

Abstract
The Fanconi anemia (FA) pathway is essential for repairing DNA inter-strand crosslinks (ICL). ICLs induce stalled DNA replication forks and trigger activation of the FA pathway by promoting recruitment of the FANCM/FAAP24/MHF complex to ICL sites. Given that stalled replication forks are proximal to ICL sites, fork-associated proteins may coordinate with FA factors to rapidly sense ICLs for activation of FA signaling. Here we report that And-1, a replisome protein, is critical for activation of the FA pathway by sensing ICL-stalled forks and recruiting the FANCM/FAAP24 complex to ICLs. In response to ICLs, And-1 rapidly accumulated at ICL-stalled forks in a manner dependent on ATR-induced phosphorylation at T826. And-1 phosphorylation triggered an intramolecular change that promoted the interaction of And-1 with FANCM/FAAP24, resulting in recruitment of the FANCM/FAAP24 complex to ICLs. Furthermore, p-T826 And-1 was elevated in cisplatin-resistant ovarian cancer cells, and activated And-1 contributed to cisplatin resistance. Collectively, these studies elucidate a mechanism by which And-1 regulates FA signaling and identify And-1 as a potential target for developing therapeutic approaches to treat platinum-resistant ovarian cancer.
INTRODUCTION
Inter-strand crosslinks (ICLs) are highly cytotoxic DNA lesions that form a covalent linkage between two complementary strands of double-stranded DNA, thereby preventing the separation of DNA strands and interfering with DNA replication or transcription. ICLs could be introduced by cytotoxic chemotherapy drugs including cisplatin, mitomycin C (MMC), etc. In addition, during normal cellular metabolism reactive aldehydes and lipid peroxidation products also generate ICLs (1). ICLs challenge DNA replication and genome integrity, and may trigger cell death if not repaired properly. Cells have evolved multiple surveillance mechanisms to repair ICLs, and the Fanconi anemia (FA) pathway is the primary mechanism to resolve ICLs during S phase. Once colliding with unrepaired ICLs, DNA replication forks are stalled, and these ICL-induced stalled replication forks (ICL-stalled forks) must be renovated immediately to avoid genomic catastrophe. The FANCM/FAAP24/MHF complex is the first group of mediators that accumulate at ICLs for the initiation of the FA signaling (2–4). Then the FA core complex (FANC-M, A, B, C, E, F, G, L along with their associated proteins) is recruited to ICLs, where it mono-ubiquitinates FANCD2 and FANCI (ID2) complex that is critical for ICL repair (5). The ID2 complex consequently engages multiple endonucleases such as XPF-ERCC1, FAN1, and MUS81-EME1 to unhook and repair ICLs (6–8).
As an early sensor of the FA signaling, FANCM has been shown to be associated with replisome factors CDC45 and MCM to restart the stalled replication fork (9). FANCM also interacts with PCNA to promote replication traverse of ICLs (10). These lines of evidence strongly suggest that replisome components play an important role in the FA signaling. However, the mechanisms by which replisome components coordinate with FANCM complex to promote the recruitment of FANCM complex to ICLs remains largely unknown.
And-1/WDHD1/Ctf4 is an acidic nucleoplasmic DNA-binding protein that contains N-terminal WD40 domains, a middle SepB domain, and a C-terminal high mobility group (HMG) domain (11,12). And-1 is essential for faithful DNA replication as a replisome protein, chromosome transmission fidelity, DNA damage repair, etc. (13–20). HMG is a DNA binding domain and proteins with HMG domain prefer to bind ICL structure on genomic DNA and function as a scaffold to recruit other proteins for maintaining genomic stability and integrity (21). Given that And-1 is a fork-associated protein with HMG domain, we hypothesize that And-1 may participate in ICL repair by rapidly sensing ICL-stalled replication forks to activate the FA pathway.
Here we found that And-1 is a unique replication fork-associated protein that is essential for the activation of the FA signaling by regulating the recruitment of the FANCM/FAAP24 complex to ICL-stalled forks. Specifically, in response to ICLs And-1 quickly accumulates at ICL-stalled forks in a manner dependent on its phosphorylation at T826 by ATR; And-1 is critical for the recruitment of the FANCM complex to ICLs; phosphorylated And-1 (p-And-1) level is elevated in cisplatin-resistant ovarian cancer cells and elevated p-And-1 contributes to cisplatin resistance in ovarian cancer; inhibition of And-1 overcomes cisplatin resistance of ovarian cancer. These results collectively reveal a novel mechanism by which And-1 coordinates with the FANCM complex for ICL repair and provide a previously unappreciated mechanism regulating cisplatin resistance in ovarian cancer.
MATERIALS AND METHODS
Cell culture
Human U2OS, HEK-293T, Hela, MCF-7 cells were purchased from ATCC. OV433 and OV433 CR are a gift from Dr. Gengsheng Wu (Wayne State University). SKOV3, SKOV3 CR, PEO1, and PEO4 cells were described previously (22). All cells were cultured in DMEM supplemented with 10% FBS at 37 °C with 5% CO2. Cells were routinely tested for Mycoplasma contamination by PCR. U2OS And-1 knockout (KO) cell, OV433 CR-And-1 KO cell, and SKOV3 CR-And-1 KO cell were generated by using CRISPR/Cas9-mediated technology. Specific guide RNAs (gRNAs) were ligated into Lenti V2 plasmid according to the protocol described previously (23). The gRNAs used in this study: (1) And-1 sgRNA1: TGGTGAGCGAGCCGAAGCGC, (2) And-1 sgRNA2: GTGGTGAGCGAGCCGAAGCG. Cells transfected with control or sgRNA were selected in medium with 2.5 μg/ml puromycin for 2–3 weeks.
Antibodies and plasmids
The antibodies against And-1, γ-H2AX, β-actin, FLAG, ATR were described previously (15); antibodies against FANCM(sc-101389), FANCD2(sc-20022), PCNA (sc-56), Histone H2B (sc-515937), XPF (sc-398032), ERCC1 (sc-17809) were from Santa Cruz; antibodies against FAAP24(ab48124), MHF1 (ab169385), MHF2 (ab104236) were from Abcam; antibodies against FANCA (#14657), FANCB (#14243), phospho-ATR/ATM substrate (#9607) were from Cell Signaling; antibodies against cisplatin-DNA adduct (#MABE416) were from Millipore We collaborated with ThermoFisher Scientific Company to develop the monoclonal antibody against And-1 at the T826 site (p-And-1 T826). The mice were immunized with the peptide C-KAAELTA(pT)QVEEE-amide. The plasmids containing Full-length And-1 (PEFF-FLAG-And-1) and And-1 mutants were described previously(24). Plasmid expressing FLAG-tagged pcDNA3.1-FANCM was a kind gift from Dr. Weidong Wang (NIH/NIA). All transfections were performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Cells were harvested for analyses 48 h after the plasmid transfection.
siRNA transfections
The information on siRNA transfection is provided in the supplementary data.
Co-immunoprecipitation
Cells were lysed with Co-IP buffer containing 20 mM Tris–HCl at pH8.0, 100 mM NaCl, 1 mM EDTA, 0.5% NP-40, 10 mM NaF and protease inhibitor cocktail (Roche) on ice for 30 min, followed by sonication 10 sec for three times. After centrifugation, the supernatants were collected and incubated with protein A/G beads coupled with the antibody against indicated proteins at 4°C overnight. The beads were then washed three times and analyzed by Western blot. For FLAG IP, cell lysates were incubated with Anti-FLAG-M2 Affinity beads overnight and IPs were analyzed by Western blot.
Immunofluorescence assay and SRB assay
The immunofluorescence assay was performed as we described previously (15), and the SRB assay was performed as described previously (25).
Mass spectrometry
Mass spectrometry was conducted as previously described (24). Briefly, HEK-293T cells transfected with FLAG-And-1 for 48 h were treated with or without MMC (100ng/ml) for 6 h before being harvested for immune precipitation (IP). IPs samples were resolved by SDS-PAGE and gels were cut into ~1mm square pieces for the LC-MS/MS. The mass spectrometry data have been deposited to the ProteomeXchange Consortium via the PRIDE (26) partner repository with the dataset identifier PXD028246.
In Situ proximity ligation assay (PLA) and In Situ analysis of proteins at DNA replication forks (SIRF) assay
In situ proximity ligation assay (PLA) were conducted in U2OS cells with Duolink II Detection Kit (Millipore Sigma, #92101) according to the manufacturer’s instructions. SIRF assay was performed as previously described (27). Briefly, cells grown on coverslips were pulse-labeled with Edu for 10 min, then clicked with biotin-azide. Cells were then incubated with primary antibodies against biotin and indicated proteins, followed by PLA protocol according to the manufacturer’s instructions. Images were captured and analyzed by immunofluorescence confocal microscopy.
