

Summary
In drug development, a major reason for attrition is the lack of understanding of cellular mechanisms governing drug toxicity. The black-box nature of conventional classification models has limited their utility in identifying toxicity pathways. Here we developed DTox (deep learning for toxicology), an interpretation framework for knowledge-guided neural networks, which can predict compound response to toxicity assays and infer toxicity pathways of individual compounds. We demonstrate that DTox can achieve the same level of predictive performance as conventional models with a significant improvement in interpretability. Using DTox, we were able to rediscover mechanisms of transcription activation by three nuclear receptors, recapitulate cellular activities induced by aromatase inhibitors and pregnane X receptor (PXR) agonists, and differentiate distinctive mechanisms leading to HepG2 cytotoxicity. Virtual screening by DTox revealed that compounds with predicted cytotoxicity are at higher risk for clinical hepatic phenotypes. In summary, DTox provides a framework for deciphering cellular mechanisms of toxicity in silico.
Keywords: drug toxicity, deep learning, model interpretation, molecular toxicology
Graphical abstract
Highlights
-
•
DTox is a deep learning model for toxicity prediction with broad applicability
-
•
It provides an interpretation framework to infer toxicity pathways of compounds
-
•
We improve the interpretability of toxicity prediction without sacrificing accuracy
-
•
DTox’s interpretation framework deciphers cellular mechanisms of toxicity in silico
The bigger picture
In drug development, a major reason for attrition is the lack of understanding of cellular mechanisms governing drug toxicity. It is challenging to explain the toxicity outcomes of newly developed compounds with limited prior knowledge. To address the challenge, we present DTox (Deep learning for Toxicology), a deep learning model incorporated with extensive knowledge from pathway ontology. DTox is a highly efficient learning model with good predictive performance. It is applicable to all compounds because it requires only chemical structure as model input. More importantly, the knowledge-guided structure of DTox enables us to identify network paths connecting query compounds to toxicity outcomes via target proteins, functional pathways, and general biological processes. Such paths can be viewed as mechanistic interpretation of toxicity and facilitate experimental investigation. We employ existing experimental datasets to validate the mechanistic interpretation by DTox and demonstrate its biological significance.
Toxicity assessment is a critical step in drug development. To overcome the black-box nature of conventional classification models, Hao et al. propose an interpretable model named DTox (deep learning for toxicology) for predicting compound response to toxicity assays and inferring toxicity pathways of individual compounds. Validation studies using experimental datasets demonstrate the effectiveness of DTox in rediscovering known mechanisms, differentiating distinctive mechanisms, and recapitulating cellular activities leading to toxicity. DTox will benefit mechanistic studies in toxicology by generating testable hypotheses for further investigation.
Introduction
With the application of quantitative high-throughput screening techniques, toxicity testing programs1,2 have generated millions of data points regarding the response of biological systems to important chemical libraries, both in vitro and in vivo. Specifically, in the Tox21 program,1 over 8,500 compounds were tested for a variety of toxicity endpoints, including stress response, genotoxicity, cytotoxicity, developmental toxicity, etc. These toxicity profiles can assist with probing how chemicals interact with proteins and pathways to trigger a certain outcome, and thus shed light on cellular mechanisms of toxicity.3 Furthermore, with the help of machine learning algorithms, researchers can identify the chemical or biological patterns of a compound that might be predictive of adverse health outcomes in humans.4,5
Previous studies have modeled toxicity endpoints from physiochemical properties of compounds using a wide range of supervised learning algorithms, including k-nearest neighbors,6,7 Bayesian matrix factorization,8 support vector machines,7,9 random forests (RFs),6,10 gradient boosting (GB),11 and more recently, deep neural networks.12, 13, 14 Even though most of these algorithms achieved decent predictive performance, none of them could overcome the trade-off between accuracy and interpretability. As algorithmic design gets more complex, it becomes challenging to interrogate how each input feature contributes to the eventual prediction.15 A few post hoc explanation techniques, such as Local Interpretable Model-agnostic Explanations (LIME)16 and deep learning important features (DeepLIFT),17 were developed to address the challenge. Nevertheless, these techniques often draw criticism in that they provide only an approximate explanation with locally fitted naive models. Thus, they may not reflect the real behavior of the original model.18 More critically, the setting of existing toxicity prediction models has limited the explanation of contributions from structural properties or target proteins while interactions with pathways remain largely uncharacterized. For toxicologists, the behavior of pathways proves crucial in deciphering the cellular activities induced by a compound and understanding how target proteins, specific pathways, and biological processes trigger the toxicity outcome as a whole.5 Therefore, a toxicity prediction model that achieves interpretability at both the gene and the pathway level is urgently needed.
Recent developments in visible neural networks (VNNs) have overcome the accuracy-interpretability trade-off. VNN is a type of neural network whose structure is guided by extensive knowledge from biological ontologies and pathways. The incorporation of ontological hierarchy in VNN forms a meaningful network structure that connects input gene features to output response via hidden pathway modules, making the model highly interpretable at both the gene and the pathway level. In a pioneering study, Ma et al.19 built a VNN with 2,526 Gene Ontology and Clique-eXtracted Ontology terms for predicting growth rate of yeast cells from gene deletion genotypes. The authors were also able to rediscover key ontology terms responsible for cell growth by examining the structure of the VNN. Subsequent studies have extended the VNN model for learning tasks regarding human cells, such as predicting drug response and synergy in cancer cell lines,20 modeling cancer dependencies,21 and stratifying prostate cancer patients by treatment-resistance state.22 It is our working hypothesis that VNNs can address the limitations of existing toxicity prediction models because of their incorporation of pathway knowledge and the resulting high interpretability. In this study, we employed the Reactome23 pathway hierarchy to develop a VNN model—namely, DTox (Deep learning for Toxicology)—for predicting compound response to 15 toxicity assays. Further, we developed a DTox interpretation framework for identifying VNN paths that can explain the toxicity outcome of compounds. We connected the identified VNN paths to cellular mechanisms of toxicity by showing their involvement in the target pathway of respective assay, their differential expression in the matched Library of Integrated Network-Based Cellular Signatures (LINCS) experiment,24 and their compliance with screening results from mechanism of action assays. We applied the DTox models of cell viability to perform a virtual screening of ∼700,000 compounds and linked predicted cytotoxicity scores with clinical phenotypes of drug-induced liver injury (DILI). We conclude with a discussion of potential discoveries made by DTox, some of which have already been validated in previous studies. Our code can be accessed openly at https://github.com/yhao-compbio/DTox. In general, the DTox interpretation framework will benefit in silico mechanistic studies and generate testable hypotheses for further investigation.
Results
Design and training of DTox for predicting compound response to toxicity assays
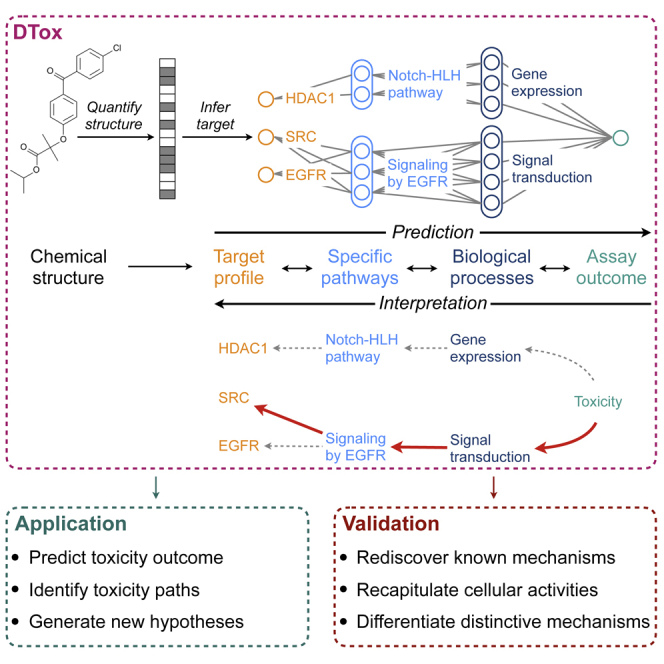
The purpose of DTox is to predict the outcome of interest from chemical structure of compounds and to explain the predicted outcome with activities of proteins and pathways. To train the model, DTox takes in a labeled dataset that specifies the 2D structural representation of each compound (in the form of SMILES string), along with the binary outcome of a screening assay (active or inactive). Because a VNN model typically starts with input layers consisting of gene or protein features, to fill in the gap, we first quantified the structure of each compound using a 166-bit MACCS fingerprint (each bit represents the answer to a yes/no question regarding chemical structure), then applied our previously developed method, named TargetTox,25 to derive a target profile of each compound (Experimental procedures). TargetTox was pre-trained on experimentally measured compound-target binding affinities to infer the target binding probability of each compound from its MACCS fingerprint. The derived profile contains 361 target proteins, spanning six functional categories: enzymes, G protein-coupled receptors (GPCRs), catalytic receptors, ion channels, nuclear hormone receptors, and transporters (Figure S1). We designed a VNN structure (Figure 1; Experimental procedures) that connects target proteins (input features) to assay outcomes (output response) via Reactome pathways (hidden modules). By our design, each pathway is represented by 1–20 neurons depending on its size. Connections between input features and the first hidden layer are constrained to follow protein-pathway annotations, while the connections among hidden layers are constrained to follow child-parent pathway relations. The incorporation of pathway hierarchy makes DTox models highly interpretable, in contrast with conventional black-box neural network models.
