

Abstract
The prime editor is a versatile tool for targeted precise editing to generate point mutations, small insertions, or small deletions in eukaryotes. However, canonical PE3 system is less efficient, notably in primary cells or pluripotent stem cells. Here, we employed RNA polymerase II promoter instead of RNA polymerase III promoter, whose application is limited by specific DNA contexts, to produce Csy4-processed intronic prime editing guide RNAs (pegRNAs) and, together with other optimizations, achieved efficient targeting with poly(T)-containing pegRNAs, as well as combinatorial and conditional genetic editing. We also found simultaneous suppression of both DNA mismatch repair and DNA damage response could achieve efficient and accurate editing in human embryonic stem cells. These findings relieve the restrictions of RNA polymerase III (RNA-Pol-III)-based base editors and broadened the applications of prime editing.
Keywords: CRISPR, prime editor, human pluripotent stem cells, p53 response
Graphical abstract
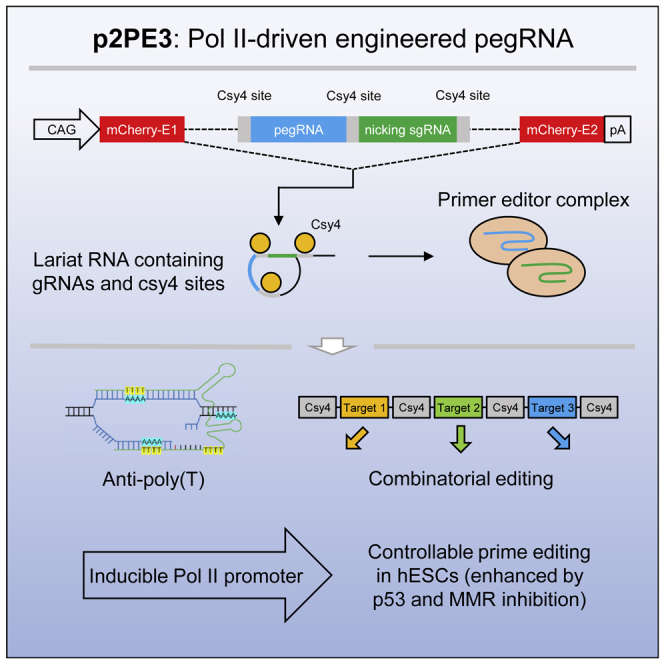
p2PE3, an prime editor using RNA-polymerase-II-driven pegRNA, was developed to achieve efficient targeting with poly(T)-containing pegRNAs, as well as combinatorial and conditional genetic editing. Simultaneous suppression of both DNA mismatch repair and p53 response could achieve efficient and accurate editing in human embryonic stem cells.
Introduction
Pluripotent stem cells are undifferentiated cells that can self-renew and differentiate into specialized cell types from all three germ layers. Genome editing of human pluripotent stem cells (hPSCs) offers great opportunities for in vitro disease modeling, drug discovery, and personalized medical treatment.1 The recently developed CRISPR-Cas9-derived genome editing agents, prime editors (PEs), can introduce precise point mutations, small insertions, or small deletions at desired sites.2 Comparing with previous agents (nucleases and base editors), PEs display advantages in precise editing without producing DNA double-strand breaks (DSBs) or Cas-independent off-target editing in DNA or RNA. In addition, PEs can introduce desired mutations at distances far from single-guide RNA (sgRNA)-targeted sites, relieving the PAM restriction. Since most of known human-disease-associated genetic variants are point mutations, small insertions, or deletions, prime editing exhibits enormous potential in therapeutic applications. However, canonical PE3 system is less efficient, especially in primary cells and in vivo, limiting its applications.3, 4, 5 Recent studies successfully employed prime editing to achieve gene correction in organoids.6,7 Nevertheless, the efficiency of prime editing remains low, suggesting an urgent need for further optimization of PEs.
To improve prime editing, we and others recently found that the stability of the 3ʹ region of the prime editing guide RNA (pegRNA) influences editing efficiency.8,9 Accordingly, we introduced multiple modifications into the pegRNA to generate ePE3. Specifically, a hairpin Csy4 recognition site was added to the 3ʹ end of the RNA polymerase III (RNA Pol III)-driven pegRNA, following by a nicking sgRNA. Stem-ring structure of Csy4 recognition site can preserve labile 3ʹ region, markedly boosting the prime editing efficiency.8 However, there are some concerns on RNA-Pol-III-based sgRNA expression in the ePE3 system. First, RNA Pol III transcription can be easily terminated by four to six contiguous U residues, making it less ideal for expressing complex RNA structures containing more than four contiguous U residues.10 Second, RNA Pol III promoter adds an extra guanine or adenine to the 5ʹ end of sgRNA, resulting in the sgRNA-DNA mismatch at the 5ʹ end, influencing the efficiency of high-fidelity SpCas9 variants.11 Third, RNA Pol III promoters are constitutively and ubiquitously expressed and therefore are not able to generate gRNAs in context-specific manners.12
RNA-Pol-II-based sgRNAs have been successfully used in a variety of CRISPR systems, including the recent application in plants to produce ribozyme-processed pegRNAs.12, 13, 14, 15, 16 Here, we upgraded ePE3 with RNA Pol II promoter to develop p2PE3, which displayed high efficiency for combinatorial genetic editing and for those sites whose targeted pegRNAs contain poly(T). We also demonstrated that, by equipping with different RNA Pol II promoters, p2PE3 is also able to perform conditional editing. Furthermore, we found p53 serves as a bottleneck impeding prime editing in human embryonic stem cells (hESCs), and transient p53 inhibition enables efficient editing in hESCs using p2PE3, suggesting p2PE3 is a versatile toolkit for PE applications.