Modified alkaline comet assay
The unhooking efficiency of ICLs lesions was assessed by the modified alkaline comet assay as previously described (28,29). Briefly, cells were first treated with 2μM cisplatin for 2h. Cells were washed with PBS and incubated in cisplatin-free medium for 0, 6, 12, or 24h. Cells were then diluted to a density of 2×104 cells/ml and irradiated with 10 Gy γ-irradiation while on ice to induce random DNA strand breaks, and processed with TREVIGEN CometAssay (#4250-050-K) according to the manufacturer’s instructions. The degree of DNA ICLs was defined as the percentage decrease in olive tail movement with the following formula: % Decrease in olive tail movement (or ICL remaining) = [1- (Cp.IR-Δ)/(IR-Δ)]×100%. where Cp.IR = mean tail moment of cisplatin-treated and irradiated cells; Δ= mean tail moment of untreated and un-irradiated cells; IR = mean tail moment of irradiated and untreated cells.
Dot blot assay
The information on dot blot assay is provided in the supplementary data.
iPOND assay
The information on iPOND assay is provided in the supplementary data.
Electrophoretic mobility shift assay (EMSA) and protein purification
The information on EMSA and protein purification is provided in the supplementary data.
Animal experiments
Six-week-old female BALB/c athymic nude mice were purchased from Jackson Laboratory. 4×106 OV433 CR or OV433 CR And-1 depleted cells, SKOV3 CR or SKOV3 CR And-1 depleted cells were subcutaneous injected into the dorsal flank of each mouse, respectively. When the tumor volume reached about 100 mm3, all mice were randomized into indicated experiment groups. Cisplatin was administrated intraperitoneally 10mg/kg every 2 days for 2 weeks. Saline was used as the control. Relative tumor volumes were calculated with the formula: V=A×(B2)/2, where A and B represent the length and width of each tumor, respectively. For immunohistochemistry (IHC) analysis, tumor samples were sliced and conducted standard IHC staining procedures, and DAB staining was utilizing SuperPicture™ Polymer Detection Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. All animal studies were carried out by the National Institutes of Health regulation concerning the care and use of experimental animals and with the approval of the Institutional Animal Care and Use Committees of the George Washington University.
Identification and analysis of And-1-FA signature genes
To identify And-1-FA signature genes, we first identified top 30 genes that are And-1 or FANCM “function” or “co-complex” related genes by PathwayNet (30), and then selected overlap genes (27 genes) of these two groups as And-1-FA signature genes. And-1-FA signature genes were then used as a bait to examine the progression-free survival (PFS) and overall survival (OS) of ovarian cancer patients by Kaplan Meier analysis via online databases (http://kmplot.com). A log-rank (Mantel-Cox) test was used in the comparison of each arm.
Statistical analysis
GraphPad Prism 7.0 software was utilized for all data analyses. Data were represented as mean ± SEM as indicated in the figure legends. Statistical analysis was performed using one-way ANOVA or Student’s t-test. For Kaplan–Meier survival analysis, a log-rank (Mantel-Cox) test was conducted to compare each of the arms. p≤0.05 was considered as statistical significance.
Data availability
All data are included in the supplemental materials, or are available from the corresponding author upon reasonable request.
RESULTS
And-1 forms complexes with the FA proteins and is required for ICL repair
To investigate the role of And-1 in ICL repair, we conducted a mass spectrometry analysis to identify And-1-associated proteins that are known to be involved in ICL repair. From this analysis, we identified multiple FA factors, including FANCD2, FANCM, and FANCA in the FLAG-And-1 immunoprecipitates (IPs) (Fig. 1A and B). To rule out the possibility that chromosome mediates protein-protein interactions, we included ethidium bromide and DNase I in the cell lysis buffer to interrupt protein-DNA interactions. Co-immunoprecipitation (co-IP) assay showed that And-1 interacted with FANCM and FAAP24 but not MHF1/2, and these interactions were increased upon MMC treatment (Fig. 1C and D). Moreover, FANCA or FANCD2 were also detected in And-1 IPs (Supplementary Fig. S1A and B). The co-localization of And-1 with FANCM was further confirmed by the Proximity Ligation Assay (PLA) (Fig. 1E).
Figure 1. And-1 interacts with the FA factors and is required for ICL repair.
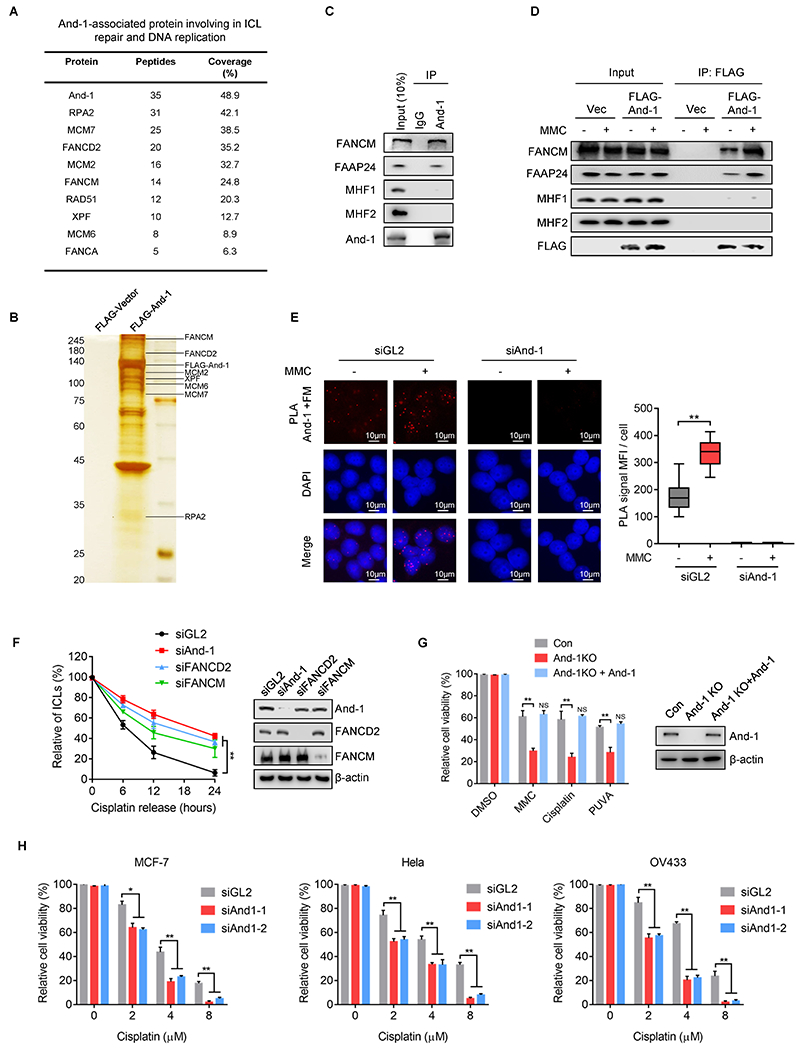
(A) Mass spectrometry analysis to identify And-1-associated proteins involved in ICL repair and DNA replication. (B) Cell extracts from 293T cells expressing FLAG-And-1 protein or FLAG-Vector with MMC treatment were subjected to immunoprecipitation (IP). FLAG IPs were resolved by SDS–PAGE followed by silver staining. (C-D) Co-immunoprecipitation (co-IP) to detect the interaction of endogenous And-1 (C) or exogenous FLAG-And-1 (D) with indicated proteins in 293T cells. (E) Proximity ligation assay (PLA) to examine the associations of And-1 with FANCM in U2OS cells with indicated treatment. The red foci represent the PLA signals. Right panel, quantification of mean fluorescence intensity (MFI) of PLA signals shown in left. (F) Modified comet assay to examine the amount of ICLs in U2OS cells with indicated treatments. (G) Cell viability was measured by SRB assay in indicated cells treated with MMC, Cisplatin, or PUVA. (H) Cell viability was measured in indicated cells transfected with indicated siRNAs and treated with cisplatin. Data represent means ± SD from three independent experiments. **, p≤0.01.
Since And-1 forms complexes with FA factors, we assumed that And-1 may involve ICL repair. To test this hypothesis, we measured the amount of ICLs using a modified comet assay. Interestingly, depletion of And-1, FANCD2 or FANCM significantly delayed ICL repair compared to siGL2-treated cells (Fig. 1F and Supplementary Fig. S1C). Consistently, removal of cisplatin-induced DNA adducts (CDAs) was also significantly impaired in And-1 KO cells compared to control cells (Supplementary Fig. S1D). And-1 depleted cells exhibited hypersensitivity towards multiple ICL-stimulus such as cisplatin, MMC, and psoralen + UVA (PUVA), while ectopic expression of And-1 restored the cell viability (Fig. 1G). Depletion of And-1 significantly attenuated the cell viability of multiple cancer cells including MCF-7 (breast cancer), OV433 (ovarian cancer), and Hela (cervical cancer) in response to cisplatin (Fig. 1H). Thus, And-1 plays an important role in ICL repair.
And-1 is required for recruitment of the FANCM/FAAP24 to ICLs
We assumed that And-1 may function as an early sensor to recognize ICL-stalled forks for the initiation of FA signaling. To test this hypothesis, we first examined whether And-1 accumulates at ICL-stalled forks by using iPOND assay. In unperturbed cells And-1 and PCNA but not FANCM/FAAP24 were detected at newly replicated DNA (Fig. 2A, lane 7), indicating that And-1 and PCNA are fork-associated proteins. Upon MMC treatment And-1 accumulated rapidly at stalled-replication forks, whereas fork-associated PCNA decreased (Fig. 2A, lane 8–9), suggesting that And-1 is a unique fork-associated protein accumulating at ICL-stalled forks. Like And-1, FANCM and FAAP24 were also detected at stalled forks (Fig. 2A, lane 8–9). Consistently, And-1 was detected at ICLs as indicated by co-localization with γ-H2AX in cells treated with various types of crosslink agents (Fig. 2B).