Figure 1.

Modeling compound response to toxicity assay with DTox
For toxicity prediction, the chemical structure of a compound is quantified using MACCS fingerprint before being converted to target profile by our previously developed method, TargetTox. The target profile is then fed into a VNN, whose structure is guided by Reactome pathway hierarchy. Specific pathways and biological processes are coded as hidden modules with a series of neurons. For model interpretation, the network output is propagated backward onto each neuron as relevance score using the layer-wise relevance propagation technique. A permutation-based strategy is then employed to identify the VNN paths of high relevance. Each path connects a compound to its toxicity outcome via the target protein, specific pathways, and biological process.
We trained DTox models on 15 datasets (Table S1; Experimental procedures) from the Tox21 high-throughput screening program.1 A DTox model was learned separately for each dataset to predict the active/inactive status of compounds (i.e., screening results of the toxicity assay). On average, each dataset contains 5,178 compounds available for DTox training, including 746 active compounds and 4,432 inactive compounds (Figure S2A). To assess model overfitting during the training process, we withheld an independent testing set from each dataset to monitor the evolution of the loss function and an early stopping criterion to conclude training when overfitting starts to occur (Experimental procedures). We discovered that while training loss continues to decrease, testing loss stops decreasing after 100–150 epochs for most datasets, a sign of model overfitting (Figure S3). Therefore, when overfitting was detected, DTox would conclude training and output the optimal model when minimal testing loss was reached. On average, the optimal DTox model was learned over 140 ± 16 epochs. We implemented hyperparameter tuning by grid search to derive an optimal model for the prediction of each assay outcome (Table S2; Experimental procedures). On average, an optimal DTox model contains 412 hidden pathway modules (Figure S2B) and 45,623 neural network parameters (Figure S2C). The average ratio between number of training samples versus number of network parameters is 0.13 ± 0.03 (Figure S2D), with the estrogen receptor (ER) agonist assay model being the highest (0.31) and the hedgehog antagonist assay model being the lowest (0.07). Compared with a conventional multi-layer perceptron (MLP) model, the DTox model has far fewer network parameters. On average, the number of network parameters for a DTox accounts for only 3% of the number for a matched MLP (Figure S2E).
To customize the network structure for prediction of each assay outcome, we made the root biological process a hyperparameter (Experimental procedures). This means through hyperparameter tuning, we can choose a branch or combination of branches from the Reactome pathway hierarchy that result in the best predictive performance for an assay of interest (Figure 2A). For instance, signal transduction pathways alone can deliver the optimal model for HEK293 cell viability assay, while additional pathways from the immune system are required for HepG2 cell viability prediction, suggesting a potential role of immune response in HepG2 cytotoxicity. In general, models built with multiple branches perform better than models built with a single branch.
Figure 2.

Prediction of compound response to 15 toxicity assays
(A) Heatmap showing the training performance of VNN built under different combinations of root biological processes (shown as upset plot at the bottom). To facilitate comparison, the model performance is normalized within each assay using Z-transform. The optimal combination for each assay is highlighted with a red star. The name of each assay is annotated on the left, with the name of the assay cell line included in parentheses.
(B) Bar plot showing the validation performance in all 15 Tox21 datasets. The performance of DTox is compared against three other models: a multi-layer perceptron with the same number of hidden layers and neurons as DTox (MLP), random forest (RF), and gradient boosting (GB). Performance is measured by two metrics: area under ROC curve and balanced accuracy, with error bar showing the 95% confidence interval.
AhR, aryl hydrocarbon receptor; AP-1, activator protein-1; ARE, antioxidant response element; AR-MDA, androgen receptor in MDA-kb2 AR-luc cell line; CAR, constitutive androstane receptor; ER-BG1, estrogen receptor in BG1 cell line, PR-BLA, progesterone receptor in PR-UAS-bla HEK293T cell line; PXR, pregnane X receptor; RAR, retinoid acid receptor; ROR, retinoid-related orphan receptor.
DTox can achieve the same level of performance as complex classification algorithms
We validated the predictive performance of DTox models on held-out validation sets, which on average contain 1,295 compounds per assay. The optimal models of all 15 assays exhibit an area under the receiver operating characteristic (ROC) curve (AUROC) greater than 0.7 (0.7–0.8: 6 models; 0.8–0.9: 9 models). Similarly, 14 models exhibit a balanced accuracy above 0.55 (0.55–0.65: 9 models; 0.65–0.75: 4 models; >0.75: 1 model) except for the optimal model of the activator protein-1 (AP-1) signaling agonist assay. We then compared the optimal performance of DTox against three other classification algorithms (Figure 2B; Table S3; Experimental procedures). Comparing DTox with a matched MLP model, we observed one assay where DTox significantly outperformed MLP in balanced accuracy (pregnane X receptor [PXR] agonist) and two assays in the opposite direction (HEK293 and HepG2 cell viability). Comparing DTox with RF and GB, we observed one assay where DTox significantly outperformed both RF and GB (constitutive androstane receptor agonist) and one assay in the opposite direction (AP-1 signaling agonist). In general, the DTox model achieved the same level of predictive performance as these well-established classification algorithms.
To evaluate the degree to which DTox benefits from the incorporation of pathway knowledge, we performed shuffling analysis (Figure S4; Table S3; Experimental procedures) and compared the predictive performance of original DTox models against alternative models built on three different layouts: shuffled ontology hierarchy (i.e., child-parent pathway relationships are perturbed), shuffled feature profile (i.e., protein-pathway annotations are perturbed), and shuffled outcome as a negative control (i.e., input data label is incorrect). We observed that shuffled feature profiles significantly impacted the predictive performance of DTox, as the resulting models exhibited random performance resembling negative controls from the shuffled outcome, suggesting the importance of correct protein-pathway annotations in DTox. By contrast, shuffled ontology hierarchy moderately impacted the predictive performance of DTox, because we observed only two assays where it resulted in a significant drop of balanced accuracy (ER agonist and retinoid-related orphan receptor gamma [RORγ] antagonist). Notably, the outcomes for both assays are directly related to a specific nuclear receptor transcription pathway (Figure 3A), as opposed to other complex outcomes that involve multiple pathways (e.g., mitochondria toxicity, cytotoxicity). That may explain why shuffled ontology hierarchy had a higher impact on these two assays given that connections to the specific pathway would be disturbed in the shuffling.
Figure 3.

Validation of identified VNN paths by known mechanisms
(A) Bar plots comparing the observed versus expected proportion of validated compounds in four nuclear receptor assays. The “ground truth” VNN path placed at the top represents the known mechanism of transcription activation by nuclear receptor. A compound is considered to be validated by the known mechanism if the ground truth path is identified by DTox. The expected proportion is computed by random sampling, with the histogram and fitted density curve showing the sampled distribution (95% confidence interval shown as error bar).
(B) Line charts comparing the proportion of validated compounds (y axis) among models. Performance of DTox interpretation framework (DTox) is compared against two other methods: Read-across (RAx) with knowledge source from ComptoxAI or DrugBank, LIME with strict or lax threshold for target feature relevance. RAx models were implemented under five different thresholds of Tanimoto coefficient (TC; x axis). A compound is considered to be validated if it can be connected to the nuclear receptor of interest.
Development of a DTox interpretation framework for explaining VNN predictions
A fundamental advantage of VNN over other classification algorithms lies in its high interpretability. The incorporation of pathway hierarchy enables us to reason through hidden layers of VNN for mechanistic interpretation. Therefore, we developed a DTox interpretation framework to identify paths from VNN that can explain the toxicity outcome of a compound (Figure 1; Experimental procedures). The framework accepts the derived target profile of each compound along with the trained DTox model that specifies learned weights for each hidden neuron. Each identified path links together a root biological process, its descendant pathway modules, and a target protein feature. The framework has two hyperparameters: γ and ε. γ controls the stability of interpretation results, while ε controls sparsity. We evaluated the effect of hyperparameter settings on identified VNN paths (Figure S5; Experimental procedures). We observed that the set of identified paths exhibits consistently high similarity across distinct hyperparameter settings, as the average Jaccard Index reaches 0.70. Due to the high similarity, we used only the VNN paths identified from one setting (γ = 0.001, ε = 0.1) for the following validation analyses (Table S4).