Results
p2PE3 exhibited high editing efficiency in HEK293T
The original PE3 system consists of a pegRNA and a nicking sgRNA driven by two U6 promoters.2 The ePE3 system uses a single U6 promoter to drive the expression of pegRNA and nicking sgRNA connected by the Csy4 recognition site.8 To obtain RNA-Pol-II-mediated transcription of pegRNA editing system, we first replaced the U6 promoter with CAG promoter for transcribing a gRNA cassette flanked by two Csy4 recognition sites to get the p2PE3-1 (Figure 1A). We next tested the editing efficiency of p2PE3-1 to make substitutions at 10 endogenous target sites in HEK293T cells. Though p2PE3-1 yielded 1.6-fold higher editing efficiency than PE3, it resulted in 1.3-fold lower editing efficiency compared with ePE3 (Figures 1B and 1C).
Figure 1.

Development of RNA-Pol-II-mediated transcription of pegRNA editing system
(A) Schemes for the different production modes of pegRNAs. p2PE3-2 utilized CAG promoter to drive expression of mCherry transcript harboring HSV-1 intron containing gRNA cassette connected by Csy4 recognition sites. E, exon. (B) Comparison of substitution edits with PE3, ePE3, p2PE3-1, and p2PE3-2 at 10 endogenous target sites in HEK293T cells is shown. (C and D) Statistical analysis of the editing frequency (C) and indels (D) induced by four editing systems in (B) is shown. (E) Comparison of editing and indel frequencies with ePE3 and p2PE3 targeting six individual sites for three deletions and three insertions in HEK293T cells is shown. (F) Statistical analysis of intended editing and indel frequencies from (E) is shown. Data were represented as the mean ± SEM (n = 3 from independent experiments). Two-tailed Student’s t tests were performed.
Given that most RNA-Pol-II-mediated transcriptions are transported to the cytoplasm, which may attribute the decline of editing efficiency, we adopted the herpes simplex virus 1 (HSV-1) latency-associated intron, which can detain the functional gRNA in the nucleus and protect it from degradation,13 to develop p2PE3-2 (Figure 1A). The functional gRNAs can be released after the lariat RNA cleavage by Csy4 (Figure S1A). Encouragingly, p2PE3-2 displayed 2.1-fold higher editing efficiency than PE3, comparable with the ePE3, although it also inherited the disadvantage of high indels of ePE3 (Figures 1D and S1B). Overall, p2PE3-2, termed p2PE3 hereafter, is proven to be a better RNA-Pol-II-driven pegRNA editing system.
We further investigated the ability of p2PE3 to install precise small insertions and deletions into the genome. Consistent with the substitutions, p2PE3 showed high editing efficiencies as ePE3 at all six loci (three insertions and three deletions; Figures 1E and 1F). To demonstrate the versatility of p2PE3, we tested the performance of p2PE3 in other cell types (HeLa and U2OS). The editing efficiencies of p2PE3 for making regular edits are comparable, despite being slightly lower, to ePE3 in U2OS and HeLa cells (44.3% versus 39.9% in U2OS and 56.1% versus 47.5% in HeLa) (Figure S2). In summary, we have successfully developed a PE system based on RNA-Pol-II-mediated transcription of pegRNA with the editing efficiency comparable to the ePE system.
p2PE3 exhibits robust editing performance with pegRNAs containing poly(T)
We next examined whether p2PE3 functions in case pegRNA sequences contain four to six deoxythymidine nucleotides in the non-template strands, which can terminate RNA-Pol-III-mediated transcription and lead to attenuated CRISPR-Cas9-based genome editing.17 We compared the efficiency of ePE3 and p2PE3 to insert four or six deoxyadenosine nucleotides at four endogenous target sites in HEK293T cells, which will result in four or six thymines in the reverse transcription template (RTT). Just as expected, poly(T) in pegRNAs seriously affected editing efficiency of ePE3 (0.1%–34.9% for 4A and 0%–2.2% for 6A), while p2PE3 still maintained high efficiency (34.3%–78.7% for 4A and 8.8%–69.5% for 6A; Figures 2A, S3A, and S3B), demonstrating that the editing efficiency of RNA-Pol-II-driven pegRNAs of p2PE3 is not affected by poly(T).
