

SUMMARY
The intestinal epithelium plays critical roles in sensing and integrating dietary and microbial signals. How microbiota and intestinal epithelial cell (IEC) interactions regulate host physiology in the proximal small intestine, particularly the duodenum, is unclear. Using single-cell RNA sequencing of duodenal IECs under germ-free (GF) and different conventional microbiota compositions, we show that specific microbiota members alter epithelial homeostasis by increasing epithelial turnover rate, crypt proliferation, and major histocompatibility complex class II (MHCII) expression. Microbiome profiling identified Faecalibaculum rodentium as a key species involved in this regulation. F. rodentium decreases enterocyte expression of retinoic acid-25 producing enzymes Adh1, Aldh1a1, and Rdh7, reducing retinoic acid signaling required to maintain certain intestinal eosinophil populations. Eosinophils suppress intraepithelial lymphocyte-mediated production of interferon-γ that regulates epithelial cell function. Thus, we identify a retinoic acid-eosinophil-interferon-γ-dependent circuit by which the microbiota modulates duodenal epithelial homeostasis.
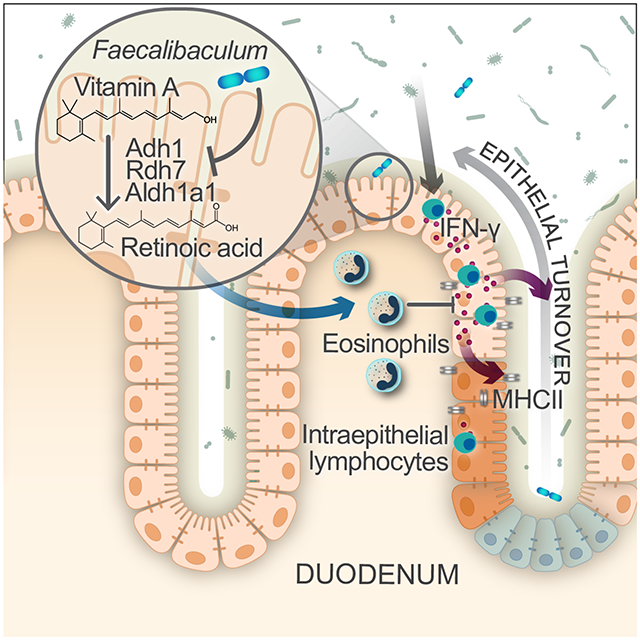
Graphical Abstract
eTOC blurb
The intestinal epithelium is a central node for communication between the microbiota and immune system. Cao et al. show that Faecalibaculum rodentium can promote epithelial proliferation and turnover by dampening retinoic acid production that supports survival of intestinal eosinophils, which in turn suppress pro-proliferative IFN-γ production.
INTRODUCTION
The intestinal epithelium consists of specialized cell types, which function in nutrient uptake, barrier integrity, and host defense (Allaire et al., 2018; Hooper, 2015; Solis et al., 2020). These cells sense dietary and microbial signals, such as short chain fatty acids, secondary bile acids, and tryptophan metabolites, and relay them to immune cells (Larsen et al., 2020; Soderholm and Pedicord, 2019). The microbiota influences intestinal epithelial cell (IEC) function by inducing the expression of antigen-presenting molecules (Tuganbaev et al., 2020), regulating the frequency of epithelial subsets, including tuft and goblet cells (Howitt et al., 2016; Nadjsombati et al., 2018; Schneider et al., 2018), and increasing the rate of epithelial turnover (Khoury et al., 1969). The microbiota also modulates the production of dietary-derived molecules such as the vitamin A metabolite retinoic acid (RA). The presence of the microbiota suppresses RA production in IECs under both steady state conditions and during dysbiosis in colon carcinogenesis, and certain microbiota members can themselves produce RA (Bhattacharya et al., 2016; Grizotte-Lake et al., 2018; Woo et al., 2021). RA in turn regulates epithelial cells, including promoting expression of serum amyloid A proteins and preventing excessive goblet and Paneth cell expansion (Gattu et al., 2019; Jijon et al., 2018).
Most host-microbiota studies of the intestinal epithelium have focused on the distal small intestine (ileum) or colon and often do not distinguish between different regions of the intestine, which vary dramatically in their physiological function and microbiota composition (Martinez-Guryn et al., 2019; Mowat and Agace, 2014). However, comparison of proximal and distal small intestinal epithelia revealed regional specialization in gene expression, particularly in absorptive enterocytes (Elmentaite et al., 2021; Haber et al., 2017). The small intestinal microbiota also regulates lipid absorption by proximal small IECs (Martinez-Guryn et al., 2018). These studies suggest that proximal small intestinal microbiota and IECs are distinct from more distal regions. In particular, the duodenum plays a pivotal role in host physiology and nutrient absorption. Given its proximity to the stomach and its inputs from the pancreas and bile ducts, the duodenum is a special ecosystem for microbes (Kastl et al., 2020). Despite the duodenum’s susceptibility to injury, infection, chronic inflammation, and malignancy, it remains surprisingly under-explored in mucosal immunity.
Herein, using single cell RNA sequencing (scRNASeq), we revealed microbiota-dependent differences in duodenal IEC gene expression, most notably in intestinal stem cells and enterocytes. Certain bacteria in specific pathogen-free (SPF) mice increased epithelial cell proliferation and turnover by suppressing enterocyte RA production. This reduction in RA led to a loss of eosinophil populations, which constrain epithelial turnover via suppression of intraepithelial lymphocyte interferon (IFN)-γ production. Thus, we have identified a mechanism by which specific microbiota members including F. rodentium regulate duodenal epithelial homeostasis via a RA-eosinophil-IFN-γ circuit.
RESULTS
Certain microbiota members regulate intestinal stem cell proliferation and epithelial turnover
To determine how the microbiota regulates the duodenal IEC compartment, we performed scRNASeq. As intestinal immune responses can differ in mice across vivaria (Howitt et al., 2016; Ivanov et al., 2009; Sivan et al., 2015), we harvested IECs from germ-free (GF) and two distinct SPF groups of C57BL6/J mice – from Jackson Labs (Jax) or bred and maintained in our in-house (IH) facility (n=1). We identified all expected IEC cell types from the duodenum in all three microbiota conditions (Figs. 1A, S1A-C), except for low Paneth cell recovery due to difficulties with recovering cells with high granularity, and assigned identities based on previously described gene signatures (Biton et al., 2018; Haber et al., 2017).
Figure 1. Certain microbiota features regulate intestinal stem cell proliferation and epithelial turnover.

(A) UMAP representation of scRNAseq of sorted live EpCAM+CD45−Ter119−CD31− IECs. n=1. TA, transit amplifying; EEC, enteroendocrine. (B) GO categories for DEGs enriched in IH vs GF and Jax ISCs (dashed line indicates p value = 0.05). (C) Top DEGs in IH vs GF and Jax ISCs ranked by significance. (D) MHCII expression in ISCs. (E) Representative images (higher magnification on right) (scale bar 50 μm) and (F) quantification of Ki67+ cells. (G) Representative images (scale bar 50 μm) and (H) quantification of epithelial turnover after 48h continuous BrdU labeling. (I) Representative plots and quantification of Ki67 expression in Lgr5-GFP ISCs. Each symbol (D, F, H, I) represents data from an individual mouse. Data are pooled from 2-3 experiments (D, F, H, I). Data are shown as mean with individual data points. *p < 0.05, **p < 0.01, ***p < 0.001, Mann-Whitney U test (D, I), one-way ANOVA with Holm-Sidak’s post-test (F, H). See also Figures S1, S2, Table S1.
Initially, we focused our investigation on intestinal stem cells (ISCs, cluster 2), which give rise to all differentiated mature IECs, as a representation of the progenitor populations which shared similar significantly differentially expressed genes (DEGs) (Table S1). IH ISCs differed more from Jax/GF ISCs overall, with 240 DEGs between IH and Jax/GF ISCs, but only 44 DEGs between Jax and GF ISCs. Gene ontology (GO) analysis of DEGs from GF and Jax vs. IH ISCs identified an enrichment in metabolic processes (including fatty acid and metal ion metabolism) in GF/Jax ISCs, while enriched categories in IH ISCs mostly related to immune responses (Fig. 1B). Most of the top DEGs, as ranked by significance, were related to major histocompatibility complex class II (MHCII) and MHCI expression in IH ISCs (Fig. 1C). This finding is in line with the “response to interferon (IFN)-γ” category in IH ISCs (Fig. 1B), as IFN-γ can induce both MHCI and MHCII expression (Skoskiewicz et al., 1985; Zhou, 2009). These differences in immune response genes were specific to IH ISCs, not simply the presence of a microbiota, as DEGs in Jax versus GF ISCs were also mainly related to lipid and metal ion metabolism (Fig. S2A-B). Furthermore, comparing Jax vs IH ISCs showed an enrichment in immune response and MHCII signatures similar to that seen in the IH vs GF/Jax comparison. These analyses support that most of the transcriptional differences attributable to the presence of a microbiota are driven by the IH microbiota_(Fig. S2C-D). To study the ISC compartment in vivo, we derived Lgr5-GFP-creERT2 mice, which express GFP in Lgr5+ ISCs, onto a Jax or IH microbiota background. We did not observe a significant difference in the proportion of ISCs in Jax and IH mice by flow cytometry or immunofluorescence (Fig. S2E-G) but confirmed increased MHCII expression in IH ISCs (Fig. 1D). We also examined gene signatures for ISC subsets identified by Biton et al., 2018 under the different microbiota conditions (Fig. S2H). We found that all microbiota conditions had more ISC-I and ISC-II compared to ISC-III and that the expression of all 3 signatures was highest in IH mice, suggesting that these ISCs are most similar to the ISCs profiled in Biton et al. and may have overall higher expression of stem-related genes. Gpx2, a gene with both pro- and anti-proliferative effects in stem cells and cancer (Florian et al., 2010; Jiao et al., 2017; Li et al., 2020), was also highly upregulated in IH ISCs, which we confirmed by qPCR (Figs. 1C, S2I), prompting us to investigate the proliferation of ISCs and epithelial turnover rate under the different microbiota conditions. We measured crypt Ki67 expression, which marks all non-G0 phase cells, and performed 48 hours of continuous bromodeoxyuridine (BrdU) labeling, which is incorporated during DNA replication, to measure epithelial turnover. IH mice had increased numbers of Ki67+ cells in the crypts as well as a higher epithelial turnover rate (the length of crypt-villus axis labeled by BrdU divided by the total axis length)_compared to GF and Jax mice (Fig. 1E-H). We also confirmed by flow cytometry that IH mice have a higher frequency of Ki67+ Lgr5-GFP ISCs than Jax mice (Fig. 1I). Cell cycle scoring of ISCs from the scRNAseq dataset also showed that IH ISCs had a slightly higher score for the S and G2/M cell cycle phases compared to GF and Jax mice (Fig. S2J). Overall, we found that microbiota composition, not just presence, is critical for regulation of epithelial phenotypes. In particular, the IH but not Jax microbiota is able to induce MHCII expression, crypt proliferation, and increased epithelial turnover rate.
The microbiota modulates intestinal immune populations via enterocyte retinoic acid production
We next examined the distinct mature IEC subsets that arise from these ISCs, focusing on absorptive enterocytes, where we observed the most pronounced differences compared to other differentiated cell types (Table S1). Unexpectedly, even though Jax and IH both possess SPF microbiota, their enterocytes had strikingly different gene expression profiles, with Jax enterocytes closely resembling those from GF mice, resulting in their categorization into a separate cluster from IH enterocytes (Fig. 1A). GO analysis of DEGs in GF/Jax vs IH enterocytes revealed a similar pattern as seen in ISCs, with GF/Jax enterocytes enriched in fatty acid metabolism and IH enterocytes enriched in immune response pathways (Fig. 2A). The top DEGs enriched in IH enterocytes as ranked by significance included MHCII-related genes and defense response genes such as the anti-microbial peptide Reg3g, the mucin Muc13, and the microbiota-epithelial cell segregation-promoting Lypd8 (Fig. 2B). IH enterocyte MHCII expression was similar or higher than ISC (Fig. S3E). MHCI genes were also upregulated in IH enterocytes (Fig. S3F). As in ISCs, the differences in immune response-related genes were specific to IH enterocytes. Comparison of Jax vs. GF enterocytes from cluster 1 did not identify activation of immune pathways (Fig. S3A-B), while comparison of IH vs Jax enterocytes revealed similar enriched genes as when comparing IH to GF and Jax (Fig. S3C-D). We confirmed high MHCII expression in total IECs from IH mice and low expression in GF and Jax IECs by flow cytometry (Fig. 2C). Among genes highly expressed in GF and Jax enterocytes, three are related to RA production (Fig. 2B): Aldh1a1, an aldehyde dehydrogenase that converts retinaldehyde to RA, as well as the alcohol dehydrogenase Adh1 and retinol dehydrogenase Rdh7, which can catalyze the upstream conversion of retinol to retinaldehyde (Kumar et al., 2012). Overall, IH enterocytes exhibited an immune activation phenotype, whereas GF and Jax enterocytes were enriched in expected metabolic processes and RA production, demonstrating that the specific composition of the microbiota can shape IEC function.