DTox can rediscover mechanisms of transcription activation by nuclear receptor
To evaluate whether DTox can rediscover known mechanisms for a toxicity outcome, we looked for “ground truth” from the VNN paths identified for four nuclear receptor assays: androgen receptor (AR) antagonist, ER agonist, retinoic acid receptor (RAR) antagonist, and RORγ antagonist. Each of the four assays measures compound response to a specific nuclear receptor transcription pathway. Therefore, we established ground truth as the VNN path that links together the root process of gene expression, nuclear receptor transcription pathway, and the specific target receptor (AR, ER⍺, RARβ, RORγ; Figure 3A). In three of the four nuclear receptor assays, our framework was able to identify the ground truth path for at least 29% of all active compounds (ER⍺: 29%, RARβ: 31%, RORγ: 54%). Comparing with the expected baseline performance from identifying by chance (Experimental procedures), our framework improved the proportion by at least 2-fold.
We also compared the interpretation performance by DTox against two state-of-the-art methods (Experimental procedures); namely, LIME, a popular interpretation method for explaining predictions of classification algorithms; and Read-across (RAx), a similarity-based inference technique commonly used in the field of toxicology. Note that neither method provides a mechanism for incorporating pathway knowledge. As a result, they can connect active compounds to toxicity outcome only via target proteins, while DTox can provide sample-level explanations linking compounds, target proteins, pathways, and toxicity outcomes. Nevertheless, in three of the four nuclear receptor assays (ER⍺, RARβ, and RORγ), DTox exhibits the best interpretation performance, while the other two methods display major methodological shortcomings (Figure 3B). Specifically, interpretation performance by LIME is dependent on the adopted threshold for feature relevance, because a stricter threshold can significantly deteriorate the performance (e.g., ER and RORγ). Inference by RAx, in contrast, is heavily dependent on the knowledge source, as well as the adopted threshold for similarity measurement. When little existing knowledge could be extracted for the target of interest, RAx would suffer from poor performance (e.g., RARβ and RORγ). Despite the strong performance in general, DTox failed to identify the ground truth path for AR antagonists (Figures 3A and 3B). Instead, DTox interpretation linked AR antagonists to target proteins such as integrins, protein kinase A, or protein tyrosine phosphates, which have been shown to regulate the function of AR.26,27 One possible explanation is that AR antagonists could interact with a variety of off-targets, making it difficult for DTox to aggregate the signal on a single receptor.
DTox can recapitulate cellular activities induced by aromatase inhibitors and PXR agonists
To evaluate whether DTox can recapitulate cellular activities induced by active compounds, we studied the differential expression of VNN paths identified for four assays: aromatase inhibitor, mitochondria toxicity, PXR agonist, and HepG2 cell viability (Experimental procedures). In total, we obtained the gene expression profile measured from 321 LINCS experiments in which an active compound was used to treat the assay cell line (121 experiments for aromatase inhibitor assay, 54 for mitochondria toxicity assay, 101 for PXR agonist assay, and 45 for HepG2 cell viability assay). Of all 321 experiments, we found 161 (50%) cases where DTox’s interpretation framework was able to identify at least one differentially expressed VNN path. On average, 3.8% ± 0.6% of VNN paths identified by our framework were found to be differentially expressed, which is significantly higher than the expected proportion by chance (2.4% ± 0.3%; p = 2.5e-3). We then performed the comparison separately by assay and dose-time combinations (Figure 4A). In the aromatase inhibitor assay, our framework outperformed the expected proportion across all three dose-time combinations. In the PXR agonist assay, our framework outperformed the expected proportion among the two groups of experiments conducted 24 h after treatment. In the HepG2 cell viability assay, although no overall difference was detected among experiments conducted 6 h after treatment, our framework was still able to identify a relatively high proportion of differentially expressed VNN paths for individual compounds such as cilnidipine (21.4%), cyclopamine (12.5%), and chloroxine (10%).
Figure 4.

Validation of identified VNN paths by differential expression
(A) A VNN path is considered to be differentially expressed (DE) if all pathways along the path are enriched for DE genes from the matched LINCS experiment. The validation analysis is performed for four assays (columns) in three different dose-time groups (rows), with each scatterplot comparing the observed versus expected proportion of DE paths for a single group. The observed proportion is computed with VNN paths identified for each compound, while the expected proportion is computed with all possible VNN paths. A Wilcoxon signed-rank test is employed to examine whether the average observed proportion of each group is significantly higher than the average expected proportion (p value and FDR shown at the bottom right). The diagonal is shown as black dashed line, with compounds in the upper triangle (observed > expected) shown in blue and compounds in the lower triangle (observed < expected) shown in gray. Compounds with the top five observed proportions in each group are annotated with their names.
(B) Bar plot showing the DE VNN paths that are recurrently identified for at least five aromatase inhibitors. Each VNN path is named after its lowest-level pathway. Paths that contain the “transcriptional regulation by TP53” pathway are highlighted in salmon, while the remaining paths are colored in cyan.
Based on the results of differential expression analysis, induced cellular activities appear to be more consistent among aromatase inhibitors compared with the other three assays, as we discovered 10 differentially expressed VNN paths that are recurrently identified for at least five aromatase inhibitors (Figure 4B). By contrast, we discovered only one such VNN path for the other three assays combined. Interestingly, “transcriptional regulation by TP53” and its descendant pathways are involved in 6 of the 10 discovered VNN paths, suggesting a potential mechanism for regulation of aromatase by p53 in the MCF-7 aro estrogen-responsive element (MCF-7aro/ERE) breast cancer cell line, a finding supported by a previous study.28 In addition to p53, interleukin-4 and interleukin-13 also appear to play an important role in regulation of aromatase, because the relevant VNN path is linked to 17 aromatase inhibitors by differential expression. This finding is worth further experimental investigation.
DTox can differentiate distinctive mechanisms leading to HepG2 cytotoxicity
Next, we sought to explain the compound-induced cytotoxicity in HepG2 cells using VNN paths identified for the HepG2 cell viability assay. A recent review paper29 summarized four major mechanisms leading to cell death in DILI: (1) tumor necrosis factor receptors 1 and 2 (TNFR1/2) mediated apoptosis via caspase activation and pro-survival inhibition, (2) MST1/2 mediated apoptosis via Hippo signaling, (3) immune response activation via MHC class II antigen presentation, and (4) TLR3/4 mediated necrosis (Figure 5A). Because the HepG2 cell line was derived from liver tissue, we can use the four mechanisms as a reference for compound-induced cytotoxicity in HepG2 cells. We identified nine Reactome pathways that participate in the four mechanisms (Figure 5A). We then mapped HepG2-cytotoxic compounds to the nine Reactome pathways via VNN paths identified by our framework (Figure S5). Of all 1,120 cytotoxic compounds, 707 (63%) compounds are mapped to at least one of the nine cell death-related pathways (Figure S6), while the remaining 413 (37%) compounds are mainly linked to HepG2 cytotoxicity via the GPCR, mTOR, and Rho GTPase signaling pathways (Figure S7). We performed hierarchical clustering on the mapping and identified two compound clusters (Figure S6). Compounds in the first cluster are linked to cytotoxicity via apoptosis, while compounds in the second cluster are linked to cytotoxicity via immune activation and necrosis. Nevertheless, we discovered a few compounds that exhibit characteristics of both clusters. For instance, according to our framework, mifepristone, a medical abortion drug, causes cytotoxicity in HepG2 cells by activating both apoptosis (via mitogen-activated protein kinase 1 [MAPK1]/MAPK3 signaling and Hippo signaling) and necrosis (via TLR3 and TLR4 cascade), a finding supported by previous studies30, 31, 32 (Figure 5D). In addition, our framework was able to link mifepristone with its therapeutic target—the glucocorticoid receptor—via PTK6 signaling (Table S4). The other therapeutic target of mifepristone—the progesterone receptor—is not in the VNN.
Figure 5.

In-depth analysis of HepG2 cytotoxicity using identified VNN paths
(A) Established mechanisms for cell death in drug-induced liver injury. Reactome pathways relevant to the mechanisms are identified and used as reference for the analysis.
(B and C) Survival plots comparing the pathway relevance scores among active (orange curve) versus inactive (gray curve) compounds of two mechanisms of action assays: caspase-3/7 induction (B) and disruption of the mitochondrial membrane potential (C). Comparisons are made for nine cell death-related pathways, with each plot showing the comparison for a single pathway. Red star at the top right denotes that the pathway is related to the respective mechanism of action. Log-rank test is employed to examine whether the two distributions in each plot are significantly different (FDR value shown at the bottom left).
(D) Network diagram showing the simplified DTox structure connecting mifepristone (triangle node) to the HepG2 cytotoxicity (rectangle node) via pathway modules (round nodes). Pathways with relevance score > 0 are colored in purple, with the scale proportional to relevance scale. The VNN paths identified for mifepristone by DTox are shown in solid lines, while the rest are shown in dashed lines.
(E and F) Heatmaps showing the enrichment of nine cell death-related pathways among compounds associated with 20 drug-induced liver injury phenotypes (E) and among compounds of 14 ATC classes (F). Cells are colored based on odds ratio. Fisher’s exact test is employed to examine the significance of enrichment (asterisk denotes FDR < 0.05).