Figure 2.

p2PE3 outperforms ePE3 with pegRNA containing poly-T
(A) Efficiency of ePE3 and p2PE3-mediated insertions of poly(A) at four endogenous target sites in HEK293T cells. (B) Schematic of a pegRNA containing poly(T) to target genome site is shown. PBS, primer binding site; RTT, reverse transcription template. (C) The proportion of pegRNA containing poly(T) to target known human pathogenic genetic variants is shown. (D) Efficiency of ePE3 and p2PE3 with pegRNA containing poly(T) at six human pathogenic sites in HEK293T cells is shown. Data and error bars in (A) and (D) indicated the mean ± SEM of three independent experiments.
Unlike normal sgRNAs, pegRNAs contain the primer binding sites (PBSs) and the downstream homology sequences in RTTs and therefore are more likely to be affected by genomic contexts (Figure 2B). To better evaluate the advantages of p2PE3, we analyzed the sequences of pegRNAs targeting human pathogenic genetic variants and found that there are 11% of pegRNAs containing at least four thymines in spacers or 3ʹ-extended sequences (Figure 2C). We used p2PE3 to model six human pathogenic mutations of which targeted pegRNAs containing poly(T). As anticipated, p2PE3 yielded 13.3-fold higher editing efficiency than ePE3 (0%–8.4% for ePE3 versus 13.0%–77.2% for p2PE3; Figures 2D, S3C, and S3D), showing that p2PE3 has stronger potential to target human pathogenic genetic variants. In order to illustrate p2PE3’s application potential in other occasions, we further analyzed the incidences of poly(T)-containing pegRNAs in pegRNAs targeting human canonical splicing sites and targeting SHP2 gene for saturation mutagenesis screening. We found that p2PE3 can better target 22.1% of RNA-splicing sites and 25.5% of amino acids of SHP2 (Figures S3E and S3F), suggesting that poly(T) pegRNAs are significantly presented in other PE applications besides correcting pathogenic variants, making p2PE3 advantageous for such scenarios.
p2PE3 can more effectively target multiple sites
Many human diseases are caused by complex genetic mechanisms. Modeling polygenic human diseases using CRISPR-Cas9-derived tools calls for simultaneous expression of multiple sgRNAs. Since the Csy4-based system can successfully express multiple gRNAs from a single transcript,18,19 we hypothesized that RNA Pol II promoter exhibits an advantage over RNA Pol III promoter in expressing long pegRNA-nicking sgRNA cassettes for multiple targets.
To verify our hypothesis, we first constructed a dual fluorescence reporter system containing two promoters to express mCherry and EGFP transcripts with 1-bp frameshift insertion, which can be repaired by the PE to restore the fluorescence of mCherry and EGFP (Figure 3A). We found that gRNA cassettes targeting frameshift inserts of mCherry and EGFP separately or simultaneously (Figure S4A) could efficiently restore the fluorescence of mCherry and EGFP (Figures S4B and S4C). The results showed that p2PE3 exhibited same efficiencies for single and dual targeting, while the efficiency of the posterior gRNA cassette targeting EGFP was decreased when being co-targeted by ePE3 (down 23%; Figure 3B).
Figure 3.

p2PE3 has an advantage in combinatorial genetic editing
(A) Diagram of the dual fluorescence reporter system for comparing combinatorial genetic editing. The prime editors can repair the 1-bp frameshift insertion and restore the fluorescence of mCherry and EGFP. (B) Statistical analysis of normalized fluorescence ratio is shown, setting the individual gRNA cassette restoring mCherry or EGFP to 1. (C) Schematic of three multiplexed arrays with pegRNA-nicking sgRNA cassettes targeting MKL2, MYH7, and NKX2-5 is shown. (D) Efficiency of ePE3 and p2PE3 to model childhood-onset cardiomyopathy mutations using individual gRNA cassettes or multiplexed arrays is shown. (E) Statistical analysis of normalized efficiency of individual gRNA cassettes or gRNA cassettes in the different position of arrays in (D) is shown, setting the individual gRNA cassettes of ePE3 to 1. Data were represented as the mean ± SEM (n = 3 from independent experiments). Two-tailed Student’s t tests were performed.
With this, we further assessed the capacity of p2PE3 in combinatorial genetic editing by modeling childhood-onset cardiomyopathy caused by three missense single-nucleotide variants in MKL2, MYH7, and NKX2-5.20 We generated three multiplexed arrays with different gRNA cassette orders (Figure 3C). ePE3 and p2PE3 exhibited similar editing efficiencies when gRNA cassettes are expressed individually or when gRNA cassettes are at the first position of arrays. Yet p2PE3 displayed a significantly higher efficiency of editing over ePE3, especially when gRNA cassettes are at the third position of arrays (1.8-fold increase; p < 0.0001; Figures 3D, 3E, and S4D). These results demonstrate the advantage of p2PE3 in combinatorial genetic editing.
