

Abstract
The complement system plays a key role in the pathogenesis of autoimmune diseases, which usually injures the kidney. More and more studies have shown the pathogenic role and indicated that abnormal activation of the complement system was highly involved in the outbreak of autoimmune diseases. This review mainly introduced recent studies of complement system activation contributing to the pathogenesis of autoimmune diseases, including systemic lupus erythematosus, antiphospholipid syndrome, antineutrophil cytoplasmic antibody‐associated vasculitides, and so on. Understanding the pathogenic roles of complement activation in various autoimmune diseases will identify potential novel therapeutic targets on complement systems.
Keywords: ANCA‐associated vasculitides, antiphospholipid syndrome, autoimmune disease, complement, systemic lupus erythematosus
Highlights
The complement system is important in the pathogenesis of a group of autoimmune diseases.
Targeting the complement system might provide us with novel target therapies in autoimmune diseases.
Complement pathways and certain targets of complement inhibitors.
1. INTRODUCTION
The complement system is an important component of innate immunity and mainly participates in antibody‐mediated immunity. The physiological functions of the complement system include defending against pathogens invasion, clearance of immune complex and apoptotic debris, and maintaining hemostatic balance. Autoimmune diseases, however, refer to body damage caused by immune responses to autoantigens. Either deficiency of early components or overactivation of complement system will induce severe autoimmune response and develop autoimmune diseases eventually. 1 Studies showed clues of the correlations between the complement system and autoimmune diseases, such as deposition of large amounts of complements in affected tissues and elevated or reduced levels of serum complements due to abnormal complement activation.
Traditional therapies, such as glucocorticoids and immunosuppressants, may achieve partial or full response in autoimmune diseases. However, the low response rate and adverse effects continue to plague rheumatologists and nephrologists. The important biologic function of the complement system makes it an important therapeutic target. Therapies targeting complement activation can reduce tissue inflammation and attenuate the adaptive immune response to exogenous and tissue antigens rapidly. This article reviews recent advances in complement activation in the pathogenesis of some autoimmune diseases and the potential therapies targeting complement components in various diseases.
2. COMPLEMENT SYSTEM
The complement system has been initially discovered in heat‐labile anti‐cholera plasma and is composed of over 30 kinds of plasma proteins and cell membrane‐bound proteins. 2 There are three pathways, including classical pathway, lectin pathway, and alternative pathway, which could be activated by immune complex, microbes, mannose, or spontaneous activated. The alternative pathway serves as an amplification loop for the lectin and classical pathways, accounting for roughly 80% of complement activation products. 3 It is noteworthy that all three pathways are merged in common pathways at the C3 level, culminating in the formation of products C5b‐9 (membranous attack complex [MAC]) (Figure 1).
Figure 1.
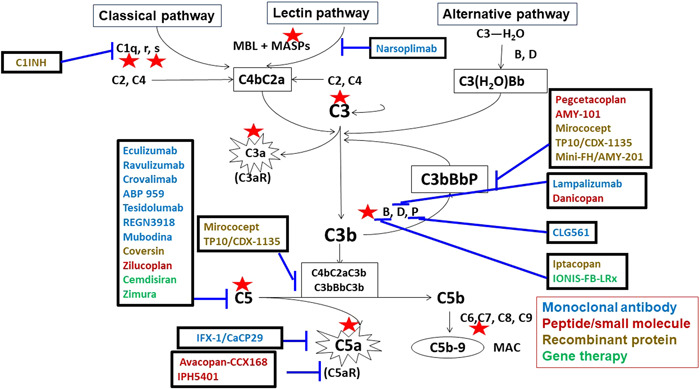
Complement pathways and certain targets of complement inhibitors. The names of the products of monoclonal antibodies are shown in blue, while the names of peptides or small molecules are shown in red, recombinant protein in yellow, and gene therapies in green. Blue lines indicate certain targets of the complement system. From left to right: ABP959, tesidolumab, REGN3918, mubodina, coversin, RA101495, cemdisiran, and zimura inhibit C5; narsoplimab inhibits mannose‐binding protein‐associated serine protease 2 (MASP‐2) of the lectin pathway; MicroCept, TP10/CDX‐1135 inhibits C3 and C5 convertases; IFX‐1 inhibits C5a; avacopan, IPH5401 inhibits C5a receptor; pegcetacoplan (formerly APL‐2), AMY‐101, and mini‐FH/AMY‐201 inhibit C3 and C3 convertase activity; lampalizumab and danicopan inhibit factor D; CLG561 inhibits properdin; iptacopan and IONIS‐FB‐LRx inhibit factor B.
