

Summary
RB’s interaction with chromatin is key to understanding its molecular functions. Here, for first time, we identify the full spectrum of chromatin-bound RB. Rather than exclusively binding promoters, as is often described, RB targets three fundamentally different types of loci (promoters, enhancers, insulators), which are largely distinguishable by the mutually exclusive presence of E2F1, c-Jun, and CTCF. While E2F/DP facilitates RB association with promoters, AP-1 recruits RB to enhancers. Although phosphorylation in CDK-sites is often portrayed to release RB from chromatin, we show that the cell-cycle redistributes RB so that it enriches at promoters in G1, and at non-promoter sites in cycling cells. RB-bound promoters include the classic E2F-targets and are similar between lineages, but RB-bound enhancers associate with different categories of genes and vary between cell types. Thus, RB has a well-preserved role controlling E2F in G1, and it targets cell type-specific enhancers and CTCF-sites when cells enter S-phase.
Graphical Abstract
eTOC:
Sanidas et al. show that, rather than exclusively targeting E2F-promoters, RB associates with specific groups of promoters, enhancers, and insulators to target different sets of genes. Cell cycle progression redistributes RB towards cell type-specific enhancers. PanChIP software confirms that RB associates with distinct transcription factors at different types of loci.
Introduction
RB was one of the first tumor suppressors to be identified; it is widely expressed in normal cells but is functionally inactivated in cancers. RB’s mechanism of action is often described using a simple model: RB associates with E2F-regulated promoters and blocks cell cycle progression in G1 by suppressing transcription of cell cycle genes (Weinberg, 1995). This property is cell cycle-dependent; active RB is generated when cells accumulate in G1, during senescence, or during checkpoint-mediated cell cycle arrest. Mitogenic signals activate cyclin-dependent kinases (CDKs) that phosphorylate RB and prevent its repression of E2F. In this way, RB phosphorylation increases E2F-mediated transcription and promotes cell cycle progression (Dyson, 1998; Kent and Leone, 2019; Knudsen and Knudsen, 2008; Rubin et al., 2020). Thus, when cells proliferate, RB is thought to oscillate between active (un-phosphorylated) and inactive (hyper-phosphorylated) states.
Sadly, this textbook description of the canonical activity of RB cannot easily explain many elements of the RB literature. In particular, this model does not explain why RB’s tumor suppressor activity is tissue-specific and context-specific. There is much evidence that RB has significant roles beyond its periodic regulation of E2F (reviewed in (Dick et al., 2018; Dyson, 2016)). In addition to E2F, multiple transcription factors (TFs) are reported to physically interact with RB (Hagemeier et al., 1993; Kim et al., 1992; Konishi et al., 1999; Nead et al., 1998; Wang et al., 1993) and signatures associated with RB loss suggest that RB can increase, or decrease, gene expression (Chen et al., 2019; Ertel et al., 2010; Markey et al., 2007). Notably, RB causes context-specific activation at genes that are important for cell differentiation (Calo et al., 2010; Thomas et al., 2001). It also appears that some activities of RB are not inactivated by CDKs (Avni et al., 2003; Ianari et al., 2009; Ishak et al., 2016; Wells et al., 2003). RB has been reported to repress the expression of repetitive genomic regions (Ishak et al., 2016), to promote genomic stability during chromosome segregation (Hernando et al., 2004; Manning et al., 2010; Manning et al., 2014), to regulate metabolic flux and mitochondrial respiration (Nicolay et al., 2015; Sanidas et al., 2019), and to associate with DNA breaks to stimulate repair (Cook et al., 2015; Velez-Cruz et al., 2016). Analysis of the impact of RB loss on the development of prostate and lung cancers shows that RB is important for cells to preserve lineage fidelity (Ku et al., 2017; Mu et al., 2017; Niederst et al., 2015; Walter et al., 2019); the loss of this activity is critical during malignant progression. Clearly, the models for RB’s mechanism of action need to be updated, and new models are needed that incorporate both canonical and non-canonical activities of RB.
A critical gap in the literature is the paucity of information about the genomic distribution of RB. ChIP-sequencing (ChIP-seq) analysis of human RB is technically difficult, with experiments giving low signal and high background. Consequently, studies of RB-associated loci have focused primarily on the promoters of E2F target genes where RB binds tightly. Here, RB-binding fluctuates with growth conditions (Chicas et al., 2010). It is evident though that the conventional E2F-regulated promoters do not give a full picture of RB action. Studies focusing on specific target genes described RB association with non-E2F regulatory elements (Gonzalez-Vasconcellos et al., 2017; Kareta et al., 2015). Interestingly, the inactivation of RB by viral proteins does not prevent RB association with chromatin but redistributes its repressor activity, in a p300/CBP-dependent manner, towards genes involved in interleukin and interferon signaling (Ferrari et al., 2014). But, beyond the highest-affinity binding sites, the full distribution of RB is unclear. The recent identification of functionally distinct, mono-phosphorylated isoforms of RB (mP-RBs) (Burke et al., 2012; Burke et al., 2014; Narasimha et al., 2014; Sanidas et al., 2019) shows that RB’s mechanism of action is more complex than previously appreciated and underscores the need for a comprehensive picture of the genomic distribution of RB.
Here, we used an inducible replacement system to circumvent the technical difficulties associated with RB ChIP and generated high-quality ChIP-seq data for wild-type RB, for unphosphorylated RB, and for the 14 previously described mP-RBs (Narasimha et al., 2014; Sanidas et al., 2019). These profiles give a detailed picture and change our understanding of where RB acts and how it is regulated. The results show that, rather than exclusively targeting promoters, RB associates with specific subsets of promoters, enhancers, and insulators and it is recruited to these different locations by distinct factors. RB’s interaction with DNA repeats is minimal, and our data does not support a report that RB targets repetitive sequences (Ishak et al., 2016). During cell cycle progression, RB is not released from chromatin, as often described, but instead re-distributes from high-affinity sites (mostly in promoters) to lower-affinity sites (mostly in enhancers and insulators). The non-promoter loci bound by RB vary greatly between cell lines. The additional binding sites are significant since RB depletion alters the expression of genes with RB-bound enhancers. We suggest that RB-binding sites in non-promoter regions do not just represent newly discovered sites of RB action and previously unappreciated pools of chromatin-associated RB, but they provide a simple mechanistic explanation for context-specific roles of RB.
Results
RB associates with both promoter and non-promoter regions in the human genome.
To obtain a detailed picture of the genomic distribution of RB, we generated ChIP-seq profiles from human retinal pigment epithelial cells (RPE1) engineered to express specific RB forms. After 48 hours of doxycycline treatment, endogenous RB was replaced in these cells by similar levels of exogenous FLAG-tagged wild-type RB, or by RBΔCDK (a constitutively active allele in which all known in vivo CDK phosphorylation sites are mutated to alanine), or by any one of 14 mP-RB alleles that contain just a single intact CDK phosphorylation site (Sanidas et al., 2019). Consistent with their ability to suppress E2F transcriptional programs (Sanidas et al., 2019), RBΔCDK and all mP-RBs associated tightly with the promoters of E2F-regulated genes (Fig. S1A). Aggregation of ChIP-seq data for all RB proteins revealed 28,115 RB peaks with fold change enrichment score >2 relative to the maximum background signal. The RB peaks were distributed almost evenly across all RB phosphorylation isoforms (Fig. S1B). To seek chromatin regions that were preferentially bound by a specific phosphorylation form, and to measure differences in the number of the peaks between isoforms, we systematically increased the peak-calling threshold. This reduced the number of peaks (for example, 23,761 peaks had an enrichment score relative to background > 4 (Fig. 1A)) but it did not reveal a substantial fraction of sites that were uniquely bound by a specific isoform, or by a subgroup of isoforms. Thus, in general, mono-phosphorylation does not target RB to unique sites.
Fig. 1 : RB associates with both promoter and non-promoter regions.
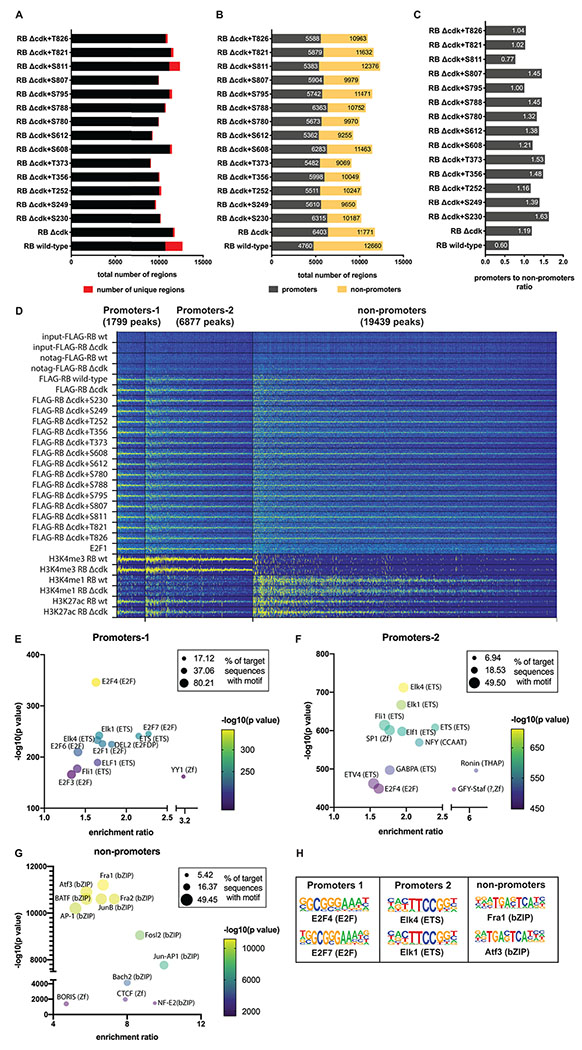
(A) Summary of RB ChIP-seq data, showing the number of peaks identified in RPE1 cells expressing each one of the 16 RB phosphorylation isoforms, counting peaks with an enrichment score of at least 4-fold above the maximum background signal from Input and No-FLAG-tag controls (see STAR Methods). Unique peaks per isoform are shown in red. (B) Breakdown of RB ChIP-seq peaks from panel A by occurrence in promoters (dark grey) or non-promoters (yellow). Left number is promoter peaks and right number is total peaks. (C) Promoter/non-promoter ratio of RB ChIP-seq peaks from panel A. (D) k-means clustering analysis (k=3) of RB ChIP-seq data from RPE1 cells. ChIP-seq profiles from RB wild-type, RBΔCDK, histone modifications (H3K4me3, H3K27ac, H3K4me1) were grouped by k-means clustering. This analysis identified two clusters of RB ChIP-seq peaks in promoter regions (H3K4me3-positive) and one cluster of RB peaks in non-promoter regions (H3K4me3-negative and H3K4me1-positive). RB ChIP-seq data from mP-RB isoforms as well as E2F1 ChIP-seq data are shown. Data is shown in a 10-kb window centered on each RB peak and includes all peaks with an enrichment score of at least 2-fold above maximum background. (E–G) Bubble plots showing the most significantly enriched HOMER motifs in the RB ChIP-seq peaks from the Promoters-1, Promoters-2 and non-promoters clusters in panel D. (H) DNA sequence motifs of the top two most significantly enriched motifs in panels E-G.
We identified RB peaks located in presumptive promoter regions (−1 kb to +100 bp from transcriptional start sites (TSS)). Interestingly, this suggested that 70% of RB peaks were not in promoters but were instead in intragenic and intergenic regions (Fig. 1B, S1C, and S1D). Focusing on 23,761 strong RB peaks (relative enrichment score >4) we noted that the distribution of RB between promoters and nonpromoter regions was phosphorylation-dependent. Wild-type RB, which can be phosphorylated at any of its 14 CDK sites, bound mostly to non-promoter regions (62%), whereas RBΔCDK and 12 of the 14 mP-RBs bound preferentially to promoters (Fig. 1B and 1C). The position of the phosphorylation site influenced the distribution of these forms of ‘active’ RB between promoters and other regions (Fig. 1C). Differential binding analysis (Ross-Innes et al., 2012) confirmed this and clustering of mP-RB’s using ChIP-seq data (Fig. S1E) gave a pattern that closely resembles the clustering of mP-RB’s based upon the proteins that they associate with (Sanidas et al., 2019). RB peaks in promoter regions were overrepresented in the top 9% strongest RB peaks (n = 2419) that had an enrichment score >10 (Fig. S1D); overall, RB associated more strongly with promoters than non-promoter regions.
To characterize the chromatin locations bound by RB, we examined the distribution of histone marks typical for promoters (H3K4me3) or enhancers (H3K4me1) and used H3K27ac to identify transcriptionally active chromatin. Histone marks were profiled in RPE1 cells expressing wild-type RB or RBΔCDK. k-means clustering analysis based on the histone marks and the peaks of wild-type RB and RBΔCDK subdivided the RB-bound regions into three distinct groups (Fig. 1D). Two groups contained RB peaks located in promoter regions that were enriched in H3K4me3 but differed in RB ChIP-sequencing signal (mean peak intensity per cluster (Fig. S1F and S1G)), with strong RB peaks in Promoters-1, and more moderate RB peaks in Promoters-2. A third group contained RB peaks that were located mostly in H3K4me1-enriched enhancer regions (Non-promoters). Wild-type RB and RBΔCDK bound to sites in each group but unphosphorylated RB showed stronger binding to promoters (Fig. S1F and S1G). Projection of the ChIP-seq data for all 14 mP-RBs into the same clusters indicated that each mono-phosphorylated form associated with similar loci in both promoters and non-promoters (Fig. 1D). E2F1 ChIP-seq in RPE1 cells expressing wild-type RB showed that most RB-bound promoters were occupied by E2F1, but RB-bound enhancers were not (Fig. 1D; Figure S1H).
HOMER software (Heinz et al., 2010) was used to identify TF binding motifs enriched in each category of RB peaks. As expected, E2F motifs were strongly enriched in Promoters-1 (Fig. 1E, 1H and Table S1). Unexpectedly, E2F motifs were not the most significantly overrepresented motifs in Promoters-2. Instead, these peaks were more significantly enriched in motifs bound by other TFs, including TFs previously reported to associate with RB, such as SP1 (Kim et al., 1992) and ETS family members (Wang et al., 1993) (Fig. 1F, 1H and Table S1). Gene-set enrichment analysis (GSEA) indicated that the two groups of RB-bound promoters regulate genes involved in distinct cellular functions (Fig. S1I and Table S2).
Strikingly, RB-bound enhancers were not significantly enriched in any of the motifs strongly enriched in Promoters-1 or -2. Instead, the non-promoter RB-binding sites were enriched for motifs bound by the Activation Protein-1 (AP-1) family of TFs, or by CTCF and the CTCF-like protein BORIS (Fig. 1G, 1H, and Table S1). An RB interaction with AP-1 proteins, including c-Jun, has been reported to enhance transcription (Nead et al., 1998; Nishitani et al., 1999), a finding consistent with reports that RB can act either as a repressor or an activator of gene expression (Markey et al., 2007; Sanidas et al., 2019). GSEA analysis of the genes in proximity to RB-bound non-promoter regions identified genes involved in signaling pathways and epithelial-to-mesenchymal transition as potential targets for RB-mediated regulation (Fig. S1I and Table S2). This list differs dramatically from the genes regulated by RB-bound promoters. Low signal-to-background ratio previously precluded the identification of many non-promoter RB-binding sites (Fig. S1J and S1K).
AP-1 and E2F/DP1 recruit RB to different genomic locations.
To better understand the RB-binding sites and to corroborate the motif analysis results therein, particularly those located outside promoter regions, we generated additional ChIP-seq profiles from RPE1 cells. We focused on two TFs implicated by the most significant motif enrichment in non-promoter RB-binding sites: c-Jun, a core component of the AP-1 complex, and CTCF. Subsequent k-means clustering analysis (k=8) of the wild-type RB and RBΔCDK peaks with histone marks, E2F1, c-Jun, and CTCF provided a greatly improved level of resolution (Fig. 2A). This confirmed that E2F1, c-Jun, and CTCF are present at RB-bound loci; indeed, these three TFs occupied 95% of RB-bound loci. Remarkably, the three TFs displayed binding at RB-bound loci that was largely mutually exclusive, suggesting that the TFs mark three different types of RB-binding sites.
