

Abstract
Aims/hypothesis
Induction of intercellular adhesion molecule-1 (ICAM-1) has been implicated in the development of macrovascular and microvascular diseases such as diabetic retinopathy. Lesions of diabetic retinopathy are unique to the retina but the reason for this is unclear, as all tissues are exposed to the same hyperglycaemic insult. We tested whether diabetes induces ICAM-1 on the luminal surface of endothelial cells to a greater extent in the retina than in other tissues and the role of vision itself in that induction.
Methods
Experimental diabetes was induced in C57Bl/6J, P23H opsin mutant and Gnat1−/− × Gnat2−/− double knockout mice using streptozotocin. The relative abundance of ICAM-1 on the luminal surface of endothelial cells in retina and other tissues was determined by conjugating anti-ICAM-1 antibodies to fluorescent microspheres (2μm), injecting them intravenously and allowing them to circulate for 30 min. After transcardial perfusion, quantification of microspheres adherent to the endothelium in tissues throughout the body was carried out by fluorescent microscopy or flow cytometry. Mice injected with lipopolysaccharide (LPS) were used as positive controls. The difference in leucostasis between retinal and non-retinal vasculature was evaluated.
Results
Diabetes significantly increased ICAM-1-mediated adherence of microspheres to retinal microvessels by almost threefold, independent of sex. In contrast, diabetes had a much smaller effect on endothelial ICAM-1 in other tissues, and more tissues showed a significant induction of endothelial ICAM-1 with LPS than with diabetes. The diabetes-induced increase in endothelial ICAM-1 in retinal vasculature was inhibited by blocking phototransduction in photoreceptor cells. Diabetes significantly increased leucostasis in the retina by threefold compared with a non-ocular tissue (cremaster).
Conclusions/interpretation
The diabetes-induced upregulation of ICAM-1 on the luminal surface of the vascular endothelium varies considerably among tissues and is highest in the retina. Induction of ICAM-1 on retinal vascular endothelial cells in diabetes is influenced by vision-related processes in photoreceptor cells. The unique presence of photoreceptors in the retina might contribute to the greater susceptibility of this tissue to vascular disease in diabetes.
Keywords: Adhesion molecule, Diabetes, Diabetic retinopathy, ICAM-1, Leucostasis, Photoreceptors, Phototransduction, Vascular inflammation
Introduction
Leucocytes have important effects in inflammatory processes and adherence of the leucocytes to the vessel wall (leucostasis) has been implicated in the occlusion of capillaries and release of superoxide and cytokines in a variety of diseases [1–7]. Adhesion of circulating leucocytes to the vascular endothelium is mediated by integrins such as intercellular adhesion molecule-1 (ICAM-1) on the luminal side of the endothelial cells [8–10].
Evidence for a role of leucocytes and inflammation in the pathogenesis of diabetes-induced vascular disease is strong for the retina; a variety of changes characteristic of inflammation (including increased vascular permeability and supranormal levels of ICAM-1, inducible nitric oxide synthase [iNOS], NF-κB, prostaglandin E2 [PGE2] and IL-1β) have been detected within the retinas of diabetic animals and patients [8, 11–17]. The diabetes-induced degeneration of retinal capillaries (a critical step in the progression to clinically significant diabetic retinopathy) has been shown to be initiated by leucocytes, because chimeric mice lacking single inflammatory proteins only from their myeloid cells are protected from the expected retinal capillary degeneration in diabetes [5, 11, 18]. Moreover, deleting ICAM-1 [12] or blocking the interaction of leucocytes with endothelial ICAM-1 [11, 19] has been found to inhibit the development of capillary degeneration in diabetic retinopathy.
As circulating leucocytes show evidence of being activated in diabetes [20–23], it would seem reasonable that the vasculature in all tissues should be equally affected by those leucocytes. On the contrary, however, not all tissues seem equally susceptible to diabetes-induced vascular disease [24, 25]. In this study, we assessed the effect of diabetes on levels of ICAM-1 on the luminal surface of the vascular endothelium (where leucocytes flowing in blood can interact) in tissues throughout the body, with a particular focus on the role of retinal photoreceptor cells in the diabetes-induced increase in ICAM-1 on retinal microvascular endothelial cells.
Methods
Ethics statement
This study was performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research, and with authorisation of the Institutional Animal Care and Use Committee at the University of California Irvine.
Experimental animals
Male and female C57Bl/6J wild-type (abbreviated as WT) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA; https://www.jax.org/strain/000664), Rhotm1.1Kpal (abbreviated as P23H) and Gnat2cpfl3; Gnat1irdr (abbreviated as Gnat1−/− × Gnat2−/−) mice were obtained from K. Palczewski at the Center for Translational Vision Research at the University of California Irvine and are also available from the Jackson Laboratory (P23H mice, https://www.jax.org/strain/017628; Gnat1−/− × Gnat2−/− mice, https://www.jax.org/strain/033163). Experimental diabetes was induced at 8-10 weeks of chronological age (male mice) or at 10-12 weeks of chronological age (female mice) by serial intraperitoneal injection of a freshly prepared solution of streptozotocin in citrate buffer (pH 4.5) at 55mg/kg body weight (male mice) or 75mg/kg body weight (female mice) once daily for 5 consecutive days [26]. Hyperglycaemia (>15.26mmol/l) was verified at least three times during the second week after streptozotocin injection and 3-4 days before the autopsy day [27]. Insulin was given as needed to achieve slow weight gain without preventing hyperglycaemia and glucosuria. Food consumption and body weight were measured weekly. For some experiments, WT non-diabetic animals were injected with lipopolysaccharide (LPS; 1mg/kg body weight; Sigma Chemical, St Louis, MO, USA) via the tail vein and euthanised 2h after. LPS administration results in well-characterised systemic inflammation and was used as a positive control [28]. Animals were studied 8 weeks after randomised assignment into experimental groups or 2h after injection of LPS.
Housing
Animals were housed in a pathogen-free facility with a standard 12h on and 12h off lighting cycle. For light-deprivation (dark-adaptation) experiments, non-diabetic and diabetic animals were placed in a room with a 48h lights off cycle (0 lux) before the autopsy day.
Generation of antibody-coated microspheres
Red fluorescent polystyrene microspheres (cat. no. R0200, Duke Scientific, Fremont, CA, USA) were coated with protein A from Staphylococcus aureus (cat. no. P3838, Sigma Aldrich, St Louis, MO, USA) by passive absorption as described previously [29, 30]. To achieve this, 1ml of the purchased solution, which contained the equivalent of 1% solids (microspheres), was incubated in a 0.1mol/l NaHCO3 buffer (pH 9.2) containing 300μg/ml of protein A at 4°C overnight in the dark. The following day, the microspheres were washed and incubated in a blocking buffer (0.1mol/l NaHCO3 [pH 9.2] supplemented with 1% bovine serum albumin) in a rotator (360° rotation at 8 rev/min) at room temperature for 1h covered from light. During the incubation time, the microspheres were quantified by flow cytometry. After incubation, the microspheres were washed and incubated with a rabbit anti-mouse polyclonal antibody to ICAM-1 (cat. no. 10020-1-AP, Proteintech, Rosemont, IL, USA) at a final concentration of 1×108 microspheres/mg rabbit polyclonal antibody. As a control, matching microspheres were coated with isotype control IgG under identical conditions. The amount of antibody in the coating solution was greater than that needed to saturate the microspheres with the IgG. After addition of the polyclonal antibody or IgG, the microspheres were incubated in a rotator for at least 2 days at 4°C in the dark and stored for a maximum of 7 days at 4°C in the dark. Before use, the microspheres were washed twice to remove unbound polyclonal antibody or IgG and shaken vigorously on a table shaker to keep them in monodispersion immediately before injection. All washes were carried out in a table centrifuge at 17,000g for 30 min.