2.1. Initiation and activation
Activation of classical pathway initiated from antibody‐antigen complexes recognized by C1q. In the activation of the classical pathway, C2 and C4 are cleaved by C1s protease, which is activated by antigen‐bind C1q and C1r. Activation of the lectin pathway is initiated from mannose‐binding lectin (MBL) binding to the carbohydrate structure on microbes. In addition, ficolins (ficolin1, 2, 3) and collectin‐11 shared a similar structure with MBL. Then C4, C2 could be further cleaved by MBL‐associated serine proteases (MASPs) after binding to MBL, ficolins or collectin‐11 and form C4bC2a. AP activation initiates from C3 spontaneous hydrolysis, C3 (H2O), which is also called “tick‐over.” C3(H2O) binds to complement factor B (CFB) and is then cleaved by factor D.
2.2. Amplification
For the classical pathway and lectin pathway, complement fragments from enzyme reactions constitute the same C3 convertase (C4bC2a). In the alternative pathway, the C3 convertase is C3bBb, which can bind properdin to stabilize itself. Both C3 convertases rapidly generate many activated C3bs, which form C5 convertases (C3bBbC3b or C2aC4bC3b) that cleave C5 to initiate the terminal pathway.
Some of the complement regulatory proteins focus on the amplification of complement activation to protect host cells from being damaged as innocent bystanders. They can be either plasma proteins, such as complement factor H (CFH) or cell membrane‐bound proteins (such as decay‐accelerating factor, CD55; membrane cofactor protein [MCP] or CD46; complement receptor 1 [CR1]; C4‐binding protein [C4BP]). CFH, composed of 20 short consensus repeats (SCR), is the major regulator of complement activation in both circulation and cell surface. It could impede the formation of the alternative pathway C3 convertase (C3bBb) by competing with CFB for binding to C3b, thereby accelerating the decay of C3bBb. Meanwhile, CFH acts as a cofactor for factor I‐mediated proteolytic inactivation of C3b.
2.3. Effector function
In the downstream cascades, C5b binds to C6 and then combines with C7, which can stick to the cell surface. After that, the C5b‐8 complex will be formed with some lytic activity. When multiple molecules of C9 comes to C5b‐8, MAC will be expressed with a more efficient lysis.
Many C3bs and C4bs deposit on foreign surfaces and serve as ligands for receptors for the opsonic function. Complement fragments C3a, C4a, and C5a are anaphylatoxins.
2.4. Additional proteases
Besides the traditional C3 convertase, there was a group of intrinsic or extrinsic proteases that could cleave C3. The intrinsic proteases include renin, kallikrein, coagulation factor IXa, Xa, and XIa. The extrinsic include proteases from Streptococcus, Neisseria meningitidis, Staphylococcus aureus, and Enterococcus faecalis. 4 , 5 , 6 Most of the intrinsic convertases could produce the same C3a, while the extrinsic proteases always resulted in the longer or shorter C3a to inactivate C3 by cleavage into nonfunctional fragments.
Recent studies have provided us with a new way of complement activation. The conformational activation of C5 adopts a C5b‐like conformation when bound to densely C3b‐opsonized surfaces and culminating in the formation of MAC in absence of C5‐activating enzymes. 7
3. COMPLEMENT AND AUTOIMMUNE DISEASES
The complement system plays a role in various rheumatic diseases, which is often unknown but always considered to contribute to tissue damage by autoantibody‐mediated immune complex formation and deposition. Here we focus on three complement activation driven autoimmune diseases systemic lupus erythematosus (SLE), antineutrophil cytoplasmic antibody (ANCA)‐associated vasculitides (AAVs), and antiphospholipid syndrome (APS). We describe the recent advances of complement activation in the pathogenesis of these diseases and the available or potential therapeutics targeting complement components in various diseases.
3.1. SLE and complement
SLE is a systemic autoimmune disease characterized by the production of multiple autoantibodies.
The role of complement in the pathogenesis of SLE is a “double‐edged sword” deficiency of early components like C1q, C1r, C1s, C4, and C2 could result in severe SLE with early onset. In addition to complete deficiency, copy number variation of the C4A and C4B is also associated with the risk of SLE.
