

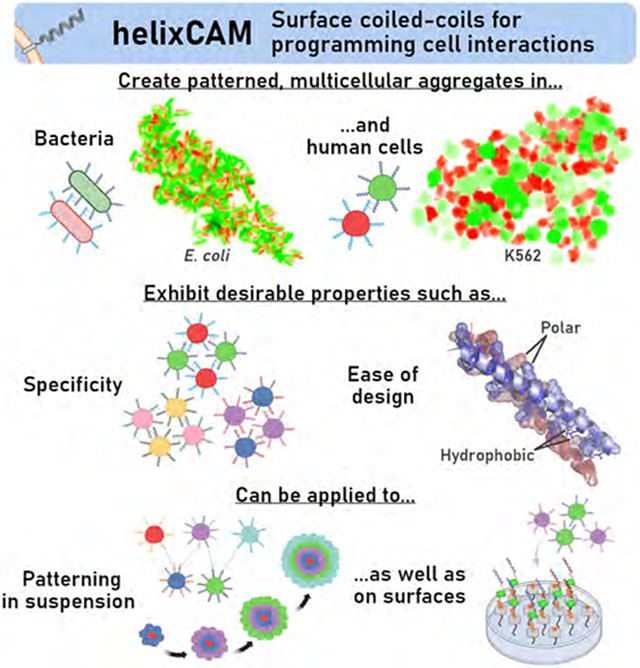
Summary
Interactions between cells are indispensable for signaling and creating structure. The ability to direct precise cell-cell interactions would be powerful for engineering tissues, understanding signaling pathways, and directing immune cell targeting. In humans, intercellular interactions are mediated by cell adhesion molecules (CAMs). However, endogenous CAMs are natively expressed by many cells and tend to have cross-reactivity, making them unsuitable for programming specific interactions. Here, we showcase “helixCAM,” a platform for engineering synthetic CAMs by presenting coiled-coil peptides on the cell surface. helixCAMs were able to create specific cell-cell interactions and direct patterned aggregate formation in bacteria and human cells. Based on coiled-coil interaction principles, we built a set of rationally designed helixCAM libraries, which led to the discovery of additional high-performance helixCAM pairs. We applied this helixCAM toolkit for various multicellular engineering applications, such as spherical layering, adherent cell targeting, and surface patterning.
Keywords: Cell Interaction, Cell Patterning, Cell Aggregation, Cell Targeting, Membrane Protein, Protein Engineering, Rational Design, Image Processing, Tissue Engineering, Mammalian Synthetic Biology
Graphical Abstract
In Brief:
Orthogonally interacting engineered pairs of coiled coils on the surface of cells enable specific cell-cell or cell-surface interactions for the controlled formation of complex multicellular structures and patterns from bacterial and human cells.
Introduction
Intercellular interactions are an indispensable part of living organisms, whether unicellular or multicellular. In bacteria, adhesins enable exchange of genetic material (Lederberg and Tatum, 1946), initiate binding to host cells for infection (Fr̈mmel et al., 2013), and mediate formation of biofilms (Anderson et al., 2003). For multicellular organisms, cell-cell interactions dictate a range of important physiological functions, including stem cell differentiation (Yim and Sheetz, 2012), morphological development and maintenance (Zeller, López-Ríos and Zuniga, 2009), and the activation of immune responses (Friedl and Storim, 2004).
The ability to program specific cell-cell interactions has numerous applications. For instance, the aggregation of bacterial cells can be used to create living biomaterials with the ability to release antibiotics (Guo et al., 2020) or undergo self-repair (H.-J. Chen et al., 2019). In human cell systems, programmed interactions between immune cells could be used to study T-cells activation (Roybal and Lim, 2017). Additionally, by using intercellular interactions to aggregate and pattern human cells, it may be possible to build synthetic tissues with potential for clinical use (Hsiao et al., 2009). Current approaches to constructing synthetic tissues composed of multiple cell types primarily rely on nozzle-based cell printing of terminally-differentiated cells (Murphy and Atala, 2014; Kolesky et al., 2016; Roohani-Esfahani, Newman and Zreiqat, 2016; Moroni et al., 2018). This method of creating cell structures faces significant obstacles from low cell viability due to shear stress and difficulty for differentiated cells to form junctions after deposition (Ozbolat and Yu, 2013; Yu et al., 2013). Rather than the top-down approach of printing cells, allowing cells to form patterned structures through selective binding could resolve both limitations and enable the synthetic construction of more complex, accurate, and viable tissues. Programmed interactions can also be extended to use for direct patterning of cells onto surfaces and targeting of immune cells to cancer (Piñero-Lambea et al., 2015).
Some approaches exist to artificially direct cell-cell interactions. By overexpressing two cadherins in mammalian cells, Cachat et al. and Shan et al. were able to assemble large cell aggregates (Shan et al., 2000; Cachat et al., 2016). Native cell adhesion molecules (CAMs) such as cadherins, however, are few in number, have significant cross-activity (Patel et al., 2003), and often can play dual roles as downstream signaling molecules(Gumbiner, 1996), limiting their use for directing multiple interactions in parallel. Another approach is to conjugate the cell membrane with single-stranded DNA (ssDNA), allowing cells to selectively interact via base-pairing (Chandra et al., 2006). In this case, limitations arise in the stability of extracellular DNA (Bagheri et al., 2019) as well as the need to chemically treat each cell population, preventing the control of cell interactions through genetic programs or small molecule inducers. A genetically-encoded mechanism to control cell-cell interactions was recently demonstrated in E. coli, using surface-mounted nanobodies and antigens (Glass and Riedel-Kruse, 2018). This groundbreaking approach enables the collection of known nanobody-antigen pairs to be repurposed for programming cell-cell interactions in E. coli and opens the door to building highly complex microbial biomaterials.
One attractive alternative for protein-mediated cell interaction is coiled-coils (CCs), well-studied protein domains with established roles in mediating protein-protein interactions across numerous native proteins (Truebestein and Leonard, 2016). Each CC consists of multiple seven amino-acid repeats, termed “heptads,” which chain together to form alpha-helical secondary structures (Hicks et al., 1997). The specific residues forming the heptads determine the orientation, binding multiplicity, and specificity of each CC (Woolfson, 2005). The potential of these short peptides to mediate interactions between micron-scale objects has been demonstrated by Obana et al., who aggregated polystyrene beads functionalized with CCs (Obana, Silverman and Tirrell, 2017). Veiga et al. (Veiga, De Lorenzo and Fernández, 2003) further demonstrated the potential of using CCs for synthetic adhesions by fusing the CC pair Jun/Fos to the β-autotransporter domain of the IgA protease and demonstrating aggregation in E. coli. Due to their well-understood pairing rules, collections of orthogonal CC peptide pairs have been engineered and characterized by numerous research groups (Thompson et al., 2012; Fink et al., 2018; Z. Chen et al., 2019; Lebar et al., 2020), making them a potential treasure trove for inducing specific cell-cell interactions.
We present an extensible framework for creating strong and programmable cell-cell interactions through cell surface presentation of CCs fused to a transmembrane domain (TMD), which we term “helixCAM” (Figure 1A). Within this work, we find that helixCAMs direct selective binding of bacteria and human cells into patterned aggregates and engineer additional CC peptides for helixCAM use, for a total of five pairs. Using these, we implement helixCAMs for a wide range of applications, such as specifying the spatial composition of spherical aggregates, targeting suspension cells to adherent cells, and patterning helixCAM cells with His-tagged CC peptides. The helixCAM platform is a foundational technology that empowers researchers with simultaneous control of multiple specific cell interactions. We expect scientists and engineers across biological disciplines to find compelling applications for helixCAMs in their work, and for the set of orthogonal helixCAMs to grow and open the door to programming increasingly complex interactions.
Figure 1. Overview of helixCAM design and applications and demonstration with Z17/Z18.

A. Schematic of the helixCAM concept and potential applications. B. Design of helixCAMs in E. coli: the coiled-coil domain is fused to the EhaA autotransporter protein for surface presentation. C. helixCAM-induced E. coli aggregation, with Z17 cells co-expressing mCherry, and Z18 cells co-expressing eGFP. Representative views are shown from several fields of view. Images were taken at 60X magnification and cropped (uncropped images and more fields of views in S2). D. Design of mammalian helixCAMs: the coiled-coil domain is fused to the PDGFR transmembrane domain. E. helixCAM-induced K562 aggregation, with Z17 cells co-expressing mCherry, and Z18 cells co-expressing eGFP. Representative views are shown from several fields of view. Image was taken at 20X magnification with 3x3 tiling and cropped (uncropped images in S3). F. 3D reconstruction of helixCAM-induced K562 aggregates of various sizes, demonstrating heterodimeric three-dimensional patterning. Images were taken at 40X magnification using a spinning disc confocal system as a Z-stack and reconstructed using ImageJ.
Results
helixCAMs induced large multicellular aggregates in E. coli
To pilot helixCAMs as a cell-cell interaction platform, the antiparallel heterodimeric CC pair Z17 and Z18 was selected from the SynZip library (Thompson et al., 2012). To generate helixCAMs in E. coli, CCs were fused to the N-terminus of the E. coli EhaA autotransporter adhesin protein (Salema et al., 2013) (Figure 1B, S1), and expressed under the IPTG-inducible T5/lac promoter (Dubendorf and Studier, 1991). We tested these bacterial helixCAMs in two distinct strains: one expressing the Z17 helixCAM along with the mCherry fluorescent protein, and another expressing the Z18 helixCAM alongside eGFP. When the two populations were mixed without arabinose induction, bacterial cells existed mostly in singlets and small aggregates of up to ten cells, whereas with arabinose induction we observed formation of large bacterial aggregates containing hundreds of cells and spanning up to 40μm in length. The images shown in Figure 1C are representative of aggregates observed across multiple fields of view (uncropped image and additional fields of views in S2). Aggregates were consistently composed of cells from both strains, primarily in an alternating pattern, demonstrating the heterodimerization effect.
HelixCAMs induced large, predictably-patterned multicellular aggregates in human K562 cells
We next sought to adapt the helixCAM system in human cells. To this end, we inserted the Z17 and Z18 CCs at the N-terminus of the platelet-derived growth factor receptor transmembrane domain and C-terminus of the secretion signaling peptide from human Immunoglobulin K, following the design from Chesnut et al. (Figure 1D)(Chesnut et al., 1996). Expression of human helixCAMs was placed under the Tet-On doxycycline-inducible promoter (Urlinger et al., 2000) along with constitutive expression of an identifying fluorescent protein under the EF1α promoter (Qin et al., 2010) and flanked with PiggyBac sites for stable line generation (Wilson, Coates and George, 2007) (S1).
We then generated stable cell lines for each helixCAM in human K562 cells, an immortalized leukemia cell line (Naumann et al., 2001) selected for their lack of innate cell adhesion and spherical shape conducive to efficient packing. Similar to our experiments in E. coli, we tested the capability of helixCAMs to aggregate human cells by mixing the Z17mCherry cells with Z18eGFP cells. Without induction, cells remained as single-cell suspensions, but after 48 hours of induction, cells formed large, tight clusters with an alternating pattern of mCherry and GFP indicative of heterodimerization (Figure 1E, S3, S4). To quantify the aggregative effect, aggregates sizes from the brightfield image was measured, showing a significant increase in the mean aggregate size (number of cells within an aggregate) over the uninduced condition (p-value = 1.64×10−16, S5). Visualizing the three-dimensional structure from confocal imaging reconstructions (Figure 1F), we observed that helixCAM proteins induced binding interfaces between cells that are junction-like in appearance. Smaller aggregates of four to ten cells formed structures in which each cell bound exclusively to cells expressing the complementary helixCAM, forming clear checkboard patterns. At higher cell counts, the alternating pattern is present but less precise, and aggregates spanned multiple cell layers in X, Y, and Z directions, forming three-dimensional aggregates spanning hundreds of microns in each direction. These data demonstrate that helixCAMs can form strong cell-cell interactions in human cell lines independently of endogenous CAM proteins.
Orthogonal helixCAM pairs enable programmable sub-aggregation within a mixed population
From the promising results of Z17/Z18, we designed four additional helixCAMs using the CC pairs P3/AP4 and P9/AP10, which were previously reported to have high affinity and orthogonality (Fink et al., 2018; Lebar et al., 2020). We first tested these helixCAMs in E. coli using a sedimentation rate assay (Trunk, Khalil and Leo, 2018) and observed faster settling when mixing E. coli expressing the intended CC pairs, indicating the formation of larger aggregates and thus higher CC affinity (S6). We also confirmed aggregate formation via microscopy (S2, S4) and induced change in aggregate size (S5). To test these helixCAMs in human cells, we generated four additional helixCAM K562 lines and denoted each helixCAM line with a corresponding fluorescent protein (P3iRFP670, AP4eBFP2, P9mPlum, and AP10mOrange). As in the case of Z17/Z18, both P3iRFP670/AP4eBFP2 cell lines (Figure 2A) and P9mPlum/AP10mOrange cell lines (Figure 2B) formed cell aggregates when induced to express helixCAMs in paired co-cultures.
Figure 2. Characterization of additional helixCAM pairs and three-pair interaction orthogonality.

A. K562 cell aggregates formed by P3/AP4 interactions. P3 cells co-express iRFP670 and AP4 cells co-express eBFP2. Image was taken at 20X magnification and cropped (uncropped image in S3). B. K562 cell aggregates formed by P9/AP10 interaction. P9 cells co-express mPlum, and AP10 cells co-express mOrange. Image was taken at 20X magnification and cropped (uncropped image in S3). C. Z17mCherry, Z18eGFP, P3iRFP670, AP4eBFP2, P9mPlum, and AP10mOrange cells were induced to express helixCAMs in a single mixed culture. Clear sub-clusters can be observed. In this image, mCherry appears more orange and mOrange appears more yellow due to the filter sets used. Image was taken at 20X with a 6x6 tile across five channels and cropped to show regions of interest (uncropped image in S8). D. Interaction frequency table was derived from the uncropped three-pair co-culture image. Analysis pipeline detailed in S9.
Next, we sought to examine the orthogonality of these interactions when all three pairs are present in a single culture. To this end, the six cell lines were seeded together at identical concentrations and helixCAM expression was induced for 48 hours. Microscopic images of the resulting multicellular assemblies showed clear binding preferences between complementary helixCAM cells (Figure 2C, uninduced image in S7). Individual aggregates comprised primarily of pairs of complementary helixCAM cells, and, while larger aggregates were composed of more cell types, indicating some promiscuous binding, distinct sub-clusters were still evident.
To quantify the orthogonality of the interactions in the structures, we performed image segmentation of a six-by-six field of view (S8) and estimated the frequency of interaction between each of the six cell types (Figure 2D). Four cell lines, Z17mCherry, Z18eGFP, P9mOrange, and AP10mPlum, showed a clear preference for binding to their complementary partners. However, while the Z17mCherry cells bound complementary Z18eGFP cells at the highest frequency, off-target interactions were also observed with P3iRFP670 and AP4eBFP2. The P9mOrange echoed this trend to a lesser extent, with the highest frequency of interactions with complementary AP10mPlum cells along with a handful of off-target interactions with P3iRFP670 and AP4eBFP2. Notably, the behavior of helixCAMs in human cells contrasted with the higher orthogonality observed in E. coli. (S6). This gap suggested that protein expression and transport may be factors in the ultimate helixCAM function and indicated a need for a eukaryotic screen when engineering CCs for use in helixCAM applications.
Rational design and two-stage screening of helixCAM library yields two additional high-performance helixCAM pairs
To build more complex cellular structures, we sought to expand the set of high-affinity and orthogonal helixCAMs by designing rationally-designed CC peptide libraries based on the four helixCAMs that exhibited high affinity and specificity in K562 cells (Z17, Z18, P9, AP10). As mentioned above, CCs are composed of heptads, within the seven amino acid positions designated as “a” through “g” (Truebestein and Leonard, 2016). As illustrated in Figure 3A, electrostatic interactions in positions “g” and “e” determine binding specificity, whereas hydrophobic residues in positions “a” and “d” interact at the binding interface to influence affinity and stability (Litowski and Hodges, 2002; Gromiha and Parry, 2004). For our CC libraries, we introduced variability in heptad interactions by placing either glutamic acid or lysine residues in the “g” and “e” positions, with the rationale that opposing charges in these positions will increase the likelihood of generating designed pairs with little specificity to the original CCs. We also placed either hydrophobic (leucine/isoleucine) or polar (asparagine) residues in the core binding region (“a” and “d”) to seed variability in baseline affinity.
Figure 3. Rational Design of helixCAM-optimized Coiled-Coil library and two-stage screen for helixCAM performance.

A. Table of amino acid substitutions for each of four template-derived helixCAM libraries. Each template consists of five heptads, within which either the “g”, “a” and “e” position or the “g”, “d”, and “e” positions are varied to form new electrostatic and hydrophobic interactions. B. Design of the tripartite split-GFP assay for CC affinity. One CC library was fused to β-strand 10 and another to β-strand 11 of the tripartite split-GFP. Interaction between CCs stabilizes a fluorescent GFP protein. C. Graph of CC candidate pairs’ frequency (in percent) in the population versus their pair score (graph uses W=0.1). The pair score represents a pair’s specificity to each other and is adjusted with a weight parameter W (Detailed in methods). The top 30 hits using three different W’s, along with several high-frequency pairs (totaling 102 individual CCs) were selected for subsequent screening. D. Design of modified yeast SynAg mating assay for helixCAM-compatible CC selection. Haploid yeast cells MATa or MATα expressing surface-presented CC candidates were mixed. CC binding induces haploid cells to mate, creating a diploid cell that survives dual auxotrophic selection. The fusion of cells also leads to the expression of the Cre recombinase (orange), which integrates the two CC constructs and their barcodes into the same DNA strand. E. Stage 2 CC candidate pairs’ enrichment versus their orthogonality. Enrichment is the log of the observed frequency of the pair as a ratio to their individual frequency in the pre-mated populations, whereas orthogonality is the log of the frequency of the pair divided by the total observations of each of the two CCs in the pair. Two pairs, sg30/sg61 and sg83/sg88, were selected from the red group for helixCAM use. F. Large aggregates of sg30mCherry and sg61eGFP K562 cells. Image was taken at 20X magnification with a 3x3 tile and subsequently cropped. G. Aggregates of sg83iRFP670 and sg88eBFP2 K562 cells. Image was taken at 20X magnification and presented without cropping.
We used Z17, Z18, P9, and AP10 as template CCs to create four distinct libraries. Each CC template was either elongated or truncated to five heptad repeats, and we combinatorially mutated three residues per repeat to one of two amino acids, leading to a library size of 215, or 32,768 members, for each template. Libraries were screened against themselves and all other libraries, resulting in 10 paired libraries, and approximately 10 billion possible interactions. To effectively screen these interactions, we employed a two-stage approach: first, a tripartite split-GFP complementation assay was performed in E. coli to select for CCs candidates exhibiting strong binding. This primary screen was followed by a yeast mating assay to evaluate CC pairs for their ability to induce eukaryotic cell aggregation.
The tripartite split-GFP method was selected for its low background and high throughput readout via FACS (Cabantous et al., 2013; Romei and Boxer, 2019). In our assay, sGFP β-strands 1-9 were constitutively expressed, β-strand 10 was fused to the C-terminus of one CC library, while β-strand 11 was fused to the N-terminus of a second CC library (Figure 3B). All three components were combined into a single plasmid and transformed into E. coli cells as pairwise mixtures. Cells with high GFP signals were sorted (S10), and the selected plasmids were sequenced with next-generation sequencing (NGS) to identify candidate CC pairs. Each pair was assigned a “pair score” (detailed in methods), which positively weighed the frequency of on-target binding (as a metric of affinity) and negatively weighed the frequency of off-target binding. Using this score, 102 candidate CC peptides were selected for the next stage of the screen (Figure 3C).