Estimation of ICAM-1 on the luminal surface of vascular endothelium in diabetes or after injection of LPS
To estimate the relative levels of ICAM-1 on the luminal surface of vascular endothelium in mice, fluorescent microspheres coated with anti-ICAM-1 or non-immune IgG were injected through the tail vein. Initial studies assessed the effect of the size (0.5, 1.0 and 2.0μm diameter) and the number (0.3×107, 1×107, 3×107 and 10×107) of microspheres injected on the results observed. We injected different sizes and amounts of microspheres into untreated control and LPS-treated male C57Bl/6J mice. Microspheres were allowed to circulate for 30 or 60 min and animals were then perfused transcardially with 5ml of 0.9% sodium chloride solution at 20ml/min to remove unbound microspheres. In all cases, the number of microspheres in a particular tissue of LPS-treated animals was compared with the number of microspheres in the same tissue from age-matched normal control animals. We concluded that 1×107 microspheres per mouse was an optimal amount to facilitate fluorescent microscopy quantification.
Mouse organs were removed and thin tissues (retina, aorta, diaphragm and cremaster) were prepared for analysis as flat mounts. The entire retina was laid out on a glass microscope slide, whereas the aorta, diaphragm and cremaster were trimmed to the same area (3×3mm2) and then laid out on glass slides. The number of adherent microspheres was determined manually by fluorescence microscopy (Nikon Eclipse Ni wide-field fluorescence microscope [Nikon, Melville, NY, USA] using a red filter – 543 nm excitation, 612 nm emission) for these tissues. The person assigned to perform the microscopy studies was masked to the origin of the samples. Liver, spleen, kidney, lung, heart, grey matter and cerebellum were trimmed to 100mg wet weight, homogenised in 500μl cell lysis buffer (cat. no. 9803S, Cell Signaling Technology, Danvers, MA, USA) and sonicated, and then a constant aliquot of 10μl was used to perform semi-quantification by flow cytometry. The person assigned to perform the flow cytometry studies did not need to be masked to the origin of the samples, as the values reported were generated by the equipment used. Because the amount of vasculature varies considerably between tissues, we quantified the diabetes- or LPS-induced increased adhesion of microspheres in each tissue as a ratio of the number of adherent microspheres in tissue from diabetic or LPS-treated mice to the number of adherent microspheres in the same tissue from age-matched non-diabetic or non-LPS treated animals of the same genetic background.
Flow cytometry
Tissues were homogenised in 500μl of cell lysis buffer (Cell Signaling Technology). Microspheres retained in each tissue after perfusion (adherent microspheres) were quantified using an Agilent NovoCyte Quanteon Flow Cytometer (Agilent, Santa Clara, CA, USA). For each tissue, 10μl of homogenised tissue was analysed using the red laser (561nm excitation, 615 nm emission). Each sample was subjected to two independent measurements. The data were processed and analysed using NovoExpress software (Agilent). Red fluorescent polystyrene microspheres of 0.5, 1.0 and 2.0μm diameter were run alone for gating.
Quantitative measurement of leucostasis
The number of leucocytes adherent to the microvasculature was determined at 8 weeks of diabetes. After cardiac catheterisation, anesthetised mice (100 mg/ml ketamine to 100 mg/ml xylazine ratio of 5:1) were exsanguinated by perfusion with 0.9% sodium chloride (Valley Vet Supply, Marysville, KS, USA) for 2min. Fluorescein-coupled concanavalin A lectin (cat. no. FL-1001-25, Vector Laboratories, Burlingame, CA, USA) at 20 μg/ml in 0.9% sodium chloride was then infused as described previously [27]. Flat-mounted retinas and 3×3mm of cremaster muscle were visualised with fluorescence microscopy, and brightly fluorescent leucocytes were counted in the entire retina and the cremaster muscle sample.
Statistical analysis
Graphs were generated using Prism 9 (GraphPad Software, San Diego, CA, USA). All mean non-diabetic values for a given tissue are expressed as 100% and the mean individual values for all diabetic animals are reported as the fold increase above the non-diabetic means. Statistical analyses were performed using unpaired Student’s t tests with Welch correction and ordinary one-way ANOVA with uncorrected Fisher’s tests as indicated. Experimental group means were considered significantly different from control group means if the p values were ≤0.05.
Results
Animals
Clinical data on non-diabetic and diabetic mouse models are provided in Table 1. The severity of diabetes was similar between WT, P23H and Gnat1−/− × Gnat2−/− diabetic mice independent of sex.
Table 1.
Clinical data for non-diabetic (N) and diabetic (D) mice with or without dark adaptation
| Genotype | Sex | Group | Body weight (g) | Non-fasted blood glucose (mmol/l) | 48h dark adaptation | LPS challenge (1mg/kg body weight) | n |
|---|---|---|---|---|---|---|---|
| WT | M | N | 30 ± 6 | 8.6 ± 2.22 | − | − | 25 |
| WT | M | N | 30 ± 7 | 8.1 ± 1.72 | − | + | 12 |
| WT | M | D | 27 ± 2 | 26.5 ± 3.9*** | − | − | 25 |
| WT | M | N | 39 ± 3 | 8.4 ± 0.94 | + | − | 10 |
| WT | M | N | 38 ± 2 | 8.5 ± 1.22 | + | + | 10 |
| WT | M | D | 27 ± 1** | 23.3 ± 2.22*** | + | − | 10 |
| P23H | M | N | 39 ± 8 | 8.0 ± 0.89 | − | − | 8 |
| P23H | M | D | 27 ± 1** | 29.0 ± 3.94*** | − | − | 8 |
| Gnat1−/− | M | N | 41 ± 8 | 8.6 ± 0.5 | − | − | 3 |
| Gnat1−/− | M | D | 29 ± 4** | 28.5 ± 2.9*** | − | − | 3 |
| Gnat1−/− × Gnat2−/− | M | N | 30 ± 2 | 8.9 ± 0.67 | − | − | 6 |
| Gnat1−/− × Gnat2−/− | M | D | 26 ± 2 | 23.2 ± 0.89*** | − | − | 6 |
| WT | F | N | 24 ± 1 | 6.6 ± 0.56 | − | − | 8 |
| WT | F | D | 21 ± 2** | 19.8 ± 2.39*** | − | − | 8 |
| WT | F | N | 30 ± 5 | 7.2 ± 1.67 | + | − | 3 |
| WT | F | N | 26 ± 1 | 7.9 ± 0.44 | + | + | 3 |
| WT | F | D | 21 ± 2** | 28.1 ± 4.33*** | + | − | 3 |
| Gnat1−/− × Gnat2−/− | F | N | 22 ± 1 | 8.7 ± 0.94 | − | − | 5 |
| Gnat1−/− × Gnat2−/− | F | D | 21 ± 0.8** | 22.5 ± 9.6*** | − | − | 5 |
Microsphere quantification and validation
We performed initial studies using microspheres with different diameters (0.5, 1.0 and 2.0μm) to determine if the fluorescent microspheres that were small enough not to occlude microvessels were easy to visualise. We concluded that microspheres with a 2.0μm diameter were suitable for our purposes. To evaluate the specificity of the anti-ICAM-1-coated microspheres, we used IgG-coated microspheres prepared under the same conditions as anti-ICAM-1-coated microspheres and found that microspheres coated with the isotype control did not attach to the vascular endothelium (data not shown).
