

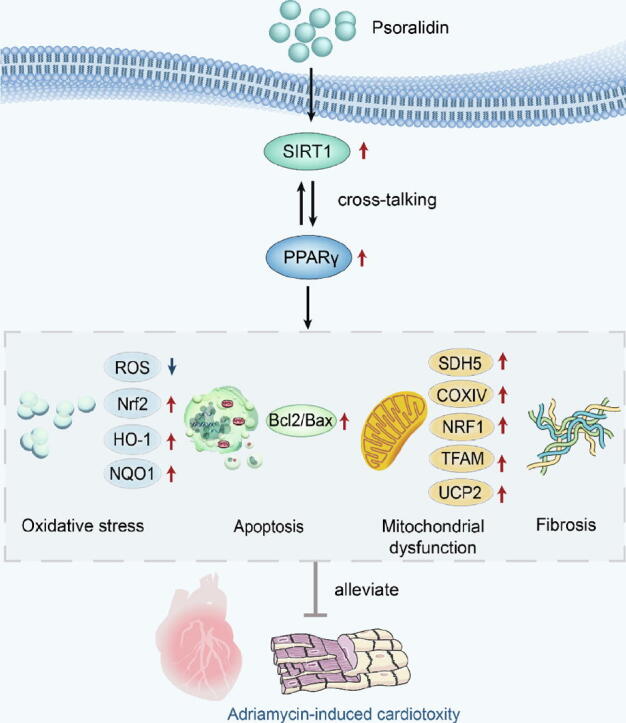
Graphical abstract
Proposed scheme depicting the mechanisms by which PSO protects against ADR-induced cardiotoxicity by activating the SIRT1/PPARγ signaling pathway.
Keywords: Adriamycin, Psoralidin, Cardiotoxicity, SIRT1, PPARγ
Research highlights
-
•
The beneficial effect of PSO on Adriamycin (ADR)-induced cardiotoxicity, which is manifested in amelioration of mitochondrial dysfunction, myocardial fibrosis, oxidative stress, and apoptosis
-
•
PSO protects ADR-induced cardiotoxicity via regulating SIRT1/PPARγ signaling pathway
-
•
PSO is proved to be a potential cardioprotective drug candidate to alleviate ADR-induced cardiotoxicity in clinical and amplify the application of ADR in oncotherapy.
Abstract
Introduction
Adriamycin (ADR) is an efficient and common broad-spectrum anticancer drug. However, the cumulative and dose-dependent toxicity induced by ADR severely limits its application in the clinic. Previous studies found that psoralidin (PSO) exhibits remarkable therapeutic effects against multiple cancers.
Objectives
The aim of this study was to determine if PSO has beneficial effects on ADR-induced cardiotoxicity and to investigate the underlying mechanisms.
Methods
ADR-induced cardiotoxicity models were established in BALB/c mice and HL-1 cardiomyocytes. A series of experimental methods were used to evaluate the effects of PSO on cardiac function indicators, blood biochemical parameters, histopathology, oxidative stress, apoptosis, mitochondrial function, fibrosis, and SIRT1/PPARγ signaling.
Results
PSO significantly improved cardiac function indicators, blood biochemical parameters, and mitochondrial function and reduced the degree of myocardial fibrosis, oxidative stress, and apoptosis in ADR-injured mice. PSO significantly increased cell viability, inhibited the release of LDH, reduced oxidative stress and apoptosis, and improved mitochondrial function in ADR-injured HL-1 cells. Moreover, we also demonstrated there was cross-talk between SIRT1 and PPARγ, as shown by SIRT1 siRNA significantly decreasing the expression of PPARγ and GW9662 (a PPARγ antagonist), which remarkably reduced the expression of SIRT1.
Conclusion
In summary, this study proved for the first time the beneficial effect of PSO on ADR-induced cardiotoxicity through activation of the SIRT1/PPARγ signaling pathway. Therefore, these findings may favor PSO as a potential cardioprotective drug candidate to alleviate ADR-induced cardiotoxicity in the clinic and improve the application of ADR in oncotherapy.
Introduction
Adriamycin (ADR), an anthracycline drug, is one of the most common and effective broad-spectrum antitumor agents used to treat malignancies, such as prostate cancer, breast cancers and childhood leukemia [1]. However, ADR-induced cumulative and dose-dependent toxicity is harmful to noncancerous tissues, especially in the myocardium, where ADR causes irreversible myocardial damage, which can result in dilated cardiomyopathy, severely limiting its clinical application [2]. Studies have found that cardiotoxicity caused by ADR is related to multiple mechanisms, such as oxidative stress, apoptosis, mitochondrial dysfunction and autophagy [3], [4]. Dexrazoxane is the only cardioprotective drug approved by the European Medicines Agency and Food and Drug Administration against anthracycline cardiotoxicity, but it increases the risk of secondary malignant neoplasms [1], [5]. However, the reports from various studies are controversial [6], [7]. Therefore, new avenues of exploration are needed to develop better pharmacotherapies and mechanisms to prevent ADR-induced cardiotoxicity.
Psoralidin (PSO), a natural phenolic coumarin (Fig. 1a) isolated from the seeds of the medicinal plant Psoralea corylifolia L., exhibits multiple biological activities, including antioxidant, antibacterial, antidepressant, anti-inflammatory, and antiallergic activities [8]. PSO is also a potent anticancer agent that inhibits cancer progression and metastasis [8]. Our previous study found that bakuchiol (BAK), another active component of Psoralea corylifolia L., can significantly alleviate ADR-induced H9c2 cell injury via activation of AMPK/PGC-1α [9]. However, whether PSO can produce beneficial effects on ADR-induced myocardial injury has not yet been elucidated.
Fig. 1.

The effects of PSO on the survival rate, body weight, and cardiac function indicators in ADR-injured mice. (a) The structure of PSO. (b) A schematic diagram of the experimental group and protocol. (c) Ten-day survival analysis, n = 12. (d) 6 days weight analysis, n = 6. (e) Blood biochemical parameters, n = 6. (f) Representative echocardiography images of the long axis, n = 6. (g) Statistical graph of stroke volume (SV) and cardiac output (CO). (h) Representative echocardiography images of the short axis, n = 6. (i) Statistical graph of SV and CO. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the control group or the ADR group; ns, nonsignificant.
Silent information regulator 1 (SIRT1) is a histone deacetylase whose activity is dependent on nicotinamide adenine dinucleotide [10]. A previous study suggested that the upregulation of SIRT1 alleviated ADR-induced cardiotoxicity in vivo and was related to the regulation of apoptosis and oxidative stress [11]. Peroxisome proliferator-activated receptor γ (PPARγ) is a member of the nuclear hormone receptor superfamily and has been shown to play a key role in cardiovascular diseases [12]. The activation of PPARγ has been demonstrated to alleviate ADR-induced cardiotoxicity, which is associated with a reduction in oxidative damage and inflammatory factors in cardiomyocytes [13]. Furthermore, the interactions between SIRT1 and PPARγ are complex and are essential for physiological and pathological function regulation in the heart [14]. In view of these results, the SIRT1/PPARγ signaling pathway has become an important target for the prevention of ADR-induced cardiotoxicity. Therefore, this study aimed to clarify the effect of PSO on the cardiotoxicity of ADR and to examine its potential mechanism, which may provide a novel strategy for the treatment of ADR-induced cardiotoxicity.
