

Graphical abstract
Keywords: Tetramic acid, Photosynthetic inhibitor, D1 protein, Docking, Homology modeling
Highlights
-
•
The model of lead molecule TeA binding to the QB site in Arabidopsis D1 protein was constructed.
-
•
A series of new derivatives were designed and docked to the QB site of by molecular simulations.
-
•
Derivatives D6, D13 and D27 with better affinities than TeA were screened out and synthesized.
-
•
D6 and D13 are promising compounds to develop new PSII herbicides with superior performance.
-
•
Model-based ligand design is a valuable tool to find new PSII inhibitors based on lead molecule TeA.
Abstract
Introduction
Computer-aided design has become an important tool to develop novel pesticides based on natural lead compounds. Tenuazonic acid (TeA), a typical representative of the natural tetramic acid family, was patented as a potential bioherbicide. However, its herbicidal efficacy is still not up to the ideal standard of commercial products.
Objectives
We aim to find new TeA’s derivatives with improved potency.
Methods
Molecular docking was used to build ligand-acceptor interaction models, design and screen new derivatives. Phytotoxicity, oxygen evolution rate, chlorophyll fluorescence and herbicidal efficacy were determined to estimate biological activity of compounds.
Results
With the aid of a constructed molecular model of natural lead molecule TeA binding to the QB site in Arabidopsis D1 protein, a series of derivatives differing in the alkyl side chain were designed and ranked according to free energies. All compounds are stabilized by hydrogen bonding interactions between their carbonyl oxygen O2 and D1-Gly256 residue; moreover, hydrogen bond distance is the most important factor for maintaining high binding affinity. Among 54 newly designed derivatives, D6, D13 and D27 with better affinities than TeA were screened out and synthesized to evaluate their photosynthetic inhibitory activity and herbicidal efficacy. Analysis of structure-activity relationship indicated that D6 and D13 with sec-pentyl and sec-hexyl side chains, respectively, were about twice more inhibitory of PSII activity and effective as herbicide than TeA with a sec-butyl side chain.
Conclusion
D6 and D13 are promising compounds to develop TeA-derived novel PSII herbicides with superior performance.
Introduction
Structure-based ligand design using computer technology is an important tool in the research and development of new drugs and pesticides. Such design usually starts with docking an interesting lead product or its derivatives into a recognized target protein [1]. Substantial structural information of many important target proteins for pesticides and drugs, such as D1 protein of photosystem II (PSII) core complex, is currently available at the Protein Data Bank (PDB) (http://www.wwpdb.org), facilitating these type of studies. The site of action of most commercial herbicides resides in the chloroplast, which is a preferred organelle for finding new herbicides because photosynthesis is a unique physicochemical process of plants and cyanobacteria [2]. The D1 protein encoded by the chloroplast gene psbA in higher plants, and its equivalent L-subunit in photosynthetic bacteria, is an important target of commercial herbicides [3]. D1 protein contains five trans-membrane α-helices and several short non-membrane helices among them [4], [5]. The secondary quinone acceptor QB or PSII herbicide-binding pocket locates in the connecting loop between the fourth (D) and the fifth (E) transmembrane helices, which starts at Phe211 and ends at Leu275 [3], [4], [6]. Although PSII inhibitor herbicides share the same action target D1 protein, there are many different orientations for individual herbicides binding to the QB niche [7], [2]. The classical urea/triazine herbicides (diuron and atrazine) with the common characteristic group N—C O or N—C N form a hydrogen bridge to D1-Ser264. The phenol-type herbicides (ioxynil and dinoseb) with an aromatic hydroxyl group orient themselves towards D1-His215 [8], [7].
The tetramic acid with the core structural unit of pyrrolidine ring is the largest family of natural products isolated from many marine and terrestrial species. Natural tetramic acids have attracted much attention due to their diverse desirable bioactivity and synthetic challenge [9], [10], [11], [12], [13]. Tenuazonic acid (TeA, 3-acetyl-5-sec-butyl-4-hydroxy-pyrrolidine-2-one) is one of the most representative natural tetramic acids. TeA is usually found among secondary metabolites of several phytopathogenic fungi including Alternaria spp., Phoma sorghina, Magnaporthe oryzae and Aspergillus spp. [14], [15], [16]. It inhibits the activity of plant p-hydroxyphenylpyruvate dioxygenase [17] and plasma membrane H+-ATPase [18]. Our previous studies demonstrated that TeA produced by A. alternata from Ageratina adenophora pathotype has the potential to be developed as a new bioherbicide and has already been patented in China and Japan because of its broad spectrum, rapid and high herbicidal activity as well as low soil persistence [19], [20]. However, an insurmountable challenge still remains for its commercialization to overcome the unsatisfactory herbicidal activity due to the variable chemical conformations of TeA under different conditions. The interconversion of various tautomers could significantly affect herbicidal efficacy [19], [21], [22]. TeA is a photosynthetic inhibitor, interrupting electron flow beyond the primary quinone acceptor QA by interacting with D1 protein at the PSII reaction center. The chloroplast-derived oxidative burst is responsible for TeA-caused cell death and leaf lesion [23], [24]. Evidence from Chlamydomonas reinhardtii mutants suggested that D1-Gly256 is a critical residue in the interaction between TeA and the QB binding site of D1 protein [23]. Analysis of structure and biological characterization of TeA and several synthetic derivative 3-acyl-5-alkyltetramic acids, indicated that the pyrrole ring containing N—C O group is a core part for photosynthetic inhibiting activity that is also affected by the length of alkyl side chain at the 5-position of pyrrole ring [23]. This means TeA is a good lead compound to design new photosynthetic herbicides.
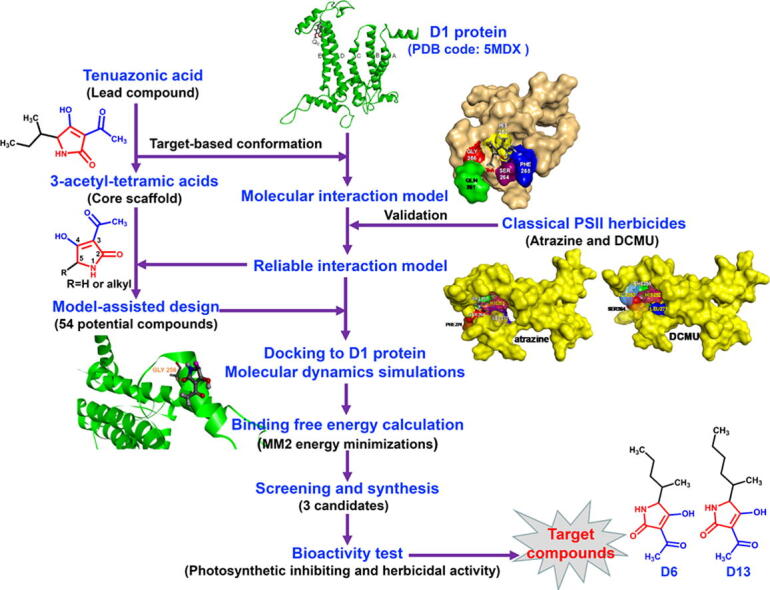
In this work, we aimed to discover new highly bioactive TeA derivatives based on a structure-based ligand design. First, a series of 3-acyl-5-alkyltetramic acid derivatives were designed on the simulated model of molecular interaction between TeA and target D1 protein of Arabidopsis by computer-aided design. Then, each derivative with high ligand affinity was screened by docking it into the D1 protein and calculating its binding free energy. Subsequently, TeA and three selected derivatives, D6 (sec-pentyl-TeA), D13 (sec-hexyl-TeA) and D27 (sec-heptyl-TeA), were synthesized by a chemical pathway. Finally, photosynthetic inhibiting and herbicidal activity of these three derivatives and TeA were evaluated to identify structure-activity relationships. Moreover, molecular docking was also performed at the QB binding site on the homology modeling of D1 protein of Ageratina adenophora and Digitaria sanguinalis to further verify the molecular interaction behavior and ligand affinity ranking. It was determined that compounds D6 and D13 with a longer alkyl side chain at 5-position indeed exhibited much higher bioactivity relative to TeA because of their higher binding affinity in the QB site. Thus, structure-based ligand design served an expeditious and reliable way to obtain new derivatives with high herbicidal activity on the basis of the core scaffolds of mycotoxin TeA. A brief description of the methodology is shown in Fig. 1.
Fig. 1.

Flow chart illustrating the structure-based ligand design and discovery of novel TeA derivatives with high herbicidal activity.
Material and methods
Plants and chemicals
Arabidopsis thaliana wild type (Columbia ecotype, Col-0) was planted in a mixture of soil (nutrition-free peat moss: vermiculite = 1: 3, v/v) in a greenhouse at 20–22 °C under 100 μmol (photons) m−2 s−1 white light (day/night, 16 h/8h) and about 80% humidity. Thirty species of plants (Table 2) including A. adenophora and D. sanguinalis were grown in substrate (peat: vermiculite: perlite, 3: 1: 0.5, v/v) at 25–28 °C under approximate 200 μmol (photons) m−2 s−1 white light (day/night, 12 h/12 h) and about 70% humidity in a greenhouse.
Table 2.