Figure 2. The microbiota modulates enterocyte phenotypes and retinoic acid production.

(A) GO categories of DEGs in GF/Jax and IH enterocytes (clusters 1 and 7, dashed line indicates p value = 0.05). (B) Top DEGs in GF/Jax and IH enterocytes ranked by significance. (C) MHCII MFI in total IECs. (D) mRNA expression in epithelial fraction. (E-F) Measurement of Aldh enzyme activity via Aldefluor assay in duodenal IECs (EpCAM+CD45−) or dendritic cells (CD45+CD11c+MHCII+CD64−). Control samples included an Aldh inhibitor (DEAB). Gating strategy as in Fig. S2E and Fig. S4A.
Each symbol (C-F) represents data from an individual mouse. Data reflect at least 2 independent experiments (C-F). Data are shown as mean with individual data points. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, one-way ANOVA with Holm-Sidak’s post-test (C-F). See also Figure S3.
Dietary retinol (vitamin A) is absorbed by epithelial cells primarily in the proximal small intestine and then processed to RA (Villablanca et al., 2011), which affects intestinal immunity via signaling through the RAR and RXR receptors (Czarnewski et al., 2017; Hall et al., 2011a). Some of these effects include: recruiting T, B, and innate lymphoid cells (ILCs), controlling the balance between different T helper cell and ILC subsets, and supporting the survival of CD11b+CD103+ dendritic cells (DCs) (Iwata et al., 2004; Kim et al., 2015; Larange and Cheroutre, 2016; Mora et al., 2006). We confirmed decreased mRNA expression of the RA-producing enzymes Adh1, Aldh1a1, and Rdh7 in IH mice by qPCR (Fig. 2D). Next, to assess RA production, we measured Aldh activity using the Aldefluor assay. We found that GF IECs have the highest Aldh activity, followed by Jax IECs, and IH IECs had the lowest activity (“Test” samples) (Fig. 2E). All IECs lacked Aldh activity in the presence of the Aldh inhibitor DEAB (“Ctrl” samples). The other major source of RA in the gut is DCs in the intestinal lamina propria (Villablanca et al., 2011). Duodenal GF, Jax, and IH DCs produced similar levels of RA (Fig. 2F), suggesting that any differential levels of RA production are likely due to epithelial-derived RA. Additionally, we found that while epithelial Aldh activity was also lower in the jejunum and ileum of IH mice than Jax mice (Fig. S3G-H), overall Aldh activity was highest in duodenal epithelial cells, consistent with previous reports (Villablanca et al., 2011). Duodenal RA levels measured by liquid chromatography-mass spectrometry (LC-MS) were slightly elevated in GF and Jax mice compared to IH mice (Fig. S3I). Jax intestinal eosinophils also had higher expression of_the RAR target gene Tgm2 (Moore et al., 1984) than IH eosinophils (Fig. S3J). Furthermore, we found higher expression of an RA target gene signature in GF and Jax mice compared to IH mice in many epithelial cell types, particularly enterocytes, with cluster 1 (GF/Jax enterocytes) having higher expression compared to cluster 7 (IH enterocytes) (Fig. S3K). Together, these results suggest that GF and Jax mice have higher RA production and signaling than IH mice.
Given RA’s importance in regulating intestinal immunity and its higher abundance in the proximal small intestine, we profiled duodenal lamina propria immune cells to determine the effect of epithelial-derived RA in Jax and IH mice (Fig. 3, Fig. S4, Fig. S5A-M). To examine which immune cell differences are specific to RA, we administered the retinoic acid receptor (RAR) inhibitor BMS493 (Germain et al., 2009). Compared to Jax mice, IH mice (with lower RA levels) had a lower proportion of CD11b+CD103+ DCs, whose expansion is dependent on RA (Klebanoff et al., 2013) (Fig. 3A). As expected, inhibiting RAR signaling also decreased CD11b+CD103+ DCs in both Jax and IH mice. We also noted a potential effect of vehicle (DMSO) treatment alone on this DC population, indicating the importance of comparing inhibitor-treated to vehicle-treated populations. Intestinal Tregs and Th17 cells are also regulated by either high or low levels of RA (Cha et al., 2010; Hall et al., 2011b; Mucida et al., 2007; Wang et al., 2010; Xiao et al., 2008). IH mice had a higher proportion of total αβT cells and Th17 cells but lower Tregs than Jax mice (Fig. 3B-D). However, these differences do not seem to mainly depend on RA, as BMS493 treatment did not cause Jax T cells to resemble IH proportions. Other cell types, including Th1, Th2, and ILCs, also differed between Jax and IH mice, but again, these differences were not directly regulated by RA as BMS493 administration did not replicate the effect on their proportions (Fig. S5A-M).
Figure 3. Differential epithelial cell production of retinoic acid in Jax and IH mice regulates intestinal immune populations.

(A-E) Flow cytometric profiling of lamina propria immune populations at steady state, or after treatment with 220μmg RAR inhibitor BMS493 or vehicle (DMSO) for 8 days. (F) Live eosinophils measured by flow cytometry after 24h culture of sorted bone marrow eosinophils with 10ng/mL IL-5, 100nM RA, or both.
Each symbol represents data from an individual mouse. Data reflect at least 2 independent experiments. Data are shown as mean with individual data points. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Mann-Whitney U test (A-E), one-way ANOVA with Holm-Sidak’s post-test (F). See also Figures S4, S5.
Eosinophils, which are regulated by RA in vitro when isolated from human peripheral blood (Ueki et al., 2008), were also significantly higher in Jax than IH mice (Fig. 3E). RAR signaling inhibition decreased the eosinophils in Jax mice to IH proportions but had no effect on IH eosinophils. Among immune cells profiled, only eosinophils exhibited this pattern of being decreased in Jax but not IH mice after treatment. We did not observe differences in the lamina propria expression of Il5, an important cytokine for eosinophil accumulation and survival (Rothenberg and Hogan, 2006), between Jax and IH mice (Fig. S5N), indicating IL-5 was not responsible for the increased eosinophils observed in Jax mice. To confirm that RAR signaling occurs in eosinophils and is blocked by BMS493, we sorted proximal small intestinal eosinophils and measured the expression of select RARs and the RAR target gene Tgm2 (Moore et al., 1984). Intestinal eosinophils express Rara and Rarb, and inhibiting RAR signaling decreased Tgm2 expression (Fig. S5O-P). Using an in vitro system with isolated bone marrow eosinophils, we directly tested the effect of RA on eosinophil survival. Incubation with RA increased eosinophil survival, though less so than interleukin (IL)-5 (Fig. 3F). These results suggest that higher RA production in Jax mice increases eosinophil levels, potentially via enhanced survival, and is the main driver behind differential eosinophil frequencies in Jax versus IH mice. Following inhibition of RAR signaling, some eosinophils remain in both groups. These data suggest a role for RA in regulating certain intestinal eosinophils and raise the possibility of different eosinophil cell states or subsets with varying dependence on RA.
Eosinophils regulate epithelial turnover and MHCII expression by suppressing IFN-γ-producing intraepithelial lymphocyte (IEL) subsets in a microbiota-dependent manner
The proximal small intestine, where we found that RA regulates eosinophils, contains the highest levels of eosinophils in the gut (Chu et al., 2014). Eosinophils in other tissues promote proliferation during the tissue repair process after injury or infection (Goh et al., 2013; Heredia et al., 2013; Vicetti Miguel et al., 2017), and the intestinal epithelium is highly proliferative at steady state. Thus, we wondered if eosinophils may be responsible for the distinct IEC proliferation phenotypes in IH and Jax mice. To test this hypothesis, we used eosinophil-deficient PHIL mice derived onto either a Jax or IH microbiota background. As we initially observed, IH wild-type (WT) mice had a higher epithelial turnover rate and more proliferating Ki67+ cells than Jax WT mice (Figs. 1E-H, 4A-B). On a Jax microbiota background, eosinophils did not affect turnover or Ki67+ cells (Fig. 4A-B). However, on an IH microbiota background, PHIL mice had increased epithelial turnover and Ki67+ cells compared to WT mice. A similar pattern was observed in MHCII expression, with very low expression in both Jax WT and PHIL mice, higher expression in IH WT mice, and highest expression in IH PHIL mice (Fig. 4C). These results suggest that eosinophils have no regulatory effect when there is little proliferative signal, as in Jax mice. However, when an increased proliferative signal is present, eosinophils act as a negative regulator to dampen excess proliferation. To determine if eosinophils also regulate the response to injury, we employed a model of small intestinal injury by injecting an anti-CD3 antibody that results in T cell-mediated epithelial damage (Merger et al., 2002). Similar to the patterns in turnover, IH (especially IH PHIL) mice had more extensive intestinal damage that was characterized by villous atrophy and crypt injury, as evaluated in histological injury scores (Fig. 4D-F). Thus, eosinophil regulation of MHCII expression, IEC proliferation, and/or IFN-γ-related signals may affect_sensitivity to inflammatory injury.
Figure 4. Eosinophils regulate epithelial cell turnover, MHCII expression, and response to injury.

Representative images (scale bar 50 μm) and quantification of (A) Epithelial turnover after 48h continuous BrdU labeling and (B) Ki67+ cells. (C) MHCII expression on epithelial cells. (D) Representative hematoxylin and eosin staining (scale bar 200 μM), (E) Histological injury score, and (F) Percent crypt injury/loss in duodenum on day 3 after injection of anti-CD3.
Each symbol represents data from an individual mouse. Data reflect at least 3 independent experiments. Data are shown as mean with individual data points. *p <= 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, two-way ANOVA with Holm-Sidak’s post-test.
IFN-γ can regulate IEC proliferation (Nava et al., 2010) and induce MHCII expression in epithelial cells (Skoskiewicz et al., 1985). Eosinophils suppress IFN-γ production by lamina propria T cells in the colon and stomach (Arnold et al., 2018) and response to IFN-γ was enriched in IH ISCs (Fig. 1B). Therefore, we measured Ifng mRNA levels in the epithelial and lamina propria fractions of Jax and IH WT and PHIL mice. In the epithelial fraction, which contains both epithelial and immune cells, IH PHIL had the highest Ifng expression (Fig. 5A), mirroring the patterns in epithelial turnover and MHCII expression. No differences were observed in the lamina propria (Fig. S6B). This epithelial Ifng likely comes from IELs, which are the majority of immune cells in the epithelium and known IFN-γ producers (McDonald et al., 2018). Epithelial cells do not produce IFN-γ under homeostatic conditions (Haber et al., 2017), and we did not detect Ifng transcripts in our epithelial scRNA-seq dataset. Thus, we next characterized IEL subsets and their IFN-γ production. IELs can be divided into TCRαβ+ IELs, which develop from naïve T cells encountering foreign antigen in the periphery, and TCRγδ+ IELs, which are more innate-like and develop after recognition of self-ligands (McDonald et al., 2018). TCRαβ+ IELs can be further sub-divided into CD8αβ+ IELs; CD4+ IELs, which depend on MHCII expression by IECs (Bilate et al., 2020; Tuganbaev et al., 2020); and CD8αα+ IELs, which are similar to TCRγδ+ IELs that share CD8αα expression. A majority of IELs in IH WT mice were TCRαβ+, whereas Jax WT mice had a majority of TCRγδ+ IELs (Figs. 5B, S6A). Jax PHIL mice had a similar IEL profile as Jax WT mice, but IH PHIL mice had even more TCRαβ+ and fewer TCRγδ+ IELs than IH WT mice. Within the TCRαβ+ IEL subsets, IH mice had fewer CD8αα+ IELs, more CD8αβ+ IELs, and more CD4+ IELs than Jax WT and PHIL mice (Fig. 5C). IH PHIL mice were similar to IH WT mice, with an additional increase in CD4+ IELs, correlated with highest MHCII expression in IH PHIL mice. Overall, these findings reflect a shift from innate-like IELs towards more conventional, peripherally educated IELs in IH and especially IH PHIL mice compared to Jax mice. Most IEL subsets in IH PHIL mice produced more IFN-γ than in Jax WT and PHIL mice (Fig. 5D). Additionally, certain subsets, including total TCRαβ+, CD8αβ+, and CD4+ IELs, produced more IFN-γ across all conditions compared to TCRγδ+ and CD8αα+ IELs. Therefore, increased IFN-γ production in IELs in IH PHIL mice is due to both a per cell increase in IFN-γ production as well as a shift in IEL populations to higher IFN-γ-producing subsets. Overall, we concluded that IH microbiota increases IFN-γ production in IELs and that eosinophils suppress this microbiota-induced increase.