Fig. 2: AP-1 and E2F/DP1 mediate RB association with distinct classes of chromatin region.
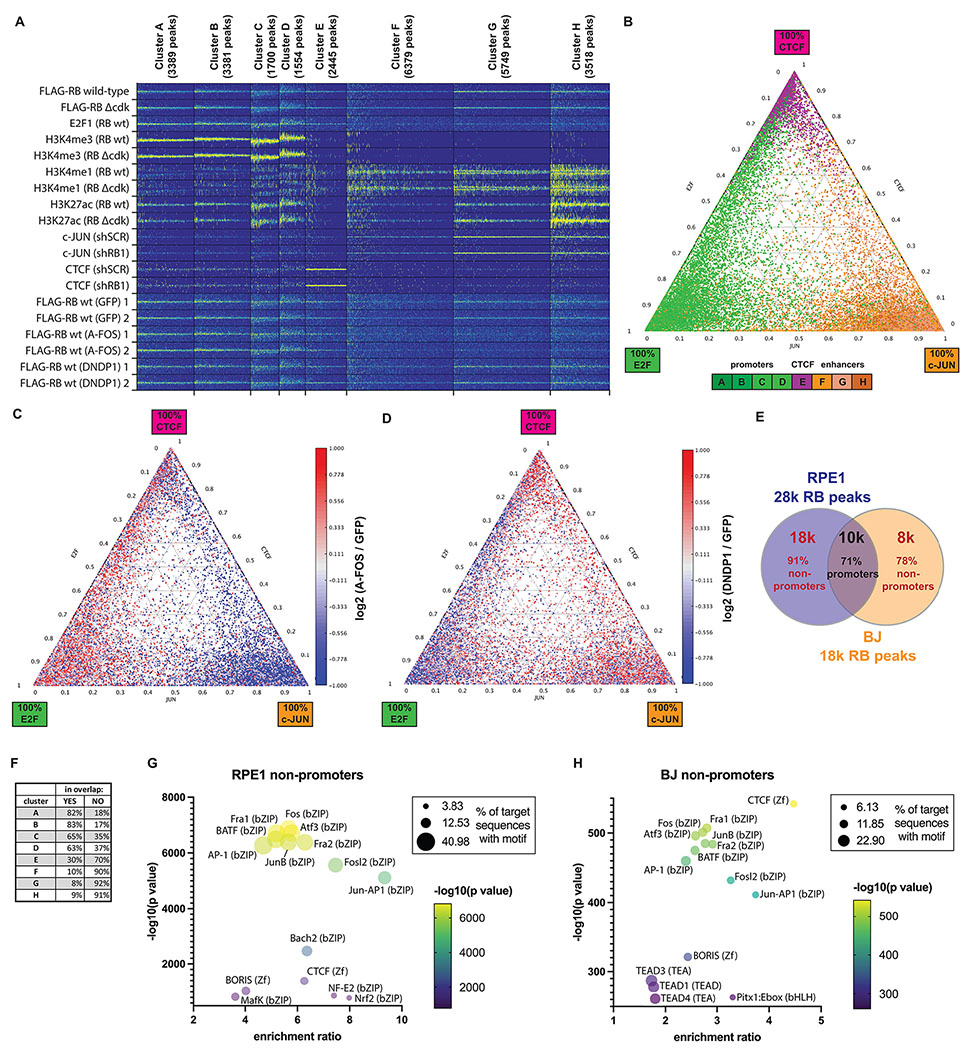
(A) k-means clustering analysis (k=8) of RB ChIP-seq data from RPE1 cells. ChIP-seq from RB wild-type, RBΔCDK, histone modifications (H3K4me3, H3K27ac, H3K4me1), E2F1, c-Jun, and CTCF were grouped by k-means clustering. ChIP-seq profiles for RB wild-type in cells expressing either dominant-negative FOS (A-Fos), dominant-negative DP1 (DNDP1), or GFP (control) are also shown. Peaks are shown in 10-kb windows centered on each RB ChIP-seq peak. (B) Ternary plot showing the 28,115 RB peaks, color-coded based on their group in panel A; green for promoter clusters A–D, orange for enhancer clusters F–H, and purple for the CTCF cluster E, respectively. The percentage of E2F1, c-Jun, and CTCF ChIP-seq signal for each RB-bound chromatin locus is shown in the axes. (C–D) Heatmaps of the log2 difference (red = increase, blue = decrease) in RB ChIP-seq signal between RPE1 cells expressing A-Fos versus GFP control (C) and DNDP1 versus GFP (D). The data is shown for each RB-bound locus presented in the ternary plot in panel B. (E) RB-binding sites conserved in both RPE1 and BJ cells are strongly associated with promoters. The number of RB ChIP-seq peaks was 28,115 for RPE1 cells and 18,237 for BJ cells. 71% of RB ChIP-seq peaks conserved in both cell lines were located in promoter regions. In contrast, RB-bound non-promoter regions were poorly conserved. (F) Conservation of each cluster of RB ChIP-seq peaks from panel A. The percentage of RB ChIP-seq peaks in BJ cells that are conserved in RPE1 cells is shown. (G–H) Bubble plots showing the most significantly enriched HOMER motifs in RB-bound non-promoter regions in RPE1 (G) and BJ (H) cells.
Clustering analysis with k=8 (Fig. 2A) revealed four groups of RB-bound promoters (clusters A–D), three groups of RB-bound enhancers (clusters F–H), and one group of RB/CTCF sites (cluster E). RB-bound promoters (A–D) were highly enriched in H3K4me3 and H3K27ac and showed extensive occupancy by E2F1, but not by c-Jun or CTCF. The differences between the four groups of RB-bound promoters stem from the shape and the directionality of the peaks (RB gave sharp peaks in clusters A and B; peaks in clusters C and D were broader and often had a shoulder (Fig. S2A, upper track)). All four groups of RB-bound promoters were associated with cell-cycle genes. Unexpectedly, the RB-bound promoters in clusters A and B were particularly enriched for genes involved in RNA metabolism and ribosomal biogenesis. Indeed, the genes associated with each cluster of RB-bound promoters had a distinct functional bias (Gene Ontology (GO) analysis is shown in Table S3).
In agreement with HOMER motif analysis, E2F1 ChIP signal was much lower at the four clusters of RB-bound non-promoter regions (E–H). c-Jun peaks overlapped with RB peaks in enhancer regions enriched in H3K4me1 and H3K27ac (F–H). These clusters of RB-bound enhancers differed in c-Jun binding, and the level of c-Jun enrichment correlated with the active state of chromatin (H3K27ac) (Fig. 2A). In contrast to RB-bound promoters, genes in proximity to RB-bound enhancers were involved in cell morphogenesis, motility, and signal transduction pathways (Table S3). CTCF was enriched at an additional cluster of RB-bound non-promoter sites (E). These RB-binding sites generally lacked E2F1 or c-Jun, and they were not consistently decorated with any of the histone marks interrogated (Fig. 2A). Overall, 18% of CTCF peaks identified in RPE1 cells were bound by RB. Many genes involved in neurogenesis and neuronal differentiation are located in proximity to RB/CTCF loci (Table S3).
To visualize the distribution of RB-binding sites we calculated the relative strength of E2F1, c-Jun, and CTCF ChIP-seq signal at each one of the 28,115 RB-bound chromatin loci and displayed these on a ternary plot in which the position of each dot indicates the relative binding signal for each of the three TFs at a single RB peak (Fig. 2B). Most dots locate close to the corners, indicating that most RB-binding sites are preferentially bound by just one of the three factors. In agreement with the clustering analysis, most RB-promoter peaks (green; clusters A–D) are primarily enriched for E2F1, most RB/CTCF peaks (purple; cluster E) are concentrated in CTCF, and most RB-enhancer peaks (orange; clusters F–H) are primarily bound by c-Jun. There are smaller numbers of sites near the edges of the distribution that sit between the corners and are bound by two TFs. This is most evident in the subset of sites in RB-bound promoters (green) that are bound by both E2F1 and CTCF. Strikingly, there are almost no sites in the center of the distribution, indicating that binding by all three TFs is extremely rare.
With this framework, we asked how the distribution of RB changes when the DNA-binding activity of AP-1 or E2F was reduced. To inactivate the DNA-binding activity of AP-1, we expressed a doxycycline-inducible dominant-negative protein, A-Fos (Olive et al., 1997), in RPE-1 cells expressing wild-type RB or RBΔCDK (Fig. S2B). A-Fos expression inhibited CCND1 expression, RB phosphorylation, and cell cycle progression (Fig. S2B). The effect of A-Fos on RB association with chromatin is shown in Fig. 2C, where the RB peaks in the ternary plot from Fig. 2B have been colored according to the change in RB ChIP-seq signal at each site in cells expressing A-Fos relative to GFP-expressing control cells (Fig. 2C). A-Fos specifically reduced RB binding to AP-1 enhancers (note a cluster of blue dots in the 100% c-Jun corner) and redistributed RB towards promoter regions (the cluster of red dots in the 100% E2F corner). These changes were confirmed in biological replicate experiments, conducted to ensure robustness to variations caused by the double infection and selection process (Fig. 2A and S2A, tracks 8–9). In contrast to A-Fos, expression of a dominant-negative DP1 protein (DNDP1) that reduces DNA binding by E2F/DP heterodimers (Wu et al., 1996), preferentially reduced RB binding to promoter sites (Fig. 2D, also Fig. 2A and S2A, tracks 10–11), and increased RB binding to enhancer regions. The reciprocal effects of A-Fos and DNDP1 on RB distribution are consistent with the differential enrichment of E2F-binding sites and AP-1 binding sites in promoters and enhancers, respectively.
Additional ChIP-seq experiments in control and RB-depleted cells showed that depletion of RB did not change the overall level of c-Jun or CTCF bound to these sites (Fig. S2A, tracks 5–6), indicating that RB is not generally required for recruitment of AP-1 or CTCF. Aggregate curves of the ChIP signal in the different RB-bound chromatin clusters showed that expression of the constitutively active RBΔCDK and the resulting accumulation of cells in G1 did not influence the levels of H3K4me3 in promoters or H3K4me1 in enhancers (Fig. S2A, tracks 3–4). In contrast, RBΔCDK significantly altered the transcriptionally active chromatin, as judged by H3K27ac. Changes in H3K27ac were seen at both promoters and enhancers but, unexpectedly, in opposite directions; RBΔCDK reduced H3K27ac at RB-bound promoters and increased H3K27ac at RB-bound enhancers (Fig. S2A, 2nd track).
Taken together, these results indicate that RB is recruited to three fundamentally different types of chromatin loci: promoter sites where RB colocalizes with E2F1, enhancer sites where RB colocalizes with c-Jun, and a third set of sites that are marked by CTCF. RB binding to distinct chromatin regions appears to be mediated by distinct TFs: E2F promotes RB binding to promoters, while AP-1 controls RB’s interaction with enhancers.
RB association with enhancer regions is cell-type-specific.
To ask how the distribution of RB compares in a different cell type, we introduced the RB replacement system into BJ human fibroblasts and replaced the endogenous RB with exogenous FLAG-tagged wild-type RB or RBΔCDK. FLAG-RB ChIP-seq profiling identified 18,237 RB peaks in BJ cells with >2 fold change enrichment score relative to the maximum background signal in input samples. Presumptive promoter regions were identified (−1 kb to +100 bp from TSS), and 52% of RB peaks in BJ cells were found to be in promoters, while 48% (8,823) were in non-promoter regions.
Fig. 2E and Fig. S2D summarize the RB-binding sites identified in the two cell types. 54% of RB peaks in BJ cells were also found in RPE1 epithelial cells (Fig. S2D, left). Interestingly, 71% of the common RB peaks were located in promoter regions. RB’s association with non-promoter regions varied far more between cell types, with 91% and 78% of unique RB peaks located in non-promoter regions in RPE1 and BJ cells, respectively (Fig. 2E). When we asked how the 9,821 RB peaks shared by BJ and RPE1 cells were distributed among the eight clusters defined in Fig. 2A (Fig. 2F and S2E; two bottom tracks) we found extensive overlap in RB-bound promoters (clusters A–D). Among the RB-bound nonpromoter regions, RB/CTCF loci (cluster E) were the best conserved (30%). The overlap of RB peaks in AP-1 enhancers (clusters F–H) was less than 10% (Fig. 3F). Similarly, classification of RB-bound promoters by E2F motif content (as described in Fig. 1D) showed that E2F-bound promoters (cluster 1) are the most conserved RB-bound loci between fibroblasts and epithelial cells (Fig. S2D, right).
Fig. 3: When cells accumulate in G1, RB redistributes towards E2F promoters.
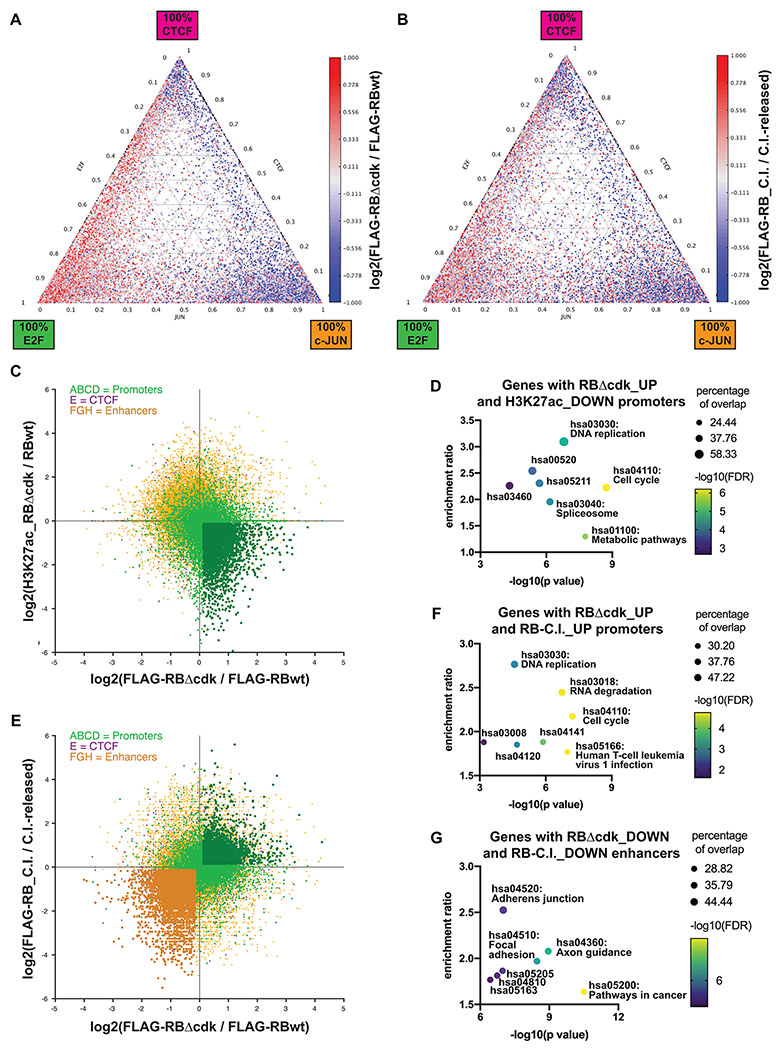
(A–B) Heatmaps of the log2 difference in RB ChIP-seq signal between RPE1 cells expressing RBΔCDK versus wild-type RB (A) and between those arrested in G1 by contact inhibition versus released into S-phase by contact inhibition relief (B). Data is shown for each RB-bound locus presented in the ternary plot in Figure 2B. (C) Scatter plot of RB ChIP-seq peaks. The axes show the log2 differences in RB (x-axis) and H3K27ac (y-axis) ChIP-seq signal in RPE1 cells expressing RBΔCDK versus wild-type RB. RB promoter peaks (clusters A–D) are indicated in green, RB enhancer peaks (clusters F–H) in orange, and RB/CTCF peaks (cluster E) in purple. (D) KEGG pathway analysis of the genes associated with RB-bound promoters identified in panel C. (E) Scatter plot of RB ChIP-seq peaks. The axes show the log2 differences in RB ChIP-seq signal between cells expressing RBΔCDK versus wild-type RB (x-axis) and between those accumulated in G1 by contact inhibition versus S-phase by contact inhibition relief (y-axis). (F–G) KEGG pathway analysis of the genes that are consistently up-regulated and associated with RB-bound promoter activity (panel F) and genes that are consistently down-regulated and associated with RB-bound enhancer activity (panel G).