Phytotoxicity of TeA and three derivatives to various plants.a
| Family | Plant species | Diameter of leaf lesion (mm) |
|||
|---|---|---|---|---|---|
| D0 (TeA) | D6 (sec-pentyl-TeA) | D13 (Sec-hexyl-TeA) | D27 (Sec-heptyl-TeA) | ||
| Gramineae | Digiteria sanguinalis | 1.35 ± 0.10 | 2.48 ± 0.13 | 2.96 ± 0.04 | 1.91 ± 0.08 |
| Setaria viridis | 1.03 ± 0.07 | 1.76 ± 0.10 | 2.03 ± 0.23 | 1.85 ± 0.23 | |
| Echinochloa crus-galli | 1.46 ± 0.05 | 2.20 ± 0.17 | 2.55 ± 0.17 | 2.31 ± 0.17 | |
| Leptochloa chinensis | 1.33 ± 0.14 | 2.53 ± 0.12 | 2.87 ± 0.05 | 2.14 ± 0.12 | |
| Zea mays | 1.05 ± 0.06 | 1.65 ± 0.04 | 1.83 ± 0.07 | 1.34 ± 0.12 | |
| Oryza sativa | 1.42 ± 0.04 | 2.00 ± 0.11 | 2.36 ± 0.04 | 1.83 ± 0.03 | |
| Compositae | Solidago canadensis | 0.73 ± 0.04 | 1.89 ± 0.08 | 2.14 ± 0.09 | 1.44 ± 0.05 |
| Conyza canadensis | 1.01 ± 0.05 | 1.78 ± 0.16 | 2.04 ± 0.13 | 1.50 ± 0.11 | |
| Conyza sumatrensis | 1.03 ± 0.08 | 2.12 ± 0.10 | 2.29 ± 0.07 | 1.64 ± 0.05 | |
| Ageratina adenophora | 2.81 ± 0.13 | 3.67 ± 0.08 | 3.98 ± 0.19 | 3.05 ± 0.11 | |
| Eclipta prostrata | 1.12 ± 0.06 | 1.76 ± 0.07 | 1.94 ± 0.07 | 1.39 ± 0.09 | |
| Cyperaceae | Cyperus iria | 0.76 ± 0.09 | 1.48 ± 0.12 | 1.65 ± 0.04 | 1.48 ± 0.20 |
| Cyperus rotundus | 1.34 ± 0.06 | 2.71 ± 0.11 | 2.91 ± 0.07 | 2.24 ± 0.12 | |
| Convolvulaceae | Ipomoea purpurea | 1.09 ± 0.02 | 2.00 ± 0.15 | 2.18 ± 0.12 | 1.56 ± 0.06 |
| Calystegia hederacea | 0.94 ± 0.02 | 1.86 ± 0.07 | 2.08 ± 0.04 | 1.43 ± 0.11 | |
| Amaranthaceae | Alternanthera philoxeroides | 1.32 ± 0.09 | 2.15 ± 0.04 | 2.38 ± 0.06 | 1.76 ± 0.07 |
| Celosia argentea | 0.73 ± 0.03 | 1.44 ± 0.05 | 1.68 ± 0.14 | 1.16 ± 0.20 | |
| Amaranthus retroflexus | 0.75 ± 0.16 | 1.82 ± 0.12 | 2.13 ± 0.08 | 1.82 ± 0.12 | |
| Portulacaceae | Portulaca oleracea | 1.13 ± 0.12 | 2.66 ± 0.11 | 2.88 ± 0.13 | 1.85 ± 0.21 |
| Malvaceae | Gossypium hirsutum | 0.32 ± 0.02 | 0.37 ± 0.04 | 0.35 ± 0.06 | 0.37 ± 0.02 |
| Cannabinaceae | Humulus scandens | 1.29 ± 0.06 | 2.02 ± 0.13 | 2.02 ± 0.13 | 2.02 ± 0.13 |
| Solanaceae | Nicotiana tabacum | 0.23 ± 0.03 | 0.26 ± 0.02 | 0.27 ± 0.05 | 0.25 ± 0.03 |
| Cruciferae | Arabidopsis thaliana | 2.56 ± 0.15 | 3.05 ± 0.12 | 3.21 ± 0.08 | 2.77 ± 0.18 |
| Plantaginaceae | Plantago asiatica | 0.85 ± 0.07 | 1.41 ± 0.08 | 1.63 ± 0.05 | 1.15 ± 0.18 |
| Euphorbiaceae | Euphorbia humifusa | 1.84 ± 0.13 | 2.91 ± 0.21 | 3.42 ± 0.10 | 2.36 ± 0.15 |
| Acalypha australis | 0.95 ± 0.10 | 1.27 ± 0.07 | 1.40 ± 0.13 | 1.12 ± 0.07 | |
| Commelinaceae | Commelina communis | 1.31 ± 0.05 | 2.09 ± 0.12 | 2.37 ± 0.04 | 1.82 ± 0.06 |
| Commelina bengalensis | 1.34 ± 0.07 | 2.63 ± 0.16 | 2.87 ± 0.11 | 1.92 ± 0.13 | |
| Nyctaginaceae | Mirabilis jalapa | 1.42 ± 0.13 | 3.03 ± 0.14 | 3.31 ± 0.08 | 2.69 ± 0.15 |
| Vitaceae | Cayratia japonica | 1.23 ± 0.10 | 2.34 ± 0.05 | 2.57 ± 0.06 | 1.91 ± 0.14 |
Detached-intact leaves from different plant species are rinsed with distilled water, dried and subsequently placed in Petri dishes with wet filter paper. The leaves were lightly punctured with a needle from leaf abaxial margin. A 20 μL solution of TeA and three derivatives with 500 μM concentration was dripped onto the punctured wound. All Petri dishes were placed in the growth chamber for 48 h at 25 °C under 100 μmol (photons) m−2 s−1 white light (day/night, 12 h/12 h). Diameter of leaf necrotic lesions was measured with calipers. Each value is the average of three independent experiments.
2-Bromopentane, 2-bromohexane, 2-bromoheptane, N-(diphenylmethylene) aminoacetonitrile, lithium diisopropylamide (LDA), L-isoleucine were purchased from Sigma-Aldrich (Shanghai, China). Tetrahydrofuran (THF), petroleum ether (boiling point range 60–90 °C), ethyl acetate (EtOAc), ether, methanol (CH3OH), diketene, sodium, thionyl chloride and primary alcohol ethoxylate were obtained from Nanjing Chemical Reagent Co. Ltd. (Nanjing, Jiangsu, China). Ammonium chloride (NH4Cl), sodium sulfate (Na2SO4), sodium hydrogen carbonate (NaHCO3) and hydrochloric acid (HCl) were got from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Tetramethylsilane (Me4Si) and methanol‑d4 were bought from Aladdin Biochemical Technology Co., Ltd. (Shanghai, China). Silica gel (200–300 mesh) was purchased from Qingdao Marine Chemical Co. Ltd. (Qingdao, China). Unless mentioned otherwise, all other chemicals were obtained from Sigma-Aldrich (Shanghai, China).
Molecular interaction modeling
Software
Molecular modeling experiments were performed using Discovery Studio (version 2016, BIOVIA, USA). The structures of different ligands were constructed using ChemDraw 18.0 software (Cambridge Soft, USA). The ligand structures were energetically minimized using MM2 energy minimizations in Chem3D Pro 14.0 (Cambridge Soft, USA).
Ligands preparation
The 2D structures of different ligands were drawn in the ChemDraw 18.0 and saved as MDL MoL files. The Chem3D Pro 14.0 was used to generate 3D structures of all ligands and energetically minimize them by the MM2 force field. The convergence criteria of the gradient of the potential energy surface was defined at a minimum RMS gradient of 0.010. After 3D structure MDL MoL files of all ligands were transformed into Discovery Studio program, quantitative estimate of drug-likeness (QED), which combines eight physicochemical properties of molecular weight, octanol-water partition coefficient, number of H-bond acceptors, number of H-bond donors, molecular polar surface area, number of rotatable bonds, number of aromatic rings and number of structural alerts, were calculated.
Protein data preparation
The crystal structure of the D1 protein of A. thaliana was obtained from the Protein Data Bank (https://rcsb.org, PDB code: 5MDX, resolution: 5.30 Å), and used as a template for the Dl protein. Its dimeric structure was simplified to a monomer by deleting water molecules and adding hydrogen atoms, and then was optimized by CHARMm force field using Discovery Studio’s tools.
Searching binding residue at the QB site
The quinone QB binding site, also known as the site of PSII-herbicide binding, falls between the helices IV and V of the D1 protein from phenylalanine (Phe211) to leucine (Leu275) [6]. TeA is a PSII inhibitor and the Gly256 amino acid plays a major role during the interaction between TeA and D1 protein of C. reinhardtii [23]. Thus, the sequence of amino acids 211 to 275 was selected as the part of TeA-, PSII herbicide- and QB-binding niche to search and identify the binding pocket.
Interaction model building between TeA and D1 protein
The LibDock tool of Discovery Studio was utilized to automate the preliminary docking process. Following the input pdb file for receptor D1 protein (PDB code: 5MDX) and MDL Mol file for ligand TeA, the docking was carried out. The QB binding site in Arabidopsis D1 crystal structure (PDB code: 5MDX) was selected as possible binding site for docking TeA. The model shows that five residues, Leu218, His252, Gly256, Gln261 and Ser264 in the D1 protein might provide bonds to TeA molecule. Thus these five residues were selected and defined as the binding site for accuracy docking of TeA using CDocker tool of Discovery Studio. The coordinate axis of binding site sphere formed by these five amino acid residues is x = 303.03 Å, y = 245.75 Å, z = 237.67 Å, and the radius of binding site sphere is 12.00 Å. After the MDL Mol file of ligand TeA was input into the QB binding site of Arabidopsis D1 protein, docking was performed. The default docking procedure was done for TeA as standards for validation in the QB binding site and then for its derivative compounds. CHARMm-based molecular dynamics (MD) scheme was performed to simulate the interaction model of TeA and QB site. Random configurations of TeA were generated by a simulated high temperature of 1000 K (1000 steps). These different conformations were evaluated according to the maximum van der Waals energy. The conformations with van der Waals energy lower than the default threshold (300) were screened out and transferred into the QB binding site. The binding poses were produced by a simulated annealing procedure with two sequential steps: 2000 steps at the temperature of 700 K and 5000 steps at the temperature of 300 K. The QB binding environment surrounding TeA was further refined through energy minimization with CHARMm force field. Subsequently, the best score was given in the software manual after the docking calculation. The top binding pose with the lowest interaction energy was opted as the favorable binding configuration between the ligand TeA and the QB binding site of Arabidopsis D1 protein. The ligand binding site atoms in molecular interaction model were demonstrated by the 2D and 3D diagram.
To validate the reliability of the constructed interaction model of TeA bound to D1 protein, the molecular models of herbicides atrazine and diuron (DCMU) and quinone QB binding to the QB binding site of Arabidopsis D1 protein (5MDX) were also established using the same procedure as well as the available experimental and theoretical data [3], [8], [25], [32]. Residues His215, Tyr262, Ser 264 and Asn266 in Arabidopsis D1 protein were selected and defined as the binding site to accurately dock atrazine and DCMU. The coordinate axis of binding site sphere formed by these four amino acid residues is x = 303.92 Å, y = 242.39 Å, z = 238.26 Å, and the radius of binding site sphere is 14.54 Å. For native quinone QB, the residues of Met214, Leu218, Ala251, Phe255 and Leu271 in Arabidopsis D1 protein were selected and defined as the binding site for docking. The coordinate axis of binding site sphere formed by these five amino acid residues is x = 298.22 Å, y = 245.83 Å, z = 240.49 Å, and the radius of binding site sphere is 10.49 Å. The molecular interaction models of atrazine, DCMU and QB binding to D1 protein were developed using the default settings previously mentioned.