Figure 2. And-1 is required for recruitment of FANCM and FAAP24 to ICLs.
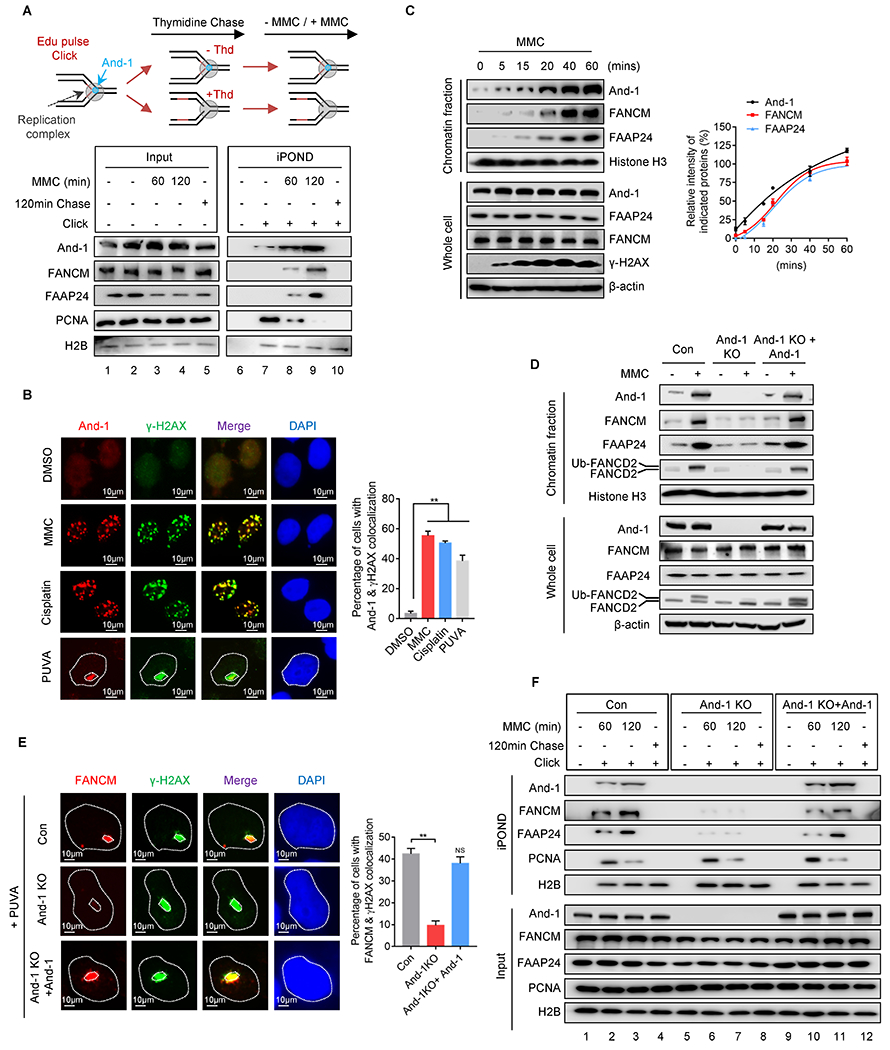
(A) Upper panel, schematic of iPOND assay. Lower panel, iPOND assay to examine the presence of indicated proteins at active or MMC-induced stalled replication forks. (B) Immunofluorescence to examine recruitment of And-1 to ICLs induced by MMC, cisplatin, and PUVA respectively. (C) Chromatin fractions were extracted at different time points after MMC treatment, followed by immunoblotting for indicated proteins. (D) U2OS cells were treated with or without MMC for 12 hr, and then chromatin fractions were immunoblotted for indicated proteins. (E) Immunofluorescence to examine recruitment of FANCM and γ-H2AX to PUVA-induced ICLs. (F) U2OS cells were labeled with EdU or chased with thymidine after EdU labeling, followed by iPOND assay to detect indicated proteins. Data represent means ± SD from three independent experiments. **, p≤0.01.
To explore the kinetics of recruitment of the And-1 and FANCM complex to ICL sites, we extracted the chromatin fractions at different time points post MMC treatment. Interestingly, And-1 has a basal level on chromatin in unperturbed cells and quickly assembled at 5 minutes, while the FANCM/FAAP24 complex began to accumulate around 15 minutes, these results were consistent with previous study that FANCM is recruited to the psoralen-induced ICLs lesion sites at 15 min after laser photoactivation (3) (Fig. 2C).
We next examined whether And-1 regulates the recruitment of the FANCM complex to ICLs. Loss of And-1 notably impaired the chromatin association of FANCM and FAAP24 in response to MMC treatment (Fig. 2D). Accordingly, loss of And-1 dramatically decreased mono-Ub of FANCD2 in whole-cell extractions (WCE), as well as its association with chromatin (Fig. 2D). Re-expression of And-1 partially restored chromatin association of FANCM/FAAP24 and mono-Ub of FANCD2 in And-1 KO cells (Fig. 2D). Although And-1 regulates recruitment of FANCM/FAAP24 to ICLs, loss of FANCM had no effects on the recruitment of And-1 to chromatin (Supplementary Fig. S2A). Moreover, depletion of And-1 also reduced FA core complex (FANCA, FANCB) and XPF-ERCC1 on chromatin (Supplementary Fig. S2B). Consistently, IF assays indicated that loss of And-1 markedly compromised ICL site recruitments of FANCM, FANCD2, and FAAP24, while ectopic expression of And-1 rescued their accumulations (Fig. 2E and Supplementary Fig. S2C–D). Interestingly, depletion of either FANCM or FAAP24 did not affect the accumulation of And-1 at ICLs (Supplementary Fig. S2E). In addition, iPOND assay showed that accumulation of FANCM and FAAP24 at ICL-stalled forks was dramatically reduced in And-1-depleted cells (Fig. 2F, lane 6–7), and ectopic expression of And-1 largely restored these defects (Fig. 2F, lane 10–11). Collectively, in response to ICL-stress, And-1 rapidly accumulates at ICL-stalled forks to initiate FA signaling by promoting recruitment of the FANCM/FAAP24 complex to ICLs.
Phosphorylation of And-1 at T826 is required for its accumulation at DNA damage sites and ICL repair
Using the protein phosphorylation prediction program GPS2.1, we identified six putative ATR/ATM phosphorylation sites on And-1. We then mutated each of them and examined And-1 phosphorylation by using a phospho-(Ser/Thr) ATM/ATR substrate antibody in cells treated with MMC. Strikingly, And-1 was found to be phosphorylated after MMC treatment, and mutation of T826A but not other putative phosphorylation sites completely abolished the phosphorylation (Fig. 3A). Thus, T826, an evolutionarily conserved site across different species (Fig. 3B), is a bona fide phosphorylation site in cells with ICLs. To further study the function of And-1 phosphorylation at T826, we generated an antibody that specifically recognized phosphorylated And-1 at T826, but not mutant And-1(T826A) in response to MMC or cisplatin in an ATR-dependent manner (Fig. 3A–3C and Supplementary Fig. S3A–3C).
Figure 3. And-1 phosphorylation at T826 is critical for ICL repair.