Motifs enriched in non-promoter RB peaks of RPE1 and BJ cells were identified by HOMER. Although RB-bound non-promoter loci were mostly cell-type-specific, the sets of TFs enriched at these RB-binding sites were strikingly similar (Fig. 2G, 2H and Table S4). AP-1 motifs were highly enriched in both RPE1 and BJ cells. Although CTCF and BORIS motifs were overrepresented in RB-bound nonpromoter regions in both cell lines, CTCF was the most significantly enriched motif in BJ cells. Interestingly, motifs for TEAD family TFs were specifically enriched in fibroblasts.
These results suggest that RB regulation of the E2F transcriptional program is well-conserved. RB-binding sites beyond E2F-bound promoters occur in both cell types; these sites are associated with similar TFs, but the locations of the sites are mostly cell-type-specific.
Accumulation in G1 phase reduces RB at enhancers.
Next, we examined the effects of cell cycle progression on the distribution of chromatin-bound RB. To do this we compared the RB ChIP-seq signal at each binding site in cells expressing RBΔCDK (arrested in G1) with the RB signal in cells expressing wild-type RB (actively proliferating). To visualize the changes, RB-bound chromatin loci were marked with red or blue to indicate increase or decrease, respectively, in the abundance of RB protein in G1-arrested cells relative to proliferating cells. The colors were projected onto the ternary plot showing the distribution of RB-binding sites according to their relative affinities for E2F1, CTCF, and c-Jun (Fig. 3A). In RBΔCDK-expressing cells, the RB ChIP-seq signal specifically increased at E2F1-bound loci (+16%; P=1.0×10−40, Welch’s t-test). In contrast, RB association with c-Jun-bound chromatin regions generally decreased when cells accumulated in G1 (−25%; P=2.5×10−40, Welch’s t-test). While the association of RB with CTCF-bound loci was less dependent on cell cycle, RB ChIP-seq signal reduced at most CTCF regions in RBΔCDK-expressing cells. Overall, the changes indicate a redistribution of RB towards promoters, and away from enhancers, in RBΔCDK G1-arrested cells. Of note, the direction of the cell cycle–dependent redistribution of RB coincided with the opposite changes in H3K27ac between RB-bound promoters and enhancers when cells express active RBΔCDK (Fig. S2A, track 2).
To validate that the re-distribution of RB reflected cell cycle and was not an artifact of the mutant RB allele, we performed a similar analysis on RB ChIP-seq data from RPE1 cells expressing wild-type RB that were arrested in G1 by contact inhibition (CI) for 6 days, compared to RB ChIP-seq data from the same cells released from CI for 24 hours and accumulated in S-phase (Fig. S3A and S3B). Again, when cells were arrested in G1 we observed reduced RB ChIP-seq signal in c-Jun-bound enhancers, and increased abundance of RB protein in E2F1-bound promoters (Fig. 3B). The redistribution of RB upon cell proliferation was confirmed by additional ChIP-quantitative PCR experiments (Fig. S3C, S3D and S3E). This finding is consistent with previous studies reporting that RB hyperphosphorylation reduced the affinity of RB for E2F1-bound promoters (Chellappan et al., 1991; Chicas et al., 2010), but reveals an important distinction: our data show that, rather than being lost from chromatin, RB redistributes to a different set of binding sites when it is hyperphosphorylated.
In RPE1 cells, RB is exclusively mono-phosphorylated in G1 (Narasimha et al., 2014). We therefore assessed the impact of mono-phosphorylation on RB distribution and compared the RB ChIP-seq signal for each of the 14 mP-RB isoforms with wild-type RB and RBΔCDK. Similar to RBΔCDK, all mP-RBs showed stronger binding to promoter regions and weaker binding to non-promoter regions, relative to wild-type RB (Fig. S3F). However, the distribution of RB was not equally affected by all 14 CDK phosphorylation sites (Fig. S3G). We conclude that mono-phosphorylation alters the distribution of RB between types of sites.
To identify genes most affected by changes in RB distribution, we selected RB-binding sites that showed the strongest increase in RB ChIP-seq signal and the strongest decrease in H3K27ac ChIP-seq signal in cells expressing RBΔCDK relative to wild-type RB (Fig. 3C). These sites were predominantly located in promoters. As expected, GO and KEGG pathway analyses of the genes regulated by these 4,290 RB-bound promoters showed an enrichment of DNA replication and cell cycle genes. The RB-regulated promoters also associated significantly with genes involved in RNA processing and spliceosome formation (Fig. 3D and Table S5). In a complementary analysis, we selected RB-bound loci that gained RB ChIP-seq signal upon G1-arrest in both RBΔCDK and CI cells (Fig. 3E), yielding a total of 3,482 promoters. GO and KEGG pathway analyses of the genes regulated by these promoters highlighted similar cellular functions (Fig. 3F and Table S5), including those involved in RNA processing. Genes to which RB redistributes after passing the G1/S checkpoint in RPE1 cells were identified by selecting enhancer sites that exhibited less RB binding upon G1-arrest (Fig. 3E). G1-arrest was performed either by RBΔCDK expression or CI, and enhancer sites implicated by both methods were selected. This yielded 5,360 enhancer sites (Fig. 3E). GO and KEGG pathway analyses of genes in proximity to these enhancers showed significant enrichment of genes involved in cell adhesion, motility, and migration and several mitogenic signaling pathways (Fig. 3G and Table S5). Thus, when RB redistributes from promoters to enhancers during cell cycle progression, it switches from the regulatory elements of one set of genes to target the regulatory elements of a completely different set of genes.
RB regulates enhancers of genes involved in the MAPK pathway.
What is the consequence of RB recruitment to enhancers? The answer is expected to be complex because genes can have multiple enhancers that exhibit varying, and often context-specific, effects on transcription (Roadmap Epigenomics et al., 2015). Moreover, the activity of AP-1-bound enhancer elements is known to vary greatly between cell lineages, and to affect multiple cellular pathways (Phanstiel et al., 2017; Vierbuchen et al., 2017).
To answer this question, we followed two strategies. First, we focused on RPE1 cells and looked for RB-bound enhancers in which the H3K4me1 enhancer mark and c-Jun were RB-dependent. We generated H3K4me1 and c-Jun ChIP-seq profiles in CRISPR/Cas9-induced RB knock-out RPE1 cells (Nicolay et al., 2015) and identified a set of RB-bound enhancers that had decreased levels of both marks in RB knock-out cells relative to wild-type. In this way, we identified 741 RB-bound enhancers that lost at least 50% of both c-Jun and H3K4me1 ChIP-seq signal in RB knock-out cells (Fig. 4A and Table S6). GO analysis of the 660 unique genes linked to these enhancers showed a very strong enrichment of genes involved in the MAPK signaling pathway (Fig. 4B and Table S7).
Fig.4: RB regulates the expression of genes located in proximity to RB-bound enhancers.

(A) Scatter plot of RB-bound enhancer sites (clusters F–H of Fig. 2A). Axes show the log2 differences in H3K4me1 (x-axis) and c-Jun (y-axis) ChIP-seq signal between RB knock-out and RB wild-type RPE1 cells. RB-bound enhancers that lost at least 50% c-Jun and 50% H3K4me1 ChIP signal in RB knock-out cells are highlighted with dark orange color. (B) Gene Ontology analysis of the 660 genes linked to 741 RB-bound enhancers identified in panel A. (C) Left: Volcano plots representing the log2 fold change in mean expression level of protein-coding genes and −log10FDR for the differences between RB1null and CDKN2Adel cell lines. Transcripts from genes with RB-bound promoters (upper plots) and RB-bound enhancers (lower plots) are shown. Right: z-score for each gene set associated with RB-bound promoters or enhancers. z-score was assessed in the combined data set consisted of RB1null and CDKN2Adel cell lines, p-values were assessed based on an unpaired two-tailed Welch’s t-test and were adjusted for multiple hypotheses by Bonferroni correction. (D) Gene Ontology analysis of genes with RB-bound enhancers that are upregulated in CDKN2Adel cell lines. (E–F) ChIP-seq tracks for the indicated proteins or histone modifications, showing the EGFR (panel E) and TGFBR2 (panel F) loci. (G–H) Expression levels of EGFR (panel G) and TGFBR2 (panel H) in four lung cancer cell lines (NCI-H2030, CALU-1, HOP62, SW1573) transduced with either shRB1 (red) or shScramble (black) following the time course of a treatment with 30 nM trametinib (n=3 per sample, unpaired two-tailed Welch’s t-test with Bonferroni correction, ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05).
Taking an independent approach, we used the cancer cell line encyclopedia (CCLE) database (Barretina et al., 2012) to examine the differential gene expression between 63 RB1null and 318 RB1 wild-type (CDKN2Adel) cell lines (Fig. S4A–D). As expected, the predominant change in the expression of genes with RB-bound promoters was an up-regulation in RB1null cells compared to RB1 wild-type (CDKN2Adell) cells (Fig. 4C, upper). Although genes in proximity to RB-bound enhancers were also differentially expressed in RB1null compared to CDKN2Adel cell lines, these changes were moderate and more varied than genes with RB-bound promoters (Fig. 4C, bottom). Nevertheless, we saw two groups of outliers, one group that is up-regulated and another group that is down-regulated in RB1null cells. GO analysis of the genes in proximity to RB-bound enhancers that were upregulated in RB1null cancer cell lines showed enrichment of genes involved in cell morphogenesis and neuronal development (Table S8). This may reflect the fact that many of the RB1null cancer cells have neuroendocrine characteristics (Dick et al., 2018). In contrast, the genes with RB-bound enhancers that are up-regulated in CDKN2Adel cells were overrepresented in gene sets related to cell adhesion and protein-kinase signaling pathways, including the MAPK pathway (Fig. 4D and Table S8).
The fact that both approaches highlighted the MAPK signaling pathway is striking; MAPK signaling activity is inhibited upon RB loss in a KRAS/p53 mouse model of lung adenocarcinoma (Walter et al., 2019). We tested several of the MAPK pathway genes identified by bioinformatics analyses (Fig. 4A–D) to identify candidates for RB-mediated enhancer activity in KRAS-mutant lung adenocarcinoma cell lines (Fig. S4E). The results show that RB can either activate or repress transcription of genes with RB-bound enhancers and that its effects are cell line-dependent (Fig. S4F). Importantly, we found that two well-known activators of MAPK pathway, EGFR and TGFBR2, are responsive to RB. In both cases, RB associates with enhancer regions in proximity to the gene, colocalizing with c-Jun, but not with E2F1 (Fig. 4E and 4F). Both genes have elevated expression in the cohort of RB wild-type (CDKN2Adel) cell lines (Fig. 4C and Table S8). The expression of both EGFR and TGFBR2 was maintained in wild-type RB cells relative to RB-depleted cells (Fig. S4F). Since most of RB is hyper-phosphorylated in KRAS-mutant cell lines, we examined the impact of RB on the expression of EGFR and TGFBR2 when cells were treated with MEK inhibitors. Trametinib induced the dephosphorylation of RB and, in an RB-dependent manner, suppressed the expression of both EGFR and TGFBR2 at multiple timepoints following drug treatment (Fig. 4G, 4H and S4G) in each of the lung cancer cell lines tested. This data supports the model that phosphorylated RB binds to AP-1-bound enhancers and alters the expression of target genes. We note, though, that the effects of RB on these targets is cell-type-specific and context-dependent.
Pan-ChIP-seq identification of proteins that colocalize with RB in promoters, enhancers, and insulators
The discovery that RB has different classes of binding sites adds a new dimension to the concept that RB interacts with diverse TFs. Three categories of RB-binding sites can be distinguished by the presence of E2F1, CTCF, and c-Jun, but additional TFs are expected to be present at each location. To identify proteins bound at RB-bound loci, we introduce PanChIP, an algorithm for pan-ChIP-seq protein colocalization analysis (https://pypi.org/project/PanChIP/). PanChIP gathers peak sets from 7,903 ChIP-seq experiments on 915 TFs (Wang et al., 2018; Zheng et al., 2019) and computes their overlap in the cis-regulatory elements. There are two modes for PanChIP: PanChIP analysis measures normalized overlap between query and bulk reference peak set, while PanChIP filter measures individual overlap between query and each individual reference peak set to provide statistical significance measures for each TF hit (STAR Methods, Methods S1). PanChIP works best for chromatin binders that yield similar ChIP-seq across cell lines, and the PanChIP filter includes an alternative setting for quantifying overlap with TFs in a cell-type-specific manner. TF hits were considered statistically significant if they satisfy the following thresholds: signal-to-noise ratio>2 and Padj<0.05 (STAR Methods; two-tailed Welch’s t-test with Bonferroni correction). PanChIP can analyze both proteins with known and unknown consensus chromatin-binding sequence motifs and supports parallelization (-t THREADS) as well as computational replicates (-r REPEATS). PanChIP produces highly correlated results for biological replicate experiments (Fig. S5E; Pearson’s r = 0.98, P<1.0×10−99) and reproduces co-localization results for multiple well-studied TFs (Methods S1).
PanChIP analysis of the k-means clusters A–H (Fig. 2A) gave lists of proteins with significant overlap with RB at each of the eight classes of RB-binding sites (Fig. 5A, 5B, S5A–D, and Table S9). For each TF hit, we corrected p-values for multiple hypotheses using Bonferroni method. The results are strong corroborative evidence that RB binds to three distinct classes of chromatin (promoters, enhancers, and insulators). RB ChIP-seq peaks in k-means clusters A–D are bound by proteins involved in the E2F1/DP pathway and promoter activity. Proteins strongly enriched at these binding sites include E2F1, E2F4, components of the RNA polymerase II preinitiation complex (TFIIB), and proteins involved in RNA polymerase II promoter-proximal pausing or termination (NELF-A, NELF-E, Xrn2) (Aoi et al., 2020; Brannan et al., 2012). PHF8, a protein reported to be enriched at E2F-regulated promoters (Liu et al., 2010), is also enriched at these groups.
Fig. 5: Colocalization of chromatin-associated proteins in RB-binding sites.