Next, we established a flow cytometry protocol to quantify adherent microspheres retained in the tissues studied, as described previously [31]. Fluorescent microspheres of different diameters (0.5, 1.0 and 2.0μm) were run alone for gating purposes (Fig. 1a). Using forward and side light scatter and fluorescent characteristics, microspheres were easily distinguished from cells and microparticles (Fig. 1b,c). The number of microspheres analysed was obtained from the corresponding microspheres’ histogram gate for the fluorescent channel (PE-Texas Red-A) and detected at an emission of 615nm when excited at 561nm (Fig. 1d).
Fig. 1.
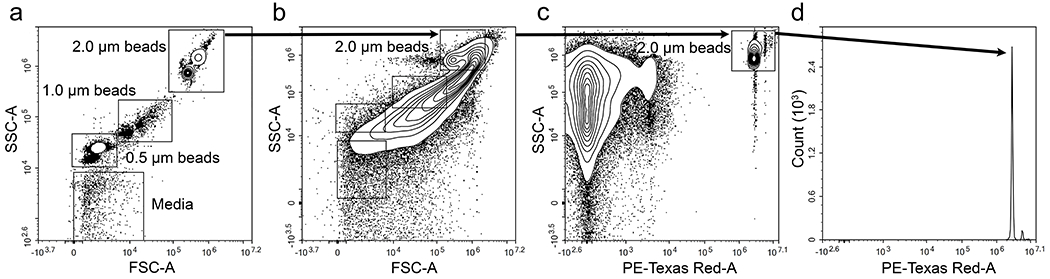
Quantification of fluorescent microspheres by flow cytometry. Gating was established using microspheres of different sizes for comparison (a). Microspheres were distinguished from cells and microparticles in forward scatter vs side scatter density plots (b) and side scatter vs fluorescence density plots (c). The number of microspheres analysed was obtained from the corresponding microspheres’ histogram gate in (c) as a reason of the fluorescent channel in (d)
After establishing a method to quantify microspheres objectively, we wanted to ensure that the number of microspheres adherent to the endothelium was not determined merely by the number of microspheres injected. We found that increasing the number of microspheres injected (0.3×107, 1×107, 3×107 and 10×107) had no effect on the number of microspheres binding to the vasculature in most tissues (Fig. 2a, b). We therefore injected 1×107 microspheres in 200μl of 1% bovine serum albumin into each mouse via the tail vein. We also compared the effect of letting the microspheres circulate in the vasculature for 30 or 60 min and found no difference in the number of microspheres that adhered to the vasculature (data not shown). Thus, a circulation duration of 30 min was used for all subsequent studies. LPS administration results in well-characterised systemic inflammation [28], so the effect of LPS on endothelial levels of ICAM-1 was also assessed in multiple tissues from WT non-diabetic mice, both as a positive control for the study and for comparison with the effects of diabetes. We observed that the number of microspheres adherent to the luminal wall of the vascular endothelium in LPS-injected non-diabetic mice was significantly greater than normal in the retina, aorta, diaphragm, spleen and lung (Fig. 2c), consistent with LPS-mediated induction of ICAM-1 on the luminal surface of vascular endothelial cells.
Fig. 2.
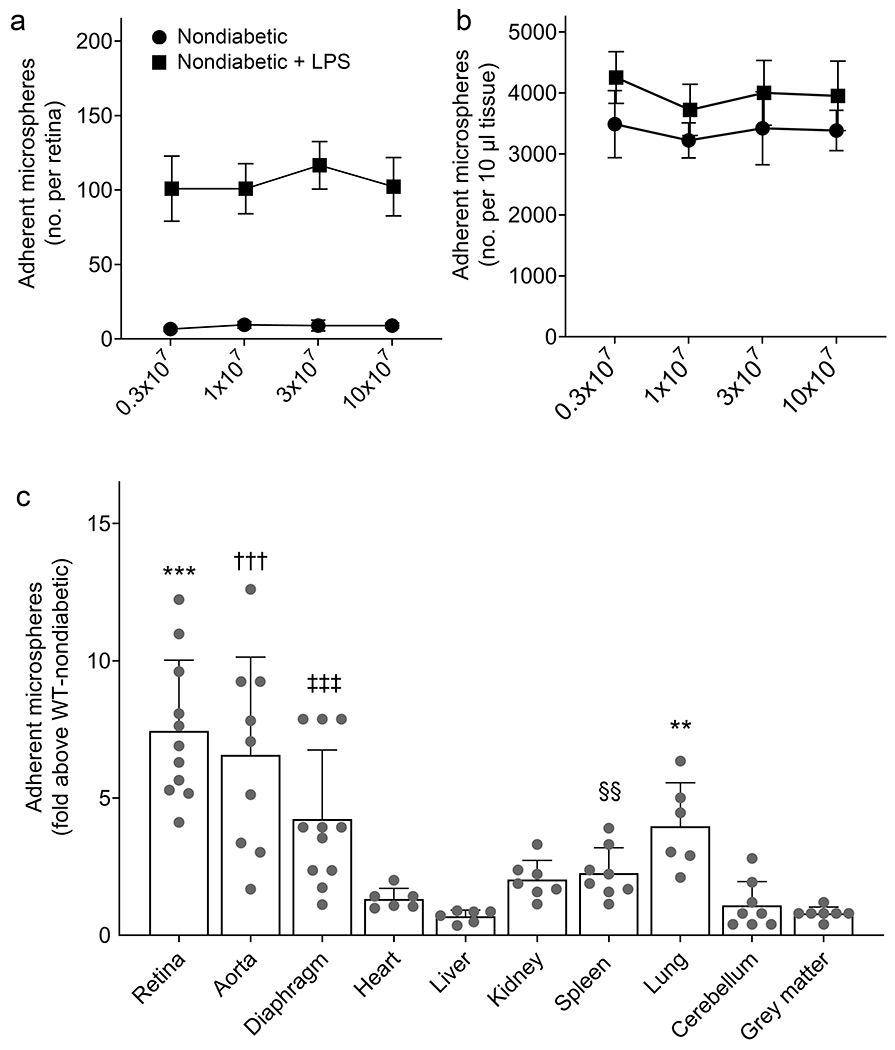
Microsphere concentration and validation. Increasing the number of injected microspheres had no effect on the number of microspheres binding to the vascular endothelium, even after LPS challenge, as shown in the retina (a) and liver (b). (c) Data represent the LPS-induced fold increase in number of adherent microspheres above that in non-diabetic animals in the vasculature of various tissues. Non-diabetic animals showed an overall increment in adherent microspheres with LPS challenge, with a statistically significant difference in the retina, aorta, diaphragm, spleen and lung. Statistical analysis was performed using an unpaired t test with Welch correction for each tissue analysed. Data are mean ± SD; n=6–11 mice. p≤0.05 was considered significant. **,§§p≤0.01 and ***,†††,‡‡‡p≤0.001 compared with WT non-diabetic mice
Finally, to determine whether some microspheres remained free (not bound to endothelial cells) in the blood after 30 min circulation (and thus might confound our assumption that microspheres found in the tissues had been retained by binding to endothelial ICAM-1), we compared the number of microspheres retained in several tissues (retina, aorta, lung, kidney and skeletal muscle) before and after perfusion (5ml of 0.9% sodium chloride solution at 20ml/min) to remove blood. These studies showed that the number of anti-ICAM-1-coated microspheres retained within tissues was significantly lower in perfused tissues than in non-perfused tissues (data not shown). Thus, all subsequent studies were performed after transcardial perfusion.