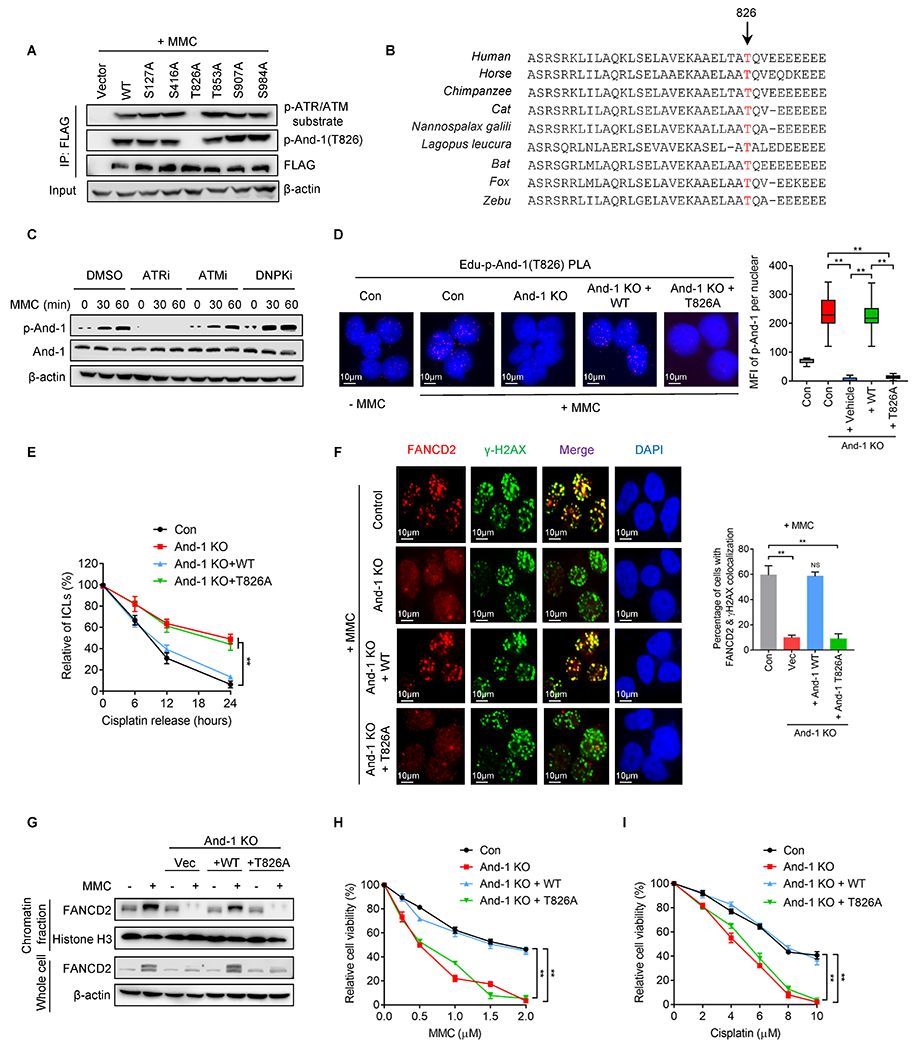
(A) 293T cells expressing indicated And-1 or its mutants were treated with MMC for 24hr, followed by FLAG-IP and immunoblotting for indicated proteins. (B) And-1 T826 site is conserved across different species. (C) U2OS cells were pretreated with ATRi, ATMi, and DNPKi for 12hr, then cells were subjected to MMC treatment for 24hr, followed by immunoblotting for indicated proteins. (D) Representative images of SIRF assay to detect the presence of p-And-1 in U2OS cells with indicated treatments. Right panel, quantification of p-And-1 signal shown in left. (E) Modified comet assay to examine relative amount of ICLs in indicated U2OS cells treated with cisplatin. (F) Immunofluorescence to detect FANCD2 in indicated cells post MMC treatment. Right panel, quantification of the percentage of cells with co-localization of FANCD2 and γ-H2AX shown in left. (G) U2OS cells were treated with or without MMC for 24 hr, then chromatin fractions were extracted and immunoblotted for indicated proteins. (H-I) Cell viability was determined by SRB assay in indicated cells treated with MMC (H) and cisplatin (I). Data represent means ± SD from three independent experiments. **, p≤0.01.
To test whether phosphorylation of And-1 at T826 regulates its accumulation at ICL-stalled forks, we conducted a sensitive single-cell analysis to measure the interactions of specific proteins with nascent DNA at replication forks using an approach called SIRF (in situ analysis of protein interactions at replication forks) (27). This assay allows us to visualize the localization of proteins at replication forks by using proximity ligation coupled with 5′-ethylene-2′-deoxyuridine click chemistry. As shown in Fig. 3D, the amount of p-And-1 at ICL-stalled forks was significantly increased in response to MMC, and the elevated p-And-1 signal was dramatically decreased in And-1-depleted cells. Moreover, only ectopic-expressed WT And-1 restored the p-And-1 signal compared with T826A mutant. Consistently, WT And-1 but not mutant T826A colocalized to the MMC-induced γ-H2AX sites (Supplementary Fig. S3D).
Reconstitution of WT but not T826A And-1 mutant largely restored ICL repair capacity and the removal efficiency of CDAs in And-1-depleted cells (Fig. 3E and Supplementary Fig. S3E), indicating that And-1 phosphorylation at T826 is essential for ICL repair. Moreover, WT but not T826A mutant restored FANCD2 focus formation, chromatin association, and cell viability in And-1-depleted cells in response to MMC or cisplatin (Fig. 3F–3I). Taken together, phosphorylation of And-1 at T826 is critical for its accumulation at ICLs and ICL repair.
And-1 phosphorylation promotes the accumulation of the FANCM/FAAP24 at ICLs
We next explored whether And-1 phosphorylation at T826 affects the accumulation of the FANCM/FAAP24 at ICL-stalled replication forks by using SIRF assay. The results indicated that loss of And-1 remarkably impaired the presence of FANCM and FAAP24 at ICL-stalled forks, whereas ectopic expression of WT-And-1 but not T826A mutant rescued the presence of both proteins at ICL-stalled forks (Fig. 4A and B). Consistently, iPOND assay also showed that WT And-1 but not And-1(T826A) restored the accumulation of FANCM and FAAP24 at the ICL-stalled forks and chromatin in And-1 depleted cells (Fig. 4C and D). Thus, the accumulation of FANCM and FAAP24 at ICLs is dependent on the phosphorylation of And-1 at T826.
Figure 4. The formation of the FANCM/FAAP24 complex and its recruitment to ICLs are dependent on And-1 phosphorylation at T826.
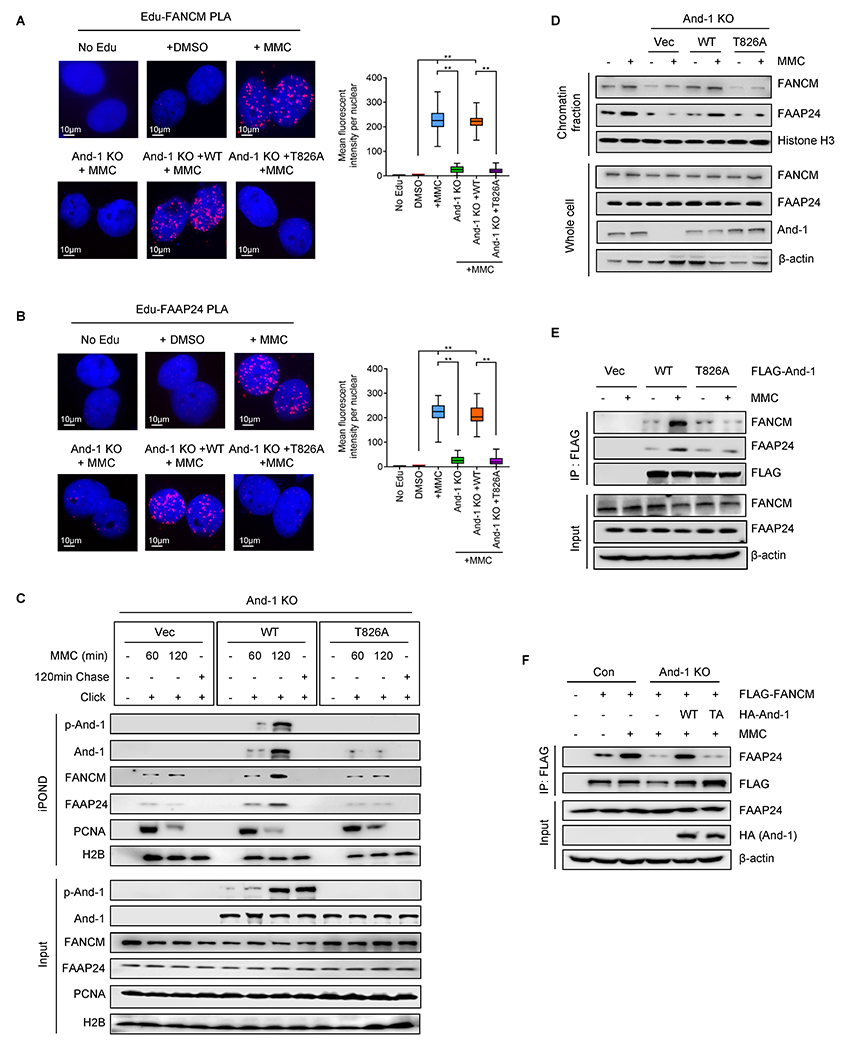
(A-B) SIRF assay to examine the presence of FANCM (A) and FAAP24 (B) at ICL-stalled replication forks in indicated U2OS cells treated with DMSO or MMC. Right panel, quantification of FANCM or FAAP24 signal. Data represent means ± SD from three independent experiments. **, p≤0.01. (C) iPOND assay to detect indicated proteins at stalled replication forks in U2OS treated with MMC. (D) Chromatin fraction assay to detect proteins in indicated U2OS cells treated with or without MMC for 24 hr. (E) 293T cells expressing indicated And-1 were treated with or without MMC for 24 hr before harvested. FLAG-IPs were immunoblotted for the indicated proteins. (F) Control U2OS cells or And-1-depleted U2OS cells with reconstituted WT-And-1 or T826A were treated with or without MMC for 24 hr before being harvested. FLAG-IP were immunoblotted for indicated proteins.
We next examined whether And-1 phosphorylation regulates its interaction with FANCM or FAAP24. As shown in Fig. 4E, T826A mutation significantly reduced the interactions of And-1 with FANCM or FAAP24 in response to MMC. Consistently, the interactions of And-1 with FANCM or FAAP24 were significantly impaired when cells were pre-incubated with ATR inhibitor or λ phosphatase (Supplementary Fig. S4A and S4B). Since FANCM-FAAP24 interaction is important for FANCM recruitment to ICLs (4,31), we examined whether And-1 phosphorylation regulates the interaction of FACNM with FAAP24. Intriguingly, loss of And-1 significantly decreased the interaction of FANCM with FAAP24, and reconstitution with WT but not T826A mutant restored this interaction (Fig. 4F). Thus, And-1 may function as a platform to facilitate the formation of FA complex in a manner dependent on And-1 phosphorylation at T826.