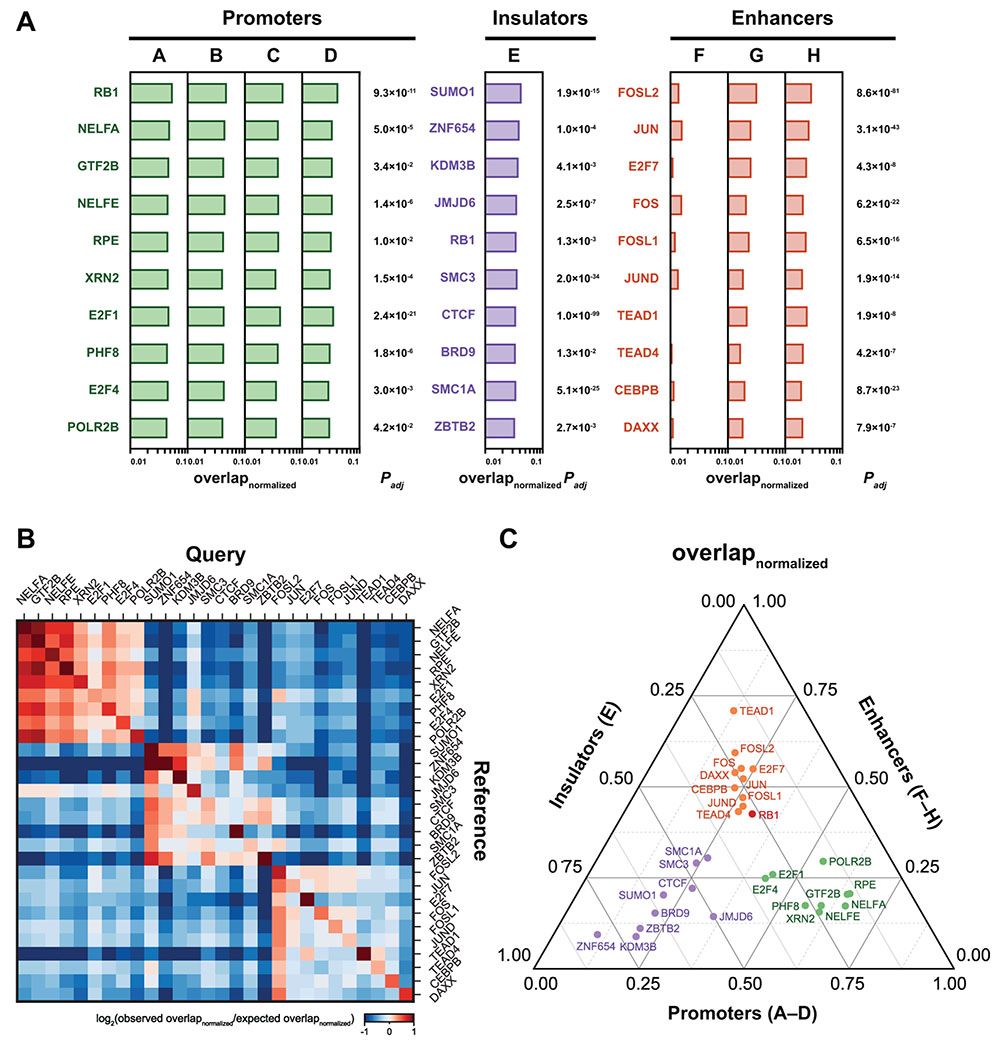
(A) Normalized overlap for the top ten most significantly enriched chromatin-associated proteins in RB-bound promoters (k-means clusters A–D, green), RB/CTCF sites (k-means cluster E, purple), and RB-bound enhancers (k-means clusters F–H, orange). Bonferroni-corrected P-values are presented on the right. (B) Heatmap of normalized overlap for the most significantly enriched chromatin-associated proteins. Normalized overlap was computed by performing PanChIP analysis using the entire PanChIP library as the input, and for the visualization of the heatmap, we assessed the log2 observed/expected values (STAR Methods). The separation of these chromatin-associated proteins into three clusters (promoters, enhancers, and insulators) was observed in this pan-ChIP-seq analysis, indicating the colocalization of these proteins in each cluster. (C) Ternary plot of normalized overlap score for each DNA-binding protein in the PanChIP library. Each axis indicates the relative fraction for the normalized overlap from three RB ChIP-seq peak sets (promoters, enhancers, and insulators). In agreement with panel B, this analysis shows that there are three distinct groups of RB-interacting proteins: E2F1 and RNA polymerase II regulators preferentially associated with RB-promoters (green), AP-1 and TEAD preferentially bound to RB-enhancers (orange), and CTCF and the cohesin complex were enriched in RB-insulators (purple).
A very different set of proteins associates with the RB ChIP-seq peaks in k-means cluster E. These sites are bound by CTCF and components of the cohesin complex (SMC1A, SMC3). This pattern suggests that these sites likely function as insulator elements and raises the possibility that functional interactions reported between RB and WapL (Manning et al., 2014) may be, at least in part, a consequence of a direct connection between RB and cohesin at a specific set of loci. Interestingly, other than RB, this analysis did not reveal any clear differences between proteins bound at RB-CTCF sites and proteins bound at CTCF sites.
The sites in RB ChIP-seq peaks in k-means clusters F–H were most frequently occupied by AP-1 TFs (c-Jun, JunD, c-Fos, Fosl1, Fosl2). Importantly, PanChIP also identified a series of additional proteins that associate with these loci, including the atypical E2F, E2F7, and the components of the YAP1/TEAD-TAZ pathway. YAP1/TEAD-TAZ are reported to associate with AP-1 in enhancers to control S-phase entry and mitosis (Zanconato et al., 2015). YAP1 also associates with B-Myb and promotes enhancer looping to promoters of mitotic genes regulated by Myb-MuvB (Pattschull et al., 2019). Moreover, RB-null tumors show compromised YAP/TEAD-TAZ transcriptional activity that supports lineage switching and drug resistance (Pearson et al., 2021). Ternary plots of the top ten most significantly enriched proteins at each cluster of binding sites (Fig. 5C) strongly support the conclusion that the three classes of RB-binding sites associate with proteins that are fundamentally different from one another. Clustering analysis of RB-binding sites using ChIP-seq data for 915 chromatin regulators (Fig. S5F–H) provides further support for this idea. We conclude that, regardless of the analytical approach used, RB-bound loci can be separated into three fundamentally different types of chromatin: promoters, enhancers, and insulators. We infer that RB’s mechanism of action, and presumably RB’s biological role, is likely to be different in these three contexts.
Discussion
Despite being one of the best-known tumor suppressors, RB remains an enigma. Beyond E2F inhibition in G1, much of RB-mediated transcriptional regulation is poorly understood. Most RB protein is associated with chromatin, but there is surprisingly little information about the number, or distribution, of RB-binding sites in the human genome. Antibodies to human RB typically give ChIP signals that have only a modest enrichment over background. As a result, existing knowledge has been restricted to RB-bound loci with the strongest RB signal. Here, using an inducible replacement system, in which endogenous human RB was replaced with FLAG-tagged protein, we succeeded in generating high-quality ChIP-seq data for wild-type RB, for constitutively active RBΔCDK, and for each mP-RB expressed in G1 (Data S1). The results give a detailed description of the mechanism of action of human RB (Fig. 6A) and a new perspective. Here we highlight five striking aspects of this data.
Fig.6: Models summarizing the multiple locations of RB and its changing role during cell cycle progression.
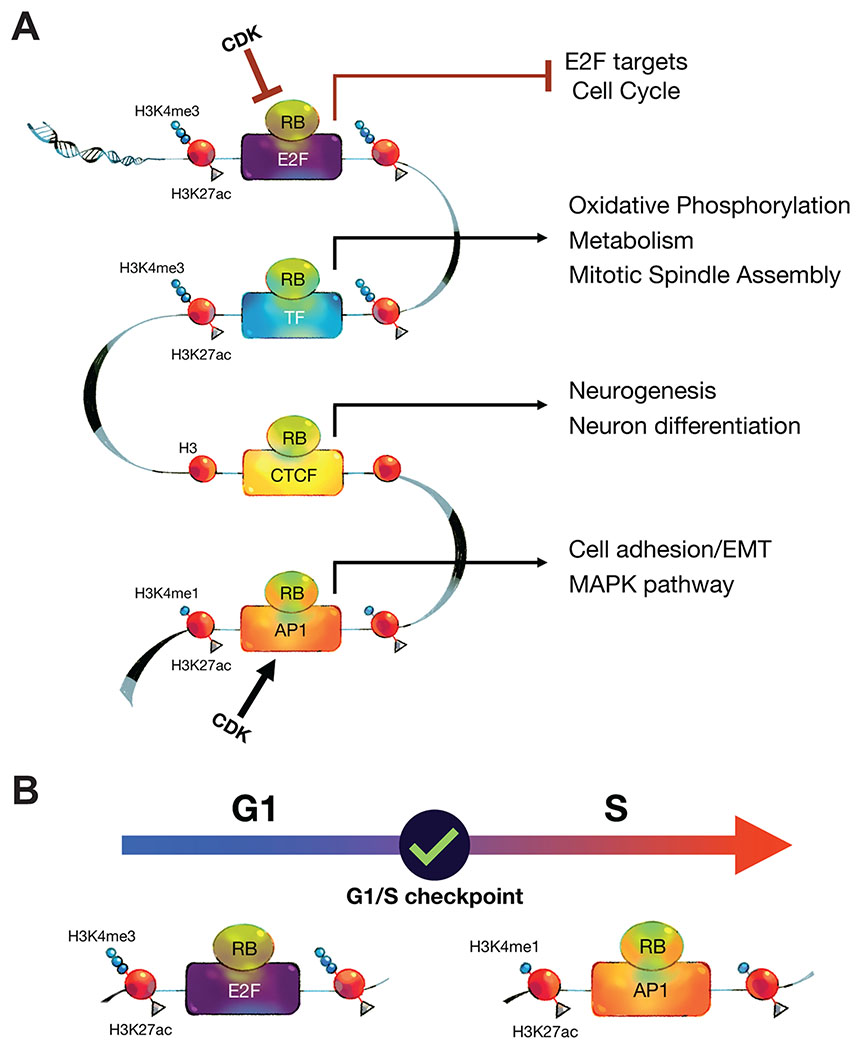
(A) RB associates with three distinct chromatin regions: promoters, insulators, and enhancers. E2F/DP1 mediates RB recruitment to E2F-promoters, an interaction that is stronger when cells accumulate in G1. These RB/E2F promoters regulate E2F target genes involved in cell cycle regulation. A subset of RB-bound promoters that is less enriched in E2F motifs but shows strong enrichment in motifs of other TFs, such as Sp1 and ETS family of TFs. This second group of RB-bound promoters regulates genes involved in metabolic pathways and mitotic spindle assembly. RB/CTCF insulator sites are CTCF-bound chromatin regions that are also bound by RB. These sites are depleted of both promoter and enhancer histone marks. Genes in proximity to RB/CTCF sites are often associated with neuronal differentiation. In enhancer regions, RB is recruited by AP-1 transcription factors, and the recruitment is inhibited when cells accumulate in G1. Genes that are linked to RB-enhancer sites are involved in cell adhesion and MAPK pathway regulation in RPE1 cells. (B) The “check ‘n go” functions of RB. RB has two separate roles during the cell cycle. In G1, RB represses E2F promoters and checks CDK activity before allowing cells to enter S-phase. RB binding to these regions is highly conserved across different cell types, enabling its “check” function in most cell types. However, when the cell makes the decision to divide, cell type-specific programs are needed to ensure the appropriate fate of the daughter cells. After the G1/S checkpoint, the role of RB changes, and it moves to enhancers, where it regulates transcriptional programs that are cell type-specific.
First, the ChIP-seq data reveals that human RB goes far beyond E2F-regulated promoters. RB does not simply bind to promoters but instead associates with three fundamentally different types of chromatin: i) promoter regions (high H3K4me3, close to TSS) where, at most of these sites, RB colocalizes with E2F1; ii) enhancers (marked by H3K4me1 and H3K27ac) which are highly enriched for AP-1 consensus sequences and overlap with c-Jun-binding sites; and iii) CTCF/BORIS-bound loci that do not consistently contain promoter or enhancer marks. ChIP-seq data for these proteins/marks reveal that E2F1, c-Jun, and CTCF generally associate with mutually exclusive subsets of RB-bound loci. As RB lacks a sequence-specific DNA-binding activity of its own, the simplest interpretation is that RB is recruited to these three locations through interactions with three different types of chromatin-binding proteins. RB association with E2F/DP1 is well established (Dick et al., 2018; Dick and Rubin, 2013; Kent and Leone, 2019). Previous studies connected RB to the Jun family of proteins (Nead et al., 1998; Nishitani et al., 1999) but the extent and significance of this interaction were not known. We found that the expression of dominantnegative DP1 reduces RB association with promoters, while RB binding to enhancer regions was specifically inhibited by the dominant negative Fos allele. The basis for RB recruitment to CTCF-bound sites is yet to be established. This may involve direct interaction with CTCF or indirect interactions via associated proteins. PanChIP, a novel algorithm for pan-ChIP-seq protein colocalization analysis, identified sets of proteins that associate with each cluster of RB binding loci. RB-bound promoter regions are enriched with proteins associated with RNA polymerase II transcriptional activity, while RB/CTCF sites are often co-occupied by components of the cohesin complex. Intriguingly, the RB-binding sites in enhancers are not only bound by AP-1 transcription factors but are also often bound by YAP1/TEAD-TAZ and E2F7. Thus, RB is recruited to several types of chromatin that play different roles, and RB intersects with completely different classes of chromatin-associated proteins at these sites. Our data provides no support for the claim that RB selectively targets sites in repetitive DNA sequences (Ishak et al., 2016). This may be because the original ChIP-seq data utilized to support this model was low in quality (Data S2).
Second, there are multiple categories of RB-bound promoters. We used several approaches to subdivide the RB-binding sites in promoters, and each analysis parsed the sites in a slightly different way. Our initial clustering analysis, based specifically on RB ChIP signal strength, subdivided the promoter binding sites into two groups: one strongly enriched for RB, presumably representing high-affinity RB-binding sites, (Promoters-1, Fig. 1D) and another with more moderate RB binding (Promoters-2). Sites with the strongest RB binding associated with classic cell cycle and replication genes and exhibit the most significant enrichment of E2F1-binding motifs. Although E2F1 was also present at many loci in Promoters-2, the strongest motif enrichment in this group was for other TFs, several of which have previously been reported to physically associate with RB (ETS, ELF, Sp1) (Kim et al., 1992; Wang et al., 1993). We hypothesize that the recruitment of RB to this set of promoters does not depend exclusively on E2Fs but occurs via combinatorial interactions with multiple TFs. In further analysis, when we included ChIP data for histone modifications, E2F1, CTCF, and c-Jun, k-means clustering revealed four types of RB-binding sites in promoters (A–D) that differed in the shape and directionality of the RB peak. Interestingly, PanChIP data indicates that RB-binding sites in Cluster A/B and Cluster C/D are occupied by different sets of promoter-binding proteins and these may confer different mechanisms of regulation. These analyses indicate heterogeneity between RB-binding sites in promoters – an heterogeneity that is not fully understood and merits further investigation.
Third, the distribution of RB between enhancers and promoters changes during cell cycle progression, with RB preferentially bound to promoters in arrested cells and to enhancers in cycling (or S-phase) cells. Interestingly the differences between arrested and cycling cells were clearer at promoters enriched in E2F motifs than at promoters enriched in other TF motifs. We note that c-Jun binds to the C-terminus domain of RB (Nead et al., 1998; Nishitani et al., 1999), a domain that is also important for RB association with E2F. We hypothesize that RB phosphorylation at specific sites inhibits binding to E2F and enhances RB interaction with AP-1 and that this antagonism contributes to the formation of distinct populations of chromatin-bound RB. When compared to wild-type RB and RBΔCDK, the mP-RB alleles distributed to varying degrees. The finding that RB redistributes away from high-affinity promoter sites to low-affinity enhancer sites upon cell cycle progression likely explains why the nuclear signal detected by RB immunofluorescence is relatively resistant to salt extraction in G1, but more easily extracted later in the cell cycle (Mittnacht and Weinberg, 1991).
Fourth, the different categories of RB-binding sites associate with strikingly different classes of genes. Whereas RB-binding sites in promoters are mainly associated with genes implicated in cell cycle, replication, oxidative phosphorylation, glycolysis, and splicing, RB-binding sites in enhancers are associated with genes involved in adhesion and MAPK signaling pathways. Intriguingly, RB/CTCF sites are associated with genes with neuronal functions. These patterns are exciting because studies of RB-mutant mice and RB-mutant cancer cells have linked RB loss with phenotypic changes that cannot easily be explained by the traditional view that RB controls only cell cycle genes. The different classes of RB-bound targets described here may give simple and direct mechanistic explanations for previous observations showing that RB loss changes regulation of the MAPK signaling pathway (Walter et al., 2019), causes defects in epithelial migration (Parisi et al., 2018), and is associated with defects in neuronal differentiation (Andrusiak et al., 2011; Ferguson et al., 2005; Ferguson et al., 2002; Ghanem et al., 2012; Vandenbosch et al., 2016). Moreover, the idea that RB is recruited to different types of targets via different TFs immediately suggests how different aspects of RB function may be controlled. We note that, whereas most promoters are repressed by RB, genes near RB-bound enhancers could either be upregulated or downregulated in RB-deficient cells. How these effects are achieved remains to be elucidated. AP-1-bound enhancers are known to recruit SWI/SNF complexes (Vierbuchen et al., 2017), of which Brg1 and hBrm were among the earliest described RB-interacting proteins (Dunaief et al., 1994; Trouche et al., 1997). This raises the fascinating possibility that RB binding may directly modulate enhancer activity.