Diabetes-induced upregulation of ICAM-1 on the endothelial surface varies among tissues
Using the anti-ICAM-1-coated microsphere method, we sought to assess the effect of diabetes on the levels of ICAM-1 on the luminal surface of the vascular endothelium in multiple tissues of mice. We found that the effect of diabetes on retention of microspheres varied considerably among tissues when compared with non-diabetic controls, indicating that diabetes did not uniformly induce endothelial ICAM-1 in all tissues. Of the tissues studied after diabetes of 8 weeks’ duration, only the retina showed a statistically significant induction of ICAM-1 on the vascular surface when compared with non-diabetic controls (Fig. 3); diabetes had a lesser (and not statistically significant) effect in the other tissues studied. Results were similar in male and female mice and so the results are combined in Fig. 3.
Fig. 3.
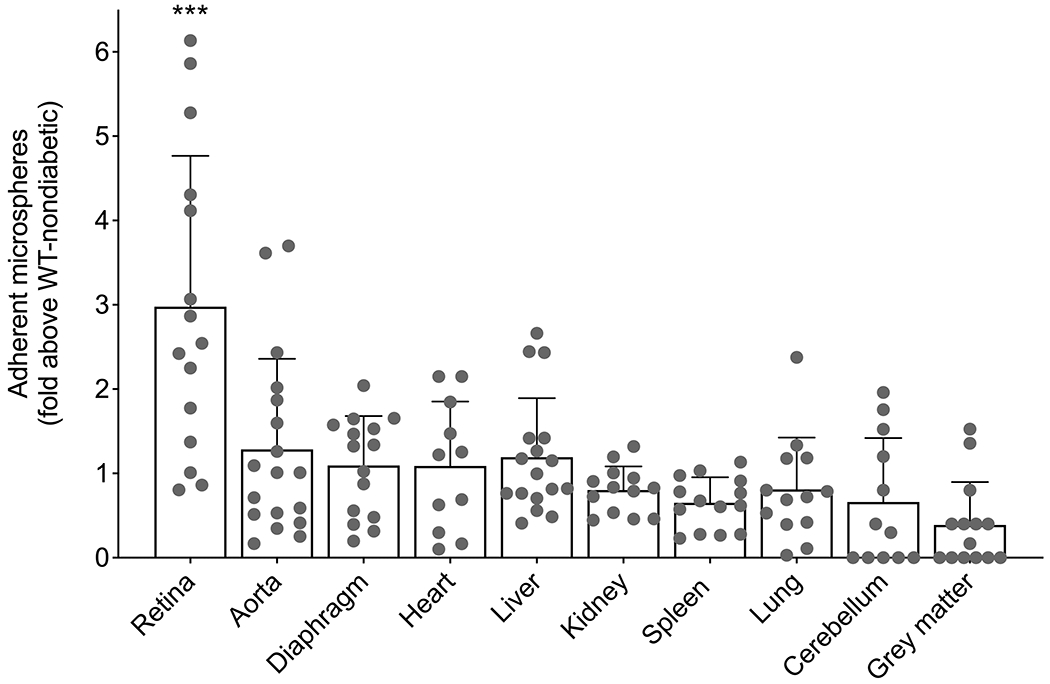
Effect of diabetes on the levels of ICAM-1 on the luminal surface of vascular endothelium throughout the body. Data represent the diabetes-induced fold increase in number of anti-ICAM-1-coated microspheres above that in the same tissue of non-diabetic animals. After 2 months of diabetes, only retina showed a statistically significant induction of ICAM-1 on the vascular surface in male and female mice. Statistical analysis was performed using an unpaired t test with Welch correction for each tissue analysed. Data are mean ± SD; n=11–18 mice. p≤0.05 was considered significant. ***p ≤0.001 compared with WT non-diabetic mice
Vision and diabetes-induced upregulation of ICAM-1 in retinal vascular endothelium
The retina contains unique cells (photoreceptors) that capture light to initiate vision through the process of phototransduction. Gnat1 and Gnat2 code for the translation of transducin, a protein critical for phototransduction in rods and cones, respectively. Previous studies have demonstrated that photoreceptors (and vision) may contribute to the diabetes-induced increase in ICAM-1 levels on retinal microvessels and so we next interrogated this hypothesis.
First, we wanted to determine if the unique metabolic processes taking place in retinal photoreceptor cells (phototransduction) are involved in the increased levels of ICAM-1 in the vasculature of diabetic animals. Compared with the number of anti-ICAM-1-coated microspheres adherent to the retinal vascular endothelium of WT mice (male and female) after diabetes of 2 months’ duration, adhesion of microspheres to the retinal endothelium in diabetic mice lacking phototransduction (Gnat1−/− × Gnat2−/− double knockout mice) was significantly lower and not different from that in non-diabetic WT mice (Fig. 4). Similarly, dark adaptation (48h) inhibited the diabetes-induced upregulation of ICAM-1 on the luminal surface of the retinal endothelium in WT diabetic male and female mice (Fig. 4). Thus, light-induced phototransduction in photoreceptor cells contributes to the activation of signalling that leads to the induction of ICAM-1 on the luminal surface of retinal endothelial cells.
Fig. 4.