And-1 directly interacts with ICL-stalled forks and promotes the association of the FANCM/FAAP24 with ICL-stalled forks in vitro
To investigate how And-1 regulates ICL repair in vitro, we generated a replication fork molecule, in which a crosslink next to the fork was created by using a specific compound SJG-136 (32), which preferentially crosslinks DNA at guanine residues of 5’ purine–GATC–pyrimidine to form an ICL (Fig. 5A, left panel). We named this DNA structure as ICL-forks. EMSA results showed that ICL-forks exhibited an up-shifted band compared to those without ICLs when resolved on a denaturing PAGE gel (Fig. 5A, right panel), suggesting that crosslinking of DNA was generated properly. We next incubated recombinant And-1 proteins with ICL-forks and found that the addition of And-1 resulted in an upshifted band, whereas including And-1 antibody to the reaction buffer leads to a super-shifted band, while incubation with an unlabeled DNA competitor abolished the upshifted bands (Fig. 5B). Moreover, the affinity of And-1 with ICL-forks was stronger than that with forks without ICLs (Fig. 5B and Supplementary Fig. S5A).
Figure 5. And-1 promotes the association of the FACNM/FAAP24 with ICLs in vitro.
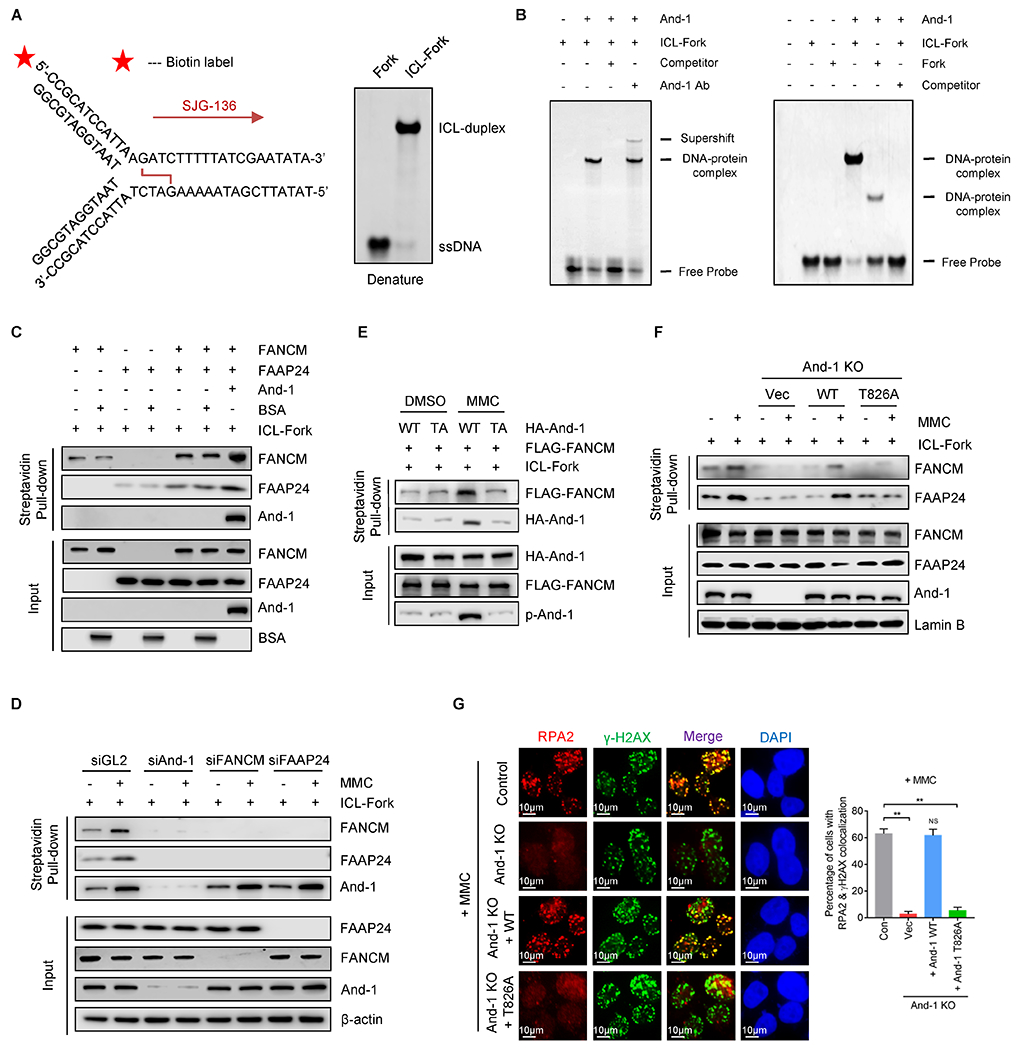
(A) Schematic of replication fork oligo with ICL induced by SJG-136. SJG-136 preferentially interacts with DNA guanine residues at 5’-GATC sequence to form ICL-forks. 5’ end of oligos was labeled with biotin as indicated with the red asterisk. Right panel, the validation of forks or ICL-fork oligos by denatured PAGE. (B) Left panel, purified And-1 protein was incubated with ICL-forks together with or without non-biotin-labeled ICL-forks (competitor), or And-1 antibody at room temperature for 2 hr, followed by EMSA. Right panel, purified And-1 was incubated with ICL-forks or fork at room temperature for 2 hr, followed by EMSA. (C) Biotin-labeled ICL-forks were incubated with purified And-1, FANCM, FAAP24, or together at room temperature for 2 hr. ICL-fork-associated proteins were resolved by SDS–PAGE. (D) Nuclear extracts from Hela cells treated with indicated siRNAs and MMC were incubated with ICL-forks. ICL-fork-associated proteins were resolved by SDS-PAGE. (E) Recombinant And-1 or And-1(T826) proteins purified from 293T cells treated with or without MMC were incubated with ICL-forks for 1 hr, followed by incubation with recombinant FANCM for an additional 2 hr. ICL-forks associated proteins were resolved by SDS–PAGE. (F) Nuclear extracts from U2OS cells with indicated treatments were incubated with biotin-labeled ICL-forks, followed by immunoblotting for the indicated proteins. (G) Immunofluorescence to detect the recruitment of RPA2 to ICLs in indicated cells treated with MMC. Data represent means ± SD from three independent experiments. **, p≤0.01.
Next, we examined how And-1 regulates the interactions of FANCM and FAAP24 with ICL-forks in vitro. To this end, purified recombinant FANCM or FAAP24 proteins were incubated with And-1 together with ICL-forks. Intriguingly, FANCM and FAAP24 mutually enhanced their association with ICL-forks, whereas the addition of And-1 proteins further enhanced the associations of FANCM and FAAP24 with ICL-forks (Fig. 5C and Supplementary Fig. S5B), while loss of And-1 significantly reduced the association of FANCM and FAAP24 with ICL-forks (Fig. 5D). We next explored how And-1 phosphorylation regulates the association of FANCM with ICL-forks. To this end, we purified recombinant WT And-1 or T826A mutant proteins from 293T cells treated with or without MMC. WT And-1 and T826A mutant proteins purified from DMSO-treated cells displayed a similar affinity to ICL-forks, whereas WT And-1 but not And-1 (T826A) proteins purified from MMC-treated cells exhibited an increased affinity to ICL-forks and significantly elevated the association of FANCM with ICL-forks (Fig. 5E). In addition, re-expression of WT but not And-1 T826A mutant markedly restored the associations of FANCM and FAAP24 with ICL-forks (Fig. 5F), and RPA accumulation in And-1-depleted cells (Fig. 5G). Thus, And-1 directly binds to ICL-forks and facilitates the association of FANCM with ICL-forks in a manner dependent on And-1 phosphorylation at T826.
Phosphorylation of And-1 at T826 induces its intramolecular conformational changes
We next investigated the molecular mechanism of how And-1 phosphorylation at T826 regulates its functional role in FA signaling. Co-IP analyses showed that FANCM and FAAP24 were only detected from IPs of full-length (FL) And-1, And-1 330-984 (SepB domain), and And-1 330-1129 (SepB + HMG domain), indicating that SepB domain is indispensable for the interactions of And-1 with FANCM and FAAP24 (Fig. 6A). Expression of FL And-1 and And-1(330-1129) but not And-1(330-984) restored the recruitment of FANCM to ICLs (Fig. 6B). Moreover, the DNA pull-down assay indicated that FL And-1 but not HMG deleted mutant And-1 (1-984) bound to ICL-forks (Fig. 6C). Consistently, the chromatin association of FANCM/FAAP24 was also reduced in the And-1 ΔHMG cells (Supplementary Fig. S6A). Together, these data suggest that And-1 binds to ICL-stalled forks via its HMG domain and interacts with FANCM complex via its SepB domain.
Figure 6. Phosphorylation at T826 changes And-1 intramolecular conformation.