Materials and methods
Animals
Male BALB/c mice (22–25 g), 8–10 weeks old, were obtained from the animal center of PLA Air Force Military Medical University (Xi’an, Shaanxi, China). All animal experiments were conducted in accordance with the guidelines of Animal Care and Use Committees at Northwest University (Approval no. 2019018, Xi’an, Shaanxi, China) and were in compliance with the Guidelines for the Care and Use of Laboratory Animals (NIH Publication No. 85–23, revised 2011). All mice had free access to food and water and were kept on a 12 h light/12 h dark cycle. After ADR (Macklin Biochemical Technology Co., Ltd, Shanghai, China) treatment, cardiac function was assessed by echocardiography. Subsequently, blood biochemical parameters were detected by an automatic blood biochemical analyzer. Mice were euthanized at the endpoint, and their heart tissues were removed for histological and molecular biological detection.
Cardiotoxicity induced by ADR
The experiment was performed two weeks after the mice had acclimatized to the environment. To determine the optimum protective effects of PSO (Chenguang Biological Technology Co., Ltd., Baoji, Shaanxi, China) on ADR (Macklin Biochemical Technology Co., Ltd., Shanghai, China)-injured mice, PSO (12.5, 25, and 50 mg/kg, i.p.), dissolved in dimethyl sulfoxide (DMSO), was injected every two days three times in total. Then, we established an ADR-induced cardiotoxicity model. Mice were treated with ADR (10 mg/kg, i.p.) dissolved in saline on the day after the completion of the PSO treatment (on the 6th day) [15]. The control group received only DMSO and saline. Five days after ADR administration (on the 11th day), cardiac function was assessed by echocardiography. Subsequently, blood biochemical parameters were detected by an automatic blood biochemical analyzer (Fig. 1b). Mice were euthanized at the endpoint, and their heart tissues were removed for histological and molecular biological detection.
Echocardiography evaluation
The main role of the heart in the circulatory system is to pump blood to meet the needs of the body's metabolism. It goes without saying that the amount of blood output by the heart is a basic indicator of heart function. The stroke volume (SV) is the volume of blood ejected by the left ventricle during systole, and the cardiac output (CO) corresponds to this volume multiplied by the heart frequency to obtain the ejected volume per minute. Five days after ADR administration, an animal-specific instrument (VisualSonics Vevo3100, VisualSonics, Toronto, ON, Canada) was used for transthoracic echocardiography. Mice were anaesthetized with 3% isoflurane and 1 L/min 100% oxygen in an induction chamber for 1–2 min. Next, the mice were laid supine on a warm platform and kept anaesthetized by 2% isoflurane until they lost their body-righting reflex. Then, a series of M-mode images at the level of papillary muscles were obtained. SV = LVEDV-LVESV (left ventricular end-diastolic volume - left ventricular end-systolic volume) and CO = SV × heart rate ÷ 1000 were measured using Vevo LAB 3.0.0. Notably, all measurements were based on three consecutive cardiac cycles.
Detection of blood biochemical parameters
Blood biochemistry is an unstable biochemical system that can reflect the state of the body and its changes under the influence of internal and external factors. There are many common blood biochemistry parameters, including liver and kidney function tests, blood lipid tests, electrolyte tests, and myocardial enzyme tests. Thus, 150 μL serum was isolated from the rest of the whole blood by centrifugation at 3000 rpm for 10 min. The levels of creatine kinase (CK), lactate dehydrogenase (LDH), aspartate aminotransferase (AST), albumin (ALB), and blood urea nitrogen (BUN) were detected by an automatic blood biochemical analyzer (XinRui Technology Co., Ltd, XR210, Zhongshan, Guangdong, China).
Histological evaluation
To analyze the histopathological changes in the cardiac tissue, the heart was excised, and one part of the myocardium was fixed overnight in 4% paraformaldehyde, embedded in paraffin, and dehydrated in an ascending series of ethanol (70, 80, 90, and 100%). The tissue samples were embedded in paraffin wax and cut into 4–5 μm thick sections. The sections were mounted on standard glass slides and stained with hematoxylin and eosin (H&E) for 2 min before histological examination. Images of the stained sections were obtained using a light microscope (EVOSM5000, Thermo Fisher Scientific, Carlsbad, CA, USA). The degree of myocardial fibrosis was examined by Masson staining (Solarbio, Co., Ltd., Beijing, China) and Sirius red staining (Servicebio, Co., Ltd., Wuhan, Hubei, China). Moreover, dihydroethidine (DHE) staining was used to detect the production of reactive oxygen species (ROS) in the myocardium (Beyotime Biotechnology, Shanghai, China). Finally, the DHE fluorescence intensity was quantified using ImageJ 5.0 software.
Cell culture, treatment, and establishment of an ADR-induced cardiotoxicity model
HL-1 cells (purchased from ATCC) were maintained in DMEM containing 10% fetal bovine serum and incubated at 37 °C under 5% CO2. The medium was renewed every 3 days. The specific operation steps of the ADR-induced cardiotoxicity model were as follows. First, HL-1 cells were seeded in 60 mm culture dishes and allowed to grow to approximately 60–70% confluence. After replacing the medium with fresh serum-free medium, ADR was given at gradient concentrations (1, 1.5, 2, and 2.5 μM) for 24 h. Then, a MUSE cell analyzer was used to detect the cell viability (Merck KGaA, Darmstadt, Germany).
To explore the protective effects of PSO on ADR-injured HL-1 cells or the effects of SIRT1 siRNA and GW9662 (GW, a PPARγ antagonist) (Aladdin Reagents Co., Ltd., Shanghai, China), cells were seeded in 60 mm culture dishes or 6-well plates as appropriate. HL-1 cells were exposed to various concentrations of PSO (10, 20, 50, or 100 μM) for 24 h to evaluate its toxic effect. Thereafter, HL-1 cells were treated with SIRT1 siRNA for 24 h and then treated with PSO for 3 h before exposure to ADR (unless otherwise indicated). Then, HL-1 cells were exposed to various concentrations of GW (10, 20, 30, 40, or 50 μM) for 12 h to evaluate its toxic effect. Cells were treated with GW for different times (3, 6, and 12 h) and then treated with PSO (3 h) before exposure to ADR. Finally, the cells were harvested for further analysis.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from HL-1 cells using the TRIzolTM total RNA Extraction Kit (TAKARA Bio Inc. Kusatsu, Shiga, Japan), and reverse transcription was performed using Prime Script RT Master Mix (TAKARA bio inc. Kusatsu, Shiga, Japan). Then, SIRT1, PGC-1α, PPARγ, Bcl2, and Bax mRNA levels were detected using quantitative real-time reverse transcriptase PCR analysis with SYBR Premix Ex Taq as shown in Table 1 (Hunan Accurate Biotechnology Co., Ltd., Changsha, Hunan, China). The reaction conditions were as follows: (1) 95 °C for 10 min, (2) 40 cycles of 95 °C for 5 s and 60 °C for 30 s, (3) 94 °C for 30 s, 60 °C for 90 s, and 94 °C for 10 s. The expression levels of the examined transcripts were compared to that of β-actin and normalized to the mean value of the controls.