Deficiency of C1q strongly predisposes to the development of SLE due to the dysfunction of removal of apoptotic cells and debris, few patients showed C1q gene mutation. However, anti‐C1q autoantibodies could be found in more than 50% of lupus nephritis. Several studies, including ours, have found an association between anti‐C1q autoantibodies and disease activity in lupus nephritis. The combination of anti‐C1q autoantibodies and anti‐dsDNA autoantibodies could predict the renal prognosis of lupus nephritis. 8 Vanhecke et al. 9 used anti‐C1q antibodies derived from SLE patients in a microarray‐based scan to identify the B‐cell epitope of C1q. They found that C1q A08 (C1q A15‐27: GRPGRRGRPGLKG) was the most important epitope of C1q. Our study further confirmed that C1q A08 antibodies were better than antibodies against intact C1q in correlation with lupus nephritis activity as well as predicting renal prognosis based on a large Chinese cohort. 10 C1q A08 was a half cryptic epitope of anti‐C1qA08 antibodies in lupus nephritis and important for the activation of classical pathways according to our in vitro study. 11 More importantly, anti‐C1qA08 antibodies accelerated the development of lupus nephritis in MRL/lpr mice, which indicated the pathogenic role of anti‐C1qA08 antibodies in lupus nephritis (unpublished data).
Recent studies raised a discussion about the role of MBL in the pathogenesis of SLE. The MBL gene polymorphism influenced susceptibility to SLE. The presence of the promotors with diverse polymorphisms and coding regions of the MBL‐2 gene led to the fluctuation of plasma levels of MBL. 12 , 13 MBL levels were variable in different clinical manifestations of SLE. Patients with musculoskeletal and cutaneous manifestations showed lower levels of MBL, while higher and intermediate MBL levels were significantly associated with lupus nephritis with other systemic manifestations. 14 Moreover, serum anti‐MBL autoantibodies can influence the functional activity of MBL and bind to MBL depositions in tissues. 15 However, the role of anti‐MBL autoantibodies in the pathogenesis of lupus is currently unclear.
Previous studies have demonstrated that activation of the alternative complement pathway could reflect disease activity and flares in patients with SLE. 16 Deficiency of complement components of the alternative pathway increased the susceptibility to SLE, such as genetic variants of CFH and CFH‐related proteins. Our previous studies showed that serum CFH levels were associated with the disease activity of lupus nephritis. Glomerular expression of CFH was stronger than in normal controls. 17 However, anti‐CFH autoantibodies were detected in 10% lupus nephritis with polyepitopes and IgG2 subclass predominance, who presented with milder renal damage. The in vitro studies showed that the purified autoantibodies could enhance the C3b binding and CFI cofactor activity of CFH, which suggested a protective role of anti‐CFH autoantibodies in the lupus nephritis. 18
Thrombotic microangiopathy (TMA) is the most severe renal vascular change with high mortality in lupus nephritis. 19 The pathogenesis of TMA in lupus nephritis is complicated and unclear. Our study found that patients with both C4d deposition and decreased serum CFH had the worse renal outcome, which indicated that complement overactivation via both classical and alternative pathways might play an important role in the pathogenesis of renal TMA in lupus nephritis. 20 Moreover, purified CFH from lupus nephritis combined TMA presented with dysfunction in binding with C3b and modified C reactive protein (mCRP), protecting abilities from the lysis of sheep erythrocytes and inducing the phagocytosis of late apoptotic cells, who had the known SNP rs1061147(SCR5, A307A), rs1061170 (SCR7, Y402H), CM050194 (SCR20, S1191W), and CM010322 (SCR20, V1197A). 21 Thus, CFH might play a role in the pathogenesis of lupus nephritis combined TMA.
3.2. APS and complement
APS is characterized by vascular thrombosis, pathogenic pregnancies, and antiphospholipid antibodies (aPL).
Increasing data indicate that the complement pathway is activated in patients with APS and acted as a cofactor in the pathogenesis of aPL‐associated clinical events. Complement activation by aPL generates C5a, which recruits neutrophils and leads to the expression of tissue factor on neutrophils, monocytes, and endothelial cells and induces activation of the extrinsic coagulation pathway. 22 Moreover, the C5a induced neutrophil activation and the respiratory burst leading to trophoblastic injury and fetal loss. 23 , 24 Beta‐2‐glycoprotein (β2GPI) belongs to a super‐family of proteins characterized by short consensus repeats (SCRs), which are frequently found in complement regulatory proteins. Studies from Gropp et al. 25 have reported that β2‐GPI acted as a complement regulator. β2‐GPI apparently changed the conformation of C3 after binding to a surface, so that the CFH attached and induced subsequent degradation by the CFI. β2‐GPI also mediated further cleavage of C3/C3b. Furthermore, aPL could induce fetal loss, fetal weight reduction, and thrombosis in wild‐type mice, but not in mice deficient in specific complement components (C3, C4, C5, and C6) or in the presence of a C5 inhibitor. 26 , 27 , 28 In addition, CFB inhibition could ameliorate aPL‐induced growth retardation and fetal resorption. 29 The above evidence indicated that the classical and alternative pathways were involved in the pathogenesis of aPL‐induced pregnancy morbidity, not only contributing to the thrombosis formation but also the complement‐mediated inflammatory process.