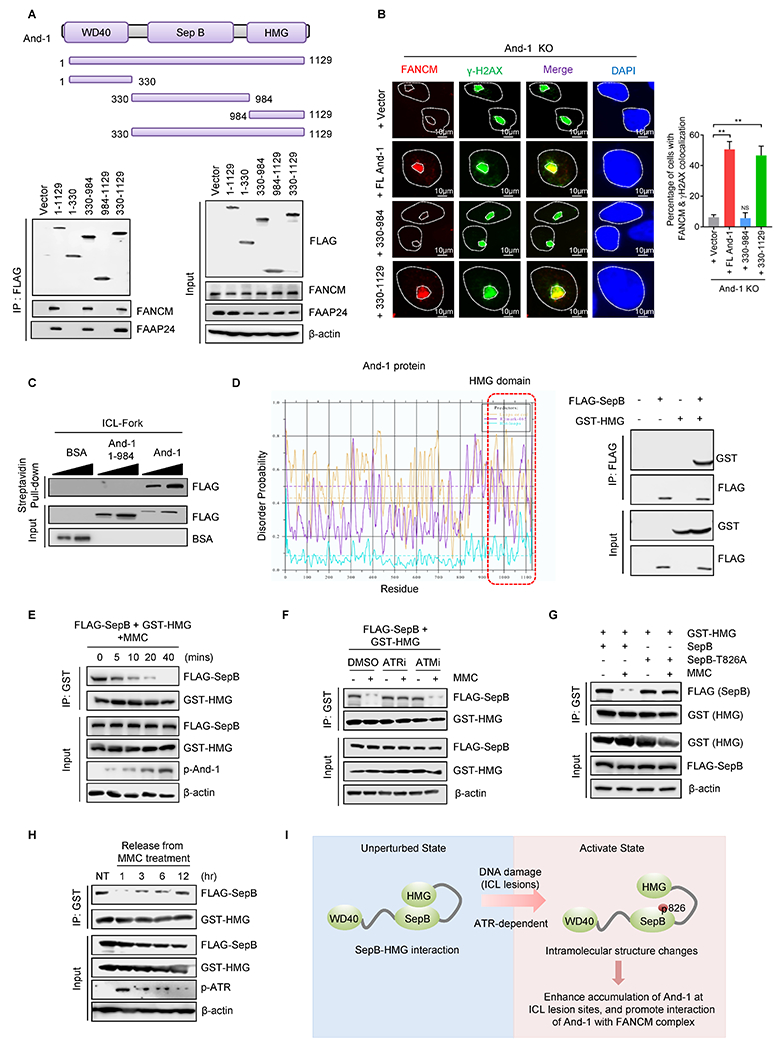
(A) Upper panel, schematic of And-1 structure. Lower panel, FLAG IPs of FLAG-And-1 and its truncated mutants expressed in 293T cells were immunoblotted for the indicated proteins. (B) Immunofluorescence to examine the recruitment of FANCM to ICLs induced by PUVA in And-1-depleted cells or And-1 depleted cells reconstituted WT-And-1 or truncated And-1 mutants. Data represent means ± SD from three independent experiments. **, p≤0.01. Right panel, quantification of the percentage of cells with co-localized FANCM and γ-H2AX. (C) And-1 or And-1(1–984) recombinant proteins purified from 293T cells were incubated with biotin-labeled ICL-forks for 2 hr, followed by immunoblotting for indicated proteins. (D) The probability prediction of the disordered region of And-1 protein by DisEMBL. The red dotted area was indicated as the disordered region in And-1. Right panel, 293T cells expressing FLAG-SepB and GST-HMG were harvested and FLAG-IPs were immunoblotted for the indicated proteins. (E) 293T cells co-transfected with FLAG-SepB and GST-HMG plasmid were treated with MMC (1μM) and cells were then harvested at indicated time points. (F) 293T cells co-transfected with FLAG-SepB and GST-HMG were pretreated with ATR inhibitor (VE-821, 10 μM) or ATM inhibitor (KU55933, 10 μM) for 12hr, followed by treatment with or without MMC (1μM) for 2 hr. FLAG-IPs were immunoblotted for indicated proteins. (G) 293T cells co-transfected with HMG-GST and FLAG-SepB or FLAG-SepB-T826A were treated with or without MMC (1μM) for 2 hr. (H) 293T cells co-transfected with HMG-GST and FLAG-SepB were treated with MMC (1μM) for 2 hr, then released into MMC-free medium. Cells were harvested at indicated time points and immunoblotted for indicated proteins. (I) Schematic of And-1 intramolecular interaction regulated by phosphorylation at T826 in response to ICLs.
Proteins with a highly flexible domain are easy to form intramolecular interactions (33). Given that the HMG domain is highly flexible (12), we speculated that And-1 may form intramolecular interactions, and phosphorylation of And-1 at T826 may alter this intra-molecular interactions. To test this hypothesis, we first analyzed the And-1 protein structure by utilizing the DisEMBL algorithm software prediction (http://dis.embl.de/) (34) and found that the HMG domain is highly disordered or flexible (Fig. 6D and Supplementary Fig. S6B), suggesting that HMG may form intramolecular interactions with SepB or WD40 domains. Indeed, co-IP assays indicated that And-1 HMG interacted with the SepB but not WD40 domains (Fig. 6D), and there was no interaction between WD40 and SepB domain (Supplementary Fig. S6C). The interaction between SepB and HMG domains was reduced upon ICL-stimulus, and the dissociation of these two domains was increased in a time- or dose-dependent pattern (Fig. 6E and Supplementary Fig. S6D).
Given that phosphorylation site T826 locates within SepB domain, we assumed that this phosphorylation may affect intramolecular interaction between SepB and HMG domains. Indeed, ATR inhibitor, but not ATM inhibitor, dramatically interfered with ICL-induced dissociation of SepB with HMG (Fig. 6F). Consistently, in response to MMC, the interaction of HMG with WT SepB but not mutant SepB (T826A) domain was significantly reduced (Fig. 6G). Intriguingly, the interaction between SepB and HMG domains was elevated in cells released from MMC or cisplatin in a time-dependent manner (Fig. 6H and Supplementary Fig. S6E). Taken together, And-1 phosphorylation at T826 prevents the intramolecular interaction between SepB and HMG domain, thereby promoting its recruitment to ICLs via HMG domain and interaction with FANCM complex through SepB domain for recruitment of FANCM to ICLs (Fig. 6I).
Targeting And-1 to overcome cisplatin resistance in ovarian cancer cells
Elevated FA signaling is a major mechanism contributing to cisplatin resistance in ovarian cancer (CROV) (35–39). We therefore examined And-1 and p-And-1 levels in three paired cisplatin-sensitive and -resistant ovarian cancer cells and found that both p-And-1 and p-ATR expression levels were significantly increased in all three resistant cells (Fig. 7A). Previously studies have demonstrated that enhanced ICL repair is critical for cisplatin resistance (40–42). As expected, OV433-CR cells have a higher ICL repair efficiency than parental cells (Supplementary Fig. S7A). Consistently, iPOND assay indicated that upon cisplatin treatment the recruitments of And-1, p-And-1, and FANCM to ICL-stalled forks in OV433-CR cells were much more robust and faster than parental cells (Supplementary Fig. S7B). Depletion of And-1 in OV433-CR cells reduced its ICL repair capacity and resensitized cells to cisplatin, which was restored by expression of WT-And-1 but not And-1(T826A) mutant (Fig. 7B–7F). Thus, p-And-1 is important for cisplatin resistance in CROV cells.
Figure 7. And-1 phosphorylation at T826 contributes to cisplatin resistance in ovarian cancer cells.
