 DB (ΔBIC of 39.7) respectively and for Halo-Smad3-QKO and Halo-Smad7-QKO the evidence was −118.9 Db (ΔBIC of −31.16) and −26.24 Db (ΔBIC of −13.24) respectively (Negative values indicate the best predictive model is given by the double exponential model while positive values indicate power-law as the best predictive model). All fits to the data were implemented with the nonlinear least square method using bisquare weights. Boxplots were generated from simulated tracks with a probability density given by the photobleaching corrected dwell time distribution of the protein of interest.
mRNA-Seq
0.5 million cultured cells were harvested, pelleted, and lysed in 100 μl of the Ambion RNAqueous lysis solution. Total RNA was extracted and treated with DNase according to the manufacturer’s protocol (RNAqueous®-Micro Kit). RNA quality and integrity was assessed using TapeStation (Agilent) and samples with RNA Integrity Number (RIN) >9 were used for further experiments. mRNA purification was performed using the NEBNext® Poly(A) mRNA Magnetic Isolation Module (NEB E7490). Libraries were prepared using the NEBNext® Ultra RNA Library Prep Kit for Illumina® (E7530). 50 cycles of sequencing data were acquired on HiSeq 2000, 2500 or 3000 (Illumina). For each sample we sequenced at least two biological replicates (three technical replicates per biological replicate). To measure global changes in the transcriptome of 4KO relative to control we included spike in controls as previously described (Kouzine et al., 2013). Transcriptomes were processed in untreated cells (10% reduction observed), or in TFGβ-treated cells for 1h (19% reduction), 6h (7% reduction) or 24h (26% reduction, shown in Figure 6D).
Spike-in mRNA-Seq normalization
To normalize transcriptome data, 1 μl of 1/10 dilution of Ambion’s ERCC RNA Spike-in Mix (Catalog number: 4456740) was added to 0.5 million cells, followed by total RNA isolation according to the mRNA-Seq protocol described above. For analysis, the read counts of spike-in RNA and mRNA were normalized by library size and exonic size of each gene to obtain RPKM (reads per kb per million aligned reads) values. A linear model was fit to the ERCC spiked-in data to correlate the spike-in RNA copy number in each cell type to the measured mRNA RPKM using the formula: lm(log10(known copy number) ~ log10(RPKM) + cell, data = counts). The linear model was then used to estimate the copy number of all expressed genes in each cell type.
MSD analysis
To analyze the motion of endogenous Halo-Smad3 and Halo-Smad7, we labeled knock-in CH12 cells with 2nM JF549. Halo-H2B was transiently expressed as a photobleaching correction. Using a short image acquisition time (10 ms), images were continuously acquired for 3000 frames in the JF549 channel at 561 nm. Perturbation Expectation Maximation (pEM) together with BIC was used to classify the trajectories of the protein into the least number of diffusive modes (sub-diffusion, diffusion and super-diffusion (Garcia et al., 2021b). The number of reinitializations were set up to 10, number of perturbations to 50, maximum number of iterations to 10000, convergence criteria for change in log-likelihood to 1e-7 and the number of features of the covariance matrix to 3. The posterior probability weighted mean-squared displacement (MSD) for each diffusive state was computed. To calculate the diffusion coefficient (D) for a diffusive state (Brownian motion), the variance of the instantaneous velocity vector ν was related to the diffusion coefficient as , Δt is the acquisition interval and D is the diffusion coefficient of the particle (Qian et al., 1991).
PCR-free Whole genome sequencing (WGS)
Genomic DNA was extracted from B cells of CAST/EiJ, BALB/cJ and C57BL/6J mice using the DNeasy Blood & Tissue kit from Qiagen. Samples were processed for whole genome sequencing using the TruSeq DNA PCR-Free High Throughput Library Prep Kit from Illumina according to manufacturer’s instructions. Paired-end sequencing (100bp) was performed on NovaSeq6000 (Illumina).
QUANTIFICATION AND STATISTICAL ANALYSIS
Processing of ATAC-Seq, DNase-Seq, and ChiP-Seq data
Raw reads were aligned to mouse (mm10) or human (hg38) genome using bowtie2 with default parameters (Langmead and Salzberg, 2012). PCR duplicates were removed using Picard, and multiple aligned reads were removed using Samtools by filtering mapping quality lower than 30 (Li et al., 2009). Mitochondria reads were also excluded for further analyses. Single-nucleotide resolution coverage track was made using bedtools and bedGraphToBigWig (UCSC utilities). For ATAC-Seq reads, the positions on the positive and negative strands were adjusted by +4 bp and −5 bp respectively (Buenrostro et al., 2013). Enriched peaks were identified using MACS2 with --nomodel --shift −75 --extsize 150 --keep-dup all --call-summits - q 0.01 (Zhang et al., 2008). For samples used to build comprehensive TF binding map, the following criterions were used for QC: signal-to-noise ratio at TSS ≥6 and 50M usable reads. For other samples used for general analysis, the following criterions were applied: signal-to-noise ratio at TSS ≥6, 25M usable reads and two or more replicates. For ATAC-Seq and DNase-Seq data, all summits detected by MACS2 were extended 100bp to each flank side, and then the fixed 201bp regions were used as the final summit regions. Differential peaks or summits were identified by DESeq2 with 2-fold change on signal and FDR ≤ 0.01 (Love et al., 2014).