Model-based ligand design
Considering that TeA’s pyrrole ring with N—C O is a core scaffold for its biological activity [23], we focused exclusively on modifying the alkyl side chain at its 5-position to find possible novel derivatives highly bioactivity based on the modeling of the molecular interaction between TeA and its target D1 protein (PDB code: 5MDX). Here, 54 TeA’s derivatives only differing in the alkyl side chain at 5-position were designed in 2D structures using ChemDraw 18.0 (see Table 1). 3D structures of these 54 ligands were constructed and energetically minimized using Chem3D Pro 14.0 according to the protocol previously explained. Molecular properties of each ligand were calculated using Discovery Studio before molecular docking.
Table 1.
Structures and quantitative estimate of drug-likeness (QED) of newly designed TeA’s derivatives, and interaction energies between different compounds and Arabidopsis D1 protein (5MDX). TeA and its three derivatives (D6, D13 and D27) show higher QED and lower interaction energy than others.
| Compound No. |
5- alkyl side chain (R-) | QED | Interaction energy (kcal/mol) | Compound No. | 5- alkyl side chain (R-) | QED | Interaction energy (kcal/mol) |
|---|---|---|---|---|---|---|---|
| D0 (TeA) | -CH(CH3)CH2CH3 | 0.57 | −29.35 | D27-D51: -C7H15 | |||
| D1 | -H | 0.36 | −22.12 | D27 | -CH(CH3)(CH2)4CH3 | 0.64 | −30.98 |
| D2 | -CH3 | 0.39 | −23.84 | D28 | -CH2CH(CH3)(CH2)3CH3 | 0.43 | −24.16 |
| D3 | -CH(CH3)CH3 | 0.52 | −27.95 | D29 | -(CH2)2CH(CH3)(CH2)2CH3 | 0.40 | −23.94 |
| D4-D5: | -C4H9 (Including D0) | D30 | -(CH2)3CH(CH3)CH2CH3 | 0.42 | −24.02 | ||
| D4 | -CH2CH(CH3)CH3 | 0.44 | −24.61 | D31 | -(CH2)4CH(CH3)CH3 | 0.46 | −25.34 |
| D5 | -CH2CH2CH2CH3 | 0.38 | –23.58 | D32 | -(CH2)6CH3 | 0.39 | −23.85 |
| D6-D11: | -C5H11 | D33 | -C(CH3)2(CH2)3CH3 | 0.32 | −20.02 | ||
| D6 | -CH(CH3)CH2CH2CH3 | 0.75 | −32.29 | D34 | -CH(CH3)CH(CH3)(CH2)2CH3 | 0.34 | −21.38 |
| D7 | -CH2CH(CH3)CH2CH3 | 0.49 | −26.14 | D35 | -CH(CH3)CH2CH(CH3)CH2CH3 | 0.34 | −20.85 |
| D8 | -CH2CH2CH(CH3)CH3 | 0.53 | −28.62 | D36 | -CH(CH3)(CH2)2CH(CH3)CH3 | 0.32 | −20.02 |
| D9 | -CH2CH2CH2CH2CH2CH3 | 0.43 | −24.18 | D37 | -CH2C(CH3)2(CH2)2CH3 | 0.35 | −21.47 |
| D10 | -CH(CH3)CH(CH3)CH3 | 0.37 | −23.53 | D38 | -CH2CH(CH3)CH(CH3)CH2CH3 | 0.33 | −20.73 |
| D11 | -C(CH2CH3)CH2CH3 | 0.34 | −21.06 | D39 | -CH2CH(CH3)CH2CH(CH3)CH3 | 0.33 | −20.26 |
| D12-D26: -C6H13 | D40 | -CH2CH(CH3)(CH2)3CH3 | 0.32 | −20.15 | |||
| D12 | -CH2CH2CH2CH2CH2CH3 | 0.48 | −25.98 | D41 | -(CH2)2C(CH3)2CH2CH3 | 0.32 | −19.54 |
| D13 | -CH(CH3)CH2CH2CH2CH3 | 0.81 | −33.41 | D42 | -(CH2)2CH(CH3)CH(CH3)CH3 | 0.31 | −19.27 |
| D14 | -CH2CH(CH3)CH2CH2CH3 | 0.49 | −26.02 | D43 | -CH(CH3)CH(CH3)CH(CH3)CH3 | 0.28 | −18.53 |
| D15 | -CH2CH2CH(CH3)CH2CH3 | 0.52 | −27.26 | D44 | -C(CH3)(CH2CH3)(CH)2CH3 | 0.27 | −18.24 |
| D16 | -CH2CH2CH2CH(CH3)CH3 | 0.53 | −28.70 | D45 | -CH(CH3)CH(CH2CH3)CH2CH3 | 0.29 | −19.07 |
| D17 | -C(CH3)2CHCH2CH3 | 0.37 | −23.57 | D46 | -CH(CH2CH3)CH(CH3)CH2CH3 | 0.28 | −18.36 |
| D18 | -CH(CH3)CH(CH3)CH2CH3 | 0.38 | −23.68 | D47 | -CH(CH2CH3)CH2CH(CH3)CH3 | 0.27 | −18.14 |
| D19 | -CH(CH3)CH2CH(CH3)CH3 | 0.44 | −24.56 | D48 | -CH2CH(CH2CH3)(CH2)2CH3 | 0.25 | −18.02 |
| D20 | -CH2CH(CH3)CH(CH3)CH3 | 0.37 | −23.14 | D49 | -(CH2)2CH(CH2CH3)2 | 0.31 | −19.18 |
| D21 | -CH(CH2CH3)CH2CH2CH3 | 0.36 | −21.82 | D50 | -CH(CH2CH2CH3)2 | 0.28 | −18.75 |
| D22 | -CH2CH(CH2CH3)CH2CH3 | 0.34 | −20.93 | D51 | -CH(CH(CH3)2)(CH2)2CH3 | 0.24 | −17.26 |
| D23 | -CH(CH2CH3)CH(CH3)CH3 | 0.35 | −21.64 | D52-D54: -CH(CH3)(CH2)nCH3 | |||
| D24 | -CH(CH3)C(CH3)3 | 0.36 | −21.97 | D52 | -CH(CH3)(CH2)5CH3 | 0.48 | −25.73 |
| D25 | -CH2C(CH3)2CH2CH3 | 0.44 | −24.33 | D53 | -CH(CH3)(CH2)6CH3 | 0.40 | −23.86 |
| D26 | -CH2CH2C(CH3)2CH3 | 0.47 | −25.68 | D54 | -CH(CH3)(CH2)7CH3 | 0.33 | −20.82 |
Molecular docking
Molecular docking of TeA derivatives
We assumed that the 54 designed TeA derivatives should bind to the QB binding site of D1 protein in a similar mode as the lead molecule TeA, since they share a common scaffold. Therefore, residues Leu218, His252, Gly256, Gln261 and Ser264 in Arabidopsis D1 protein (5MDX) were selected and defined as the binding site for TeA and its derivatives. The conformation of natural compound TeA was taken as a template. Following preparation of 3D structure MDL MoL input files of all ligands, CDocker tool was utilized to accurately simulate the molecular docking of these 54 compounds. For each compound docking, the number of diverse top poses (Top Hits) was set to 10, and the radius of pose clustering (Pose Cluster Radius) was set to 0.5 Å. Other docking parameters were set to the default values. Based on the energy minimization and molecular dynamics simulations, each model of molecular interaction between ligand and the QB binding site of Arabidopsis D1 protein was individually optimized. The energy of the interaction between each ligand and the QB binding site of Arabidopsis D1 protein was calculated (Table 1).
Homology model building of Dl protein of A. adenophora and D. sanguinalis
The amino acid sequence of target Dl protein of A. adenophora (Reference Seq. No. YP_004564352.1) and D. sanguinalis (Reference Seq. No. AIY26621.1) were obtained from NCBI. Both sequences served as a query in searching for evolutionary related proteins with available structures by the BLAST program through the SWISS-MODEL Template Library (SMTL) [26]. The searching templates of D1 protein were estimated by Global Model Quality Estimate (GMQE) and Quaternary Structure Quality Estimate (QSQE) and ranked according to the model quality. The top five ranked D1 protein templates with about 90% in the value of both GMQE and sequence identity were chosen relative to target protein sequences of A. adenophora and D. sanguinalis, including Spinacia oleracea (PDB code: 3JCU), A. thaliana (PDB code: 5MDX), Pisum sativum (PDB code: 5XNL), Chaetoceros gracilis (PDB code: 6JLU), C. reinhardtii (PDB code: 6KRC), Cyanidium caldarium (PDB code: 4YUU) and Thermosynechococcus elongates BP-1 (PDB code: 4IXQ). Data of sequence alignments and protein structures of the top four ranked templates were downloaded in the PDB, and then used to build the homology model of target D1 protein of A. adenophora and D. sanguinalis by the Protein Modeling Module of Discovery Studio. The accuracy and quality of homology models were estimated according to PDF (probability density function) total energy, PDF physical energy, DOPE (discrete optimized protein energy) and RMSD (root mean square deviation). The lowest energy and best DOPE and RMSD scored model of D1 protein of A. adenophora (YP_004564352.1.M0001) or D. sanguinalis (AIY26621.1.M0001) based on the top four ranked templates was chosen as the initial structure for further docking. The already explained docking protocol was used for performing docking of compound D0 (TeA), D6, D13 and D27. The preliminary docking model based on LibDock showed that amino acid residues Leu218, His252, Gly256, Gln261 and Ser264 in D1 protein of A. adenophora or D. sanguinalis should be defined as the biding site for the four compounds. Their accurate molecular docking was performed with CDocker.