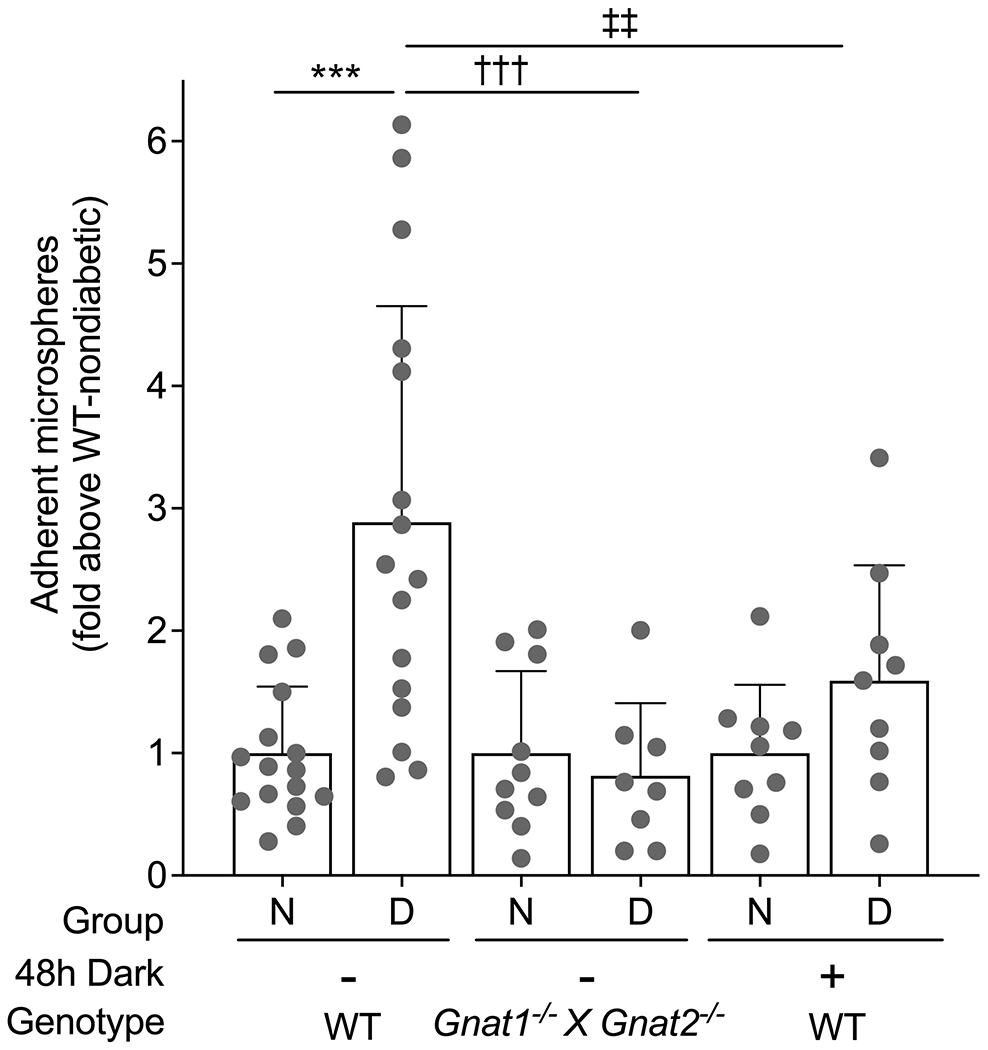
Phototransduction inhibition in rods and cones and induction of ICAM-1 in the retinal vascular endothelium. Genetic inhibition of phototransduction (Gnat1−/− × Gnat2−/− double knockout mice) or 48h of dark adaptation of WT animals inhibited the diabetes-induced upregulation of ICAM-1 on the luminal surface of the retinal endothelium in male and female mice. Statistical analysis was performed using one-way ANOVA with an uncorrected Fisher’s test. Data are mean ± SD; n=8–16 mice. p≤0.05 is considered significant. ‡‡p ≤ 0.01 and ***,†††p ≤ 0.001 as shown. D, diabetic; N, non-diabetic
Of the two types of retinal photoreceptor cells, available evidence suggests that rod photoreceptor cells play a particularly important role in the development of the vascular lesions of diabetic retinopathy. We next induced diabetes in a small group of Gnat1−/− mice (in which phototransduction was blocked only in rods) and generated preliminary evidence that inhibition of phototransduction only in rods was sufficient to inhibit the induction of ICAM-1 on the luminal surface of retinal endothelial cells in diabetes (data not shown). P23H mice, which express a mutant opsin that leads to impaired phototransduction and visual function [32], were similarly protected from the diabetes-induced increase in ICAM-1 on retinal endothelial cells (Fig. 5).
Fig. 5.
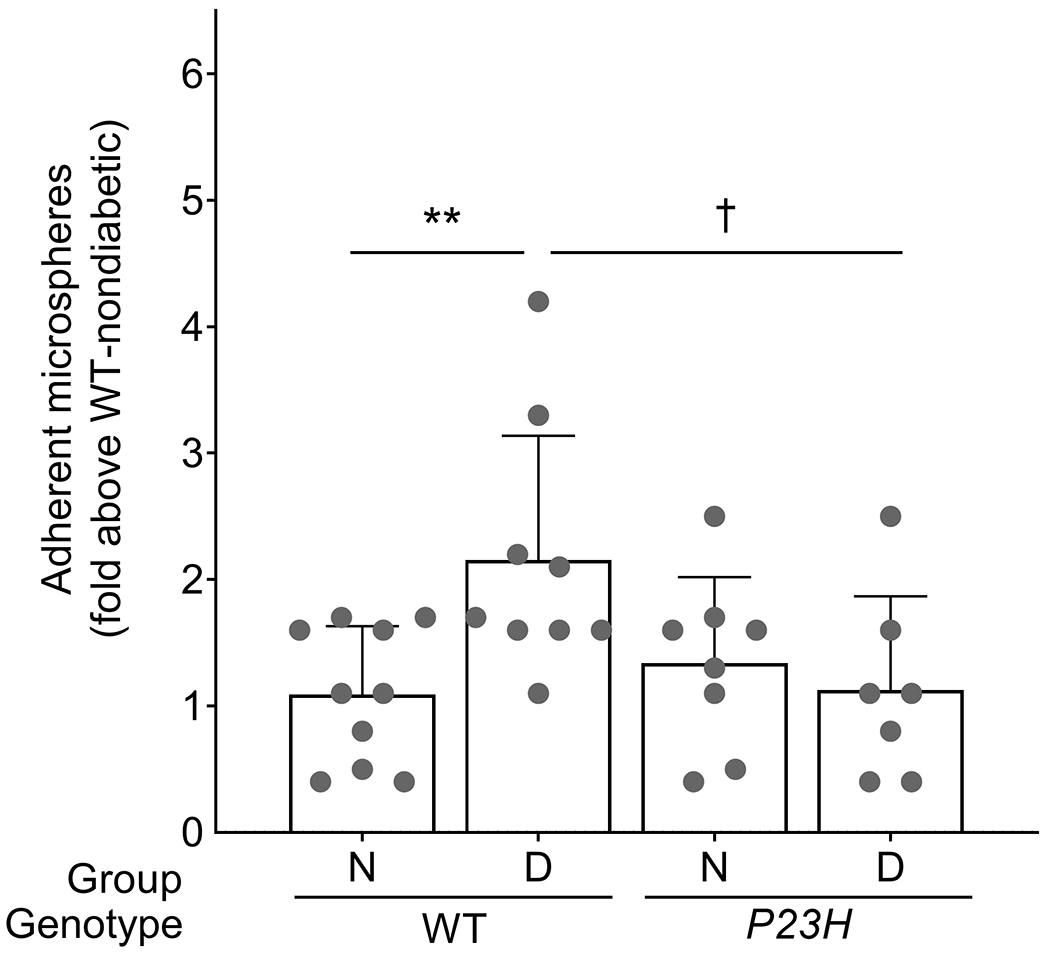
Induction of ICAM-1 in the retinal vascular endothelium of P23H opsin mutant mice. The expression of a mutant opsin (P23H) is sufficient to inhibit the diabetes-induced upregulation of ICAM-1 on the luminal surface of the retinal endothelium in male mice. Statistical analysis was performed using one-way ANOVA with an uncorrected Fisher’s test. Data are mean ± SD; n=7–10. p≤0.05 is considered significant. †p≤0.05, **p ≤ 0.01 as shown. D, diabetic; N, non-diabetic
Induction of ICAM-1 and leucostasis in diabetes
To investigate if the levels of ICAM-1 in different vascular beds had biological significance, we compared the number of leucocytes adhering to the vascular endothelium with the relative level of ICAM-1 on the luminal surface (as assessed using our anti-ICAM-1-coated microsphere method) of retina and cremaster muscle, a tissue that showed little induction of ICAM-1 in diabetes. For these studies we used WT diabetic animals and compared them with WT non-diabetic animals. We found that diabetes of 2 months’ duration resulted in a significant increase in leucostasis in the retinal vasculature but not in the cremaster vasculature (Fig. 6a); these results paralleled the increased level of ICAM-1 in the retinal vasculature compared with that in the cremaster vasculature (Fig. 6b).