VOD/SOS, veno-occlusive disease and sinusoidal obstruction syndrome.
To evaluate whether DTox can differentiate distinctive mechanisms leading to HepG2 cytotoxicity, we looked for concordance between the assigned pathway relevance and screening results from two mechanism of action assays (included in the Tox21 datasets). The first assay we studied measures caspase-3/7 induction in HepG2 cells. Caspase-3 and caspase-7 are key executioners of apoptosis.33 They are involved in TNFR1/2-mediated apoptosis and YAP/TAZ phosphorylation of Hippo signaling34 (Figure 5A). Accordingly, we compared the assigned relevance scores between caspase-3/7+ and caspase-3/7− compounds regarding TNFR1-induced proapoptotic signaling and Hippo signaling (Figure 5B). Overall, we did not observe significantly higher relevance among caspase-3/7+ compounds regarding the two signaling pathways (false discovery rate [FDR] = 0.31 and 0.42, respectively). However, for Hippo signaling, we did observe higher pathway relevance among caspase-3/7+ compounds above the 90th percentile of two distributions (highlighted in Figure 5B), hence a partial agreement between assigned pathway relevance and caspase-3/7 induction screening. By contrast, the pattern among top-ranked compounds was not observed in other cell death-related pathways except for MHC class II antigen presentation (FDR = 0.02; Figure 5B), suggesting a potential role of caspase-3/7 in MHC class II antigen presentation, a finding worth further investigation.
The second assay we studied measures disruption of the mitochondrial membrane potential (MMP). MMP is a key indicator of mitochondrial activity because it is required for ATP synthesis. Disruption of MMP can lead to release of cytochrome c, which in turn amplifies the apoptosis signal.35 The downstream effectors of TNFR1/2, including caspase activation and inhibition of nuclear factor κB (NF-κB) activation, can cause disruption of MMP35,36 (Figure 5A). Accordingly, we compared the assigned relevance scores between MMP-disruptive and nondisruptive compounds regarding TNFR1-induced proapoptotic signaling, TNFR1-induced NF-κB signaling, and TNFR2-induced NF-κB signaling (Figure 5C). We observed significantly higher relevance among MMP-disrupting compounds regarding TNFR1-induced proapoptotic and TNFR1-induced NF-κB signaling (FDR = 5e−6 and 3e−6, respectively). Also, the pattern of higher relevance is consistent across all percentiles of two distributions, hence an agreement between assigned pathway relevance and MMP disruption screening. By contrast, the pattern was not observed in other cell death-related pathways except for MHC class II antigen presentation (FDR = 4e−6; Figure 5C), suggesting the potential involvement of mitochondria in antigen presentation, a finding supported by previous work.37
Interpretation of HepG2 cytotoxicity links clinical phenotypes of DILI to TLR3/4-mediated necrosis
We also sought to explain 20 clinical phenotypes of DILI using the derived mapping between compounds and nine cell death-related pathways. For each DILI phenotype, we identified the enriched pathways among its associated compounds (Figure 5E; Experimental procedures). We observed a disproportionate prevalence of high odds ratio (OR) in the two necrosis-related pathways (TLR3 and TLR4 cascade signaling) across almost all DILI phenotypes, with hepatic necrosis, hepatitis, and hepatic fibrosis being the three highest. In total, nine phenotypes are significantly enriched for TLR3/4-mediated necrosis (FDR < 0.05). By contrast, only four phenotypes are significantly enriched for immune activation via MHC class II antigen presentation, while no phenotype is significantly enriched for Hippo signaling or TNFR1/2-mediated apoptosis. These results suggest that TLR3/4-mediated necrosis is a common cause for clinical phenotypes of DILI, a finding supported by previous studies.38,39
Similarly, we identified the enriched pathways among compounds of 14 Anatomical Therapeutic Chemical (ATC) classes (Figure 5F). Each ATC class represents a group of drugs that act on a specific organ or system. We found three classes (hormonal, sensory, and dermatological) significantly enriched for TLR3/4-mediated necrosis and two classes (hormonal and dermatological) significantly enriched for immune activation via MHC class II antigen presentation.
DTox can be applied to a wide range of chemicals other than drugs
Finally, to demonstrate the applicability of DTox among a broader spectrum of chemicals, we implemented the optimal DTox models of two cell viability assays (HepG2 and HEK293) to predict the probability of cytotoxicity for 708,409 compounds from distributed structure-searchable toxicity (DSSTox)40 (Table S5). These compounds provide considerable coverage of the chemical landscape of interest to toxicological and environmental researchers and have not been screened by the Tox21 project. We first analyzed the predicted HepG2 cytotoxicity by compiled compound lists from DrugBank regarding drug approval status (Figure 6A; Table S6; Experimental procedures). We discovered that regardless of approval status, compounds in all six lists exhibited significantly lower predicted HepG2 cytotoxicity than positive controls (active in Tox21 screening). However, only compounds in the nutraceutical list exhibited no significant difference from negative controls (inactive in Tox21 screening). We discovered the same result when analyzing the predicted HEK293 cytotoxicity (Figure S8A; Table S6). These nutraceutical compounds are mostly dietary supplements and food additives that can be taken daily, and thus are expected to appear less toxic to the human body.
Figure 6.

Application of predicted cytotoxicity score among DSSTox compounds
(A) Boxplot showing the distribution of predicted HepG2 cytotoxicity scores among positive controls (leftmost box in red), 10 EPA chemical lists (boxes in light red), six DrugBank lists (boxes in light blue), and negative controls (rightmost box in blue). Mann-Whitney U test is employed to examine whether the cytotoxicity scores of each list exhibit no significant difference from the positive controls (red star above list name), or no significant difference from the negative controls (blue star above list name).
(B) Boxplot on the right compares the predicted HepG2 cytotoxicity scores among drugs associated with clinical hepatic phenotypes (green box) versus negative controls (yellow box), while bar plot on the left shows the odds ratio between HepG2 cytotoxicity and each phenotype (95% confidence interval shown as error bar). Results for 10 phenotypes with odds ratio > 1 are shown in the plot. Mann-Whitney U test is employed to examine whether the drugs associated with each phenotype are predicted with higher cytotoxicity scores than the negative controls (red star next to the phenotype name denotes p < 0.05).
See also Figure S8.
We then analyzed the predicted HepG2 cytotoxicity by compound lists from the Environmental Protection Agency (EPA) regarding their chemical properties (Table S6; Experimental procedures). Among the 265 chemical lists compiled by the EPA, we discovered 12 lists in which compounds exhibited significantly higher predicted HepG2 cytotoxicity than negative controls (inactive in Tox21 screening) and no significant difference from positive controls (active in Tox21 screening). In 10 of the 12 lists (shown in Figure 6A), compounds share a common function. Compounds in the other two lists (“casmi2017” and “tscawp”) were compiled together because of joint appearance in contest datasets. Similarly, we discovered 12 such functional lists (shown in Figure S8A) when analyzing the predicted HEK293 cytotoxicity. These compounds are either industrial manufacturing products (e.g., bisphenols, dioxins) or lethal to a certain species (e.g., insecticides, rodenticides), and thus are expected to appear more toxic to the human body. These results also demonstrate that DTox can be applied to a wide range of chemicals other than drugs, including food ingredients, environmental chemicals, industrial chemicals, etc.
HepG2 cytotoxicity scores predicted by DTox can differentiate hepatic cyst compounds from negative controls
To demonstrate the clinical application of DTox, we sought to differentiate DSSTox compounds associated with DILI phenotypes from negative controls using the predicted HepG2 cytotoxicity score (Figure 6B). We were able to detect significantly higher predicted scores among the compounds associated with hepatic cyst (p = 0.015), because hepatic cyst is the only DILI phenotype showing a significant association with HepG2 cytotoxicity (OR = 1.90, 95% confidence interval [CI]: 1.04–3.45). Among the remaining 19 DILI phenotypes showing weak or no association with HepG2 cytotoxicity (9 phenotypes with OR > 1, 10 with OR < 1), we were able to detect only a significant difference for one phenotype: hepatic steatosis (p = 0.008). Similarly, we sought to differentiate DSSTox compounds associated with drug-induced kidney injury (DIKI) phenotypes from negative controls using predicted HEK293 cytotoxicity score (Figure S8B). Unfortunately, we were not able to detect a significant difference for any of the 24 DIKI phenotypes, because none of them exhibit a significant association with HEK293 cytotoxicity (lower bound of 95% CI < 1).