Increasing the prime editing efficiency in hESCs
The outperformance of p2PE3 encouraged us to assess its performance in non-transformed cells, such as hESCs. We first tested p2PE3-mediated base substitution and small targeted deletion in H1 hESCs. p2PE3 still showed significant but lower editing efficiencies in hESCs (the frequency: 4.3%–17.3%; Figure S5A). A recent study revealed that DNA mismatch repair (MMR) impedes prime editing.21 In addition, p53 has been reported to inhibit CRISPR-Cas9 editing in hPSCs.22,23 Based on those results, we tested whether transient MMR inhibition by co-expression of a dominant negative MMR protein (hMLH1NTD-NLS, PE5 strategy)21 and/or transient p53 inhibition by SV40 large T antigen (SV40LT strategy)24 can improve the editing efficiency of PEs in hESCs. Excitingly, both strategies significantly increased the editing efficiency of RNA-Pol-II-based PE in hESCs (1.5-fold for PE5 strategy [p2PE5] and 1.7-fold for SV40LT strategy [p2PE3+SV40LT]), while the combined use of the two strategies maximized the editing efficiency (2.6-fold, p2PE5+SV40LT; Figures 4A and 4B). We then compared editing efficiencies of ePE3 and p2PE3 at six loci (two substitutions, two insertions, and two deletions) in the presence of hMLH1NTD-NLS and SV40LT (ePE5+SV40LT and p2PE5+SV40LT). Interestingly, the editing efficiency of p2PE3 is still lower than that of ePE3 in hESCs (59.3% versus 24.3%) (Figures S5B and S5C), which could be attributed to the activity of CAG promoter in hESCs. Those results, together with previous ones, suggest that p2PE3 (RNA Pol II) and ePE3 (RNA Pol III) system, as well as efficiency-boosting methods, should be chosen based on pegRNA properties, applications, and cell types.
Figure 4.

Controllable prime editing achieved by p2PE3 in hESCs
(A) Comparison of the different strategies to improve editing efficiency of base substitution and small targeted deletion in hESCs. Co-expression of p2PE3 with dominant negative MLH1 (p2PE5), SV40 large T antigen (p2PE3+SV40LT), or both (p2PE5+SV40LT) was tested. (B) Statistical analysis of the editing frequency in (A) is shown. Two-tailed Student’s t tests were performed. (C) Editing and indel frequencies of inducible p2PE3 system combining with PE5 and SV40LT strategies are shown, comparing with or without doxycycline (Dox). Data were represented as the mean ± SEM (n = 3 from independent experiments).
As PE5 strategy only enhances small (<15 bp) targeted indels,21 we next evaluated whether SV40LT strategy can increase the efficiency of insertion 27-bp hemagglutinin (HA) tag in stemness- or lineage-specific genes (SOX2, TROP2, and SOX17) in hESCs. We first tested the editing efficiency of ePE3 and p2PE3 for insertion HA tag in HEK293T (Figure S6A) and then applied them to hESCs. There is no significant difference between ePE3 and p2PE3 for insertion HA tag, and their efficiencies could be boosted to ∼10% by co-expression with SV40LT (5.6-fold increase; Figures S6B–S6D). The increased corresponding edit:indel ratios also suggest that p53 enhances the editing efficiency to the greater extent than induces indels (Figure S6E). Finally, given that p53 inhibitor might affect the functional features of hESCs, we evaluated the effect of transient expression of SV40LT on cell proliferation and stemness gene expression and found these features were minimally affected (Figure S7). Together, these results indicate that transient inhibition of p53 is a valid strategy to improve the editing efficiency of primer editors in hESCs.
Controllable prime editing achieved by p2PE3 in hESCs
As mentioned above, RNA Pol III promoters are constitutively expressed and therefore are not able to generate gRNAs in conditional manners. Since the expression of RNA Pol II promoters can be inducible or specific in different cell types, we constructed an inducible p2PE3 system (i-p2PE3) in TetON-3G-expressing hESCs by replacing CAG promoter with doxycycline-inducible TRE3GS promoter. The i-p2PE5+SV40LT system achieved high editing efficiency upon the treatment of doxycycline in hESCs (up to 47.3%), while the editing was hardly monitored in the absence of doxycycline (Figure 4C). This result suggests that p2PE3 is able to perform conditional editing using specific promoters.