Chemical synthesis of precursor amino acids, TeA and its derivatives
Three precursor amino acids 2-amino-3-methylhexanoic acid, 2-amino-3-methylheptanoic acid and 2-amino-3-methyloctanoic acid were prepared according to Qiang et al. [27]. Briefly, the catalyst LDA (3 mL, 6 mmol) was added to 30 mL THF solution of N-(diphenylmethylene)aminoacetonitrile (1.1 g, 5 mmol) in a three-neck glass flask (250 mL) and stirred for 5 min, and then 0.74 mL 2-bromopentane, 2-bromohexane or 2-bromoheptane was slowly added under a given nitrogen atmosphere at 0 °C. The mixture was stirred for 30 h at room temperature, and 20 mL saturated aqueous NH4Cl (7 M) was added to stop the reaction. The organic layer was dried over anhydrous Na2SO4, filtered and evaporated under vacuum at 65 °C in a rotary evaporator (EYELA, Japan). The crude extract was purified by column chromatography on silica gel (200–300 mesh, Qingdao Marine Chemical Co. Ltd., Qingdao, China) (petroleum ether : EtOAc = 20 : 1) to afford yellow-green oil product of 2-((diphenylmethylene)amino)-3-methylhexanenitrile, 2-((diphenylmethylene)amino)-3-methylheptanenitrile or 2-((diphenylmethylene)amino)-3-methyloctanenitrile. Subsequently, the oil product 2-((diphenylmethylene)amino)-3-methylhexanenitrile (0.87 g, 3 mmol), 2-((diphenylmethylene)amino)-3-methylheptanenitrile (0.91 g, 3 mmol) or 2-((diphenylmethylene)amino)-3-methyloctanenitrile (0.96 g, 3 mmol) was dissolved in 20 mL ether in a three-neck glass flask (250 mL) and 4 mL HCl (1 M) was added. The mixture was stirred for 24 h at room temperature and washed with ether (2 × 10 mL) in a separating funnel, and the aqueous layer was evaporated under vacuum at 100 °C in a rotary evaporator (EYELA, Japan) to leave white solid of 2-amino-3-methylhexanenitrile, 2-amino-3-methylheptanenitrile or 2-amino-3-methyloctanenitrile. Then, 2-amino-3-methylhexanenitrile (0.63 g, 5 mmol), 2-amino-3-methylheptanenitrile (0.70 g, 5 mmol) or 2-amino-3-methyloctanenitrile (0.77 g, 5 mmol) were dissolved in 10 mL HCl (6 M) and stirred for 48 h at 100 °C in a three-neck glass flask (250 mL). The mixture was cooled down to room temperature and washed with ether (2 × 10 mL) in a separating funnel, and the aqueous layer was evaporated under vacuum at 100 °C in a rotary evaporator (EYELA, Japan) to get the precursor amino acid of 2-amino-3-methylhexanoic acid, 2-amino-3-methylheptanoic acid or 2-amino-3-methyloctanoic acid.
Compounds D0, D6, D13 and D27 (purity greater than 94%) were synthesized by a straightforward five-step synthesis approach including esterification, neutralization, acidylation, cyclization and acidification as previously described with some modifications (Fig. 4; [28]. After SOCl2 (1.42 g, 12 mmol) was added in 50 mL CH3OH in a three-neck glass flask (250 mL) and stirred for 1 h at 0 °C, the starting material L-isoleucine (1.31 g, 10 mmol), 2-amino-3-methylhexanoic acid (1.45 g, 10 mmol), 2-amino-3-methylheptanoic acid (1.59 g, 10 mmol) or 2-amino-3-methyloctanoic acid (1.73 g, 10 mmol) was added. Subsequently, the mixture was stirred for 3 h at room temperature, and then heated for 4 h at 65 °C. The solvent in the reaction mixture was evaporated under vacuum at 65 °C in a rotary evaporator (EYELA, Japan) to get the esterification product, which was re-dissolved in 50 mL CH3OH in a three-neck glass flask (250 mL). CH3ONa (0.11 g sodium metal in 10 mL CH3OH) was added in the CH3OH solution of the esterification product to get the neutralization product of L-isoleucine methyl ester, 2-amino-3-methylhexanoic acid methyl ester, 2-amino-3-methylheptanoic acid methyl ester or 2-amino-3-methyloctanoic acid methyl ester. And then, (CH2CO)2 (0.42 g, 5 mmol) was added and stirred for 16 h at room temperature. After the solvent in the reaction mixture was evaporated under vacuum at 65 °C in a rotary evaporator (EYELA, Japan), the crude extract was dissolved in 50 mL EtoAc and washed, respectively, with HCl (1 M, 2 × 5 mL) and aqueous NaHCO3 (0.5 M, 2 × 5 mL) in a separating funnel. The organic layer was dried over anhydrous Na2SO4, filtered and evaporated at 65 °C in a rotary evaporator (EYELA, Japan) to get the acidylation product of N-acetoacetyl-L-isoleucine methyl ester, N-acetoacetyl-2-amino-3-methylhexanoic acid methyl ester, N-acetoacetyl-2-amino-3-methylheptanoic acid methyl ester or N-acetoacetyl-2-amino-3-methyloctanoic acid methyl ester. After the acidylation product (5 mmol) was re-dissolved in 50 mL CH3OH, CH3ONa (0.11 g sodium metal in 10 mL CH3OH) was added and stirred for 3 h at 65 °C. The solvent in the reaction mixture was evaporated under vacuum at 65 °C in a rotary evaporator (EYELA, Japan) to get the cyclization product. Finally, the cyclization product was dissolved in 50 mL water and acidified with HCl (6 M) at pH 2. After twenty mL of ether was added in the mixture in a separating funnel and shaken at room temperature, and the organic layer was dried over anhydrous Na2SO4, filtered and evaporated at 65 °C in a rotary evaporator (EYELA, Japan) to get D0, D6, D13 or D27.
Fig. 4.

The chemical synthetic pathway for TeA (D0) and its three derivatives D6 (sec-butyl-TeA), D13 (sec-pentyl-TeA) and D27 (sec-heptyl). Here, THF: tetrahydrofuran; LDA: lithium diisopropylamide; r.t.: room temperature; R: alkyl (sec-butyl, sec-pentyl, sec-hexyl or sec-heptyl).
Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker Avance 400 spectrometer (Bruker, Switzerland), operating at 400 MHz for 1H and 100 MHz for 13C. Tetramethylsilane (Me4Si) was used for internal standard. Chemical shifts are given in parts per million (δ). Coupling constants (J) are given in Hz. D0, D6, D13 and D27 were dissolved in 0.5 mL methanol‑d4 for NMR, respectively.
Biological activity evaluation
Phytotoxicity assay in vitro
The detached-intact healthy leaves from 30 plant species (Table 2) were rinsed with distilled water, blotted-dry with sterile paper and then placed in petri dishes lined with moistened (distilled water) filter paper. Leaves were lightly punctured with a needle from their margin on the abaxial side. A 20 μL aliquot of each compound at 500 μM was dripped onto each de punctured area. All Petri dishes were placed in a growth chamber for 48 h at 25 °C under approximately 100 μmol (photons) m−2 s−1 white light (day/night,12 h/12 h). Diameter of leaf necrotic lesions was measured with a vernier caliper (ROHS HORM 2002/95/EC, Xifeng, China). Each mean value was obtained from at least fifteen leaf samples.
Analysis photosynthesis inhibiting activity
Thylakoids of Arabidopsis were prepared as described in Lu et al. [29]. The rate of oxygen evolution of PSII was measured using a Clark type oxygen electrode (Hansatech Instruments Ltd., King’s Lynn, UK) according to Chen et al. [23]. Prior to measurement, 250 μL thylakoid suspensions (200 μg Chl mL−1) were dark incubated for 30 min at 4 °C in the presence of each compound D0, D6, D13 or D27 at final concentrations of 0 (0.1% MeOH), 50, 100, 200, 400 and 800 μM. The treated-thylakoids containing 40 µg chlorophylls were added into the reaction medium including 50 mM Hepes-KOH buffer (pH = 7.6), 4 mM K3Fe(CN)6, 5 mM NH4Cl, 1 mM p-phenylenediamine. Thylakoids were illuminated with 200 µmol (photons) m−2 s−1 red actinic light. The rate of oxygen evolution was measured during the first three minutes after onset of illumination. The I50 (the concentration producing 50% inhibition) value was calculated from plots of oxygen evolution rate in the presence of various concentrations of each compound.
Chlorophyll (Chl) a fluorescence rise kinetics was measured by 1 s pulses of red light (650 nm, 3500 μmol m−2 s−1) with a plant efficiency analyzer (Handy PEA fluorometer, Hansatech Instruments Ltd., UK). Before fluorescence measurement, 7-mm diameter leaf discs of Arabidopsis plants were prepared and subsequently exposed for 6 h to 0.1% MeOH (mock), D0, D6, D13 or D27 at different cited concentrations in the dark at room temperature. The basic parameters from the original measurements were used as follows: FO (the minimal fluorescence intensity at 20 μs), FJ (the fluorescence intensity at 2 ms), FI (the fluorescence intensity at 30 ms), and the maximal fluorescence intensity FM is equal to FP. To further analyze the kinetics OJIP properties, the fluorescence curves double normalized by FO and FM were presented as relative variable fluorescence Vt = (Ft − FO)/(FM − FO) and ΔVt versus logarithmic time scale. The expressions of fluorescence parameters VJ, φEo, ψEo, PIABS, QA-reducing centers and RJ were referred to as previous references [30], [31].
Herbicidal efficacy
The seedlings of A. thaliana, A. adenophora and D. sanguinalis were sprayed at room temperature to runoff with 1.8 mL of each compound D0, D6, D13 or D27 solution containing 0.04% primary alcobol ethoxylate at the indicated concentrations using a black polypropylene fine mist sprayer (SKS Bottle & Packaging Inc., West Watervliet, NY, USA). After 5 days of treatment, herbicidal efficacy was visually evaluated according to percentage foliage damage, calculated as percentage of damage rate = ∑(injury rate × individuals) /(5 × total of individuals observed) [20]. One-way ANOVA was carried out and means were separated by Duncan LSD at 95% using SPSS Statistics 20.0.