Fifth, rather than being simply a regulator of cell cycle genes, RB has a very extensive and variable “reach”. The large number of binding sites in the human genome demonstrates that RB has the potential to directly affect many expression programs. Furthermore, the comparison between RPE1 and BJ cells shows that although RB is recruited to chromatin by similar TFs in different cell types, the precise location of RB-binding sites varies between lineages, with the greatest variation seen in non-promoter regions. In the past, when comparing profiles of RB-mutant cells, investigators focused on changes that were common between cell types. We suggest that RB likely acts on different targets in different cells. As a consequence, there is no single signature for RB loss, but rather its effects on gene expression are specific for each cell type. Chromatin-bound profiles of RB in additional cell lines or different tissues may elucidate RB association with cell-type-specific TFs and its role in cell differentiation (Calo et al., 2010; Thomas et al., 2001). The idea that RB localizes to different enhancers in different cell types fits well with previous studies showing that AP-1 is essential for the selection of enhancers that define cell identity (Li et al., 2017; Vierbuchen et al., 2017). A study that investigated the epigenetic changes induced by CDK4/6 inhibition in breast cancer cells found that long-term exposure to the CDK4/6 inhibitor abemaciclib increased the accessibility of AP-1-bound enhancers in an RB-dependent manner (Watt et al., 2021). There is remarkably little overlap between the RB and AP-1-bound enhancers described in our study and the sites identified by Watt et al. (4% and 11% between RPE1 and MDA-MB-453 or MCF7 cells respectively). Nevertheless, as RB-binding sites vary between cell lines, it is quite possible that the described abemaciclib-induced changes in enhancer accessibility occurred at RB-bound loci.
Taken together, these results paint a fascinating picture of RB in action. An important message from this data is that there is no single RB-binding domain that reproduces the full spectrum of RB action. This has major implications for studies using E2F reporters to give a readout of RB function (Kwon et al., 2017; Yao et al., 2008). The true picture is complex and variable. Indeed, one wonders why RB needs to be so complicated. We hypothesize that, at its core, RB is a master regulator of cell cycle progression. In a multicellular organism, a cell’s decision to divide must integrate with context-specific signals, so that daughter cells are used purposefully. However, the activated programs in each cell type are different. Our ChIP-seq data leads us to hypothesize that RB has a “check ’n go” property (Fig. 6B). In G1, RB repression of E2F represents a check against inappropriate cell cycle progression and helps to ensure that a high level of CDK activity is generated before cells enter S-phase. Once the cell has initiated replication, the role of RB changes: it then redistributes to enhancers and insulators and helps to enforce programs that are cell-type-specific. In this way, RB helps to promote lineage-specific functions. This dual nature may explain much of the biology of RB, particularly its variety of loss-of-function phenotypes.
Limitations of the Study
1. ChIP-seq data was generated using exogenous FLAG-tagged protein and this method may detect some RB-binding sites better than others. Endogenous RB ChIP-seq displayed specific binding to each of the 8 classes of RB-binding sites reported here, albeit with lower signal-to-background ratio, supporting the idea that these are authentic binding sites (Fig. S6).
2. The distribution of RB-binding sites varies between cell types, and it is possible that the redistribution of RB is also controlled differently in other contexts. To glimpse the variety of RB-binding patterns we assembled a cluster map (Fig. S6) using the data from RPE and BJ cells described here, similarly generated ChIP-seq data from T47D and MCF10A cells, published RB ChIP-seq data from IMR90 fibroblasts (Chicas et al., 2010), and unpublished RB ChIP-seq data for K562 and GM12878 cells available from ENCODE. Clusters of RB-binding sites enriched in E2F, AP-1, and CTCF/BORIS-binding motifs exist in multiple cell lines, and most cell lines also have clusters of private RB peaks. It has not escaped our attention that the largest numbers of RB-bound enhancers are seen in non-transformed cells, and some cancer cell lines have far fewer.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the lead contact, Nicholas J. Dyson (dyson@helix.mgh.harvard.edu).
Materials Availability
Plasmids and cell lines generated by this study are available upon request.
Data and Code Availability
ChIP sequencing data have been deposited at GEO and are publicly available as of the date of publication. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The original code for PanChIP has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| anti-DYKDDDDK Tag (Binds to same epitope as Sigma’s Anti-FLAG® M2 Antibody) | Cell Signaling Technology | Cat# 14793, RRID:AB_2572291 |
| anti-E2F1 | Cell Signaling Technology | Cat# 3742, RRID:AB_2096936 |
| anti-Histone H3 (Tri-Methyl-Lys4) | Cell Signaling Technology | Cat# 9751, RRID:AB_2616028 |
| anti-Histone H3 (Mono-Methyl-Lys4) | Abcam | Cat# ab8895, RRID:AB_306847 |
| anti-Histone H3 (Acetyl-Lys27) | Active Motif | Cat# 39034 |
| anti-c-Jun | Cell Signaling Technology | Cat# 9165, RRID:AB_2130165 |
| anti-CTCF | Cell Signaling Technology | Cat# 3418, RRID:AB_2086791 |
| anti-RB | Cell Signaling Technology | Cat# 9309S, RRID:AB_10696874 |
| anti-Phospho-RB S608 | Cell Signaling Technology | Cat# 8147S, RRID:AB_10949974 |
| anti-Phospho-RB S780 | Cell Signaling Technology | Cat# 8180S, RRID:AB_10950972 |
| anti-Phospho-RB S795 | Cell Signaling Technology | Cat# 9301P, RRID:AB_10830074 |
| anti-Phospho-RB S807/S811 | Cell Signaling Technology | Cat# 8516S, RRID:AB_11178658 |
| anti-Phospho-RB T373 | Abcam | Cat# ab52975, RRID:AB_2177344 |
| anti-a-Tubulin | Sigma-Aldrich | Cat# T9026, RRID:AB_477593 |
| anti-FLAG (M5) | Sigma-Aldrich | Cat# F4042, RRID:AB_439686 |
| anti-HA-Tag | Cell Signaling Technology | Cat# 3724, RRID:AB_1549585 |
| anti-p44/42 MAPK (Erk1/2) | Cell Signaling Technology | Cat# 9102, RRID:AB_330744 |
| anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling Technology | Cat# 4370, RRID:AB_2315112 |
| anti-Cyclin D1 | Santa Cruz Biotechnology | Cat# sc-753, RRID:AB_2070433 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 |
| Trametinib (GSK1120212) | Selleckchem | Cat# S2673 |
| Gateway LR Clonase II Enzyme mix | Invitrogen | Cat# 11791-020 |
| X-tremeGENE 9 DNA Transfection Reagent | Roche | Cat# 6365787001 |
| Polybrene | Sigma-Aldrich | Cat# 107689 |
| Geneticin | Gibco | Cat# 10131035 |
| Puromycin | Sigma-Aldrich | Cat# P9620 |
| EdU (5-ethynyl-2’-deoxyuridine) | Invitrogen | Cat# A10044 |
| Alexa Fluor 647 Azide, Triethylammonium Salt | Invitrogen | Cat# A10277 |
| DAPI (4′,6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | Cat# D1306 |
| FastStart Universal SYBR Green Master | Roche | Cat# 4913914001 |
| EGS (ethylene glycol bis(succinimidyl succinate)) | Thermo Scientific | Cat# 21565 |
| RNase A | Roche | Cat# 11119915001 |
| Proteinase K | Thermo Scientific | Cat# 25530049 |
| Agencourt AMPure XP | Beckman Coulter | Cat# A63881 |
| Protein G-Dynabeads | Life Technologies | Cat# 10004D |
| Critical Commercial Assays | ||
| pENTR™/D-TOPO Cloning Kit | Invitrogen | Cat# K240020 |
| Direct-zol RNA Kit | Zymo Research | Cat# R2073 |
| TaqMan Reverse Transcription Reagents | Thermo Fisher Scientific | Cat# N8080234 |
| Qubit dsDNA HS Assay kit | Thermo Fisher Scientific | Cat# Q32851 |
| Ovation Ultralow System V2 | TECAN | Cat# 0344NB-A01 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | Cat# 20024906 |
| Deposited Data | ||
| ChIP sequencing data | This paper | GEO: GSE176035 |
| Original images for Western Blots | This paper | DOI: 10.17632/p2x5xr4vt2.1 |
| RB ChIP-seq data from K562 cell line | ENCODE Project | ENCODE accession: ENCSR670JDQ GEO accession: GSE105638 |
| RB ChIP-seq data from GM12878 cell line | ENCODE Project | ENCODE accession: ENCSR785OKZ GEO accession: GSE106046 |
| Experimental Models: Cell Lines | ||
| hTERT-RPE1 human epithelial cells | Dr. David Pellman’s laboratory, Dana Farber Cancer Institute | RRID:CVCL_4388 |
| hTERT-BJ1 human fibroblasts | ATCC | Cat# CRL-4001, RRID:CVCL_6573 |
| MCF10A human mammary epithelial cells | Dr. Lee Zou’s laboratory, MGH | RRID:CVCL_0598 |
| T47D human breast cancer | Center for Molecular Therapeutics - MGH | RRID:CVCL_0553 |
| SW1573 human lung adenocarcinoma | Center for Molecular Therapeutics - MGH | RRID:CVCL_1720 |
| NCI-H2030 human lung adenocarcinoma | Center for Molecular Therapeutics - MGH | RRID:CVCL_1517 |
| CALU-1 human lung squamous cell carcinoma | Center for Molecular Therapeutics - MGH | RRID:CVCL_0608 |
| HOP62 human lung adenocarcinoma | Center for Molecular Therapeutics - MGH | RRID:CVCL_1285 |
| Oligonucleotides | ||
| Primers for cloning A-FOS and DNDP1 cDNA in pENTR/D-TOPO, see Table S8 | This paper | N/A |
| Real Time RT-PCR primers, see Table S8 | MGH Primer Bank database | N/A |
| Recombinant DNA | ||
| pINDUCER11 | (Meerbrey et al., 2011) | Addgene plasmid# 44363 |
| pINDUCER20 | (Meerbrey et al., 2011) | Addgene plasmid# 44012 |
| pLenti-CMVtight-puro-Dest | gift from Eric Campeau | Addgene plasmid# 26430 |
| pLenti-CMVtight-eGFP-puro | gift from Eric Campeau | Addgene plasmid# 26431 |
| pINDUCER11-shRB1 (5’-CAGAGATCGTGTATTGAGATT-3’) | This paper | N/A |
| pINDUCER11-shScramble (5’-ACTAAGGTTAAGTCGCCCTCGA -3’) | This paper | N/A |
| pINDUCER20-RB wild-type | (Sanidas et al., 2019) | N/A |
| pINDUCER20-FLAG-RB wild-type | (Sanidas et al., 2019) | N/A |
| pINDUCER20-RB Δcdk | (Sanidas et al., 2019) | N/A |
| pINDUCER20-FLAG-RB Δcdk | (Sanidas et al., 2019) | N/A |
| pINDUCER20-FLAG-mP-RB mutant alleles | (Sanidas et al., 2019) | N/A |
| pLenti-CMVtight-HA-A-FOS-puro | This paper | N/A |
| pLenti-CMVtight-HA-DNDP1-puro | This paper | N/A |
| Software and Algorithms | ||
| Geneset enrichment analysis (GSEA) | (Subramanian et al., 2005) | N/A |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| WebGestalt | (Liao et al., 2019) | http://www.webgestalt.org |
| Prism 6 | GraphPad | https://www.graphpad.com |
| OriginPro | Origin Lab | https://www.originlab.com/ |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Python | N/A | https://www.python.org/ |
| gdown (Python package) | Kentaro Wada | https://github.com/wkentaro/gdown |
| argparse (Python package) | Ben West | https://github.com/bewest/argparse |
| R | N/A | https://www.r-project.org/ |
| ggplot2 (R package) | N/A | https://cran.r-project.org/web/packages/ggplot2/index.html |
| leaflet (R package) | N/A | https://cran.r-project.org/web/packages/leaflet/index.html |
| RColorBrewer (R package) | N/A | https://cran.r-project.org/web/packages/RColorBrewer/index.html |
| DiffBind (R package) | N/A | https://doi.org/doi:10.18129/B9.bioc.DiffBind |
| BEDTools | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| ENCODE | (An integrated encyclopedia of DNA elements in the human genome, 2012) | https://www.encodeproject.org/ |
| Cistrome Data Browser | (Zheng et al., 2019) | http://cistrome.org/db/ |
| BART2 | (Wang et al., 2018) | https://github.com/zanglab/bart2 |
| MARGE | (Wang et al., 2016) | http://cistrome.org/MARGE |
| Cromwell | (Voss et al., 2017) | https://doi.org/10.7490/f1000research.1114634.1 |
| ENCODE ChIP-seq Pipeline | (Landt et al., 2012) | https://github.com/ENCODE-DCC/chip-seq-pipeline2 |
| PanChIP | This paper | DOI: 10.5281/zenodo.6787997 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture:
The telomerase-expressing non-transformed human retina epithelial cells RPE1 and skin fibroblasts BJ cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 5% Fetal Bovine Serum (FBS) and antibiotics (100 units/ml Penicillin and 100 μg/ml Streptomycin; P/S). The human lung cancer cell lines SW1573, NCI-H2030, CALU-1 and HOP62 were cultured in RPMI 1640 medium supplemented with 5% FBS and antibiotics (P/S). The MCF10A human breast epithelial cell line were cultured in DMEM/Nutricient mixture F12 medium supplemented with 5% horse serum (Gibco, Cat. No. 16050122), 20 ng/ml human EGF (ThermoFisher, Cat. No. PHG0311), 0.5 μg/ml hydrocortisone (Sigma, Cat. No. H-0888), 100 ng/ml cholera toxin (Sigma, Cat. No. C-8052), 10 μg/ml insulin (Sigma, Cat. No. I-1882) and antibiotics (P/S). The human breast cancer cell line T47D was cultured in RPMI 1640 medium supplemented with 10% FBS and antibiotics (P/S).The human embryonic kidney 293T cells were cultured in DMEM supplemented with 10% FBS and antibiotics (P/S). RB1 knock down or replacement of the endogenous by exogenous RB protein in cells transduced with pINDUCER11 and/or pINDUCER20 constructs were induced by addition of 0.5 μg/ml DOX (Sigma, Cat. No. D9891) for 48 hours. Expression of the dominant negative A-FOS or the dominant negative DP1_aa127-410 in cells transduced with pLenti-CMVtight puro constructs were induced by addition of 0.5 μg/ml DOX for 48 hours. Contact inhibition experiments performed by maintaining RPE1 cells in confluent culture for 6 days. To inhibit Erk1/2 kinase activity, cells were treated with 30 nM trametinib (GSK1120212) (Selleckchem; Cat.No. S2673) for the indicating time.
METHODS DETAILS
Cloning:
HA-tagged dominant negative A-FOS was amplified by PCR from CMV500-A-FOS (Addgene; 33353), using the oligos described in Table S8 and it was transferred to the pENTR/D-TOPO cloning vector (Invitrogen, Cat. No. 45-0218). HA-tagged dominant negative TFDP1 (DNDP1; DP1_aa127-410) was amplified by PCR from CMV-TFDP1 that was kindly provided by Dr. Fred Dick, using the oligos described in Table S8 and it was transferred to the pENTR/D-TOPO cloning vector. A-FOS and DNDP1 cDNA were subsequently transferred into pLenti-CMVtight-puro-Dest (Addgene; 26430) with LR clonase reaction (Invitrogen, cat. No. 11791-020). shRNA that targets the 3’-UTR of RB1 mRNA at the sequence 5’ - CAGAGATCGTGTATTGAGATT −3’ and shScramble control 5’- ACTAAGGTTAAGTCGCCCTCGA −3’ were subcloned in XhoI – MluI sites in pINDUCER11 (Addgene; 44363), as it was described before (Meerbrey et al., 2011).
Transfections and infections:
pINDUCER11, pINDUCER20 and pLenti-CMVtight-puro lentiviral constructs were packaged in 293T cells by transient transfection, in combination with the envelope plasmid pCMV-VSV-G (Addgene plasmid #8454) and the packaging plasmid pCMV-dR8.2 dvpr (Addgene plasmid #8455). Transfections were carried out using X-tremeGENE™ 9 DNA Transfection Reagent (Sigama, Cat. No. 6365787001). All cell lines were infected with the lentiviral particles in the presence of 5 μg/ml polybrene (Sigma, Cat. No. 107689). Stable cell lines were selected for expression of the green fluorescence protein by fluorescence-activated cell sorting or for resistance to specific antibiotics; 400μg/ml G-418 (Gibco, Cat. No. 10131035) for pINDUCER20 and 6μg/ml puromycin (Sigma, Cat. No. P9620) for pLenti-CMVtight-puro.