Catastrophic APS refers to rapidly progressing APS with the development of thrombotic events in at least three different organs within a week. This occurs in 1% of patients with APS but has a high mortality rate of 50%. A recent study suggests that 60% of patients with CAPS have underlying mutations in complement regulatory genes, 30 which share almost the same frequency in aHUS. The C5b‐9 deposition could be inhibited by anti‐C5 antibody induced by APS sera. However, factor D inhibitor did not prevent the C5b‐9 deposition induced by APS sera, suggesting that complement activation in APS sera primarily occurs through the classical pathway. While C5b‐9 deposition was partially blocked by factor D inhibitor and was completely blocked by anti‐C5 monoclonal antibody‐induced by CAPS sera, which suggested complement activation in CAPS sera through classical and alternative pathways. Thus, mutations in complement regulatory genes serve as a “second hit,” leading to uncontrolled complement activation and more severe thrombotic events in CAPS.
Eculizumab has successfully been used to treat catastrophic APS refractory to standard therapies in case studies. 31 , 32 , 33 Müller‐Calleja et al. 34 reported three patients undergoing a renal transplant. They failed in prednisone, rituximab, and anticoagulation. They were treated with eculizumab and were under successful engraftment for up to 4 years without any relapse of APS, which suggests eculizumab prevents relapse of CAPS even in the setting of renal transplant. However, the potential of complement inhibitors as a therapeutic target in APS require further study.
3.3. AAV and complement
AAV comprises a group of autoimmune disorders, including granulomatosis with polyangiitis (GPA), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA). 35 Various evidence implicates the complement system as a key player in the pathogenesis of this disease.
Animal studies from Xiao et al. 36 first discovered the role of complement activation in the pathogenesis of AAV by the mouse model of MPO‐ANCA vasculitis. C5‐deficient mice, factor‐B‐deficient, C5aR‐deficient (also known as CD88) mice or wild‐type mice pretreated with cobra venom factor to deplete complement, failed to develop AAV. AAV development was comparable in wild‐type and C4‐deficient or C6‐deficient mice, which indicated that the classical pathway and terminal pathway were not as important as C5a in the development of AAV. Knockout of C5a‐like receptor 2 (C5L2) resulted in more severe disease, which suggested a different role of C5L2 compared with C5aR. 37 Moreover, pretreatment with anti‐C5 antibodies before passive transfer of anti‐MPO IgG could also prevent the development of AAV in mice. 38 Oral administration CCX168 (avacopan, a small molecule antagonist of human C5aR) could ameliorate anti‐MPO‐induced AAV in the human C5a receptor knocked‐in mice. These data suggest that complement activation via the alternative pathway and C5a/C5aR are critical in the pathogenesis of AAV.
Our study investigated various components of complement deposited in renal specimens of patients with pauci‐immune MPO‐AAV. C5b‐9, C3d, and CFB could be detected in the specimens but not C4d. Moreover, C3d and CFB colocalized with C5b‐9 in active glomerular lesions. 39 Expression of C5aR and C5L2 was mainly on infiltrating neutrophils and macrophages. The downregulation of C5aR was observed in these renal specimens. It might be a result of C5a‐mediated internalization to alleviate C5a‐mediated inflammation. 40 These data further proved the complement activation of AAV from animal studies.
Platelets counts always elevated correlate with disease activity in active AAV. Platelets express receptors for C3a and C5a and could be induced by sC5b−9 to release α‑granules and microparticles. 41 In AAV, activated platelets triggered the alternative complement pathway activation, which is partially attributed to the thrombin‐PARs pathway. 42 The role of platelet and complement interactions in the pathogenesis of AAV requires further investigation to determine the novel therapies targeting the interactions between the platelet and complements.
ADVOCATE was a Phase III randomized trial that compared avacopan with a tapering schedule of prednisone in patients with AAV concurrently treated with immunosuppressive drugs. 43 The results turned out that avacopan was superior to prednisone tapered in terms of sustained remission at Week 52, which suggested that avcopan might have a better therapeutic effect for prednisone.