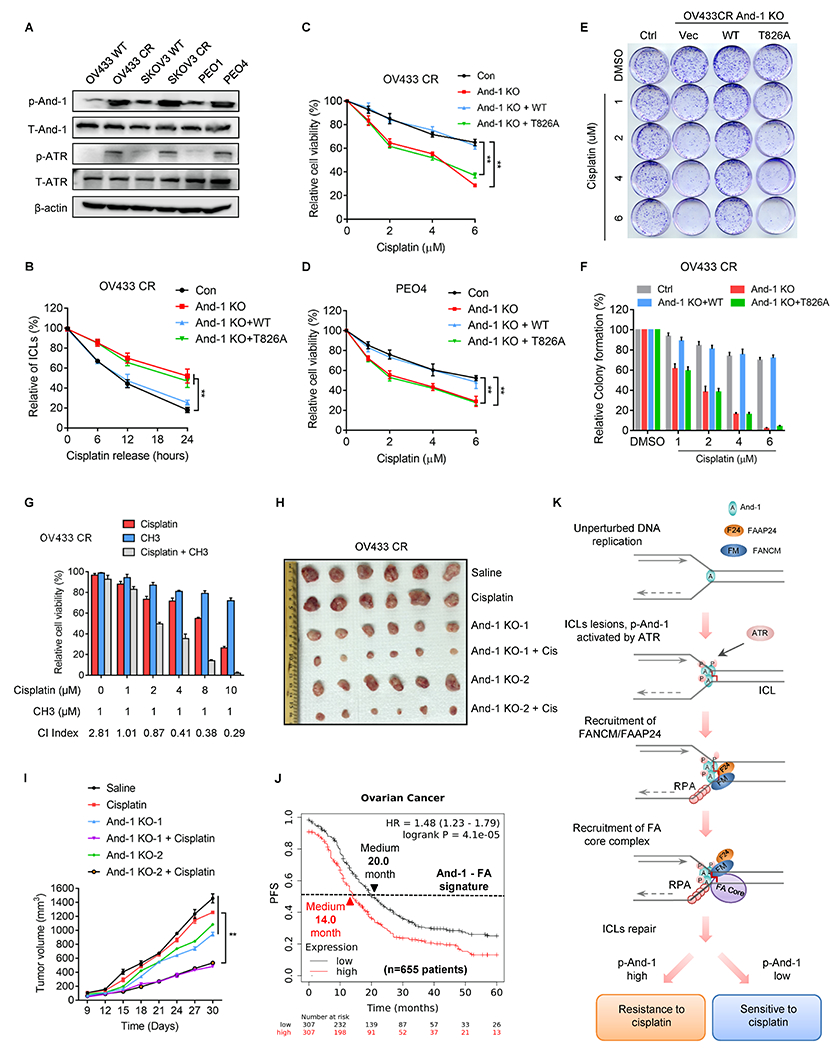
(A) Cell lysates from three paired parental and cisplatin-resistant (CR) ovarian cancer cells were immunoblotted for indicated proteins. (B) Modified comet assay to examine ICLs in OV433-CR or OV433 CR-And-1 depleted cells with constituted WT-And-1 or And-1 T826A. (C-D) Cell viability was examined in OV433-CR cells (C) and PEO4 cells (D) with reconstituted WT-And-1 or And-1 T826A in response to cisplatin. Data represent means ± SD from three independent experiments. **, p≤0.01. (E-F) Representative images of colony formation (E) and quantification results (F) in OV433 CR cells with indicated treatments. (G) The synergistic effects of cisplatin and CH3 in OV433 CR cells. CI values <1.0 were represented as good synergy. (H-I) Representative images (H) and growth curves (I) of OV433 CR or OV433 CR And-1 depleted xenograft tumors treated with vehicle or cisplatin (2mg/kg intraperitoneal) for 2 weeks. Data are represented as means ± SEM, n = 6 mice/group. **, p≤0.01. (J) Kaplan–Meier analyses of 5-year PFS based on clinical and molecular data from ovarian cancer patients (n=655 patients) with cisplatin drug treatment history. The patients were stratified by the expression levels of And-1-FA signature genes in tumors. Medium survival, log-rank (Mantel-Cox), p values, and HRs were shown as indicated. (K) Working model of And-1 in ICL repair.
We recently discovered a specific And-1 inhibitor CH3 (43). Interestingly, the combination of cisplatin and CH3 displayed an excellent synergy (CI < 1.0) in all three CROV cells (Fig. 7G and Supplementary Fig. S7C and S7D). We next subcutaneously implanted these cells into nude mice to form tumors, then followed by multiple treatments. Similarly, the combinatorial treatment with cisplatin and And-1 suppression dramatically impaired tumor growth compared to treatment with cisplatin alone (Fig. 7H–7I and Supplementary Fig. S7E–S7F). A similar synergy effect was also observed in SKOV3-CR tumors (Supplementary Fig. S7G–S7H).
To further explore the role of And-1 in the regulation of cisplatin resistance in ovarian cancer, we identified 27 genes that are involved in both And-1 and FA-mediated pathways by using PathwayNet analysis (http://pathwaynet.princeton.edu/) and named this group of genes as And-1-FA signature genes (Supplementary Table. S1). Strikingly, ovarian cancer patients with cisplatin drug treatment history exhibited a worse 5-year PFS and OS when And-1-FA signature gene expression in the tumors was higher (Fig. 7J and Supplementary Fig. S7I). Thus, the activated And-1-FA pathway is highly correlated with worse survival after cisplatin drug-based therapy. These results collectively suggested that And-1 and its phosphorylation are necessary for cisplatin resistance in ovarian cancer.
DISCUSSION
FANCM is considered as a key FA factor to initiate the FA pathway for repairing ICLs. Here, for the first time, we reported that And-1, a replication fork-associated protein, is essential for sensing ICL-stalled replication forks and coordinates with the FANCM complex for activation of the FA signaling by regulating the recruitment of the FANCM/FAAP24 to ICLs (Fig. 7K). We also elucidated the molecular detail of how ATR-mediated phosphorylation of And-1 leads to a favorable intramolecular change, thereby resulting in its activation and accumulation at ICL-forks for ICL repair.
The FA pathway is rapidly activated when the replication forks stall upon colliding with ICLs during S phase, indicating that fork-associated protein(s) may participate in sensing stalled forks for initiation of the FA pathway. Due to the spatial proximity of ICLs with stalled forks, fork-associated protein(s) could provide a quick and efficient response to stalled forks by facilitating the FA activation. However, such a protein linker at ICL-stalled forks has yet to be identified. Our studies demonstrate that And-1 appears to be such a fork-associated protein that quickly senses and accumulates at ICL-stalled forks, where it links ICL-stalled forks with activation of the FA signaling. The evidence that And-1 quickly accumulates at ICL-stalled forks (Fig. 2A–2C) strongly supports our model (Fig. 7K). Since And-1 binds to ICLs from early stage (5 min) until 60 min after MMC treatment (Fig. 2C), we are not surprised to observe the interaction of And-1 with the FANCM complex (FANCM/FAAP24), FANCA, and FANCD2 (Fig. 1 and Supplementary Fig. 1). In response to MMC treatment, the chromatin association of And-1 persists up to 720 minutes, which is much longer than the FANCM/FAFP24 (Supplementary Fig. S2F), suggesting that the role of And-1 in the FA signaling may not only sense ICL-stalled forks for initiation of the FA signaling, but also affect the activation and recruitment of the FA core complex. To support this notion, loss of And-1 has more robust effects on Ub-FANCD2 levels than the loss of FANCM (Supplementary Fig. S2A).
The evidence that And-1 phosphorylation at T826 is elevated in CROV cells and activated And-1 contributes to cisplatin resistance provides the clinical relevance of And-1 activity in ovarian cancer. Given that And-1 inhibition re-sensitizes CROV cells to cisplatin, targeting And-1 could be a powerful approach to treating CROV patients. Both yeast genetic study and CRISPR/Cas9 screening in human cells identified And-1 as a promising cancer therapeutic target (44,45). Moreover, it was reported that And-1 plays an important role in tumorigenesis of breast cancer, esophageal cancer, and cholangiocarcinoma (46–48). The accumulating shred of evidence collectively supports the notion that targeting And-1 is a promising strategy to treat cancers. The FA pathway has been shown to play a major role in cisplatin resistance, targeting the FA pathway is an important approach to overcoming cisplatin resistance, and many efforts have been invested to identify FA inhibitors (49–53). Recently we have discovered two potent And-1 inhibitors for cancer treatments (43). The future work to combine And-1 inhibitors and chemotherapy drugs may benefit the treatment of cisplatin resistant ovarian cancer, as well as other cancers.
Supplementary Material
Significance:
This work shows that phosphorylation of And-1 by ATR activates Fanconi anemia signaling at inter-strand crosslink-stalled replication forks by recruiting the FANCM/FAAP24 complex, revealing And-1 as a potential therapeutic target in cancer.