Western Blot analysis:
Cells were washed with phosphate-buffered saline (PBS) and cell lysates were collected in lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 1mM PMSF) supplement with protease (Roche, Cat. No. 04693159001) and phosphatase (Roche, Cat. No. 04906837001) inhibitor-cocktail. Lysates were passed 10 times through a 27-gauge needle, clarified by centrifugation at 16,000 x g for 10 min at 4°C, analyzed on Criterion TGX gels (BioRad, Cat. No. 5671084) and transferred on PVDF membranes (BioRad, Cat. No. 1704273). Primary antibodies were used in 5% bovine serum albumin (BSA; Boston BioProducts, Cat. No. P-753) in Tris-buffered saline (TBS) containing 0.1% Tween 20 (Santa Cruz, Cat. No. sc-362311). Cell Signaling Technology secondary antibodies were used at 1:5,000 dilutions.
ChIP-seq:
The chromatin immunoprecipitation protocol was customized based on previously described methods (Boulay et al., 2017; Chicas et al., 2010) and it was optimized to increase the RB ChIP signal relative to background. ChIP assays were carried out on RPE1, BJ, MCF10A and T47D cell cultures using approximately 5 million cells per sample and per epitope. Cells were washed once with PBS and fixed with 1.5 mM ethylene glycol bis(succinimidyl succinate) (Thermo Scientific, Cat. No. 21565) for 30 min at 25°C and 1% formaldehyde (Sigma, Cat. No. F8775) for 10 min at 37°C. 20mM Tris-HCl pH7.5 and 150 mM Glycine were used to quench cross-linking. Cell pellets were washed twice with ice-cold PBS and lysates were prepared using 300 μl lysis buffer (50mM Tris-HCl pH 7.4, 1% SDS, 0.25% DOC) supplement with protease and phosphatase inhibitors cocktail (PPI) (Thermo Scientific, Cat. No. 78442) for 10 min on ice. Cell lysates were diluted with 900 μl ChIP dilution buffer (50mM Tris-HCl pH 7.4, 0.1% SDS, 150mM NaCl, 1.84% Triton-X) supplement with PPI and chromatin was fragmented to a size of 200 – 700 bases with Branson 250 sonifier. Solubilized chromatin was further diluted with 1800 μl ChIP dilution buffer and immunoprecipitated with the indicated antibodies overnight at 4°C. Anti-DYKDDDDK Tag (Cell Signaling, Cat. No. 14793) was used for FLAG-RB ChIP and anti-RB (Cell Signaling, Cat. No. 9309) was used for endogenous RB ChIP. Antibody-chromatin complexes were pulled down with protein G-Dynabeads (Life Technologies, Cat. No. 10004D) for 2 hours at 4°C and washed 3 times with RIPA 150 Wash Buffer (0.1% SDS, 0.1% DOC 1% Triton X-100, 1mM EDTA, 10mM Tris-HCl pH 8, 150mM NaCl), 3 times with RIPA 500 Wash Buffer (0.1% SDS, 0.1% DOC 1% Triton X-100, 1mM EDTA, 10mM Tris-HCl pH 8, 500mM NaCl), 3 times with LiCl wash buffer (10mM Tris-HCl, pH 8, 250mM LiCl, 0.5% Triton X-100, 0.5% DOC) and once with ice-cold 10mM Tris-HCl, pH 8. Chromatin complexes were eluted from the beads using elution buffer (10mM Tris-HCl pH 8, 0.1% SDS, 150mM NaCl, 5 mM DTT) for 1 hour at 65°C and treated with RNase A (Roche, Cat. No. 11119915001) for 30 min at 37°C. Proteinase K (Thermo Scientific, Cat. No. 25530049) treatment and reverse cross-linking was performed for 3 hours at 65°C. After crosslink reversal, immunoprecipitated DNA was extracted with AMP Pure XP beads (Beckman Coulter, Cat. No. A63881). ChIP DNA was quantified with Qubit dsDNA HS Assay kit (Thermo Scientific, Cat. No. Q32851). 1-2 ng ChIP DNA samples were used to prepare sequencing libraries, using the Ovation Ultralow System V2 kit (TECAN, Cat. No. 0344NB-A01). ChIP DNA and input controls were sequenced using the NextSeq 500/550 High Output Kit v2.5 (75 Cycles) (Illumina, Cat. No. 20024906) with the Nextseq 500 Illumina genome analyzer.
Bioinformatics processing:
Sequencing reads were trimmed using trim-galore, aligned to the human genome (build hg19) using bwa-mem, then duplicate-marked using sambamba, and filtered to remove reads that were duplicates, had mapping quality <30, had >2 mismatches, or had >1/3 of their sequence soft-clipped. Filtered BAMs were sorted and indexed, and deeptools-bamCoverage was used to generate CPM-normalized bigwig files for peak visualization, and bedtools-coverage was used to count reads in 200bp genomic windows tiling the genome. Binned readcounts were normalized, and RB peaks were identified on the basis of having at least twice the normalized readcounts in any of the sixteen FLAG-RB samples, compared to the maximum normalized readcounts across the four control samples (two Input samples and two No-tag samples), yielding a set of 28,115 RB peaks that were subjected to k-means clustering by two procedures. The first clustering procedure (Fig. 1) used k=3 and included eight data tracks: the FLAG-RB, H3K4me3, H3K4me1, and K3K27ac ChIP data from cells expressing either RB wild-type or RBΔcdk. Clustering was performed on the peak strength and peak shape, represented by the 10Kb neighborhood (50 bins) centered on the peak summit, using Euclidean distance metric. The second clustering procedure (Fig. 3) used k=8, and included twelve data tracks: the same eight as above, plus CTCF and c-Jun ChIP data from cells treated with either an shRNA (“D5”) targeting RB1, or a scramble version of it. Clustering was performed on peak shape only, using cosine distance metric. Total RB ChIP signal in each cluster of peaks was compared between cells expressing RB wild-type, RBΔcdk, or the 14 mono-phosphorylated forms by calculating the total normalized reads in that cluster in each sample, and then calculating the percent change in this number between the two samples being compared, with significance calculated by t-test and Bonferroni-corrected for the total number of tests performed. DNA sequences of the peaks in each cluster were analyzed using the findMotifsGenome function (-size given - gc) of the HOMER software package (Heinz et al., 2010). Genes overlapping each peak were analyzed for enrichment (hypergeometric test) in the MSigDB Hallmark genesets (Subramanian et al., 2005). To assess the library complexity and the overall reproducibility of peak sets, ENCODE ChIP-seq pipeline (v2.1.4) was utilized on top of the Cromwell (v76) scientific workflow engine. Sequencing reads were directly used as the input for the ENCODE ChIP-seq pipeline and were aligned to the human genome (build hg19). The encode blacklist was utilized to filter genomic loci. The mode for the ENCODE ChIP-seq pipeline was set as “tf.” The alignment statistics was directly measured from raw unfiltered BAM files using SAMstat, which included the statistics for the fraction of mapped reads. The library complexity of the ChIP-seq experiment was assessed by analyzing the PBC1, PBC2, and NRF values of filtered non-mitochondrial BAM files. PBC1 greater than or equal to 0.5, PBC2 greater than or equal to 1, and NRF greater than or equal to 0.5 was considered acceptable per the ENCODE guidelines. Comparison of RB wild-type and RBΔcdk ChIP-seq peak sets was performed using two different methods. The first method involved IDR (Irreproducible Discovery Rate), which is a default method utilized in the ENCODE ChIP-seq pipeline. IDR plots comparing the RB wild-type to RBΔcdk as well as between pooled replicates are presented for each cell line (RPE1, BJ, and MCF10A). The second method involved DiffBind, which is an R package that analyzes the reproducibility of peak sets. All 16 RPE1 RB ChIP-seq peak sets were used as an input for DiffBind analysis. Of note, due to the great number of samples utilized in the analysis, BED files rather than BAM files were utilized as inputs for the DiffBind analysis. Correlation heatmap as well as dendrograms from the hierarchical clustering are presented.
Cell Cycle analysis:
48h after DOX-induction or 6 days after contact inhibition and release RPE1 cells were incubated with 20 μM 5-ethynyl-2’-deoxyuridine (EdU; Life technologies, Cat. No. A10044) for 2h, fixed with 3.7% formaldehyde in PBS for 15 min, blocked with 3% BSA in PBS for 1 min, permeabilized with 0.5% Triton X-100 in PBS for 30 min and stained with ClickIT reaction (100mM Tris-HCl pH 7.5, 3mM CuSO4, 50 mM Ascorbic Acid, 2.5 μM Alexa Fluor-647 azide; Life Technologies, Cat. No. A10277) for 30 minutes and 3μM 4’,6-Diamidino-2-Phenylindole Dihydrochloride (DAPI; Life Technologies, Cat. No. D1306) in staining buffer (100mM Tris-HCl pH 7.5, 150mM NaCl, 1mM CaCl2, 0.5mM MgCl2, 0.1% NP-40), for 15 minutes. FACS analysis was performed with LSR II flow cytometer (BD Biosciences).
Real-time RT-PCR:
Total cell RNA was extracted using the Direct-zol RNA MiniPrep Plus kit (Zymo Research, Cat. No. R2073). cDNA was synthesized from 0.5 μg total RNA, using oligo-dT priming and the TaqMan Reverse Transcription kit (Applied biosystems, Cat. No. N8080234). Relative expression of transcripts was quantified by real-time PCR, using the FastStart Universal SYBR Green Master mix (Roche, Cat. No. 4913914001) and the LightCycler 480 System (Roche). mRNA levels were normalized to ACTB and GAPDH. Primer sets are listed on the Table S10.
ChIP-quantitative PCR:
ChIP DNA was purified as it was described in ChIP-seq paragraph. The enrichment of the different genomic loci was quantified by real-time PCR, using the ChIP-qPCR primers described in Table S10.
Gene enrichment analysis:
Gene enrichment in figures 4, 5 and 6 was performed using Over-Representation analysis (ORA) for the indicated groups of genes. ORA was done using the WebGestalt toolkit (http://www.webgestalt.org) for Biological Process and KEGG pathways. The set of all mapped entrezgene IDs of the protein-coding genome was used as reference list. Top 10 significant Biological Processes or KEGG pathways were identified, and False Discovery Rate (FDR) was calculated by Benjamini-Hochberg method.
Normalized Overlap of ChIP-seq peak sets:
A library consisted of 7,903 ChIP-seq experiments on 915 DNA-bound proteins is prepared by aggregating data generated in this paper with those from the Cistrome Data Browser (Zheng et al., 2019). For statistical analysis, we filtered the library such that every DNA-bound protein has at least two ChIP-seq experiments in the filtered library. The filtered library featured 639 DNA-bound proteins and 7,619 ChIP-seq experiments. Histone ChIP-seq experiments were excluded from both the original and the filtered library, since the peak calling mode of histone ChIP-seq experiments differs from that of standard transcription factor ChIP-seq experiments. On average, the dataset for each DNA-bound protein in the original library is consisted of 9 ChIP-seq experiments with a standard deviation of 41. Only peaks that locate at one of the union DNase I hypersensitivity sites (Wang et al., 2016) were assessed. Normalized overlap between the library and the query was assessed following these three steps: 1) preliminary normalization via data permutation, 2) overlap calculation, and 3) normalization of the overlap. The normalization of the overlap is required for a library-wide analysis of ChIP-seq experiments, since the number of peaks as well as the bin size vary throughout the library. The normalized overlap between two ChIP-seq peak sets is defined as
Reference indicates a DNA-bound protein from the original library consisted of 915 DNA-bound proteins. Query indicates an input peak set provided by the user. In this study, the input peak set was the RB ChIP-seq peak set generated from 16 FLAG-tagged RPE1 RB ChIP-seq experiments. For cluster-wise analysis, the peak set for each k-means cluster (k=8) was utilized as the input. The following three steps illustrate the design of the algorithm that performs the calculation of this normalized overlap between the reference and the query.
1). preliminary normalization
To ensure that values from the denominator do not significantly vary library-wide (reference) and input-wide (query), a preliminary normalization was performed by permutating the peak set with target values. The permutation process was performed using the shuf function of bash, and the process allowed repeats. The target values were set as follows:
The permutation process allows comparison between datasets, even if the datasets have different number of peaks and bin sizes. The weighting formula for the reference peak set was set as the likelihood of a given region showing up as a peak in a ChIP-seq experiment for the DNA-bound protein (i.e., weightreference = P(peak ∈ experiment | peak ∈ ∪referenceexperiment)), while the weighting formula for the query peak set was set as the score between 0 and 1000 in a standard BED6 format.
2). overlap calculation
The overlap between the query and the reference was measured as , which is the numerator of the formula for the normalized overlap. The selection of peaks that satisfy the peak ∈ (reference ∩ query) condition was performed by utilizing the intersect functionality of bedtools.
3). normalization of the overlap
Finally, the normalized overlap between the reference and the query was measured by dividing the calculated overlap by the denominator . While we had implemented a preliminary normalization step that permutates the library and the input until they reach a target value by approximation, there still exists a slight variation in the exact values. In order to remove the bias created by this variation, the final value for the normalized overlap is acquired by dividing the calculated overlap by this reference- and query-specific denominator.
The measurement of the normalized overlap was done in triplicates and their mean values were utilized for further analysis in the study.
In order to ensure the reproducibility of the calculation, we made our algorithm publicly available through the PyPI repository (accession code: PanChIP; https://pypi.org/project/PanChIP/). The source code is available for download at GitHub (https://github.com/hanjunlee21/PanChIP). The library can be accessed via https://drive.google.com/uc?id=17L0c1pv8dx2906O7WMfWwcqgDLMJvq7T&confirm=t or through the panchip init function. The PanChIP version v.3.0.9 was utilized for the generation of plots.
Statistical analysis of the overlap:
To assess the statistical significance of the overlap and to remove any DNA-bound protein hits that are due to a mere noise, we have performed a statistical analysis for the overlap in the filtered library consisted of 639 DNA-bound proteins and 7,619 ChIP-seq experiments. Instead of doing a bulk analysis on a specific DNA-bound protein, we measured the individual overlap between the peak set from each of the 7,619 ChIP-seq experiments and the query. The mathematical representation of the individual overlap is as follows:
No permutation step was utilized for the measurement of individual overlaps. The weighting formula for the peak set from each ChIP-seq experiment was set as a uniform distribution (i.e., weightexperiment = 1), while the weighting formula for the query peak set was set as the score between 0 and 1000 in a standard BED6 format. Since all DNA-bound proteins have at least two experiments in the filtered library, the standard deviation as well as the mean value were calculated for each of the 639 DNA-bound proteins. The signal-to-noise ratio, which is defined as
was measured for each of the 639 DNA-bound proteins to assess the statistical significance of the overlap. We further preformed unpaired two-tailed Welch’s t-test to assess the statistical significance, and the adjusted p-values that were corrected for multiple hypotheses using the Bonferroni method are provided. Only DNA-bound proteins that exhibited signal-to-noise ratio greater than 2 and adjusted p-value less than 0.05 were utilized for further examination. Individual overlap scores between E2F1 and RB-Promoters (k-means clusters A–D), CTCF and RB-Insulators (k-means cluster E), and c-Jun and RB-Enhancers (k-means clusters F–H) were significantly greater compared to the individual overlap scores generated from the entire filtered library (Fig. S5D, Padj<0.05). Similarly, unpaired two-tailed Welch’s t-test on the individual overlap scores of each cluster’s top ten TF hits validated that each overlap is statistically significant (Fig. 5A, Padj<0.05). Histograms are presented to support this statistical analysis (Fig. S5D).
In order to ensure the reproducibility of the calculation, we have incorporated the signal-to-noise ratio as well as the adjusted p-value calculation algorithm in the PanChIP package.
Using the PanChIP software:
PanChIP is a python- and bash-based algorithm that can be downloaded from the PyPI repository using the following command:
The PanChIP algorithm is designed to ensure the reproducibility of the calculation of overlaps presented in this study. There are three functions in the PanChIP software: init, analysis, and filter.
1). $ panchip init [-h] library_directory
This function requires a single positional argument, which is the path to the target directory wherein the library would be saved. Both the relative and the absolute paths are allowed as input. This initialization process is required after every version upgrade of the algorithm in order to ensure that the version of the library matches the version of the algorithm. The target directory should contain a minimum of 14 GB of available storage for downloading the library. After running panchip init, the target directory would contain three subdirectories: source codes for python and bash scripts (PanChIP-v.x.x.x), the original library for the calculation of the normalized overlap (v.x.x/Analysis), and the filtered library for the calculation of the individual overlap (v.x.x/Experiment). Once the library directory is set, it can be utilized for all subsequent PanChIP runs.