Figure 5. Eosinophils suppress IFN-γ-producing IEL subsets to regulate epithelial cell turnover and MHCII expression in a microbiota-dependent manner.

(A) Ifng mRNA expression in epithelial fraction. (B-C) Flow cytometric profiling of IEL populations. (D) IFN-γ production by IEL populations. (E) IEC MHCII expression, (F) Epithelial turnover after 48h continuous BrdU labeling, and (G-H) BrdU+ cells after 2h short-term labeling in mice treated every other day for 8 days with 200μg anti-IFN-γ neutralizing Ab or PBS.
Each symbol represents data from an individual mouse. Data reflect at least 2 independent experiments. Data are shown as mean with individual data points. *p<0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, two-way ANOVA with Holm-Sidak’s post-test. See also Figure S6.
To determine if the increased IFN-γ production in IH and IH PHIL mice was a driver of differences in epithelial phenotype, we used an IFN-γ neutralizing antibody (Ab). There was no effect in Jax WT and Jax PHIL mice injected with the Ab, as expected based on the low IFN-γ production in Jax mice (Figs. 5E-F, S6C). However, IFN-γ blockade abrogated the difference in IEC turnover between IH WT and IH PHIL mice without affecting Ki67+ crypt cells and also decreased MHCII expression. This difference in cell turnover versus Ki67+ cells could be because Ki67 marks all non-G0 phase cells. ISCs often remain in a quiescent G1 phase (Carroll et al., 2018), so a change in IFN-γ-dependent proliferation could occur without changes in Ki67. As a complementary method to track proliferation, we measured BrdU+ cells in the crypt 2 hours after BrdU injection. We found that anti-IFN-γ Ab reduced BrdU+ cells in IH WT and PHIL mice, indicating reduced proliferation (Fig. 5G-H). Additionally, we observed very few TUNEL+ apoptotic IECs under any condition (Fig. S6D), suggesting that IFN-γ-mediated effects are not due to changes in apoptosis. Overall, these findings demonstrate that eosinophil restraint of epithelial turnover depends on suppression of IFN-γ.
Faecalibaculum rodentium regulates RA-eosinophil-epithelial cell circuit
After observing that decreased RA production by IECs in IH mice results in fewer eosinophils, increased IFN-γ production, and increased IEC turnover rates and MHCII expression, we investigated if these differences could be modulated by microbiota transfer. Because IH IECs differed more from both Jax and GF IECs, we chose to transfer IH microbiota to Jax mice. A one-time gavage of IH stool or cecal contents in Jax mice (without any antibiotic pre-treament of Jax mice) resulted in reduced IEC Adh1, Aldh1a1, and Rdh7 expression, decreased eosinophil proportion, and shifts in IEL composition toward more TCRαβ+, CD8αβ+, and CD4+ IELs and increased IFN-γ production (Fig. 6A-D). As a result, MHCII expression and Ki67+ crypt cells also increased after transfer, with a greater increase in Jax PHIL mice receiving IH contents compared to Jax WT recipients (Fig. 6E-G). The turnover rate was only slightly different between WT and PHIL post-transfer, possibly because the newly-introduced IH microbiota induced a very high turnover rate reminiscent of baseline IH PHIL mice in both groups. Pre-treatment of IH mice with broad-spectrum antibiotics prior to transfer of their microbiota to Jax mice eliminated the regulatory activity of the transfer (Fig. S6E). Co-housing Jax and IH mice in the same cage also resulted in Jax mice taking on an IH phenotype (Fig. S6F). Collectively, these results indicate that antibiotic-sensitive member(s) of the IH microbiota responsible for RA-eosinophil-epithelial regulation can be transferred to Jax mice and confer immunomodulatory effects.
Figure 6. IH microbiota regulates epithelial-eosinophil crosstalk.

Jax mice received one transfer of IH microbiota contents via gavage. (A) mRNA expression in epithelial fraction 3 wks post-transfer. (B) Eosinophil levels 2 wks post-transfer. (C) IEL subsets and (D) IEL IFN-γ production 3 wks post-transfer. (E) MHCII expression on IECs 3 wks post-transfer. (F) Epithelial turnover after 48h continuous BrdU labeling and (G) Ki67+ cells 4 wks post-transfer.
Each symbol represents data from an individual mouse. Data reflect at least 2 independent experiments. Data are shown as mean with individual data points. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, Mann-Whitney U test (A-B), two-way ANOVA with Holm-Sidak’s post-test (C-G). See also Figure S6.
To pinpoint which component of the IH microbiota is responsible for the microbiota’s regulatory capacity, we performed 16S rRNA gene amplicon sequencing of stool from Jax and IH mice under the previously tested experimental conditions. Since both cohousing and direct microbiota transfer recapitulated the IH-like RA-eosinophil-epithelial regulatory capacity in Jax mice, we focused on bacterial species absent in Jax mice and Jax mice receiving antibiotic-treated IH microbiota but present in the other groups. Microbiome communities clustered into two groups, with one group comprising samples from high RA/high eosinophil/low epithelial turnover mice (Jax and Jax with Abx-treated IH transfer) and the second comprising low RA/low eosinophil/high epithelial turnover mice (IH, IH cohoused, Jax cohoused, and Jax with IH transfer) (Fig. S7A). Differentially abundant bacterial genera that distinguish these two groups were identified using linear discriminant analysis effect size (LEfSe) analysis (Segata et al., 2011) (Fig. 7A). In particular, Bifidobacterium, Faecalibaculum, and Lachnospiraceae A2 genera were enriched in IH mice and Jax mice with IH cohousing/transfer compared to Jax, which was confirmed by qPCR on stool samples (Fig. 7B). Bifidobacterium and Faecalibaculum were also detected in the duodenal lumenal contents of IH mice (Fig. 7B), raising the possibility of a direct local effect on the epithelial-immune interplay we observed. To further investigate this interaction, we performed fluorescence in situ hybridization (FISH) using fluorochrome-tagged 16S rRNA gene probes specific for F. rodentium. We detected F. rodentium in the duodenal crypts (Fig. 7C). In contrast, when we examined F. rodentium localization in the colon, we observed it principally in the colonic contents (Fig. S7B). Although there are relatively few bacteria in the duodenum, as expected given the overall lower bacterial density, this observation suggests that F. rodentium could adhere to or directly interact with duodenal IECs.
Figure 7. 16S rRNA gene amplicon survey analysis identifies Faecalibaculum as a regulator of a retinoic acid-eosinophil-epithelial circuit.

(A) Heatmap of differentially abundant genera identified by LefSE analysis of 16S rRNA gene amplicon sequencing of stool from different microbiota conditions. (B) Colonization levels in gut regions determined by qPCR. (C) 16S FISH for F. rodentium in IH duodenum. (D-G) GF mice were mono-colonized for 2 wks. (D) mRNA expression in epithelial fraction. (E) Eosinophil levels measured by flow cytometry. (F) Epithelial turnover after 48h continuous BrdU labeling. (G) Ki67+ cells. Each symbol (B, D-G) represents data from an individual mouse. Data reflect at least 2 independent experiments. Data are shown as mean with individual data points. *p < 0.05, Mann-Whitney U test (B), one-way ANOVA with Holm-Sidak’s post-test (D-G). See also Figure S7.
To investigate if any of these identified bacteria can regulate epithelial cell phenotypes, we monocolonized GF mice with strains of these genera that were isolated from IH mice. B. pseudolongum and F. rodentium but not Lachnospiraceae A2 successfully monocolonized GF mice (Fig. S7C). F. rodentium alone reduced Aldh1a1 expression and eosinophil proportions compared to GF mice, thereby increasing epithelial turnover and Ki67+ crypt cells (Fig. 7D-G). B. pseudolongum reduced Adh1 expression but did not affect eosinophils or epithelial turnover, perhaps because other enzymes can compensate for Adh1 activity, whereas only Aldh1a1 in IECs can convert retinaldehyde to RA. However, we did not observe an effect of F. rodentium or B. pseudolongum on MHCII or IFN-γ production (Fig. S7D-F). Thus, we posit that a different bacterial component(s) in the IH microbiota acts as a separate signal for IFN-γ induction in IELs. F. rodentium suppresses RA to regulate eosinophils, which then modulate IFN-γ production induced by this second source. The observation that F. rodentium still increases proliferation and turnover, despite not inducing IFN-γ, also indicates that both IFN-γ-dependent and independent mechanisms are involved in controlling this process. F. rodentium also only slightly increased anti-CD3 induced injury (Fig. S7G), suggesting that while heightened proliferation may increase susceptibility, other factors likely including IFN-γ signaling also regulate the response to injury or initial damage severity. Thus, F. rodentium is one IH microbiota member sufficient to recapitulate its RA-eosinophil-epithelial regulation, though other members also play a role in a full SPF microbiota, particularly in inducing IFN-γ.
DISCUSSION
We have uncovered a retinoic acid-eosinophil-IFN-γ circuit by which the microbiota can regulate duodenal IEC turnover and MHCII expression. Specific microbiota members including F. rodentium can activate this circuit, thereby altering both intestinal epithelial and immune homeostasis. Thus, we identify multiple points of regulation for epithelial turnover rate, modulation of which is critical for maintaining barrier function and allowing repair after injury or infection. Furthermore, this finding emphasizes the interconnected nature of microbiota-epithelial-immune interactions, with IECs as both sensors and readouts of changes in microbiota composition and function.
Our work highlights the impact of different SPF microbiota and the importance of certain microbiota members. Although both Jax and IH mice possess complex microbiota with many bacterial species, in many aspects of epithelial and immune cell function, we found that Jax mice more closely resemble GF mice than IH mice. GF mice have long been known to have a slower small intestinal epithelial turnover rate compared to SPF mice (Khoury et al., 1969). However, we further demonstrate that a specific species present in many mouse microbiotas, F. rodentium (Chang et al., 2015; Cox et al., 2017; Zagato et al., 2020), can accelerate epithelial turnover. In terms of RA regulation, IECs from SPF mice produce less RA than GF mice due to suppression of Rdh7 expression (Grizotte-Lake et al., 2018). In our study, we extend this work to show that only IH microbiota, and specifically F. rodentium, was able to suppress IEC RA production. In addition to Rdh7, we also found that Adh1 and Aldh1a1 expression was reduced in IH IECs, perhaps because of different microbiota compositions. Recent work has also shown that certain microbiota members, such as segmented filamentous bacteria (SFB, called Candidatus savagella), can produce RA (Woo et al., 2021). Although our mice lack SFB (data not shown), there may be RA-producing species that could represent another RA source.
Additionally, our observations reveal an immunomodulatory role off F. rodentium, an understudied microbiota member. F. rodentium is a Gram-positive, anaerobic member of the family Erysipelotrichaceae (Chang et al., 2015; Cox et al., 2017). It is found predominantly in the murine gut, though a closely related species, Holdemanella biformis, is present in humans (Zagato et al., 2020). F. rodentium in the ileum (where it is found in the mucus layer) and colon reduces tumor growth in mice through production of short-chain fatty acids (Zagato et al., 2020). We also found that F. rodentium can colonize duodenal crypts. Further work is needed to determine if F. rodentium regulates IEC RA production through metabolites or direct contact with IECs.