Processing of RNA-Seq data and identification of active genes
Raw reads were aligned to the mouse genome (mm10) or human genome (hg38) using STAR (Dobin et al., 2013). Gene expression levels were quantified using RSEM (Li and Dewey, 2011), and only protein-coding genes were considered and used for normalization and quantification. Differential expressed genes across conditions or cell types were identified using DESeq2 with 2-fold change on signal and FDR ≤ 0.01 (Love et al., 2014).
To identify active genes in each RNA-Seq sample, gene expression level was transformed to zFPKM as shown in (Hart et al., 2013). The approach was as follows: a) Gene expression levels were normalized to log2 (FPKM). Typically, log2 transformed gene expression values follow a bimodal distribution; b) Gene expression values falling on the right half-Gaussian curve were extracted, and then the two parameters of the Gaussian distribution were estimated (μ,σ) by mirroring extracted values. c) Expression value of all protein-coding genes were transformed with the following formula:
In this study, genes with zFPKM ≥-2 were treated as active genes.
Estimating the size of potential accessible regions of human and mouse genome
For all human or mouse samples, samples from similar tissue or cell type were clustered into the same group. For each group, all regulatory elements from different samples were combined and merged as non-overlapped regulatory elements. Meanwhile, we also combined regulatory elements from all samples, and merged them as super non-overlapped regulatory elements (pan-element), and then calculated average length of pan-element. Further, we made a binary matrix based on overlapping information between pan-element and non-overlapped regulatory elements in each group. In matrix, each row represents a pan-element, while each column represents a group. Xij represents the value on ith row (pan-element) and jth column (group):
Based on this matrix, we utilized an approach used for bacterial pan-genome profile (Zhao et al., 2014) to estimate the relationship between element number and group number (number of different tissue/cell types). For example, if the matrix includes G columns and S rows, the approach could be summarized as follows: we created a new sub-matrix by randomly selecting n (1, 2, 3, … G) columns from the original matrix, and then counted the number of rows with 1. To avoid bias in sampling, we replicated 2,000 times for each n with the same value. Based on this rule, we estimated the relationship between pan-element number and group number. The size of accessible genome was calculated by multiplying the number of pan-element and average length of pan-element.
Assignment of transcription factor to regulatory elements
Position weight matrix (PWMs) of human and mouse transcription factors (TFs) were extracted from TRANSFAC (commercial version), JASPAR and CIS-BP with manual curation (Fornes et al., 2020). For mouse and human genome, we created a redundant TF binding sites pool separately by aligning all mouse or human PWMs respectively to their reference sequences using fimo with a p-value of 1e-5 (Grant et al., 2011). Based on the redundant TF binding sites pool, we subsequently created a non-redundant TF binding sites pool by merging overlapped TF binding sites of the same TF. For each ATAC-Seq or DNase-Seq sample, we annotated each regulatory element (also mentioned as enriched peaks in previous context) and summit with non-redundant TFBS pool, followed by removal of binding sites for TFs with expression level < −2 (measured by zFPKM).
Identification and annotation of footprints
Footprints on ATAC-Seq and DNase-Seq data were identified using rgt-hint (Gusmao et al., 2016). For human samples, we also downloaded footprint data from Vierstra et al. paper (Vierstra et al., 2020). Redundant TF binding sites pool was used to assign TFs into footprints with the following standards: 90% of motif region overlaps with footprint region or 90% of footprint region overlaps with motif region. According to the expression level in the same or matched sample, TFs with zFPKM < −2 were removed.
Colocalization of pairwise TFs
As mentioned above, we identified all potential TF motifs in all extended summits (201bp) in each tissue and cell type. If TF A was assigned to a summit, we considered this summit as TF A positive. If %x of TF A positive summits was also TF B positive, the colocalization score of TF A to TF B was considered x%. Based on this method, we also calculated the colocalization score of TF B to TF A as y%. Of note, x% is not equal to y% in almost all cases, since these two scores are completely different indicators. Based on this strategy, we calculated pairwise TFs colocalization score for all active transcription factors tissue by tissue. To visualize the profile of pairwise TF colocalization, we generated a matrix with colocalization scores for each tissue. In the matrix, all TF As are listed as rows and all TF Bs are listed as columns. The matrix was further clustered using Morpheus (https://software.broadinstitute.org/morpheus) with hierarchical clustering method. In a tissue or cell type, if x% was found to be larger or equal to 10%, we defined TF A to be colocalized by TF B. If over 30% TFs in a tissue or cell type were colocalized by TF B, we defined TF B as a stripe factor. Typically, the column where TF B co-localizes is seen as a white stripe in the heatmap. We note that overlapping motifs were only counted once in these analyses.
To evaluate whether a pair of TFs is preferentially colocalized, or preferentially excluded by each other, we used the following formulae to compute p-values for each pair of TFs: pc indicates the probability of TF pair being preferentially colocalized in the same summit, while pe indicates the probability of TF pair being preferentially excluded by each other. P values were further corrected by Benjamini-Hochberg procedure.
In above formulae, N is the total number of summits in a specific tissue or cell type, Na is the number of TF A positive summits, Nb is the number of TF B positive summits, M is the number of summits positive for both TF A and TF B.
Statistical analysis of TF motif colocalization
To determine whether GC content influenced TF colocalization, we first extracted DNA sequences for all ATAC-Seq summits (201bp) from each cell type. Next, we performed 1,000 simulations on scrambled summit sequences and determined PWM hits for all expressed TFs using Fimo (p < 1e-5). The p value for each TF pair was computed based on their colocalization score in the original sequences and the distribution scores in scrambled sequences. The Benjamini-Hochberg method was then used to estimate the False Discovery Rate (FDR). For LPS + IL4 activated B cells the analysis showed that of 96,721 TF pairs, 11,500 had a colocalization score >= 10%, and 10,188 had the same colocalization score with an FDR of <=0.01, which represents 88.6% of total TF pairs with that score. Similar results were obtained for the analysis of CH12 B cells (89.4%). This means that about 90% of all colocalized TF pairs can be explained by motif distribution rather than by chance or GC content.