Specificity of co-expression strategy in hESCs
Though it has been proven that PEs exhibit high specificity in human cells, plants, and hESCs,25, 26, 27 we asked whether transient inhibiting of p53 and MMR will lead to off-target effects. To this end, plasmids expressing the editor, hMLH1NTD-NLS, SV40LT, and p2PE3 gRNA cassette targeting GPSM2 site were co-transfected into hESCs and the transfected cells were sorted for whole-genome sequencing (WGS) and RNA sequencing (RNA-seq) 48 h later. hESCs transfected with EGFP-expressing plasmid were used as control, and untreated cells were used to eliminate the background (Figures S8A and S9A). With the high efficiency of on-target editing (GPSM2 +1 C del) observed by WGS (Figure S8B), we investigated the magnitudes and frequencies of DNA variants and found that the use of p2PE5 plus SV40LT did not trigger DNA random mutations; its level was indistinguishable from control (Figures 5A and 5B). We also confirmed there is no sgRNA-dependent off targeting by comparing with predicted off-target sites (Figure 5C). Finally, we evaluated the influence of p2PE5 plus SV40LT on the transcriptome. Transient inhibiting of p53 and MMR caused nuanced changes in gene expression (Figure S9B), with differentially expressed genes enriched in the p53 signaling pathway (Figure S9C). Those genes were down-regulated in the treatment group (Figure S9D), confirming the effect of expressing SV40LT. In addition, co-transfection did not cause redundant mutations or changes in mutation types in the transcriptome (Figures 5D and 5E).
Figure 5.

Evaluating the specificity of co-expression strategy through WGS and RNA-seq
(A) Summary of total unique variants detected by WGS in control and edited samples. Control: H1 hESCs were transfected with EGFP plasmid; edited: H1 hESCs were transfected with pEF1a-PE2-P2A-Csy4, p2PE3 gRNA cassette targeting GPSM2 site, and pEF1a-SV40LT-P2A-hMLH1NTD-NLS plasmids. (B) Mutation frequency of total variants is shown. Two-tailed Student’s t tests were performed. (C) Potential off-target sites of pegRNA and nicking sgRNA were predicted by Cas-OFFinder considering up to 5-bp mismatch in protospacer with NRG PAM. (D) Summary of single-nucleotide variants and indels detected by RNA-seq in control and edited samples is shown. (E) Distribution of RNA editing types in control and edited samples is shown.
Discussion
With its great potential in precise editing, PE optimization is extremely desired. In this study, we utilized RNA Pol II promoter to produce Csy4-processed intronic pegRNAs to develop p2PE3 and demonstrated its advantage to target human pathogenic genetic variants and model polygenic human diseases. We also achieved controllable prime editing in hESCs, suggesting that the p2PE3 system with changeable RNA Pol II promoters could be a versatile toolkit for PE applications.
Although PEs do not make blunt DNA DSBs theoretically, PE3 nicks both strands of DNA, which may also trigger p53 responses. A previous study suggested that transient p53 inhibition may facilitate genome editing recovery in primary and p53+/+ cell lines.28 More recently, other studies have confirmed the selection of correctly edited colonies led to the accumulation of p53-inactivating mutations by CRISPR-Cas9.29,30 We found that, by transient expression of SV40 large T-antigen, editing efficiencies of PE-mediated base substitution, small targeted deletion, and longer targeted insertion (HA-tag insertion, 27 bp) could be enhanced in hESCs. Those observations strengthen the speculation that p53 limits prime editing in non-transformed cells. It merits further investigations for the detailed molecular and cellular mechanisms.
Transient inhibition of p53 has been employed in clinical-relevant applications, such as the generation of human induced pluripotent stem cells.31 In hESCs, although inhibition of p53 enhances the editing efficiency to a greater extent, it still induces indels. This problem could be alleviated by sequencing hESC clones. We also confirmed that p2PE5 and SV40LT did not trigger DNA or RNA mutations at both genome-wide and transcriptome-wide level. These findings support the strategy of incorporation of transient p53 inhibition into CRISPR-Cas9-based genome-engineering applications. Therefore, the SV40LT strategy could be a reliable method to further increase the editing efficiency of PE5 system in primary and wild-type p53 cells.
In summary, our results suggest that p2PE3 shows an impressive editing efficiency on targeting pathogenic genetic sites, making combinatorial genetic edits, and performing conditional genomic editing. Transient inhibition of p53 significantly could further enhance the efficiency of PEs in non-transformed cells. The RNA-Pol-II-based PE would therefore expand the reach of RNA-Pol-III-based PE for applications in vitro and in vivo.