DTox offers flexibility in balancing between model efficiency and performance
Lastly, we compared the efficiency of the DTox model against other classification algorithms. We reported the central processing unit (CPU) time and run time of all algorithms in Table S7. On average, it took 1.72 h for a DTox model to complete training on a single Tox21 dataset (∼5,000 samples with mini-batch size of 32), 3 times the duration for MLP and 12 times the duration for RF and GB. As mentioned above, DTox employs an early stopping criterion to conclude training when an optimal model can be detected. In this study, we adopted a conservative stopping criterion for maximizing the predictive performance of derived DTox models. We further tuned the stopping hyperparameter to investigate the flexibility in model efficiency and performance (Figure S9). We discovered that, on average, the run time of DTox can be saved by 30% with a 2% sacrifice in model performance. Further, the run time of DTox can be cut in half with a 5% sacrifice in model performance. These statistics offer DTox users some flexibility in balancing model efficiency and performance, especially when implementing the model on larger datasets.
Discussion
Biologically informed VNNs provide a solution to the dilemma posed by conventional supervised learning models: whether to achieve good predictive performance or high model interpretability. Here, we have explored the implementation of VNNs for predicting and explaining compound response to toxicity assays. Compared with previous efforts, our DTox model uniquely stands out in four aspects. First, the structure of DTox can be customized for an outcome of interest according to the underlying biological processes, making the model flexible toward various toxicity outcomes. It also provides a molecular overview of each toxicity outcome regarding which pathway categories are relevant to the outcome (Figure 2A). The molecular overview can help prioritize areas for future research in drug discovery because many toxicity outcomes we analyzed are linked to complex diseases. For instance, the AR, ER, and progesterone receptor all exhibit aberrant activities in various cancer types and play a central role in the progression and metastasis of these types, including breast cancer, prostate cancer, and ovarian cancer. Both constitutive androstane receptor and PXR can regulate drug-metabolizing enzymes and transporters, and thus play a critical role in resistance to cancer therapy and other adverse drug reactions. Second, trimming of network hierarchy can remove unrelated pathways from the network and significantly reduce the number of trainable parameters in VNNs, which in turn prevents overfitting. Through comparisons with well-established classification algorithms, we have demonstrated that DTox is a highly efficient learning model with good predictive performance. For instance, DTox achieved the same level of performance as a matched MLP with only 3% of the network parameters. Through shuffling analysis, we have demonstrated that DTox benefits from the incorporation of Reactome pathway knowledge, including protein-pathway annotations and child-parent pathway relationships. Shuffling child-parent pathway relationships (higher hierarchy) exhibits a moderate impact on model performance compared with shuffling protein-pathway annotations (lower hierarchy), because there are fewer alternative pathways to sample from in the higher hierarchy. It also implies undocumented relationships other than child-parent, such as crosstalk between pathways from different branches, may play a critical role in some toxicity outcomes. Future investigation should be conducted in how to train a VNN to recognize these interactions. Third, the introduction of an early stopping criterion combined with a relatively small network size makes DTox a fast-learning model. Last, and most importantly, DTox advances an interpretation framework that identifies high-relevance VNN paths for explaining the toxicity outcome of compounds. The framework builds on top of layer-wise relevance propagation (LRP)41 and assigns a relevance score to each VNN path. The innovation of our framework resides in its ability to statistically assess the significance of each path, with an empirical p value computed from permutation testing. With the help of existing experimental datasets, we have validated the mechanistic interpretation by our framework and demonstrated the biological significance of DTox. For instance, we showed that DTox was able to consistently identify the corresponding “ground truth” VNN path representing mechanisms of transcription activation by three nuclear receptors. We employed Mechanism of Assay screening data to show that DTox was able to differentiate distinctive mechanisms leading to HepG2 cytotoxicity. We employed drug-induced transcriptome profiling data to show that DTox was able to disproportionately identify VNN paths representing the cellular activities induced by aromatase inhibitors and PXR agonists, implying its potential to detect mechanisms of action. In addition to mechanistic assay and transcriptome profiling, DTox interpretation can be validated by other types of experiment in the future. For instance, knockdown and overexpression experiments can be performed to evaluate the inferred causality between toxicity phenotypes and target proteins/pathways. For toxicity outcomes that can be linked to clinical phenotypes in patients, observation data from electronic health records can be employed to perform survival analysis, evaluating the inferred causality between the phenotype and lab measurements (e.g., enzyme level, cell count) that can inform the activities of certain proteins/pathways.
Besides the expected results, DTox also generated new mechanistic hypotheses along model interpretation, some of which are supported by previous studies. For instance, our framework suggested a potential role for p53 in the regulation of aromatase in MCF-7aro/ERE, a breast cancer cell line. It has been revealed that p53 can directly bind to proximal promoter PII in breast adipose stromal cells, which in turn inhibits aromatase expression.28 In another case, our framework suggested three signaling pathways, including MAPK/ERK (i.e., MAPK1/MAPK3 Reactome pathway), Hippo, and TLR3/4, contribute to the HepG2 cytotoxicity of mifepristone. Accordingly, recent studies have pointed out the effect of mifepristone on ERK activation,30 YAP (a core factor of Hippo) activation,31 and TLR4 regulation.32 Particularly, ERK activation by mifepristone can lead to cytotoxicity in uterine natural killer cells,30 while YAP activation by mifepristone can induce hepatomegaly in mice.31 Two additional findings from our cytotoxicity analysis have been corroborated by previous studies: (1) the involvement of mitochondria in antigen presentation via ATP synthase and mitochondrial calcium uniporter,37 and (2) the disruption of TLR3/4 signaling in DILI.38,39 In addition, some unexpected findings by DTox are worth further investigation, such as the role of immune response in HepG2 cytotoxicity, the role of interleukin-3/14 in regulation of aromatase, the role of caspase-3/7 in MHC class II antigen presentation, etc.
Despite the highlights mentioned above, DTox in its current form bears some limitations from both technical and methodological perspectives. In terms of technical limitations, as with all deep learning models, DTox requires a time-consuming hyperparameter tuning process before an optimal model can be reached. As we observed in the analysis (Figure 2A), an optimal setting may greatly improve the predictive performance of DTox. However, the issue can be resolved with implementation of graphics processing unit (GPU) computing. In terms of methodological limitations, to start with, DTox did not significantly outperform other well-established classification algorithms, because most differences are within the 95% CI of performance metrics. In the interpretation analyses, DTox was not able to identify the ground truth path for a particular assay: AR antagonist. It also failed to identify more differentially expressed paths in general for two assays: mitochondria toxicity and HepG2 cell viability. These results imply that additional factors unaccounted for in DTox may also play a critical role leading to toxicity, such as pharmacodynamic and pharmacokinetic profile, compound-induced gene expression profile, etc. In this study, we emphasized the applicability of DTox such that it can be adopted to study any compounds with or without additional profiling information. Therefore, we limited the model input to the 2D structural representation of compounds. In the future, we expect the performance of DTox to be enhanced after incorporation of additional profiles (e.g., pharmacodynamic, pharmacokinetic, gene expression) once they become available for more compounds. We also acknowledge the recent development of toxicology-focused graph databases, such as ComptoxAI, which provide extensive knowledge on relations among chemicals, genes, assays, and many other entities.42 Such a database may help researchers generate a more comprehensive feature profile for model training and thus improve the performance of DTox. In addition to better feature profiling, we think the incorporation of context-specific knowledge may further enhance the performance of DTox. As we have discussed above, undocumented interactions between pathways may play a critical role in some toxicity outcomes. These interactions can be specific to the outcome of interest. Therefore, the issue can be addressed by using context-specific gene networks (e.g., tissue-specific, disease-specific) to inform connections between pathways during VNN construction, or incorporating stochastic connections between pathways of distinct branches during VNN training.
In the future, we anticipate the application of DTox in two distinct directions. The first direction is concerned with efficacy or toxicity prediction for virtual screening. As with what we have accomplished in the screening of ∼700,000 DSSTox compounds for cytotoxicity, DTox can quickly go through large-scale chemical libraries and prioritize compounds for further experimental testing. The second direction is concerned with outcome explanation for generating new hypotheses. As we have shown throughout the study, DTox’s interpretation framework may detect a new mechanism of action for compounds, uncover cellular mechanism for outcomes of interest, and identify new therapeutic targets for diseases.
Experimental procedures
Resource availability
Lead contact
Requests for information should be directed to the lead contact, Jason Moore (jason.moore@csmc.edu).
Materials availability
This study did not generate any new materials.
Processing Tox21 datasets and inferring feature profile for DTox model training
The Tox21 datasets1 contain screening results describing the response of in vitro toxicity assays to compounds of interest, including approved drugs, experimental drugs, small molecules, and environmental chemicals. We extracted active and inactive compounds from the screening results of each assay, then removed compounds with inconclusive or ambiguous results. We further removed assays with fewer than 5,000 available compounds and focused our analyses on the remaining 15 assays. To quantify structural properties of compounds, we used rcdk package to compute a 166-bit binary MACCS fingerprint that covers most of the interesting physicochemical features for drug discovery.43 We then implemented TargetTox25 to infer the target-binding probability of each compound from its MACCS fingerprint. TargetTox comprises binding prediction models that were pre-trained on hundreds of thousands of compound-target binding affinity data points that were experimentally measured in EC50/IC50/Kd/Ki. It first employs a feature selection pipeline to identify the fingerprint features that are predictive of the binding outcome for each target protein, then fits an RF classification model using the predictive features. We selected 361 target proteins of which the binding outcome can be well predicted by TargetTox (model AUROC > 0.85 on held-out validation set). The derived target-binding profile containing 361 proteins was then used as input feature data for assay outcome modeling.