To confirm this result, we also picked 150,000 random 201bp regions from the genome and measured the colocalization score for all TF pairs in LPS + IL4 activated B cells and CH12 cells. The scores were compared to those obtained from the analysis of DHS+ regions. The p-value for each TF pair was computed based on the distribution of colocalization scores from 1,000 simulations. The Benjamini-Hochberg procedure was then used to estimate the False Discovery Rate (FDR). For LPS + IL4 activated B cells, the analysis showed that of 11,500 TF pairs with a colocalization score >= 10%, 9, 913 (86.2%) have higher colocalization scores in the DHS summits than in the random regions (see Figure below). In addition, 9,074 (78.9%) of those colocalizations show a statistically significant (FDR <= 0.01) enrichment in the DHS+ elements. Similar results were obtained with CH12 cells (87% of TF pairs showed higher colocalization in DHS+ and 79.2% of those were significant). This means that in mammalian cells most TF pairs are most significantly colocalized in DHS+ elements than in the rest of the genome.
Annotation with the protein sequences of universal stripe factors
For all genes of interest, we downloaded annotation of amino acid sequences from Swiss-Prot database of UniProt (UniProt, 2021). Map of C2H2 Zinc finger domains were visualized using in-house python script.
Validation of stripe factors in human ChIP-Seq
Human ChIP-Seq samples for HEK293, MCF7, GM12878, K562 lines were downloaded from ENCODE database. Raw reads were aligned and peaks were called using MACS2. All ChIP-Seq summits were extended to 500bp. TF colocalization scores were calculated on all pair-wised ChIP-Seq samples as done for mouse samples. Stripe factors were also defined as done for mouse cells.
Analysis of ATAC-Seq and ChIP-Seq data from F1 hybrid mouse
F1 genomes were processed by aligning raw reads to the mm10 genome using bowtie2. Genetic variants were identified using Bcftools (https://samtools.github.io/bcftools/) and the F1 reference genomes were then built by masking nucleotide variations as Ns. ATAC-Seq and TF ChIP-Seq reads were then aligned to each allele based on SNPsplit (https://github.com/FelixKrueger/SNPsplit). Differential peaks or summits were called whenever the number of reads were at least 4-fold different and 2) no less than 10 reads for the allele with highest number of reads.
The impact of TF loss binding on chromatin accessibility was assessed as follows. First, we extracted all TF ChIP-Seq peaks which harbor a single SNP that was associated with a defined TF motif. Based on the forementioned standards, only ChIP-Seq peaks with differential reads count on the two alleles were kept for further analysis.
Allele-specific ChIP-Seq and ATAC-Seq tracks were created based on pseudo allele-specific reads as follows: 1) reads carrying SNPs were assigned to the exact allele; 2) those without allelic information was randomly assigned to any allele with 50% vs 50% probability.
Supplementary Material
Supplementary Table S1. Bioinformatic analysis and sample details. (A) Related to Figure 1: ATAC-Seq and DNase-Seq samples processed. (B) Related to Figure 1: list of mouse samples analyzed by ATAC-Seq or DNase-Seq and RNA-Seq. (C), related to Figure 1: list of human samples analyzed by ATAC-Seq or DNase-Seq and RNA-Seq. (D) Related to Figure 2: "Excluded" TF motifs from mouse activated B cells (LPS+IL-4). (E) Related to Figure 2: Mouse and human stripe factors. (F) Related to Figure 3: Stripe factors identified in HEK293, MCF7, GM12878, and K562 cell lines based on ChIP-Seq assays. (G) Related to Figure 3: Universal Stripe Factors in various human and mouse tissues. (H) Related to STAR Methods: Accession numbers for publicly available data analyzed.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD180 (RP105) | BD PharMingen | Cat# 552128 |
| SP1 | Abcam | Cat# ab13370 |
| MAZ | Bethyl Laboratories | Cat # A301-652A |
| E2F4 | ENCODE | ENCSR000ERU |
| IRF4 | Santa Cruz Biotech. | Cat# sc-6059 |
| NRF1 | Abcam | Cat# ab34682 |