Table 1.
Sequence of primers used in PCR amplification.
| Gene | Sequence | |
|---|---|---|
| SIRT1 | Forward | GTTACTGCCACAGGAACTAGAGG |
| Reverse | GGAGCAGATTAGTAAGCGGCTTG | |
| PGC-1α | Forward | TCTGGGTGGATTGAAGTGG |
| Reverse | GGTCGCTACACCACTTCAATC | |
| PPARγ | Forward | GTGATGGAAGACCACTCGC |
| Reverse | CAAAGGAATGCGAGTGGTC | |
| Bcl2 | Forward | CCTGTGGATGACTGAGTACCTG |
| Reverse | AGCCAGGAGAAATCAAACAGAGG | |
| Bax | Forward | AGGATGCGTCCACCAAGAAGC |
| Reverse | TCCGTGTCCACGTCAGCAATCA |
Western blotting
Isolated heart tissues and cells were homogenized in RIPA buffer containing protease and phosphatase inhibitors (Beyotime Biotechnology, Shanghai, China). Thirty-fifty μg of total protein extract was separated on SDS–PAGE (8% or 10%) and transferred onto PVDF membranes (Millipore, Billerica, MA, USA), which were blocked with 5% skim milk and then incubated with primary antibodies at 4 °C for 24 h. In this study, the primary antibodies were as follows: anti-SIRT1 (1:1000, bs-0921R, Bioss Biotechnology Co., Ltd., Beijing, China), anti-PGC-1α (1:1000, GB11912, Servicebio, Wuhan, Hubei, China), anti-UCP2 (1:1000, GB11377, Servicebio, Wuhan, Hubei, China), anti-PPARγ (1:1000, sc-271392, Santa Cruz Biotechnology, Dallas, TX, USA), anti-Bcl2 (1:1000, BA0412, Boster Biological Technology Co., Ltd., CA, USA), anti-Nrf2 (1:1000, PB9290, Boster Biological Technology Co., Ltd., CA, USA), anti-Bax (1:1000, 2772s, Cell Signaling Technology, Inc, Danvers, Massachusetts, USA), anti-NQO1 (1:1000, sc-393736, Santa Cruz Biotechnology, Dallas, TX, USA), anti-HO-1 (1:1000, ab189491, Abcam, Cambridge, UK), anti-NRF1 (1:1000, 46743s, Cell Signaling Technology, Inc, Danvers, Massachusetts, USA), anti-TFAM (1:1000, sc-166965, Santa Cruz Biotechnology, Dallas, TX, USA), anti-SDH5 (1:000, #45849, Cell Signaling Technology, Inc., Danvers, Massachusetts, USA), anti-COXIV (1:000, #3E11, Cell Signaling Technology, Inc., Danvers, Massachusetts, USA), anti-β-actin (1:2000, GB11001, Servicebio, Wuhan, Hubei, China), and anti-GAPDH (1:2000, GB11002, Servicebio, Wuhan, Hubei, China). Then, the membranes were washed three times with TBST and incubated with the appropriate secondary antibodies (goat anti-mouse IgG, 1:5000, 7076s, Cell Signaling Technology, Inc., Danvers, Massachusetts, USA; goat anti-rabbit IgG, 1:5000, BA1054, Boster Biological Technology Co., Ltd., CA, USA) at room temperature for 1 h. Subsequently, the fluorescence signal was used to visualize the proteins on the membrane, and a MiNiChemi610 imaging system was used for detection (Sagecreation Co., Ltd., Beijing, China). Finally, the signal was quantified by ImageJ 5.0 software (National Institutes of Health, Bethesda, MD, USA).
Cell viability and intracellular ROS generation assessment
HL-1 cells were digested with trypsin and suspended in PBS, and then 100 μL of the cell suspension was collected. Subsequently, the cells were mixed with MuseTM Count & Viability Regent for 5 min in dark conditions and then tested on a Muse cell analyzer. ROS generation in the HL-1 cells was examined by 2′,7′-dichlorofluorescin diacetate (DCF-DA, Beyotime, Shanghai, China) staining at 37 °C for 20 min in the dark. Then, the cells were washed three times with PBS. Fluorescence images were obtained with a confocal microscope (EVOSM5000, Thermo Fisher Scientific, Carlsbad, CA, USA), and the fluorescent signal was quantified using Image J 5.0 Software.
LDH release and apoptosis assessment
Then, 500 μL supernatant from the HL-1 cells was collected, and the level of LDH was detected by an LDH detection kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, Jiangsu, China). The Muse Cell Analyzer was used following the manufacturer’s recommendations. In short, the treated cells were collected and stained under dark conditions using a Muse™ Annexin V & Dead Cell Kit. Live cells, early (and late) apoptotic cells, and dead cells were tested. Under standard conditions, untreated populations were used to determine the baseline levels of apoptosis and the number of dead cells. There were three repetitions for each experiment.
SIRT1 knockdown by small interfering RNA (siRNA)
The optimal sequence and concentration of siRNA were selected by cell viability and mRNA and protein expression. The sequences of siRNA targeting murine SIRT1-1269 (sense, 5′-GCACUAAUUCCAAGUUCUATT-3′; antisense, 5′-UAGAACUUGGAAUUAGUGCTT-3′) were synthesized by Sangon Biotechnology (Shanghai, China). The siRNA was used to knockdown SIRT1 in HL-1 cells. For transfection, cells were seeded in 60 mm culture dishes to grow to approximately 60–70% confluence. As recommended by the manufacturer, Lipofectamine 2000 was used to transfect cells with siRNA molecules targeting SIRT1 (Invitrogen, Thermo Fisher Scientific, Carlsbad, CA, USA) for at least 20 min in medium without antibiotics, and then the cells were incubated for 24 h. The knockdown efficiency was confirmed by qRT-PCR.
Statistical analysis
Data were analyzed with GraphPad Prism 9.0.0 (GraphPad Software Inc., San Diego, CA, USA). The data conformed to a normal distribution. All values are presented as the mean ± standard deviation (SD). For the analysis of experimental data, a t-test was used to compare any two groups, and one-way ANOVA was used for comparing more than two groups.