Constructing VNN with Reactome pathway hierarchy
We designed the VNN structure based on the Reactome pathway hierarchy that comprises root biological processes, child-parent pathway relations, and protein-pathway annotations (downloaded in August 2019).23 To trim the scale of the neural network and prevent overfitting, we adopted two hyperparameters to filter Reactome pathways: (1) minimal pathway size (values for tuning: 5, 20) and (2) root biological process (values for tuning: “gene expression,” “immune system,” “metabolism,” “signal transduction,” and all possible combinations among the four, 15 values in total). We selected the four processes because of their broad coverage and direct involvement in cellular mechanism of toxicity. Each pathway is coded as a hidden module with fixed number of neurons. For a pathway p, the number is defined by ,
where Sp denotes the size of p, Smin and Smax denote the minimal and maximal size of a pathway in the VNN, respectively, and Nmax (= 20) denotes the maximal number of neurons for a hidden module. As a result, hidden modules of larger pathways are assigned with more neurons to capture potentially more complex responses.
Under the Reactome hierarchy, the VNN model of DTox starts from an input layer containing 361 protein features, which are connected to lowest-level hidden modules by protein-pathway annotations. The connections to a hidden module of pathway p are encoded by a weight matrix Wp with dimensions Np∗Nprotein, where Np denotes the hidden module size and Nprotein denotes the number of input proteins annotated with p. With Wp, input vector xp is transformed to output vector yp via , where bp is a bias vector. The hidden modules are then interconnected by child-parent pathway relations until root biological processes are reached. Finally, the root biological processes are connected to an output layer containing the assay outcome. The connections to the output layer are encoded by a weight matrix Wr with dimensions 1∗Nr, where Nr denotes the sum of root hidden module sizes. The final output yr is computed as , where the Logistic Sigmoid function converts layer inputs to an output score between 0 and 1 (i.e., the predicted outcome probability). In addition, we adopted the idea of auxiliary layers from DCell19 to prevent gradients from vanishing in the lower hierarchy and to facilitate the learning of new patterns from individual pathways. Specifically, output vector of hidden module yp is transformed to an auxiliary scalar via , where denotes the weight matrix with dimensions 1∗Np. The auxiliary scalars from all hidden modules are then evaluated in a loss function along with the final output:. The auxiliary factor ⍺ is a hyperparameter of the VNN model (values for tuning: 0.1, 0.5, 1), balancing between root and auxiliary loss terms. βp serves as the adjustment factor for auxiliary loss terms from pathway p, being computed as the inverse number of pathway count within the corresponding hidden layer. Therefore, pathways higher in the hierarchy exhibit greater contribution to the loss function as pathway count decreases dramatically along the hierarchy. λ (= 1e−4) is the coefficient for L2 regularization.
Learning optimal DTox model for Tox21 assay outcome prediction
Each dataset is split into learning and validation sets by ratio of 4:1. During model training, the learning set is further split into training and testing sets by ratio of 7:1. The purpose of the split is to set aside an independent testing set for overfitting assessment during model training. At every epoch, forward and backward propagation are performed on the training set for deriving gradients of model parameters. The parameters are then optimized by Adam algorithm with mini-batch size of 32. At the end of every epoch, loss function is evaluated on the testing set for assessing overfitting and determining whether the early stopping criterion has been met (testing loss has not decreased for P epochs, where P represents the “patience” hyperparameter and is set at 20 in this study). Model training stops after 200 epochs or if the early stopping criterion has been met (in our experience, the early stopping criterion is often met long before 200 epochs).
As mentioned above, the VNN model of DTox has three hyperparameters: minimal pathway size, root biological process, and the auxiliary factor ⍺. To find the optimal setting for each assay, we adopted a grid search and implemented all possible hyperparameter combinations to train DTox models (90 combinations in total, listed in Table S8). We evaluated each trained model by computing the loss function on the whole learning set, then identified the optimal model that minimizes learning loss. Finally, the held-out validation set was used to evaluate the performance of the optimal DTox model and compare with other machine learning models. We adopted two performance metrics for the task: AUROC and balanced accuracy. We computed the 95% CI of metrics using bootstrapped samples from predicted outcome probabilities. On average, the bootstrapped samples contain 63.3% of unique original samples. The performance of two methods is significantly different if their CIs do not overlap. Three machine learning models were considered for performance comparison: (1) a fully connected MLP model with the same number of hidden layers and neurons as optimal DTox model, (2) an optimal RF model derived from tuning of six hyperparameters (“n_estimators,” “criterion,” “max_features,” “min_samples_split,” “min_samples_leaf,” and “boostrap”) by grid search (2800 combinations in total, listed in Table S8), and (3) an optimal GB model derived from tuning of five hyperparameters (“n_estimators,” “max_depth,” “learning_rate,” “subsample,” and “min_child_weight”) by grid search (3,000 combinations in total, listed in Table S8).
In addition, shuffling analysis was performed to assess the influence of pathway knowledge and hierarchy on DTox performance. Three distinctive layouts were considered for performance comparison. First, an alternative DTox model built under shuffled Reactome ontology hierarchy while the shuffle preserves the number of children for each parent pathway and the number of connections between hidden layers (suppose a parent pathway is connected to three children in the original DTox, two in layer i and one in layer j). By hierarchy shuffling, the parent will be connected to two pathways sampled from layer i and one pathway sampled from layer j. This shuffling strategy ensures that the resulting DTox model is still consecutively connected from input to output layer.) Second is an alternative DTox model built with shuffled input target profile (the input values are shuffled among features). The third layout is an alternative DTox model built with shuffled assay outcome as negative control (the outcome labels are shuffled among compounds within the learning set).
Interpretating optimal DTox model by LRP
LRP41 is a model interpretation tool for deep neural networks. Through backward propagation, LRP assigns each neuron a share of the network output and redistributes it to its predecessors in equal amounts until the input layer is reached. The propagation procedure ensures that relevance conservation is an inherent property of LRP. To implement LRP, we adopted two local propagation rules: generic rule and input-layer rule.44
Generic rule was applied to relevance propagation of the hidden neurons. For two connected neurons, j and k, from a child-parent pathway pair, the forward propagation of VNN follows , where ak denotes the activation of neuron k. The generic rule propagates relevance between them as , where γ and ε are two hyperparameters of the rule. γ (values for tuning: 0.001, 0.01, 0.1) controls the contribution of positive weights in relevance propagation. Increasing the value of γ can marginalize neurons with negative weights and decrease the variance of relevance across neurons, and thus may lead to more stable interpretation results. ε (values for tuning: 0.001, 0.01, 0.1) absorbs relevance from neurons with weak or contradictory weights. Increasing the value of ε can give prominence to a few neurons with high weights, and thus may lead to more sparse interpretation results.
Input-layer rule was applied only to relevance propagation of the input protein features. For a protein feature i and its connected neuron j from a lowest-level pathway, the input-layer rule propagates relevance between them as , where li (= 0) and hi (= 1) are the lower and upper bound of input feature values.
Identifying significant VNN paths for explaining toxicity outcome of compounds
After relevance of each neuron is assigned via LRP, a relevance score is computed for each pathway by summing the relevance scores of its neurons. An observed score is then computed for each VNN path connecting input protein feature to output assay outcome as , where p denotes a protein or pathway along the path. The relevance scores are converted to non-negative values, as we are interested in only the proteins or pathways that are more likely to result in a toxicity outcome. The log transformation is adopted to adjust the scale of relevance scores from different layers, because the number of pathways decreases dramatically along the hierarchy.
To assess the significance of each observed path score, we employed a permutation-based strategy to derive the null distribution. Specifically, we shuffled the outcome label of each Tox21 dataset, then re-trained random DTox models using the same hyperparameter setting as a previously trained optimal model. The procedure was repeated n = 200 times, a balance between sample size and running time. Scores derived from the random DTox models comprise the null distribution for each observed path score, and thus the empirical p value can be computed as . We used the FDR to perform multiple testing correction on all VNN paths, then identified the significant paths (FDR < 0.05) for each active compound.
As mentioned above, DTox’s interpretation framework has two hyperparameters: γ and ε from the generic rule. To study the effect of hyperparameter settings on model interpretation, we implemented all possible (nine in total) hyperparameter combinations to identify significant VNN paths for active compounds. We measured the similarity between each pair of settings by the median Jaccard Index among active compounds regarding their identified significant paths.
Comparing DTox against existing interpretation methods regarding rediscovering mechanisms of transcription activation by nuclear receptor
Three interpretation methods were considered for performance comparison regarding the task. The first method serves as a baseline for DTox interpretation framework, in which we randomly sampled the same number of VNN paths for each compound as identified by DTox from the pool of all possible paths in the network. The performance metric was computed as the proportion of active compounds that were sampled with the “ground truth” VNN path (linking together root process of gene expression, nuclear receptor transcription pathway, and the specific target receptor). The procedure was repeated 1,000 times to account for the stochastic nature of sampling. The average performance and 95% CI were computed and adopted as baseline for DTox.