Considering the potential disconnect between prokaryotic and eukaryotic helixCAM characteristics, the next stage of CC screening was designed to select for the ability to induce aggregation in eukaryotes while maintaining a reasonable screening throughput; the yeast-based SynAg mating assay was a good fit (Younger et al., 2017). Briefly, MATa and MATα haploid yeast cells, respectively carrying lysine or leucine auxotrophic markers, were transformed with the yeast helixCAM library (S11) comprised of the 102 CC candidates. The two mating types are then incubated in a shaking culture. Yeast cells displaying interacting helixCAMs were promoted to mate, merging the two haploid cells into a diploid cell. Diploid cells gain both auxotrophic markers and are positively selected. Merged diploid yeast cells also began to express the Cre recombinase, which recombined the two helixCAM DNA constructs into a single strand (Figure 3D). After using NGS to examine the barcodes of yeasts that survive selection, we assigned an “enrichment” and an “orthogonality” score for each helixCAM pair and, using these metrics, selected two top candidate CC pairs (sg30/sg61 and sg83/sg88) that were likely to be capable of directing strong and specific cell aggregation (Figure 3E).
To test the library-derived CC pairs as helixCAMs, we built four K562 cell lines that inducibly expressed each and repeated the pairwise aggregation experiment (Figure 3F, 3G, S4). We found that both engineered helixCAM pairs robustly generated patterned cell aggregates by quantifying aggregate size change (S5). In particular, the sg30/sg61 pairing consistently yielded aggregates containing thousands of cells, and some aggregates spanned millimeters in length (S12) and were visible by eye (S13).
“Human cell sedimentation assay” reveals high affinity and specificity in library-derived helixCAMs
With the additional of two library-derived helixCAM pairs, we had ten distinct helixCAMs to test for binding orthogonality. As it is challenging to distinguish beyond eight fluorescent proteins through conventional fluorescent microscopy, an imaging-independent assay was required to determine pairwise affinity across all helixCAMs. Using the same principles of the E. coli sedimentation rate assay, we developed a human cell sedimentation rate assay (HCSRA) to assess aggregation in K562 cells. For this assay, helixCAM-presenting cells were co-cultured in ultra-low-adhesion V-bottom plates for 48 hours and evaluated for settling by measuring cell settling over time through optical density (Figure 4A). The difference between the time at which 50% of the max density is reached for the uninduced cells and the induced cells, Δt50, is used as a metric that positively correlates with aggregate size. A detailed overview of this analysis workflow is shown in S14.
Figure 4. HCSRA measurement of helixCAM affinity and optimized three-pair aggregation.

A. Cartoon of Human Cell Sedimentation Rate Assay (HCSRA). B. Heatmap showing Δt50 values from HCSRA for helixCAM affinity for self, pairwise, and wild-type conditions. Each square represents the mean of three replicates. C. Comparison of Δt50 values from HCSRA to pairwise interaction frequencies from Figure 2D. A linear correlation was observed, with an R2 of 0.624 and p-value of 0.0005, using the F-statistic. D. Bar graph of Δt50 for complementary helixCAM pairs, demonstrating a range of affinity. Error bars represent S.D., N=3. E. Dose curve of helixCAM-induced binding strength for the top four helixCAM pairs. Δt50 for each pair is normalized to each pair’s maximum measured Δt50 value. Positions along the x-axis are slightly shifted for ease of interpretation. Error bars represent S.D., N=4. Absolute Δt50 values and statistical comparison are in S15. F. P9mPlum, AP10mOrange, sg30mCherry, sg61eGFP, sg83iRFP670, and sg88eBFP2 cells were induced to express helixCAM within a single mixed culture. This subset of helixCAMs was selected as the most orthogonal set from HCSRA results. Clear sub-clusters can be observed. Widefield image was taken at 20X with a 5x5 tile across five channels and cropped to show regions of interest (Uncropped image in S17). Confocal image was taken at 40X magnification with a spinning disc confocal system as a Z-stack and reconstructed using Nikon Elements. Due to slight movement of cells during Z-stack acquisition across channels, mOrange cells can appear half yellow and half red. G. Interaction frequency table derived from the optimized three-pair co-culture image. Designed co-localization can be observed in all three helixCAM pairs. Analysis pipeline detailed in S9.
Using the HCSRA, the Δt50 for all self and pairwise helixCAM interactions were measured, along with interaction with wild-type K562 cells (Figure 4B). It was noted that the Δt50 values for interactions between the original six helixCAMs correlated with interaction frequencies derived from microscopy (Figure 4C), with an R2 of 0.624 and a linear model p-value of 0.0005. The agreement of the HCSRA Δt50 values to the microscopy-derived interaction frequency supports the intended function of HCSRA as a readout for the affinity between cells. Additionally, we observed that, in agreement with microscopy observations, both library-derived helixCAM pairs demonstrated high affinity and specificity (Figure 4D). Indeed, the mean HCSRA values measured for the sg30/sg61 pair surpasses that of the previously highest-affinity pair, Z17/Z18 (3.43 to 2.38 respectively, p-value = 0.19).
Using the most orthogonal set of helixCAM pairs (P9/AP10, sg30/sg61, sg83/sg88), we repeated the three-pair co-culture experiment. Similar to the previous set, striking segregation was observed in cell aggregates (Figure 4F, uninduced image in S7). In this case, quantification of interaction frequencies revealed that the new set had notably fewer inter-pair interactions, leading to frequent complementary interactions and few off-target interactions (Figure 4G, S17). These results show that, by utilizing the HCSRA results, it is possible to select multiple compatible helixCAM for directing parallel cell adhesion events within a single culture.
Leveraging the higher throughput enabled by the HCSRA, we further investigated further into the characteristics of the four highest-affinity helixCAM pairs. First, we investigated whether the aggregative effect can be adjusted by changing helixCAM expression levels. Affinity across a range of doxycycline induction concentrations was tested (Figure 4E, S15), revealing that helixCAM cell pairs could be tuned to form aggregates of varying sizes solely by controlling helixCAM expression. These findings suggest that helixCAM-induced affinity could be easily calibrated for individual applications through controlling helixCAM expression with small-molecule inducers. Next, the kinetics of helixCAM protein expression and cell aggregation were measured by HCSRA across 72 hours, with each time point cultured on a separate plate to avoid disturbance (S16). The time course experiment revealed that aggregates start to form around 8 hours post-induction, with aggregate sizes plateauing between 36 to 48 hours post-induction, a time window which aligned with previously reported expression kinetics of the Tet-On protein expression system (Campbell, Willoughby and Jensen, 2012). Accordingly, helixCAM cells pre-induced with doxycycline for 48 hours was used for subsequent experiments demonstrating various applications of helixCAM.
helixCAMs enable additive construction of sophisticated three-dimensional cell structures
Specific spatial patterning of different specialized cell types is a hallmark of human tissue, where the organization enables functions such as nutrient exchange and cell signaling (Heller and Fuchs, 2015). We hypothesized that, by using a “core” cell expressing a single helixCAM followed by sequential incubation with “layer” cells expressing two helixCAMs, it would be possible to replicate complex layers of cells, similar to those found in human tissues. To this end, we selected the sg30mCherry cell as the “core” cell and created three intermediate “layer” cell types, each expressing two orthogonal helixCAMs (sg61+sg88eBFP2, SG83+P9iRFP670, P10+Z18eGFP). The goal was to form stable inner layers using the most orthogonal helixCAMs while relegating the more promiscuous Z17 for the outer layer (Z17eCFP) (Figure 5A).
Figure 5. helixCAMs enable additive construction of sophisticated three-dimensional cell structures.

A. Schematic of spherical layering workflow. Five K562 lines were built, expressing either one or two helixCAMs and an identifying fluorescent protein. Layer 1 cells were mixed with core cells at an 8:1 ratio, and subsequent layers were then mixed at a 2:1 ratio. B. Three select aggregates resembling the desired patterning are shown. Arrows overlaid on Aggregate 1 indicate the location of each cell type. Aggregates were imaged at 20X magnification and cropped for emphasis. C. Confocal imaging of patterned spherical structures. Core and layer cells are visible and near intended locations. Confocal images were taken at 40X magnification with a spinning disc confocal system as a Z-stack and reconstructed using Nikon Elements. D. Interaction frequency of cells within Aggregate 1. Analysis pipeline detailed in S9.
The affinity of the dual helixCAM-expressing “layer” lines to the previously-made single helixCAM-expressing lines as well as pairwise to each other was tested by HCSRA (S18) and confirmed to have the intended affinities. All five cell types were then first induced to express helixCAM for 48 hours. Core sg30mCherry cells were then incubated with sg61+sg88eBFP2 cells in a 1:8 ratio for 12 hours, followed by addition of subsequent layers of cells at a 1:2 ratio (total prior cells:new cells), each followed by a 12 hour incubation (Figure 5A). The dense cell mixture is then allowed to settle briefly and supernatant removed along with unbound cells. The precipitated cellular aggregates are then imaged using widefield and confocal microscopy.
A number of aggregates formed through this process closely resembled the envisioned structural layout. We present three select aggregates imaged in widefield in Figure 5B and as confocal 3D reconstructions in Figure 5C. In particular, Aggregate 1 was composed of 135 cells, regularly patterned, and demonstrated a number of features representative of other aggregates, so it was selected for imaging-based interaction analysis (Figure 5D). Unsurprisingly, the most consistent interactions were the SG30/SG61-induced interactions between sg30mCherry “cores” and sg61+sg88eBFP2 “layer 1” cells. We also observed a layer of magenta SG83+P9iRFP670 “layer 2” cells coating the blue “layer 1 cells,” induced by SG83/SG88 interactions. The next expected layer would be the P10+Z18eGFP “layer 3” cells, which are present to some degree. However, a significant number of green “layer 3” cells were also found intermixed with Z17eCFP “layer 4” cells rather than forming clearly delineated layers, likely a consequence of the high affinity of the Z17/Z18 interaction. Additionally, we observed that, whereas initial layers formed spherical structures, unevenness in these seed aggregates appear to be amplified by subsequent cell layers, often leading to cells from layers 2-4 organizing on opposite sides of the core and forming a hamburger-like presentation. The striking complexity formed with four helixCAM pairs, and the resemblance of aggregates to the intended multicellular structure blueprint, alludes to the potential for helixCAMs to guide the patterning of sophisticated multicellular structures.
helixCAMs enable targeting of suspension cells to adherent cells
In addition to tissue construction, cell-cell interactions are critical to the targeting of immune cells to areas of active infection or malignancy (Zhu, Yao and Chen, 2011; Waldman, Fritz and Lenardo, 2020). We hypothesized that, if adherent cells of interest can be made to express one member of a helixCAM pair, it would be possible to recruit suspension cells expressing a complimentary helixCAM to that region. To start, we generated adherent HEK293 cell lines capable of expressing helixCAMs. After allowing them to grow to confluency, we added corresponding K562 helixCAM-expressing cells, then washed off unbound cells. While few suspension cells remained bound to the adherent wild-type and uninduced helixCAM cells, copious suspension cells were bound to the induced adherent helixCAM cells throughout multiple washes (S19). A similar binding experiment was performed between adherent HEK293 helixCAM cells and a trypsinized suspension of complementary HEK293 helixCAM cells (Figure 6A). As with K562 cells, strong helixCAM-dependent enrichment of bound suspension HEK293 cells was observed, and, by 24 hours, the bound cells had re-established their adherent morphology (Figure 6B). These experiments indicate that helixCAM is a viable tool for targeting suspension cells and adherent cells alike to pre-established adherent cells of interest.
Figure 6. helixCAMs enable targeting of suspension cells to adherent cells and can impact adherent cell morphology.

A. Workflow for targeting suspended HEK293 cells to adherent HEK293 cells. After cells were grown to confluency, one population is trypsinized and added to the other population, after which unbound cells are washed off. Bound cells are then allowed to re-establish an adherent morphology. B. Panel images show three helixCAM pairs uninduced, induced for 48 hours and immediately post-wash, or induced for 48 hours and then incubated for an additional 24 hours post-wash. The 24h post-wash condition is shown as slice view, with horizontal cross-sections of a z-stack shown on the bottom and right panels. Uninduced and induced 0h conditions were imaged using a widefield microscope at 20X magnification with 2x2 tiling, and induced 24h images were taken using a confocal microscope as a Z-stack, also at 20X magnification with 2x2 tiling. C. Workflow for joint seeding of complementary helixCAM-expressing HEK293 cells. Cells were seeded and induced, followed by trypsinization and re-seeding as a single mixture. D. Panels show the distribution and morphology of co-cultured helixCAM HEK293 cells for three helixCAM pairs. Uninduced cells establish normal cell morphology, whereas induced cells form long, stretched bundles.
helixCAM interactions impact adherent cell migration and morphology
Adherent cells, such as HEK293s, express various adhesion proteins that mediate strong interactions with the plate surface and with each other. To examine whether the strength of helixCAM interactions was capable of competing with endogenous adherence, pairs of HEK293 helixCAM cell lines were co-cultured and examined for preferential interactions by widefield microscopy (Figure 6C). While the two cell types in uninduced controls were evenly distributed and displayed normal cell morphology, induced cells were much more locally concentrated, left larger areas of the well empty, and formed elongated bundles (Figure 6D). Within each bundle, it is possible to identify the cell bodies through alternating fluorescence, and rather than the rotund morphology found in the uninduced conditions, the induced cells appear stretched, with some cell bodies spanning over 100μm (uninduced cell lengths were closer to 30μm). The notable change in morphology from HEK293 helixCAM cells indicates that helixCAM interactions can compete with endogenous interactions to influence adherent cells’ migration and morphology.
helixCAMs enable tunable and simultaneous patterning of multiple cells types on CC-patterned surfaces
While the experiments above focus on helixCAMs’ capabilities to bind cells to one another, the affinity and specificity of helixCAMs could also be leveraged to pattern multiple cell types onto a surface, enabling purification of cells from a mixed population or patterning of microfluidic devices. To test helixCAM’s capacity to pattern cells, we expressed and purified both His-tagged CC peptides as well as His-tagged GFP-CC fusion proteins for CCs sg30, sg61, sg83, and sg88 and confirmed the quality of the purified protein species with LC-MS (S20). The protein solutions were applied to nickel-coated plates to create the CC-coated surface, followed by the addition of helixCAM-expressing cells for patterning. After removing unbound cells, the remaining cells were then imaged and quantified (Figure 7A).
Figure 7. helixCAMs enable tunable and simultaneous patterning of multiple cell types onto CC-patterned surfaces.

A. Workflow for CC surface patterning across a gradient and downstream automated image segmentation and analysis. Wells are coated with His-tagged CCs or His-tagged CC-GFP fusion protein solutions, then cells are added. Unbound cells were washed off, and the entire well was imaged at 4X magnification as a 2x2 tile. Images were segmented and cells were counted by CellProfiler. B. Normalized bound cell count is plotted against CC coating concentration for each of four CC-His peptides tested. Bound cell counts were normalized to the maximum in each channel to reduce variance from segmentation (raw counts in S21). Reported values are medians with N=4 and error bars are S.D. C. Normalized bound cell count is plotted against CC coating concentration for each of four CC-GFP-His peptides tested. All four proteins demonstrated strong capability for binding complementary helixCAM cells. Reported values are medians with N=4 and error bars are S.D. D. Workflow for simultaneous patterning of two cell types within one well. Two distinct CC-GFP-His protein solutions were added in a “G” or a “C” pattern. The two complementary helixCAM cell populations are added as a single mixture to cover the well surface. Unbound cells were then washed off and the bound cells were imaged at 4X magnification as a 2x2 tile. E. Dual-CC-patterned wells with two cell populations pre- and post-wash. Pre-wash, cell populations are evenly distributed and fully cover the plate surface. Post-wash, helixCAM cells complementary to the CC patterned at the G or C locations remain bound, but the non-complementary cells are not bound to those locations, and outside of patterned regions, few cells are observed (quantification of cells in each region in S22).
Using this method, we tested five orders of magnitude of protein concentrations and an untreated condition for all eight CC fusions. Results from four replicates were normalized by channel to control for variability due to image segmentation, and the median and standard deviation reported in Figure 7B for CC-His coating and Figure 7C for CC-GFP-His (raw counts can be found in S21). For all conditions but sg30-His, the CC-coated surface led to helixCAM cell binding, and the number of cells bound strongly correlated to the concentration of protein coating applied. Notably, at the same coating concentration, CC-GFP-His-coated wells bound markedly more cells than for CC-His-coated wells (p-value=5.51×10−10, ANOVA), and, at higher concentrations, led to cells covering the entirety of the plate surface.
Next, we tested whether it would be possible to simultaneously bind two distinct helixCAM cell populations to the plate in a specified spatial pattern. To test this, we pre-patterned each well with two distinct CC-GFP-His proteins, either in a “G” shape on the left or a “C” shape on the right, added a mixture of the two corresponding K562 helixCAM cells, then removed unbound cells (Figure 7E). With four CC options, there are four compatible 2-CC combinations: sg30+sg83, sg30+sg88, sg61+sg83, and sg61+sg8. Combinations between complementary CCs, such as sg30+sg61, are not compatible for this purpose, as the corresponding helixCAM cells would form aggregates in suspension. Figure 7E shows the pre- and post- washed CC-GFP-His-patterned, helixCAM cell-coated plates. The pre-wash images exhibit uniform distribution of the two helixCAM cell types across the well surface. After washing, dense helixCAM cells can be found localized to the corresponding “G” and “C” CC-patterned regions, whereas untreated regions are almost entirely free of cells. Image-based quantitative comparisons of cell localization demonstrated a clear enrichment of cell localization to their intended region over other regions of the well (S22). The same experiment, using dots instead of letters as patterns, replicates this result (S23). The strong and programmable binding of helixCAM cells to CC-patterned surfaces demonstrates its potential as a method for highly specific cellular pull-down and patterning, with the number of simultaneous cell types patterned determined by the number of orthogonal CC-pairs available.
Discussion
In this study, we presented helixCAM, an extensible platform for engineering programmable cell-cell interactions in both bacteria and human cells. helixCAMs were demonstrated to induce formation of large, patterned cell aggregates containing thousands of cells and spanning multiple cell layers. From helixCAMs composed of previously established CC domains, we rationally designed CC peptide libraries and, after screening for affinity and specificity, identified two engineered CC pairs with favorable helixCAM properties. We then tested high-affinity helixCAM pairs across a range of cell types and applications, demonstrating their versatility in guiding multicellular patterns and morphologies.
The set of helixCAMs augments the existing work on synthetic protein adhesins. Similar to previous work by Foty et al., Cachat et al., and Shan et al. using cadherins, helixCAMs are capable of forming large cell aggregates, with the difference being the heterodimeric nature of the CCs compared to the homodimeric cadherins. Heterodimeric and specific affinity is required for more complex pattern formation, such as the five-layer aggregate (Figure 5) and simultaneous patterning (Figure 7). The programmability of helixCAM is comparable to the nanobody-antigen-based adhesion platform from Glass and Riedel-Kruse, and the two may work complementarily to allow for further expansion of orthogonal adhesins. Pinero-Lambea et al. had previously used surface-mounted immunoglobulin domains to pattern E. coli onto surfaces and target cancer cells. With helixCAMs, these applications can utilize human cells in place of E. coli, with intriguing implications in cell therapy (Roybal et al., 2016).