Results
The effects of PSO on survival rate, body weight, blood biochemical parameters, and cardiac function indicators in ADR-injured mice
First, the survival rate of ADR-injured mice pretreated with PSO was investigated. The results showed that PSO significantly increased the survival rate in ADR-injured mice (Fig. 1c, compared with the ADR group, P < 0.05). The protective effect was most obvious with 25 mg/kg PSO, so this concentration of PSO was adopted for further study. Then, the body weight of the mice was observed daily. ADR injury remarkably decreased the body weight of the mice; however, pretreatment with PSO significantly increased the body weight of ADR-injured mice (compared with the ADR group, Fig. 1d, P < 0.05). ADR injury significantly increased the levels of blood biochemical parameters, such as LDH, CK, AST, and BUN, and decreased the levels of ALB. However, PSO pretreatment markedly reversed these alterations, except for serum LDH levels (compared with the ADR group, Fig. 1e, P < 0.05). Cardiac function indicators were detected by echocardiography. As shown in Fig. 1f-i, ADR resulted in severe cardiac dysfunction, as demonstrated by decreased SV and CO. However, PSO pretreatment restored cardiac dysfunction induced by ADR (P < 0.05). In addition, H&E staining showed irregular ADR-injured cell structures, sparse muscle bundles, interstitial edema, and a large number of muscle fiber breaks. Consistently, PSO pretreatment remarkably alleviated the damage to the myocardium morphology caused by ADR (Fig. 2a).
Fig. 2.

The effects of PSO on fibrosis, oxidative stress, apoptosis, and mitochondrial function in ADR-injured mice. (a) H&E staining. (b) Masson staining. (c) Sirius red staining. (d) DHE staining. (e) Statistical graph of DHE staining. (f) Representative images of SIRT1, PPARγ, PGC-1α, Bcl2, Bax, Nrf2, NQO1, HO-1, SDH5, and COXIV detected by western blots. (g) Quantitative analysis of western blots normalized to GAPDH. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the control group or the ADR group. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The effects of PSO on fibrosis, oxidative stress, apoptosis, and mitochondrial function in ADR-injured mice
To monitor the effects of PSO on fibrosis, Masson staining and Sirius red staining assays were performed. Obvious collagen fiber deposition in the myocardium was observed in ADR-injured mice, and PSO pretreatment alleviated the degree of myocardial fibrosis (Fig. 2b and c). Using in situ DHE staining, we found that ADR increased intracellular ROS production and that PSO remarkably reversed this effect (Fig. 2d and e, P < 0.05). Western blotting was used to detect the effects of PSO on the antioxidant-related proteins Nrf2, HO-1 and NQO1. As shown in Fig. 2f and g, ADR injury downregulated the expression of Nrf2, NQO1, and upregulated the expression of HO-1, whereas PSO pretreatment upregulated the expression of Nrf2, HO-1, and NQO1 (compared with the ADR group, P < 0.05). Regarding apoptosis, ADR injury decreased Bcl2 (an antiapoptotic protein) expression and increased Bax (a proapoptotic protein) expression. However, PSO pretreatment reversed these effects (Fig. 2f and g, P < 0.05). Moreover, PSO pretreatment significantly improved mitochondrial function, as demonstrated by its upregulating the expression of SDH5 and COXIV (Fig. 2f and g, compared with the ADR group, P < 0.05).
Toxic and protective effects of PSO on murine cardiomyocytes
Cell viability assays showed that 2 μM ADR reduced cell viability to approximately 50%, so this concentration was used for further experiments (Fig. 3a and b). Then, the toxic effects of PSO were assayed. HL-1 cells were exposed to different concentrations of PSO (10, 20, 50, or 100 μM) for 24 h. Notably, the concentration of PSO was equal to or less than 50 μM, which did not decrease the cell viability or change the cell morphology (Fig. 3c and d). As shown in Fig. 3e and f, the levels of SIRT1, PGC-1α, PPARγ, Bcl2/Bax, HO-1, Nrf2, UCP2, NRF1, NQO1, and TFAM were upregulated by PSO pretreatment (compared with the control group, P < 0.05).
Fig. 3.

Toxic and protective effects of PSO on murine cardiomyocytes. (a) An ADR-induced cardiotoxicity model was constructed in HL-1 cells. (b) Statistical graph of cell viability. (c) Toxicity exploration of PSO on HL-1 cells. (d) Statistical graph of cell viability. (e) Representative images of SIRT1, PPARγ, PGC-1α, Bcl2, Bax, Nrf2, NQO1, HO-1, NRF1, TFAM, and UCP2 detected by western blot. (f) Quantitative analysis of western blots normalized to β-actin. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the control group; ns, nonsignificant.
PSO protected cardiomyocytes against ADR injury
According to the toxicity test, 5, 10, and 20 μM PSO were selected for further study. The results showed that ADR injury decreased cell viability and increased the apoptotic rate, intracellular ROS generation, and LDH release (Fig. 4a-g, compared with the control group, P < 0.05). Pretreatment with PSO, especially at 20 μM, had the most significant protective effects, which manifested as increased cell viability and a decreased apoptotic rate, intracellular ROS generation, and LDH release (Fig. 4a-g, compared with the ADR group, P < 0.05). Therefore, this concentration of PSO (20 μM) was used for further study.
Fig. 4.

The effects of PSO on ADR-injured cardiomyocytes. (a) Cell viability of ADR-injured HL-1 cells. (b) Statistical graph of cell viability. (c) Apoptotic analysis of ADR-injured HL-1 cells. (d) Apoptotic rates of ADR-injured HL-1 cells. (e) Intracellular ROS of ADR-injured HL-1 cells. (f) Statistical graph of intracellular ROS. (g) LDH level of ADR-injued HL-1 cells. (h) Representative images of PPARγ, SIRT1, PGC-1α, Bcl2, Bax, Nrf2, NQO1, HO-1, NRF1, TFAM, and UCP2 detected by western blots. (i) Quantitative analysis of western blots normalized to β-actin. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the control group or ADR group; ns, nonsignificant.
The effects of PSO on apoptosis, oxidative stress and mitochondrial function in ADR-injured cardiomyocytes
HL-1 cells were treated with the specified PSO concentration (20 μM), and then the cells were collected. Bcl2/Bax, Nrf2, NQO1, HO-1, NRF1, TFAM, and UCP2 levels were detected by western blotting. Compared with the control group, ADR injury decreased the levels of Bcl2/Bax, Nrf2, NRF1, TFAM, UCP2, and increased the level of HO-1. However, PSO pretreatment upregulated the expression levels of these proteins (Fig. 4h and i, P < 0.05).
The effects of PSO on SIRT1/PPARγ signaling in ADR-injured cardiomyocytes and the myocardium
To explore the protective mechanisms of PSO in cardiomyocytes and myocardium injured by ADR, the key molecules of SIRT1/PPARγ signaling were detected. In normal HL-1 cells, PSO pretreatment upregulated SIRT1, PGC-1α, and PPARγ expression (Fig. 3e and f, compared with the control group, P < 0.05). Moreover, ADR injury downregulated the expression of SIRT1, PGC-1α, and PPARγ in HL-1 cells and the myocardium (Figs. 2f, g, 4h, and i, compared with the control group, P < 0.05). However, pretreatment with PSO remarkably reversed the downregulation of SIRT1, PGC-1α, and PPARγ (Figs. 2f, g, 4h, and i, compared with the ADR group, P < 0.05).