Materials and methods
Plasmid construction
Using ClonExpress MultiS Cloning Kit (Vazyme), we modified the pCMV-PE2 (Addgene plasmid no. 132775) and pGL3-U6-sgRNA-EGFP (Addgene plasmid no. 107721) plasmids with several different transgenes. We replaced promoter of pCMV-PE2 with EF1a promoter and added Csy4 into PE2 downstream to create pEF1a-PE2-P2A-Csy4. To construct RNA-Pol-II-mediated transcription of pegRNA-editing system, we first replaced U6-sgRNA of pGL3-U6-sgRNA-EGFP with mCherry fragment under control of CAG promoter. The HSV-1 latency-associated intron harboring two Csy4 recognition sites, two BsaI restriction sites, and one gRNA scaffold was synthesized and inserted between K52 and V53 of mCherry. For construction of pegRNA-nicking sgRNA cassettes, oligos were synthesized, annealed, and cloned into BsaI site of the sgRNA expression vector according to the methods described in our previous study.8 pegRNA and nicking sgRNA were designed by PrimeDesign32 and listed in Table S1. pEF1a-hMLH1NTD-NLS was a gift from David Liu (Addgene plasmid no. 174826). The sequence of SV40LT was synthesized and inserted into hMLH1NTD-NLS upstream or replaced it. To construct inducible RNA-Pol-II-mediated transcription of pegRNA-editing system, TRE3GS promoter was synthesized and replaced the CAG promoter. The expression plasmid of TetON-3G protein was a gift from David Vereide (Addgene plasmid no. 104543).
Cell culture and transfection
HEK293T, HeLa, and U2OS cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Hyclone), supplemented with 10% fetal bovine serum (FBS) (v/v) (Gemini) and 1% penicillin streptomycin (v/v) (Gibco). Cells were seeded on 24-well plates and transfected at approximately 70% confluence using Lipofectamine 2000 (Thermo Scientific) according to the manufacturer’s protocol. H1 hESCs was routinely cultured in mTeSR1 (STEMCELL Technologies) on Matrigel (STEMCELL Technologies)-coated plates. Cells were passaged every 4 days using Gentle Cell Dissociation Reagent (STEMCELL Technologies). The day before transfection, cells were dissociated with Accutase (Thermo Scientific) and plated 50,000–75,000 cells/well in the precoated 24-well plate with 10 μM Y-27632 (Tocris). Lipofectamine Stem reagent (Thermo Scientific) was used to transfect H1 hESCs according to the manufacturer’s protocol. A total of 400 ng pEF1a-PE2-P2A-Csy4, 300 ng pegRNA, 100 ng corresponding nick sgRNA (where indicated), 300 ng hMLH1NTD-NLS or SV40 Large T Antigen or SV40LT-hMLH1NTD-NLS plasmid (where indicated), and 200 ng TetOn-3G plasmid (where indicated) were transfected into cells per well. Forty-eight hours (for H1 hESCs) or seventy-two hours (for HEK293T, HeLa, and U2OS cells) after transfection, EGFP-positive cells were collected from fluorescence-activated cell sorting (FACS). Cell counting was performed using Countess II Automated Cell Counter (Invitrogen) by trypan blue staining.
Genomic DNA extraction and amplification
Genomic DNA of cells was extracted using QuickExtract DNA Extraction Solution (Lucigen) or DNA extraction reagent (solarbio) according to manufacturer’s protocols. The isolated DNA was PCR amplified with Phanta Max Super-Fidelity DNA Polymerase (Vazyme). Primers used were listed in Table S2.
Targeted deep sequencing
Target sites were amplified from extracted genomic DNA using Phanta Max Super-Fidelity DNA Polymerase (Vazyme). PCR products with different barcodes were pooled together for deep sequencing using Illumina NovaSeq (PE150) at the Annoroad Gene Technology, Beijing, China. Primers used for deep sequencing were listed in Table S2. Sequencing reads were demultiplexed using AdapterRemoval (v.2.2.2), and the pair-end reads with 11 bp or more alignments were combined into a single consensus read. All processed reads were then mapped to the target sequences using the BWA-MEM algorithm (BWA v.0.7.16). Prime editing efficiency was calculated as percentage of (number of reads with the desired edit that do not contain indels)/(total mapped reads). Indel frequency was calculated as number of indel-containing reads/total mapped reads.
Whole-genome sequencing
DNA extracted from harvested cells was sequenced using Illumina NovaSeq (PE150) at the Annoroad Gene Technology, Beijing, China. All cleaned reads were mapped to the human reference genome (GRCh38/hg38) using BWA v.0.7.17 with default parameters. Sequence reads were removed for duplicates using Sambamba v.0.6.7. Variants were identified by GATK (v.4.1.8.1) MuTect2 and filtered with FilterMutectCalls. The depth for a given variant for each individual should be between one-third to 3-fold of corresponding mean depth, and these variants were required to have at least three of reads supporting the variant. Potential off-target sites were predicted by Cas-OFFinder.33
RNA analysis
RNA was immediately extracted from harvested cells using Total RNA Extraction Reagent (Vazyme, and sequenced using Illumina NovaSeq (PE150) at the Annoroad Gene Technology, Beijing, China. All cleaned reads were mapped to the human reference genome (GRCh38/hg38) by STAR software (v.2.7.9a), using annotation from GENCODE v.30. After removing duplications, variants were identified by GATK (v.4.1.8.1) MuTect2 and filtered with FilterMutectCalls. The depth for a given variant should be at least 10×, and these edits were required to have at least three of reads supporting the variant. Differential gene expression analysis was performed using DESeq2_1.30.0. For qRT-PCR, cDNA was generated using the HiScript II Q RT SuperMix (Vazyme), RNA expression was measured with ChamQ SYBR qPCR Master Mix kit (Vazyme), and GAPDH expression was used as control.