One of the most complex applications tested was the construction of sequentially layered spherical aggregates using five engineered cell types (Figure 5), from which a number of interesting observations arose. Based on pairwise affinity data, we expected the Z17-eCFP cells to be the least predictable due to their promiscuity and placed them last in the coating sequence to mitigate its impact. Interestingly, the strength of the Z17/Z18 interaction appeared to overpower previously formed P9/AP10 interactions, pulling the green “layer 3” cells away from “layer 2” cells to form sub-clusters rather than clearly delineated layers. This observation is consistent with the “differential adhesion hypothesis” of how cells in an aggregate naturally self-segregate based on binding affinity – a key element of biological development (Foty and Steinberg, 2005). The potential for subsequent helixCAM interactions to disrupt previously-formed ones indicates a need for independent tuning of the expression level of each helixCAM. It may also be possible to guard against disruption by subsequent cells by solidifying interactions between additions with an intermediary step of homodimer CAM expression, such as cadherins. Relatedly, the oval structures that formed instead of the desired spherical structure may be due to a combination of the initial core-layer1 aggregates not forming perfectly spherical structures, and the strength of subsequent layer interactions accentuating the imperfections.
An important consideration to any protein effector designed for sustained expression in human cells is its impact on the expressing cells’ viability. We investigated this in the ten helixCAM lines we built by inducing helixCAM expression and measuring cell number and viability over four days (S24). We found a range of responses, from slowed growth with sustained viability, as in the case of sg61eGFP and AP10mOrange, to an observable decrease in both growth rate and viability in the case of sg30mCherry and sg83iRFP670. As sg30mCherry and sg61eGFP comprise our best-performing pair, the impact of helixCAM expression on growth rate warrants discussion. Of note, while some lines exhibit significantly reduced growth, other lines do not, leading us to hypothesize that the reduced growth is not caused by a direct biological response to the helixCAM proteins. As the goal of this work was to demonstrate the aggregative effect of helixCAMs, we selected for cell lines that expressed helixCAMs at the highest level, leading to lines that saturated in aggregative effect well below maximum induction dosage (Figure 4E). The high level of protein overexpression is likely a key contributor to the observed reduction in growth rate and viability, as it has been previously shown that even overexpression of the commonly-utilized and well-folded protein GFP can lead to cell growth defects (Kintaka, Makanae and Moriya, 2016) and toxicity (Ansari et al., 2016). It would be interesting to further characterize the magnitude and mechanistic cause of helixCAM expression on cell growth in future works, as well as whether this effect can be attenuated by reducing helixCAM expression while maintaining aggregative effect. We recommend that researchers seeking to utilize helixCAM in their preferred cell types to prioritize lines that retain high viability and tune the level of helixCAM expression induction to fit their aggregative requirements without reaching saturation.
We envision several potential biological applications enabled by the helixCAM in immunology. For instance, intercellular signaling is known to be critical to the activation of immune cells (Zhu, Yao and Chen, 2011; Waldman, Fritz and Lenardo, 2020), but few synthetic methods for directing or augmenting cell-cell interactions exist beyond the use of endogenous CAMs. By using helixCAMs, small molecule inducers could be used to adjust the binding strength of immune cells to their targets in conjunction with B- or T-cell receptors and other co-stimulatory signals, leading to a more refined understanding of the physical role of cell interactions in immune cell activation. In addition, helixCAMs can augment activation events, such as those of chimeric antigen receptors or SynNotch binding (Morsut et al., 2016), either through constitutive expression or triggered to be expressed by other binding events. helixCAMs may even be used to directly localize immune cells to infected or malignant tissues, given that those cells can be transduced to express helixCAMs or functionalized with targeting CCs (for instance, with a CC-antibody fusion).
Tissue engineers may also find compelling applications for helixCAMs. Current paradigms for engineering tissues for therapeutic purposes, such as 3D cell printing (Murphy and Atala, 2014; Kolesky et al., 2016; Roohani-Esfahani, Newman and Zreiqat, 2016; Moroni et al., 2018), are constrained by printing resolution and limited survival of printed cells (Ozbolat and Yu, 2013; Yu et al., 2013). As demonstrated in Figure 5B, helixCAMs can be used to manufacture complex spherical structures with single-cell level accuracy. By expressing helixCAMs in physiologically-relevant cells, it may be possible to create smaller high-resolution building blocks that are subsequently arranged into more complex tissues (Todhunter et al., 2015). Furthermore, helixCAMs can be integrated naturally into existing tissue engineering workflows. For instance, for platforms that grow (Song et al., 2018) or 3D print vasculature (Chen et al., 2021), by utilizing helixCAM, it should be possible to functionalize the synthetic vasculature with additional cell types to enhance its resemblance to physiological vasculature.
The helixCAM platform’s versatility extends beyond cell-cell interactions to cell-surface interactions, opening the door to opportunities to augment conventional cell purification and patterning methods. For instance, by expressing helixCAMs in the desired cell population, those cells can be isolated using CC-coated magnetic beads. Another area benefiting from programmable cell-surface interactions is organ-on-a-chip platforms (Kim et al., 2012; Bhatia and Ingber, 2014; Stevens et al., 2017), where it may be desirable to culture multiple cell types in specific spatial orientation. By pre-patterning the microfluidic device with multiple CC species, complex cell distribution patterns could be designed to further mimic the physiological environment.
The helixCAM platform offers a simple but powerful way to control cell-cell interactions, a ubiquitous feature found from prokaryotes to eukaryotes. Beyond the set of five helixCAM pairs characterized in this work, there are likely many more orthogonal helixCAMs to be discovered to further expand potential applications. We believe that the toolkit of helixCAMs presented here will assist biologists across multiple fields to better understand the roles cell interactions play in various contexts, as well as enable engineers to better control these interactions for therapeutic applications.
Limitations of the Study
While the goal of this study was to demonstrate the platform design, expandability, and showcase proof-of-concept applications for the helixCAM platform, a number of considerations should be taken into account. First, the mammalian section of this work explored the characteristics of helixCAMs expressed in K562 and HEK293 cancer cell lines. As such, the expression, behavior, and viability measured were in the context of robust and growth-oriented cancer cells and are likely to differ when utilized in primary or stem cells. Second, while the design of the HCSRA for measuring cell aggregation had throughput and reproducibility benefits over imaging-based approaches, it ultimately represented a measurement of the rate of cell sedimentation. As such, HCSRA negative controls should be designed to be as close to the treatment conditions as possible, as the HCSRA cannot distinguish between “intended” causes of aggregation (such as through synthetic CAMs expression) versus “unintended” causes (such as from media conditions or endogenous CAMs). Finally, the higher-order cell patterning work presented were all sampled within 48-72 hours of formation. For longer-term maintenance of these structures, growth rate post-induction will likely be a key consideration and should be measured in the context of each application.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, George Church (gchurch@genetics.med.harvard.edu).
Material availability
Plasmids for expressing helixCAM in E. coli and for constructing mammalian helixCAM cell lines have been deposited to Addgene under Addgene IDs 186303-186318.
Data and code availability
Images used for analysis of aggregate size, helixCAM affinity, and CC-His affinity, along with all raw data used to calculate HCSRA values for all helixCAM pairs are publicly available as of the date of publication. The DOI is listed in the key resource table.
All original code necessary for the imaging-based interaction analysis pipeline and for calculating HCSRA have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| DH5α | ThermoFisher | Cat# 18265017 |
| DH10β | ThermoFisher | Cat# EC0113 |
| T7 Express | New England Biolab | Cat# C2566 |
| E cloni 10G Supreme | Lucigen | Cat# 60107-1 |
| BL21(DE3) | Novagen | Cat# 69450 |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 2000 | ThermoFisher | Cat# 11668030 |
| Blasticidin | ThermoFisher | Cat# A1113903 |
| Puromycin | ThermoFisher | Cat# A1113803 |
| Doxycycline | ThermoFisher | Cat#J67043-AE |
| Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Gold Bio | Cat# I2481C |
| phenylmethylsulfonyl fluoride (PMSF) | Millipore Sigma | Cat#P7626 |
| Imidazole | MP Biomedicals | Cat# 102033 |
| Tris base | Fisher Chemical | Cat#BP152-5 |
| Glucose | Fisher Chemical | Cat#D16-500 |
| Critical commercial assays | ||
| Pierce™ BCA Protein Assay Kit | ThermoFisher | Cat#23225 |
| Deposited data | ||
| Stitched brightfield images of uninduced and induced cell aggregate for aggregate size analysis | This paper | 10.17632/5j93mffst8.1 |
| Stitched 3-pair co-culture images used for imaging-based interaction analysis | This paper | 10.17632/5j93mffst8.1 |
| Raw OD650 data for HCSRA | This paper | 10.17632/5j93mffst8.1 |
| Images used for analyzing CC-His affinity | This paper | 10.17632/5j93mffst8.1 |
| Experimental models: Cell lines | ||
| K562 | ATCC | CCL-243 |
| HEK293AAV | Agilent | 240073 |
| Hek293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| S. Cerevisiae SynAg MATa | Kavins Lab | N/A |
| S. Cerevisiae SynAg MAT□ | Kavins Lab | N/A |
| Recombinant DNA | ||
| Plasmid: PB_TRE_Z17-PDGFR_EF1a_mCherry | This Paper | Addgene 186303 |
| Plasmid: PB_TRE_Z18-PDGFR_EF1a_eGFP | This Paper | Addgene 186304 |
| Plasmid: PB_TRE_P3-PDGFR_EF1a_iRFP670 | This Paper | Addgene 186305 |
| Plasmid: PB_TRE_AP4-PDGFR_EF1a_eBFP2 | This Paper | Addgene 186306 |
| Plasmid: PB_TRE_P9-PDGFR_EF1a_mPlum | This Paper | Addgene 186307 |
| Plasmid: PB_TRE_AP10-PDGFR_EF1a_mOrange | This Paper | Addgene 186308 |
| Plasmid: PB_TRE_sg30-PDGFR_EF1a_mCherry | This Paper | Addgene 186309 |
| Plasmid: PB_TRE_sg61-PDGFR_EF1a_eGFP | This Paper | Addgene 186310 |
| Plasmid: PB_TRE_sg83-PDGFR_EF1a_iRFP670 | This Paper | Addgene 186311 |
| Plasmid: PB_TRE_sg88-PDGFR_EF1a_eBFP2 | This Paper | Addgene 186312 |
| Plasmid: pQE80_T5_Z17-ehaA_LacI | This Paper | Addgene 186313 |
| Plasmid: pQE80_T5_Z18-ehaA_LacI | This Paper | Addgene 186314 |
| Plasmid: pQE80_T5_P3-ehaA_LacI | This Paper | Addgene 186315 |
| Plasmid: pQE80_T5_AP4-ehaA_LacI | This Paper | Addgene 186316 |
| Plasmid: pQE80_T5_P9-ehaA_LacI | This Paper | Addgene 186317 |
| Plasmid: pQE80_T5_AP10-ehaA_LacI | This Paper | Addgene 186318 |
| Plasmid: pET22b_His-sg30 | This Paper | N/A |
| Plasmid: pET22b_His-sg61 | This Paper | N/A |
| Plasmid: pET22b_His-sg83 | This Paper | N/A |
| Plasmid: pET22b_His-sg88 | This Paper | N/A |
| Plasmid: pCAG-sg30-TEVs-GFP-His | This Paper | N/A |
| Plasmid: pCAG-sg61-TEVs-GFP-His | This Paper | N/A |
| Plasmid: pCAG-sg83-TEVs-GFP-His | This Paper | N/A |
| Plasmid: pCAG-sg88-TEVs-GFP-His | This Paper | N/A |
| Plasmid: PB-sg61-PDGFR-blast | This Paper | N/A |
| Plasmid: PB-sg88-PDGFR-blast | This Paper | N/A |
| Library general plasmid: pQE80X_Z17-autotransporter | This Paper | N/A |
| Library general plasmid: pSYNAGaV3_sgX_SUMO_kan | This Paper | N/A |
| Library general plasmid: pETDEST_sGFP_sgX | This Paper | N/A |
| Software and algorithms | ||
| CellProfiler | Stirling, et al. 2021 | https://cellprofiler.org/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Software and algorithms | ||
| CellProfiler | Stirling, et al. 2021 | https://cellprofiler.org |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Zen Blue | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Nikon Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| A Plasmid Editor (ApE) | M. Wayne Davis | https://jorgensen.biology.utah.edu/wayned/ape/ |
| Agilent Mass Hunter | Agilent | https://www.agilent.com/en/promotions/masshunter-mass-spec |
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab.html |
| Python | Python Software Foundation | https://www.python.org/ |
| BioPython | Cock, et al. 2009 | https://biopython.org/ |
| Sickle | N/A | https://github.com/najoshi/sickle |
| Code for Image-based Frequency Analysis and HCSRA | This paper | 10.5281/zenodo.6323728 |
| ProtParam | ExPasy | https://web.expasy.org/protparam/ |
| Other | ||
| Ultra-Low-Adherence v-bottom plates | S-Bio | Cat# MS-9096VZ |
| Coiled-Coil and helixCAM Amino Acid Sequences | This Paper | S26 |
EXPERIMENTAL MODELS AND SUBJECT DETAILS
E. coli:
Standard cloning strains DH5a and DH10B were used for most cloning and DNA preparation purposes. For bacterial adhesion assays, the T7 Express Competent E. coli cells were used (NEB C2566). Libraries were transformed using E. cloni® 10G Supreme Electrocompetent Bacterial Cells (Lucigen 60107-1). CC-His proteins were expressed in BL21(DE3) cells (Novagen 69450). All cultures were grown in LB Miller media at 37°C.
S. Cerevisiae:
Yeast strains for each haploid yeast strain (MATa/ɑ) were gifted by David Younger and the Klavins Lab. Yeast strains were grown at 30°C in YPD or Yeast SDO media.
Cell Lines:
K562 cells (ATCC CCL-243, female) were used for most human helixCAM line generation. They were cultured in either shaking or standing suspension and passaged at a concentration of 2x106/mL to a seed concentration of 2x105/mL. Adherent cells used for targeting experiments were the AAV-293 cells (Agilent 240073) for their superior adhesion. They were seeded at 10% confluency and allowed to grow to 90% confluency before passaging. All cells were cultured at 37°C and 5% CO2 in DMEM+GlutaMax (ThermoFisher 10566016) supplemented with 10% Fetal Bovine Serum (Corning 35-010-CV) and 1% Penicillin-Streptomycin (ThermoFisher 15070063)
METHOD DETAILS
Plasmid Design and Construction
For E. coli helixCAM construction, existing coiled-coil sequences were found via literature search and private communications and synthesized (Twist). Coils-encoding DNA fragments were cloned into a pQE protein expression vector (Qiagen pQE-80L) with in-frame with a flanking leader sequence and an EhaA coding sequence via Gibson Assembly (NEB E2611L). Plasmids were transformed in T7 Express Competent E. coli cells (NEB C2566), miniprepped (Qiagen 27104), and verified via Sanger sequencing (Genewiz).
To construct the coiled-coil-sGFP libraries, DNA fragments containing the full sGFP expression cassette with flanking Golden Gate Assembly overhangs were synthesized (IDT). The plasmids were then cloned into the pET-DEST T7 expression vector (ThermoFisher 12276010), creating a final single plasmid containing all three subunits of GFP (S1). Coiled-coil sequences were PCR-amplified from gene fragments using Q5 Polymerase (NEB M0492L) with BsaI recognition sites and unique 4 base pair overhangs to either fuse the fragment 3’ of GFP10 or 5’ of GFP11 using Golden Gate Assembly (NEB E1601L). Assembled plasmids were then transformed into E. cloni® 10G Supreme Electrocompetent Bacterial Cells (Lucigen 60107) Cloned plasmids were miniprepped and transformed into T7 Express Competent E. coli strain (NEB C2566) for expression and screening.
To construct coiled-coil libraries for yeast SynAg screening, the SynAg plasmid vectors were used (gifted from the Klavin Lab). The carbenicillin resistance cassette was replaced with kanamycin resistance to prevent pQE plasmid contamination. Additionally, silent mutations were made to the SUMO coding region in the SynAg-a cassette via site-directed-mutagenesis (NEB E0554S) to distinguish from SynAg-alpha during amplification. Coiled-coil sequences were amplified from the previously-described E. coli pQE vectors. Correct amplification was determined through individual qPCR reactions and representative PCR products were checked via gel electrophoresis. PCR products were pooled and assembled into the SynAg vectors via Gibson Assembly. Assembled plasmids were purified and electroporated into E. cloni® 10G Supreme Electrocompetent Bacterial Cells (Lucigen 60107). Cloning efficiency was quantified via plating of dilutions onto LB+Kanamycin and counting colonies. Cultures were grown with selection overnight and then miniprepped to produce plasmid DNA for transformations. Library scale lithium acetate transformations of S. cerevisiae were performed as described in Younger et al. (Gietz and Schiestl, 2007; Younger et al., 2017), with the adjustment of 10μg of PmeI digested plasmid DNA and fresh salmon sperm DNA. Serial dilutions of the transformed yeast culture was plated onto selective yeast Synthetic Drop-Out -trp agarose plates were performed, and transformation efficiency was confirmed to be >300-fold coverage of the coiled-coil library size.
To construct plasmids for human helixCAM experiments, the Thermo pDisplay was used (ThermoFisher V66920). Coiled-coil sequences were flanked with Gibson Assembly overhangs and inserted in-frame between the IgK leader peptide and PDGFR sequences through Gibson Assembly (NEB E2611L). The full-length human helixCAM construct was amplified through PCR and inserted behind the pTet promoter of the PiggyBac Tet-On vector (Takara 631168). An identifying fluorescent protein sequence with a T2A sequence was placed between the T2A sequence and the puromycin resistance cassette of the Tet-On vector. For the blasticidin-resistance plasmid (for dual-helixCAMs), the fluorescent protein sequence and the puromycin resistance gene were replaced with a blasticidin resistance gene, which was cloned from Addgene Plasmid 74918 (gifted from Jose Silva Lab).
Plasmids for E. coli expression of His-tagged coiled-coil peptides was done by first synthesizing codon-optimized gBlocks (IDT) comprising each Coiled-Coil with a C-terminal 6xHis tag. The DNA fragments were inserted into a pET22b expression vector (Novagen 69744) via Gibson assembly (New England Biolabs E2611S). Plasmids for expression of His-tagged CC-GFP fusion protein were done by replacing the CMV promoter of the pcDNA3.1 vector (ThermoFisher V79020) with the CAG promoter, followed by insertion of the coiled-coil gBlock (codon-optimized for mammalian expression) N-terminally to a TEV cleavage sequence, GFP, and 6xHis fusion (S1).
helixCAM K562 Cell Line Construction
PiggyBac-flanked donor plasmids were packaged with the Super PiggyBac transposase vector (SBIPB210PA-1) using Lipofectamine 2000 (ThermoFisher 11668019) at a 1:1 ratio in Opti-MEM (ThermoFisher 31985062). For K562 cells, the liposome-DNA solution was first added to a well, then K562 cells were added in culture media on top. For HEK293 cells, cells were allowed to first grow to 70% confluency, then the liposome-DNA solution was added dropwise to the media. 48 hours after transfection, cells were changed into selection media containing either puromycin (ThermoFisher A1113802) or blasticidin (ThermoFisher A1113903). For K562 cells, the selection concentration used was 1μg/mL for puromycin and 6 μg/mL for blasticidin, and for HEK293, it was 2μg/mL for puromycin and 10 ug/mL for blasticidin. After four days of selection, cells were passaged to recover for 48 hours, then subsequently single-cell sorted (BD FACSAria), either with fluorescence selection (for puromycin lines) or only through FSC/SSC discrimination (for blasticidin lines). Subsequently, surviving lines were imaged and scored for fluorescence and viability, and the ones scoring highly in both were selected.