The second method is widely used for explaining predictions of classification algorithms, namely, LIME.16 LIME explains predictions by fitting local linear models to approximate the behavior of the original model. For each nuclear receptor of interest, we implemented LIME on the optimal RF model (derived previously from hyperparameter tuning) to explain the predicted outcome of each compound by target feature relevance (our implementation was based on the tutorials in https://github.com/marcotcr/lime). The performance metric was computed as the proportion of active compounds that were explained with high relevance regarding the specific target receptor. We adopted two thresholds for defining “high relevance”: (1) feature relevance for the target receptor is positive (lax threshold), and (2) feature relevance for the target receptor is above average (strict threshold).
The third method is commonly used for inferring toxicity profile of new compounds, namely, RAx. RAx does not rely on classification algorithms. Instead, it assigns existing knowledge on source compounds to the query compounds with similar chemical structure. For each nuclear receptor of interest, we extract compounds with known connections (source compounds) from two resources: DrugBank45 and ComptoxAI.42 The performance metric was computed as the proportion of active compounds (query) that exhibit similar structure to at least one source compound. Five thresholds of Tanimoto coefficient were adopted to define structural similarity between source and query compound: 0.8, 0.85, 0.9, 0.95, and 1.
Processing LINCS dataset for validation of DTox interpretation results
The LINCS dataset24 contains gene expression profiles derived from genetic and small-molecule perturbation experiments on a number of cell lines, including MCF-7 (which was used in Tox21’s aromatase assay) and HepG2 (used in Tox21’s mitochondria toxicity assay, PXR agonist assay, and HepG2 cell viability assay). We extracted the profiles induced by active compounds of the four assays in their respective cell line. We removed the profiles that did not pass quality control, then separated the remaining ones into three groups based on dose and time of perturbation (1.11 μM/24 h, 10 μM/6 h, 10 μM/24 h). We used the LINCS level 5 data, which consist of moderated differential expression Z scores, for the validation analysis.
To assess the differential expression of VNN paths identified for each compound, we first identified differentially expressed genes (DEGs) from the corresponding profile by |Z| > 2, as suggested by LINCS. Then, we used Fisher’s exact test to examine whether the pathways along each VNN path are enriched for DEGs. A test p value was computed for each pathway. We used FDR to perform multiple testing correction on all pathways along each path. A VNN path is differentially expressed if all the pathways involved are significantly enriched for DEGs (FDR < 0.05). Finally, we calculated the proportion of differentially expressed paths among the paths identified by DTox (observed proportion) and among all possible paths in VNN (expected proportion).
Processing datasets for analyzing DTox results on HepG2- and HEK293-cytotoxic compounds
We obtained six DrugBank lists from https://go.drugbank.com/releases/latest#external-links.45 Each list contains a number of compounds sharing a particular approval status. We obtained 265 EPA chemical lists from https://comptox.epa.gov/dashboard/chemical-lists. Each list contains a number of compounds sharing a particular property. Descriptions of these lists can be found in Table S6.
The NSIDES dataset46 contains drug-adverse event relations that are derived from US Food and Drug Administration (FDA) reports after adjusting for confounding factors. Each drug-adverse event pair is assigned with a proportional reporting ratio (PRR) score along with its 95% CI, which measures the extent to which the adverse event is disproportionately reported among individuals taking the drug. We manually curated a list of 20 clinical phenotype terms associated with DILI (Table S9) and a list of 24 clinical phenotype terms associated with DIKI (Table S9). Drugs associated with each phenotype of interest are identified by the lower bound of 95% CI (>1). Drugs not associated with each phenotype of interest (negative controls) are identified by both the lower (<1) and the upper (>1) bound of 95% CI.
To measure the association between each DILI phenotype and HepG2 cytotoxicity, we calculated the OR and its 95% CI based on a 2 × 2 contingency table. The same procedure was performed to measure the association between each DIKI phenotype and HEK293 cytotoxicity. We also used Fisher’s exact test to evaluate the enrichment of nine cell death-related pathways among the drugs associated with DILI phenotypes. The OR and test p value were computed for each phenotype-pathway pair. We used FDR to perform multiple testing correction on all phenotype-pathway pairs.
Acknowledgments
This work was supported by NIH grants P30 ES013508, R01 LM010098, R01 AG066833, and K99 LM013646.
Author contributions
J.H.M. and Y.H. conceived the DTox project. J.H.M. and Y.H. designed the DTox model and data analysis workflow. Y.H. and J.D.R performed the analysis. J.H.M. and Y.H. interpreted the results and wrote the paper with editing by J.D.R. All authors read and approved the final manuscript.
Declaration of interests
J.H.M. is a member of the advisory board of Patterns.
Published: August 24, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.patter.2022.100565.
Supplemental information
Data and code availability
The processed datasets used in our study (including Tox21, LINCS, and NSIDES) are available at https://github.com/yhao-compbio/tox_data. DTox source code is available at https://github.com/yhao-compbio/DTox. The code has also been deposited at Zenodo under https://doi.org/10.5281/zenodo.6808324.
References
- 1.Richard A.M., Huang R., Waidyanatha S., Shinn P., Collins B.J., Thillainadarajah I., Grulke C.M., Williams A.J., Lougee R.R., Judson R.S., et al. The Tox21 10K compound library: collaborative chemistry advancing toxicology. Chem. Res. Toxicol. 2021;34:189–216. doi: 10.1021/acs.chemrestox.0c00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kleinstreuer N.C., Yang J., Berg E.L., Knudsen T.B., Richard A.M., Martin M.T., Reif D.M., Judson R.S., Polokoff M., Dix D.J., et al. Phenotypic screening of the ToxCast chemical library to classify toxic and therapeutic mechanisms. Nat. Biotechnol. 2014;32:583–591. doi: 10.1038/nbt.2914. [DOI] [PubMed] [Google Scholar]
- 3.Huang R., Xia M., Sakamuru S., Zhao J., Shahane S.A., Attene-Ramos M., Zhao T., Austin C.P., Simeonov A. Modelling the Tox21 10 K chemical profiles for in vivo toxicity prediction and mechanism characterization. Nat. Commun. 2016;7:10425. doi: 10.1038/ncomms10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chan H.C.S., Shan H., Dahoun T., Vogel H., Yuan S. Advancing drug discovery via artificial intelligence. Trends Pharmacol. Sci. 2019;40:592–604. doi: 10.1016/j.tips.2019.06.004. [DOI] [PubMed] [Google Scholar]
- 5.Hemmerich J., Ecker G.F. In silico toxicology: from structure–activity relationships towards deep learning and adverse outcome pathways. WIREs Comput. Mol. Sci. 2020;10:e1475. doi: 10.1002/wcms.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sedykh A., Zhu H., Tang H., Zhang L., Richard A., Rusyn I., Tropsha A. Use of in vitro HTS-derived concentration-response data as biological descriptors improves the accuracy of QSAR models of in vivo toxicity. Environ. Health Perspect. 2011;119:364–370. doi: 10.1289/ehp.1002476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J., Mansouri K., Judson R.S., Martin M.T., Hong H., Chen M., Xu X., Thomas R.S., Shah I. Predicting hepatotoxicity using ToxCast in vitro bioactivity and chemical structure. Chem. Res. Toxicol. 2015;28:738–751. doi: 10.1021/tx500501h. [DOI] [PubMed] [Google Scholar]
- 8.Ammad-ud-din M., Georgii E., Gönen M., Laitinen T., Kallioniemi O., Wennerberg K., Poso A., Kaski S. Integrative and personalized QSAR analysis in cancer by kernelized Bayesian matrix factorization. J. Chem. Inf. Model. 2014;54:2347–2359. doi: 10.1021/ci500152b. [DOI] [PubMed] [Google Scholar]
- 9.Yamane J., Aburatani S., Imanishi S., Akanuma H., Nagano R., Kato T., Sone H., Ohsako S., Fujibuchi W. Prediction of developmental chemical toxicity based on gene networks of human embryonic stem cells. Nucleic Acids Res. 2016;44:5515–5528. doi: 10.1093/nar/gkw450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Capuzzi S.J., Politi R., Isayev O., Farag S., Tropsha A. QSAR modeling of Tox21 challenge stress response and nuclear receptor signaling toxicity assays. Front. Environ. Sci. 2016;4 doi: 10.3389/fenvs.2016.00003. [DOI] [Google Scholar]