The effects of SIRT1 siRNA on cell viability and SIRT1/PPARγ signaling in ADR-injured cardiomyocytes pretreated with PSO
To confirm the effects of SIRT1/PPARγ signaling in the process of PSO against ADR-injured HL-1 cells, SIRT1 was silenced by 3 siRNAs, and the most effective interference fragment, SIRT1 siRNA 1269, was selected (data not shown). First, SIRT1 siRNA treatment (100 pM, 24 h) showed no toxic effect on HL-1 cells but effectively decreased SIRT1 expression (Fig. 5a and b). The qRT-PCR assay demonstrated that SIRT1 siRNA significantly decreased SIRT1, PGC-1α, PPARγ, and Bcl2 mRNA expression but increased Bax mRNA expression in HL-1 cells (Fig. 5c, compared with the control siRNA group, P < 0.05). After being treated with SIRT1 siRNA for 24 h, HL-1 cells were pretreated with PSO and then exposed to ADR injury. SIRT1 siRNA prominently reversed the effect of PSO against ADR-injured HL-1 cell death (Fig. 5d and e, compared with the control siRNA + PSO + ADR group, P < 0.05). Additionally, compared with the control siRNA + PSO + ADR group, SIRT1 siRNA reversed the increase in SIRT1, PPARγ, Bcl2, Nrf2, NQO1, HO-1, and UCP2 in HL-1 cells pretreated with PSO (Fig. 5f and g, P < 0.05).
Fig. 5.

The effects of SIRT1 siRNA on cell viability and SIRT1/PPARγ signaling in ADR-injured cardiomyocytes treated with PSO. (a) Toxicity exploration of SIRT1 siRNA on HL-1 cells. (b) Statistical graph of cell viability. (c) qRT–PCR analysis of SIRT1, PPARγ, PGC-1α, Bcl2, and Bax mRNA by normalizing to β-actin. (d) Cell viability of ADR-injured HL-1 cells. (e) Statistical graph of cell viability. (f) Representative images of PPARγ, SIRT1, PGC-1α, Bcl2, Nrf2, NQO1, HO-1, and UCP2 detected by western blots. (g) Quantitative analysis of western blots normalized to β-actin. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the control siRNA group (a-c)vs. the control siRNA + ADR group or the control siRNA + PSO + ADR group (d-g); ns, nonsignificant.
The effects of GW on cell viability and SIRT1/PPARγ signaling in ADR-injured cardiomyocytes pretreated with PSO
The toxic effect of GW (10, 20, 30, 40, or 50 μM) was evaluated in HL-1 cells. We found that GW had no significant decrease in cell viability at concentrations of 40 μM and below, so 40 μM GW was used for further study (Fig. 6a and b, compared with the control group, P < 0.05). The mRNA levels of SIRT1, PGC-1α, and PPARγ were evaluated at different time points after GW treatment. The results showed that the expression of these molecules changed most significantly at 12 h after GW treatment, so this time point was adopted for subsequent experiments (compared with the control group, Fig. 6c P < 0.05). However, GW significantly reversed the effects of PSO against ADR-injured HL-1 cell death (compared with the control + PSO + ADR group, Fig. 6d and e, P < 0.05). In addition, PSO pretreatment upregulated the levels of PPARγ, PGC-1α, SIRT1, Bcl2, UCP2, NQO1, HO-1 and Nrf2 in ADR-injured HL-1 cells. However, GW reversed the upregulation of some proteins, including SIRT1, PPARγ, UCP2, and Nrf2. Unexpectedly, GW treatment did not reverse the levels of NQO1, HO-1, and Bcl2 and instead it upregulated the levels of these proteins (compared with the GW + PSO + ADR group, Fig. 6f and g, P < 0.05).
Fig. 6.

The effects of GW on cell viability and SIRT1/PPARγ signaling in ADR-injured cardiomyocytes treated with PSO. (a) Toxicity exploration of GW on HL-1 cells. (b) Statistical graph of cell viability. (c) qRT–PCR analysis of SIRT1, PPARγ, and PGC-1α mRNA by normalizing to β-actin. (d) Cell viability of ADR-injured HL-1 cells. (e) Statistical graph of cell viability. (f) Representative images of PPARγ, SIRT1, PGC-1α, Bcl2, Nrf2, NQO1, HO-1, and UCP2 detected by western blots. (g) Quantitative analysis of Western blots normalized to β-actin. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs. the control group (a-c)vs. the control + ADR group or the control + PSO + ADR group (d-g); ns, nonsignificant.
Discussion
ADR is an efficient and common broad-spectrum anticancer drug used to treat various cancers as a basic chemotherapy agent in clinical practice [1]. However, ADR is a double-edged sword, as it can induce acute and chronic myocardial injury during or after treatment [2]. According to previous reports, oxidative stress, apoptosis, fibrosis and mitochondrial function of cardiomyocytes play important roles in ADR induction cardiotoxicity [3], [4]. As a natural phenolic coumarin, PSO has been proven to have a significant therapeutic effect on many diseases, such as depression, osteoporosis, and cancer. [8]. A variety of biological activities, such as antioxidative, antibacterial, antidepressant, anti-inflammatory, and antiallergic activities, are responsible for its extensive therapeutic effects [8]. In this study, PSO exerted significant protective effects against ADR-induced cardiotoxicity by scavenging ROS, reducing apoptosis, alleviating fibrosis, and improving mitochondrial function. Moreover, SIRT1/PPARγ signaling also played a positive role in the process of PSO against ADR injury.
Previous studies have demonstrated that ADR-induced cardiotoxicity can be attributed to a variety of mechanisms, whereas increased cardiomyocyte apoptosis is the most extensively accepted mechanism [16]. Ghosh et al. confirmed that both excessive ROS production and ADR itself could induce apoptosis of cardiomyocytes, even causing dilated cardiomyopathy and heart failure [16]. Accumulating evidence suggests that PSO, in a dose-dependent manner, has marked antitumor activity, as demonstrated by its inhibiting proliferation and inducing apoptosis in various cancer cells [17], [18]. Anti-apoptotic Bcl2 and pro-apoptotic Bax play an important role in cell survival or death [19]. Consistent with these findings, increased apoptosis was observed in response to ADR injury. Importantly, this study showed that PSO significantly increased the level of Bcl2/Bax. These results suggested that PSO pretreatment remarkably reduced ADR-induced cardiotoxicity by inhibiting cardiomyocyte apoptosis.
Oxidative stress is a kind of cellular stress and damage caused by the imbalance between the production of ROS and antioxidant defense [20]. Excessive accumulation of ROS could cause dozens of diseases, including myocardial infarction, coronary heart disease, heart failure and so on [21], [22]. Oxidative stress is also an important factor in cardiotoxicity caused by ADR. NF-E2-related factor 2 (Nrf2) is an extremely critical transcription factor whose main role is to induce endogenous antioxidant enzymes to resist oxidative stress. Meanwhile, Nrf2 is regarded as a regulator that maintains redox homeostasis in the body [23]. Heme oxygenase-1 (HO-1) and NADPH:quinone oxidoreductase-1 (NQO1) have been proven to be downstream target genes of Nrf2 [24]. In this study, DHE staining showed that PSO significantly reduced ADR-induced excessive accumulation of ROS in cardiomyocytes. PSO conspicuously upregulated Nrf2, NQO1, and HO-1 levels. The above results indicated that PSO might inhibit oxidative stress and ultimately attenuate ADR-induced cardiotoxicity.