Pairwise helixCAM Aggregate Formation and Sedimentation Assays
helixCAM interactions in E. coli were tested as follows. A single colony was picked into 3mL of LB+carbenicillin, and grown overnight. 150uL of culture was diluted into 30mL LB+carbenicillin and grown for 75 minutes. 60mg of arabinose was added to induce, and culture was grown for an additional 3 hours. 2mL from two separate cultures, each expressing a distinct helixCAM, were mixed, and after four hours, a pipette is placed a quarter of the depth into the culture, and 100uL of cells are taken. This was either measured as an OD600 or plated onto a #1.5 glass coverslip for imaging.
helixCAM interactions in K562 cells were tested as follows. 2x105 of two distinct K562 helixCAM cell lines were co-cultured in a low-adherence 24-well plate (Corning 3473) in culture media with 500μg/mL doxycycline added. Cells were incubated for 48 hours, then either mounted using a #1.5 glass coverslip for widefield imaging or embedded in low melting point agarose (ThermoFisher 16520050) for confocal imaging using wide-bore pipette tips (Rainin 30389191). For multi-pair co-cultures, the same process, including the number of cells of each line, was followed.
For the Human Cell Sedimentation Rate Assay (HCSRA), 1.75x105 of each cell line to be tested (or 3.5x105 for self-interaction) was added to 150uL of culture media with or without doxycycline. The cells were cultured in a specific ultra-low-adhesion v-bottom plate (SBio MS-9096VZ), which has the right v-bottom curvature allowing this assay to work. The cells were allowed to interact for 48 hours on an orbital shaker at 150rpm. To measure, the cells were resuspended using 15 seconds of shaking at 900rpm, then immediately placed into a plate reader (Molecular Devices M5) on the same bench, and kinetic OD650 was measured over 15 minutes. The data is then exported and analyzed in MATLAB.
Coiled-Coil Library Screening
For stage one of the CC library screen, a tripartite split-GFP method was used. Colonies were picked from strains containing each paired library combination and grown in 3mL LB+Carbenicillin (50 ng/μl) overnight. The culture was diluted 1:100 in 100μL of LB carbenicillin and grown in shaking culture in a BioTek Synergy H1 plate at 567 cycles per minute and 37°C for 1hr. Next, the cells were induced at 1:1000 with IPTG (Teknova I3431) and grown for 45 minutes. Cells were then diluted 1:100 into PBS (ThermoFisher 10010023) and sorted on a BioRad S3e Cell Sorter. From each library, cells were sorted into for the top 0.5% GFP intensity, and the sorted cells were grown to a high density and sorted again, selecting for the top 2% of GFP intensity. Plasmids were miniprepped from the resulting population and sequenced through NGS. 102 coiled-coil candidates demonstrating the highest affinity and orthogonality were selected for the next stage of the screen.
The next stage of the screen used the yeast SynAg assay. Haploid MATa and MAT□ strains expressing the SynAg cassette were picked from fresh colonies into 3mL of YPD and grown in a drum rotator for 24 hours. 2.5μL of MATa and 5μL MAT□ saturated cultures were added to a fresh 3mL of YPD and grown for 24 hours. For optimization of the SynAg system, cells were diluted 1:200 in 1X PBS for assay through flow cytometry (Miltenyi MACSQuant VYB) of the expression of mCherry and mTurquoise. For screening CC candidates, diploid selection was done through complementary lysine and leucine auxotrophic markers, then enriched for an additional 24 hours before genomic DNA preparation and NGS. We noticed that the original aga1p/aga2p yeast display system (Boder and Wittrup, 1997) led to low surface presentation of the CCs, which we attributed to protein degradation. By inserting a SUMO-tag (Kuo, Nie and Courey, 2014) between the coiled-coil and the ga2p domain, the membrane protein was stabilized, which increased surface presentation (S11) and led to successful yeast mating events.
Next-Generation Sequencing of screened CC candidates
Plasmid DNA was extracted from each library and amplified via PCR (NEB M0492L) with primers containing homology to the binding region of the indexing primers. PCR reactions were monitored via qPCR (SigmaMillipore S4438) on a Roche LightCycler 96. Indexing of the library member DNA proceeded with a global primer (Pi5) and a unique primer (Pi7) for sample barcoding. Sequencing was performed at the Biopolymers Facility at Harvard Medical School on an Illumina NextSeq 500 sequencer, yielding 300bp forward and reverse reads containing both coils when combined.
helixCAM Additive Spherical Patterning
To build the spherical structure, all five cell types (core and layers 1-4) were pre-induced for 48 hours with 500ng/mL doxycycline in culture media. Core cells were added to layer 1 cells in a 1:8 ratio, followed by a 12-hour incubation. After incubation, aggregates were resuspended through gentle vortexing, and a pellet was allowed to form before removing the unbound cell supernatant. Subsequent cell populations are added at a 1:2 ratio, following the same steps. After the last 12-hour incubation and unbound cell removal, aggregates were diluted in culture media and either mounted using a #1.5 glass coverslip for widefield imaging or embedded in low melting point agarose (ThermoFisher 16520050) for confocal imaging.
HEK293-HEK293 and HEK293-K562 Cell Targeting
Complementary helixCAM lines of either two HEK293 cell types or one HEK293 and one K562 are grown to 80% confluency on a 24-well glass-bottom plate (Mattek P24G-1.5-10-F). Doxycycline is then added at 500ng/mL and cells were induced to express helixCAMs for 72 hours. In the case of HEK293-HEK293 targeting, one population of HEK293 cells were trypsinized to be temporarily suspension cells, then added to the second still-adherent population of helixCAM-expressing HEK293 cells. For HEK293-K562 targeting, the K562 cells were directly added to the adherent HEK293 cells. The coated cells were allowed to incubate for 30 minutes then imaged (pre-wash). Unbound cells were then aspirated and washed with culture media+doxycycline twice. The cells were then imaged again (post-wash). For HEK293-HEK293 targeting, cells were returned to the incubator for an additional 24 hours of growth before imaging again to observe the re-establishment of adherent cell morphology.
Expression and Purification of His-tagged CC and His-tagged CC-GFP
To express His-tagged CCs, E. coli cells (BL21 DE3) were grown in Terrific Broth (TB) overnight at 25°C with 200 μg/mL ampicillin and glucose to 0.5% till they reached an OD of ~0.5. They were then induced with 1mM Isopropyl ß-D-1-thiogalactopyranoside (IPTG) and grown overnight at 16°C. The cultures were harvested via centrifugation at 6000xg for 15min at 4°C, pellets resuspended in 20mM Tris, 500mM NaCl, 20mM Imidazole, 500μM PMSF at a pH of 8.0, then lysed via sonication. The resulting lysate was then spun at 30,000xg at 4°C for 90 minutes to remove insoluble proteins and cell debris. The supernatant was passed through a 0.45μm filter then run on a HisTrap HP 1mL column (Cytiva 17-5247-01) and fractions were collected using an Akta Pure FPLC. The fractions were run on an SDS-PAGE gel (BioRad 4569033) and stained via Coomassie (BioRad 1610786) to select fractions with significant protein. Pooled fractions were then dialyzed using Slide-a-Lyzer mini dialysis cassettes with a 3.5k MWCO (Thermo Fischer, Cat#88403) into 50mM Tris, 100mM NaCl, pH 7.6. Some contaminating proteins precipitated during dialysis so the solutions were clarified by spinning at 15,000xg for 15min at 4°C. Yields ranged from 6-25 mg of protein per liter of culture.
To express His-tagged CC-GFP fusion protein, HEK293T cells were cultured to 70% confluency in a T75 flask, then transfected with 15μg of CC-GFP-His expression plasmid using PEI. Cells were grown for 72 hours, then lysed in 2mL of Triton buffer (50mM Tri pH8, 150mM NaCl, 0.1% Triton X-100). His-tagged proteins were bound to Ni-NTA Resin Cartridges (ThermoFisher 90098), washed with 20mL of wash buffer (50mM Tris pH 8, 500mM NaCl, 10mM imidazole), followed by elution into 2mL of elution buffer (0mM Tris pH 8, 500mM NaCl, 300mM imidazole). The protein was then dialyzed and concentrated using a size-selection spin concentrator (MilliporeSigma UFC201024).
Protein concentration was determined using a BCA (ThermoFisher 23225) and constructs were analyzed via LC-MS (S20). The liquid chromatography was done using an Agilent 1260 Infinity II system on a PLRP-S column (Agilent PL1912-1500) over an acetonitrile gradient of 5-95% connected directly to an Agilent 6530 QTOF. Predicted molecular weights were determined using ExPasy ProtParam without the N-terminal Methionine.
helixCAM Cell Patterning with Coiled-Coil Coated Plates
First, K562 helixCAM cells were pre-induced for 48 hours with 500ng/mL doxycycline. For testing CC coating concentrations, Ni-coated 96-well plate wells were coated with 100μL of either his-tagged CCs or his-tagged CC-GFP fusions at a range of protein concentrations then incubated for 30 minutes. The CC protein solution was then aspirated and washed with PBS+20mM imidazole (ThermoFisher 88229), then blocked with PBS+20mM imidazole for 30 minutes. The blocking solution was then aspirated, and the pre-induced HelixCAM K562 cells were added in culture media+20mM imidazole+500ng/mL doxycycline for 2 hours. We then washed the plate with culture media twice, followed by imaging and image segmentation to count the number of bound cells. For both dot-based and letter-based dual-CC patterning, the two CC-GFP-His protein solutions used were patterned using a 10uL pipette tip, carefully avoiding contact with the other solution or with the well edge. Subsequent steps are identical to the single CC patterning described above.
Quantification of helixCAM cell viability post-induction
To explore the effect of helixCAM expression on cell viability, each of the ten single-helixCAM lines was individually seeded and sampled over four days to measure cell count and viability. For each line, 4x105 cells were seeded into wells in 24-well plates along with 750uL of culture media either with or without 500ng/mL of doxycycline, in triplicates. At five separate time points (0, 1, 2, 3, and 4 days after seeding), 10uL of cells from each well was mixed with 10uL of 0.4% Trypan Blue Stain (NanoEntek EBT-001), from which 10uL was taken out and loaded into a disposable cell counting slide (NanoEntek E1020). The cell count and viability are then measured with a ThermoFisher Countess II automated cell counter.
We elected to measure the effect of helixCAM expression using individual helixCAM lines over co-cultured helixCAM lines for multiple reasons. First, the formation of large, tightly-bound cell aggregates reduces the accuracy of cell viability measurements. Secondly, we wanted to avoid conflating the effect of expressing helixCAM protein on growth and viability with the added effect of nutrient and oxygen diffusion in large cell aggregates. As other have previously established, aggregates greater than 100μm in diameter start to observe viability changes due to limitations in nutrient and oxygen diffusion(Griffith et al., 2005; Jain et al., 2005), and helixCAM aggregates can span over 2,000μm (S12). By measuring each line in independent cultures, we avoid the confounding effect of nutrient and oxygen diffusion on growth and viability measurements.
Imaging
Below is a table summarizing the scopes and setups used for imaging in this work:
| Scope Body | Objectives | Excitation | Ex/Em Setup | Confocal Setup |
Camera |
|---|---|---|---|---|---|
| Zeiss Axios Observer Z1 | 5X 20X 63X |
Colibri LED (full spectrum) | DAPI 365/445 GFP 470/525 YFP 550/605 mCherry 560/630 Cy5 640/690 |
N/A | Hamamatsu Image EM-1K, Hamamatsu Orca-R2 |
| Nikon Eclipse Ti2 | 4X 20X 60X |
Lumencor SpectraX LED: 395nm 440nm 470nm 510nm 550nm 575nm 640nm |
DAPI 395/460 CFP 440/480 GFP 470/525 YFP 510/535 mCherry 575/630 Cy5 640/700 |
N/A | Andor Zyla |
| Nikon Eclipse Ti2 | 20X 40X |
Laser Lines: 405nm 408nm 561nm 640nm |
DAPI 405/455 CFP 405/480 GFP 488/525 mCh 561/605 Cy5 640/705 |
W1 Yokogawa Spinning Disk (50μm pinholes) | Andor Zyla |
Widefield fluorescent microscopy of E. coli helixCAM aggregates was conducted using a Nikon Eclipse Ti2 microscope at 60X magnification or on the Zeiss Axio Observer Z1 at 63X magnification. Images were taken in the GFP and mCherry channels. Samples were wet-mounted onto a #1.5 coverslip without prior fixation.
Widefield imaging of human cell aggregates (K562, HEK293) was done on either a Zeiss Axios Observer Z1 microscope at 4X or a Nikon Eclipse Ti2 microscope at 20X. For K562 cells, samples were wet-mounted onto a #1.5 coverslip without prior fixation. HEK293 cells were directly grown on a glass-bottom 24-well plate (Mattek P24G-1.5-10-F) and directly imaged. For tiled images composing multiple fields of views, images were taken with a 10% overlap and stitched using the Nikon Elements software.
Widefield imaging of CC-patterned helixCAM K562 cells was done at 4X on a Nikon Eclipse Ti2 microscope. The objective was oriented to the center of the well, and a 2x2 tile was captured and stitched with Nikon Elements, capturing the entire 96-well well surface.
Confocal imaging of K562 cell aggregates and HEK293 cell targeting was done with a Nikon Eclipse Ti2 microscope coupled with a Yokogawa W1 Spinning Disc. In the case of K562 cell aggregates, aggregates were allowed to settle, media was removed, then cells were gently resuspended in melted low melting point agarose (ThermoFisher 16520050) dissolved into culture media and pre-equilibrated to 37°C, then gently dropped onto the glass coverslip of a 24-well glass-bottom plate (Mattek P24G-1.5-10-F). The agarose was allowed to solidify at room temperature for 15 minutes, followed by confocal imaging. 3D reconstruction for 2-color images was done in ImageJ. 3D reconstructions for 3+ color imaging and slice view were created in Nikon Elements.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Summaries and Tests
In Figures 4B, each square represents a mean Δt50 value with three replicates. In Figure 4C, the same mean values from Figure 4B are used for the y-axis. R2 is the coefficient of determination, and the p-value was determined via an F-statistic test against a constant model. Both values are obtained through MATLAB’s fitlm function. In Figure 4D, the values are the same as in Figure 4B, and error bars are standard deviations. Statistical significance was calculated using two-sample t-test in MATLAB. In Figure 4E, values are Δt50 values normalized by the max observed Δt50 value of that set. The mean of two reads is reported along with standard deviation as error bars. In Figure 7B and Figure 7C, the values are cell counts normalized by the maximum cell counted in that channel and run. This normalization is used to control for variability in cell viability and exposure differences that may confound the absolute count, which is reported in S21 and does not change the interpretation of the findings. The values are reported as medians over four replicates, due to the tendency of small changes in the washing process of producing outliers, and error bars are standard deviations. Significance of the three conditions (CC type, Concentration, and with or without GFP tag) was performed using cell counts from shared concentration values (0.01, 0.1, and 1nM) using N-way ANOVA in Matlab with sequential sum of squares (Type I), with p-values of 1.95×10−5, 3.55×10−11, and 5.51×10−10 respectively.
Next-Generation Sequencing Data Analysis and CC Candidate Selection
FASTQ files from NGS were inspected for quality and then trimmed using Sickle (https://github.com/najoshi/sickle). Trimmed FASTQ files were read into Python via the SeqIO function from the BioPython package (https://biopython.org/) (Cock et al., 2009). For the E. coli screen, CC candidate sequences were identified, extracted from the read, and paired via an in-house script.
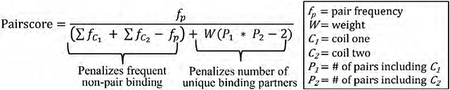
CC pairs were then assigned a pair score which evaluates their frequency of on-target interactions against the frequency of off-target interactions and tuned with a weight factor W. Top 30 hits using a W of 0, 0.01, and 0.1, along with the highest frequency CC candidates, were selected for a total of 102 candidate pairs. From the yeast SynAg screen, fused barcodes representing the two interacting CCs were mapped. CC pairs were then evaluated for enrichment (a function of frequency) and orthogonality (the percentage of on-target interactions divided by all observed interactions), and two pairs (four CCs) with the highest enrichment and orthogonality were selected for characterization in human cells.
Image segmentation of Brightfield for Aggregate Size Distribution
3x3 tiles of brightfield images taken at 20X magnification was used for all analysis. Brightfield images were analyzed using the pixel classification software ilastik(Berg et al., 2019), which creates probability maps of locations of cells in the image. Probability maps were then segmented and quantified in CellProfiler (Carpenter et al., 2006; Lamprecht, Sabatini and Carpenter, 2007; McQuin et al., 2018; Stirling, Carpenter and Cimini, 2021), where object overlay images were also generated for transparency. The list of object sizes were then imported into MATLAB, where a “single cell” size was calculated using the median of the uninduced image, and pixel-based aggregate sizes were then converted into the number of cells. Each aggregate was then split into component cells, each of which was given a label corresponding to the total size of the aggregate, The distribution of aggregate sizes for each cell was then graphed as boxplots, and statistical comparisons were performed using the 2-sample t-test.
Image segmentation for measuring interaction frequency and patterning
All image processing and analysis in this work were done downstream of CellProfiler (Carpenter et al., 2006; Lamprecht, Sabatini and Carpenter, 2007; McQuin et al., 2018; Stirling, Carpenter and Cimini, 2021) image segmentation and quantification. For stitched 3-pair fluorescent imaging panels, cells are segmented using raw 12-bit tiff files from each channel using the IdentifyPrimaryObjects module. Exact settings were tuned manually, but primarily using the Otsu 3-Class classification (with the middle class as foreground). For stitched cell patterning images, a circular mask is first applied to remove the signal from the walls of the well. Illumination correction (CorrectIlluminationCalculate and CorrectIlluminationApply) was applied to reduce the difference in the absolute background due to stitching of 4X images, and IdentifyPrimaryObjects was subsequently used to count the number of cells. Despite best efforts, cells in the overlap region of the stitching were often missed for channels with lower signal intensity, leading to our presentation of the data as normalized cell counts.
Image-based Interaction Frequency Analysis Workflow
A detailed explanation of this workflow can be found in S9. In short, cells were segmented and quantified using CellProfiler then imported into MATLAB. The cells in individual channels are merged into a consensus list of cells based on an empirically selected distance threshold. The mean fluorescence of each cell in the list across the five measured channels was calculated, then used as parameters for supervised classification of the cells into the six cell types. Another distance threshold was empirically selected by comparing centroid distances against cell diameters, and this threshold was applied to determine cells that are interacting. Interactions between pairs of cell types are then tallied, then normalized by the sum of the number of interactions in each row and column to obtain the frequency table.
HCSRA Processing Workflow
A detailed explanation of this workflow can be found in S14. In short, kinetic OD650 read data over 15 minutes is first imported into MATLAB. For each well, the OD650 is normalized to between 0 to 1, which removes the variance from changes in growth rate affecting the raw OD650 value. The time it took for each sample to reach 0.5 of the normalized OD650 was recorded, which we termed t50. For each experiment, both an uninduced and an induced well containing identical cell types and quantity were seeded, and the t50 measured for the induced well was subtracted from that of the uninduced well to arrive at a Δt50, a measurement of the magnitude of the effect of inducing helixCAM expression in the cells that positively corresponds with increased affinity.
Supplementary Material
S25 – Table of Key Protein Sequences, Related to Figures 1-7
Table of key protein sequences used in this study.