2). $ panchip analysis [-h] [-t THREADS] [-r REPEATS] library_directory input_directory output_directory
This function requires three positional arguments and accepts two optional arguments. The three positional arguments are: the path to the library directory, the path to the input directory, and the path to the output directory. The input directory should contain the list of BED files of query peak sets in the standard BED6 format. The input directory should not contain files other than the BED files. The output directory is where the output files would be generated. The output directory will be automatically generated if the directory is non-existent. When panchip analysis is done, the output directory should contain the primary.output.tsv file, which is a matrix file that contains the normalized overlap values. The two optional arguments are: the number of threads and the number of permutational iterations. The number of threads enables parallelization of the panchip analysis algorithm. If the number of permutational iterations is set at a value greater than one, the primary.output.tsv file would contain the mean normalized overlap values. The default for all optional arguments is set at one.
3). $ panchip filter [-h] [-t THREADS] library_directory input_file output_directory
This function requires three positional arguments and accepts a single optional argument. The three positional arguments are: the path to the library directory, the path to the input file, and the path to the output directory. The input file would be the BED file in the standard BED6 format. The individual overlap would be calculated between each ChIP-seq experiment from the library and the input peak set. The output directory is where the output files would be generated and will be automatically generated if the directory is non-existent. When panchip filter is done, the output directory should contain the primary.output.tsv file, which is a matrix file that contains the individual overlap values. Statistical analysis (i.e., calculation of the signal-to-noise ratio and the adjusted p-value) is done using these individual overlap values. This function accepts the number of threads as an optional argument, of which default is set at one.
Detailed description on the usage of the algorithm as well as the troubleshooting guidelines are available at Methods S1 or at the PanChIP manual available for download at https://github.com/hanjunlee21/PanChIP/tree/v.3.0.9/PanChIP_manual.pdf.
Heatmap representation of normalized overlap:
For the heatmap representation of normalized overlap presented in Fig. 5B, we assessed the log2 observed/expected values for the normalized overlap score measured between the query and the reference dataset. The log2 observed/expected values were mathematically defined as follows:
where the function E(x) is defined as the expected value of x.
Non-negative Matrix Factorization:
To enable pan-ChIP-seq unbiased clustering of peaks, we generated a union matrix of all peak sets from the PanChIP library. The union DNase I hypersensitivity sites from the MARGE software (Wang et al., 2016) were utilized to filter all peak sets, and the dimension of the resultant union matrix was given by 2,723,010-by-915. We then extracted a submatrix of this 2,723,010-by-915 union matrix that has an overlap with RB binding sites. Since the 28,115 RB binding sites span 84,932 distinct union DNase I hypersensitivity sites, the submatrix had a dimension of 84,932-by-915. For the assessment of the cophenetic correlation coefficient, we randomly pooled 200 different submatrices of 84,932-by-915 matrix that has the dimension of 1,000-by-915. From k values of 2 to 8, non-negative matrix factorization was performed using the nnmf function of MATLAB and their 200-iteration average of cophenetic correlation coefficient as well as their 95% confidence intervals were assessed. The distance was measured based on a Euclidean distance measured from the pdist function of MATLAB. The cophenetic correlation coefficient formed a plateau when k≥3 and showed local maximum at k=5. Hence, k=5 was selected for the pan-ChIP-seq unbiased clustering of peaks using non-negative matrix factorization (we note that the cophenetic correlation coefficient did not form a peak but rather formed a plateau in our analysis). We then performed non-negative matrix factorization using the entire 84,932-by-915 matrix and repeated the analysis 200 times to assess the cluster membership. MATLAB algorithm for the non-negative matrix factorization is available for download at https://github.com/hanjunlee21/Sanidas-et-al-2021/tree/v.1.0.0.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of the real-time PCR data was performed using the GraphPad Prism 6 software. Statistical comparison among groups was carried out using unpaired two-tailed Welch’s t-test with Bonferroni correction. For the statistical significance of a quantitative PCR result in FLAG-RB chromatin immunoprecipitates from RPE1 cells expressing FLAG-tagged wild-type RB or RBΔCDK, one-way ANOVA relative to wild-type RB was assessed. For GSEA, Benjamini-Hochberg correction was utilized to correct for multiple hypotheses. Cluster membership of each peak in non-negative matrix factorization was assessed by 200 iterations of the clustering analysis. The calculated statistical significance is indicated as ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05.
Supplementary Material
Methods S1: PanChIP manual v3.0.10. Related to Figure 5 and Star Methods.
Table S1: Homer motif analysis of Promoters-1, Promoters-2 and non-promoters k-means clusters of RB peaks. Related to Figure 1.
Table S2: GSEA analysis of the genes with RB-bound promoters (promoters-1 and promoters-2 clusters) or genes in proximity to RB peaks located in non-promoter regions. Related to Figure 1.
Table S3: Over-Representation analysis of genes in proximity to RB peaks located in the different k-means clusters presenting in Fig. 2A. The top ten most significant Biological Processes and KEGG pathways are shown for each set of genes. Related to Figure 2.
Table S4: Homer motif analysis of non-promoters RB peaks in BJ and RPE1 cells. Related to figure 2.
Table S5: Over-Representation analysis of genes in proximity to RB peaks that are highlighted in Fig. 3C and 3E. The top 10 most significant Biological Processes and KEGG pathways are shown for each set of genes. Related to Figure 3.
Table S6: Co-ordinates of the RB peaks in the enhancers that are highlighted in Fig. 4A. The gene that it is in proximity to each RB-bound enhancer is shown. Related to Figure 4.
Table S7: Over-Representation analysis of the genes with RB-dependent enhancers that identified in Fig. 4A and Table S6. The top 10 most significant Biological Processes are shown. Related to Figure 4.
Table S8: Over-Representation analysis of the different sets of genes with RB-bound enhancers that are highlighted in Fig. 4C. The top 10 most significant Biological Processes and KEGG pathways are shown for each set of genes. Related to Figure 4.
Table S9: Normalized overlap scores of 915 chromatin-associated proteins in different groups of RB ChIP-seq peaks (k-means clusters A–H). Statistical significance measures, including the p-value and the signal-to-noise ratio, are presented for RB-bound promoters, RB/CTCF sites, and RB-bound enhancers. Raw data for the generation of heatmap in Fig. 5B are also shown. Related to Figure 5.
Highlights:
RB associates with E2F-promoters, AP-1 enhancers and CTCF-insulators.
RB-bound promoters and enhancers target different sets of genes.
Cell cycle progression redistributes chromatin-bound RB towards enhancers.
RB-bound promoters are conserved while enhancers are cell type-specific.
Acknowledgements
We thank L. Zou, Y.H. Xing, and B. Krishnan for helpful discussions and technical help; F.A. Dick for critical reading of the manuscript. This work was supported by NIH grants CA236538 and GM117413 to N.J.D.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References:
- Andrusiak MG, McClellan KA, Dugal-Tessier D, Julian LM, Rodrigues SP, Park DS, Kennedy TE, and Slack RS (2011). Rb/E2F regulates expression of neogenin during neuronal migration. Mol Cell Biol 31, 238–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoi Y, Smith ER, Shah AP, Rendleman EJ, Marshall SA, Woodfin AR, Chen FX, Shiekhattar R, and Shilatifard A (2020). NELF Regulates a Promoter-Proximal Step Distinct from RNA Pol II Pause-Release. Molecular Cell 78, 261–274.e265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avni D, Yang H, Martelli F, Hofmann F, ElShamy WM, Ganesan S, Scully R, and Livingston DM (2003). Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol Cell 12, 735–746. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulay G, Sandoval GJ, Riggi N, Iyer S, Buisson R, Naigles B, Awad ME, Rengarajan S, Volorio A, McBride MJ, et al. (2017). Cancer-Specific Retargeting of BAF Complexes by a Prion-like Domain. Cell 171, 163–178 e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannan K, Kim H, Erickson B, Glover-Cutter K, Kim S, Fong N, Kiemele L, Hansen K, Davis R, Lykke-Andersen J, et al. (2012). mRNA Decapping Factors and the Exonuclease Xrn2 Function in Widespread Premature Termination of RNA Polymerase II Transcription. Molecular Cell 46, 311–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JR, Hura GL, and Rubin SM (2012). Structures of inactive retinoblastoma protein reveal multiple mechanisms for cell cycle control. Genes Dev 26, 1156–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JR, Liban TJ, Restrepo T, Lee HW, and Rubin SM (2014). Multiple mechanisms for E2F binding inhibition by phosphorylation of the retinoblastoma protein C-terminal domain. J Mol Biol 426, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, and Lees JA (2010). Rb regulates fate choice and lineage commitment in vivo. Nature 466, 1110–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chellappan SP, Hiebert S, Mudryj M, Horowitz JM, and Nevins JR (1991). The E2F transcription factor is a cellular target for the RB protein. Cell 65, 1053–1061. [DOI] [PubMed] [Google Scholar]
- Chen WS, Alshalalfa M, Zhao SG, Liu Y, Mahal BA, Quigley DA, Wei T, Davicioni E, Rebbeck TR, Kantoff PW, et al. (2019). Novel RB1-Loss Transcriptomic Signature Is Associated with Poor Clinical Outcomes across Cancer Types. Clin Cancer Res 25, 4290–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, and Lowe SW (2010). Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17, 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium, E.P. (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook R, Zoumpoulidou G, Luczynski MT, Rieger S, Moquet J, Spanswick VJ, Hartley JA, Rothkamm K, Huang PH, and Mittnacht S (2015). Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep 10, 2006–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, Goodrich DW, Sage J, and Dyson NJ (2018). Non-canonical functions of the RB protein in cancer. Nat Rev Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick FA, and Rubin SM (2013). Molecular mechanisms underlying RB protein function. Nat Rev Mol Cell Biol 14, 297–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K, Luban J, Begemann M, Crabtree GR, and Goff SP (1994). The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell 79, 119–130. [DOI] [PubMed] [Google Scholar]
- Dyson N (1998). The regulation of E2F by pRB-family proteins. Genes Dev 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- Dyson NJ (2016). RB1: a prototype tumor suppressor and an enigma. Genes Dev 30, 1492–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertel A, Dean JL, Rui H, Liu C, Witkiewicz AK, Knudsen KE, and Knudsen ES (2010). RB-pathway disruption in breast cancer: differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 9, 4153–4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KL, McClellan KA, Vanderluit JL, McIntosh WC, Schuurmans C, Polleux F, and Slack RS (2005). A cell-autonomous requirement for the cell cycle regulatory protein, Rb, in neuronal migration. EMBO J 24, 4381–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, et al. (2002). Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J 21, 3337–3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari R, Gou D, Jawdekar G, Johnson SA, Nava M, Su T, Yousef AF, Zemke NR, Pellegrini M, Kurdistani SK, et al. (2014). Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 16, 663–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanem N, Andrusiak MG, Svoboda D, Al Lafi SM, Julian LM, McClellan KA, De Repentigny Y, Kothary R, Ekker M, Blais A, et al. (2012). The Rb/E2F pathway modulates neurogenesis through direct regulation of the Dlx1/Dlx2 bigene cluster. J Neurosci 32, 8219–8230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Vasconcellos I, Schneider R, Anastasov N, Alonso-Rodriguez S, Sanli-Bonazzi B, Fernandez JL, and Atkinson MJ (2017). The Rb1 tumour suppressor gene modifies telomeric chromatin architecture by regulating TERRA expression. Sci Rep 7, 42056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagemeier C, Bannister AJ, Cook A, and Kouzarides T (1993). The activation domain of transcription factor PU.1 binds the retinoblastoma (RB) protein and the transcription factor TFIID in vitro: RB shows sequence similarity to TFIID and TFIIB. Proc Natl Acad Sci U S A 90, 1580–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Molecular Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando E, Nahle Z, Juan G, Diaz-Rodriguez E, Alaminos M, Hemann M, Michel L, Mittal V, Gerald W, Benezra R, et al. (2004). Rb inactivation promotes genomic instability by uncoupling cell cycle progression from mitotic control. Nature 430, 797–802. [DOI] [PubMed] [Google Scholar]
- Ianari A, Natale T, Calo E, Ferretti E, Alesse E, Screpanti I, Haigis K, Gulino A, and Lees JA (2009). Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell 15, 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishak CA, Marshall AE, Passos DT, White CR, Kim SJ, Cecchini MJ, Ferwati S, MacDonald WA, Howlett CJ, Welch ID, et al. (2016). An RB-EZH2 Complex Mediates Silencing of Repetitive DNA Sequences. Mol Cell 64, 1074–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF, Cecchini MJ, Spacek D, Batista LF, O’Brien M, et al. (2015). Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent LN, and Leone G (2019). The broken cycle: E2F dysfunction in cancer. Nat Rev Cancer 19, 326–338. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Onwuta US, Lee YI, Li R, Botchan MR, and Robbins PD (1992). The retinoblastoma gene product regulates Sp1-mediated transcription. Mol Cell Biol 12, 2455–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, and Knudsen KE (2008). Tailoring to RB: tumour suppressor status and therapeutic response. Nat Rev Cancer 8, 714–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi Y, Tominaga M, Watanabe Y, Imamura F, Goldfarb A, Maki R, Blum M, De Robertis EM, and Tominaga A (1999). GOOSECOID inhibits erythrocyte differentiation by competing with Rb for PU.1 binding in murine cells. Oncogene 18, 6795–6805. [DOI] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbe DP, Gomez EC, Wang J, et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JS, Everetts NJ, Wang X, Wang W, Della Croce K, Xing J, and Yao G (2017). Controlling Depth of Cellular Quiescence by an Rb-E2F Network Switch. Cell Rep 20, 3223–3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, et al. (2012). ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res 22, 1813–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu J, Yang X, Zhou C, Guo J, Wu C, Qin Y, Guo L, He J, Yu S, et al. (2017). Chromatin Accessibility Dynamics during iPSC Reprogramming. Cell Stem Cell 21, 819–833 e816. [DOI] [PubMed] [Google Scholar]
- Liao Y, Wang J, Jaehnig EJ, Shi Z, and Zhang B (2019). WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res 47, W199–W205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT, Ohgi KA, Benner C, Garcia-Bassets I, Aggarwal AK, et al. (2010). PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature 466, 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Longworth MS, and Dyson NJ (2010). Loss of pRB causes centromere dysfunction and chromosomal instability. Genes Dev 24, 1364–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning AL, Yazinski SA, Nicolay B, Bryll A, Zou L, and Dyson NJ (2014). Suppression of genome instability in pRB-deficient cells by enhancement of chromosome cohesion. Mol Cell 53, 993–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markey MP, Bergseid J, Bosco EE, Stengel K, Xu H, Mayhew CN, Schwemberger SJ, Braden WA, Jiang Y, Babcock GF, et al. (2007). Loss of the retinoblastoma tumor suppressor: differential action on transcriptional programs related to cell cycle control and immune function. Oncogene 26, 6307–6318. [DOI] [PubMed] [Google Scholar]