4. TARGET THERAPIES OF COMPLEMENT SYSTEM
The complement‐mediated disorders or so‐called “complementopathies” consist of complement‐mediated and complement‐associated diseases. The former include paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome, C3 nephropathy, and so on, among which the complement system plays a critical role in the pathogenesis. The complement‐associated diseases refer to a group of diseases that complement system dysfunction combined with other known causes resulted in the onset of the diseases, like AAV, SLE, and APS, even COVID‐19, and so on. As the increasing recognition of complement‐mediated disorders is widespread, precision medicine has been expanded in complement therapeutics (Figure 1).
Eculizumab is a recombinant fully humanized IgG2/IgG4 monoclonal antibody that binds to C5 and consequently, prevents the formation of the terminal complement complex. Eculizumab is the first approved complement inhibitor for PNH associated with sustained improvements in intravascular hemolysis, anemia, thrombotic events, transfusion independence, survival, and quality of life. 44 Now, it has been used to explore the application feasibility on other types of glomerulonephritis induced by complement dysfunction, including IgA nephropathy, Shigatoxin‐producing Escherichia coli and even for lupus nephritis. Eculizumab could sustain the suppression of complement protein C5 for approximately 2 weeks. Recently, four amino acid substitutions in the Fc regions of eculizumab resulted in ravulizumab. It showed enhanced endosomal dissociation of C5 and could be recycled to the circulation through the neonatal Fc receptor pathway. 45 Ravulizumab could completely inhibit C5 for approximately 8 weeks with side effects similar to eculizumab. So, it was approved by FDA in 2018 for PNH treatment.
Except for the monoclonal antibodies already in the market, many more were undergoing clinical trials. Crovalimab shows a long half‐life and can be injected subcutaneously, which is also effective in patients with C5 polymorphism. The Phase I/II COMPOSER trial demonstrated superior efficacy and safety data, enabling crovalimab to proceed to Phase III clinical trials. 46 Besides, ABP 959 is a proposed biosimilar monoclonal antibody to eculizumab. Tesidolumab, an all‐human antibody targeting C5, had also been studied in dry age‐related macular degeneration (AMD).
Lampalizumab is an antigen‐binding fragment of a humanized monoclonal antibody against complement factor D. Another monoclonal antibody targeted MASP‐2 termed Narsoplimab, is a humanized IgG4 monoclonal antibody that was explored in IgAN in Phase III study. 47
Compared with monoclonal antibodies, small molecular was easier to be recognized and absorbed by the lining of the intestine. Zilucoplan, a small (3.5 kDa), 15‐amino acid macrocyclic peptide, was a representative small molecular drug binding to C5 with high affinity and specificity. 48 Avacopan (CCX168), an orally administered, selective C5a receptor inhibitor, could replace oral glucocorticoids used in AAV. 49 Besides, zilucopan could bind to C5. Pegcetacoplan, AMY‐101, a peptidic inhibitor of C3 based on the third‐generation compstatin analog Cp40.
Some prodrugs or drugs could achieve gene therapy by binding to nucleic acid. Cemdisiran, an N‐acetylgalactosamine (GalNAc) conjugated RNA interference (RNAi) therapeutic, was currently under development for the treatment of complement‐mediated diseases by suppressing liver production of C5. 50 IONIS‐FB‐LRx was developed by ligand conjugate antisense technology. It could decrease the production of CFB with clinical trials ongoing in geographic atrophy. More and more complement targeted therapies are being discovered and evaluated for efficacy and safety in clinical trials (Figure 1).
5. CONCLUSION
Deficiency or overactivation of the complement system will induce severe autoimmune response and develop autoimmune diseases. It is of vital importance to elucidate the role of complement in the pathogenesis and progress of autoimmune diseases. As the understanding of complement‐mediated autoimmune diseases becomes more widespread, complement targeted therapies will shed a light on precision and personalized medicine. More clinical trials with complement inhibitors/modulators in complement‐mediated autoimmune diseases still need further investigation.
AUTHOR CONTRIBUTIONS
Changhao Jia and Ying Tan are the main contributors. Ying Tan designed the study and Minghui Zhao revised it.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
ETHICS STATEMENT
None.
ACKNOWLEDGEMENT
None.
Jia C, Tan Y, Zhao M. The complement system and autoimmune diseases. Chronic Dis Transl Med. 2022;8:184‐190. 10.1002/cdt3.24
Edited by Yi Cui
DATA AVAILABILITY STATEMENT
Data available on request.