S1 – Detailed construct design and plasmid map of bacterial, yeast, and human helixCAM, Related to STAR Methods
a. Left: Generic plasmid design for E. coli helixCAM expression. The helixCAM consists of a PelB leader sequence, followed by a His-tag, the coiled-coil flanked with glycine-serine linkers, and the ehaA autotransporter protein. The E. coli helixCAM expression is controlled by a T5/lac promoter. Right: Generic plasmid design for the all-in-one tripartite split-GFP plasmid used for stage 1 of coiled-coil library screening. The three components are linked polycistronically under the T7/lac promoter, each with its own ribosomal binding site. The first component is composed of the first coiled-coil fused to the C-terminus of the split GFP β-strand 10. The second component is composed of the second coiled-coil fused to the N-terminus of the split GFP β-strand 11. Finally, the third component is the split GFP β-barrel consisting of strands 1-9. The three components are built on a single plasmid to simplify downstream sequencing characterization of positive hits. b. Generic plasmid design for expression of aga2-SUMO-coiled-coil for yeast SynAg assay. The coiled-coil is presented on the cell wall through the fusion to SUMO and aga2 and constitutively expressed under the strong GPD promoter. On the TEF1 promoter, a corresponding fluorescent protein (mCherry or mTurquoise) is expressed. A lox site is present to allow Cre to fuse the two distinct plasmids in mated yeast cells. For the library screen, a similar design is used, but rather than a fluorescent protein, either a lysine or a leucine auxotrophic marker was used to allow for complementary auxotrophic selection. c. Left: Generic plasmid design used for most human helixCAM lines made in this work. The helixCAM consists of the Ig K-chain leader peptide fused to the coiled-coil and flanked on either side by an HA tag on the 5’ and either a myc or a His tag on the 3’, followed by the transmembrane domain of PDGFR. This is driven by the pTRE promoter. The Tet-On activator, fluorescent protein, and puromycin resistance genes are all constitutively expressed polycistronically under a single EF1a promoter. Right: The plasmid design for blasticidin-selection helixCAM for constructing dual-helixCAM lines. The general design resembles that of the left design, aside from the removal of the fluorescent protein and replacement of the puromycin resistance gene with a blasticidin resistance gene, still polycistronically expressed under EF1a. d. Left: Design for histidine-tagged coiled-coil plasmid for expression in E coli. The coiled-coil is tagged with a 12 amino acid glycine-serine linker (3xGGGS) followed by six histidine residues, is expressed under a T7 promoter, and tuned with a lac operator. Right: Design for an eGFP and histidine-tagged coiled-coil plasmid for expression in human cells. The coiled-coil is fused to a Tobacco Etch Virus cleavage site flanked by short glycine-serine linkers (GGSGGG), followed by a full-length eGFP protein, and terminated C-terminally with six histidine residues. The fusion protein is expressed under a CAG promoter for maximal expression.
S2 - Uncropped images of E. coli Z17+Z18, P3+AP4, and P9+AP10, Related to Figure 1
a. Three uncropped fields of view showing uninduced vs. induced Z17mCherry and Z18eGFP expression in E. coli. Images were taken at 60X using a Nikon Eclipse Ti2 inverted microscope with a Hamamatsu Orca-R2. For uninduced and induced cultures, identical culture conditions and mounting volumes were used. The apparent higher concentration in induced conditions may be due to the shedding of cells from the large aggregates during the mounting process. b. Uninduced and induced images of E. coli P3mCherry+AP4eGFP co-cultures. Images were taken at 63X on a Zeiss Axio Explorer Z1 inverted microscope. Uninduced images were captured using the Hamamatsu Image-EM camera, which provides higher sensitivity and larger field of view (from a larger sensor) but lacks resolution. Induced images were captured using the Hamamatsu Orca-R2 camera to allow for more resolved images of aggregates.
S3 – Uncropped images of K562 Z17+Z18, P3+AP4, P9+AP10, sg30+sg61, and sg83+sg88 co-cultures, Related to Figures 1-4
Uncropped images of pairwise cultures for all K562 helixCAM lines are shown here. All images were taken as 3x3 tiles at 20X magnifications. Cells for the uninduced condition were mixed, DMSO was added, then allowed to incubate for 48 hours before imaging. Cells for the induced condition were mixed, then doxycycline was added at 500ng/mL, then allowed to incubate for 48 hours before imaging.
S4 – Image-based Analysis of Interaction Frequencies for All K562 pairwise helixCAM co-culture, Related to Figures 2 and 4
Image-based interaction frequencies for all five pairwise helixCAM co-cultures. For each image, the full uncropped image (as seen in S3) was used for analysis, and the left panel represents a representative, cell-dense region that was cropped to demonstrate proper segmentation and interaction detection. The right panel represents the frequency of each pairwise interaction, as analyzed by the pipeline detailed in S9.
S5 – Distribution of Cells by Aggregate Size for WT and all pairwise co-cultures for uninduced and induced conditions, Related to Figure 1
a. Brightfield DIC images of WT and paired co-cultures of helixCAM cells were taken at 20X magnification with 3x3 tiling either with or without doxycycline. The images were then processed using ilastik to create a probability mask of cell vs. background, which is then subsequently segmented using CellProfiler. Colored masks designate CellProfiler assignment of each cell into aggregates. b. Boxplots representing the distribution of cells by aggregate size for WT and paired helixCAM cells. “Aggregate size” here refers to the number of cells comprising each aggregate. A “Cells by Aggregate Size” value was then computed, representing the total number of cells within the image that belonged to an aggregate of size X, and these values were plotted as a boxplot. While wild-type did not show significant change from doxycycline additional, all pairwise helixCAM cultures increased significantly higher in size compared to the uninduced conditions (p values: WT+WT, 0.27; Z17+Z18, 1.64×10−16; P3+AP4, 1.50×10−37; P9+AP10, 1.28×10−43; sg30+sg61, 4.89×10−78; sg83+sg88, 9.51×10−156). P-values were computed by 2-sample t-tests.
S6 – Sedimentation rate assay for original three helixCAM pairs (Z17+Z18, P3+AP4, P9+AP10) in E coli. and statistical tests for induced and uninduced samples, Related to Figure 1
a. Sedimentation rate assay of helixCAM-expressing E. coli for self- and pairwise interactions. The above heatmap shows the average OD600 across three replicates for each pairwise helixCAM interaction in E. coli after four hours of induction and incubation. For the E. coli sedimentation assay, the top quarter of the 5mL culture is collected, and the OD600 is immediately measured. Unlike the HCSRA (Figure 4B), lower values indicate faster sedimentation, and thus higher aggregate size and affinity. b. Bar graph of uninduced vs. induced settling results for paired helixCAMs in E. coli. Lower OD600 values indicate faster settling, which is interpreted to indicate larger aggregate formation. N=3 for all bars, and p-values were obtained through the two-sample t-test, with 2.22×10−4 for Z17+Z18, 3.11×10−5 for P3+AP4, and 7.59×10−6 for P9+AP10.
S7 – Full stitched image of uninduced and induced 3-pair helixCAM co-cultures, Related to Figures 2 and 4
Stitched microscopy images of uninduced and induced 3-pair helixCAM co-culture experiments. Cells for the uninduced condition were mixed, DMSO was added, then allowed to incubate for 48 hours before imaging. Cells for the induced condition were mixed, then doxycycline was added at 500ng/mL, then allowed to incubate for 48 hours before imaging. All images are shown as composites across five fluorescent channels (Cy5, mCherry, YFP, GFP, and DAPI). For the 3 pairs from Figure 2C (Z17mCherry, Z18eGFP, P3iRFP670, P4eBFP2, P9mPlum, P10mOrange), images were taken using the Zeiss Axio Observer Z1 microscope at 20X magnification and captured using an Orca R2 camera. The uninduced image was captured with a 3x3 tiling, and the induced image was taken with a 5x5 tiling to capture more aggregates for downstream quantification. For the 3 pairs from Figure 4F (P9mPlum, P10mOrange, sg30mCherry, sg61eGFP, sg83iRFP670, sg88eBFP2), images were taken using a Nikon Eclipse Ti2 microscope at 20X magnification and captured using an Andor Zyla camera. Similar to the first set, the uninduced image was captured with a 3x3 tiling, and the induced image was taken with a 5x5 tiling. Despite imaging at the same magnification and tiling settings, due to the difference in sensor size between the cameras of the two scopes, the images taken with the Zeiss microscope cover a smaller field of view and therefore appear more magnified than the Nikon-captured images.
S8 – Full stitched image and channel images of Z17/Z18/P3/AP4/P9/AP10 co-culture, Related to Figure 2
The full stitched image of the induced co-culture of Z17mCherry, Z18eGFP, P3iRFP670, AP4eBFP2, P9mPlum, and AP10mOrange used to calculate the imaging-based interaction frequency (Figure 2D) is shown here. The image was taken using a 6x6 tiling at 20X using a Zeiss Axio Observer Z1 across the five channels shown (DAPI, GFP, YFP, mCherry, and Cy5) with a 10% overlap, followed by stitching in Nikon Elements.
S9 – Processing Pipeline for Imaging-based cell interaction quantification, Related to Figures 2 and 4
A. Schematic of our image-segmentation-based method for characterizing cell-cell interaction frequency, consisting of five general components. First, cells are segmented using CellProfiler. Next, the object data is imported into MATLAB. At this point, we imported the raw image data from each channel and measured average intensities for each object in each channel. This was followed by supervised classification of the objects into the six fluorescent protein-expressing cell types. Finally, interactions are assigned based on distance and assigned to pairs. B. Step1: Image segmentation and Object Identification in CellProfiler. The stitched .tif image in each of the five channels (DAPI, GFP, YFP, mCherry, Cy5) is first segmented through CellProfiler, primarily through the IdentifyPrimaryObject module. The exact values change from image to image, but generally a diameter of 30-100 along with adaptive thresholding using Otsu 3-class (middle intensity class assigned to foreground), worked well for most channels (Left). On the right panel, it is possible to see an example of the segmentation obtained for the GFP channel (image is zoomed in for clarity). C. Step 2: Object import into MATLAB. Objects from each channel are first imported into MATLAB. The left pane shows the objects, outlined by channel color, overlaid on the fluorescent image. However, as some fluorescent proteins have signal across multiple channels, it is necessary to identify those cells as a single object during import. To do this, we set a distance threshold based on a histogram of pairwise distances, then sequentially merge the list of objects into a master list from the highest to lowest signal-to-noise ratio (GFP->DAPI->mCherry->YFP->Cy5). The center pane shows the process of merging Cy5 objects into the list. Black circles show existing master list objects, green circles show newly accepted objects, and red circles show rejected objects. The right pane shows the outline of all master list objects on the fluorescent image. D. Step 3. Measure object Intensity. We calculated the fluorescence of each accepted object by determining cell boundaries using their imported X/Y coordinates and diameters to measure the mean intensity from the raw channel image data. The histograms on the left show the distribution of intensities in each channel. It is possible to identify a bimodal distribution for each channel corresponding to cells either positive or negative to that channel. E. Step 4. Supervised Classification of objects by fluorescent protein. To identify six FP-expressing cell types using five channels, certain cells will be visible in multiple channels. However, PCA of the mean intensities reveals distinct clusters, allowing us to categorize each cell by fluorescence (a). First, we look at the two FPs most distinct in their channels: eBFP2 and eGFP. For these two cell types, a simple intensity threshold can be used for classification. Removing these two populations, we visualize the remaining four cell populations in the YFP, mCherry, and Cy5 channels alongside manually annotated cells to identify which population expresses which FP. We then sequentially classify cells and remove them from the population using an inequality hyperplane defined by two channels (iRFP670: Cy5, mCherry; mPlum: Cy5, YFP; mOrange: mCherry, YFP). F. Circles plotted to represent classified cells by fluorescent protein color. G. Step 5. Identification of cell-cell interaction and tallying of self- and pairwise interactions. Graphing a heatmap scatterplot of all distances between pairs of cells against the sum of the pairs’ radii, it is possible to see a cluster form near the y=x diagonal (a). We define this cluster as “interacting cells,” which we can visualize by overlaying on top of the fluorescent image (b). With a purely distanced-based approach, however, the method would also consider cells that were brought next to each other due to shared binding to a third cell, as a self-interaction. However, this is likely not reflective of the true interactions occurring. To address this, we prune interactions by removing “interactions” between cells that share a third interaction partner. In practice, this filter does not remove many interactions (<10%), and its effects can be seen by comparing (b) and (c). H. After determining interacting cells, we tally the number of observed interactions for each self and paired interactions as a table (a), then divide each square by the sum of the number of interactions in its row and column to obtain a frequency matrix, which is mirrored across the diagonal (b). We present this data as a black/red heatmap in Figure 2D.
S10 – FACS distributions of GFP intensity across paired sGFP libraries, Related to Figure 3
As the higher throughput first stage of our two-stage coiled-coil library screen, we used the tripartite split-GFP approach in E. coli. eGFP is split into β-strands 1-9, and β-strands 10 and 11. These three components have low affinity to each other unless β-strands 10 and 11 are brought together by a separate domain, leading to the formation of the intact GFP β-barrel and green fluorescence. We built four coiled-coil libraries based on Z17, Z18, P9, and AP10, and fused them to either the C-terminus of strand 10 or the N-terminus of strand 11. We then mixed these libraries in pairs, transformed them into competent cells, and sorted for GFP-positive cells. All mixed populations demonstrated a bimodal distribution, demonstrating both a functional screen as well as interacting coiled-coil pairs in each pool. Note that in the legends, the library on the left indicates the one that is fused to β-strands 10, and the library on the right is fused to β-strands 11.
S11 – Design of yeast helixCAM pre- and post- SUMO addition, Related to Figure 3
For stage two of our coiled-coil screening approach, we looked to enrich for helixCAM-compatible candidates – coiled-coils amenable to binding when presented on the cell surface. To do this at high throughput, we used the yeast SynAg assay, which uses surface protein-induced mating of haplotype-α and haplotype-a yeast cells into diploid, leading to cells expressing two markers (either two fluorescent proteins or two auxotrophic markers). We first tested the standard SynAg using Z17 and Z18 with the mCherry and mTurquoise fluorescent proteins. However, we did not observe the expected dual-positive population that successful mating should create. We hypothesized that this was due to trouble with expressing the coiled-coil-Aga2p fusion and localizing it to the yeast cell wall. To resolve this, we tried inserting a SUMO protein domain between the coiled-coil and Aga2p to help improve stability and solubility. This resulted in the formation of a third, dual-fluorescent yeast population, indicating successful yeast mating, so we adopted this design for stage two of our coiled-coil screening process.
S12 – Large sg30mCherry+sg61eGFP aggregate image with scale bar, Related to Figure 3
Measurement of large sg30+sg61 aggregation. Sg30mCherry and sg61eGFP K562 helixCAM cells were induced with doxycycline and incubated for 48 hours in a 24-well nonstick plate. At the 48-hour point, large aggregates were visible by eye. 20uL of these aggregates were transferred with a wide-bore pipetted and mounted onto a glass coverslip, followed by widefield imaging at 20X magnification. This image is stitched from 6x6 tiling with 10% overlap, and distance overlay was done in the imaging software (Nikon Elements). When compressed by the coverslip, the largest aggregate size observed was over 2.8 millimeters in length. The ability for helixCAMs to form large aggregates indicates strong potential for use in tissue engineering.
S13 – helixCAM-induced aggregation visible without magnification, Related to Figure 3
Image of 12-well plate well containing a mixture of sg30mCherry and sg61eGFP cells after 48 hours of induction. Cell aggregates could be seen by eye, and the image was captured with a cell phone camera at 1X magnification.
S14 – Analysis workflow for HCSRA, Related to Figure 4
Measurement and analysis workflow for Human Cell Sedimentation Rate Assay. K562 helixCAM cells are mixed together at 1.875e5 cells per population for paired interactions or 3.75e5 for self-interaction in 150uL of DMEM+10% FBS+1% P/S+1ug/mL doxycycline in ultra-low-adhesion v-bottom 96-well plates. The plate is incubated at 37 °C and 5% CO2 with shaking for 48 hours. Plates are then shaken at 900rpm for 15 seconds, immediately followed by OD650 measurement for 15 minutes at 37 °C. The plate is then shaken again and the OD650 is read again, resulting in two reads. The raw data can be seen in the top row. Due to some variability in growth rate between induction and cell types, the OD650 values are normalized by subtracting the minimum, then divided by the maximum to span a range of 0 to 1 (row 2). From this data, the time point at which the OD surpasses 0.5 is taken, which we refer to as t50 (row 3). The t50 for the induced condition is then subtracted from the t50 from the uninduced condition, leading to a positive value if doxycycline induction led to faster sedimentation and thus larger aggregates, and a negative value if doxycycline induction led to slower sedimentation. Finally, the values are averaged to obtain a final representative t50 value for each pairwise- and self-interaction. As seen in Figure 4c, the values from the HCSRA correlate to interaction frequencies observed through microscopy, indicating that the HCSRA is a simple and effective method for measuring cell-cell affinity.
S15 – helixCAM Induction Curve Absolute Δt50 values and significance tests, Related to Figure 4
Absolute Δt50 values from the induction curves of the top four helixCAM pairs across various doxycycline concentrations (normalized figure shown in Figure 4E). Each point represents the mean of N=4 and error bars are S.D. Statistical tests compare conditions from 5ng/mL onwards to that of the 2.5ng/mL condition. Comparisons to the uninduced conditions were not feasible due to their use to calculate the Δt50 values. P-values were derived using the two-sample t-test. At the 5ng/mL condition, the sg30+sg61 helixCAM pair already demonstrated significant binding, and the other pairs reach significance at 10 and 20ng/mL conditions.
S16 – helixCAM Induction Kinetics, Related to Figure 4
Absolute and normalized Δt50 values of the top four helixCAM pairs, induced at 500ng/mL and recorded at 0, 4, 8, 16, 24, 36, 48, and 72 hours after induction. Each point represents the mean of N=4 and error bars are S.D. Cells for each time point were cultured in distinct 96-well plates to minimize disturbance from reads, which requires vigorous shaking prior to read acquisition. Overall, aggregate sizes, as measured by Δt50 values, appear to increase up to the 36-hour time point. For sg30+sg61 and sg83+sg88 pairs, it appears to plateau at this point, whereas Δt50 values continue to increase for Z17+Z18 and P9+AP10 paired cultures.
S17 – Full stitched image and channel images of P9/AP10/sg30/sg61/sg83/sg88 co-culture, Related to Figure 4
The full stitched image of the induced co-culture of P9mPlum, AP10mOrange, sg30mCherry, sg61eGFP, sg83iRFP670, and sg88eBFP2 used to calculate the imaging-based interaction frequency (Figure 4G) is shown here. The image was taken using 5x5 tiling at 20X using a Nikon Eclipse Ti2 microscope across the five channels shown (DAPI, GFP, YFP, mCherry, and Cy5) with a 10% overlap, followed by stitching in Nikon Elements.
S18 – HCSRA Values for Dual-helixCAM lines, Related to Figure 5
HCSRA measurements were taken for dual helixCAM-expressing cell lines designed for sequential cell layering. In the above graph, the affinities between single helixCAM-expressing lines are first shown, followed by the affinity between single helixCAM-expressing line and dual helixCAM-expressing lines for each helixCAM pair. This is then followed by the affinity of two dual helixCAM-expressing lines, if applicable. We found that all dual helixCAM-expressing lines demonstrate intended affinities with their binding partners. All bars represent the mean of N=2, and error bars are S.D.