Myocardial fibrosis can cause significant negative remodeling of the myocardium, resulting in decreased myocardial contraction and diastolic tension and eventually heart failure [25]. Myocardial fibrosis can affect the conduction system of the heart and cause arrhythmia and even the occurrence of sudden cardiac death [26]. Early research has shown that intraperitoneal injection of ADR can lead to obvious myocardial fibrosis and suppress heart function in mice [27]. Moreover, resveratrol, as an activator of SIRT1, mitigates diastolic dysfunction by decreasing myocardial fibrosis in ADR-injured mice [28]. In this study, Masson staining and Sirius red staining were performed. Obvious collagen fiber deposition occurred in ADR-injured myocardium, and PSO pretreatment alleviated the collagen fiber deposition, which suggested that PSO pretreatment may decrease myocardial fibrosis in ADR-injured mice.
Mitochondria are the main organelles that produce ROS. Mitochondrial dysfunction stimulates the production of ROS, which in turn affects its metabolic function [29], [30]. Uncoupling protein 2 (UCP2), nuclear respirator factor 1 (NRF1) and mitochondrial transcription factor A (TFAM) are the main molecular conductors of the intergenomic regulation of mitochondrial biogenesis [31]. A recent study proved that the protein expression levels of PINK1 and Parkin are increased in human adult ventricular cardiomyocytes treated with ADR. Then, the increase results in PGC‐1α, NRF‐1, and TFAM inhibition and mitochondrial damage [32]. Moreover, melatonin can inhibit ADR-induced mitochondrial dysfunction, apoptosis, and oxidative stress by activating AMPK and upregulating PGC-1α with its downstream signals NRF1, TFAM, and UCP2 [33]. Cytochrome c oxidase subunit IV (COXIV) and succinate dehydrogenase 5 (SDH5) are mitochondrial proteins and main components of the respiratory chain [34]. Our study suggested that PSO significantly improved mitochondrial function, as evidenced by upregulating the expression of SDH5 and COXIV in ADR-injured mice. Moreover, PSO pretreatment increased the levels of NRF1, TFAM, and UCP2 in ADR-injured HL-1 cells. Notably, in addition to participating in oxidative stress, Nrf2 also participates in mitochondrial biogenesis in cardiomyocytes by activating HO-1 and NRF1 [35], [36]. Therefore, the dominant regulatory cross-talk between these molecules in the cardiotoxicity of ADR needs to be further investigated.
SIRT1 is a multifunctional adenine dinucleotide (NAD+) nicotinamide deacetylase protein that plays a key role in coordinating cell functions by deacetylating its substrate PGC-1α. Stimulate [37], [38]. Dihydromyricetin can inhibit the NLRP3 inflammasome by activating SIRT1, thereby alleviating ADR-induced cardiotoxicity [39]. Zhang et al. confirmed that oroxylin A can mitigate ADR-induced cardiotoxicity by activating SIRT1 signaling [40]. This evidence indicates that the activation of SIRT1 plays a positive role in ADR-induced cardiotoxicity. This study showed that PSO treatment significantly increased the ADR-induced reduction in SIRT1 and PGC-1α expression. SIRT1 siRNA also significantly reduced the mRNA levels of SIRT1, PGC-1α, PPARγ, and Bcl2 and increased the mRNA level of Bax. Moreover, SIRT1 siRNA remarkably reversed the role of PSO against cell death and upregulated SIRT1, PPARγ, Bcl2, Nrf2, HO-1, and UCP2 in ADR-injured HL-1 cells.
PPARγ is activated by ligand transcription factors and members of the nuclear hormone receptor superfamily [41]. PPARγ agonists and antagonists have been used in many pharmacological and pathological studies. Studies have shown that PPARγ plays an important role in regulating free radicals, anti-inflammatory drugs and immune responses [42], [43]. Moreover, knockout or inhibition of PPARγ in mice increased sensitivity to oxidative stress. The expression of PPARγ is reduced in myocardial ischemia–reperfusion and myocardial hypertrophy. Furthermore, the process is considered harmful to the body, accompanied by the production of ROS and the reduction of ATP [44], [45], [46]. In addition, Yan et al. found that piperine can reduce oxidative stress, inflammation and apoptosis in mice by activating PPARγ signaling and ultimately attenuating ADR-induced cardiotoxicity[47]. Consistent with the results described in the literature, our study found that PSO increased the expression of PPARγ in ADR-injured myocardium and cardiomyocytes. GW, as an inhibitor of PPARγ, did not affect cell viability within the safe range of concentrations. In addition, PSO upregulated the levels of PPARγ, PGC-1α, SIRT1, Bcl2, UCP2, NQO1, HO-1, and Nrf2 in ADR-injured HL-1 cells. However, GW reversed the upregulation of some proteins, including SIRT1, PPARγ, and Nrf2. Unexpectedly, GW treatment did not reverse the levels of NQO1, HO-1, and Bcl2 but upregulated the levels of these proteins. Therefore, we deduced that the possible reason is that GW9662, as a small molecule compound, may exert its biological function through other pathways rather than affecting the expression of these molecules by regulating PPARγ directly.
In addition, this study also has certain limitations. First, we used a clinical model of acute ADR cardiotoxicity, which is accordance with the results of the previously published research by Shioji et al. [48]. However, in clinical situations, ADR-induced cardiotoxicity can also occur many years after chemotherapy, particularly after treatment for cancer of the breast in young women [49]. The acute injury model of ADR was used in this study. The accumulation of ADR in the body can also lead to chronic injury. We will study the protective effect and mechanism of PSO on chronic myocardial injury caused by ADR in our future work.
Conclusion
In conclusion, this study demonstrates for the first time the beneficial effect of PSO on ADR-induced cardiotoxicity, which is manifested in the amelioration of mitochondrial dysfunction, myocardial fibrosis, oxidative stress, and apoptosis, mainly by activating the SIRT1/PPARγ signaling pathway. Notably, we also proved the cross-talk between SIRT1 and PPARγ, as evidenced by SIRT1 siRNA significantly decreasing the expression of PPARγ and GW remarkably reducing the expression of SIRT1 (shown in Graphical abstract). Therefore, these findings may favor PSO as a potential cardioprotective drug candidate to alleviate ADR-induced cardiotoxicity in the clinic and amplify the application of ADR in oncotherapy.
Funding
This work was supported by the National Natural Science Foundation of China (81660210, 81871607, 82070422, and 81600306), Natural Science Foundation of Shaanxi Province (2020JM-386 and 2018JM3042), Innovation Capability Strong Foundation Plan of Xi'an City (Medical Research Project, 21YXYJ0037), Key Research and Development Program of Shaanxi (2020ZDLSF04-03), and Major Research Projects of Xi'an Science and Technology Plan (201805104YX12SF38(2)).