Results and discussion
Molecular model of TeA binding to the QB niche of Arabidopsis D1 protein
Prior report demonstrated that TeA binds to D1 protein and inhibits PSII electron flow beyond QA [23], 2014). Mutational studies on D1-Gly256 of C. reinhardtii has implicated its key role during the interaction between TeA and D1 protein [23]. However, the protein binding environment for TeA remains unclear because the crystal structure of D1 protein with bound TeA is not available. Here, the crystal structure information of Arabidopsis D1 protein (PDB: 5MDX) was selected to model the position of TeA in the QB site using Discovery Studio (Fig. 2A-2C). In the model, the residues that are identified to be responsible for TeA binding are Leu218, Ala251, His252, Gly253, Phe255, Gly256, Gln261, Try262, Ala263, Ser264, Phe265, Asn266 and Leu271 (Fig. 2B-2C). Gly256 contributes to TeA binding by providing a hydrogen bond to the O2 carbonyl oxygen atom of its pyrrole ring. The modeled bond distance is 3.66 Å. Ser264 residue is likely to provide an alkyl hydrophobic interaction to the O6 acetyl oxygen atom at the 3-position of TeA with the modeled bond distance at 3.24 Å (Fig. 2C, Table S1). This is consistent with early evidence from C. reinhardtii D1 mutations. The single mutations of Gly256Asp and Ser264Ala could cause around 37- and 8-fold resistance to TeA, respectively, based on PSII oxygen evolution measurements [23]. Additionally, Gln261 is also identified as responsible for interacting with the C9 of the 5-position sec-butyl tail, and Phe265 forms π hydrophobic interaction with the O4 hydroxyl oxygen atom (Fig. 2C, Table S1).
Fig. 2.

The simulated modeling of TeA, quinone QB, atrazine and DCMU in the QB binding site of D1 protein of A. thaliana (5MDX). (A) Surface representation of the QB binding site with quinone QB. (B) Stereo view of TeA binding environment in the QB binding site. (C) Hydrogen bonding interactions for TeA binding to the QB binding site. Here, carbon atoms are shown in grey, nitrogen atoms in blue, oxygen in red and hydrogen atoms in white. The possible hydrogen bond is indicated by dashed line. (D) Surface representation of the QB binding site with bound TeA. (E) Surface representation of the QB binding site with bound atrazine. (F) Surface representation of the QB binding site with bound DCMU. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To validate the reliability of above generated molecular interaction model, native quinone QB, atrazine and DCMU were also modeled to the QB-binding site of Arabidopsis D1 protein based on the available experimental and theoretical structure information of QB, atrazine and DCMU binding in a bacterial reaction center (Fig. 2D-2F). Binding of quinone QB to the QB site was simulated according to the available crystal structure information of QB binding in a cyanobacterial PSII reaction center [32] and Arabidopsis D1 protein crystals (5MDX) (Fig. 2D). It is proposed that potential hydrogen-bonds are formed between D1-His215 and the O1 carbonyl oxygen of QB, as well as between D1-Ser264 and the O4 carbonyl oxygen of QB (Fig. S1 A, Table S1). In T. elongatus PSII, QB binds deep into a cavity lined with hydrophobic residues, where the O1 of QB head is likely to be hydrogen bonded to the δ-N of D1-His215, the O4 of QB head may form hydrogen bonds with the side chain γ-O of D1-Ser264 and the amide nitrogen of D1-Phe265 [32], [33]. The isoprenoid tail of QB is also vital for QB stabilization in the QB-binding site, which is responsible for more than 60% of the total molecule interaction energy [34]. Our modeling is essentially in agreement with prior studies [32], [33]. As shown in Fig. 2E-2F, modeling of herbicide atrazine and DCMU to the QB site of D1 protein (5MDX) was based on the crystal structure information of complexes of Rhodopseudomonas viridis RC with atrazine (5PRC) and R. viridis mutant T4 RC with DCMU [25]. Atrazine interacts with residues D1-Ser264, D1-Leu271 and D1-Phe274. The N11 hydrogen and N1 atoms of atrazine are hydrogen bonded to D1-Ser264 (Fig. 2E, Fig. S1B, Table S1). In modeling the binding of DCMU to the QB site, it appears that Phe255, Ala263 and Ser264 play a major role. A hydrogen bound is formed between the N9 amide hydrogen of DCMU and the oxygen atom of D1-Ser264 with the bond distance at 3.72 Å (Fig. 2F, Fig. S1C, Table S1). These results support early crystallographic investigations and studies with resistant mutants [8], [7]. In the context of the atrazine binding site of bacterial R. viridis RC with atrazine complex (5PRC), the N7 and N11 hydrogen atom of atrazine donate hydrogen bonds to the residues L-Tyr222 and L-Ser223 [8]. The counterparts of Tyr222 and Gly223 in the L-subunit of bacterial RC are just D1-Tyr262 and D1-Ser264 in higher plant, respectively. Previous studies had demonstrated that residue D1-Ser264 may provide a hydrogen bond through its hydroxyl oxygen to the N9 amide hydrogen of DCMU. Another weak hydrogen bridge is formed between the carbonyl group of DCMU and the side chain of D1-His215 [35], [25], [3]. Furthermore, there are numerous experimental observations on mutants implying that hydrogen bonding to D1-Ser264 is the favoured orientation for atrazine and DCMU [7]. Undoubtedly, our modeling is consistent with most existing data of the classical PSII herbicides, which has high reliability and accuracy.
Consequently, the residues that appear to anchor TeA binding in the QB site are different from those responsible for the binding of urea and triazine herbicides, although the protein-binding environment of TeA overlaps with theirs. TeA orients itself preferentially towards D1-Gly256 by a hydrogen bond, however, atrazine and DCMU form a main hydrogen bond with D1-Ser264. In our modeling, it is also notable that QB, atrazine and DCMU are totally nestled in the cavity formed by the QB-binding pocket. However, for TeA only the pyrrole ring head group enters inside the cavity, its tail of sec-butyl alkyl side chain lies along the wall of the cavity and exposes partly outer the QB pocket (Fig. S2). The energy of TeA according to the model is −29.35 kcal/mol. The energy of atrazine and DCMU model is −37.51 kcal/mol and −35.27 kcal/mol, respectively. It is indicated that TeA has a lower binding affinity (higher interaction energy) for the QB site compared with atrazine and DCMU. This might be the reason that TeA exhibits a weaker PSII inhibitory activity compared with those commercial herbicides.
Design and generation of novel TeA derivatives
Our docking model and early studies [23] have demonstrated that the pyrrole ring containing N—C O group is a core scaffold of TeA for inhibiting photosynthetic activity, and that the length and hydrophobicity of alkyl side chain at the 5-position also affects inhibition. Therefore, modifying the chemical structure of lead compound TeA should allow finding novel bioactive derivatives. An attractive option is to maintain the template scaffold of pyrrole ring while changing the alkyl side chain at 5-position to improve binding affinity. Fifty-four TeA’s derivatives with modified alkyl side chain were designed based on the modeling of TeA binding to the QB site in Arabidopsis D1 protein (Table 1). Three of them, D1, D2 and D3, have the 5-position alkyl chain substituted by hydrogen, methyl or sec-propyl. Compounds D4, D5 and D0 (TeA) have a four-carbon atom hydrophobic alkyl side chain. Compounds D6 to D11, D12 to D26, and D27 to D51 families have an alkyl side chain of 5, 6 and 7 carbon atoms with varying configurations. Compounds D52, D53 and D54 have a sec-alkyl side chain of 8, 9 and 10 carbon atoms, respectively. Notably, only compounds D6, D13 and D27 reveal higher QED than TeA among this series of compounds (Table 1). The QED integrating eight key physicochemical properties of a drug candidate provides an efficient approach to assess druglikeness and chemical attractiveness [36]. Druglikeness is a key consideration for screening of compounds during the early stages of drug discovery. A greater QED represents a higher druglikeness, meaning a compound is more active [37]. So, it was concluded that D6, D13 and D27 are probably the most potential compounds.
Molecular docking of the designed compounds and virtual screening
Molecular docking is one of the modeling tools most widely used for predicting the orientation and conformation of candidate ligands with the active site of the target molecule, performing virtual screening based on a ranking scores or binding energy of forming the ligand-receptor interaction complex [38], [39]. While 54 newly designed 3-acyl-5-alkyltetramic acid compounds were aligned on the template TeA in the same way and docked to the QB site of Arabidopsis D1 protein, the substitutions at 5-position in the ligands were minimized but the QB site and the common pyrrole ring moiety of the ligand were fixed for the energy minimization. The predicted interaction energy values for the reliable model of TeA and its 54 derivatives binding to the QB site are listed in Table 1. All 3-acyl-5-alkyltetramic acids demonstrated negative binding energies, indicating that the interaction between the ligand and the target is possible and thermodynamically favorable. Compared to TeA, compounds D6, D13 and D27 were predicted to have the lowest interaction energies; the rest had higher estimated binding energies than TeA. It’s well recognized that interaction energy reflects the ligand binding affinity for target receptor, allowing evaluating the ligand inhibitory potency [39]. Compounds D1 to D3 with short alkyl side chains at 5-position had higher predicted interaction energies (Table 1), having been proven to be weaker inhibitors in PSII electron transfer activity compared with TeA [23]. Compound D3 (3-acetyl-5-isopropyltetramic acid) with a binding affinity of −27.95 kcal/mol was about half as active as TeA at inhibiting the rate of PSII O2 evolution relative to TeA (Table 1, [40]. Previous hypotheses suggested that a longer alkyl side chain at 5-position is better suited for tight binding at the QB site [23]. However, an increase in the length of alkyl side chain was not sufficient for strengthening binding affinity of newly design derivatives. Among compounds D6 to D51 with an alkyl side chain of 5 to 7 carbon atoms, only those with sec-pentyl (D6), sec-hexyl (D13) and sec-heptyl (D27) side chains, had higher binding affinity than TeA with its sec-butyl side chain. D6, D13 and D27 are also the compounds with the highest affinity within their own derivative family with an alkyl side chain of same carbon atoms. The position shift in branched methyl group of sec-alkyl side chain or increase in the number of branched methyl group would decrease the binding affinity (Table 1). This might be attributed to a nonnegligible stabilization of ligand molecule due to the interaction of the C9 methyl carbon atom of the sec-alkyl tail with amino acid residues in the QB site. Clearly, the structure of sec-alkyl at 5-position is of utmost importance for the high affinity of compound binding to the QB site. However, it was also observed that compounds D52, D53 and D54 with long sec-alkyl side chain of over 7 carbon atoms lost affinity for the QB site compared with TeA (Table 1). Therefore, D6, D13 and D27 were identified as potential candidates with stronger photosynthetic inhibiting ability than the lead TeA. This result is in complete agreement with the QED analysis.