S19 – helixCAM-induced K562 binding to HEK293 cells, Related to Figure 6
In order to demonstrate the capability of helixCAMs to selectively target natively suspension cells to adherent cells for applications such as immuno-oncology, we tested our sg61/sg88eBFP2 K562 cells against either wild-type HEK293 cells and sg30mCherry HEK293 cells, both uninduced and induced. The K562 cell line is particularly suitable for this demonstration, as it stems from a lymphoblast lineage (immune cell-like), and HEK293 represents an interesting target – stemming from an embryonic adrenal lineage (hard tumor-like). The two cell types for each experiment are grown separately to 80% confluency, followed by either no induction (center column) or 48 hours of induction (right column). The suspension cells are then added to the adherent cells, followed by an additional 12 hours of incubation. The cells are imaged prior to wash, then washed twice with culture medium with gentle shaking. The cells are then imaged again. Brightfield is only shown for wild-type HEK293 conditions to demonstrate complete coverage of the field of view. While there are some remnant cells in both the wild-type and uninduced sg30-mCherry cell conditions, the induced condition has a denser coat of eBFP2-expressing K562 cells covering the surface of the mCherry-expressing HEK293 cells, demonstrating the ability of helixCAMs to increase binding of suspension cells to adherent cells.
S20 – Liquid Chromatography and Mass Spectrometry Analysis of CC-His and CC-GFP-His, Related to Figure 7
Coiled-coils tagged with either 6 histidine residues or both GFP and 6 histidine residues were expressed using E. coli or HEK293 cells respectively and subsequently analyzed by liquid chromatography (LC, top trace) and mass spectrometry (MS, bottom trace). The CC-His proteins (left) were reasonably pure, with only one major peak in the LC trace and likewise in the MS trace corresponding to the expected molecular weight. The CC-GFP-His proteins (right) had a side peak that eluted later in LC, which, when run through MS, shows the same molecular weight, leading us to conclude that it represents the unfolded CC-GFP-His protein. The resulting MS measurements of molecular weight for CC-GFP-His proteins were all higher than expected by 22 Da, which we believe to be an addition of a sodium ion from the PBS buffer used for purification.
S21 – Absolute Counts for CC-His and CC-GFP-His patterning, Related to Figure 7
Absolute cell counts for gradient of CC-His and CC-GFP-His cell patterning. Concentrations used for CC-His were 0, 0.01, 0.1, 1, 10, and 100nM, and for CC-GFP-His were 0, 0.0001, 0.001, 0.01, 0.1, and 1nM (5 order of magnitude each). CC-GFP-His concentrations were constrained by expression level. Significantly more cells bound for CC-GFP-His compared with the CC-His conditions for all CCs. Additionally, sg61eGFP cells, corresponding to the sg30-His and sg30-GFP-His conditions, had higher background binding, even for the no CC condition. This is likely due to the His tag on the sg61 construct binding to the Ni2+ coating, despite the presence of 20mM imidazole to inhibit this interaction. N=4, error bars are S.D.
S22 – Quantitative Comparison of Cell Localization for Dual-Pair CC-GFP-His patterning, Related to Figure 7
a. Quantification of helixCAM-expressing cell localization to either the “G” region, the “C” region, or unpatterned regions of the well. Images from Figure 7E were segmented using CellProfiler to obtain spatial locations for each cell, then analyzed by MATLAB. For each row, in the left panel, gray indicates the region patterned with the first CC-GFP-His protein, and yellow indicates the region patterned with the second CC-GFP-His protein. Cell locations are overlaid on these regions using their respective fluorescent protein colors. In the right panel, the portion of the cells for “Color 1” (paired to the CC patterned to G) and “Color 2” (paired to the CC patterned to C) in each region of the well is presented as bar graphs. Across all four wells, cells were primarily localized to the well region patterned with their paired coils. Cells found outside of the patterned regions were mostly around the edge of the well, which is likely due to the reduced shear force leading to incomplete cell removal during washes. b. Bar graph of cell localization percentages aggregated across the CC-GFP-His protein used for patterning. Similar to individual results, most cells localized to the intended region. Statistical comparison between the intended vs. opposite regions as well as between the intended vs. unpatterned regions was taken using the two-sample T-test, with an N of 2. *** indicates p-values of less than 0.005, ** indicates p-values of less than 0.01, and * indicates p-value of less than 0.05, above which the comparison is marked “not significant” (n.s.).
S23 – Dot-shaped patterning of K562 helixCAM cells using CC-GFP-His, Related to Figure 7
Similar to Figure 7c, Nickel-coated plates were pre-patterned with two distinct Coiled-Coil-GFP-His protein solutions. In this case, 1uL of each protein solution was dotted on either the top or the bottom of the well. The solution was incubated for 30 minutes, washed with PBS+imidazole, then blocked for 30 minutes (also with PBS+imidazole). After removing the blocking buffer, a mixture of the two corresponding K562 helixCAM cells was added into a dual induction/blocking media (1e5 of each cell type in 150uL of DMEM+10%FBS+1%P/S+1ug/mL doxycycline+20mM imidazole). The wells were incubated with the cells for 2 hours, followed by two washes with the same media, and the well was imaged as a 2x2 stitching at 4X magnification. Prior to the wash, it was already possible to see cells localizing densely to the dotted location. Post-wash, there was distinct localization of densely bound cells to the two patterned dots, with few cells in the surrounding region. Since all aspiration and addition of media were done along the bottom of the well, the pipette tips appear to have removed areas of bound cells, but the differential patterning is still clearly visible. It should be noted that addition and removal of media were done along the bottom edge of the wells, which likely led to consistently more dissociation of the bottom dot.
S24 – Viability of helixCAM cells post-induction, Related to Figures 1-4
The viabilities of wild-type and helixCAM K562 cells, with and without doxycycline induction, were measured over four days. Cells were mixed 1:1 with Trypan Blue, then loaded onto measurement slides. Measurements were taken using a Countess II FL, which reads out both cell concentrations as well as percent viabilities based on Trypan Blue exclusion. Data are reported as the mean of N=3, and error bars are S.D. Across all helixCAM lines, growth rate was reduced by induction of helixCAM expression. Most lines maintained high cell viability with the exception of sg30mCherry and sg83iRFP670. Due to the similarity in structure of the helixCAMs and that most cell lines maintain high viability across the four days, it is likely that variable expression of helixCAM can contribute to viability changes. For sensitive cell types, it may be worth optimizing expression level to use the lowest expression of helixCAM needed for the desired cell affinity to minimize viability effects.
Highlights.
helixCAM interactions induce large, patterned aggregates in E. coli and human cells
Known principles of coiled-coil interactions can inform design of novel helixCAMs
Multiple helixCAMs can be simultaneously used to form complex cell architecture
helixCAMs can pattern cells onto other cells and coiled-coil-coated surfaces
Acknowledgements
We are extremely grateful to Dr. Roman Jerala for sharing the engineered coiled-coil pairs P3/AP4 and P9/AP10. We are grateful to Dr. Bridget Baumgartner, Dr. Justin Gallivan, Dr. Jesse Dill, and Dr. Joseph Pomerening for helping to fund and shape the direction of our work. Thank you to Dr. Ron Raines at MIT for the use of LC-MS equipment for characterizing the CC-His and CC-GFP-His proteins. We thank Dr. Paula Montero Llopis and Ryan Stephansky from the HMS MicRoN core facility for their assistance in microscopy techniques and maintenance of microscopes. We also thank the HMS Immunology Flow Cytometry facility, particularly Chad Araneo, Jeff Nelson, and Meegan Sleeper for training and maintenance of the FACS machines. Thank you to Songlei Liu for providing the v-bottom ULA plates for HCSRA experiments. We are extremely grateful to Tiffany Dill and John Aach help to edit the manuscript. Thank you to Emma Taddeo for all of her help with the lab’s administrative work, and to Nicole D’Aleo for managing the grants. Plasmid design and map image generation was done in A Plasmid Editor (ApE). A majority of schematic figures made in this work were created with BioRender.
This work was supported in large part by the DARPA Engineered Living Materials program under contract W911NF-17-2-0079. G.C. was also supported by the NHGRI Centers of Excellence in Genomic Science (RM1HG008525) as well as a general gift from the Zhijun Yang Research Fund. T.W. was also supported by the DoE under the award DE-FG02-02ER63445.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
G.C., T.W., and G.M.C. are inventors on a patent application filed by the President and Fellows of Harvard College. The authors declare no other competing interests.
References
- Anderson GG et al. (2003) ‘Intracellular bacterial biofilm-like pods in urinary tract infections’, Science. American Association for the Advancement of Science, 301(5629), pp. 105–107. doi: 10.1126/SCIENCE.1084550/SUPPL_FILE/ANDERSON.SOM.PDF. [DOI] [PubMed] [Google Scholar]
- Ansari AM et al. (2016) ‘Cellular GFP Toxicity and Immunogenicity: Potential Confounders in in Vivo Cell Tracking Experiments’, Stem Cell Reviews and Reports. Humana Press Inc., 12(5), pp. 553–559. doi: 10.1007/S12015-016-9670-8/TABLES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagheri Y et al. (2019) ‘A quantitative assessment of the dynamic modification of lipid–DNA probes on live cell membranes’, Chemical Science. The Royal Society of Chemistry, 10(48), pp. 11030–11040. doi: 10.1039/C9SC04251B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg S et al. (2019) ‘ilastik: interactive machine learning for (bio)image analysis’, Nature Methods 2019 16:12. Nature Publishing Group, 16(12), pp. 1226–1232. doi: 10.1038/s41592-019-0582-9. [DOI] [PubMed] [Google Scholar]
- Bhatia SN and Ingber DE (2014) ‘Microfluidic organs-on-chips’, Nature Biotechnology. Nature Publishing Group, 32(8), pp. 760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- Boder ET and Wittrup KD (1997) ‘Yeast surface display for screening combinatorial polypeptide libraries’, Nature Biotechnology. Nature Publishing Group, 15(6), pp. 553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- Cabantous S et al. (2013) ‘A new protein-protein interaction sensor based on tripartite split-GFP association’, Scientific Reports. Sci Rep, 3. doi: 10.1038/srep02854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cachat E et al. (2016) ‘2- and 3-dimensional synthetic large-scale de novo patterning by mammalian cells through phase separation’, Scientific Reports. Nature Publishing Group, 6. doi: 10.1038/srep20664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LJ, Willoughby JJ and Jensen AM (2012) ‘Two Types of Tet-On Transgenic Lines for Doxycycline-Inducible Gene Expression in Zebrafish Rod Photoreceptors and a Gateway-Based Tet-On Toolkit’, PLOS ONE. Public Library of Science, 7(12), p. e51270. doi: 10.1371/JOURNAL.PONE.0051270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE et al. (2006) ‘CellProfiler: Image analysis software for identifying and quantifying cell phenotypes’, Genome Biology. BioMed Central, 7(10), pp. 1–11. doi: 10.1186/GB-2006-7-10-R100/FIGURES/4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra RA et al. (2006) ‘Programmable cell adhesion encoded by DNA hybridization’, Angewandte Chemie - International Edition. John Wiley & Sons, Ltd, 45(6), pp. 896–901. doi: 10.1002/anie.200502421. [DOI] [PubMed] [Google Scholar]
- Chen EP et al. (2021) ‘3D Bioprinting of Vascularized Tissues for in vitro and in vivo Applications’, Frontiers in Bioengineering and Biotechnology. Frontiers Media S.A., 9, p. 326. doi: 10.3389/FBIOE.2021.664188/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-J et al. (2019) ‘Self-Healing Concrete by Biological Substrate’, Materials. MDPI AG, 12(24), p. 4099. doi: 10.3390/ma12244099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z et al. (2019) ‘Programmable design of orthogonal protein heterodimers’, Nature. Nature Publishing Group, 565(7737), pp. 106–111. doi: 10.1038/s41586-018-0802-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesnut JD et al. (1996) ‘Selective isolation of transiently transfected cells from a mammalian cell population with vectors expressing a membrane anchored single- chain antibody’, J.Immunol.Methods, 193(1), pp. 17–27. [DOI] [PubMed] [Google Scholar]
- Cock PJA et al. (2009) ‘Biopython: freely available Python tools for computational molecular biology and bioinformatics’, Bioinformatics. Oxford Academic, 25(11), pp. 1422–1423. doi: 10.1093/BIOINFORMATICS/BTP163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubendorf JW and Studier FW (1991) ‘Controlling basal expression in an inducible T7 expression system by blocking the target T7 promoter with lac repressor’, Journal of molecular biology. J Mol Biol, 219(1), pp. 45–59. doi: 10.1016/0022-2836(91)90856-2. [DOI] [PubMed] [Google Scholar]
- Fink T et al. (2018) ‘Design of fast proteolysis-based signaling and logic circuits in mammalian cells’, Nature Chemical Biology 2018 15:2. Nature Publishing Group, 15(2), pp. 115–122. doi: 10.1038/s41589-018-0181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foty RA and Steinberg MS (2005) ‘The differential adhesion hypothesis: A direct evaluation’, Developmental Biology. Academic Press Inc., 278(1), pp. 255–263. doi: 10.1016/j.ydbio.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Friedl P and Storim J (2004) ‘Diversity in immune-cell interactions: States and functions of the immunological synapse’, Trends in Cell Biology. Elsevier, 14(10), pp. 557–567. doi: 10.1016/J.TCB.2004.09.005/ATTACHMENT/2FFB1FB6-FB8A-4924-89EB-D61C2C82D420/VIDEO1.MOV. [DOI] [PubMed] [Google Scholar]
- Fr̈mmel U et al. (2013) ‘Adhesion of human and animal escherichia coli strains in association with their virulence-associated genes and phylogenetic origins’, Applied and Environmental Microbiology. American Society for Microbiology, 79(19), pp. 5814–5829. doi: 10.1128/AEM.01384-13/SUPPL_FILE/ZAM999104720SO1.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD and Schiestl RH (2007) ‘High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method’, Nature protocols. Nat Protoc, 2(1), pp. 31–34. doi: 10.1038/NPROT.2007.13. [DOI] [PubMed] [Google Scholar]
- Glass DS and Riedel-Kruse IH (2018) ‘A Synthetic Bacterial Cell-Cell Adhesion Toolbox for Programming Multicellular Morphologies and Patterns’, Cell. Cell Press, 174(3), pp. 649–658.e16. doi: 10.1016/j.cell.2018.06.041. [DOI] [PubMed] [Google Scholar]
- Griffith CK et al. (2005) ‘Diffusion Limits of an in Vitro Thick Prevascularized Tissue’, https://home.liebertpub.com/ten. Mary Ann Liebert, Inc. 2 Madison Avenue Larchmont, NY 10538 USA , 11(1–2), pp. 257–266. doi: 10.1089/TEN.2005.11.257. [DOI] [PubMed] [Google Scholar]
- Gromiha MM and Parry DAD (2004) ‘Characteristic features of amino acid residues in coiled-coil protein structures’, Biophysical chemistry. Biophys Chem, 111(2), pp. 95–103. doi: 10.1016/J.BPC.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM (1996) ‘Cell adhesion: The molecular basis of tissue architecture and morphogenesis’, Cell. Elsevier, pp. 345–357. doi: 10.1016/S0092-8674(00)81279-9. [DOI] [PubMed] [Google Scholar]
- Guo S et al. (2020) ‘Engineered Living Materials Based on Adhesin-Mediated Trapping of Programmable Cells’, ACS Synthetic Biology. American Chemical Society, 9(3), pp. 475–485. doi: 10.1021/acssynbio.9b00404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heller E and Fuchs E (2015) ‘Tissue patterning and cellular mechanics’, The Journal of Cell Biology. The Rockefeller University Press, 211(2), p. 219. doi: 10.1083/JCB.201506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks MR et al. (1997) ‘Coiled-coil assembly by peptides with non-heptad sequence motifs’, Folding and Design. Cell Press, 2(3), pp. 149–158. doi: 10.1016/S1359-0278(97)00021-7. [DOI] [PubMed] [Google Scholar]
- Hsiao SC et al. (2009) ‘Direct cell surface modification with DNA for the capture of primary cells and the investigation of myotube formation on defined patterns’, Langmuir, 25(12), pp. 6985–6991. doi: 10.1021/la900150n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain RK et al. (2005) ‘Engineering vascularized tissue’, Nature Biotechnology 2005 23:7. Nature Publishing Group, 23(7), pp. 821–823. doi: 10.1038/nbt0705-821. [DOI] [PubMed] [Google Scholar]
- Kim HJ et al. (2012) ‘Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow.’, Lab on a chip, 12(12), pp. 2165–74. doi: 10.1039/c2lc40074j. [DOI] [PubMed] [Google Scholar]
- Kintaka R, Makanae K and Moriya H (2016) ‘Cellular growth defects triggered by an overload of protein localization processes’, Scientific Reports 2016 6:1. Nature Publishing Group, 6(1), pp. 1–11. doi: 10.1038/srep31774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolesky DB et al. (2016) ‘Three-dimensional bioprinting of thick vascularized tissues’, Proceedings of the National Academy of Sciences of the United States of America. National Academy of Sciences, 113(12), pp. 3179–3184. doi: 10.1073/pnas.1521342113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo D, Nie M and Courey AJ (2014) ‘SUMO as a Solubility Tag and In Vivo Cleavage of SUMO Fusion Proteins with Ulp1’, in, pp. 71–80. doi: 10.1007/978-1-4939-1034-2_6. [DOI] [PubMed] [Google Scholar]
- Lamprecht MR, Sabatini DM and Carpenter AE (2007) ‘CellProfiler: free, versatile software for automated biological image analysis’, BioTechniques. Biotechniques, 42(1), pp. 71–75. doi: 10.2144/000112257. [DOI] [PubMed] [Google Scholar]
- Lebar T et al. (2020) ‘A tunable orthogonal coiled-coil interaction toolbox for engineering mammalian cells’, Nature Chemical Biology. Nature Research, 16(5), pp. 513–519. doi: 10.1038/s41589-019-0443-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederberg J and Tatum EL (1946) ‘Gene Recombination in Escherichia Coli’, Nature 1946 158:4016. Nature Publishing Group, 158(4016), pp. 558–558. doi: 10.1038/158558a0. [DOI] [PubMed] [Google Scholar]
- Litowski JR and Hodges RS (2002) ‘Designing Heterodimeric Two-stranded α-Helical Coiled-coils: EFFECTS OF HYDROPHOBICITY AND α-HELICAL PROPENSITY ON PROTEIN FOLDING, STABILITY, AND SPECIFICITY’, Journal of Biological Chemistry. Elsevier, 277(40), pp. 37272–37279. doi: 10.1074/JBC.M204257200. [DOI] [PubMed] [Google Scholar]
- McQuin C et al. (2018) ‘CellProfiler 3.0: Next-generation image processing for biology’, PLOS Biology. Public Library of Science, 16(7), p. e2005970. doi: 10.1371/JOURNAL.PBIO.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni L et al. (2018) ‘Biofabrication strategies for 3D in vitro models and regenerative medicine’, Nature Reviews Materials. Nature Publishing Group, 3(5), pp. 21–37. doi: 10.1038/s41578-018-0006-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsut L et al. (2016) ‘Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors’, Cell. Elsevier Inc., 164(4), pp. 780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy SV and Atala A (2014) ‘3D bioprinting of tissues and organs’, Nature Biotechnology. Nature Publishing Group, 32(8), pp. 773–785. doi: 10.1038/nbt.2958. [DOI] [PubMed] [Google Scholar]
- Naumann S et al. (2001) ‘Complete karyotype characterization of the K562 cell line by combined application of G-banding, multiplex-fluorescence in situ hybridization, fluorescence in situ hybridization, and comparative genomic hybridization’, Leukemia Research, 25(4), pp. 313–322. doi: 10.1016/S0145-2126(00)00125-9. [DOI] [PubMed] [Google Scholar]
- Obana M, Silverman BR and Tirrell DA (2017) ‘Protein-Mediated Colloidal Assembly’, Journal of the American Chemical Society, 139(40), pp. 14251–14256. doi: 10.1021/jacs.7b07798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozbolat IT and Yu Y (2013) ‘Bioprinting toward organ fabrication: Challenges and future trends’, IEEE Transactions on Biomedical Engineering, 60(3), pp. 691–699. doi: 10.1109/TBME.2013.2243912. [DOI] [PubMed] [Google Scholar]