Our work also provides insight into regulation of MHCII expression in ISCs by both the microbiota and eosinophils. The IH microbiota can induce epithelial MHCII expression, which has also been attributed to SFB and their attachment properties (Tuganbaev et al., 2020). Our IH mice lack SFB, and future work is needed to identify IFN-γ-inducing bacteria that regulate MHCII expression in our system. This higher MHCII expression is associated with lower levels of RA, whereas RA has previously been shown to promote MHCI expression on colon tumor cells (Bhattacharya et al., 2016). This apparently divergent function of RA could be due to RA signaling through eosinophils to regulate MHCII in our system compared to the direct action on tumor cells, as well as the difference between normal and cancerous cells, as RA did not promote MHCI expression on normal epithelium (Bhattacharya et al., 2016). Functionally, ISC MHCII can regulate CD4+ T helper cell subsets that in turn modulate ISC fates, and ablation of MHCII on epithelial cells can increase Lgr5+ ISC numbers (Biton et al., 2018). We did not find a difference in Jax and IH ISC frequency, perhaps due to the difference in genetic ablation used in that study versus the lack of inducing signal we observed. Suppression of ISC MHCII by high-fat diet can promote intestinal turnorigenesis, and ISC sternness also influences antigen-presentation machinery expression and the immune environment in colorectal cancer (Beyaz et al., 2021; Chen et al., 2021). Thus, our work demonstrating the role of microbiota, eosinophils, and IEL-derived IFN-γ in regulating MHCII expression could aid in mechanistic understanding of how MHCII is controlled in relevant disease settings.
We also uncovered a pivotal role for the microbiota and RA in regulating intestinal eosinophils. Jax mice with high RA had more intestinal eosinophils than IH mice with low RA, and inhibiting RAR signaling reduced Jax eosinophils to IH levels. This finding is in accordance with observations that SPF mice have fewer intestinal eosinophils than GF mice (Jiménez-Saiz et al., 2020) and provides a mechanism for that regulation. Another recent study found that eosinophils regulate the host response to microbial colonization, but the microbiota did not control eosinophil abundance (Ignacio et al., 2022). Our finding that only specific bacteria regulate eosinophil abundance provides an explanation for the discrepancies in these previous findings. Furthermore, the eosinophils remaining in Jax mice after inhibiting RAR signaling and the lack of an effect in IH mice suggest that there are different eosinophil functional states or subsets in the duodenum with distinct requirements for RA. Consistent with prior observations in human eosinophils (Ueki et al., 2008), we found that eosinophils express RAR and RXR receptors and that RAR signaling is active in intestinal eosinophils. Future work will also determine if RA-dependent and RA-independent eosinophils have different functional properties and if this contributes to differences in Jax and IH eosinophil function.
Additionally, we demonstrated that intestinal eosinophils suppress production of IFN-γ by IELs to restrict excessive epithelial turnover. This anti-proliferative role contrasts with the pro-proliferative function of eosinophils in injury and repair contexts (Goh et al., 2013; Heredia et al., 2013), which may be due to the homeostatic nature of the intestinal interaction and presence of resident eosinophils, whereas pro-proliferative eosinophils are rapidly recruited after an insult. Gastric and colonic eosinophils can modulate lamina propria IFN-γ production by T cells via a PD-L1-dependent pathway (Arnold et al., 2018). It remains to be determined if intestinal eosinophils directly interact with IELs to regulate IFN-γ production and if a similar pathway mediates that regulation. We found that neutralization of IFN-γ abrogates increased epithelial turnover in IH WT and PHIL mice. Both type I (IFN-α/β) and type II (IFN-γ) IFNs can promote epithelial proliferation (Eriguchi et al., 2018; Sun et al., 2015). We further show that specific SPF microbiota can alter IFN-γ production by IELs, similar to what is known for lamina propria CD8+ T cells (Tanoue et al., 2019). This second microbiota signal in addition to suppression of RA production and eosinophil levels is required to increase IEC turnover, as the absence of eosinophils in Jax PHIL mice does not itself result in faster turnover rates as seen in IH PHIL mice. These findings point to eosinophils as an critical regulator of intestinal homeostasis by fine-tuning microbiota inputs, adding to growing work showing that eosinophils have homeostatic functions beyond allergic and parasitic responses (Ignacio et al., 2022; Shah et al., 2020).
In summary, we identified a mechanism by which microbiota members including F. rodentium can promote epithelial proliferation through a RA-eosinophil-IFN-γ-dependent pathway. This role for the microbiota in controlling duodenal IECs illustrates the importance of deeper exploration of microbiota-host interactions in the proximal small intestine, which could open up therapeutic avenues for upper gastrointestinal tract diseases like celiac disease, eosinophilic esophagitis, and environmental enteric dysfunction. Further study of how eosinophils contribute to intestinal epithelial homeostasis could also identify targets for modulating epithelial proliferation and preventing tumor initiation.
STAR METHODS
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Wendy S. Garrett (wgarrett@hsph.harvard.edu).
Materials Availability
Bacterial strains isolated from our facility mice are available upon request.
Data and Code Availability
Single-cell RNA-seq data and 16S rRNA amplicon sequencing data have been deposited at SRA as BioProject PRJNA763366.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental Model and Subject Details
Mice
Wild-type C57BL/6J mice (called in-house, IH) were bred and housed in microisolator cages in the specific-pathogen-free (SPF) barrier facility at the Harvard T.H. Chan School of Public Health. These mice are distinct from other mice in our facility that we have previously described in Howitt et al., 2016, in that the IH mice in this paper are maintained free of the protozoan Tritrichomonas muris. C57BL/6 WT (called Jax) and Lgr5-GFP-creERT2 mice were obtained from the Jackson Laboratory, Bar Harbor, Maine. PHIL mice (Lee et al., 2004) were obtained from Dr. Elizabeth Jacobsen at the Mayo Clinic (Scottsdale, Arizona). All genetically modified mouse lines were bred on a C57BL6/J background and were bred as heterozygotes to generate littermate controls. Both male and female mice were used. Mice were used experimentally between 6-12 weeks of age.
Germ-free WT C57BL/6J mice were bred and maintained in semi-rigid gnotobiotic isolators in the Harvard T. H. Chan Gnotobiotic Center for Mechanistic Microbiome Studies.
To rederive genetically modified lines onto a Jax microbiota background, pups were taken on the day of birth or one day after and raised by a dam of the desired microbiota background. These Jax mice were maintained in SPF facility caging but all SPF cages were changed by the investigators to prevent mixing of microbiota. Microbiota status was monitored by qPCR and Jax background mice were regularly rederived onto freshly ordered Jax mice.
Animal studies and experiments were approved and carried out in accordance with Harvard Medical School's Standing Committee on Animals and the National Institutes of Health guidelines for animal use and care.
Bacterial strains and gnotobiotic colonization
Bifidobacterium pseudolongum was isolated from BIH mice from our facility by plating stool on selective Bifidobacterium iodoacetate medium agar plates (Sasajima et al., 2010). Lachnospiraceae A2 was isolated from BIH mice by plating stool on brain-heart infusion (BHI) plates supplemented with 1 g/L inulin. 48 colonies of each condition were screened by colony polymerase chain reaction (PCR) using Bifidobacterium genus or Lachnospiraceae A2 primers. Faecalibaculum rodentium was obtained from the DSMZ collection (#103405) and was also isolated from our IH mice by plating on Eggerth-Gagnon plates (ATCC medium 2840) and screening by colony PCR with F. rodentium primers. All of these strains were cultivated under anaerobic conditions at 37°C.
For gnotobiotic experiments, B. pseudolongum was grown in MRS medium (BD) supplemented with 0.5 g/L L-cysteine for 24 hours, F. rodentium in DSMZ medium 104 (modified PYG medium) for 24 hours, and Lachnospiraceae A2 in BHI (BD) supplemented with 5 g/L yeast extract, L-cysteine, and hemin (Sigma) for 48 hours. Cultures were observed to be turbid and concentrated 10x for gavage in GF mice. Monocolonization was confirmed by Sanger sequencing of stool DNA, and germ-free status was confirmed by qPCR with universal bacterial 16S primers.
Method Details
scRNAseq
Cell isolation and sorting
Epithelial cells for sequencing were isolated similarly to as described in Biton et al., 2018. The duodenum was excised, opened longitudinally, rinsed in PBS, and sliced into fragments about 2-5mm in length. The tissue was incubated in 20 mM EDTA (VWR) in PBS on ice for 45 min with occasional inversion, then shaken vigorously. The tissue was then transferred to subsequent EDTA-PBS solutions for a total of 4 times, for 5 minutes each, and then shaken. Fractions were examined under a light microscope and crypt-enriched fractions were collected and combined. This crypt fraction was passed through a 70 μM filter, centrifuged, dissociated with pre-warmed TrypLE express (Invitrogen) for 1 min at 37 °C, centrifuged, and resuspended in 1% BSA (Roche), 5 mM HEPES (Corning), 1 mM EDTA. Cells from each condition were stained with a distinct barcoded antibody (Cell-Hashing antibody, TotalSeq-A, Biolegend). Epithelial (live EpCAM+CD45−Ter119−CD31−) cells were sorted on a FACSAria (BD).
Sequencing and analysis
Single cell RNA-seq experiments were performed by the Brigham and Women’s Hospital Single Cell Genomics Core. ~7,500 cells from each condition were resuspended in 0.4% BSA in PBS at a concentration of 2,000 cells per μl, pooled together, then loaded onto a single lane (Chromium Next GEM Chip G, 10X Genomics) followed by encapsulation in a lipid droplet (Chromium Next GEM Single Cell 3’ GEM, Library & Gel Bead Kit v3.1, 10X Genomics) followed by cDNA and library generation according to the manufacturer’s protocol.
mRNA libraries were sequenced to an average of 30,000 reads per cell and HTO (Cell Hashing antibodies) libraries sequenced to an average of 5,000 reads per cell on an Illumina Novaseq. scRNA-seq reads were processed with Cell Ranger v3.1, which demultiplexed cells from different samples and quantified transcript counts per putative cell. Quantification was performed using the STAR aligner against the mm10 transcriptome.
Further processing and visualization was performed using the Seurat v3 R package (Stuart et al., 2019). Cells with either less than 200 or more than 6,000 detected genes or >0.12 mitochondrial fraction were excluded from further analysis. Cell expression was normalized followed by selection of highly variable features, data scaling and cell clustering. We identified genes that are differentially expressed (had an adjusted P value lower than 0.05 and fold change >1.5 or <0.5) using the MAST test (Finak et al., 2015) implemented in Seurat v3.
Clusters were manually annotated using gene signatures described in Haber et al., 2017 and Biton et al., 2018. Genes used for cluster identification and annotation are included in Fig. S1A-B. In particular, cluster 3 was defined as immature enterocytes based on high expression of Ccl25 and Nfe2l2. Cluster 9 was identified as goblet and Paneth because the majority of cells expressed goblet cell markers Agr2 and Fcgbp, while a small number highly expressed Paneth cell markers Lyz1 and Defa26. Cluster 10 was identified as stem (Aqp1) because of the expression of Aqp1, Pdgfa, and Snhg8, as well as low Olfm4 expression. Cluster 12 was identified as tuft because of Trpm5, Dclk1, and Il25 expression.
ISC subset analysis was performed using gene signatures from Biton et al., 2018. RA gene signature (Tgm2, Isx, Rara, Rarb, Rarg, Rbp1, Rbp2, Cd38, Egr1, Cdx1, Oat) was chosen from previously described RA target genes (Balmer and Blomhoff, 2002; Dekaney et al., 2008; Nakshatri and Chambon, 1994). Cell cycle analysis was performed using the Seurat CellCycleScoring function and built-in gene lists. Enrichment of differentially expressed genes in gene ontology categories was determined using the topGO package (Alexa et al., 2006). P values were calculated using Fisher’s exact test.