Fig. 6.
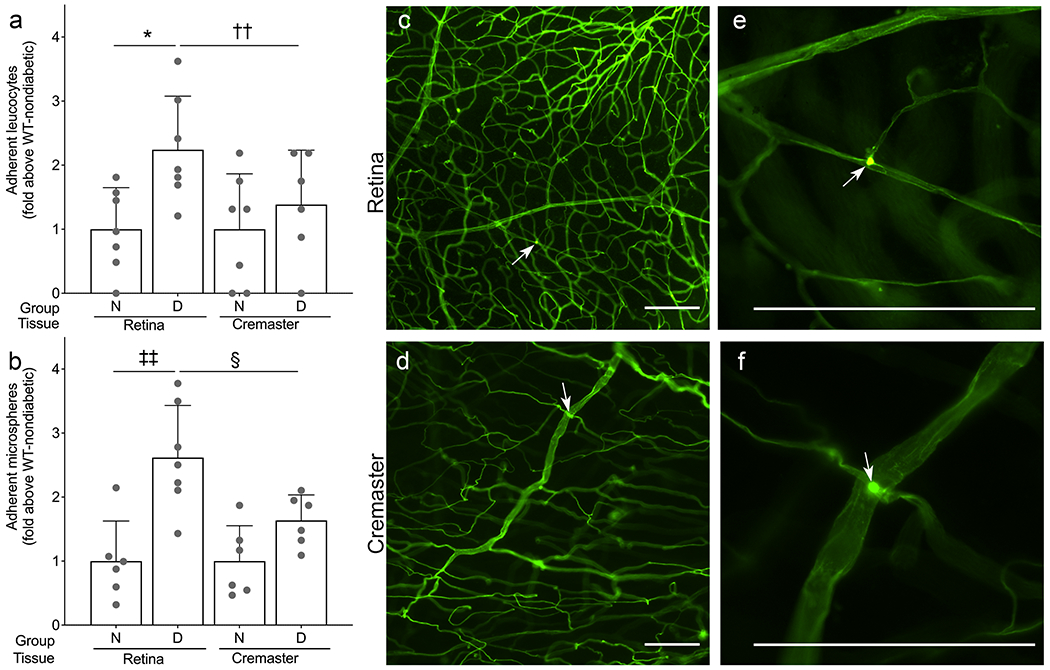
The number of leucocytes adherent to the vascular endothelium is consistent with the effect of diabetes on ICAM-1 expression. Leucostasis is greater in the retinal vasculature (a, c, e) than in the cremaster vasculature (a, d, f) of animals with diabetes of 2 months’ duration. The induction of ICAM-1 in the retinal vasculature compared with the cremaster vasculature correlates with the number of adherent microspheres (b). (c, e) Representative images of retinal leucostasis (white arrows); (e) is a magnification of (c). (d, f) Representative images of cremaster leucostasis (white arrows); (f) is a magnification of (d). Scale bars are 100μm. Statistical analysis was performed using one-way ANOVA with an uncorrected Fisher’s test. Data are mean ± SD; n=6–7. p≤0.05 is considered significant. *,§p≤0.05 and ††,‡‡p ≤ 0.01 as shown. D, diabetic; N, non-diabetic
Discussion
Inflammation and leucocytes have previously been implicated in the pathogenesis of diabetic retinopathy in diabetic patients. A critical step in inflammation is the adhesion of activated leucocytes to ICAM-1 and other adhesion molecules present on the luminal surface of vascular endothelium [33, 34]. Diabetes induces proinflammatory proteins in the retina, including ICAM-1 [17], and deletion of ICAM-1 (or its ligand on leucocytes) significantly inhibits diabetes-induced leucostasis and damage to the retinal vasculature, including increased permeability and degeneration of retinal capillaries [12]. Likewise, blocking the interaction of leucocytes with ICAM-1 via neutrophil inhibitory factor (NIF) has been shown to significantly inhibit the development of the retinal histopathology seen in early diabetic retinopathy [5, 12, 19].
In the present study, the effect of diabetes on the distribution of ICAM-1 on the luminal surface of the vascular endothelium was compared among multiple tissues. The data indicate that diabetes increases the level of ICAM-1 on the luminal surface of endothelial cells, but it does so to different extents in different tissues. The relative increase in endothelial cell levels of ICAM-1 as a result of diabetes was greatest in the retina compared with other tissues. In contrast, diabetes caused less of an increase in endothelial ICAM-1 in many other tissues; the observed changes did not achieve statistical significance compared with non-diabetic animals. Significant variability in ICAM-1 levels was observed in the WT diabetic control groups, probably in part because of the substantial daily fluctuations in blood glucose and insulin [35, 36].
The non-uniform induction of ICAM-1 in the tissues analysed was not restricted to diabetes; it also occurred in a model of systemic inflammation (LPS), suggesting that hyperglycaemia is not a unique initiator of endothelial inflammation or injury. Consistent with the effect of diabetes, LPS administration in non-diabetic animals resulted in upregulation of vascular ICAM-1 to a greater extent in some tissues than in others; the retinal vasculature again showed a large increase in ICAM-1 levels following LPS administration. Unlike diabetes, however, LPS caused a significant induction of ICAM-1 on the luminal surface of the vascular endothelium in a number of tissues not significantly affected by diabetes. Toll-like receptor 4 (TLR4) is the receptor for LPS [37], suggesting that the induction of endothelial ICAM-1 after LPS administration (and perhaps also in diabetes) might be mediated via TLR/MyD88/NF-κB signalling. As LPS was injected systemically, we would expect the LPS-mediated induction of ICAM-1 to be similar among all tissues; however, we observed significantly increased levels of ICAM-1 after LPS challenge in the aorta, diaphragm, spleen and lung, in addition to the retina. The difference among tissues in the LPS-mediated induction of ICAM-1 might be due in part to the tissue distribution of TLR4. The highest levels of Tlr4 mRNA detected by PCR are in the spleen and cells of myeloid origin, followed by the colon, ovary, lungs, small intestine and placenta, whereas Tlr4 expression is low in the brain, heart, kidneys, liver, prostate, pancreas, testes, muscle and thymus [38–41].
The function of the retina—to convert light to a neural signal—is unique among tissues. Rod and cone photoreceptors are the most prevalent cells in the retina and are extremely metabolically active [42, 43]. Signals sent out from these photoreceptors affect the function of other cells in the retina, and mutation or deletion of photoreceptor cell opsin has been shown to lead to dysfunction and degeneration of the retinal vasculature [44]. Responses of photoreceptor cells to the abnormal milieu of diabetes have been reported to cause the oxidative stress and inflammation that are found in the retina in diabetes [45, 46] and we now report that the diabetes-induced increase in ICAM-1 on the luminal surface of retinal endothelial cells is also influenced by photoreceptors and, specifically, by the molecular processes that lead to vision.