Availability of data and material
High-throughput sequencing data were deposited in the NCBI Sequence Read Archive database under accession code NCBI: PRJNA789416. All other data are available upon reasonable request.
Acknowledgments
We thank members of Lin lab and Huang lab for helpful discussions. We thank the Molecular and Cell Biology Core Facility (MCBCF) at the School of Life Science and Technology, ShanghaiTech University for providing technical support. This work is supported by National Key R&D Program of China (no. 2020YFA0710800), National Natural Science Foundation of China (no. 31871487), Shanghai Science and Technology Commission (no. 19JC1414200), the ShanghaiTech University start-up fund, and the Leading Talents of Guangdong Province Program (2016LJ06S386).
Author contributions
C.-P.L. and X.H. conceived, designed, and supervised the project. S.H., Z.Z., and W.T. performed most experiments with the help of Y.L., X.L., X.W., J.H., and P.-Y.W. S.H., Z.Z., X.H., and C.-P.L. wrote the original manuscript. All authors reviewed and proofread the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.07.002.
Contributor Information
Xingxu Huang, Email: huangxx@shanghaitech.edu.cn.
Chao-Po Lin, Email: linzhb@shanghaitech.edu.cn.
Supplemental information
References
- 1.Hendriks D., Clevers H., Artegiani B. CRISPR-cas tools and their application in genetic engineering of human stem cells and organoids. Cell Stem Cell. 2020;27:705–731. doi: 10.1016/j.stem.2020.10.014. [DOI] [PubMed] [Google Scholar]
- 2.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu Y., Li X., He S., Huang S., Li C., Chen Y., Liu Z., Huang X., Wang X. Efficient generation of mouse models with the prime editing system. Cell Discov. 2020;6:27. doi: 10.1038/s41421-020-0165-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu P., Liang S.Q., Zheng C., Mintzer E., Zhao Y.G., Ponnienselvan K., Mir A., Sontheimer E.J., Gao G., Flotte T.R., et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 2021;12:2121. doi: 10.1038/s41467-021-22295-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhi S., Chen Y., Wu G., Wen J., Wu J., Liu Q., Li Y., Kang R., Hu S., Wang J., et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 2021;30:283–294. doi: 10.1016/j.ymthe.2021.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schene I.F., Joore I.P., Oka R., Mokry M., van Vugt A.H.M., van Boxtel R., van der Doef H.P.J., van der Laan L.J.W., Verstegen M.M.A., van Hasselt P.M., et al. Prime editing for functional repair in patient-derived disease models. Nat. Commun. 2020;11:5352. doi: 10.1038/s41467-020-19136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Geurts M.H., de Poel E., Pleguezuelos-Manzano C., Oka R., Carrillo L., Andersson-Rolf A., Boretto M., Brunsveld J.E., van Boxtel R., Beekman J.M., Clevers H. Evaluating CRISPR-based prime editing for cancer modeling and CFTR repair in organoids. Life Sci. Alliance. 2021;4:e202000940. doi: 10.26508/lsa.202000940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y., Yang G., Huang S., Li X., Wang X., Li G., Chi T., Chen Y., Huang X., Wang X. Enhancing prime editing by Csy4-mediated processing of pegRNA. Cell Res. 2021;31:1134–1136. doi: 10.1038/s41422-021-00520-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson J.W., Randolph P.B., Shen S.P., Everette K.A., Chen P.J., Anzalone A.V., An M., Newby G.A., Chen J.C., Hsu A., Liu D.R. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022;40:402–410. doi: 10.1038/s41587-021-01039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Song J., Pang S., Lu Y., Chiu R. Poly(U) and polyadenylation termination signals are interchangeable for terminating the expression of shRNA from a pol II promoter. Biochem. Biophys. Res. Commun. 2004;323:573–578. doi: 10.1016/j.bbrc.2004.08.128. [DOI] [PubMed] [Google Scholar]
- 11.Kulcsár P.I., Tálas A., Tóth E., Nyeste A., Ligeti Z., Welker Z., Welker E. Blackjack mutations improve the on-target activities of increased fidelity variants of SpCas9 with 5'G-extended sgRNAs. Nat. Commun. 2020;11:1223. doi: 10.1038/s41467-020-15021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao Y., Zhao Y. Self-processing of ribozyme-flanked RNAs into guide RNAs in vitro and in vivo for CRISPR-mediated genome editing. J. Integr. Plant Biol. 2014;56:343–349. doi: 10.1111/jipb.12152. [DOI] [PubMed] [Google Scholar]