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
Compliance with Ethics Requirements
This study was approved by the ethics committee of Northwest University. The informed consent was obtained from each participant. All animal experiment protocols were performed in accordance with the guidelines of Animal Care and Use Committees at Northwest University (Approval no. 2019018, Xi’an, Shaanxi, China) and were in compliance with the Guidelines for the Care and Use of Laboratory Animals (NIH Publication No.85-23, revised 2011). Extensive efforts were made to ensure minimal suffering of the animals used during the study.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2021.12.007.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Vejpongsa P., Yeh E.T.H. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J Am Coll Cardiol. 2014;64(9):938–945. doi: 10.1016/j.jacc.2014.06.1167. [DOI] [PubMed] [Google Scholar]
- 2.Singal P.K., Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339(13):900–905. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 3.Rochette L., Guenancia C., Gudjoncik A., Hachet O., Zeller M., Cottin Y., et al. Anthracyclines/trastuzumab: new aspects of cardiotoxicity and molecular mechanisms. Trends Pharmacol Sci. 2015;36(6):326–348. doi: 10.1016/j.tips.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 4.Zhang S., Liu X., Bawa-Khalfe T., Lu L.-S., Lyu Y.L., Liu L.F., et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. 2012;18(11):1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 5.Cvetković R.S., Scott L.J. Dexrazoxane: a review of its use for cardioprotection during anthracycline chemotherapy. Drugs. 2005;65(7):1005–1024. doi: 10.2165/00003495-200565070-00008. [DOI] [PubMed] [Google Scholar]
- 6.Sawicki K.T., Sala V., Prever L., Hirsch E., Ardehali H., Ghigo A. Preventing and treating anthracycline cardiotoxicity: new insights. Annu Rev Pharmacol Toxicol. 2021;61(1):309–332. doi: 10.1146/annurev-pharmtox-030620-104842. [DOI] [PubMed] [Google Scholar]
- 7.Barry E.V., Vrooman L.M., Dahlberg S.E., Neuberg D.S., Asselin B.L., Athale U.H., et al. Absence of secondary malignant neoplasms in children with high-risk acute lymphoblastic leukemia treated with dexrazoxane. J Clin Oncol. 2008;26(7):1106–1111. doi: 10.1200/JCO.2007.12.2481. [DOI] [PubMed] [Google Scholar]
- 8.Xin Z., Wu X., Yu Z., Shang J., Xu B., Jiang S., et al. Mechanisms explaining the efficacy of psoralidin in cancer and osteoporosis, a review. Pharmacol Res. 2019;147:104334. doi: 10.1016/j.phrs.2019.104334. [DOI] [PubMed] [Google Scholar]
- 9.Lu C., Han Y., Xin Z., Yan J., Li T., Han M. Protective effect and mechanism of bakchiol on adriamycin-inuced myocardial injury. J Northwest Univ (Nat Sci Ed) 2018;48:769–775. [Google Scholar]
- 10.Yang Y., Duan W., Li Y., Jin Z., Yan J., Yu S., et al. Novel role of silent information regulator 1 in myocardial ischemia. Circulation. 2013;128(20):2232–2240. doi: 10.1161/CIRCULATIONAHA.113.002480. [DOI] [PubMed] [Google Scholar]
- 11.Hu C., Zhang X., Song P., Yuan Y.-P., Kong C.-Y., Wu H.-M., et al. Meteorin-like protein attenuates doxorubicin-induced cardiotoxicity via activating cAMP/PKA/SIRT1 pathway. Redox Biol. 2020;37:101747. doi: 10.1016/j.redox.2020.101747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robinson E., Grieve D.J. Significance of peroxisome proliferator-activated receptors in the cardiovascular system in health and disease. Pharmacol Ther. 2009;122(3):246–263. doi: 10.1016/j.pharmthera.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 13.Pakravan G, Foroughmand AM, Peymani M, Ghaedi K. Downregulation of miR-130a, antagonized doxorubicin-induced cardiotoxicity via increasing the PPARγ expression in mESCs-derived cardiac cells. 2018;9:758. [DOI] [PMC free article] [PubMed]
- 14.Kalliora C., Kyriazis I.D., Oka S.I., Lieu M.J., Yue Y., Area-Gomez E. Dual peroxisome-proliferator-activated-receptor-α/γ activation inhibits SIRT1-PGC1α axis and causes cardiac dysfunction. JCI Insight. 2019;5 doi: 10.1172/jci.insight.129556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo D., Carter K.A., Razi A., Geng J., Shao S., Lin C., et al. Porphyrin-phospholipid liposomes with tunable leakiness. J Control Release. 2015;220:484–494. doi: 10.1016/j.jconrel.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh J., Das J., Manna P., Sil P.C. The protective role of arjunolic acid against doxorubicin induced intracellular ROS dependent JNK-p38 and p53-mediated cardiac apoptosis. Biomaterials. 2011;32(21):4857–4866. doi: 10.1016/j.biomaterials.2011.03.048. [DOI] [PubMed] [Google Scholar]
- 17.Yu B., Wang A.-H., Zhou K., Chai L.-J., Liu L.u. Molecular pathway of psoralidin-induced apoptosis in HepG2 cell line. Chin J Integr Med. 2019;25(10):757–762. doi: 10.1007/s11655-016-2251-5. [DOI] [PubMed] [Google Scholar]
- 18.Jin Z., Yan W., Jin H., Ge C., Xu Y. Psoralidin inhibits proliferation and enhances apoptosis of human esophageal carcinoma cells via NF-κB and PI3K/Akt signaling pathways. Oncol Lett. 2016;12:971–976. doi: 10.3892/ol.2016.4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dong J.W., Zhu H.F., Zhu W.Z., Ding H.L., Ma T.M., Zhou Z.N. Intermittent hypoxia attenuates ischemia/reperfusion induced apoptosis in cardiac myocytes via regulating Bcl-2/Bax expression. Cell Res. 2003;13(5):385–391. doi: 10.1038/sj.cr.7290184. [DOI] [PubMed] [Google Scholar]
- 20.Sies H., Jones D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol. 2020;21(7):363–383. doi: 10.1038/s41580-020-0230-3. [DOI] [PubMed] [Google Scholar]
- 21.Butterfield D.A., Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019;20(3):148–160. doi: 10.1038/s41583-019-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Münzel T., Gori T., Bruno R.M., Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur Heart J. 2010;31:2741–2748. doi: 10.1093/eurheartj/ehq396. [DOI] [PubMed] [Google Scholar]
- 23.Sies H., Berndt C., Jones D.P. Oxidative stress. Annu Rev Biochem. 2017;86(1):715–748. doi: 10.1146/annurev-biochem-061516-045037. [DOI] [PubMed] [Google Scholar]
- 24.Osama A., Zhang J., Yao J., Yao X., Fang J. Nrf2: a dark horse in Alzheimer's disease treatment. Ageing Res Rev. 2020;12:64. doi: 10.1016/j.arr.2020.101206. [DOI] [PubMed] [Google Scholar]