Intestinal lamina propria and epithelial cell isolation
The small intestine and/or colon were removed and flushed with ice-cold sterile PBS using a 19-gauge feeding needle. The duodenum (defined as the first 7 cm following the stomach), jejunum (the 10 cm following the duodenum), and ileum (the last 10 cm of the small intestine, immediately proximal to the cecum) were excised and Peyer’s patches were removed. Intestines were then opened longitudinally and gently agitated at 4°C in PBS, 2% FBS (Gibco), 5mM HEPES (Corning), 1mM DTT (Sigma) for 10 min. The tissue was then transferred into prewarmed PBS, 2% FBS, 5mM HEPES, 5mM EDTA and rotated at 37°C for 15 minutes followed by vigorous shaking to remove epithelial cells. This was repeated and epithelial cells from both fractions were combined and washed with PBS prior to epithelial digestion. The remaining non-epithelial tissue was used for lamina propria cell isolation. The tissue was rinsed twice in PBS, placed into an Eppendorf tube with RPMI, minced with scissors, and digested in RPMI containing 5% FBS, 5mM HEPES, 1% penicillin/streptomycin (Corning), 0.5 units/ml Dispase II (StemCell Technologies), 50 μg/ml DNase (Roche), and 0.25 mg/mL collagenase A (Roche) for 45 minutes at 37°C. For epithelial cell isolation, the epithelial fraction was digested in RPMI containing 5% FBS, 5mM HEPES, 1% penicillin/streptomycin, 0.5 units/ml Dispase II, and 50 μg/ml DNase for 10 minutes at 37°C. Both the epithelial and lamina propria fraction were then passed through 40 μm filters and washed with PBS, 2% FBS, 1mM EDTA.
Flow cytometry
Single cell suspensions were initially Fc blocked with anti-CD16/CD32 (clone 93, Biolegend) and then stained with surface antibodies and viability dye (20 min on ice). For transcription factor staining, after surface staining, cells were fixed for 45 min with eBioscience Transcription Factor Staining Set and then stained intracellularly for 45 min at room temperature. For Ki67+ staining of Lgr5-GFP cells, after surface staining, cells were fixed for 20 min with BD Cytofix, followed by fixation for 45 min with eBioscience Transcription Factor Staining Set and then stained intracellularly for 45 min at room temperature. For cytokine (IFN-γ) staining, 1 million isolated epithelial fraction cells were stimulated with eBioscience Cell Stimulation Cocktail (with protein transport inhibitors) for 4h at 37 ° C in RPMI with 10% fetal bovine serum and penicillin/streptomycin. Cells were then washed, blocked, surface stained, and fixed in BD Cytofix/Cytoperm for 20 minutes. Intracellular staining was performed for 45 min on ice. Samples were acquired on an LSRII or FACS Symphony (BD). Antibodies used are listed in the Key Resources Table.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD45 APC-Cy7 (30-F11) | BioLegend | Cat#:103116; RRID:AB_312981 |
| Anti-mouse CD45 Pacific Blue (30-F11) | BioLegend | Cat#:103126; RRID:AB_493535 |
| Anti-mouse CD45 APC (30-F11) | BioLegend | Cat#:103112; RRID:AB_312977 |
| Anti-mouse CD45 BUV395 (30-F11) | BD Biosciences | Cat#:564279; RRID:AB_2651134 |
| Anti-mouse CD90.2 (53-2.1) PE/Cy7 | BioLegend | Cat#:140310; RRID:AB_10643586 |
| Anti-mouse CD90.2 (30-H12) BUV805 | BD Biosciences | Cat#:741909; RRID:AB_2871223 |
| Anti-mouse Lineage markers Pacific Blue (17A2/RB6-8C5/RA3-6B2/Ter-119/M1/70) | BioLegend | Cat#:133310; RRID:AB_11150779 |
| Anti-mouse Lineage markers FITC (145-2c11/RB6-8C5/RA3-6B2/Ter-119/M1/70) | BioLegend | Cat#:133302; RRID:AB_10697030 |
| Anti-mouse CD64 (X54-5/7.1) PE/Cy7 | BioLegend | Cat#:139314; RRID:AB_2563904 |
| Anti-mouse SiglecF (E50-2440) AlexaFluor 647 | BD Biosciences | Cat#:562680; RRID:AB_2687570 |
| Anti-mouse SiglecF (E50-2440) PE | BD Biosciences | Cat#:552126; RRID:AB_394341 |
| Anti-mouse/human CD11b (M1/70) FITC | BioLegend | Cat#:101206; RRID:AB_312789 |
| Anti-mouse CD11c (N418) Pacific Blue | BioLegend | Cat#:117322; RRID:AB_755988 |
| Anti-mouse CD103 (2E7) PE | eBioscience | Cat#:12-1031-81; RRID:AB_465798 |
| Anti-mouse I-A/I-E (MHCII) (M5/114.15.2) PerCP | BioLegend | Cat#:107624; RRID:AB_2191073 |
| Anti-mouse EpCAM APC (G8.8) | BioLegend | Cat#:118214; RRID:AB_1134102 |
| Anti-mouse EpCAM PE/Cy7 (G8.8) | BioLegend | Cat#:118216; RRID:AB_1236471 |
| Anti-mouse CD3 (17A2) FITC | BioLegend | Cat#:100204; RRID:AB_312661 |
| Anti-mouse CD4 (GK1.5) BUV496 | BD Biosciences | Cat#:612952; RRID:AB_2813886 |
| Anti-mouse CD4 (GK1.5) PE/Cy7 | BioLegend | Cat#:100422; RRID:AB_312707 |
| Anti-mouse CD4 (RM4-5) AlexaFluor 488 | BioLegend | Cat#:100529; RRID:AB_389303 |
| Anti-mouse CD4 (RM4-5) PerCP-Cy5.5 | BioLegend | Cat#:100540; RRID:AB_893326 |
| Anti-mouse CD8a (53-6.7) APC | BioLegend | Cat#:100712; RRID:AB_312751 |
| Anti-mouse CD8a (53-6.7) APC/Cy7 | BioLegend | Cat#:100714; RRID:AB_312753 |
| Anti-mouse CD8b (YTS156.7.7) PE | BioLegend | Cat#:126607; RRID:AB_961300 |
| Anti-mouse TCR gamma delta (GL3) APC/Fire 750 | BioLegend | Cat#:118136; RRID:AB_2650828 |
| Anti-mouse TCR beta (H57-597) Pacific Blue | BioLegend | Cat#:109226; RRID:AB_1027649 |
| Anti-mouse TCR beta (H57-597) PerCP-Cy5.5 | BioLegend | Cat#:109228; RRID:AB_1575173 |
| Anti-mouse/rat RORgt (B2D) APC | eBioscience | Cat#:17-6981-82; RRID:AB_2573254 |
| Anti-mouse/rat RORgt (B2D) PE | eBioscience | Cat#:12-6981-82; RRID:AB_10807092 |
| Anti-mouse GATA3 (L50-823) Alexa Fluor 488 | BD Biosciences | Cat#:560077; RRID:AB_1645303 |
| Anti-mouse GATA3 (L50-823) BV711 | BD Biosciences | Cat#:565449; RRID:AB_2739242 |
| Anti-mouse T-bet (4B10) PE/Cy7 | BioLegend | Cat#:644824; RRID:AB_2561761 |
| Anti-mouse/rat/human Foxp3 (150D) PE | BioLegend | Cat#:320008; RRID:AB_492980 |
| Anti-mouse Foxp3 (MF-14) BV421 | BioLegend | Cat#:126419; RRID:AB_2565933 |
| Anti-mouse IFN gamma (XMG1.2) PE/Cy7 | BioLegend | Cat#:505826; RRID:AB_2295770 |
| Anti-mouse I-A/I-E (MHCII) (M5/114.15.2) PE | BioLegend | Cat#:107608; RRID:AB_313323 |
| Anti-mouse Ki-67 (SolA15) PE/Cy7 | Thermo Fisher Scientific | 25-5698-82; RRID:AB_11220070 |
| TotalSeq-A0309 anti-mouse Hashtag 9 Antibody (M1/42; 30-F11) | BioLegend | Cat#:155817; RRID:AB_2750042 |
| TotalSeq-A0310 anti-mouse Hashtag 10 Antibody (M1/42; 30-F11) | BioLegend | Cat#:155819; RRID:AB_2750043 |
| TotalSeq-A0311 anti-mouse Hashtag 11 Antibody (M1/42; 30-F11) | BioLegend | Cat#:155821; RRID:AB_2750136 |
| TotalSeq-A0312 anti-mouse Hashtag 12 Antibody (M1/42; 30-F11) | BioLegend | Cat#:155823; RRID:AB_2750137 |
| InVivoMab anti-mouse IFN gamma (XMG1.2) | Bio X Cell | Cat#:BE0055; RRID:AB_1107694 |
| Mouse monoclonal anti-E-cadherin primary antibody (36/E-Cadherin) | BD Biosciences | Cat#:610181; RRID:AB_397580 |
| Rat monoclonal anti-Ki-67 primary antibody (SolA15) | eBioscience | Cat#:14-5698-82; RRID:AB_10854564 |
| Rat monoclonal anti-BrdU primary antibody (BU1/75 (ICR1)) | Abcam | Cat#:ab6326; RRID:AB_305426 |
| Goat polyclonal anti-GFP primary antibody | Abcam | Cat#:ab6673; RRID:AB_305643 |
| Rat monoclonal anti-EpCAM primary antibody (G8.8) | Invitrogen | Cat#:14-5791-81; RRID:AB_953624 |
| Donkey anti-rat IgG AlexaFluor 647 secondary antibody | Invitrogen | Cat#: A78947; RRID:AB_2910635 |
| Donkey anti-mouse IgG AlexaFluor 647 secondary antibody | Abcam | Cat#:ab150111; RRID:AB_2890625 |
| Donkey anti-rat IgG AlexaFluor 594 secondary antibody | Jackson Immunoresearch | Cat#:712-585-153; RRID:AB_2340689 |
| Anti-mouse CD3ε (145-2C11) | BioLegend | Cat#:100340; RRID:AB_11149115 |
| Armenian Hamster IgG Isotype Ctrl Antibody | BioLegend | Cat#:400940; RRID:AB_11203529 |
| Bacterial and Virus Strains | ||
| Faecalibaculum rodentium | DSMZ | #103405 |
| Lachnospiraceae A2 | This paper | N/A |
| Bifidobacterium pseudolongum | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| BMS493 | Tocris | 3509 |
| TrypLE Express | Invitrogen | 12-604-013 |
| HEPES | Corning | 25-060-CI |
| Fetal bovine serum | Gibco | 10438-026 |
| Sodium pyruvate | Corning | 25-000-CI |
| 2-Mercaptoethanol | Sigma | M3148 |
| Dithiothreitol | Sigma | 00-5523-00 |
| Penicillin/streptomycin | Corning | 30-002-CI |
| Collagenase D | Roche | 11088882001 |
| Collagenase A | Roche | 10103586001 |
| DNase I | Roche | 10104159001 |
| Dispase | StemCell Technologies | 07913 |
| Ammonium Chloride Solution | StemCell Technologies | 07850 |
| Cell Stimulation Cocktail (plus protein transport inhibitors) | eBioscience | 00-4975-03 |
| Paraformaldehyde | Sigma | 441244 |
| Bromodeoxyuridine | Sigma | B5002 |
| QIAzol | Qiagen | 79306 |
| Bovine serum albumin fraction V | Roche | 03116956001 |
| Normal donkey serum | Jackson Immunoresearch | 017-000-121 |
| Triton-X 100 | Sigma | T8787 |
| Saponin | Sigma | S4521 |
| Aqua-Poly/Mount | Polysciences | 18606-20 |
| Target Retrieval Solution, pH 6.1 | Agilent | S169984-2 |
| RPMI + GlutaMAX | Gibco | 61870-036 |
| EDTA | VWR | E522 |
| Phenol – chloroform – isoamyl alcohol mixture | Sigma | 77617 |
| UltraPure Buffer-Saturated Phenol | Thermo Fisher Scientific | 15513039 |
| DMSO | Corning | 25-950-CQC |
| Glucose | Sigma | G7021 |
| Brain Heart Infusion | BD Biosciences | 237500 |
| MRS Broth | BD Biosciences | 288130 |
| Hemin | Sigma | H9039 |
| Yeast extract | BD Biosciences | 212750 |
| Vancomycin hydrochloride | Alfa Aesar | J62790-06 |
| Metronidazole | Sigma | M1547 |
| Neomycin sulfate | Alfa Aesar | J61499-14 |
| Amphotericin B | Alfa Aesar | J61491-03 |
| Ampicillin sodium salt | Corning | 61-238-RM |
| Retinoic acid | Sigma | R2625 |
| Retinoic acid-d5 | Toronto Research Chemicals | R250202 |
| Hexane | Sigma | 32293 |
| Hydrochloric acid | Sigma | 258148 |
| Recombinant mouse IL-5 (carrier free) | BioLegend | 581502 |
| Critical Commercial Assays | ||
| Live/Dead Fixable yellow dead cell stain kit | Invitrogen | L34959 |
| Live/Dead Fixable Near-IR dead cell stain kit | Invitrogen | L10119 |
| ALDEFLUOR Kit | STEMCELL Technologies | 01700 |
| BD Cytofix/Cytoperm Fixation Buffer | BD Bioscience | 554714 |
| Foxp3 / Transcription Factor Staining Buffer Set Kit | eBioscience | 00-5523-00 |
| iScript Reverse Transcription Supermix | Bio-Rad | 1708891 |
| SYBR FAST Universal qPCR Master Mix | KAPA Biosystems | KK4619 |
| DNA-free DNA Removal Kit | Invitrogen | AM1906 |
| Direct-zol RNA Miniprep Kit | Zymo Research | R0251 |
| Anti-PE Microbeads | Miltenyi Biotec | 130-048-801 |
| Anti-Siglec-F Microbeads | Miltenyi Biotec | 130-118-513 |