| JUND | ENCODE | ENCSR000ERR |
| YY1 | Abcam | Cat# ab109237 |
| RREB1 | Bethyl Laboratories | Cat# A303-129A |
| EGR1 | Cell Signaling | Cat# 4153S |
| PU.1 | Santa Cruz Biotech. | Cat# sc-352 X |
| RELA | Santa Cruz Biotech. | Cat# sc-372 |
| Bacterial and virus strains | ||
| One shot Stbl3 E. coli | Thermo Fisher | C737303 |
| 10-beta competent E. coli | NEB | C3019H |
| Chemicals, peptides, and recombinant proteins | ||
| IL-4 from mouse | Sigma | Cat# SRP3211 |
| LPS | Sigma | Cat# L2630 |
| FBS | Gemini | Cat# 100-500 |
| RPMI 1640 | Invitrogen (Gibco) | Cat#R7388 |
| penicillin/streptomycin | Invitrogen (Gibco) | Cat#15070063 |
| 2-beta mercaptoethanol | Invitrogen (Gibco) | Cat# 21985-023 |
| puromycin | Sigma | Cat#P8833 |
| 37% formaldehyde | Sigma | Cat# F1635 |
| Agencourt AMPure XP | Beckman Coulter | A63882 |
| DMEM (Dulbecco~s Modified Eagle Medium) | Thermo Fisher | 11960-044 |
| DMEM, low glucose pyruvate | Thermo Fisher | 11885084 |
| Dynabeads MyOne Streptavidin T1 | Thermo Fisher | 65601 |
| Dynabeads Protein A | Thermo Fisher | 10002D |
| Dynabeads Protein G | Thermo Fisher | 10004D |
| Dynabeads M-280 streptavidin | Thermo Fisher | 11205D |
| EDTA 0.5M | Thermo Fisher | 15575020 |
| Fetal Bovine Serum, BenchMark | Gemini - Bio Products | 100-106 |
| Fetal Bovine Serum, ES Cell qualified | ATCC | SCRR-30-2020 |
| GlutaMAX | Thermo Fisher | 35050061 |
| HEPES | Thermo Fisher | 15630-080 |
| Janelia Fluor 549 HaloTag Ligand | Promega | GA1111 |
| Lipofectamine™ LTX Reagent with PLUS™ Reagent | Thermo Fisher | 15338100 |
| MEM Non-Essential Amino Acids | Thermo Fisher | 11140-050 |
| NucSpot Live 488 Nuclear Stain | Biotium | 40081 |
| Opti-MEM® I Reduced Serum Medium | Thermo Fisher | 31985-070 |
| PA Janelia Fluor 549 HaloTag Ligand | Jiji Chen | N/A |
| Penicillin-Streptomycin | Thermo Fisher | 15140122 |
| Proteinase K | Thermo Fisher | 26160 |
| Puromycin | LifeTechnologies | A11138-03 |
| Q5 High-Fidelity DNA polymerase | New England Biolabs | M0491L |
| Sodium Dodecyl Sulfate Solution, 10X | Thermo Fisher | 27730020 |
| Sodium Pyruvate | Thermo Fisher | 11360-070 |
| Glycine | Sigma | Cat#50046 |
| Mithramycin analogue | Francisco Moris | EC-8042 |
| Critical commercial assays | ||
| EasySep™ Mouse B Cell Isolation Kit | Stemcell technologies | 19854 |
| Genomic extraction kit | Biotool™ | B4001 |
| Ovation Ultralow Library System V2 | Nugen | 344 |
| ChIP DNA clean and concentrator | ZymoResearch | Cat # 11379 |
| RNAeasy kit | Qiagen | Cat# 74104 |
| DNeasy blood and tissue kit | Qiagen | Cat #69504 |
| MinElute PCR purification kit | Qiagen | Cat# 28004 |
| Nucleofector Kit V | Lonza | Cat# VCA-1003 |
| Tagment DNA Enzyme and Buffer (ATAC-seq) | Illumina | Cat3 20034210 |
| CloneAmp HiFi PCR Premix | TaKaRa | 639298 |
| NEBNext Ultra II Directional RNA Library Prep Kit for Illumina | New England Biolabs | E7760 |
| NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | E5520S |
| Qubit ds DNA HS assay kit | Thermo Fisher | Q32851 |
| RNAqueous™-Micro Total RNA Isolation Kit | Thermo Fisher | AM1931 |
| SF Cell Line 4D-Nucleofector™ X Kit | Lonza | V4XC-2032 |
| SuperScript III First-Strand Synthesis System | Life Technologies | 18080-051 |
| ZR Plasmid Miniprep | Zymo Research | D4015 |
| NEBNext poly(A) mRNA Magnetic Isolation Module | New England Biolabs | E7490L |
| Ultralow DNAseq kits | Nugen | 0344NB-A01 |
| Tn5 transposase; Nextera® DNA Library Preparation Kit | Illumina | FC-121-1030 |
| Deposited data | ||
| Raw and analyzed data | This paper | GSE164906 |
| Imaging data | This paper |
https://data.mendelev.com/datasets/4k2xrcmjwg/1
https://data.mendeley.com/datasets/9fmpt74b6y/1 |
| Experimental models: Cell lines | ||
| Mouse: primary splenic B cells | This paper | N/A |
| Mouse: CH12B lymphoma cell line | Tasuku Honjo | N/A |
| Human: PlatA retroviral packaging cell line | Cell Biolabs Inc. | RV-102 |
| Human: PlatE retroviral packaging cell line | Cell Biolabs Inc. | RV-101 |
| CH12 Halotag-Smad3 cell line | This paper | N/A |
| CH12 Halotag-Smad7 cell line | This paper | N/A |