- 13.Nissim L., Perli S.D., Fridkin A., Perez-Pinera P., Lu T.K. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Mol. Cell. 2014;54:698–710. doi: 10.1016/j.molcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Port F., Bullock S.L. Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nat. Methods. 2016;13:852–854. doi: 10.1038/nmeth.3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knapp D.J.H.F., Michaels Y.S., Jamilly M., Ferry Q.R.V., Barbosa H., Milne T.A., Fulga T.A. Decoupling tRNA promoter and processing activities enables specific Pol-II Cas9 guide RNA expression. Nat. Commun. 2019;10:1490. doi: 10.1038/s41467-019-09148-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin Q., Zong Y., Xue C., Wang S., Jin S., Zhu Z., Wang Y., Anzalone A.V., Raguram A., Doman J.L., et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 2020;38:582–585. doi: 10.1038/s41587-020-0455-x. [DOI] [PubMed] [Google Scholar]
- 17.Dang Y., Jia G., Choi J., Ma H., Anaya E., Ye C., Shankar P., Wu H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015;16:280. doi: 10.1186/s13059-015-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsai S.Q., Wyvekens N., Khayter C., Foden J.A., Thapar V., Reyon D., Goodwin M.J., Aryee M.J., Joung J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014;32:569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y., Gao Y., Gao Y., Zhang Q. Targeted deletion of floral development genes in Arabidopsis with CRISPR/Cas9 using the RNA endoribonuclease Csy4 processing system. Hortic. Res. 2019;6:99. doi: 10.1038/s41438-019-0179-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gifford C.A., Ranade S.S., Samarakoon R., Salunga H.T., de Soysa T.Y., Huang Y., Zhou P., Elfenbein A., Wyman S.K., Bui Y.K., et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science. 2019;364:865–870. doi: 10.1126/science.aat5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen P.J., Hussmann J.A., Yan J., Knipping F., Ravisankar P., Chen P.F., Chen C., Nelson J.W., Newby G.A., Sahin M., et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell. 2021;184:5635–5652.e29. doi: 10.1016/j.cell.2021.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haapaniemi E., Botla S., Persson J., Schmierer B., Taipale J. CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018;24:927–930. doi: 10.1038/s41591-018-0049-z. [DOI] [PubMed] [Google Scholar]
- 23.Ihry R.J., Worringer K.A., Salick M.R., Frias E., Ho D., Theriault K., Kommineni S., Chen J., Sondey M., Ye C., et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018;24:939–946. doi: 10.1038/s41591-018-0050-6. [DOI] [PubMed] [Google Scholar]
- 24.An P., Sáenz Robles M.T., Pipas J.M. Large T antigens of polyomaviruses: amazing molecular machines. Annu. Rev. Microbiol. 2012;66:213–236. doi: 10.1146/annurev-micro-092611-150154. [DOI] [PubMed] [Google Scholar]
- 25.Kim D.Y., Moon S.B., Ko J.H., Kim Y.S., Kim D. Unbiased investigation of specificities of prime editing systems in human cells. Nucleic Acids Res. 2020;48:10576–10589. doi: 10.1093/nar/gkaa764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin S., Lin Q., Luo Y., Zhu Z., Liu G., Li Y., Chen K., Qiu J.L., Gao C. Genome-wide specificity of prime editors in plants. Nat. Biotechnol. 2021;39:1292–1299. doi: 10.1038/s41587-021-00891-x. [DOI] [PubMed] [Google Scholar]
- 27.Habib O., Habib G., Hwang G.H., Bae S. Comprehensive analysis of prime editing outcomes in human embryonic stem cells. Nucleic Acids Res. 2022;50:1187–1197. doi: 10.1093/nar/gkab1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geisinger J.M., Stearns T. CRISPR/Cas9 treatment causes extended TP53-dependent cell cycle arrest in human cells. Nucleic Acids Res. 2020;48:9067–9081. doi: 10.1093/nar/gkaa603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Enache O.M., Rendo V., Abdusamad M., Lam D., Davison D., Pal S., Currimjee N., Hess J., Pantel S., Nag A., et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020;52:662–668. doi: 10.1038/s41588-020-0623-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinha S., Barbosa K., Cheng K., Leiserson M.D.M., Jain P., Deshpande A., Wilson D.M., 3rd, Ryan B.M., Luo J., Ronai Z.A., et al. A systematic genome-wide mapping of oncogenic mutation selection during CRISPR-Cas9 genome editing. Nat. Commun. 2021;12:6512. doi: 10.1038/s41467-021-26788-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K.i., et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 32.Hsu J.Y., Grünewald J., Szalay R., Shih J., Anzalone A.V., Lam K.C., Shen M.W., Petri K., Liu D.R., Joung J.K., Pinello L. PrimeDesign software for rapid and simplified design of prime editing guide RNAs. Nat. Commun. 2021;12:1034. doi: 10.1038/s41467-021-21337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
High-throughput sequencing data were deposited in the NCBI Sequence Read Archive database under accession code NCBI: PRJNA789416. All other data are available upon reasonable request.