REFERENCES
- 1. Dijkstra DJ, Joeloemsingh JV, Bajema IM, Trouw LA. Complement activation and regulation in rheumatic disease. Semin Immunol. 2019;45:101339. 10.1016/j.smim.2019.101339 [DOI] [PubMed] [Google Scholar]
- 2. Walport MJ. Complement. First of two parts. N Engl J Med. 2001;344:1058‐1066. 10.1056/NEJM200104123441506 [DOI] [PubMed] [Google Scholar]
- 3. Sacks S, Zhou W. New boundaries for complement in renal disease. J Am Soc Nephrol. 2008;19:1865‐1869. 10.1681/ASN.2007101121 [DOI] [PubMed] [Google Scholar]
- 4. Del Tordello E, Vacca I, Ram S, Rappuoli R, Serruto D. Neisseria meningitidis NalP cleaves human complement C3, facilitating degradation of C3b and survival in human serum. Proc Natl Acad Sci USA. 2014;111:427‐432. 10.1073/pnas.1321556111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Park SY, Shin YP, Kim CH, et al. Immune evasion of Enterococcus faecalis by an extracellular gelatinase that cleaves C3 and iC3b. J Immunol. 2008;181:6328‐6336. 10.4049/jimmunol.181.9.6328 [DOI] [PubMed] [Google Scholar]
- 6. Lynskey NN, Reglinski M, Calay D, et al. Multi‐functional mechanisms of immune evasion by the streptococcal complement inhibitor C5a peptidase. PLoS Pathog. 2017;13:e1006493. 10.1371/journal.ppat.1006493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mannes M, Dopler A, Zolk O, et al. Complement inhibition at the level of C3 or C5: mechanistic reasons for ongoing terminal pathway activity. Blood. 2021;137:443‐455. 10.1182/blood.2020005959 [DOI] [PubMed] [Google Scholar]
- 8. Yang XW, Tan Y, Yu F, Zhao MH. Combination of anti‐C1q and anti‐dsDNA antibodies is associated with higher renal disease activity and predicts renal prognosis of patients with lupus nephritis. Nephrol Dial Transplant. 2012;27(9):3552‐3559. 10.1093/ndt/gfs179 [DOI] [PubMed] [Google Scholar]
- 9. Vanhecke D, Roumenina LT, Wan H, Osthoff M, Schaller M, Trendelenburg M. Identification of a major linear C1q epitope allows detection of systemic lupus erythematosus anti‐C1q antibodies by a specific peptide‐based enzyme‐linked immunosorbent assay. Arthritis Rheum. 2012;64:3706‐3714. 10.1002/art.34605 [DOI] [PubMed] [Google Scholar]
- 10. Pang Y, Tan Y, Li Y, et al. Serum A08 C1q antibodies are associated with disease activity and prognosis in Chinese patients with lupus nephritis. Kidney Int. 2016;90:1357‐1367. 10.1016/j.kint.2016.08.010 [DOI] [PubMed] [Google Scholar]
- 11. Wu WJ, Tan Y, Liu XL, Yu F, Zhao MH. C1q A08 is a half‐cryptic epitope of anti‐C1q A08 antibodies in lupus nephritis and important for the activation of complement classical pathway. Front Immunol. 2020;11:848. 10.3389/fimmu.2020.00848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takahashi R, Tsutsumi A, Ohtani K, et al. Association of mannose binding lectin (MBL) gene polymorphism and serum MBL concentration with characteristics and progression of systemic lupus erythematosus. Ann Rheum Dis. 2005;64:311‐314. 10.1136/ard.2003.020172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mahto H, Pati A, Sahu SK, Sharma HP, Padhi A, Panda AK. Association of MBL‐2 gene polymorphisms with systemic lupus erythematosus: an updated meta‐analysis and trial sequential analysis. Lupus. 2020;29:1227‐1237. 10.1177/0961203320939156 [DOI] [PubMed] [Google Scholar]
- 14. Singh SS, Cheung RC, Wong JH, Ng TB. Mannose binding lectin: a potential biomarker for many human diseases. Curr Med Chem. 2016;23:3847‐3860. 10.2174/0929867323666160817162208 [DOI] [PubMed] [Google Scholar]
- 15. Pradhan V, Mahant G, Rajadhyaksha A, et al. A study on anti‐mannose binding lectin (anti‐MBL) antibodies and serum MBL levels in Indian systemic lupus erythematosus patients. Rheumatol Int. 2013;33:1193‐1199. 10.1007/s00296-012-2519-9 [DOI] [PubMed] [Google Scholar]
- 16. Buyon JP, Tamerius J, Ordorica S, Young B, Abramson SB. Activation of the alternative complement pathway accompanies disease flares in systemic lupus erythematosus during pregnancy. Arthritis Rheum. 1992;35:55‐61. 10.1002/art.1780350109 [DOI] [PubMed] [Google Scholar]