| CH12 Zbtb7a−/− cell line | This paper | N/A |
| CH12 Maz−/− cell line | This paper | N/A |
| CH12 Klf16−/− cell line | This paper | N/A |
| CH12 Sp1−/− cell line | This paper | N/A |
| CH12 Sp1−/− Zbtb7a−/− double KO cell line | This paper | N/A |
| CH12 Klf16−/− Zbtb7a−/− double KO cell line | This paper | N/A |
| CH12 Sp1−/− Klf16−/− Zbtb7a−/− triple KO cell line | This paper | N/A |
| CH12 Sp1−/− Klf16−/− Zbtb7a−/− Maz−/− quadruple KO cells | This paper | N/A |
| Experimental models: Organisms/strains | ||
| C57BL6 | Jackson Laboratory | JAX:000664 |
| CAST/EiJ | Jackson Laboratory | JAX:000928 |
| BALB/cJ | Jackson Laboratory | JAX:000651 |
| CAST/EiJ X BALB/cJ F1 progeny | This paper | N/A |
| C57BL/6J X BALB/cJ F1 progeny | This paper | N/A |
| Oligonucleotides | ||
| Klf16_sgRNA 1: CGTAAGAACAAGCCAAACGGAGG | Sigma | N/A |
| Klf16_sgRNA 2: ATCCATAGCCCGATGGCCGCAGG | Sigma | N/A |
| Maz_sgRNA 1: TCTTGGGGGTCTCGCGCTCGGGG | Sigma | N/A |
| Maz_sgRNA 2: AGGGCCCAATAGGGGATCGCAGG | Sigma | N/A |
| Sp1_sgRNA 1: GAACAGCCAATTACGCGCCGAGG | Sigma | N/A |
| Sp1_sgRNA 2: AAGTAACATCGGTATTCACAAGG | Sigma | N/A |
| Zbtb7a_sgRNA 1: AAGTACGGTTCAATCGGGCGTGG | Sigma | N/A |
| Zbtb7a_sgRNA 2: TAGGGGGCCTTAGCCTATCCAGG | Sigma | N/A |
| Klf16_screen F: AGTGCCTGAATGTGGAGTGGGAG | Sigma | N/A |
| Klf16_screen R: CTACTGTAGACTCTCCTGAGACCTTCC | Sigma | N/A |
| Maz_screen F: AATAAACCACTGGAATGGGGAGG | Sigma | N/A |
| Maz_screen R: AGTACCTATAGGGAGAAACACCAAGG | Sigma | N/A |
| Sp1_screen F: CTCTGACACTTGTGGAGGGCTC | Sigma | N/A |
| Sp1_screen R: ACCAGCAGAAGGCAGGTAAATC | Sigma | N/A |
| Zbtb7a_screen F: CATCTTCTTGGCTAGTGTCACTGC | Sigma | N/A |
| Zbtb7a_screen R: GTGGCAGTCACCTGTGAATAGAC | Sigma | N/A |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-GFP (PX458) | Addgene | Cat #48138 |
| pMy-BirA-T2A-mOrange | Nakahashi et al. 2013 | N/A |
| pMy-BirA-T2A-eGFP | Nakahashi et al. 2013 | N/A |
| pMy-Biotag-SMAD3-T2A-mOrange | This paper | N/A |
| pMy-Biotag-SMAD7-T2A-mOrange | This paper | N/A |
| pMy-Biotag-ZBTB7A-T2A-eGFP | This paper | N/A |
| pMy-Biotag-KLF13-P2A-eGFP | This paper | N/A |
| pMy-PAX5-Biotag-T2A-mOrange | This paper | N/A |
| pCR-Blunt II-Topo-halotag-Smad3 | This paper | N/A |
| pCR-Blunt II-Topo-halotag-Smad7 | This paper | N/A |
| Software and algorithms | ||
| sgRNA CRISPR Design software | https://crispr.zhaopage.com | N/A |
| bowtie2 | Version 2.3.4.1, http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | N/A |
| SAMtools | Version 1.9, http://www.htslib.org/ | N/A |
| bedtools | Version 2.29, https://github.com/arq5x/bedtools2 | N/A |
| UCSC utilities | http://hgdownload.soe.ucsc.edu/admin/exe/ | N/A |
| MACS2 | https://github.com/macs3-project/MACS | N/A |
| DESeq2 | Version 1.22.2, https://bioconductor.org/packages/release/bioc/html/DESeq2.html | N/A |
| STAR | Version 020201, https://github.com/alexdobin/STAR | N/A |
| RSEM | Version 1.3.0, https://deweylab.github.io/RSEM/ | N/A |
| MEME/FIMO | Version 5.0, https://meme-suite.org/meme/doc/download.html | N/A |
| RGT-Hint | https://www.regulatory-genomics.org/hint/introduction/ | N/A |
| Other | ||
| Publicly Available Data Analyzed | Please refer to Suppl. Table 1H for accession numbers of published data analyzed. | N/A |
Highlights.
“Stripe” TFs colocalize with most expressed factors at promoters and enhancers
Lineage-specific stripe factors regulate the transcriptome by binding most elements
Universal stripe factors (USFs) recognize overlapping GC sequences in all tissues
USFs provide accessibility and increase residence time of co-binding factors
Acknowledgments
This work was funded by the division of intramural research of NIAMS, NCI, NHGRI, and NIAID within the NIH, and supported by NIH Helix Systems (http://helix.nih.gov). M.T.W. was supported by the following grants R01 AR073228, U01 AI130830, R01 AI024717, and CCHMC ARC Award 53632. We thank Remy Bosselut for providing samples and useful comments. All animal related procedures were performed by following NIAMS ACUC protocol. We thank Rose Hurlbut (JAX) for providing the mouse photos used in Figure 5.