Increased levels of ICAM-1 on the luminal surface of the retinal endothelium in diabetes could be interpreted as a marker of the proinflammatory status of the retinal vasculature in diabetes and could also be a therapeutic target in diabetes. Use of blocking antibodies against ICAM-1 or other adhesion molecules and their ligands on white blood cells seems undesirable, as immune surveillance could also be impaired [47, 48]. We previously reported that a selective antagonist of integrin αmβ2 on leucocytes (NIF) inhibited the diabetes-induced degeneration of retinal capillaries in mice by blocking the excessive interaction between leucocytes and retinal endothelial cells [19]. Importantly, it did not significantly impair the ability of mice to clear an opportunistic bacterial challenge, suggesting that it did not compromise immune surveillance.
In summary, our studies demonstrate that diabetes-induced upregulation of endothelial ICAM-1 does not occur uniformly in all tissues, and the increase in ICAM-1 levels in retinal endothelial cells is due at least in part to photoreceptor cell metabolic activity related to vision. Appreciable induction of ICAM-1 on the endothelial surface leads to increased adhesion of leucocytes to these endothelial cells, with secondary leakage and degeneration of the retinal microvasculature.
Acknowledgements
We thank G. Li for his contribution to the initial studies related to this project. G. Li was a student in the laboratory when we initiated the studies on ICAM-1 distribution and is now a staff pathologist in the Community Healthcare System in Chicago, IL. We also thank J. Atwood at the Institute for Immunology Flow Core facility at the University of California Irvine for assistance in protocol development for flow cytometry analysis of the microspheres.
Funding
This work was supported by National Institutes of Health (NIH) grants EY033002, EY022938, EY022938-S1 and R24 EY024864, and by the Department of Veterans Affairs (grant no. BX003604). The authors acknowledge an unrestricted grant from Research to Prevent Blindness to the Gavin Herbert Eye Institute at the University of California Irvine.
Abbreviations
- ICAM-1
Intercellular adhesion molecule-1
- LPS
Lipopolysaccharide
- NIF
Neutrophil inhibitory factor
- TLR4
Toll-like receptor 4
- WT
Wild-type
Footnotes
Authors’ relationships and activities The authors declare that there are no relationships or activities that might bias, or be perceived to bias, their work.
Data availability
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.
References
- [1].Harris NR, Russell JM, Granger DN (1994) Mediators of endotoxin-induced leukocyte adhesion in mesenteric postcapillary venules. Circ Shock 43(4): 155–160 [PubMed] [Google Scholar]
- [2].Hartl R, Medary MB, Ruge M, Arfors KE, Ghajar J (1997) Early white blood cell dynamics after traumatic brain injury: effects on the cerebral microcirculation. J Cereb Blood Flow Metab 17(11): 1210–1220. 10.1097/00004647-199711000-00010 [DOI] [PubMed] [Google Scholar]
- [3].Kaneider NC, Leger AJ, Kuliopulos A (2006) Therapeutic targeting of molecules involved in leukocyte-endothelial cell interactions. FEBS J 273(19): 4416–4424. 10.1111/j.1742-4658.2006.05441.x [DOI] [PubMed] [Google Scholar]
- [4].Adamis AP, Berman AJ (2008) Immunological mechanisms in the pathogenesis of diabetic retinopathy. Semin Immunopathol 30(2): 65–84. 10.1007/s00281-008-0111-x [DOI] [PubMed] [Google Scholar]
- [5].Talahalli R, Zarini S, Tang J, et al. (2013) Leukocytes regulate retinal capillary degeneration in the diabetic mouse via generation of leukotrienes. J Leukoc Biol 93(1): 135–143. 10.1189/jlb.0112025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Rodrigues SF, Granger DN (2014) Leukocyte-mediated tissue injury in ischemic stroke. Curr Med Chem 21(19): 2130–2137. 10.2174/0929867321666131228192119 [DOI] [PubMed] [Google Scholar]
- [7].Herdade AS, Silva IM, Calado A, et al. (2020) Effects of Diabetes on Microcirculation and Leukostasis in Retinal and Non-Ocular Tissues: Implications for Diabetic Retinopathy. Biomolecules 10(11). 10.3390/biom10111583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Miyamoto K, Khosrof S, Bursell SE, et al. (1999) Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc Natl Acad Sci U S A 96(19): 10836–10841. 10.1073/pnas.96.19.10836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hopkins AM, Baird AW, Nusrat A (2004) ICAM-1: targeted docking for exogenous as well as endogenous ligands. Adv Drug Deliv Rev 56(6): 763–778. 10.1016/j.addr.2003.10.043 [DOI] [PubMed] [Google Scholar]
- [10].Lawson C, Wolf S (2009) ICAM-1 signaling in endothelial cells. Pharmacol Rep 61(1): 22–32. 10.1016/s1734-1140(09)70004-0 [DOI] [PubMed] [Google Scholar]
- [11].Joussen AM, Murata T, Tsujikawa A, Kirchhof B, Bursell SE, Adamis AP (2001) Leukocyte-mediated endothelial cell injury and death in the diabetic retina. Am J Pathol 158(1): 147–152. 10.1016/S0002-9440(10)63952-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Joussen AM, Poulaki V, Le ML, et al. (2004) A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 18(12): 1450–1452. 10.1096/fj.03-1476fje [DOI] [PubMed] [Google Scholar]
- [13].Kern TS (2007) Contributions of inflammatory processes to the development of the early stages of diabetic retinopathy. Exp Diabetes Res 2007: 95103. 10.1155/2007/95103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kern TS, Miller CM, Du Y, et al. (2007) Topical administration of nepafenac inhibits diabetes-induced retinal microvascular disease and underlying abnormalities of retinal metabolism and physiology. Diabetes 56(2): 373–379. 10.2337/db05-1621 [DOI] [PubMed] [Google Scholar]
- [15].Vincent JA, Mohr S (2007) Inhibition of caspase-1/interleukin-1beta signaling prevents degeneration of retinal capillaries in diabetes and galactosemia. Diabetes 56(1): 224–230. 10.2337/db06-0427 [DOI] [PubMed] [Google Scholar]
- [16].Zheng L, Du Y, Miller C, et al. (2007) Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 50(9): 1987–1996. 10.1007/s00125-007-0734-9 [DOI] [PubMed] [Google Scholar]
- [17].Tang J, Kern TS (2011) Inflammation in diabetic retinopathy. Prog Retin Eye Res 30(5): 343–358. 10.1016/j.preteyeres.2011.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tang J, Allen Lee C, Du Y, et al. (2013) MyD88-dependent pathways in leukocytes affect the retina in diabetes. PLoS One 8(7): e68871. 10.1371/journal.pone.0068871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Veenstra AA, Tang J, Kern TS (2013) Antagonism of CD11b with neutrophil inhibitory factor (NIF) inhibits vascular lesions in diabetic retinopathy. PLoS One 8(10): e78405. 10.1371/journal.pone.0078405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ocaranza MP, Valderas P, Moya J, et al. (2020) Rho kinase cascade activation in circulating leukocytes in patients with diabetes mellitus type 2. Cardiovasc Diabetol 19(1): 56. 10.1186/s12933-020-01027-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chen M, Obasanmi G, Armstrong D, et al. (2019) STAT3 activation in circulating myeloid-derived cells contributes to retinal microvascular dysfunction in diabetes. J Neuroinflammation 16(1): 138. 10.1186/s12974-019-1533-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sampson MJ, Davies IR, Brown JC, Ivory K, Hughes DA (2002) Monocyte and neutrophil adhesion molecule expression during acute hyperglycemia and after antioxidant treatment in type 2 diabetes and control patients. Arterioscler Thromb Vasc Biol 22(7): 1187–1193. 10.1161/01.atv.0000021759.08060.63 [DOI] [PubMed] [Google Scholar]