To further probe the properties of potential candidates, the molecular models of compound D6, D13 and D27 binding to the QB site of Arabidopsis D1 protein (5MDX) were constructed. The docked poses with binding energies of −32.29 kcal/mol, −33.41 kcal/mol and −30.98 kcal/mol for D6, D13 and D27, respectively, are displayed in Fig. 3. The residue D1-Gly256 forms a hydrogen bond with the O2 carbonyl oxygen atom of pyrrole ring of the three compounds, whereas hydrophobic interactions occur with several other residues (Fig. 3B, 3D, 3F; Fig. S3). The modeled distances of hydrogen bonds are 3.39 Å, 3.24 Å and 3.51 Å for D6, D13 and D27, respectively, which is greater than that between the residue and the carbonyl oxygen O2 of D0 (TeA) at 3.66 Å (Table S1). In fact, there is a distinct negative linear relationship between the binding affinity and hydrogen bond distance in the models of TeA and its 54 derivative compounds binding to the QB site (Fig. S4A). Therefore, a small hydrogen bond distance may be most crucial for the predicted binding stability of the three selected candidates (D6, D13 and D27). A notable linear relationship between the QED of all compounds and their hydrogen bond distance (Fig. S4B) or binding affinity (Fig. S4C) was also observed. This indicates that molecular physicochemical properties of a compound are the primary cause of deciding its binding affinity for target.
Fig. 3.

Docking of the QB binding site of D1 protein of A. thaliana (5MDX) with TeA derivatives. Surface representation of the QB binding site with compounds D6 (A), D13 (C) and D27 (E) is shown in left panel, respectively; stereo view of compounds D6 (B), D13 (D) and D27 (F) binding environment at the QB binding site is shown in right panel, respectively.
Similar to TeA, it was also found that there is an important hydrophobic interaction between the O6 acetyl oxygen atom of D6 and D13 and the residue D1-Ser264. D1-Gln261 and D1-Ala263 also forms, respectively, a hydrophobic interaction with the methyl carbon C9 and the hydroxyl oxygen O4 of D6. Additionally, D1-Ala263 appears to make further hydrophobic contact with the acetyl oxygen O6 of D6 (Fig. 3B, Fig. S3A, Table S1). In the model of D13, hydrophobic interactions between the C7, C9 or C10 carbon atoms and the residues D1-Phe255, D1-Gln263 or D1-His252 also contributes to the stability of the complexes (Fig. 3D, Fig. S3B, Table S1). Additional stability is provided by the interaction of the C7 and C10 carbon atoms with electron clouds of residues Phe255 and His252 for the complex of D13 binding to the QB site, explaining its highest binding affinity of −33.41 kcal/mol. Unlike compound D6 and D13, the acetyl oxygen O6 does not participate in the complex stabilization of D27 binding to the QB site. Such difference may explain partly why the binding affinity of D27 at the QB site is much lower than that of compounds D6 and D13. For compound D27, hydrophobic interactions occur mainly between the hydroxyl oxygen O4 and D1-Phe255, as well as the methyl carbon C9 and C14 and two residues D1-Gln263 and D1-Phe265, respectively. (Fig. 3F, Fig. S3C, Table S1). However, the π hydrophobic interaction formed between D1-Phe265 and the terminal unit C14 with the bond distance at 3.18 Å led to a bend in the sec-heptyl tail of D27 (Fig. S3C). The energy of D27 model is −30.98 kcal/mol that is lower than TeA, but higher than D6 and D13. The change in spatial conformation of TeA derivatives due to a very-long sec-alkyl side chain at 5-positon decreased the binding affinity for the QB pocket. In this case, the cavity formed by the QB-binding pocket might be too small to tightly hold the ligand molecule. This explains perhaps that compounds D52, D53 or D54 having longer sec-alkyl side chains than TeA depict a weaker stabilization of intermolecular bonds in the QB site. Considering the binding possibilities and energy scores, D6, D13 and D27 were designated as the most potent candidates.
Chemical synthesis and phytotoxicity analysis of selected candidate compounds
To prove the predictive accuracy of molecular docking, compound D0 (TeA) and its three selected derivatives, D6, D13 and D27 were synthesized using L-isoleucine, 2-amino-3-methylhexanoic acid, 2-amino-3-methylheptanoic acid and 2-amino-3-methyloctanoic acid as the starting materials, respectively (Fig. 4). The structures of the four synthesized compounds were identified by proton and carbon NMR (Figs. S5-S8) before evaluation of their phytotoxicity on 30 different plant species. At 48 h after treatment, the size of leaf lesions was determined as a criterion for herbicidal activity (Table 2). Among all species, tobacco (Nicotiana tobacum) from the Solanaceae family and cotton (Gossypium hirsutum) from the Malvaceae family were the most tolerant to TeA and its derivatives. Conversely, several species, including A. adenophora, A. thaliana, D. sanguinalis, Euphorbia humifusa, Mirabilis jalapa were highly sensitive to TeA, D6, D13 and D27. Chlorotic or necrotic lesions were observed in these susceptible plant leaves, indicating cell damage of photosynthetic tissues or death. This is consistent with our previous report indicating that TeA purified from A. alternata cultures was phytotoxic to several mono- and dicotyledonous plants, but that tobacco, cotton and Abutilon theophrasti, among 46 tested species, were highly tolerant to it [41]. Interestingly, D6, D13 and D27 were more phytotoxic than TeA to all species tested. Moreover, the bioactivity was enhanced with the increase in sec-alkyl side chain length (sec-hexyl > sec-pentyl > sec- butyl). Nevertheless, the hypothesis was inapplicable to compound D27 with the longest alkyl side chain of sec-heptyl group. Among the four compounds, D13 with its sec-hexyl side chain was the most herbicidal. These bioassay results are in full agreement with those about the binding affinity of the compounds for the QB site.
Evaluation of photosynthetic inhibitory activity and herbicidal efficacy
To confirm the relationship between biological inhibitory potency and binding affinity of TeA and its three selected derivatives, PSII oxygen evolving rate and fast chlorophyll fluorescence rise kinetics were measured in treated and untreated Arabidopsis plants. The rate of oxygen evolution revealed a distinct concentration-dependent decrease after compounds treatment (Fig. 5A). In general, extending the sec-alkyl side chain improved PSII inhibiting activity. The I50-value of compounds D0 (TeA), D6, D13 and D27 for the rate of PSII oxygen evolution was 201 μM, 105 μM, 87 μM and 162 μM, respectively. Thus the three derivatives were more active inhibitors of PSII oxygen evolution than TeA, particularly D6 and D13, that were about twice as potent as TeA.
Fig. 5.

Effect of active compounds D0 (TeA) and derivatives D6, D13 and D27 on photosynthetic activity of A. thaliana. (A) The rate of O2 evolution in thylakoids of A. thaliana at increasing concentrations of D0, D6, D13 or D27·H2O and p-phenylenediamine are the electron donor and acceptor, respectively. Data shown are mean values ± SD of three independent biological replicates. (B) Chl a fluorescence rise kinetics of leaf discs after 6 h treatment with 0.1% MeOH (mock), 200 μM D0, D6, D13 or D27. The top panel shows fluorescence kinetics normalized by FO and FM as Vt = (Ft - FO)/(FM-FO), and the bottom panel shows ΔVt = Vt(treated) - Vt(mock). Each data point is the average of 30 measurements from three independent biological replicates. (C-H) The concentration-dependent effect of TeA and derivatives on fluorescence parameter VJ (relative variable fluorescence at the J-step), φEo (quantum yield for PSII electron transport), ψEo (probability that an electron moves further than QA-), QA-reducing centers, RJ (number of PSII RCs with QB-site filled with compounds), and PIABS (PSII performance index). Each parameter value is the average of 30 measurements from (B). (I) Analysis of the correlation for PIABS versus φEo.
Chlorophyll fluorescence rise kinetics is an excellent indicator to determine the precise effect of different stresses on photosynthesis. To further evaluate the difference in photosynthetic inhibiting activity, fluorescence rise OJIP curves were determined after Arabidopsis leaves were exposed to TeA or its three derivatives for 6 h (Fig. S9). The major impact on the OJIP curves is a concentration-dependent fast rise of the J-step at 2 ms in the presence of any of the compounds. At 200 μM, the three derivatives led to a bigger rise of the J-step than TeA (Fig. 5B). This matches well the results of VJ as a parameter for quantization of the fluorescence level of the J-step (Fig. 5C). Occurrence of a rapid rise of the J-step is most distinctive of the OJIP curves of DCMU- and TeA-treated plants that is attributed to a large accumulation of QA reduced state (QA-) in reaction centers of PSII due to the blockage of the electron flow beyond QA [30], [23], 2015). Concomitantly, the derivatives decreased the value of φEo and ψEo in a concentration-dependent pattern at a greater extent than TeA (Fig. 5D-5E). The same tendency was observed at QA-reducing centers (Fig. 5F) and PIABS (Fig. 5H). The parameter φEo expresses the quantum yield for PSII electron transport; ψEo refers to the probability that a trapped exciton moves an electron into the electron transport chain beyond QA; PIABS reflects the overall photosynthetic activity of PSII [30]. The data in Fig. 5D-5E indicated that three derivatives (D6, D13 and D27) have higher inhibitory ability of PSII electron transport compared with that of TeA. Compound D13 is the strongest inhibitor, and D27 is a weaker inhibitor than D6. The results are further supported by the observed significant increase in the RJ parameter (Fig. 5G) of plants treated with D6, D13 and D27 compared with that of TeA-treated plants as RJ provides a measurement of the number of PSII reaction centers with their QB site filled by a PSII inhibitor [42], [31], thus corroborating their likely higher binding ability to the QB site (Fig. 5G, Table S2). A stronger inhibition of PSII electron transport activity would certainly result in a faster decrease in fraction of QA-reducing centers (Fig. 5D-5F, [31], and also led ultimately to a greater decrease in the overall PSII activity (Fig. 5H). The calculated I50 values for PIABS of compound D0 (TeA), D6, D13 and D27 were 197 μM, 102 μM, 80 μM and 168 μM, which closely correspond to those for PSII oxygen evolution rate (Fig. 5A, 5H; Table S2). A distinct linear relationship between PIABS and φEo provides strong evidence that the inhibition of electron flow beyond QA is the major determinant of the loss of overall PSII activity (Fig. 5I). The absolute value of the slope, denoted by k, represents the inhibiting potency of compounds on PSII activity. The k-value of D6 and D13 is approximately 4.4 and 4.5, which is greater than that for D27 and TeA with a k-values of 2.9 and 2.6, respectively. A greater k-value means a stronger PSII inhibitory activity. The above analyses on fluorescence data demonstrate that compounds D6 and D13 possess much stronger inhibitory ability of PSII electron transport, and D27 has a slightly higher PSII inhibiting activity relative to TeA.