Footnotes
Competing interests
S.P. and L.V. are employees of AstraZeneca and may own stock or stock options. All other authors do not have competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barozzi I, Simonatto M, Bonifacio S, Yang L, Rohs R, Ghisletti S, and Natoli G (2014). Coregulation of transcription factor binding and nucleosome occupancy through DNA features of mammalian enhancers. Mol Cell 54, 844–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassuk AG, and Leiden JM (1995). A direct physical association between ETS and AP-1 transcription factors in normal human T cells. Immunity 3, 223–237. [DOI] [PubMed] [Google Scholar]
- Biddie SC, John S, Sabo PJ, Thurman RE, Johnson TA, Schiltz RL, Miranda TB, Sung MH, Trump S, Lightman SL, et al. (2011). Transcription factor AP1 potentiates chromatin accessibility and glucocorticoid receptor binding. Mol Cell 43, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourges C, Groff AF, Burren OS, Gerhardinger C, Mattioli K, Hutchinson A, Hu T, Anand T, Epping MW, Wallace C, et al. (2020). Resolving mechanisms of immune-mediated disease in primary CD4 T cells. EMBO Mol Med 12, e12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, and Groudine M (2010). Enhancers: the abundance and function of regulatory sequences beyond promoters. Dev Biol 339, 250–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano-Gamez E, and Trynka G (2020). From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases. Front Genet 11, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Fulcoli FG, Tang S, and Baldini A (2009). Tbx1 regulates proliferation and differentiation of multipotent heart progenitors. Circ Res 105, 842–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Glover JN, Hogan PG, Rao A, and Harrison SC (1998). Structure of the DNA-binding domains from NFAT, Fos and Jun bound specifically to DNA. Nature 392, 42–48. [DOI] [PubMed] [Google Scholar]
- Chronis C, Fiziev P, Papp B, Butz S, Bonora G, Sabri S, Ernst J, and Plath K (2017). Cooperative Binding of Transcription Factors Orchestrates Reprogramming. Cell 168, 442–459 e420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comings OE, Kovacs BW, Avelino E, and Harris DC (1975). Mechanisms of chromosome banding. V. Quinacrine banding. Chromosoma 50, 111–114. [DOI] [PubMed] [Google Scholar]
- Consortium, E.P., Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, and Snyder M (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplancke B, Alpern D, and Gardeux V (2016). The Genetics of Transcription Factor DNA Binding Variation. Cell 166, 538–554. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farley EK, Olson KM, Zhang W, Brandt AJ, Rokhsar DS, and Levine MS (2015). Suboptimization of developmental enhancers. Science 350, 325–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico A, Steinfass T, Larribere L, Novak D, Moris F, Nunez LE, Umansky V, and Utikal J (2020), Mithramycin A and Mithralog EC-8042 Inhibit SETDB1 Expression and Its Oncogenic Activity in Malignant Melanoma. Mol Ther Oncolytics 18, 83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fornes O, Castro-Mondragon JA, Khan A, van der Lee R, Zhang X, Richmond PA, Modi BP, Correard S, Gheorghe M, Baranasic D, et al. (2020). JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res 48, D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DA, Fettweis G, Presman DM, Paakinaho V, Jarzynski C, Upadhyaya A, and Hager GL (2021a). Power-law behavior of transcription factor dynamics at the single-molecule level implies a continuum affinity model. Nucleic Acids Res 49, 6605–6620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia DA, Johnson TA, Presman DM, Fettweis G, Wagh K, Rinaldi L, Stavreva DA, Paakinaho V, Jensen RAM, Mandrup S, et al. (2021b). An intrinsically disordered region-mediated confinement state contributes to the dynamics and function of transcription factors. Mol Cell 81, 1484–1498 e1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, and Plagnol V (2014). Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giral H, Landmesser U, and Kratzer A (2018). Into the Wild: GWAS Exploration of Non-coding RNAs. Front Cardiovasc Med 5, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giroux M, Delisle JS, O'Brien A, Hebert MJ, and Perreault C (2010). T cell activation leads to protein kinase C theta-dependent inhibition of TGF-beta signaling. J Immunol 185, 1568–1576. [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, and Noble WS (2011). FIMO: scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm JB, English BP, Choi H, Muthusamy AK, Mehl BP, Dong P, Brown TA, Lippincott-Schwartz J, Liu Z, Lionnet T, et al. (2016). Bright photoactivatable fluorophores for single-molecule imaging. Nat Methods 13, 985–988. [DOI] [PubMed] [Google Scholar]
- Gusmao EG, Allhoff M, Zenke M, and Costa IG (2016). Analysis of computational footprinting methods for DNase sequencing experiments. Nat Methods 13, 303–309. [DOI] [PubMed] [Google Scholar]
- Hammelman J, Krismer K, Banerjee B, Gifford DK, and Sherwood RI (2020). Identification of determinants of differential chromatin accessibility through a massively parallel genome-integrated reporter assay. Genome Res 30, 1468–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Komori HK, LaMere S, Podshivalova K, and Salomon DR (2013). Finding the active genes in deep RNA-seq gene expression studies. BMC genomics 14, 778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C, Weidenbach S, Cano KE, Wang Z, Mitra P, Ivanov DN, Rohr J, and Tsodikov OV (2016). Structures of mithramycin analogues bound to DNA and implications for targeting transcription factor FLI1. Nucleic Acids Res 44, 8990–9004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Zhang Z, Kashiwagi M, Yoshida T, Joshi I, Jena N, Somasundaram R, Emmanuel AO, Sigvardsson M, Fitamant J, et al. (2016). Superenhancer reprogramming drives a B-cell-epithelial transition and high-risk leukemia. Genes Dev 30, 1971–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Hatzi K, and Melnick A (2013). Lineage-specific functions of Bcl-6 in immunity and inflammation are mediated by distinct biochemical mechanisms. Nat Immunol 14, 380–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindal GA, and Farley EK (2021). Enhancer grammar in development, evolution, and disease: dependencies and interplay. Dev Cell 56, 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SR, Palfy M, Hilbert L, Kumar M, Karschau J, Zaburdaev V, Shevchenko A, and Vastenhouw NL (2017). Competition between histone and transcription factor binding regulates the onset of transcription in zebrafish embryos. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczynski J, Cook T, and Urrutia R (2003). Sp1- and Kruppel-like transcription factors. Genome Biol 4, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampmann M (2020). CRISPR-based functional genomics for neurological disease. Nat Rev Neurol 16, 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerppola TK, and Curran T (1991). Fos-Jun heterodimers and Jun homodimers bend DNA in opposite orientations: implications for transcription factor cooperativity. Cell 66, 317–326. [DOI] [PubMed] [Google Scholar]
- Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Cheneby J, Kulkarni SR, Tan G, et al. (2018). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer-Kwon KR, Nimura K, Rao SSP, Xu J, Jung S, Pekowska A, Dose M, Stevens E, Mathe E, Dong P, et al. (2017). Myc Regulates Chromatin Decompaction and Nuclear Architecture during B Cell Activation. Mol Cell 67, 566–578 e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, An H, Shearer RS, Sharif M, Fan C, Choi JO, Ryu S, and Park Y (2019). A principled strategy for mapping enhancers to genes. Sci Rep 9, 11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzine F, Wojtowicz D, Yamane A, Resch W, Kieffer-Kwon KR, Bandle R, Nelson S, Nakahashi H, Awasthi P, Feigenbaum L, et al. (2013). Global regulation of promoter melting in naive lymphocytes. Cell 153, 988–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kregel S, Kiriluk KJ, Rosen AM, Cai Y, Reyes EE, Otto KB, Tom W, Paner GP, Szmulewitz RZ, and Vander Griend DJ (2013). Sox2 is an androgen receptor-repressed gene that promotes castration-resistant prostate cancer. PLoS One 8, e53701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson AJM, Johnsson P, Hagemann-Jensen M, Hartmanis L, Faridani OR, Reinius B, Segerstolpe A, Rivera CM, Ren B, and Sandberg R (2019). Genomic encoding of transcriptional burst kinetics. Nature 565, 251–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ran C, Li E, Gordon F, Comstock G, Siddiqui H, Cleghorn W, Chen HZ, Kornacker K, Liu CG, et al. (2008). Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev Cell 14, 62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Lacroix L, Gamot A, Cuddapah S, Queille S, Lhoumaud P, Lepetit P, Martin PG, Vogelmann J, Court F, et al. (2014). Chromatin immunoprecipitation indirect peaks highlight long-range interactions of insulator proteins and Pol II pausing. Mol Cell 53, 672–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link VM, Duttke SH, Chun HB, Holtman IR, Westin E, Hoeksema MA, Abe Y, Skola D, Romanoski CE, Tao J, et al. (2018). Analysis of Genetically Diverse Macrophages Reveals Local and Domain-wide Mechanisms that Control Transcription Factor Binding and Function. Cell 173, 1796–1809 e1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, et al. (2008). HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol 3, 373–382. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luwor RB, Baradaran B, Taylor LE, Iaria J, Nheu TV, Amiry N, Hovens CM, Wang B, Kaye AH, and Zhu HJ (2013). Targeting Stat3 and Smad7 to restore TGF-beta cytostatic regulation of tumor cells in vitro and in vivo. Oncogene 32, 2433–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F, Lopez-Rodriguez C, and Rao A (2001). Partners in transcription: NFAT and AP-1. Oncogene 20, 2476–2489. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Falvo JV, Kim TH, Kim TK, Lin CH, Parekh BS, and Wathelet MG (1998). Structure and function of the interferon-beta enhanceosome. Cold Spring Harb Symp Quant Biol 63, 609–620. [DOI] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. (2012). Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirny LA (2010). Nucleosome-mediated cooperativity between transcription factors. Proc Natl Acad Sci U S A 107, 22534–22539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgunova E, and Taipale J (2017), Structural perspective of cooperative transcription factor binding. Curr Opin Struct Biol 47, 1–8. [DOI] [PubMed] [Google Scholar]
- Murphy TL, Tussiwand R, and Murphy KM (2013). Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol 13, 499–509. [DOI] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. (2012). An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez LE, Nybo SE, Gonzalez-Sabin J, Perez M, Menendez N, Brana AF, Shaaban KA, He M, Moris F, Salas JA, et al. (2012). A novel mithramycin analogue with high antitumor activity and less toxicity generated by combinatorial biosynthesis. J Med Chem 55, 5813–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Fairfax KA, and Kallies A (2007). BLIMP1 guides the fate of effector B and T cells. Nat Rev Immunol 7, 923–927. [DOI] [PubMed] [Google Scholar]
- Oeckinghaus A, and Ghosh S (2009). The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1, a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paakinaho V, Presman DM, Ball DA, Johnson TA, Schiltz RL, Levitt P, Mazza D, Morisaki T, Karpova TS, and Hager GL (2017). Single-molecule analysis of steroid receptor and cofactor action in living cells. Nat Commun 8, 15896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panne D, Maniatis T, and Harrison SC (2007). An atomic model of the interferon-beta enhanceosome. Cell 129, 1111–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor WA, Liu W, Chen D, Ho J, Kim R, Hunt TJ, Lukianchikov A, Liu X, Polo JM, Jacobsen SE, et al. (2018). TFAP2C regulates transcription in human naive pluripotency by opening enhancers. Nat Cell Biol 20, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach KJ, and Widom J (1996). A model for the cooperative binding of eukaryotic regulatory proteins to nucleosomal target sites. J Mol Biol 258, 800–812. [DOI] [PubMed] [Google Scholar]