- [23].de Vries MA, Alipour A, Klop B, et al. (2015) Glucose-dependent leukocyte activation in patients with type 2 diabetes mellitus, familial combined hyperlipidemia and healthy controls. Metabolism 64(2): 213–217. 10.1016/j.metabol.2014.10.011 [DOI] [PubMed] [Google Scholar]
- [24].Gilbert RE (2013) Endothelial loss and repair in the vascular complications of diabetes: pathogenetic mechanisms and therapeutic implications. Circ J 77(4): 849–856. 10.1253/circj.cj-13-0236 [DOI] [PubMed] [Google Scholar]
- [25].Mota RI, Morgan SE, Bahnson EM (2020) Diabetic vasculopathy: macro and microvascular injury. Curr Pathobiol Rep 8(1): 1–14. 10.1007/s40139-020-00205-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Saadane A, Lessieur EM, Du Y, Liu H, Kern TS (2020) Successful induction of diabetes in mice demonstrates no gender difference in development of early diabetic retinopathy. PLoS One 15(9): e0238727. 10.1371/journal.pone.0238727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Veenstra A, Liu H, Lee CA, Du Y, Tang J, Kern TS (2015) Diabetic Retinopathy: Retina-Specific Methods for Maintenance of Diabetic Rodents and Evaluation of Vascular Histopathology and Molecular Abnormalities. Curr Protoc Mouse Biol 5(3): 247–270. 10.1002/9780470942390.mo140190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Seemann S, Zohles F, Lupp A (2017) Comprehensive comparison of three different animal models for systemic inflammation. J Biomed Sci 24(1): 60. 10.1186/s12929-017-0370-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Goetz DJ, Greif DM, Ding H, et al. (1997) Isolated P-selectin glycoprotein ligand-1 dynamic adhesion to P- and E-selectin. J Cell Biol 137(2): 509–519. 10.1083/jcb.137.2.509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Shinde Patil VR, Campbell CJ, Yun YH, Slack SM, Goetz DJ (2001) Particle diameter influences adhesion under flow. Biophys J 80(4): 1733–1743. 10.1016/s0006-3495(01)76144-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Montes M, Jaensson EA, Orozco AF, Lewis DE, Corry DB (2006) A general method for bead-enhanced quantitation by flow cytometry. J Immunol Methods 317(1–2): 45–55. 10.1016/j.jim.2006.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sakami S, Maeda T, Bereta G, et al. (2011) Probing mechanisms of photoreceptor degeneration in a new mouse model of the common form of autosomal dominant retinitis pigmentosa due to P23H opsin mutations. J Biol Chem 286(12): 10551–10567. 10.1074/jbc.M110.209759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wee H, Oh HM, Jo JH, Jun CD (2009) ICAM-1/LFA-1 interaction contributes to the induction of endothelial cell-cell separation: implication for enhanced leukocyte diapedesis. Exp Mol Med 41(5): 341–348. 10.3858/emm.2009.41.5.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Bui TM, Wiesolek HL, Sumagin R (2020) ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J Leukoc Biol 108(3): 787–799. 10.1002/JLB.2MR0220-549R [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Takami S, Yamashita S, Kihara S, Kameda-Takemura K, Matsuzawa Y (1998) High concentration of glucose induces the expression of intercellular adhesion molecule-1 in human umbilical vein endothelial cells. Atherosclerosis 138(1): 35–41. 10.1016/s0021-9150(97)00286-4 [DOI] [PubMed] [Google Scholar]
- [36].Gao Y, Meng D, Sun N, et al. (2014) Clock upregulates intercellular adhesion molecule-1 expression and promotes mononuclear cells adhesion to endothelial cells. Biochem Biophys Res Commun 443(2): 586–591. 10.1016/j.bbrc.2013.12.022 [DOI] [PubMed] [Google Scholar]
- [37].Lu YC, Yeh WC, Ohashi PS (2008) LPS/TLR4 signal transduction pathway. Cytokine 42(2): 145–151. 10.1016/j.cyto.2008.01.006 [DOI] [PubMed] [Google Scholar]
- [38].Vaure C, Liu Y (2014) A comparative review of toll-like receptor 4 expression and functionality in different animal species. Front Immunol 5: 316. 10.3389/fimmu.2014.00316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Asahina Y, Yoshioka N, Kano R, Moritomo T, Hasegawa A (2003) Full-length cDNA cloning of Toll-like receptor 4 in dogs and cats. Vet Immunol Immunopathol 96(3-4): 159–167. 10.1016/s0165-2427(03)00159-4 [DOI] [PubMed] [Google Scholar]
- [40].Hornung V, Rothenfusser S, Britsch S, et al. (2002) Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol 168(9): 4531–4537. 10.4049/jimmunol.168.9.4531 [DOI] [PubMed] [Google Scholar]
- [41].Garay-Malpartida HM, Mourão RF, Mantovani M, Santos IA, Sogayar MC, Goldberg AC (2011) Toll-like receptor 4 (TLR4) expression in human and murine pancreatic beta-cells affects cell viability and insulin homeostasis. BMC Immunol 12: 18. 10.1186/1471-2172-12-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fu Z, Kern TS, Hellstrom A, Smith LEH (2021) Fatty acid oxidation and photoreceptor metabolic needs. J Lipid Res 62: 100035. 10.1194/jlr.TR120000618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Ahmed J, Braun RD, Dunn R Jr., Linsenmeier RA (1993) Oxygen distribution in the macaque retina. Invest Ophthalmol Vis Sci 34(3): 516–521 [PubMed] [Google Scholar]
- [44].Liu H, Tang J, Du Y, et al. (2016) Photoreceptor Cells Influence Retinal Vascular Degeneration in Mouse Models of Retinal Degeneration and Diabetes. Invest Ophthalmol Vis Sci 57(10): 4272–4281. 10.1167/iovs.16-19415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Liu H, Tang J, Du Y, et al. (2015) Retinylamine Benefits Early Diabetic Retinopathy in Mice. J Biol Chem 290(35): 21568–21579. 10.1074/jbc.M115.655555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Liu H, Tang J, Du Y, et al. (2019) Transducin1, Phototransduction and the Development of Early Diabetic Retinopathy. Invest Ophthalmol Vis Sci 60(5): 1538–1546. 10.1167/iovs.18-26433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Furuya K, Takeda H, Azhar S, et al. (2001) Examination of several potential mechanisms for the negative outcome in a clinical stroke trial of enlimomab, a murine anti-human intercellular adhesion molecule-1 antibody: a bedside-to-bench study. Stroke 32(11): 2665–2674. 10.1161/hs3211.098535 [DOI] [PubMed] [Google Scholar]
- [48].Uettwiller F, Rigal E, Hoarau C (2011) Infections associated with monoclonal antibody and fusion protein therapy in humans. MAbs 3(5): 461–466. 10.4161/mabs.3.5.16553 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated and/or analysed during the current study are available from the corresponding author on reasonable request.