To further estimate herbicidal activity of derivatives, seedlings of Arabidopsis and the two susceptible species, A. adenophora and D. sanguinalis were sprayed with the four compounds at increasing doses. Treated seedlings quickly developed typical lesions from mild to severe necrosis and wilting to death of the whole plant (Fig. 6A). At 5 days after treatment with 500 μM, the herbicidal efficacy of TeA on the three species was 25%-35%, which was significantly lower than that observed with D6 (44%-64%), D13 (53%-73%) and D27 (34–46%). At twice the dose (1000 μM), D6 and D13 caused 93%-100% mortality, followed by D27 (74%-92%) and TeA (63%-74%). When treated with D6 or D13 at the highest concentration of 2000 μM, all plants died (Fig. 6B, Table S3). The calculated ED90 values for the derivatives were much lower than that of TeA (Table S3); D13 was the most active herbicide in accordance to the results of PSII oxygen evolution and fast chlorophyll fluorescence rise kinetics.
Fig. 6.

Comparison of whole plant phytotoxicity of active compounds D0 (TeA), D6, D13 and D27 on A. thaliana, A. adenophora and D. sanguinalis. Plants were sprayed to runoff with a solution containing 0 (0.5% MeOH, mock), 250, 500 1000, 2000 μM of each D0, D6, D13 or D2 and maintained for 5 d in a growth chamber at 25 °C under around 200 μmol photons m−2 s−1 white light. (A) Photographs were taken after 5 d treatment. Results shown are representative of three independent biological replicates. (B) Control efficacy in A. thaliana, A. adenophora and D. sanguinalis seedlings after 5 d treatment of TeA, D6, D13 or D27. Data are mean values ± SD of efficacy ratings based on percentage foliage damage from three independent biological replicates from (A).
It is clear that the three derivatives (D6, D13 and D27) with longer sec-alkyl side chain of 5–7 carbon atoms are more active as herbicides given their stronger ability to inhibit photosynthesis, compared with lead TeA with sec-butyl side chain of 4 carbon atoms.
Validation of structure-activity relationship
The differential susceptibility of the three species tested to TeA and its derivatives is concordant with early evidence from chlorophyll fluorescence [41]. A. adenophora as host plant of A. alternata producing TeA was highly susceptible, while the model plant Arabidopsis was intermediate, and D. sanguinalis was more tolerant (Fig. 6, Table S3). To further clarify the relationship between herbicidal activity and chemical structure, TeA and three selected derivatives were also respectively docked to the QB site of A. adenophora and D. sanguinalis. It’s known that homology modeling technology based on amino acid sequence can reasonably simulate the three-dimensional structure of the target protein [43]. Here, utilizing the data of top ranked evolutionary related proteins with available structures from the SWISS-MODEL Template Library (Fig. 7A), the homology modeling of D1 protein structure of A. adenophora (Fig. 7B) and D. sanguinalis (Fig. 7C) were constructed using the Protein Modeling Module of Discovery Studio. Subsequently, D0 (TeA), D6, D13 and D27 were respectively modeled in the QB site of both species according to the constructed homology model. Additionally, the modeling of atrazine and DCMU at the QB site of A. adenophora (Fig. S10) and D. sanguinalis (Fig. S11) were simulated to further assess the reliability of docking procedures. Results revealed that potential location of main hydrogen bond formed between atrazine or DCMU and D1 protein of A. adenophora or D. sanguinalis is just the same as that of Arabidopsis (Figs. S1B-C), being strongly supported by previous x-ray crystallography evidence [35], [25], [8]. Fig. 7D and 7E illustrate the docked pose of four compounds at the QB binding site of A. adenophora and D. sanguinalis, respectively. For these two plants, as in Aradidopsis model, D1-Gly256 was also suggested to provide a key ligand to the carbonyl oxygen O2 of the four compounds by forming a hydrogen bond (Figs. S12-13). However, there is noticeable difference in the hydrogen bound distance and predicted interaction energy for the homology model of every compound (D0, D6, D13 and D27) binding to the QB site of Aradidopsis, A. adenophora and D. sanguinalis (Table 3). For these models, A. adenophora shows the lowest interaction energy and the shortest bond distance from D1-Gly256 to the O2 carbonyl oxygen atom, resulting in the highest herbicidal efficacy with the lowest ED90 value. On the contrary, D. sanguinalis with the lowest herbicidal efficacy for its maximum ED90 has the highest energy and the longest hydrogen bond distance. The performance of Aradidopsis belongs to the middle level of herbicidal efficacy for its medium interaction energy and bond distance in the model. Such data also provide strong support for the hypothesis that the three derivatives with longer sec-alkyl side chain of 5 to 7 carbon atoms have higher binding affinities, leading to higher herbicidal efficacy, compared with TeA with its sec-alkyl side chain of 4 carbon atoms. Further analysis indicates that there is a significant linear correlation for binding affinity versus hydrogen bond distance and for the ED90 value versus binding affinity or hydrogen bond distance in these four compounds (Fig. S14). Considering results from photosynthetic effect (Fig. 5), it is deduced that the binding affinities for the QB site should also be responsible for higher photosynthetic inhibitory activities of these three designed molecules (D6, D13 and D27). A significant linear negative relationship between the ED90 value of three plants and QED of four compounds (D0, D6, D13 and D27) (Fig. S15) indicated that herbicidal activity of a compound is directly associated with key physicochemical properties. Thus, the most plausible explanation for the improvement of inhibitory and herbicidal potency of three candidates is that extending the length of sec-alkyl side chain caused significant increase of binding ability at the QB site of the D1 protein due to optimizing their molecular physicochemical properties.
Fig. 7.

Docking of the QB binding site of D1 protein of A. adenophora and D. sanguinalis with active compounds D0 (TeA), D6, D13 or D27. (A) Search results of evolutionary related proteins with available structures by the BLAST program through the SWISS-MODEL Template Library (SMTL) for target D1 protein of A. adenophora and D. sanguinalis. The top five ranked templates are listed. The homology modeling of D1 protein structure of A. adenophora (B) and D. sanguinalis (C) based on the top 4 ranked templates. For D1 protein model of A. adenophora, PDF total energy is 9917.77 kcal/mol, PDF physical energy is 918.535 kcal/mol, DOPE Score is −36553.9, RMSD is 1.63; for D1 protein model of D. sanguinalis, PDF total energy is 11056.2 kcal/mol, PDF physical energy is 869.134 kcal/mol, DOPE Score is −36696.8, RMSD is 0.709. Here, α-helix is illustrated in red, β-pleated sheet is illustrated in blue, β-turn is illustrated in green, random coil is illustrated in white. (D) Docked poses of compounds D0, D6, D13 and D27 inside the QB binding site of A. adenophora. (E) Docked poses of compounds D0, D6, D13 and D27 inside the QB binding site of D. sanguinalis. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Table 3.
Interaction energies and distances of hydrogen bonds in the QB site of TeA and its three derivatives and their herbicidal efficacy.
| Compounds | D0 (TeA) |
D6 (sec-pentyl-TeA) |
D13 (sec-hexyl-TeA) |
D27 (sec-heptyl-TeA) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Interaction Energy (kcal/mol) | Hydrogen bond (Å) | ED90 (μM) | Interaction Energy (kcal/mol) | Hydrogen bond (Å) | ED90 (μM) | Interaction Energy (kal/mol) | Hydrogen bond (Å) | ED90 (μM) | Interaction energy (kcal/mol) | Hydrogen bond (Å) | ED90 (μM) | |
| A. thaliana | −29.35 | 3.66 | 2241.54 | −32.29 | 3.39 | 1226.53 | −33.41 | 3.24 | 1069.21 | −30.98 | 3.51 | 1644.53 |
| A. adenophora | −29.40 | 3.27 | 1705.24 | −34.06 | 3.14 | 1069.87 | −36.83 | 3.05 | 934.93 | −32.40 | 3.20 | 1321.35 |
| D. sanguinalis | −27.86 | 3.72 | 2530.86 | −30.15 | 3.51 | 1549.28 | −31.64 | 3.37 | 1366.47 | −28.83 | 3.65 | 2361.04 |
Conclusion
Structure-based ligand design in this study is a valuable tool to find new PSII inhibitors based on lead natural compound TeA. The validated molecular interaction model of TeA binding to the QB site directed the design of TeA derivatives. These newly designed derivatives were screened according to the predicted binding affinity ranking and then synthesized successfully to assess for their bioactivity. Compounds D6 and D13 were identified as stronger inhibitors of photosynthetic activity and potent herbicides than its parental TeA because of their higher binding affinity for the QB site (lower interaction energy values). The findings could be further exploited to develop novel and more potent PSII herbicides derived from TeA and even other tetramic acid.
Compliance with Ethical Requirements
This article does not contain any studies with human or animal subjects.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This project was supported by the National Key Research and Development Program of China (2017YFD0201300), Jiangsu Agriculture Science and Technology Innovation Fund (CX(21)3093) and Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX19_0617). The authors also wish to thank Prof. Hailiang Zhu (Nanjing University, China) for helpful suggestions on the synthesis of chemicals.
Footnotes
Peer review under responsibility of Cairo University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jare.2021.12.001.
Contributor Information
Shiguo Chen, Email: chenshg@njau.edu.cn.