ACKNOWLEDGMENTS
This work was partially supported by funding from the National Institutes of Health (CA247684, CA258357 and CA184717 to W.Z.), and a grant from the McCormick Genomic and Proteomic Center. We thank Dr. Weidong Wang and Dr. Lei Li for reagents.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011;11:467–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Y, Leung JW, Jiang Y, Lowery MG, Do H, Vasquez KM, et al. FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol Cell 2013;49:997–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, et al. A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol Cell 2010;37:865–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh TR, Saro D, Ali AM, Zheng XF, Du CH, Killen MW, et al. MHF1-MHF2, a histone-fold-containing protein complex, participates in the Fanconi anemia pathway via FANCM. Mol Cell 2010;37:879–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim H, D’Andrea AD. Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev 2012;26:1393–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosselli F, Briot D, Pichierri P. The Fanconi anemia pathway and the DNA interstrand cross-links repair. Biochimie 2003;85:1175–84 [DOI] [PubMed] [Google Scholar]
- 7.Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, et al. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009;326:1698–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res 2009;668:4–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang J, Zhang J, Bellani MA, Pokharel D, Gichimu J, James RC, et al. Remodeling of Interstrand Crosslink Proximal Replisomes Is Dependent on ATR, FANCM, and FANCD2. Cell Rep 2019;27:1794–808 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rohleder F, Huang J, Xue Y, Kuper J, Round A, Seidman M, et al. FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res 2016;44:3219–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohler A, Schmidt-Zachmann MS, Franke WW. AND-1, a natural chimeric DNA-binding protein, combines an HMG-box with regulatory WD-repeats. J Cell Sci 1997;110 ( Pt 9):1051–62 [DOI] [PubMed] [Google Scholar]
- 12.Guan C, Li J, Sun D, Liu Y, Liang H. The structure and polymerase-recognition mechanism of the crucial adaptor protein AND-1 in the human replisome. J Biol Chem 2017;292:9627–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu W, Ukomadu C, Jha S, Senga T, Dhar SK, Wohlschlegel JA, et al. Mcm10 and And-1/CTF4 recruit DNA polymerase alpha to chromatin for initiation of DNA replication. Genes Dev 2007;21:2288–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka H, Kubota Y, Tsujimura T, Kumano M, Masai H, Takisawa H. Replisome progression complex links DNA replication to sister chromatid cohesion in Xenopus egg extracts. Genes Cells 2009;14:949–63 [DOI] [PubMed] [Google Scholar]
- 15.Li Y, Li Z, Wu R, Han Z, Zhu W. And-1 is required for homologous recombination repair by regulating DNA end resection. Nucleic Acids Res 2017;45:2531–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abe T, Kawasumi R, Giannattasio M, Dusi S, Yoshimoto Y, Miyata K, et al. AND-1 fork protection function prevents fork resection and is essential for proliferation. Nat Commun 2018;9:3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Liu H, Zhang H, Sun C, Hu Z, Tian Q, et al. And-1 coordinates with CtIP for efficient homologous recombination and DNA damage checkpoint maintenance. Nucleic Acids Res 2017;45:2516–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simon AC, Zhou JC, Perera RL, van Deursen F, Evrin C, Ivanova ME, et al. A Ctf4 trimer couples the CMG helicase to DNA polymerase alpha in the eukaryotic replisome. Nature 2014;510:293–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samora CP, Saksouk J, Goswami P, Wade BO, Singleton MR, Bates PA, et al. Ctf4 Links DNA Replication with Sister Chromatid Cohesion Establishment by Recruiting the Chl1 Helicase to the Replisome. Mol Cell 2016;63:371–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hao J, de Renty C, Li Y, Xiao H, Kemp MG, Han Z, et al. And-1 coordinates with Claspin for efficient Chk1 activation in response to replication stress. EMBO J 2015;34:2096–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grosschedl R, Giese K, Pagel J. HMG domain proteins: architectural elements in the assembly of nucleoprotein structures. Trends Genet 1994;10:94–100 [DOI] [PubMed] [Google Scholar]
- 22.Zhou W, Sun W, Yung MMH, Dai S, Cai Y, Chen CW, et al. Autocrine activation of JAK2 by IL-11 promotes platinum drug resistance. Oncogene 2018;37:3981–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanjana NE, Shalem O, Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 2014;11:783–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Xiao H, de Renty C, Jaramillo-Lambert A, Han Z, DePamphilis ML, et al. The involvement of acidic nucleoplasmic DNA-binding protein (And-1) in the regulation of prereplicative complex (pre-RC) assembly in human cells. J Biol Chem 2012;287:42469–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc 2006;1:1112–6 [DOI] [PubMed] [Google Scholar]
- 26.Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019;47:D442–D50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy S, Luzwick JW, Schlacher K. SIRF: Quantitative in situ analysis of protein interactions at DNA replication forks. J Cell Biol 2018;217:1521–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spanswick VJ, Hartley JM, Ward TH, Hartley JA. Measurement of drug-induced DNA interstrand crosslinking using the single-cell gel electrophoresis (comet) assay. Methods Mol Med 1999;28:143–54 [DOI] [PubMed] [Google Scholar]
- 29.Zhang H, Chen Z, Ye Y, Ye Z, Cao D, Xiong Y, et al. SLX4IP acts with SLX4 and XPF-ERCC1 to promote interstrand crosslink repair. Nucleic Acids Res 2019;47:10181–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park CY, Krishnan A, Zhu Q, Wong AK, Lee YS, Troyanskaya OG. Tissue-aware data integration approach for the inference of pathway interactions in metazoan organisms. Bioinformatics 2015;31:1093–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell 2007;25:331–43 [DOI] [PubMed] [Google Scholar]
- 32.Hartley JA, Spanswick VJ, Brooks N, Clingen PH, McHugh PJ, Hochhauser D, et al. SJG-136 (NSC 694501), a novel rationally designed DNA minor groove interstrand cross-linking agent with potent and broad spectrum antitumor activity: part 1: cellular pharmacology, in vitro and initial in vivo antitumor activity. Cancer Res 2004;64:6693–9 [DOI] [PubMed] [Google Scholar]
- 33.Andrews AM, McCartney HJ, Errington TM, D’Andrea AD, Macara IG. A senataxin-associated exonuclease SAN1 is required for resistance to DNA interstrand cross-links. Nat Commun 2018;9:2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Linding R, Jensen LJ, Diella F, Bork P, Gibson TJ, Russell RB. Protein disorder prediction: implications for structural proteomics. Structure 2003;11:1453–9 [DOI] [PubMed] [Google Scholar]
- 35.Liu W, Palovcak A, Li F, Zafar A, Yuan F, Zhang Y. Fanconi anemia pathway as a prospective target for cancer intervention. Cell Biosci 2020;10:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rocha CRR, Silva MM, Quinet A, Cabral-Neto JB, Menck CFM. DNA repair pathways and cisplatin resistance: an intimate relationship. Clinics (Sao Paulo) 2018;73:e478s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Damia G, Broggini M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers (Basel) 2019;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.D’Andrea AD. The Fanconi Anemia/BRCA signaling pathway: disruption in cisplatin-sensitive ovarian cancers. Cell Cycle 2003;2:290–2 [PubMed] [Google Scholar]
- 39.Dicks E, Song H, Ramus SJ, Oudenhove EV, Tyrer JP, Intermaggio MP, et al. Germline whole exome sequencing and large-scale replication identifies FANCM as a likely high grade serous ovarian cancer susceptibility gene. Oncotarget 2017;8:50930–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wynne P, Newton C, Ledermann JA, Olaitan A, Mould TA, Hartley JA. Enhanced repair of DNA interstrand crosslinking in ovarian cancer cells from patients following treatment with platinum-based chemotherapy. Br J Cancer 2007;97:927–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson SW, Swiggard PA, Handel LM, Brennan JM, Godwin AK, Ozols RF, et al. Relationship between platinum-DNA adduct formation and removal and cisplatin cytotoxicity in cisplatin-sensitive and -resistant human ovarian cancer cells. Cancer Res 1994;54:5911–6 [PubMed] [Google Scholar]
- 42.Johnson SW, Perez RP, Godwin AK, Yeung AT, Handel LM, Ozols RF, et al. Role of platinum-DNA adduct formation and removal in cisplatin resistance in human ovarian cancer cell lines. Biochem Pharmacol 1994;47:689–97 [DOI] [PubMed] [Google Scholar]
- 43.Li J, Zhang Y, Sun J, Chen L, Gou W, Chen CW, et al. Discovery and characterization of potent And-1 inhibitors for cancer treatment. Clin Transl Med 2021;11:e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Pel DM, Stirling PC, Minaker SW, Sipahimalani P, Hieter P. Saccharomyces cerevisiae genetics predicts candidate therapeutic genetic interactions at the mammalian replication fork. G3 (Bethesda) 2013;3:273–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Behan FM, Iorio F, Picco G, Goncalves E, Beaver CM, Migliardi G, et al. Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 2019;568:511–6 [DOI] [PubMed] [Google Scholar]
- 46.Liu B, Hu Y, Qin L, Peng XB, Huang YX. MicroRNA-494-dependent WDHDI inhibition suppresses epithelial-mesenchymal transition, tumor growth and metastasis in cholangiocarcinoma. Dig Liver Dis 2019;51:397–411 [DOI] [PubMed] [Google Scholar]
- 47.Sato N, Koinuma J, Fujita M, Hosokawa M, Ito T, Tsuchiya E, et al. Activation of WD repeat and high-mobility group box DNA binding protein 1 in pulmonary and esophageal carcinogenesis. Clin Cancer Res 2010;16:226–39 [DOI] [PubMed] [Google Scholar]
- 48.Ertay A, Liu H, Liu D, Peng P, Hill C, Xiong H, et al. WDHD1 is essential for the survival of PTEN-inactive triple-negative breast cancer. Cell Death Dis 2020;11:1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sharp MF, Murphy VJ, Twest SV, Tan W, Lui J, Simpson KJ, et al. Methodology for the identification of small molecule inhibitors of the Fanconi Anaemia ubiquitin E3 ligase complex. Sci Rep 2020;10:7959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chirnomas D, Taniguchi T, de la Vega M, Vaidya AP, Vasserman M, Hartman AR, et al. Chemosensitization to cisplatin by inhibitors of the Fanconi anemia/BRCA pathway. Mol Cancer Ther 2006;5:952–61 [DOI] [PubMed] [Google Scholar]
- 51.Landais I, Hiddingh S, McCarroll M, Yang C, Sun A, Turker MS, et al. Monoketone analogs of curcumin, a new class of Fanconi anemia pathway inhibitors. Mol Cancer 2009;8:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jacquemont C, Simon JA, D’Andrea AD, Taniguchi T. Non-specific chemical inhibition of the Fanconi anemia pathway sensitizes cancer cells to cisplatin. Mol Cancer 2012;11:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, et al. Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med 2003;9:568–74 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are included in the supplemental materials, or are available from the corresponding author upon reasonable request.