- 25.Chen S., Zhang Y., Lighthouse J.K., Mickelsen D.M., Wu J., Yao P. Novel role of cyclic nucleotide phosphodiesterase 10A in pathological cardiac remodeling and dysfunction. Circulation. 2020;141:217–233. doi: 10.1161/CIRCULATIONAHA.119.042178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zegard A., Okafor O., de Bono J., Kalla M., Lencioni M., Marshall H., et al. Myocardial fibrosis as a predictor of sudden death in patients with coronary artery disease. J Am Coll Cardiol. 2021;77(1):29–41. doi: 10.1016/j.jacc.2020.10.046. [DOI] [PubMed] [Google Scholar]
- 27.Hamed S., Barshack I., Luboshits G., Wexler D., Deutsch V., Keren G., et al. Erythropoietin improves myocardial performance in doxorubicin-induced cardiomyopathy. Eur Heart J. 2006;27:1876–1883. doi: 10.1093/eurheartj/ehl044. [DOI] [PubMed] [Google Scholar]
- 28.Cappetta D., Esposito G., Piegari E., Russo R., Ciuffreda L.P., Rivellino A., et al. SIRT1 activation attenuates diastolic dysfunction by reducing cardiac fibrosis in a model of anthracycline cardiomyopathy. Int J Cardiol. 2016;205:99–110. doi: 10.1016/j.ijcard.2015.12.008. [DOI] [PubMed] [Google Scholar]
- 29.Wallace K.B., Sardão V.A., Oliveira P.J. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ Res. 2020;126(7):926–941. doi: 10.1161/CIRCRESAHA.119.314681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahin E., Colla S., Liesa M., Moslehi J., Müller F.L., Guo M., et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470(7334):359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfanner N., Warscheid B., Wiedemann N. Mitochondrial proteins: from biogenesis to functional networks. Nat Rev Mol Cell Biol. 2019;20(5):267–284. doi: 10.1038/s41580-018-0092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osataphan N, Phrommintikul A, Chattipakorn SC, Chattipakorn N. Effects of doxorubicin-induced cardiotoxicity on cardiac mitochondrial dynamics and mitochondrial function: Insights for future interventions. 2020:6534–57. [DOI] [PMC free article] [PubMed]
- 33.Liu D., Ma Z., Di S., Yang Y., Yang J., Xu L., et al. AMPK/PGC1α activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic Biol Med. 2018;129:59–72. doi: 10.1016/j.freeradbiomed.2018.08.032. [DOI] [PubMed] [Google Scholar]
- 34.Khader A., Yang W.-L., Godwin A., Prince J.M., Nicastro J.M., Coppa G.F., et al. Sirtuin 1 stimulation attenuates ischemic liver injury and enhances mitochondrial recovery and autophagy. Crit Care Med. 2016;44(8):e651–e663. doi: 10.1097/CCM.0000000000001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paiva C.N., Feijó D.F., Dutra F.F., Carneiro V.C., Freitas G.B., Alves L.S., et al. Oxidative stress fuels Trypanosoma cruzi infection in mice. J Clin Invest. 2012;122(7):2531–2542. doi: 10.1172/JCI58525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li Q., Xiang Y., Chen Y.u., Tang Y., Zhang Y. Ginsenoside Rg1 protects cardiomyocytes against hypoxia/reoxygenation injury via activation of Nrf2/HO-1 signaling and inhibition of JNK. Cell Physiol Biochem. 2017;44(1):21–37. doi: 10.1159/000484578. [DOI] [PubMed] [Google Scholar]
- 37.Feng J., Yang Y., Zhou Y., Wang B., Xiong H., Fan C., et al. Bakuchiol attenuates myocardial ischemia reperfusion injury by maintaining mitochondrial function: the role of silent information regulator 1. Apoptosis. 2016;21(5):532–545. doi: 10.1007/s10495-016-1225-6. [DOI] [PubMed] [Google Scholar]
- 38.Hu M.Z., Zhou B., Mao H.Y., Sheng Q., Du B., Chen J.L. Exogenous hydrogen sulfide postconditioning protects isolated rat hearts from ischemia/reperfusion injury through Sirt1/PGC-1α signaling pathway. Int Heart J. 2016;57:477–482. doi: 10.1536/ihj.15-506. [DOI] [PubMed] [Google Scholar]
- 39.Sun Z., Lu W., Lin N.a., Lin H., Zhang J., Ni T., et al. Dihydromyricetin alleviates doxorubicin-induced cardiotoxicity by inhibiting NLRP3 inflammasome through activation of SIRT1. Biochem Pharmacol. 2020;175:113888. doi: 10.1016/j.bcp.2020.113888. [DOI] [PubMed] [Google Scholar]
- 40.Zhang WB, Zheng YF. Protective effects of oroxylin A against doxorubicin-induced cardiotoxicity via the activation of sirt1 in mice. 2021;2021:6610543. [DOI] [PMC free article] [PubMed]
- 41.Ahmadian M., Suh J.M., Hah N., Liddle C., Atkins A.R., Downes M., et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19(5):557–566. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown J.D., Plutzky J. Peroxisome proliferator-activated receptors as transcriptional nodal points and therapeutic targets. Circulation. 2007;115(4):518–533. doi: 10.1161/CIRCULATIONAHA.104.475673. [DOI] [PubMed] [Google Scholar]
- 43.Szanto A., Nagy L. The many faces of PPARgamma: anti-inflammatory by any means? Immunobiology. 2008;213:789–803. doi: 10.1016/j.imbio.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 44.Qian J., Chen H., Birnbaum Y., Nanhwan M.K., Bajaj M., Ye Y. Aleglitazar, a balanced dual PPARα and -γ agonist, protects the heart against ischemia-reperfusion injury. Cardiovasc Drugs Ther. 2016;30(2):129–141. doi: 10.1007/s10557-016-6650-9. [DOI] [PubMed] [Google Scholar]
- 45.Kilter H., Werner M., Roggia C., Reil J.C., Schäfers H.J., Kintscher U. The PPAR-gamma agonist rosiglitazone facilitates Akt rephosphorylation and inhibits apoptosis in cardiomyocytes during hypoxia/reoxygenation. Diabetes Obes Metab. 2009;11:1060–1067. doi: 10.1111/j.1463-1326.2009.01097.x. [DOI] [PubMed] [Google Scholar]
- 46.Fang X., Stroud M.J., Ouyang K., Fang L., Zhang J., Dalton N.D., Gu Y. Adipocyte-specific loss of PPARγ attenuates cardiac hypertrophy. JCI Insight. 2016;1 doi: 10.1172/jci.insight.89908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan J, Xu SC, Kong CY. Piperine alleviates doxorubicin-induced cardiotoxicity via activating PPAR-γ in mice. 2019;2019:2601408. [DOI] [PMC free article] [PubMed]
- 48.Shioji K., Kishimoto C., Nakamura H., Masutani H., Yuan Z., Oka S.-I., et al. Overexpression of thioredoxin-1 in transgenic mice attenuates adriamycin-induced cardiotoxicity. Circulation. 2002;106(11):1403–1409. doi: 10.1161/01.cir.0000027817.55925.b4. [DOI] [PubMed] [Google Scholar]
- 49.Quagliariello V., De Laurentiis M., Rea D., Barbieri A., Monti M.G., Carbone A., et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. 2021;20(1) doi: 10.1186/s12933-021-01346-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.