| Click-iT™ Plus TUNEL Assay for In Situ Apoptosis Detection, Alexa Fluor™ 594 dye | Thermo Fisher Scientific | C10618 |
| Deposited Data | ||
| scRNA-seq | This paper | SRA BioProject PRJNA763366 |
| 16S rRNA amplicon sequencing | This paper | SRA BioProject PRJNA763366 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Lgr5-EGFP-IRES-creERT2 | Jackson Laboratory | 008875 |
| Mouse: PHIL | The Mayo Clinic, Dr. Elizabeth Jacobsen | N/A |
| Mouse: C57BL/6J | Jackson Laboratory or bred in own facility | 000664, from Jackson room MP15 |
| Oligonucleotides | ||
| See STAR methods for list of quantitative RT-PCR primers | This paper | N/A |
| Software and Algorithms | ||
| BD FACSDiva v6.2 | BD Biosciences | N/A |
| Prism v9 | GraphPad | N/A |
| STAR aligner version 2.7 | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| phyloseq | (McMurdie & Holmes, 2013) | https://github.com/joey711/phyloseq |
| QIIME v1.9 | (Caporaso et al., 2010) | http://qiime.org/ |
| LefSE | (Segata et al., 2011) | https://huttenhower.sph.harvard.edu/lefse/ |
| Seurat v3 | (Stuart et al., 2019) | https://satijalab.org/seurat/ |
| topGO | (Alexa et al., 2006) | https://bioconductor.org/packages/release/bioc/html/topGO.html |
| NSI-Element Basic Research software | Nikon | N/A |
| ImageJ v1.53c | ImageJ | https://imagej.nih.gov/ij/ |
| FlowJo v10.7.1 | TreeStar | N/A |
Histology and fluorescence microscopy
The duodenum was opened longitudinally and rolled before overnight fixation in 4% paraformaldehyde (Sigma). The tissue was then embedded in paraffin and cut into 5μm thick sections at the Harvard Medical School Rodent Histopathology Core. For immunofluorescence staining, sections were initially deparaffinized and rehydrated. Heat-mediated antigen retrieval was performed in Target Retrieval Solution, Citrate pH 6.1 (Agilent) for 20 minutes. Afterwards, the slides were washed in PBS and blocked in PBS containing 3% BSA (Roche), 3% donkey serum (Jackson Immunoresearch), 0.1% Triton X-100 (Sigma), 0.1% saponin (Sigma) for 1 hour at room temperature. Primary antibodies were incubated overnight at 4°C and secondary antibodies were applied for 1.5 hours at room temperature. Primary antibodies included: rat anti-BrdU (1:200 dilution, ab6326, Abcam), rat anti-Ki-67 (1:300 dilution, 14-5698-82, ThermoFisher Scientific), mouse anti-E-Cadherin (1:400 dilution, 36/E-Cadherin, BD Biosciences). DNA was labeled with DAPI (0.5 μg/ml). TUNEL staining was performed using the Click-iT™ Plus TUNEL Assay for In Situ Apoptosis Detection, Alexa Fluor™ 594 dye (ThermoFisher) per the manufacturer’s instructions.
For Lgr5-GFP visualization, the duodenum was opened longitudinally and rolled, fixed overnight in 4% paraformaldehyde, placed in 20% sucrose for 6h, and then placed in 30% sucrose overnight until the tissue sank. The tissue was then embedded in OCT and frozen at −80°C. 8μm frozen sections were cut and labeled with goat anti-GFP (Abcam ab6673) and rat anti-EpCAM (Invitrogen 14-5791-81).
For 16S rRNA gene FISH, intestinal tissue was fixed overnight in methanol-Carnoy’s fixative followed by routine paraffin embedding and sectioning. After deparaffinization, fluorescence in situ hybridization was performed at 50°C for 90 min in 5% formamide-0.1% SDS-TBS buffer with 2.5 ng/μl of each F. rodentium-specific probes 5’-Cy3- GCCAACCAACTAATGCACCG-3’ and 5’-Cy3- CCGGGAATACGCTCTGGAAA-3’ (Zagato et al., 2020), and 5 ng/μl of a eubacterial 16S RNA sequence specific probe EUB338 (5’-AF488-GCTGCCTCCCGTAGGAGT-3’). Slides were then washed in pre-warmed washing buffer at 50°C and stained with DAPI to visualize nuclei.
Image analysis was performed in Fiji (a version of ImageJ). Epithelial turnover was quantified by dividing the length of the crypt-villus axis labeled by BrdU after 48h of treatment by the total crypt-villus length labeled by DAPI. Ki67+ and BrdU+ cells after 2h BrdU pulse were counted in the crypts. For both epithelial turnover and Ki67+ cell quantification, 15-20 villi/crypts were usually counted per mouse and averaged for a final value.
Eosinophil isolation and sorting
For intestinal eosinophils, the lamina propria was processed as described for flow cytometry and cells were sorted with a two-step MACS process according to the manufacturer’s instructions, first by labeling with MHCII-PE antibody (Biolegend) and using Anti-PE Microbeads (Miltenyi Biotech) to deplete MHCII+ cells, and then sorting SiglecF+ cells with Anti-Siglec-F MicroBeads (mouse) (Miltenyi Biotech). The purity of SiglecF+MHCII− eosinophils was usually >95%. For bone marrow eosinophils, the tibia and femur were harvested, cleaned of muscle, one end of the bone cap was cut off, and bone marrow was collected by centrifuging at 12,000 rpm for 2 minutes. Red blood cells were removed with Ammonium Chloride solution (STEMCELL Technologies). SiglecF+ cells were sorted from the remaining bone marrow cells Anti-Siglec-F MicroBeads (mouse) (Miltenyi Biotech). The purity of SiglecF+ eosinophils was usually >75%.
In vitro eosinophil culture
Sorted bone marrow eosinophils were cultured in RPMI with Glutamax, 10% FBS, penicillin/streptomycin, sodium pyruvate, and 50 μM beta-mercaptoethanol for 24h. In some conditions, 10 ng/mL IL-5 (Biolegend) and/or 100 nM all-trans retinoic acid (Sigma) was included in the culture. After 24h, cell viability was measured by flow cytometry.
RNA/DNA isolation and RTq-PCR
For RNA isolation from total intestinal tissue, tissue was snap frozen and lysed in Qiazol (Qiagen) for RNA extraction following manufacturer’s instructions. For RNA isolation from eosinophils, sorted cells were lysed in Qiazol and processed with the Direct-zol RNA Miniprep Kit (Zymo Research). cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad). Bacterial DNA isolation from stool or intestinal contents was performed using the QIAamp Fast DNA Stool Mini Kit (Qiagen). qPCR was performed using the Kapa SybrFast mastermix. Primers used are listed in Table S2.
BrdU
Mice were injected intraperitoneally (i.p.) with 100 μl of a 3mg/mL BrdU (Sigma) solution in PBS and simultaneously started on drinking water containing 0.8 mg/mL BrdU and 0.5% glucose. Drinking water was continued for 48 hours to measure epithelial turnover.
For short term BrdU incorporation, mice were injected i.p. with 100 μl of a 3mg/mL BrdU and sacrificed after 2 hours.
Aldefluor assay
Epithelial and lamina propria cells were isolated as described. The assay was performed using the ALDEFLUOR Kit (StemCell Technologies) according to the manufacturer’s instructions, at a concentration of 500,000 cells/mL with 30 min incubation time.
Retinoic acid quantification
RA in intestinal tissue was measured by liquid chromatography mass spectrometry via a method adapted from (Kane and Napoli, 2010) and (Grizotte-Lake et al., 2018). Briefly, RA was extracted from intestine under yellow light to prevent retinoid isomerization and degradation using a two-step liquid-liquid extraction method. Glass tubes and pipettes were used to minimize RA adherence to plastic. 60-120 mg tissue was snap frozen in liquid nitrogen and homogenized with 1000 μL of 0.9% saline on ice using a dounce homogenizer and 50 μL of internal standard (RA-d5 at a concentration of 200pg/μL) in ethanol was added. To extract retinoids 2 mL of ethanol containing 0.025 M KOH was added, the sample was briefly vortexed and 10 mL hexane was added. After centrifugation at 800 x g for 2 min the hexane layer was removed. To the remaining aqueous layer, 120 μL of 4 M HCl was added, the sample vortexed, and 10 mL hexane was added to the acidified sample to extract atRA. After centrifugation at 800 x g for 2 min, the top hexane layer containing RA was collected and evaporated to dryness at 25-30°C under a gentle stream of nitrogen. The atRA containing residue was resuspended in 60 μL DMSO. The resuspended extracts were transferred to amber glass vials with glass inserts.
LC-MS analysis was performed by the Harvard Center for Mass Spectrometry. RA was measured by LC-MS on an Agilent 6460 LCMS triple-quad system. 5 μl of sample was injected and separated on a Phenomenex Luna C18 (150 x 2mm) column. Mobile phase A consisted of 10mM ammonium acetate and mobile phase B was methanol. A linear gradient was generated over 15 min with a flow rate of 0.3 mLs/min: 0 min, 70% B; 10 min, 95% B; 12 min 95% B; 12.1 min, 70% B; 15 min 70% B. The MS was operated in multiple reaction monitoring mode with negative ionization. The gas temperature was 350°C, gas flow rate was 12 liters/min, nebulizer pressure was 35 psi, sheath gas temperature was 400°C, and sheath gas flow rate was 12 liters/min.
Retinoic acid receptor inhibitor
Mice were injected i.p. with 220 μg of the pan-retinoic acid receptor inverse agonist BMS493 (Tocris) or vehicle control DMSO (Corning) daily for 8 days, in a volume of 25 μl.
Anti-CD3 injury
Mice were injected i.p. with 50 μg anti-CD3ε or Armenian hamster IgG isotype control (Biolegend). On day 3 after injection, the duodenum was excised, opened longitudinally, and prepared for paraffin embedding as described above in the histology section. Hematoxylin and eosin stained slides were evaluated by a blinded pathologist and histological injury was scored on a scale of 0-4 for the parameters of villus to crypt ratio, crypt injury/loss, and monocyte and polymorphonuclear cell infiltration. The summed score was multiplied by a conversion factor of 1-4 based on the percent of intestine length involved by injury/abnormality.
IFN-γ neutralization
Mice were injected i.p. with 200 μg of anti-IFN-γ (BioXCell clone XMG1.2) neutralizing antibody or PBS every other day for 8 days. BrdU treatment started on day 6, 48h before the endpoint.
Transfer of intestinal contents
Stool or cecal contents were collected into 1 mL of sterile PBS. Usually 2 stool pellets (approximately 10-20% of total liquid volume) were used. Contents were homogenized by mashing with a pestle and vortexing, and 100 μl of liquid was orally gavaged into recipient mice.
Antibiotic treatment
IH mice were provided with 0.5g/L vancomycin (Alfa Aesar), 1 g/L metronidazole (Sigma), 1 g/L neomycin (Alfa Aesar), 1 g/L ampicillin (Corning), and 0.2 g/L amphotericin B (Alfa Aesar) in their drinking water ad libitum with water changes every 3 days. For experiments with antibiotic pre-treatment of donor mice prior to cecal content transfer, antibiotic treatment was for 2 weeks.