- Patel SD et al. (2003) ‘Cadherin-mediated cell-cell adhesion: Sticking together as a family’, Current Opinion in Structural Biology. Elsevier Ltd, 13(6), pp. 690–698. doi: 10.1016/j.sbi.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Piñero-Lambea C et al. (2015) ‘Programming controlled adhesion of E. coli to target surfaces, cells, and tumors with synthetic adhesins’, ACS Synthetic Biology. American Chemical Society, 4(4), pp. 463–473. doi: 10.1021/SB500252A/SUPPL_FILE/SB500252A_SI_003.PDF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin JY et al. (2010) ‘Systematic comparison of constitutive promoters and the doxycycline-inducible promoter’, PLoS ONE, 5(5), pp. 3–6. doi: 10.1371/journal.pone.0010611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romei MG and Boxer SG (2019) ‘Split Green Fluorescent Proteins: Scope, Limitations, and Outlook’, Annual Review of Biophysics. Annual Reviews Inc., pp. 19–44. doi: 10.1146/annurev-biophys-051013-022846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roohani-Esfahani SI, Newman P and Zreiqat H (2016) ‘Design and Fabrication of 3D printed Scaffolds with a Mechanical Strength Comparable to Cortical Bone to Repair Large Bone Defects’, Scientific Reports. Nature Publishing Group, 6(1), pp. 1–8. doi: 10.1038/srep19468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT et al. (2016) ‘Precision Tumor Recognition by T Cells with Combinatorial Antigen-Sensing Circuits’, Cell. Cell Press, 164(4), pp. 770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT and Lim WA (2017) ‘Synthetic immunology: Hacking immune cells to expand their therapeutic capabilities’, Annual Review of Immunology. Annual Reviews Inc., 35, pp. 229–253. doi: 10.1146/annurev-immunol-051116-052302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salema V et al. (2013) ‘Selection of Single Domain Antibodies from Immune Libraries Displayed on the Surface of E. coli Cells with Two β-Domains of Opposite Topologies’, PLoS ONE. Edited by Sturtevant J. Public Library of Science, 8(9), p. e75126. doi: 10.1371/journal.pone.0075126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan WS et al. (2000) ‘Functional cis-heterodimers of N- and R-cadherins’, Journal of Cell Biology. The Rockefeller University Press, 148(3), pp. 579–590. doi: 10.1083/jcb.148.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song HHG et al. (2018) ‘Vascular Tissue Engineering: Progress, Challenges, and Clinical Promise’, Cell Stem Cell. Elsevier Inc., 22(3), pp. 340–354. doi: 10.1016/j.stem.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens KR et al. (2017) ‘In situ expansion of engineered human liver tissue in a mouse model of chronic liver disease’, Science Translational Medicine. American Association for the Advancement of Science, 9(399), pp. eaah5505–eaah5505. doi: 10.1126/scitranslmed.aah5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DR, Carpenter AE and Cimini BA (2021) ‘CellProfiler Analyst 3.0: accessible data exploration and machine learning for image analysis’, Bioinformatics. Oxford Academic, 37(21), pp. 3992–3994. doi: 10.1093/BIOINFORMATICS/BTAB634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson KE et al. (2012) ‘Synzip protein interaction toolbox: In vitro and in vivo specifications of heterospecific coiled-coil interaction domains’, ACS Synthetic Biology, 1(4), pp. 118–129. doi: 10.1021/sb200015u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todhunter ME et al. (2015) ‘Programmed synthesis of three-dimensional tissues’, Nature Methods. Nature Publishing Group, 12(10), pp. 975–981. doi: 10.1038/nmeth.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truebestein L and Leonard TA (2016) ‘Coiled- coils: The long and short of it’, Bioessays. Wiley-Blackwell, 38(9), p. 903. doi: 10.1002/BIES.201600062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trunk T, Khalil HS and Leo JC (2018) ‘Bacterial autoaggregation’, AIMS Microbiology. AIMS Press, 4(1), p. 140. doi: 10.3934/MICROBIOL.2018.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urlinger S et al. (2000) ‘Exploring the sequence space for tetracycline-dependent transcriptional activators: Novel mutations yield expanded range and sensitivity’, Proceedings of the National Academy of Sciences of the United States of America. National Academy of Sciences, 97(14), pp. 7963–7968. doi: 10.1073/pnas.130192197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga E, De Lorenzo V and Fernández LA (2003) ‘Autotransporters as Scaffolds for Novel Bacterial Adhesins: Surface Properties of Escherichia coli Cells Displaying Jun/Fos Dimerization Domains’, Journal of Bacteriology. American Society for Microbiology (ASM), 185(18), p. 5585. doi: 10.1128/JB.185.18.5585-5590.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldman AD, Fritz JM and Lenardo MJ (2020) ‘A guide to cancer immunotherapy: from T cell basic science to clinical practice’, Nature Reviews Immunology 2020 20:11. Nature Publishing Group, 20(11), pp. 651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MH, Coates CJ and George AL (2007) ‘PiggyBac transposon-mediated gene transfer in human cells.’, Molecular therapy: the journal of the American Society of Gene Therapy, 15(1), pp. 139–145. doi: 10.1038/sj.mt.6300028. [DOI] [PubMed] [Google Scholar]
- Woolfson DN (2005) ‘The design of coiled-coil structures and assemblies’, Advances in Protein Chemistry. Academic Press Inc., 70, pp. 79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- Yim EKF and Sheetz MP (2012) ‘Force-dependent cell signaling in stem cell differentiation’, Stem Cell Research and Therapy. BioMed Central, 3(5), pp. 1–12. doi: 10.1186/SCRT132/TABLES/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younger D et al. (2017) ‘High-throughput characterization of protein–protein interactions by reprogramming yeast mating’, Proceedings of the National Academy of Sciences of the United States of America. National Academy of Sciences, 114(46), pp. 12166–12171. doi: 10.1073/pnas.1705867114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y et al. (2013) ‘Evaluation of cell viability and functionality in vessel-like bioprintable cell-laden tubular channels’, Journal of Biomechanical Engineering. American Society of Mechanical Engineers Digital Collection, 135(9). doi: 10.1115/1.4024575/371049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeller R, López-Ríos J and Zuniga A (2009) ‘Vertebrate limb bud development: moving towards integrative analysis of organogenesis’, Nature Reviews Genetics 2009 10:12. Nature Publishing Group, 10(12), pp. 845–858. doi: 10.1038/nrg2681. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Yao S and Chen L (2011) ‘CELL SURFACE SIGNALING MOLECULES IN THE CONTROL OF IMMUNE RESPONSES: A TIDE MODEL’, Immunity. NIH Public Access, 34(4), p. 466. doi: 10.1016/J.IMMUNI.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S25 – Table of Key Protein Sequences, Related to Figures 1-7
Table of key protein sequences used in this study.
S1 – Detailed construct design and plasmid map of bacterial, yeast, and human helixCAM, Related to STAR Methods
a. Left: Generic plasmid design for E. coli helixCAM expression. The helixCAM consists of a PelB leader sequence, followed by a His-tag, the coiled-coil flanked with glycine-serine linkers, and the ehaA autotransporter protein. The E. coli helixCAM expression is controlled by a T5/lac promoter. Right: Generic plasmid design for the all-in-one tripartite split-GFP plasmid used for stage 1 of coiled-coil library screening. The three components are linked polycistronically under the T7/lac promoter, each with its own ribosomal binding site. The first component is composed of the first coiled-coil fused to the C-terminus of the split GFP β-strand 10. The second component is composed of the second coiled-coil fused to the N-terminus of the split GFP β-strand 11. Finally, the third component is the split GFP β-barrel consisting of strands 1-9. The three components are built on a single plasmid to simplify downstream sequencing characterization of positive hits. b. Generic plasmid design for expression of aga2-SUMO-coiled-coil for yeast SynAg assay. The coiled-coil is presented on the cell wall through the fusion to SUMO and aga2 and constitutively expressed under the strong GPD promoter. On the TEF1 promoter, a corresponding fluorescent protein (mCherry or mTurquoise) is expressed. A lox site is present to allow Cre to fuse the two distinct plasmids in mated yeast cells. For the library screen, a similar design is used, but rather than a fluorescent protein, either a lysine or a leucine auxotrophic marker was used to allow for complementary auxotrophic selection. c. Left: Generic plasmid design used for most human helixCAM lines made in this work. The helixCAM consists of the Ig K-chain leader peptide fused to the coiled-coil and flanked on either side by an HA tag on the 5’ and either a myc or a His tag on the 3’, followed by the transmembrane domain of PDGFR. This is driven by the pTRE promoter. The Tet-On activator, fluorescent protein, and puromycin resistance genes are all constitutively expressed polycistronically under a single EF1a promoter. Right: The plasmid design for blasticidin-selection helixCAM for constructing dual-helixCAM lines. The general design resembles that of the left design, aside from the removal of the fluorescent protein and replacement of the puromycin resistance gene with a blasticidin resistance gene, still polycistronically expressed under EF1a. d. Left: Design for histidine-tagged coiled-coil plasmid for expression in E coli. The coiled-coil is tagged with a 12 amino acid glycine-serine linker (3xGGGS) followed by six histidine residues, is expressed under a T7 promoter, and tuned with a lac operator. Right: Design for an eGFP and histidine-tagged coiled-coil plasmid for expression in human cells. The coiled-coil is fused to a Tobacco Etch Virus cleavage site flanked by short glycine-serine linkers (GGSGGG), followed by a full-length eGFP protein, and terminated C-terminally with six histidine residues. The fusion protein is expressed under a CAG promoter for maximal expression.
S2 - Uncropped images of E. coli Z17+Z18, P3+AP4, and P9+AP10, Related to Figure 1
a. Three uncropped fields of view showing uninduced vs. induced Z17mCherry and Z18eGFP expression in E. coli. Images were taken at 60X using a Nikon Eclipse Ti2 inverted microscope with a Hamamatsu Orca-R2. For uninduced and induced cultures, identical culture conditions and mounting volumes were used. The apparent higher concentration in induced conditions may be due to the shedding of cells from the large aggregates during the mounting process. b. Uninduced and induced images of E. coli P3mCherry+AP4eGFP co-cultures. Images were taken at 63X on a Zeiss Axio Explorer Z1 inverted microscope. Uninduced images were captured using the Hamamatsu Image-EM camera, which provides higher sensitivity and larger field of view (from a larger sensor) but lacks resolution. Induced images were captured using the Hamamatsu Orca-R2 camera to allow for more resolved images of aggregates.
S3 – Uncropped images of K562 Z17+Z18, P3+AP4, P9+AP10, sg30+sg61, and sg83+sg88 co-cultures, Related to Figures 1-4
Uncropped images of pairwise cultures for all K562 helixCAM lines are shown here. All images were taken as 3x3 tiles at 20X magnifications. Cells for the uninduced condition were mixed, DMSO was added, then allowed to incubate for 48 hours before imaging. Cells for the induced condition were mixed, then doxycycline was added at 500ng/mL, then allowed to incubate for 48 hours before imaging.
S4 – Image-based Analysis of Interaction Frequencies for All K562 pairwise helixCAM co-culture, Related to Figures 2 and 4
Image-based interaction frequencies for all five pairwise helixCAM co-cultures. For each image, the full uncropped image (as seen in S3) was used for analysis, and the left panel represents a representative, cell-dense region that was cropped to demonstrate proper segmentation and interaction detection. The right panel represents the frequency of each pairwise interaction, as analyzed by the pipeline detailed in S9.
S5 – Distribution of Cells by Aggregate Size for WT and all pairwise co-cultures for uninduced and induced conditions, Related to Figure 1
a. Brightfield DIC images of WT and paired co-cultures of helixCAM cells were taken at 20X magnification with 3x3 tiling either with or without doxycycline. The images were then processed using ilastik to create a probability mask of cell vs. background, which is then subsequently segmented using CellProfiler. Colored masks designate CellProfiler assignment of each cell into aggregates. b. Boxplots representing the distribution of cells by aggregate size for WT and paired helixCAM cells. “Aggregate size” here refers to the number of cells comprising each aggregate. A “Cells by Aggregate Size” value was then computed, representing the total number of cells within the image that belonged to an aggregate of size X, and these values were plotted as a boxplot. While wild-type did not show significant change from doxycycline additional, all pairwise helixCAM cultures increased significantly higher in size compared to the uninduced conditions (p values: WT+WT, 0.27; Z17+Z18, 1.64×10−16; P3+AP4, 1.50×10−37; P9+AP10, 1.28×10−43; sg30+sg61, 4.89×10−78; sg83+sg88, 9.51×10−156). P-values were computed by 2-sample t-tests.
S6 – Sedimentation rate assay for original three helixCAM pairs (Z17+Z18, P3+AP4, P9+AP10) in E coli. and statistical tests for induced and uninduced samples, Related to Figure 1
a. Sedimentation rate assay of helixCAM-expressing E. coli for self- and pairwise interactions. The above heatmap shows the average OD600 across three replicates for each pairwise helixCAM interaction in E. coli after four hours of induction and incubation. For the E. coli sedimentation assay, the top quarter of the 5mL culture is collected, and the OD600 is immediately measured. Unlike the HCSRA (Figure 4B), lower values indicate faster sedimentation, and thus higher aggregate size and affinity. b. Bar graph of uninduced vs. induced settling results for paired helixCAMs in E. coli. Lower OD600 values indicate faster settling, which is interpreted to indicate larger aggregate formation. N=3 for all bars, and p-values were obtained through the two-sample t-test, with 2.22×10−4 for Z17+Z18, 3.11×10−5 for P3+AP4, and 7.59×10−6 for P9+AP10.
S7 – Full stitched image of uninduced and induced 3-pair helixCAM co-cultures, Related to Figures 2 and 4
Stitched microscopy images of uninduced and induced 3-pair helixCAM co-culture experiments. Cells for the uninduced condition were mixed, DMSO was added, then allowed to incubate for 48 hours before imaging. Cells for the induced condition were mixed, then doxycycline was added at 500ng/mL, then allowed to incubate for 48 hours before imaging. All images are shown as composites across five fluorescent channels (Cy5, mCherry, YFP, GFP, and DAPI). For the 3 pairs from Figure 2C (Z17mCherry, Z18eGFP, P3iRFP670, P4eBFP2, P9mPlum, P10mOrange), images were taken using the Zeiss Axio Observer Z1 microscope at 20X magnification and captured using an Orca R2 camera. The uninduced image was captured with a 3x3 tiling, and the induced image was taken with a 5x5 tiling to capture more aggregates for downstream quantification. For the 3 pairs from Figure 4F (P9mPlum, P10mOrange, sg30mCherry, sg61eGFP, sg83iRFP670, sg88eBFP2), images were taken using a Nikon Eclipse Ti2 microscope at 20X magnification and captured using an Andor Zyla camera. Similar to the first set, the uninduced image was captured with a 3x3 tiling, and the induced image was taken with a 5x5 tiling. Despite imaging at the same magnification and tiling settings, due to the difference in sensor size between the cameras of the two scopes, the images taken with the Zeiss microscope cover a smaller field of view and therefore appear more magnified than the Nikon-captured images.
S8 – Full stitched image and channel images of Z17/Z18/P3/AP4/P9/AP10 co-culture, Related to Figure 2
The full stitched image of the induced co-culture of Z17mCherry, Z18eGFP, P3iRFP670, AP4eBFP2, P9mPlum, and AP10mOrange used to calculate the imaging-based interaction frequency (Figure 2D) is shown here. The image was taken using a 6x6 tiling at 20X using a Zeiss Axio Observer Z1 across the five channels shown (DAPI, GFP, YFP, mCherry, and Cy5) with a 10% overlap, followed by stitching in Nikon Elements.
S9 – Processing Pipeline for Imaging-based cell interaction quantification, Related to Figures 2 and 4
A. Schematic of our image-segmentation-based method for characterizing cell-cell interaction frequency, consisting of five general components. First, cells are segmented using CellProfiler. Next, the object data is imported into MATLAB. At this point, we imported the raw image data from each channel and measured average intensities for each object in each channel. This was followed by supervised classification of the objects into the six fluorescent protein-expressing cell types. Finally, interactions are assigned based on distance and assigned to pairs. B. Step1: Image segmentation and Object Identification in CellProfiler. The stitched .tif image in each of the five channels (DAPI, GFP, YFP, mCherry, Cy5) is first segmented through CellProfiler, primarily through the IdentifyPrimaryObject module. The exact values change from image to image, but generally a diameter of 30-100 along with adaptive thresholding using Otsu 3-class (middle intensity class assigned to foreground), worked well for most channels (Left). On the right panel, it is possible to see an example of the segmentation obtained for the GFP channel (image is zoomed in for clarity). C. Step 2: Object import into MATLAB. Objects from each channel are first imported into MATLAB. The left pane shows the objects, outlined by channel color, overlaid on the fluorescent image. However, as some fluorescent proteins have signal across multiple channels, it is necessary to identify those cells as a single object during import. To do this, we set a distance threshold based on a histogram of pairwise distances, then sequentially merge the list of objects into a master list from the highest to lowest signal-to-noise ratio (GFP->DAPI->mCherry->YFP->Cy5). The center pane shows the process of merging Cy5 objects into the list. Black circles show existing master list objects, green circles show newly accepted objects, and red circles show rejected objects. The right pane shows the outline of all master list objects on the fluorescent image. D. Step 3. Measure object Intensity. We calculated the fluorescence of each accepted object by determining cell boundaries using their imported X/Y coordinates and diameters to measure the mean intensity from the raw channel image data. The histograms on the left show the distribution of intensities in each channel. It is possible to identify a bimodal distribution for each channel corresponding to cells either positive or negative to that channel. E. Step 4. Supervised Classification of objects by fluorescent protein. To identify six FP-expressing cell types using five channels, certain cells will be visible in multiple channels. However, PCA of the mean intensities reveals distinct clusters, allowing us to categorize each cell by fluorescence (a). First, we look at the two FPs most distinct in their channels: eBFP2 and eGFP. For these two cell types, a simple intensity threshold can be used for classification. Removing these two populations, we visualize the remaining four cell populations in the YFP, mCherry, and Cy5 channels alongside manually annotated cells to identify which population expresses which FP. We then sequentially classify cells and remove them from the population using an inequality hyperplane defined by two channels (iRFP670: Cy5, mCherry; mPlum: Cy5, YFP; mOrange: mCherry, YFP). F. Circles plotted to represent classified cells by fluorescent protein color. G. Step 5. Identification of cell-cell interaction and tallying of self- and pairwise interactions. Graphing a heatmap scatterplot of all distances between pairs of cells against the sum of the pairs’ radii, it is possible to see a cluster form near the y=x diagonal (a). We define this cluster as “interacting cells,” which we can visualize by overlaying on top of the fluorescent image (b). With a purely distanced-based approach, however, the method would also consider cells that were brought next to each other due to shared binding to a third cell, as a self-interaction. However, this is likely not reflective of the true interactions occurring. To address this, we prune interactions by removing “interactions” between cells that share a third interaction partner. In practice, this filter does not remove many interactions (<10%), and its effects can be seen by comparing (b) and (c). H. After determining interacting cells, we tally the number of observed interactions for each self and paired interactions as a table (a), then divide each square by the sum of the number of interactions in its row and column to obtain a frequency matrix, which is mirrored across the diagonal (b). We present this data as a black/red heatmap in Figure 2D.