- 11.Zhang J., Mucs D., Norinder U., Svensson F. LightGBM: an effective and scalable algorithm for prediction of chemical toxicity-application to the Tox21 and mutagenicity data sets. J. Chem. Inf. Model. 2019;59:4150–4158. doi: 10.1021/acs.jcim.9b00633. [DOI] [PubMed] [Google Scholar]
- 12.Mayr A., Klambauer G., Unterthiner T., Hochreiter S. DeepTox: toxicity prediction using deep learning. Front. Environ. Sci. 2016;3 doi: 10.3389/fenvs.2015.00080. [DOI] [Google Scholar]
- 13.Idakwo G., Thangapandian S., Luttrell J., 4th, Zhou Z., Zhang C., Gong P. Deep learning-based structure-activity relationship modeling for multi-category toxicity classification: a case study of 10K Tox21 chemicals with high-throughput cell-based androgen receptor bioassay data. Front. Physiol. 2019;10:1044. doi: 10.3389/fphys.2019.01044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuzaka Y., Uesawa Y. Molecular image-based prediction models of nuclear receptor agonists and antagonists using the DeepSnap-deep learning approach with the Tox21 10K library. Molecules. 2020;25:E2764. doi: 10.3390/molecules25122764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Polishchuk P. Interpretation of quantitative structure-activity relationship models: past, present, and future. J. Chem. Inf. Model. 2017;57:2618–2639. doi: 10.1021/acs.jcim.7b00274. [DOI] [PubMed] [Google Scholar]
- 16.Ribeiro M.T., Singh S., Guestrin C. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining - KDD '16. 2016. Why should I trust you? [Google Scholar]
- 17.Shrikumar A., Greenside P., Kundaje A. PMLR); 2017. Learning Important Features through Propagating Activation Differences; pp. 3145–3153. [Google Scholar]
- 18.Du M., Liu N., Hu X. Techniques for interpretable machine learning. Commun. ACM. 2019;63:68–77. doi: 10.1145/3359786. [DOI] [Google Scholar]
- 19.Ma J., Yu M.K., Fong S., Ono K., Sage E., Demchak B., Sharan R., Ideker T. Using deep learning to model the hierarchical structure and function of a cell. Nat. Methods. 2018;15:290–298. doi: 10.1038/nmeth.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuenzi B.M., Park J., Fong S.H., Sanchez K.S., Lee J., Kreisberg J.F., Ma J., Ideker T. Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer Cell. 2020;38:672–684.e6. doi: 10.1016/j.ccell.2020.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin C.H., Lichtarge O. Using interpretable deep learning to model cancer dependencies. Bioinformatics. 2021;37:2675–2681. doi: 10.1093/bioinformatics/btab137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elmarakeby H.A., Hwang J., Arafeh R., Crowdis J., Gang S., Liu D., AlDubayan S.H., Salari K., Kregel S., Richter C., et al. Biologically informed deep neural network for prostate cancer discovery. Nature. 2021;598:348–352. doi: 10.1038/s41586-021-03922-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jassal B., Matthews L., Viteri G., Gong C., Lorente P., Fabregat A., Sidiropoulos K., Cook J., Gillespie M., Haw R., et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020;48:D498–D503. doi: 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subramanian A., Narayan R., Corsello S.M., Peck D.D., Natoli T.E., Lu X., Gould J., Davis J.F., Tubelli A.A., Asiedu J.K., et al. A next generation connectivity map: L1000 Platform and the first 1, 000, 000 profiles. Cell. 2017;171:1437–1452.e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hao Y., Moore J.H. TargetTox: a feature selection pipeline for identifying predictive targets associated with drug toxicity. J. Chem. Inf. Model. 2021;61:5386–5394. doi: 10.1021/acs.jcim.1c00733. [DOI] [PubMed] [Google Scholar]
- 26.Sarwar M., Sandberg S., Abrahamsson P.-A., Persson J.L. Elsevier; 2014. Protein Kinase A (PKA) Pathway is Functionally Linked to Androgen Receptor (AR) in the Progression of Prostate Cancer; p. 25.e1-e12. [DOI] [PubMed] [Google Scholar]
- 27.Lamb L.E., Zarif J.C., Miranti C.K. The androgen receptor induces integrin α6β1 to promote prostate tumor cell survival via NF-κB and Bcl-xL Independently of PI3K signaling. Cancer Res. 2011;71:2739–2749. doi: 10.1158/0008-5472.CAN-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X., Docanto M.M., Sasano H., Kathleen Cuningham Foundation Consortium for Research into Familial Breast Cancer. Lo C., Simpson E.R., Brown K.A. Prostaglandin E2 inhibits p53 in human breast adipose stromal cells: a novel mechanism for the regulation of aromatase in obesity and breast cancer. Cancer Res. 2015;75:645–655. doi: 10.1158/0008-5472.CAN-14-2164. [DOI] [PubMed] [Google Scholar]
- 29.Iorga A., Dara L. Cell death in drug-induced liver injury. Adv. Pharmacol. 2019;85:31–74. doi: 10.1016/bs.apha.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y., Wang Y., Zhuang Y., Zhou F., Huang L. Mifepristone increases the cytotoxicity of uterine natural killer cells by acting as a glucocorticoid antagonist via ERK activation. PLoS One. 2012;7:e36413. doi: 10.1371/journal.pone.0036413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yao X.P., Jiao T.Y., Jiang Y.M., Fan S.C., Zhao Y.Y., Yang X., Gao Y., Li F., Zhou Y.Y., Chen P.P., et al. PXR mediates mifepristone-induced hepatomegaly in mice. Acta Pharmacol. Sin. 2022;43:146–156. doi: 10.1038/s41401-021-00633-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srivastava M.D., Thomas A., Srivastava B.I.S., Check J.H. Expression and modulation of progesterone induced blocking factor (PIBF) and innate immune factors in human leukemia cell lines by progesterone and mifepristone. Leuk. Lymphoma. 2007;48:1610–1617. doi: 10.1080/10428190701471999. [DOI] [PubMed] [Google Scholar]
- 33.Brentnall M., Rodriguez-Menocal L., De Guevara R.L., Cepero E., Boise L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013;14:32. doi: 10.1186/1471-2121-14-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yosefzon Y., Soteriou D., Feldman A., Kostic L., Koren E., Brown S., Ankawa R., Sedov E., Glaser F., Fuchs Y. Caspase-3 regulates YAP-dependent cell proliferation and organ size. Mol. Cell. 2018;70:573–587.e4. doi: 10.1016/j.molcel.2018.04.019. [DOI] [PubMed] [Google Scholar]
- 35.Wang C., Youle R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009;43:95–118. doi: 10.1146/annurev-genet-102108-134850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Albensi B.C. What is nuclear factor kappa B (NF-kappaB) doing in and to the mitochondrion? Front. Cell Dev. Biol. 2019;7:154. doi: 10.3389/fcell.2019.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bonifaz L.C., Cervantes-Silva M.P., Ontiveros-Dotor E., López-Villegas E.O., Sánchez-García F.J. A role for mitochondria in antigen processing and presentation. Immunology. 2014;144:461–471. doi: 10.1111/imm.12392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin S., Gao B. Toll-like receptor 3 in liver diseases. Gastroenterol. Res. Pract. 2010;2010:750904. doi: 10.1155/2010/750904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo J., Friedman S.L. Toll-like receptor 4 signaling in liver injury and hepatic fibrogenesis. Fibrogenesis Tissue Repair. 2010;3:21. doi: 10.1186/1755-1536-3-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grulke C.M., Williams A.J., Thillanadarajah I., Richard A.M. EPA's DSSTox database: History of development of a curated chemistry resource supporting computational toxicology research. Comput. Toxicol. 2019;12:100096. doi: 10.1016/j.comtox.2019.100096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bach S., Binder A., Montavon G., Klauschen F., Müller K.R., Samek W. On Pixel-wise explanations for non-linear classifier decisions by layer-wise relevance propagation. PLoS One. 2015;10:e0130140. doi: 10.1371/journal.pone.0130140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romano J.D., Hao Y., Moore J.H. Improving QSAR modeling for predictive toxicology using publicly aggregated semantic graph data and graph neural networks. Pac. Symp. Biocomput. 2022;27:187–198. [PMC free article] [PubMed] [Google Scholar]
- 43.Cereto-Massagué A., Ojeda M.J., Valls C., Mulero M., Garcia-Vallvé S., Pujadas G. Molecular fingerprint similarity search in virtual screening. Methods. 2015;71:58–63. doi: 10.1016/j.ymeth.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 44.Montavon G., Binder A., Lapuschkin S., Samek W., Müller K.R. Layer-wise relevance propagation: an overview. Explainable AI: interpreting, explaining and visualizing deep learning. 2019:193–209. [Google Scholar]
- 45.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatonetti N.P., Ye P.P., Daneshjou R., Altman R.B. Data-driven prediction of drug effects and interactions. Sci. Transl. Med. 2012;4:125ra131. doi: 10.1126/scitranslmed.3003377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The processed datasets used in our study (including Tox21, LINCS, and NSIDES) are available at https://github.com/yhao-compbio/tox_data. DTox source code is available at https://github.com/yhao-compbio/DTox. The code has also been deposited at Zenodo under https://doi.org/10.5281/zenodo.6808324.