Cohousing experiments
In general, unless otherwise noted, mice were cohoused with littermates at weaning and all mice on a Jax background were maintained in cages separate from in-house mice. For experiments in which Jax and IH mice were cohoused, equal numbers of Jax and IH mice were placed in the same cage for 2 weeks.
16S rRNA sequencing
DNA was isolated from stool pellets by phenol-chloroform extraction after bead beating as previously described (Lobel et al., 2020). The 16S rRNA gene sequencing protocol was adapted from the Earth Microbiome Project (Thompson et al., 2017). The 16S rRNA V4 region was amplified from the extracted DNA by PCR and sequenced by using the 2 150–base pair paired-end reading on a MiSeq instrument (Illumina, San Diego, CA). Analysis of 16S rRNA sequence data was performed using Microbiome Helper scripts and QIIME v1.9 (Caporaso et al., 2010; Comeau et al., 2017). Sequences were clustered into operational taxonomic units (OTUs) at a similarity threshold of 97% by using the sortmernasumaclust method of open-reference OTU picking. OTUs were subsequently mapped to a subset of the SILVA 132 database containing only sequences from the V4 region of the 16S rRNA gene to determine taxonomies (Quast et al., 2013). To account for variations in sequencing depth, OTU tables were ratified to the lowest sequence depth among samples. Data were visualized in R using the phyloseq package (McMurdie and Holmes, 2013). Differentially abundant OTUs between groups at the genus level were identified using LefSE (Segata et al., 2011).
Quantification and Statistical Analysis
Data were analyzed with GraphPad Prism (version 9.0). Data are shown as mean with individual data points as noted. For comparison between two independent experimental groups, a two-tailed Mann-Whitney U test was used. For comparison between more than two groups, one-way ANOVA followed by Holm-Sidak’s test was performed. For comparison in experiments with two independent variables, two-way ANOVA followed by Holm-Sidak’s test was performed. Details of statistical analysis and sample size are provided in the figure legends. No samples were excluded from any experiments performed in this study unless in the case of technical failure. Experimenters were not blinded to experimental conditions. Differences of P < 0.05 were considered statistically significant.
Supplementary Material
Table S1. Related to Figure 1. Differentially expressed genes between GF/Jax and IH cells in epithelial cells. Tabs contain top differentially expressed genes between group 1 (GF/Jax) and group 2 (IH) cells in each cell type cluster for clusters 2, 5, 6, 8, 9, 10, 11, 12, 13, 14, 15. For enterocytes, as GF/Jax and IH enterocytes formed different clusters, the comparison in these tabs are between clusters: cluster 1 (GF/Jax mature enterocytes) vs cluster 7 (IH mature enterocytes); and cluster 3 (GF/Jax immature enterocytes) vs cluster 4 (IH immature enterocytes). The following columns are provided for each tab: Gene symbol, p value (based on MAST test), avg_logFC (group 1 vs group 2) (positive values indicate higher expression in group 1), fraction of cells in group 1 with non-zero expression, fraction of cells in group 1 with non-zero expression, and adjusted p value (based on Bonferroni correction).
HIGHLIGHTS.
Microbiota promotes epithelial turnover and suppresses retinoic acid production
Retinoic acid supports intestinal eosinophil survival
Eosinophils suppress IFN-γ production by intra-epithelial lymphocytes
IFN-γ increases epithelial turnover and MHCII expression
ACKNOWLEDGMENTS
This work is supported by NIH grant R24 DK110499 to W.S.G and NIH T32 AI118692, F31 DK121375, and the Karen Doreen Keim Scholarship to Y.G.C. We thank L. Ricci for preparing the graphical abstract. We thank the Garrett Laboratory and Dr. A. Pawluk for helpful discussions and manuscript review.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial interests. WSG is on the SAB of Senda Biosciences, Freya Biosciences and Artizan Biosciences, all outside the current work.
REFERENCES
- Alexa A, Rahnenführer J, and Lengauer T (2006). Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607. [DOI] [PubMed] [Google Scholar]
- Allaire JM, Crowley SM, Law HT, Chang S-Y, Ko H-J, and Vallance BA (2018). The Intestinal Epithelium: Central Coordinator of Mucosal Immunity. Trends Immunol. 39, 677–696. [DOI] [PubMed] [Google Scholar]
- Arnold IC, Artola-Borán M, Tallón de Lara P, Kyburz A, Taube C, Ottemann K, van den Broek M, Yousefi S, Simon H-U, and Müller A (2018). Eosinophils suppress Th1 responses and restrict bacterially induced gastrointestinal inflammation. J. Exp. Med. jem 20172049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer JE, and Blomhoff R (2002). Gene expression regulation by retinoic acid. J. Lipid Res 43, 1773–1808. [DOI] [PubMed] [Google Scholar]
- Beyaz S, Chung C, Mou H, Bauer-Rowe KE, Xifaras ME, Ergin I, Dohnalova L, Biton M, Shekhar K, Eskiocak O, et al. (2021). Dietary suppression of MHC class II expression in intestinal epithelial cells enhances intestinal tumorigenesis. Cell Stem Cell 28, 1922–1935.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya N, Yuan R, Prestwood TR, Penny HL, DiMaio MA, Reticker-Flynn NE, Krois CR, Kenkel JA, Pham TD, Carmi Y, et al. (2016). Normalizing Microbiota-Induced Retinoic Acid Deficiency Stimulates Protective CD8 + T Cell-Mediated Immunity in Colorectal Cancer. Immunity 45, 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilate AM, London M, Castro TBR, Mesin L, Bortolatto J, Kongthong S, Harnagel A, Victora GD, and Mucida D (2020). T Cell Receptor Is Required for Differentiation, but Not Maintenance, of Intestinal CD4 + Intraepithelial Lymphocytes. Immunity 53, 1001–1014.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biton M, Haber AL, Rogel N, Burgin G, Beyaz S, Schnell A, Ashenberg O, Su CW, Smillie C, Shekhar K, et al. (2018). T Helper Cell Cytokines Modulate Intestinal Stem Cell Renewal and Differentiation. Cell 175, 1307–1320.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll TD, Newton IP, Chen Y, Blow JJ, and Näthke I (2018). Lgr5+ intestinal stem cells reside in an unlicensed G1 phase. J. Cell Biol 217, 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha H-R, Chang S-Y, Chang J-H, Kim J-O, Yang J-Y, Kim C-H, and Kweon M-N (2010). Downregulation of Th17 Cells in the Small Intestine by Disruption of Gut Flora in the Absence of Retinoic Acid. J. Immunol 184, 6799–6806. [DOI] [PubMed] [Google Scholar]
- Chang D-H, Rhee M-S, Ahn S, Bang B-H, Oh JE, Lee HK, and Kim B-C (2015). Faecalibaculum rodentium gen. nov., sp. nov., isolated from the faeces of a laboratory mouse. Antonie Van Leeuwenhoek 108, 1309–1318. [DOI] [PubMed] [Google Scholar]
- Chen B, Scurrah CR, McKinley ET, Simmons AJ, Ramirez-Solano MA, Zhu X, Markham NO, Heiser CN, Vega PN, Rolong A, et al. (2021). Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 184, 6262–6280.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu DK, Jimenez-Saiz R, Verschoor CP, Walker TD, Goncharova S, Llop-Guevara A, Shen P, Gordon ME, Barra NG, Bassett JD, et al. (2014). Indigenous enteric eosinophils control DCs to initiate a primary Th2 immune response in vivo. J. Exp. Med 211, 1657–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeau AM, Douglas GM, and Langille MGI (2017). Microbiome Helper: a Custom and Streamlined Workflow for Microbiome Research. MSystems 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LM, Sohn J, Tyrrell KL, Citron DM, Lawson PA, Patel NB, Iizumi T, Perez-Perez GI, Goldstein EJC, and Blaser MJ (2017). Description of two novel members of the family Erysipelotrichaceae: Ileibacterium valens gen. nov., sp. nov. and Dubosiella newyorkensis, gen. nov., sp. nov., from the murine intestine, and emendation to the description of Faecalibacterium rodentium. Int. J. Syst. Evol. Microbiol 67, 1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnewski P, Das S, Parigi SM, and Villablanca EJ (2017). Retinoic acid and its role in modulating intestinal innate immunity. Nutrients 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekaney CM, Wu G, Yin YL, and Jaeger LA (2008). Regulation of ornithine aminotransferase gene expression and activity by all-transretinoic acid in Caco-2 intestinal epithelial cells. J. Nutr. Biochem 19, 674–681. [DOI] [PubMed] [Google Scholar]
- Elmentaite R, Kumasaka N, Roberts K, Fleming A, Dann E, King HW, Kleshchevnikov V, Dabrowska M, Pritchard S, Bolt L, et al. (2021). Cells of the human intestinal tract mapped across space and time. Nature 597, 250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriguchi Y, Nakamura K, Yokoi Y, Sugimoto R, Takahashi S, Hashimoto D, Teshima T, Ayabe T, Selsted ME, and Ouellette AJ (2018). Essential role of IFN-γ in T cell–associated intestinal inflammation. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finak G, McDavid A, Yajima M, Deng J, Gersuk V, Shalek AK, Slichter CK, Miller HW, McElrath MJ, Prlic M, et al. (2015). MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol. 2015 161 16, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florian S, Krehl S, Loewinger M, Kipp A, Banning A, Esworthy S, Chu FF, and Brigelius-Flohé R (2010). Loss of GPx2 increases apoptosis, mitosis, and GPx1 expression in the intestine of mice. Free Radic. Biol. Med 49, 1694–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattu S, Bang YJ, Pendse M, Dende C, Chara AL, Harris TA, Wang Y, Ruhn KA, Kuang Z, Sockanathan S, et al. (2019). Epithelial retinoic acid receptor β regulates serum amyloid A expression and Vitamin A-dependent intestinal immunity. Proc. Natl. Acad. Sci. U. S. A 166, 10911–10916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain P, Gaudon C, Pogenberg V, Sanglier S, Van Dorsselaer A, Royer CA, Lazar MA, Bourguet W, and Gronemeyer H (2009). Differential Action on Coregulator Interaction Defines Inverse Retinoid Agonists and Neutral Antagonists. Chem. Biol 16, 479–489. [DOI] [PubMed] [Google Scholar]
- Goh YPS, Henderson NC, Heredia JE, Red Eagle A, Odegaard JI, Lehwald N, Nguyen KD, Sheppard D, Mukundan L, Locksley RM, et al. (2013). Eosinophils secrete IL-4 to facilitate liver regeneration. Proc. Natl. Acad. Sci 110, 9914–9919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grizotte-Lake M, Zhong G, Duncan K, Kirkwood J, Iyer N, Smolenski I, Isoherranen N, and Vaishnava S (2018). Commensals Suppress Intestinal Epithelial Cell Retinoic Acid Synthesis to Regulate Interleukin-22 Activity and Prevent Microbial Dysbiosis. Immunity 49, 1103–1115.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, et al. (2017). A single-cell survey of the small intestinal epithelium. Nature 551, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Grainger JR, Spencer SP, and Belkaid Y (2011a). The Role of Retinoic Acid in Tolerance and Immunity. Immunity 35, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JA, Cannons JL, Grainger JR, Santos L.M. Dos, Hand TW, Naik S, Wohlfert EA, Chou DB, Oldenhove G, Robinson M, et al. (2011b). Essential role for retinoic acid in the promotion of CD4+ T cell effector responses via retinoic acid receptor alpha. Immunity 34, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia JE, Mukundan L, Chen FM, Mueller A. a, Deo RC, Locksley RM, Rando T. a, and Chawla A (2013). Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV (2015). Epithelial Cell Contributions to Intestinal Immunity. In Advances in Immunology, (Academic Press; ), pp. 129–172. [DOI] [PubMed] [Google Scholar]