Sheng Qiang, Email: wrl@njau.edu.cn.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Sun H. In: A Practical Guide to Rational Drug Design. Sun H., editor. Elsevier; Amsterdam-Boston-Cambridge-Heidelberg-London-New York-Oxford-Paris-San Diego-San Francisco-Singapore-Sydney-Tokyo: 2016. Homology modeling and ligand-based molecule design; pp. 109–160. [Google Scholar]
- 2.Fedtke C., Duke S.O. In: Plant Toxicology. 4th ed. Hock B., Elstner E.F., editors. Marcel Dekker; New York: 2005. Herbicides; pp. 247–330. [Google Scholar]
- 3.Xiong J., Jee G., Subramaniam S. Modeling of the D1/D2 proteins and cofactors of the photosystem II reaction center: Implications for herbicide and bicarbonate binding. Protein Sci. 1996;5:2054–2073. doi: 10.1002/pro.5560051012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kamiya N., Shen J.R. Crystal structure of oxygen-evolving photosystem II from Thermosynechococcus vulcanus at 3.7-Å resolution. P Natl Acad Sci USA. 2003;100:98–103. doi: 10.1073/pnas.0135651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shen J.R. The structure of photosystem II and the mechanism of water oxidation in photosynthesis. Annu Rev Plant Biol. 2015;66:23–48. doi: 10.1146/annurev-arplant-050312-120129. [DOI] [PubMed] [Google Scholar]
- 6.Trebst A. In: The Triazine Herbicides 50 Years Revolutionizing Agriculture. LeBaron H.M., McFarland J.E., Burnside O.C., editors. Elsevier; San Diego: 2008. The mode of action of triazine herbicides in plants; pp. 101–110. [Google Scholar]
- 7.Oettmeier W. Herbicide resistance and supersensitivity in photosystem II. CMLS, Cell Mol Life Sci. 1999;55:1255–1277. doi: 10.1007/s000180050370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lancaster C.R.D., Michel H. Refined crystal structures of reaction centres from Rhodopseudomonas viridis in complexes with the herbicide atrazine and two chiral atrazine derivatives also lead to a new model of the bound carotenoid. J Mol Biol. 1999;286:883–898. doi: 10.1006/jmbi.1998.2532. [DOI] [PubMed] [Google Scholar]
- 9.Royles B.J.L. Naturally occurring tetramic acids: structure, isolation, and synthesis. Chem Rev. 1995;95:1981–2001. [Google Scholar]
- 10.Schobert R. Domino syntheses of bioactive tetronic and tetramic acids. Naturwissenschaften. 2007;94:1–11. doi: 10.1007/s00114-006-0152-8. [DOI] [PubMed] [Google Scholar]
- 11.Schobert R., Schlenk A. Tetramicand tetronic acids: an update on new derivatives and biological aspects. Bioorg Med Chem. 2008;16:4203–4221. doi: 10.1016/j.bmc.2008.02.069. [DOI] [PubMed] [Google Scholar]
- 12.Lou L., Qian G., Xie Y., Hang J., Chen H., Zaleta-Rivera K., et al. Biosynthesis of HSAF, a tetramic acid-containing macrolactam from Lysobacter enzymogenes. J Am Chem Soc. 2011;133:643–645. doi: 10.1021/ja105732c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stock F., Syrpas M., van Creveld S.G., Backx S., Blommaert L., Dow L., et al. N-acyl homoserine lactone derived tetramic acids impair photosynthesis in Phaeodactylum tricornutum. ACS Chem Biol. 2019;14:198–203. doi: 10.1021/acschembio.8b01101. [DOI] [PubMed] [Google Scholar]
- 14.Montemurro N., Visconti A. In: Alternaria Biology, Plant Diseases and Metabolites. Chelkowski J., Visconti A., editors. Elsevier Science; Amsterdam-London-New York-Tokyo: 1992. Alternaria metabolites-chemical and biological data; pp. 449–541. [Google Scholar]
- 15.Ebbole D.J. Magnaporthe as a model for understanding host-pathogen interactions. Annu Rev Phytopathol. 2007;45:437–456. doi: 10.1146/annurev.phyto.45.062806.094346. [DOI] [PubMed] [Google Scholar]
- 16.Yun C.-S., Motoyama T., Osada H. Biosynthesis of the mycotoxin tenuazonic acid by a fungal NRPS–PKS hybrid enzyme. Nat Commun. 2015;6:8758. doi: 10.1038/ncomms9758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meazza G., Scheffler B.E., Tellez M.R., Rimando A.M., Romagni J.G., Duke S.O., et al. The inhibitory activity of natural products on plant p-hydroxyphenylpyruvate dioxygenase. Phytochemistry. 2002;59:281–288. doi: 10.1016/s0031-9422(02)00121-8. [DOI] [PubMed] [Google Scholar]
- 18.Bjørk P.K., Rasmussen S.A., Gjetting S.K., Havshøi N.W., Petersen T.I., Ipsen J.Ø., et al. Tenuazonic acid from Stemphylium loti inhibits the plant plasma membrane H+-ATPase by a mechanism involving the C-terminal regulatory domain. New Phytol. 2020;226:770–784. doi: 10.1111/nph.16398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen S., Qiang S. Recent advances in tenuazonic acid as a potential herbicide. Pest Biochem Phys. 2017;143(9):252–257. doi: 10.1016/j.pestbp.2017.01.003. [DOI] [PubMed] [Google Scholar]
- 20.Zhou B., Wang H., Meng B., Wei R., Wang L., An C., et al. An evaluation of tenuazonic acid, a potential biobased herbicide in cotton. Pest Manag Sci. 2019;75:2482–2489. doi: 10.1002/ps.5402. [DOI] [PubMed] [Google Scholar]
- 21.Nolte M.J., Steyn P.S., Wessels P.L. Structural investigations of 3-acylpyrrolidine-2,4-diones by nuclear magnetic resonance spectroscopy and X-ray crystallography. J Chem Soc, Perkin Trans. 1980;1:1057–1065. [Google Scholar]
- 22.Schobert R, Jagusch C, Melanophy C, Mullen G. Synthesis and reactions of polymer-bound Ph3P=C=C=O: a quick route to tenuazonic acid and other optically pure 5-substituted tetramates. Org Biomol Chem. 2004;2:3524–3529. doi: 10.1039/b412779j. [DOI] [PubMed] [Google Scholar]
- 23.Chen S., Xu X., Dai X., et al. Identification of tenuazonic acid as a novel type of natural photosystem II inhibitor binding in QB-site of Chlamydomonas reinhardtii. BBA. 2007;1767(4):306–318. doi: 10.1016/j.bbabio.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Chen S., Ying C., Qiang S., Zhou F., Dai X. Chloroplastic oxidative burst induced by tenuazonic acid, a natural photosynthesis inhibitor, triggers cell necrosis in Eupatorium adenophorum Spreng. BBA. 2010;1797:391–405. doi: 10.1016/j.bbabio.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 25.Sinning I. Herbicide binding in the bacterial photosynthetic reaction center. Trends Biochem Sci. 1992;17:150–154. doi: 10.1016/0968-0004(92)90324-3. [DOI] [PubMed] [Google Scholar]
- 26.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiang S, Zhang Q, Wang Z, Zhu H, Chen S. Synthesis of alkyl glycine manufacturing method, China Patent; 2018. CN108358797A.
- 28.Yang C, Qiang S, Huang L, Zhang P, Zhu Z. Synthesis of tenuazonic acid and isotenuazonic acid manufacturing method, China Patent; 2008. ZL200610038765.X.
- 29.Lu Y., Hall D.A., Last R.L. A small zinc finger thylakoid protein plays a role in maintenance of photosystem II in Arabidopsis thaliana. Plant Cell. 2011;23:1861–1875. doi: 10.1105/tpc.111.085456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strasser RJ, Tsimilli-Michael M, Srivastava A. Analysis of the chlorophyll a fluorescence transient. In: Papageorgiou GC, Govindjee, editors. Chlorophyll a Fluorescence: A Signature of Photosynthesis. Netherlands: Springer Press; 2004. p. 321–62.
- 31.Chen S., Strasser R.J., Qiang S. In vivo assessment of effect of phytotoxin tenuazonic acid on PSII reaction centers. Plant Physiol Bioch. 2014;84:10–21. doi: 10.1016/j.plaphy.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Ferreira K.N., Iverson T.M., Maghlaoui K., Barber J., Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. [DOI] [PubMed] [Google Scholar]
- 33.Loll B., Kern J., Saenger W., Zouni A., Biesiadka J. Towards complete cofactor arrangement in the 3.0Å resolution structure of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. [DOI] [PubMed] [Google Scholar]
- 34.Lambreva M.D., Russo D., Polticelli F., Scognamiglio V., Antonacci A., Zobnina V., et al. Structure/function/dynamics of photosystem II plastoquinone binding sites. Curr Protein Pept Sc. 2014;15:285–295. doi: 10.2174/1389203715666140327104802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trebst A. The three-dimensional structure of the herbicide binding niche on the reaction center polypeptides of photosystem II. Z Naturforsch. 1987;42c:742–750. [Google Scholar]
- 36.Bickerton G.R., Paolini G.V., Besnard J., Muresan S., Hopkins A. Quantifying the chemical beauty of drugs. Nat Chem. 2012;4:90–98. doi: 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meier S., Hanif M., Adhireksan Z., Pichler V., Novak M., Jirkovsky E., et al. Novel metal(II) arene 2-pyridinecarbothioamides: a rationale to orally active organometallic anticancer agents. Chem Sci. 2013;4:1837–1846. [Google Scholar]
- 38.Ribeiro F.F., Júnior F.J.B.M., Silva M.S., da Scotti M.T., Scotti L. Computational and investigative study of flavonoids active against Trypanosoma cruzi and Leishmania spp. Nat Prod Commun. 2015;10:917–920. [PubMed] [Google Scholar]
- 39.Scotti L., Júnior F.J.B.M., Ishiki H.M., Ribeiro F.F., Duarte M.C., Santana G.S., et al. In: Natural and Artificial Flavoring Agents and Food Dyes. Grumezescu A.M., Holban A.M., editors. Academic Press; 2018. Computer-aided drug design studies in food chemistry; pp. 261–297. [Google Scholar]
- 40.Chen S., Zhou F., Yin C., Strasser R.J., Qiang S., Yang C. Application of fast chlorophyll a fluorescence kinetics to probe action target of 3-acetyl-5-isopropyltetramic acid. Environ Exp Bot. 2011;71:269–279. [Google Scholar]
- 41.Chen S., Kang Y., Zhang M., et al. Differential sensitivity to the potential bioherbicide tenuazonic acid probed by the JIP-test based on fast chlorophyll fluorescence kinetics. Environ Exp Bot. 2015;112(112):1–15. [Google Scholar]
- 42.Lazár D., Brokeš M., Nauš J., Dvořák L. Mathematical modeling of 3-(3’,4’-dichlorophenyl)-1,1-dimethylurea action in plant leaves. J Theor Biol. 1998;191:79–86. doi: 10.1006/jtbi.1997.0566. [DOI] [PubMed] [Google Scholar]
- 43.Fiser A., Sali A. Modeller: generation and refinement of homology-based protein structure models. Method Enzymol. 2003;374:461–491. doi: 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.