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, et al. (2011). The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc Natl Acad Sci U S A 108, 3665–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittnacht S, and Weinberg RA (1991). G1/S phosphorylation of the retinoblastoma protein is associated with an altered affinity for the nuclear compartment. Cell 65, 381–393. [DOI] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, Wongvipat J, Ku SY, Gao D, Cao Z, et al. (2017). SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, and Dowdy SF (2014). Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nead MA, Baglia LA, Antinore MJ, Ludlow JW, and McCance DJ (1998). Rb binds c-Jun and activates transcription. EMBO J 17, 2342–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolay BN, Danielian PS, Kottakis F, Lapek JD Jr., Sanidas I, Miles WO, Dehnad M, Tschop K, Gierut JJ, Manning AL, et al. (2015). Proteomic analysis of pRb loss highlights a signature of decreased mitochondrial oxidative phosphorylation. Genes Dev 29, 1875–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, et al. (2015). RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun 6, 6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitani J, Nishinaka T, Cheng CH, Rong W, Yokoyama KK, and Chiu R (1999). Recruitment of the retinoblastoma protein to c-Jun enhances transcription activity mediated through the AP-1 binding site. J Biol Chem 274, 5454–5461. [DOI] [PubMed] [Google Scholar]
- Olive M, Krylov D, Echlin DR, Gardner K, Taparowsky E, and Vinson C (1997). A dominant negative to activation protein-1 (AP1) that abolishes DNA binding and inhibits oncogenesis. J Biol Chem 272, 18586–18594. [DOI] [PubMed] [Google Scholar]
- Parisi T, Balsamo M, Gertler F, and Lees JA (2018). The Rb tumor suppressor regulates epithelial cell migration and polarity. Mol Carcinog 57, 1640–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattschull G, Walz S, Grundl M, Schwab M, Ruhl E, Baluapuri A, Cindric-Vranesic A, Kneitz S, Wolf E, Ade CP, et al. (2019). The Myb-MuvB Complex Is Required for YAP-Dependent Transcription of Mitotic Genes. Cell Rep 27, 3533–3546 e3537. [DOI] [PubMed] [Google Scholar]
- Pearson JD, Huang K, Pacal M, McCurdy SR, Lu S, Aubry A, Yu T, Wadosky KM, Zhang L, Wang T, et al. (2021). Binary pan-cancer classes with distinct vulnerabilities defined by pro- or anti-cancer YAP/TEAD activity. Cancer Cell 39, 1115–1134 e1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanstiel DH, Van Bortle K, Spacek D, Hess GT, Shamim MS, Machol I, Love MI, Aiden EL, Bassik MC, and Snyder MP (2017). Static and Dynamic DNA Loops form AP-1-Bound Activation Hubs during Macrophage Development. Mol Cell 67, 1037–1048 e1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics, C., Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin SM, Sage J, and Skotheim JM (2020). Integrating Old and New Paradigms of G1/S Control. Mol Cell 80, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanidas I, Morris R, Fella KA, Rumde PH, Boukhali M, Tai EC, Ting DT, Lawrence MS, Haas W, and Dyson NJ (2019). A Code of Mono-phosphorylation Modulates the Function of RB. Mol Cell 73, 985–1000 e1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, and Hinds PW (2001). The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell 8, 303–316. [DOI] [PubMed] [Google Scholar]
- Trouche D, Le Chalony C, Muchardt C, Yaniv M, and Kouzarides T (1997). RB and hbrm cooperate to repress the activation functions of E2F1. Proc Natl Acad Sci U S A 94, 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbosch R, Clark A, Fong BC, Omais S, Jaafar C, Dugal-Tessier D, Dhaliwal J, Lagace DC, Park DS, Ghanem N, et al. (2016). RB regulates the production and the survival of newborn neurons in the embryonic and adult dentate gyrus. Hippocampus 26, 1379–1392. [DOI] [PubMed] [Google Scholar]
- Velez-Cruz R, Manickavinayaham S, Biswas AK, Clary RW, Premkumar T, Cole F, and Johnson DG (2016). RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev 30, 2500–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vierbuchen T, Ling E, Cowley CJ, Couch CH, Wang X, Harmin DA, Roberts CWM, and Greenberg ME (2017). AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol Cell 68, 1067–1082 e1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss K, Van der Auwera G, and Gentry J (2017). Full-stack genomics pipelining with GATK4 + WDL + Cromwell. In F1000Research ( 10.7490/f1000research.1114634.1). [DOI] [Google Scholar]
- Walter DM, Yates TJ, Ruiz-Torres M, Kim-Kiselak C, Gudiel AA, Deshpande C, Wang WZ, Cicchini M, Stokes KL, Tobias JW, et al. (2019). RB constrains lineage fidelity and multiple stages of tumour progression and metastasis. Nature 569, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Petryniak B, Thompson CB, Kaelin WG, and Leiden JM (1993). Regulation of the Ets-related transcription factor Elf-1 by binding to the retinoblastoma protein. Science 260, 1330–1335. [DOI] [PubMed] [Google Scholar]
- Wang S, Zang C, Xiao T, Fan J, Mei S, Qin Q, Wu Q, Li X, Xu K, He HH, et al. (2016). Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Res 26, 1417–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Civelek M, Miller CL, Sheffield NC, Guertin MJ, and Zang C (2018). BART: a transcription factor prediction tool with query gene sets or epigenomic profiles. Bioinformatics 34, 2867–2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AC, Cejas P, DeCristo MJ, Metzger-Filho O, Lam EYN, Qiu X, BrinJones H, Kesten N, Coulson R, Font-Tello A, et al. (2021). CDK4/6 inhibition reprograms the breast cancer enhancer landscape by stimulating AP-1 transcriptional activity. Nature Cancer 2, 34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA (1995). The retinoblastoma protein and cell cycle control. Cell 81, 323–330. [DOI] [PubMed] [Google Scholar]
- Wells J, Yan PS, Cechvala M, Huang T, and Farnham PJ (2003). Identification of novel pRb binding sites using CpG microarrays suggests that E2F recruits pRb to specific genomic sites during S phase. Oncogene 22, 1445–1460. [DOI] [PubMed] [Google Scholar]
- Wu CL, Classon M, Dyson N, and Harlow E (1996). Expression of dominant-negative mutant DP-1 blocks cell cycle progression in G1. Mol Cell Biol 16, 3698–3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G, Lee TJ, Mori S, Nevins JR, and You L (2008). A bistable Rb-E2F switch underlies the restriction point. Nat Cell Biol 10, 476–482. [DOI] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, and Piccolo S (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 17, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, Chen C-H, Brown M, Zhang X, Meyer CA, et al. (2019). Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Research 47, D729–D735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods S1: PanChIP manual v3.0.10. Related to Figure 5 and Star Methods.
Table S1: Homer motif analysis of Promoters-1, Promoters-2 and non-promoters k-means clusters of RB peaks. Related to Figure 1.
Table S2: GSEA analysis of the genes with RB-bound promoters (promoters-1 and promoters-2 clusters) or genes in proximity to RB peaks located in non-promoter regions. Related to Figure 1.
Table S3: Over-Representation analysis of genes in proximity to RB peaks located in the different k-means clusters presenting in Fig. 2A. The top ten most significant Biological Processes and KEGG pathways are shown for each set of genes. Related to Figure 2.
Table S4: Homer motif analysis of non-promoters RB peaks in BJ and RPE1 cells. Related to figure 2.
Table S5: Over-Representation analysis of genes in proximity to RB peaks that are highlighted in Fig. 3C and 3E. The top 10 most significant Biological Processes and KEGG pathways are shown for each set of genes. Related to Figure 3.
Table S6: Co-ordinates of the RB peaks in the enhancers that are highlighted in Fig. 4A. The gene that it is in proximity to each RB-bound enhancer is shown. Related to Figure 4.
Table S7: Over-Representation analysis of the genes with RB-dependent enhancers that identified in Fig. 4A and Table S6. The top 10 most significant Biological Processes are shown. Related to Figure 4.
Table S8: Over-Representation analysis of the different sets of genes with RB-bound enhancers that are highlighted in Fig. 4C. The top 10 most significant Biological Processes and KEGG pathways are shown for each set of genes. Related to Figure 4.
Table S9: Normalized overlap scores of 915 chromatin-associated proteins in different groups of RB ChIP-seq peaks (k-means clusters A–H). Statistical significance measures, including the p-value and the signal-to-noise ratio, are presented for RB-bound promoters, RB/CTCF sites, and RB-bound enhancers. Raw data for the generation of heatmap in Fig. 5B are also shown. Related to Figure 5.
Data Availability Statement
ChIP sequencing data have been deposited at GEO and are publicly available as of the date of publication. Original western blot images have been deposited at Mendeley and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
The original code for PanChIP has been deposited at Zenodo and is publicly available as of the date of publication. DOI is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| anti-DYKDDDDK Tag (Binds to same epitope as Sigma’s Anti-FLAG® M2 Antibody) | Cell Signaling Technology | Cat# 14793, RRID:AB_2572291 |
| anti-E2F1 | Cell Signaling Technology | Cat# 3742, RRID:AB_2096936 |
| anti-Histone H3 (Tri-Methyl-Lys4) | Cell Signaling Technology | Cat# 9751, RRID:AB_2616028 |
| anti-Histone H3 (Mono-Methyl-Lys4) | Abcam | Cat# ab8895, RRID:AB_306847 |
| anti-Histone H3 (Acetyl-Lys27) | Active Motif | Cat# 39034 |
| anti-c-Jun | Cell Signaling Technology | Cat# 9165, RRID:AB_2130165 |
| anti-CTCF | Cell Signaling Technology | Cat# 3418, RRID:AB_2086791 |
| anti-RB | Cell Signaling Technology | Cat# 9309S, RRID:AB_10696874 |
| anti-Phospho-RB S608 | Cell Signaling Technology | Cat# 8147S, RRID:AB_10949974 |
| anti-Phospho-RB S780 | Cell Signaling Technology | Cat# 8180S, RRID:AB_10950972 |
| anti-Phospho-RB S795 | Cell Signaling Technology | Cat# 9301P, RRID:AB_10830074 |
| anti-Phospho-RB S807/S811 | Cell Signaling Technology | Cat# 8516S, RRID:AB_11178658 |
| anti-Phospho-RB T373 | Abcam | Cat# ab52975, RRID:AB_2177344 |
| anti-a-Tubulin | Sigma-Aldrich | Cat# T9026, RRID:AB_477593 |
| anti-FLAG (M5) | Sigma-Aldrich | Cat# F4042, RRID:AB_439686 |
| anti-HA-Tag | Cell Signaling Technology | Cat# 3724, RRID:AB_1549585 |
| anti-p44/42 MAPK (Erk1/2) | Cell Signaling Technology | Cat# 9102, RRID:AB_330744 |
| anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) | Cell Signaling Technology | Cat# 4370, RRID:AB_2315112 |
| anti-Cyclin D1 | Santa Cruz Biotechnology | Cat# sc-753, RRID:AB_2070433 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Doxycycline hyclate | Sigma-Aldrich | Cat# D9891 |
| Trametinib (GSK1120212) | Selleckchem | Cat# S2673 |
| Gateway LR Clonase II Enzyme mix | Invitrogen | Cat# 11791-020 |
| X-tremeGENE 9 DNA Transfection Reagent | Roche | Cat# 6365787001 |
| Polybrene | Sigma-Aldrich | Cat# 107689 |
| Geneticin | Gibco | Cat# 10131035 |
| Puromycin | Sigma-Aldrich | Cat# P9620 |
| EdU (5-ethynyl-2’-deoxyuridine) | Invitrogen | Cat# A10044 |
| Alexa Fluor 647 Azide, Triethylammonium Salt | Invitrogen | Cat# A10277 |
| DAPI (4′,6-Diamidino-2-Phenylindole, Dihydrochloride) | Invitrogen | Cat# D1306 |
| FastStart Universal SYBR Green Master | Roche | Cat# 4913914001 |
| EGS (ethylene glycol bis(succinimidyl succinate)) | Thermo Scientific | Cat# 21565 |
| RNase A | Roche | Cat# 11119915001 |
| Proteinase K | Thermo Scientific | Cat# 25530049 |
| Agencourt AMPure XP | Beckman Coulter | Cat# A63881 |
| Protein G-Dynabeads | Life Technologies | Cat# 10004D |
| Critical Commercial Assays | ||
| pENTR™/D-TOPO Cloning Kit | Invitrogen | Cat# K240020 |
| Direct-zol RNA Kit | Zymo Research | Cat# R2073 |
| TaqMan Reverse Transcription Reagents | Thermo Fisher Scientific | Cat# N8080234 |
| Qubit dsDNA HS Assay kit | Thermo Fisher Scientific | Cat# Q32851 |
| Ovation Ultralow System V2 | TECAN | Cat# 0344NB-A01 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | Cat# 20024906 |
| Deposited Data | ||
| ChIP sequencing data | This paper | GEO: GSE176035 |
| Original images for Western Blots | This paper | DOI: 10.17632/p2x5xr4vt2.1 |
| RB ChIP-seq data from K562 cell line | ENCODE Project | ENCODE accession: ENCSR670JDQ GEO accession: GSE105638 |
| RB ChIP-seq data from GM12878 cell line | ENCODE Project | ENCODE accession: ENCSR785OKZ GEO accession: GSE106046 |
| Experimental Models: Cell Lines | ||
| hTERT-RPE1 human epithelial cells | Dr. David Pellman’s laboratory, Dana Farber Cancer Institute | RRID:CVCL_4388 |
| hTERT-BJ1 human fibroblasts | ATCC | Cat# CRL-4001, RRID:CVCL_6573 |
| MCF10A human mammary epithelial cells | Dr. Lee Zou’s laboratory, MGH | RRID:CVCL_0598 |
| T47D human breast cancer | Center for Molecular Therapeutics - MGH | RRID:CVCL_0553 |
| SW1573 human lung adenocarcinoma | Center for Molecular Therapeutics - MGH | RRID:CVCL_1720 |
| NCI-H2030 human lung adenocarcinoma | Center for Molecular Therapeutics - MGH | RRID:CVCL_1517 |
| CALU-1 human lung squamous cell carcinoma | Center for Molecular Therapeutics - MGH | RRID:CVCL_0608 |
| HOP62 human lung adenocarcinoma | Center for Molecular Therapeutics - MGH | RRID:CVCL_1285 |
| Oligonucleotides | ||
| Primers for cloning A-FOS and DNDP1 cDNA in pENTR/D-TOPO, see Table S8 | This paper | N/A |
| Real Time RT-PCR primers, see Table S8 | MGH Primer Bank database | N/A |
| Recombinant DNA | ||
| pINDUCER11 | (Meerbrey et al., 2011) | Addgene plasmid# 44363 |
| pINDUCER20 | (Meerbrey et al., 2011) | Addgene plasmid# 44012 |
| pLenti-CMVtight-puro-Dest | gift from Eric Campeau | Addgene plasmid# 26430 |
| pLenti-CMVtight-eGFP-puro | gift from Eric Campeau | Addgene plasmid# 26431 |
| pINDUCER11-shRB1 (5’-CAGAGATCGTGTATTGAGATT-3’) | This paper | N/A |
| pINDUCER11-shScramble (5’-ACTAAGGTTAAGTCGCCCTCGA -3’) | This paper | N/A |
| pINDUCER20-RB wild-type | (Sanidas et al., 2019) | N/A |
| pINDUCER20-FLAG-RB wild-type | (Sanidas et al., 2019) | N/A |
| pINDUCER20-RB Δcdk | (Sanidas et al., 2019) | N/A |
| pINDUCER20-FLAG-RB Δcdk | (Sanidas et al., 2019) | N/A |
| pINDUCER20-FLAG-mP-RB mutant alleles | (Sanidas et al., 2019) | N/A |
| pLenti-CMVtight-HA-A-FOS-puro | This paper | N/A |
| pLenti-CMVtight-HA-DNDP1-puro | This paper | N/A |
| Software and Algorithms | ||
| Geneset enrichment analysis (GSEA) | (Subramanian et al., 2005) | N/A |
| HOMER | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/ |
| WebGestalt | (Liao et al., 2019) | http://www.webgestalt.org |
| Prism 6 | GraphPad | https://www.graphpad.com |
| OriginPro | Origin Lab | https://www.originlab.com/ |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| Python | N/A | https://www.python.org/ |
| gdown (Python package) | Kentaro Wada | https://github.com/wkentaro/gdown |
| argparse (Python package) | Ben West | https://github.com/bewest/argparse |
| R | N/A | https://www.r-project.org/ |
| ggplot2 (R package) | N/A | https://cran.r-project.org/web/packages/ggplot2/index.html |
| leaflet (R package) | N/A | https://cran.r-project.org/web/packages/leaflet/index.html |
| RColorBrewer (R package) | N/A | https://cran.r-project.org/web/packages/RColorBrewer/index.html |
| DiffBind (R package) | N/A | https://doi.org/doi:10.18129/B9.bioc.DiffBind |
| BEDTools | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| ENCODE | (An integrated encyclopedia of DNA elements in the human genome, 2012) | https://www.encodeproject.org/ |
| Cistrome Data Browser | (Zheng et al., 2019) | http://cistrome.org/db/ |
| BART2 | (Wang et al., 2018) | https://github.com/zanglab/bart2 |
| MARGE | (Wang et al., 2016) | http://cistrome.org/MARGE |
| Cromwell | (Voss et al., 2017) | https://doi.org/10.7490/f1000research.1114634.1 |
| ENCODE ChIP-seq Pipeline | (Landt et al., 2012) | https://github.com/ENCODE-DCC/chip-seq-pipeline2 |
| PanChIP | This paper | DOI: 10.5281/zenodo.6787997 |