- Presman DM, Ball DA, Paakinaho V, Grimm JB, Lavis LD, Karpova TS, and Hager GL (2017). Quantifying transcription factor binding dynamics at the single-molecule level in live cells. Methods 123, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Sheetz MP, and Elson EL (1991). Single particle tracking. Analysis of diffusion and flow in two-dimensional systems. Biophysical journal 60, 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Ritchie RP, Fu Z, Cao D, Cumming J, Miano JM, Wang DZ, Li HJ, and Li L (2005). Myocardin enhances Smad3-mediated transforming growth factor-beta1 signaling in a CArG box-independent manner: Smad-binding element is an important cis element for SM22alpha transcription in vivo. Circ Res 97, 983–991. [DOI] [PubMed] [Google Scholar]
- Reiter F, Wienerroither S, and Stark A (2017). Combinatorial function of transcription factors and cofactors. Curr Opin Genet Dev 43, 73–81. [DOI] [PubMed] [Google Scholar]
- Rickels R, and Shilatifard A (2018). Enhancer Logic and Mechanics in Development and Disease. Trends Cell Biol 28, 608–630. [DOI] [PubMed] [Google Scholar]
- Rowan S, Siggers T, Lachke SA, Yue Y, Bulyk ML, and Maas RL (2010). Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev 24, 980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer T, and Lengerke C (2020). SOX2 protein biochemistry in stemness, reprogramming, and cancer: the PI3K/AKT/SOX2 axis and beyond. Oncogene 39, 278–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, and Karin M (2002). AP-1 as a regulator of cell life and death. Nat Cell Biol 4, E131–136. [DOI] [PubMed] [Google Scholar]
- Spitz F, and Furlong EE (2012). Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13, 613–626. [DOI] [PubMed] [Google Scholar]
- Stavreva DA, Garcia DA, Fettweis G, Gudla PR, Zaki GF, Soni V, McGowan A, Williams G, Huynh A, Palangat M, et al. (2019). Transcriptional Bursting and Co-bursting Regulation by Steroid Hormone Release Pattern and Transcription Factor Mobility. Mol Cell 75, 1161–1177 e1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- UniProt, C. (2021), UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierstra J, Lazar J, Sandstrom R, Halow J, Lee K, Bates D, Diegel M, Dunn D, Neri F, Haugen E, et al. (2020). Global reference mapping of human transcription factor footprints. Nature 583, 729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Rubin EM, and Pennacchio LA (2009). Genomic views of distant-acting enhancers. Nature 461, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino C, Nunez LE, Moris F, and Portugal J (2014). Genome-wide modulation of gene transcription in ovarian carcinoma cells by a new mithramycin analogue. PLoS One 9, e104687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, Schiltz RL, Sung MH, Yen PM, Stamatoyannopoulos JA, Biddie SC, Johnson TA, Miranda TB, John S, and Hager GL (2011). Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 146, 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingender E (2008). The TRANSFAC project as an example of framework technology that supports the analysis of genomic regulation. Brief Bioinform 9, 326–332. [DOI] [PubMed] [Google Scholar]
- Yao L, Wang S, Westholm JO, Dai Q, Matsuda R, Hosono C, Bray S, Lai EC, and Samakovlis C (2017). Genome-wide identification of Grainy head targets in Drosophila reveals regulatory interactions with the POU domain transcription factor Vvl. Development 144, 3145–3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandvakili A, Campbell I, Gutzwiller LM, Weirauch MT, and Gebelein B (2018). Degenerate Pax2 and Senseless binding motifs improve detection of low-affinity sites required for enhancer specificity. PLoS Genet 14, e1007289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J (2020). Seven myths of how transcription factors read the cis-regulatory code. Curr Opin Syst Biol 23, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Jia X, Yang J, Ling Y, Zhang Z, Yu J, Wu J, and Xiao J (2014). PanGP: a tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 30, 1297–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, Wei B, Dodonova SO, Nitta KR, Morgunova E, Taipale M, et al. (2018). The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Bioinformatic analysis and sample details. (A) Related to Figure 1: ATAC-Seq and DNase-Seq samples processed. (B) Related to Figure 1: list of mouse samples analyzed by ATAC-Seq or DNase-Seq and RNA-Seq. (C), related to Figure 1: list of human samples analyzed by ATAC-Seq or DNase-Seq and RNA-Seq. (D) Related to Figure 2: "Excluded" TF motifs from mouse activated B cells (LPS+IL-4). (E) Related to Figure 2: Mouse and human stripe factors. (F) Related to Figure 3: Stripe factors identified in HEK293, MCF7, GM12878, and K562 cell lines based on ChIP-Seq assays. (G) Related to Figure 3: Universal Stripe Factors in various human and mouse tissues. (H) Related to STAR Methods: Accession numbers for publicly available data analyzed.
Data Availability Statement
RNA-seq, ChIP-seq, ATAC-seq, whole genome sequencing and other deep-sequencing data reported in this paper have been deposited at GEO and are publicly available as of the date of publication. Accession numbers can be found at GSE164906. This paper also analyzes existing, publicly available data. These accession numbers for the datasets are listed in the supplementary table 1H.
Original code to determine TF colocalization has been archived at Zenodo (DOI: 10.5281/zenodo.6642964) and is publicly available as of the date of publication.
Any additional information required to reanalyze the data reported in this paper is available from Yongbing Zhao upon request (im@ybzhao.com).