- Howitt MR, Lavoie S, Michaud M, Blum AM, Tran SV, Weinstock JV, Gallini CA, Redding K, Margolskee RF, Osborne LC, et al. (2016). Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science (80-. ). 351, 1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignacio A, Shah K, Bernier-Latmani J, Köller Y, Coakley G, Moyat M, Hamelin R, Armand F, Wong NC, Ramay H, et al. (2022). Small intestinal resident eosinophils maintain gut homeostasis following microbial colonization. Immunity 55, 1250–1267.e12. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee C. a, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata M, Hirakiyama A, Eshima Y, Kagechika H, Kato C, and Song SY (2004). Retinoic Acid Imprints Gut-Homing Specificity on T Cells. Immunity 21, 527–538. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Wang Y, Guo S, and Wang G (2017). Glutathione peroxidases as oncotargets. Oncotarget 8, 80093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jijon HB, Suarez-Lopez L, Diaz OE, Das S, De Calisto J, Yaffe MB, Pittet MJ, Mora JR, Belkaid Y, Xavier RJ, et al. (2018). Intestinal epithelial cell-specific RARα depletion results in aberrant epithelial cell homeostasis and underdeveloped immune system. Mucosal Immunol. 11, 703–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Saiz R, Anipindi VC, Galipeau H, Ellenbogen Y, Chaudhary R, Koenig JF, Gordon ME, Walker TD, Mandur TS, Abed S, et al. (2020). Microbial Regulation of Enteric Eosinophils and Its Impact on Tissue Remodeling and Th2 Immunity. Front. Immunol 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MA, and Napoli JL (2010). Quantification of Endogenous Retinoids. In Methods in Molecular Biology (Clifton, N.J.), (Methods Mol Biol; ), pp. 1–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastl AJ, Terry NA, Wu GD, and Albenberg LG (2020). The Structure and Function of the Human Small Intestinal Microbiota: Current Understanding and Future Directions. Cell. Mol. Gastroenterol. Hepatol 9, 33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury KA, Floch MH, and Hersh T (1969). Small Intestinal Mucosal Cell Proliferation and Bacterial Flora in the Conventionalization of the Germfree Mouse. J. Exp. Med 130, 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Taparowsky EJ, and Kim CH (2015). Retinoic Acid Differentially Regulates the Migration of Innate Lymphoid Cell Subsets to the Gut. Immunity 43, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, Spencer SP, Torabi-Parizi P, Grainger JR, Roychoudhuri R, Ji Y, Sukumar M, Muranski P, Scott CD, Hall JA, et al. (2013). Retinoic acid controls the homeostasis of pre-cDC–derived splenic and intestinal dendritic cells. J. Exp. Med 210, 1961–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Sandell LL, Trainor PA, Koentgen F, and Duester G (2012). Alcohol and Aldehyde Dehydrogenases: Retinoid Metabolic Effects in Mouse Knockout Models. Biochim. Biophys. Acta 1821, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larange A, and Cheroutre H (2016). Retinoic Acid and Retinoic Acid Receptors as Pleiotropic Modulators of the Immune System. Annu. Rev. Immunol 34, 369–394. [DOI] [PubMed] [Google Scholar]
- Larsen SB, Cowley CJ, and Fuchs E (2020). Epithelial cells: liaisons of immunity. Curr. Opin. Immunol 62, 45–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JJ, Dimina D, Macias MMP, Ochkur SI, McGarry MP, O’Neill KR, Protheroe C, Pero R, Nguyen T, Cormier SA, et al. (2004). Defining a link with asthma in mice congenitally deficient in eosinophils. Science (80-. ). 305, 1773–1776. [DOI] [PubMed] [Google Scholar]
- Li F, Dai L, and Niu J (2020). GPX2 silencing relieves epithelial–mesenchymal transition, invasion, and metastasis in pancreatic cancer by downregulating Wnt pathway. J. Cell. Physiol 235, 7780–7790. [DOI] [PubMed] [Google Scholar]
- Lobel L, Cao YG, Fenn K, Glickman JN, and Garrett WS (2020). Diet posttranslationally modifies the mouse gut microbial proteome to modulate renal function. Science 369, 1518–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Guryn K, Hubert N, Frazier K, Urlass S, Musch MW, Ojeda P, Pierre JF, Miyoshi J, Sontag TJ, Cham CM, et al. (2018). Small Intestine Microbiota Regulate Host Digestive and Absorptive Adaptive Responses to Dietary Lipids. Cell Host Microbe 23, 458–469.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Guryn K, Leone V, and Chang EB (2019). Regional Diversity of the Gastrointestinal Microbiome. Cell Host Microbe 26, 314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald BD, Jabri B, and Bendelac A (2018). Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol 18, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merger M, Viney JL, Borojevic R, Steele-Norwood D, Zhou P, Clark D. a, Riddell R, Maric R, Podack ER, and Croitoru K (2002). Defining the roles of perforin, Fas/FasL, and tumour necrosis factor alpha in T cell induced mucosal damage in the mouse intestine. Gut 51, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore WT, Murtaugh MP, and Davies PJA (1984). Retinoic Acid-induced Expression of Tissue Transglutaminase in Mouse Peritoneal Macrophages. J. Biol. Chem 259, 12794–12802. [PubMed] [Google Scholar]
- Mora JR, Iwata M, Eksteen B, Song S-Y, Junt T, Senman B, Otipoby KL, Yokota A, Takeuchi H, Ricciardi-Castagnoli P, et al. (2006). Generation of Gut-Homing IgA-Secreting B Cells by Intestinal Dendritic Cells. Science (80-. ). 314, 1157–1160. [DOI] [PubMed] [Google Scholar]
- Mowat AM, and Agace WW (2014). Regional specialization within the intestinal immune system. Nat. Rev. Immunol 2014 1410 14, 667–685. [DOI] [PubMed] [Google Scholar]
- Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, and Cheroutre H (2007). Reciprocal TH17 and Regulatory T Cell Differentiation Mediated by Retinoic Acid. Science (80-. ). 317, 256–260. [DOI] [PubMed] [Google Scholar]
- Nadjsombati MS, McGinty JW, Lyons-Cohen MR, Jaffe JB, DiPeso L, Schneider C, Miller CN, Pollack JL, Nagana Gowda GA, Fontana MF, et al. (2018). Detection of Succinate by Intestinal Tuft Cells Triggers a Type 2 Innate Immune Circuit. Immunity 49, 33–41.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakshatri H, and Chambon P (1994). The directly repeated RG(G/T)TCA motifs of the rat and mouse cellular retinol-binding protein II genes are promiscuous binding sites for RAR, RXR, HNF-4, and ARP-1 homo- and heterodimers. J. Biol. Chem 269, 890–902. [PubMed] [Google Scholar]
- Nava P, Koch S, Laukoetter MG, Lee WY, Kolegraff K, Capaldo CT, Beeman N, Addis C, Gerner-Smidt K, Neumaier I, et al. (2010). Interferon-γ Regulates Intestinal Epithelial Homeostasis through Converging β-Catenin Signaling Pathways. Immunity 32, 392–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, and Glöckner FO (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg ME, and Hogan SP (2006). The eosinophil. Annu. Rev. Immunol 24, 147–174. [DOI] [PubMed] [Google Scholar]
- Sasajima N, Ogasawara T, Takemura N, Fujiwara R, Watanabe J, and Sonoyama K (2010). Role of intestinal Bifidobacterium pseudolongum in dietary fructo-oligosaccharide inhibition of 2,4-dinitrofluorobenzene-induced contact hypersensitivity in mice. Br. J. Nutr 103, 539–548. [DOI] [PubMed] [Google Scholar]
- Schneider C, O’Leary CE, von Moltke J, Liang HE, Ang QY, Turnbaugh PJ, Radhakrishnan S, Pellizzon M, Ma A, and Locksley RM (2018). A Metabolite-Triggered Tuft Cell-ILC2 Circuit Drives Small Intestinal Remodeling. Cell 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah K, Ignacio A, McCoy KD, and Harris NL (2020). The emerging roles of eosinophils in mucosal homeostasis. Mucosal Immunol. 1–10. [DOI] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino-Michaels K, Earley ZM, Benyamin FW, Man Lei Y, Jabri B, Alegre M-L, et al. (2015). Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science (80-. ). 350, 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoskiewicz MJ, Colvin RB, Schneeberger EE, and Russell PS (1985). Widespread and selective induction of major histocompatibility complex-determined antigens in vivo by gamma interferon. J. Exp. Med 162, 1645–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderholm AT, and Pedicord VA (2019). Intestinal epithelial cells: at the interface of the microbiota and mucosal immunity. Immunology 158, 267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis AG, Klapholz M, Zhao J, and Levy M (2020). The bidirectional nature of microbiome-epithelial cell interactions. Curr. Opin. Microbiol 56, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Miyoshi H, Origanti S, Nice TJ, Barger AC, Manieri NA, Fogel LA, French AR, Piwnica-Worms D, Piwnica-Worms H, et al. (2015). Type I Interferons Link Viral Infection to Enhanced Epithelial Turnover and Repair. Cell Host Microbe 17, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, Narushima S, Vlamakis H, Motoo I, Sugita K, et al. (2019). A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature 565, 600–605. [DOI] [PubMed] [Google Scholar]
- Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, et al. (2017). A communal catalogue reveals Earth’s multiscale microbial diversity. Nat. 2017 5517681 551, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuganbaev T, Mor U, Bashiardes S, Liwinski T, Nobs SP, Leshem A, Dori-Bachash M, Thaiss CA, Pinker EY, Ratiner K, et al. (2020). Diet Diumally Regulates Small Intestinal Microbiome-Epithelial-Immune Homeostasis and Enteritis. Cell 1–19. [DOI] [PubMed] [Google Scholar]
- Ueki S, Mahemuti G, Oyamada H, Kato H, Kihara J, Tanabe M, Ito W, Chiba T, Takeda M, Kayaba H, et al. (2008). Retinoic Acids Are Potent Inhibitors of Spontaneous Human Eosinophil Apoptosis. J. Immunol 181, 7689–7698. [DOI] [PubMed] [Google Scholar]
- Vicetti Miguel RD, Quispe Calla NE, Dixon D, Foster RA, Gambotto A, Pavelko SD, Hall-Stoodley L, and Cherpes TL (2017). IL-4–secreting eosinophils promote endometrial stromal cell proliferation and prevent Chlamydia -induced upper genital tract damage. Proc. Natl. Acad. Sci 114, E6892–E6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villablanca EJ, Wang S, De Calisto J, Gomes DCO, Kane MA, Napoli JL, Blaner WS, Kagechika H, Blomhoff R, Rosemblatt M, et al. (2011). MyD88 and retinoic acid signaling pathways interact to modulate gastrointestinal activities of dendritic cells. Gastroenterology 141, 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Kang SG, Esch HH, Love PE, and Kim CH (2010). Retinoic acid determines the precise tissue tropism of inflammatory Th17 cells in the intestine. J. Immunol. 184, 5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo V, Eshleman EM, Hashimoto-Hill S, Whitt J, Wu S, Engleman L, Rice T, Karns R, Qualls JE, Haslam DB, et al. (2021). Commensal segmented filamentous bacteria-derived retinoic acid primes host defense to intestinal infection. Cell Host Microbe 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, and Kuchroo VK (2008). Retinoic acid increases Foxp3+ regulatory T cells and inhibits development of Th17 cells by enhancing TGF-β-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J. Immunol 181, 2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagato E, Pozzi C, Bertocchi A, Schioppa T, Saccheri F, Guglietta S, Fosso B, Melocchi L, Nizzoli G, Troisi J, et al. (2020). Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat. Microbiol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F (2009). Molecular Mechanisms of IFN-γ to Up-Regulate MHC Class I Antigen Processing and Presentation. Int. Rev. Immunol 28, 239–260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 1. Differentially expressed genes between GF/Jax and IH cells in epithelial cells. Tabs contain top differentially expressed genes between group 1 (GF/Jax) and group 2 (IH) cells in each cell type cluster for clusters 2, 5, 6, 8, 9, 10, 11, 12, 13, 14, 15. For enterocytes, as GF/Jax and IH enterocytes formed different clusters, the comparison in these tabs are between clusters: cluster 1 (GF/Jax mature enterocytes) vs cluster 7 (IH mature enterocytes); and cluster 3 (GF/Jax immature enterocytes) vs cluster 4 (IH immature enterocytes). The following columns are provided for each tab: Gene symbol, p value (based on MAST test), avg_logFC (group 1 vs group 2) (positive values indicate higher expression in group 1), fraction of cells in group 1 with non-zero expression, fraction of cells in group 1 with non-zero expression, and adjusted p value (based on Bonferroni correction).
Data Availability Statement
Single-cell RNA-seq data and 16S rRNA amplicon sequencing data have been deposited at SRA as BioProject PRJNA763366.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.