S10 – FACS distributions of GFP intensity across paired sGFP libraries, Related to Figure 3
As the higher throughput first stage of our two-stage coiled-coil library screen, we used the tripartite split-GFP approach in E. coli. eGFP is split into β-strands 1-9, and β-strands 10 and 11. These three components have low affinity to each other unless β-strands 10 and 11 are brought together by a separate domain, leading to the formation of the intact GFP β-barrel and green fluorescence. We built four coiled-coil libraries based on Z17, Z18, P9, and AP10, and fused them to either the C-terminus of strand 10 or the N-terminus of strand 11. We then mixed these libraries in pairs, transformed them into competent cells, and sorted for GFP-positive cells. All mixed populations demonstrated a bimodal distribution, demonstrating both a functional screen as well as interacting coiled-coil pairs in each pool. Note that in the legends, the library on the left indicates the one that is fused to β-strands 10, and the library on the right is fused to β-strands 11.
S11 – Design of yeast helixCAM pre- and post- SUMO addition, Related to Figure 3
For stage two of our coiled-coil screening approach, we looked to enrich for helixCAM-compatible candidates – coiled-coils amenable to binding when presented on the cell surface. To do this at high throughput, we used the yeast SynAg assay, which uses surface protein-induced mating of haplotype-α and haplotype-a yeast cells into diploid, leading to cells expressing two markers (either two fluorescent proteins or two auxotrophic markers). We first tested the standard SynAg using Z17 and Z18 with the mCherry and mTurquoise fluorescent proteins. However, we did not observe the expected dual-positive population that successful mating should create. We hypothesized that this was due to trouble with expressing the coiled-coil-Aga2p fusion and localizing it to the yeast cell wall. To resolve this, we tried inserting a SUMO protein domain between the coiled-coil and Aga2p to help improve stability and solubility. This resulted in the formation of a third, dual-fluorescent yeast population, indicating successful yeast mating, so we adopted this design for stage two of our coiled-coil screening process.
S12 – Large sg30mCherry+sg61eGFP aggregate image with scale bar, Related to Figure 3
Measurement of large sg30+sg61 aggregation. Sg30mCherry and sg61eGFP K562 helixCAM cells were induced with doxycycline and incubated for 48 hours in a 24-well nonstick plate. At the 48-hour point, large aggregates were visible by eye. 20uL of these aggregates were transferred with a wide-bore pipetted and mounted onto a glass coverslip, followed by widefield imaging at 20X magnification. This image is stitched from 6x6 tiling with 10% overlap, and distance overlay was done in the imaging software (Nikon Elements). When compressed by the coverslip, the largest aggregate size observed was over 2.8 millimeters in length. The ability for helixCAMs to form large aggregates indicates strong potential for use in tissue engineering.
S13 – helixCAM-induced aggregation visible without magnification, Related to Figure 3
Image of 12-well plate well containing a mixture of sg30mCherry and sg61eGFP cells after 48 hours of induction. Cell aggregates could be seen by eye, and the image was captured with a cell phone camera at 1X magnification.
S14 – Analysis workflow for HCSRA, Related to Figure 4
Measurement and analysis workflow for Human Cell Sedimentation Rate Assay. K562 helixCAM cells are mixed together at 1.875e5 cells per population for paired interactions or 3.75e5 for self-interaction in 150uL of DMEM+10% FBS+1% P/S+1ug/mL doxycycline in ultra-low-adhesion v-bottom 96-well plates. The plate is incubated at 37 °C and 5% CO2 with shaking for 48 hours. Plates are then shaken at 900rpm for 15 seconds, immediately followed by OD650 measurement for 15 minutes at 37 °C. The plate is then shaken again and the OD650 is read again, resulting in two reads. The raw data can be seen in the top row. Due to some variability in growth rate between induction and cell types, the OD650 values are normalized by subtracting the minimum, then divided by the maximum to span a range of 0 to 1 (row 2). From this data, the time point at which the OD surpasses 0.5 is taken, which we refer to as t50 (row 3). The t50 for the induced condition is then subtracted from the t50 from the uninduced condition, leading to a positive value if doxycycline induction led to faster sedimentation and thus larger aggregates, and a negative value if doxycycline induction led to slower sedimentation. Finally, the values are averaged to obtain a final representative t50 value for each pairwise- and self-interaction. As seen in Figure 4c, the values from the HCSRA correlate to interaction frequencies observed through microscopy, indicating that the HCSRA is a simple and effective method for measuring cell-cell affinity.
S15 – helixCAM Induction Curve Absolute Δt50 values and significance tests, Related to Figure 4
Absolute Δt50 values from the induction curves of the top four helixCAM pairs across various doxycycline concentrations (normalized figure shown in Figure 4E). Each point represents the mean of N=4 and error bars are S.D. Statistical tests compare conditions from 5ng/mL onwards to that of the 2.5ng/mL condition. Comparisons to the uninduced conditions were not feasible due to their use to calculate the Δt50 values. P-values were derived using the two-sample t-test. At the 5ng/mL condition, the sg30+sg61 helixCAM pair already demonstrated significant binding, and the other pairs reach significance at 10 and 20ng/mL conditions.
S16 – helixCAM Induction Kinetics, Related to Figure 4
Absolute and normalized Δt50 values of the top four helixCAM pairs, induced at 500ng/mL and recorded at 0, 4, 8, 16, 24, 36, 48, and 72 hours after induction. Each point represents the mean of N=4 and error bars are S.D. Cells for each time point were cultured in distinct 96-well plates to minimize disturbance from reads, which requires vigorous shaking prior to read acquisition. Overall, aggregate sizes, as measured by Δt50 values, appear to increase up to the 36-hour time point. For sg30+sg61 and sg83+sg88 pairs, it appears to plateau at this point, whereas Δt50 values continue to increase for Z17+Z18 and P9+AP10 paired cultures.
S17 – Full stitched image and channel images of P9/AP10/sg30/sg61/sg83/sg88 co-culture, Related to Figure 4
The full stitched image of the induced co-culture of P9mPlum, AP10mOrange, sg30mCherry, sg61eGFP, sg83iRFP670, and sg88eBFP2 used to calculate the imaging-based interaction frequency (Figure 4G) is shown here. The image was taken using 5x5 tiling at 20X using a Nikon Eclipse Ti2 microscope across the five channels shown (DAPI, GFP, YFP, mCherry, and Cy5) with a 10% overlap, followed by stitching in Nikon Elements.
S18 – HCSRA Values for Dual-helixCAM lines, Related to Figure 5
HCSRA measurements were taken for dual helixCAM-expressing cell lines designed for sequential cell layering. In the above graph, the affinities between single helixCAM-expressing lines are first shown, followed by the affinity between single helixCAM-expressing line and dual helixCAM-expressing lines for each helixCAM pair. This is then followed by the affinity of two dual helixCAM-expressing lines, if applicable. We found that all dual helixCAM-expressing lines demonstrate intended affinities with their binding partners. All bars represent the mean of N=2, and error bars are S.D.
S19 – helixCAM-induced K562 binding to HEK293 cells, Related to Figure 6
In order to demonstrate the capability of helixCAMs to selectively target natively suspension cells to adherent cells for applications such as immuno-oncology, we tested our sg61/sg88eBFP2 K562 cells against either wild-type HEK293 cells and sg30mCherry HEK293 cells, both uninduced and induced. The K562 cell line is particularly suitable for this demonstration, as it stems from a lymphoblast lineage (immune cell-like), and HEK293 represents an interesting target – stemming from an embryonic adrenal lineage (hard tumor-like). The two cell types for each experiment are grown separately to 80% confluency, followed by either no induction (center column) or 48 hours of induction (right column). The suspension cells are then added to the adherent cells, followed by an additional 12 hours of incubation. The cells are imaged prior to wash, then washed twice with culture medium with gentle shaking. The cells are then imaged again. Brightfield is only shown for wild-type HEK293 conditions to demonstrate complete coverage of the field of view. While there are some remnant cells in both the wild-type and uninduced sg30-mCherry cell conditions, the induced condition has a denser coat of eBFP2-expressing K562 cells covering the surface of the mCherry-expressing HEK293 cells, demonstrating the ability of helixCAMs to increase binding of suspension cells to adherent cells.
S20 – Liquid Chromatography and Mass Spectrometry Analysis of CC-His and CC-GFP-His, Related to Figure 7
Coiled-coils tagged with either 6 histidine residues or both GFP and 6 histidine residues were expressed using E. coli or HEK293 cells respectively and subsequently analyzed by liquid chromatography (LC, top trace) and mass spectrometry (MS, bottom trace). The CC-His proteins (left) were reasonably pure, with only one major peak in the LC trace and likewise in the MS trace corresponding to the expected molecular weight. The CC-GFP-His proteins (right) had a side peak that eluted later in LC, which, when run through MS, shows the same molecular weight, leading us to conclude that it represents the unfolded CC-GFP-His protein. The resulting MS measurements of molecular weight for CC-GFP-His proteins were all higher than expected by 22 Da, which we believe to be an addition of a sodium ion from the PBS buffer used for purification.
S21 – Absolute Counts for CC-His and CC-GFP-His patterning, Related to Figure 7
Absolute cell counts for gradient of CC-His and CC-GFP-His cell patterning. Concentrations used for CC-His were 0, 0.01, 0.1, 1, 10, and 100nM, and for CC-GFP-His were 0, 0.0001, 0.001, 0.01, 0.1, and 1nM (5 order of magnitude each). CC-GFP-His concentrations were constrained by expression level. Significantly more cells bound for CC-GFP-His compared with the CC-His conditions for all CCs. Additionally, sg61eGFP cells, corresponding to the sg30-His and sg30-GFP-His conditions, had higher background binding, even for the no CC condition. This is likely due to the His tag on the sg61 construct binding to the Ni2+ coating, despite the presence of 20mM imidazole to inhibit this interaction. N=4, error bars are S.D.
S22 – Quantitative Comparison of Cell Localization for Dual-Pair CC-GFP-His patterning, Related to Figure 7
a. Quantification of helixCAM-expressing cell localization to either the “G” region, the “C” region, or unpatterned regions of the well. Images from Figure 7E were segmented using CellProfiler to obtain spatial locations for each cell, then analyzed by MATLAB. For each row, in the left panel, gray indicates the region patterned with the first CC-GFP-His protein, and yellow indicates the region patterned with the second CC-GFP-His protein. Cell locations are overlaid on these regions using their respective fluorescent protein colors. In the right panel, the portion of the cells for “Color 1” (paired to the CC patterned to G) and “Color 2” (paired to the CC patterned to C) in each region of the well is presented as bar graphs. Across all four wells, cells were primarily localized to the well region patterned with their paired coils. Cells found outside of the patterned regions were mostly around the edge of the well, which is likely due to the reduced shear force leading to incomplete cell removal during washes. b. Bar graph of cell localization percentages aggregated across the CC-GFP-His protein used for patterning. Similar to individual results, most cells localized to the intended region. Statistical comparison between the intended vs. opposite regions as well as between the intended vs. unpatterned regions was taken using the two-sample T-test, with an N of 2. *** indicates p-values of less than 0.005, ** indicates p-values of less than 0.01, and * indicates p-value of less than 0.05, above which the comparison is marked “not significant” (n.s.).
S23 – Dot-shaped patterning of K562 helixCAM cells using CC-GFP-His, Related to Figure 7
Similar to Figure 7c, Nickel-coated plates were pre-patterned with two distinct Coiled-Coil-GFP-His protein solutions. In this case, 1uL of each protein solution was dotted on either the top or the bottom of the well. The solution was incubated for 30 minutes, washed with PBS+imidazole, then blocked for 30 minutes (also with PBS+imidazole). After removing the blocking buffer, a mixture of the two corresponding K562 helixCAM cells was added into a dual induction/blocking media (1e5 of each cell type in 150uL of DMEM+10%FBS+1%P/S+1ug/mL doxycycline+20mM imidazole). The wells were incubated with the cells for 2 hours, followed by two washes with the same media, and the well was imaged as a 2x2 stitching at 4X magnification. Prior to the wash, it was already possible to see cells localizing densely to the dotted location. Post-wash, there was distinct localization of densely bound cells to the two patterned dots, with few cells in the surrounding region. Since all aspiration and addition of media were done along the bottom of the well, the pipette tips appear to have removed areas of bound cells, but the differential patterning is still clearly visible. It should be noted that addition and removal of media were done along the bottom edge of the wells, which likely led to consistently more dissociation of the bottom dot.
S24 – Viability of helixCAM cells post-induction, Related to Figures 1-4
The viabilities of wild-type and helixCAM K562 cells, with and without doxycycline induction, were measured over four days. Cells were mixed 1:1 with Trypan Blue, then loaded onto measurement slides. Measurements were taken using a Countess II FL, which reads out both cell concentrations as well as percent viabilities based on Trypan Blue exclusion. Data are reported as the mean of N=3, and error bars are S.D. Across all helixCAM lines, growth rate was reduced by induction of helixCAM expression. Most lines maintained high cell viability with the exception of sg30mCherry and sg83iRFP670. Due to the similarity in structure of the helixCAMs and that most cell lines maintain high viability across the four days, it is likely that variable expression of helixCAM can contribute to viability changes. For sensitive cell types, it may be worth optimizing expression level to use the lowest expression of helixCAM needed for the desired cell affinity to minimize viability effects.
Data Availability Statement
Images used for analysis of aggregate size, helixCAM affinity, and CC-His affinity, along with all raw data used to calculate HCSRA values for all helixCAM pairs are publicly available as of the date of publication. The DOI is listed in the key resource table.
All original code necessary for the imaging-based interaction analysis pipeline and for calculating HCSRA have been deposited at Zenodo and are publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| DH5α | ThermoFisher | Cat# 18265017 |
| DH10β | ThermoFisher | Cat# EC0113 |
| T7 Express | New England Biolab | Cat# C2566 |
| E cloni 10G Supreme | Lucigen | Cat# 60107-1 |
| BL21(DE3) | Novagen | Cat# 69450 |
| Chemicals, peptides, and recombinant proteins | ||
| Lipofectamine 2000 | ThermoFisher | Cat# 11668030 |
| Blasticidin | ThermoFisher | Cat# A1113903 |
| Puromycin | ThermoFisher | Cat# A1113803 |
| Doxycycline | ThermoFisher | Cat#J67043-AE |
| Isopropyl β- d-1-thiogalactopyranoside (IPTG) | Gold Bio | Cat# I2481C |
| phenylmethylsulfonyl fluoride (PMSF) | Millipore Sigma | Cat#P7626 |
| Imidazole | MP Biomedicals | Cat# 102033 |
| Tris base | Fisher Chemical | Cat#BP152-5 |
| Glucose | Fisher Chemical | Cat#D16-500 |
| Critical commercial assays | ||
| Pierce™ BCA Protein Assay Kit | ThermoFisher | Cat#23225 |
| Deposited data | ||
| Stitched brightfield images of uninduced and induced cell aggregate for aggregate size analysis | This paper | 10.17632/5j93mffst8.1 |
| Stitched 3-pair co-culture images used for imaging-based interaction analysis | This paper | 10.17632/5j93mffst8.1 |
| Raw OD650 data for HCSRA | This paper | 10.17632/5j93mffst8.1 |
| Images used for analyzing CC-His affinity | This paper | 10.17632/5j93mffst8.1 |
| Experimental models: Cell lines | ||
| K562 | ATCC | CCL-243 |
| HEK293AAV | Agilent | 240073 |
| Hek293T | ATCC | CRL-3216 |
| Experimental models: Organisms/strains | ||
| S. Cerevisiae SynAg MATa | Kavins Lab | N/A |
| S. Cerevisiae SynAg MAT□ | Kavins Lab | N/A |
| Recombinant DNA | ||
| Plasmid: PB_TRE_Z17-PDGFR_EF1a_mCherry | This Paper | Addgene 186303 |
| Plasmid: PB_TRE_Z18-PDGFR_EF1a_eGFP | This Paper | Addgene 186304 |
| Plasmid: PB_TRE_P3-PDGFR_EF1a_iRFP670 | This Paper | Addgene 186305 |
| Plasmid: PB_TRE_AP4-PDGFR_EF1a_eBFP2 | This Paper | Addgene 186306 |
| Plasmid: PB_TRE_P9-PDGFR_EF1a_mPlum | This Paper | Addgene 186307 |
| Plasmid: PB_TRE_AP10-PDGFR_EF1a_mOrange | This Paper | Addgene 186308 |
| Plasmid: PB_TRE_sg30-PDGFR_EF1a_mCherry | This Paper | Addgene 186309 |
| Plasmid: PB_TRE_sg61-PDGFR_EF1a_eGFP | This Paper | Addgene 186310 |
| Plasmid: PB_TRE_sg83-PDGFR_EF1a_iRFP670 | This Paper | Addgene 186311 |
| Plasmid: PB_TRE_sg88-PDGFR_EF1a_eBFP2 | This Paper | Addgene 186312 |
| Plasmid: pQE80_T5_Z17-ehaA_LacI | This Paper | Addgene 186313 |
| Plasmid: pQE80_T5_Z18-ehaA_LacI | This Paper | Addgene 186314 |
| Plasmid: pQE80_T5_P3-ehaA_LacI | This Paper | Addgene 186315 |
| Plasmid: pQE80_T5_AP4-ehaA_LacI | This Paper | Addgene 186316 |
| Plasmid: pQE80_T5_P9-ehaA_LacI | This Paper | Addgene 186317 |
| Plasmid: pQE80_T5_AP10-ehaA_LacI | This Paper | Addgene 186318 |
| Plasmid: pET22b_His-sg30 | This Paper | N/A |
| Plasmid: pET22b_His-sg61 | This Paper | N/A |
| Plasmid: pET22b_His-sg83 | This Paper | N/A |
| Plasmid: pET22b_His-sg88 | This Paper | N/A |
| Plasmid: pCAG-sg30-TEVs-GFP-His | This Paper | N/A |
| Plasmid: pCAG-sg61-TEVs-GFP-His | This Paper | N/A |
| Plasmid: pCAG-sg83-TEVs-GFP-His | This Paper | N/A |
| Plasmid: pCAG-sg88-TEVs-GFP-His | This Paper | N/A |
| Plasmid: PB-sg61-PDGFR-blast | This Paper | N/A |
| Plasmid: PB-sg88-PDGFR-blast | This Paper | N/A |
| Library general plasmid: pQE80X_Z17-autotransporter | This Paper | N/A |
| Library general plasmid: pSYNAGaV3_sgX_SUMO_kan | This Paper | N/A |
| Library general plasmid: pETDEST_sGFP_sgX | This Paper | N/A |
| Software and algorithms | ||
| CellProfiler | Stirling, et al. 2021 | https://cellprofiler.org/ |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Software and algorithms | ||
| CellProfiler | Stirling, et al. 2021 | https://cellprofiler.org |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| Zen Blue | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html |
| Nikon Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| A Plasmid Editor (ApE) | M. Wayne Davis | https://jorgensen.biology.utah.edu/wayned/ape/ |
| Agilent Mass Hunter | Agilent | https://www.agilent.com/en/promotions/masshunter-mass-spec |
| MATLAB | Mathworks | https://www.mathworks.com/products/matlab.html |
| Python | Python Software Foundation | https://www.python.org/ |
| BioPython | Cock, et al. 2009 | https://biopython.org/ |
| Sickle | N/A | https://github.com/najoshi/sickle |
| Code for Image-based Frequency Analysis and HCSRA | This paper | 10.5281/zenodo.6323728 |
| ProtParam | ExPasy | https://web.expasy.org/protparam/ |
| Other | ||
| Ultra-Low-Adherence v-bottom plates | S-Bio | Cat# MS-9096VZ |
| Coiled-Coil and helixCAM Amino Acid Sequences | This Paper | S26 |