- 17. Zhao J, Wu H, Khosravi M, et al. Association of genetic variants in complement factor H and factor H‐related genes with systemic lupus erythematosus susceptibility. PLoS Genet. 2011;7:e1002079. 10.1371/journal.pgen.1002079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li LL, Tan Y, Song D, et al. Anti‐complement factor H autoantibodies may be protective in lupus nephritis. Clin Chim Acta. 2020;508:1‐8. 10.1016/j.cca.2020.05.005 [DOI] [PubMed] [Google Scholar]
- 19. Wu LH, Yu F, Tan Y, et al. Inclusion of renal vascular lesions in the 2003 ISN/RPS system for classifying lupus nephritis improves renal outcome predictions. Kidney Int. 2013;83:715‐723. 10.1038/ki.2012.409 [DOI] [PubMed] [Google Scholar]
- 20. Song D, Wu LH, Wang FM, et al. The spectrum of renal thrombotic microangiopathy in lupus nephritis. Arthritis Res Ther. 2013;15:R12. 10.1186/ar4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang FM, Song D, Pang Y, Song Y, Yu F, Zhao MH. The dysfunctions of complement factor H in lupus nephritis. Lupus. 2016;25:1328‐1340. 10.1177/0961203316642307 [DOI] [PubMed] [Google Scholar]
- 22. Ritis K, Doumas M, Mastellos D, et al. A novel C5a receptor‐tissue factor cross‐talk in neutrophils links innate immunity to coagulation pathways. J Immunol. 2006;177:4794‐4802. 10.4049/jimmunol.177.7.4794 [DOI] [PubMed] [Google Scholar]
- 23. Redecha P, Tilley R, Tencati M, et al. Tissue factor: a link between C5a and neutrophil activation in antiphospholipid antibody induced fetal injury. Blood. 2007;110:2423‐2431. 10.1182/blood-2007-01-070631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Redecha P, Franzke CW, Ruf W, Mackman N, Girardi G. Neutrophil activation by the tissue factor/factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome. J Clin Invest. 2008;118:3453‐3461. 10.1172/JCI36089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gropp K, Weber N, Reuter M, et al. β‐glycoprotein I, the major target in antiphospholipid syndrome, is a special human complement regulator. Blood. 2011;118:2774‐2783. 10.1182/blood-2011-02-339564 [DOI] [PubMed] [Google Scholar]
- 26. Holers VM, Girardi G, Mo L, et al. Complement C3 activation is required for antiphospholipid antibody‐induced fetal loss. J Exp Med. 2002;195:211‐220. 10.1084/jem.200116116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Girardi G, Berman J, Redecha P, et al. Complement C5a receptors and neutrophils mediate fetal injury in the antiphospholipid syndrome. J Clin Invest. 2003;112:1644‐1654. 10.1172/JCI18817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pierangeli SS, Girardi G, Vega‐Ostertag M, Liu X, Espinola RG, Salmon J. Requirement of activation of complement C3 and C5 for antiphospholipid antibody‐mediated thrombophilia. Arthritis Rheum. 2005;52:2120‐2124. 10.1002/art.21157 [DOI] [PubMed] [Google Scholar]
- 29. Thurman JM, Kraus DM, Girardi G, et al. A novel inhibitor of the alternative complement pathway prevents antiphospholipid antibody‐induced pregnancy loss in mice. Mol Immunol. 2005;42:87‐97. 10.1016/j.molimm.2004.07.043 [DOI] [PubMed] [Google Scholar]
- 30. Chaturvedi S, Braunstein EM, Yuan X, et al. Complement activity and complement regulatory gene mutations are associated with thrombosis in APS and CAPS. Blood. 2020;135:239‐251. 10.1182/blood.2019003863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chaturvedi S, Brodsky RA, McCrae KR. Complement in the pathophysiology of the antiphospholipid syndrome. Front Immunol. 2019;10:449. 10.3389/fimmu.2019.00449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Shapira I, Andrade D, Allen SL, Salmon JE. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719‐2723. 10.1002/art.34440 [DOI] [PubMed] [Google Scholar]
- 33. Zikos TA, Sokolove J, Ahuja N, Berube C. Eculizumab induces sustained remission in a patient with refractory primary catastrophic antiphospholipid syndrome. J Clin Rheumatol. 2015;21:311‐313. 10.1097/RHU.0000000000000290 [DOI] [PubMed] [Google Scholar]
- 34. Müller‐Calleja N, Ritter S, Hollerbach A, Falter T, Lackner KJ, Ruf W. Complement C5 but not C3 is expendable for tissue factor activation by cofactor‐independent antiphospholipid antibodies. Blood Adv. 2018;2:979‐986. 10.1182/bloodadvances.2018017095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65:1‐11. 10.1002/art.37715 [DOI] [PubMed] [Google Scholar]
- 36. Xiao H, Heeringa P, Hu P, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955‐963. 10.1172/JCI15918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xiao H, Dairaghi DJ, Powers JP, et al. C5a receptor (CD88) blockade protects against MPO‐ANCA GN. J Am Soc Nephrol. 2014;25:225‐231. 10.1681/ASN.2013020143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huugen D, van Esch A, Xiao H, et al. Inhibition of complement factor C5 protects against anti‐myeloperoxidase antibody‐mediated glomerulonephritis in mice. Kidney Int. 2007;71:646‐654. 10.1038/sj.ki.5002103 [DOI] [PubMed] [Google Scholar]
- 39. Xing GQ, Chen M, Liu G, et al. Complement activation is involved in renal damage in human antineutrophil cytoplasmic autoantibody associated pauci‐immune vasculitis. J Clin Immunol. 2009;29:282‐291. 10.1007/s10875-008-9268-2 [DOI] [PubMed] [Google Scholar]
- 40. Yuan J, Gou SJ, Huang J, Hao J, Chen M, Zhao MH. C5a and its receptors in human anti‐neutrophil cytoplasmic antibody (ANCA)‐associated vasculitis. Arthritis Res Ther. 2012;14:R140. 10.1186/ar3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sims PJ, Wiedmer T. Induction of cellular procoagulant activity by the membrane attack complex of complement. Semin Cell Biol. 1995;6:275‐282. 10.1006/scel.1995.0037 [DOI] [PubMed] [Google Scholar]
- 42. Miao D, Li DY, Chen M, Zhao MH. Platelets are activated in ANCA‐associated vasculitis via thrombin‐PARs pathway and can activate the alternative complement pathway. Arthritis Res Ther. 2017;19:252. 10.1186/s13075-017-1458-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tesar V, Hruskova Z. Avacopan in the treatment of ANCA‐associated vasculitis. Expert Opin Investig Drugs. 2018;27:491‐496. 10.1080/13543784.2018.1472234 [DOI] [PubMed] [Google Scholar]
- 44. Kulasekararaj AG, Hill A, Rottinghaus ST, et al. Ravulizumab (ALXN1210) vs eculizumab in C5‐inhibitor‐experienced adult patients with PNH: the 302 study. Blood. 2019;133:540‐549. 10.1182/blood-2018-09-876805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sheridan D, Yu ZX, Zhang Y, et al. Design and preclinical characterization of ALXN1210: a novel anti‐C5 antibody with extended duration of action. PLoS One. 2018;13:e0195909. 10.1371/journal.pone.0195909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Röth A, Nishimura JI, Nagy Z, et al. The complement C5 inhibitor crovalimab in paroxysmal nocturnal hemoglobinuria. Blood. 2020;135:912‐920. 10.1182/blood.2019003399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lafayette RA, Rovin BH, Reich HN, Tumlin JA, Floege J, Barratt J. Safety, tolerability and efficacy of narsoplimab, a novel MASP‐2 inhibitor for the treatment of IgA nephropathy. Kidney Int Rep. 2020;5:2032‐2041. 10.1016/j.ekir.2020.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Howard JE, Jr. , Nowak RJ, Wolfe GI, et al. Clinical effects of the self‐administered subcutaneous complement inhibitor zilucoplan in patients with moderate to severe generalized myasthenia gravis: results of a phase 2 randomized, double‐blind, placebo‐controlled, multicenter clinical trial. JAMA Neurol. 2020;77:582‐592. 10.1001/jamaneurol.2019.5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jayne DRW, Bruchfeld AN, Harper L, et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA‐associated vasculitis. J Am Soc Nephrol. 2017;28:2756‐2767. 10.1681/ASN.2016111179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Badri P, Jiang X, Borodovsky A, et al. Pharmacokinetic and pharmacodynamic properties of cemdisiran, an RNAi therapeutic targeting complement component 5, in healthy subjects and patients with paroxysmal nocturnal hemoglobinuria. Clin Pharmacokinet. 2021;60:365‐378. 10.1007/s40262-020-00940-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request.