

Abstract
Approved drugs for the treatment of osteoporosis can prevent further bone loss but do not stimulate bone formation. Approaches that improve bone density in metabolic diseases are needed. Therapies that take advantage of the ability of mesenchymal stem cells (MSCs) to differentiate into various osteogenic lineages to treat bone disorders are of particular interest. Here we examine the ability of small interfering RNA (siRNA) to enhance osteoblast differentiation and bone formation by silencing the negative suppressor gene GNAS in bone MSCs. Using clinically validated lipid nanoparticle (LNP) siRNA delivery systems, we show that silencing the suppressor gene GNAS in vitro in MSCs leads to molecular and phenotypic changes similar to those seen in osteoblasts. Further, we demonstrate that these LNP-siRNAs can transfect a large proportion of mice MSCs in the compact bone following intravenous injection. Transfection of MSCs in various animal models led to silencing of GNAS and enhanced differentiation of MSCs into osteoblasts. These data demonstrate the potential for LNP delivery of siRNA to enhance the differentiation of MSCs into osteoblasts, and suggests that they are a promising approach for the treatment of osteoporosis and other bone diseases.
Keywords: lipid nanoparticles, siRNA, GNAS, bone formation, mesenchymal stem cells, gene silencing
Graphical abstract
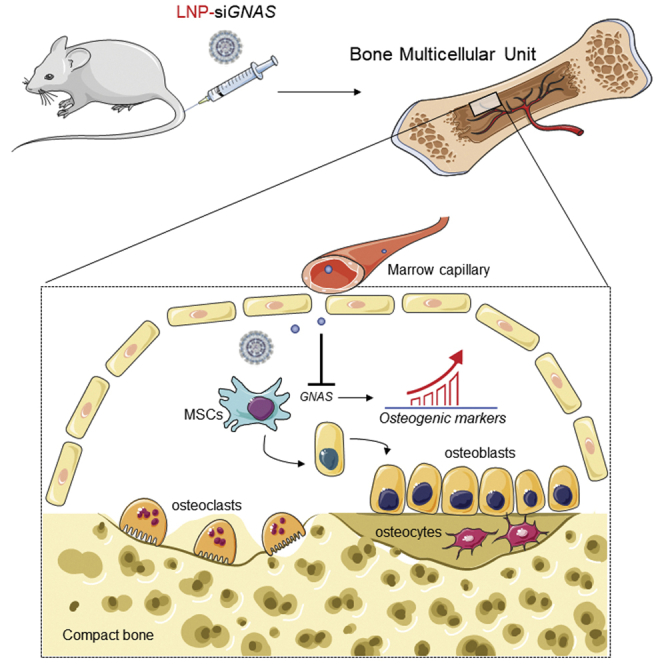
Basha and colleagues demonstrate that LNP-siRNAs can transfect a large proportion of mesenchymal stem cells (MSCs) in the compact bone following systemic administration, leading to silencing of GNAS gene, subsequently enhancing the differentiation of these MSCs into cells that exhibit molecular characteristics of osteoblasts.
Introduction
Bone homeostasis is critical to quality of life as healthy bones are vital for mobility, while fragile bones (e.g., in osteoporosis) are susceptible to fractures resulting in long-term disability and mortality. Currently approved drugs for the prevention and treatment of osteoporosis include bisphosphonates, calcitonin, and selective estrogen receptor modulators that work by inhibiting bone resorption.1, 2, 3, 4, 5 However, these compounds can have many adverse effects and do not stimulate new bone formation.6 Parathyroid hormone, the first approved compound capable of stimulating new bone formation, is limited to 24 months use due to potential side effects, including activation of bone resorption, and has shown a limited efficacy on nonvertebral fractures.7,8 Understanding the molecular details of the pathways that control bone formation is critical for the development of novel approaches to reverse osteoporosis, including the development of new anabolic agents to improve bone quality. Presently, the most successful anabolic drug is romosozumab, a monoclonal antibody that binds sclerostin, increases bone formation, and decreases bone resorption.9 However, follow-up observations show increased risk for infections, and currently applications are limited to osteoporosis. There is a significant need for new anabolic drugs with broader scope that can be used to modulate the molecular pathways that control bone formation. This approach could be used not only to treat osteoporosis but also to accelerate the healing of fractures that fail to resolve quickly.
The ability of human mesenchymal stem cells (hMSCs) to differentiate into chondrogenic and osteogenic lineages10 suggests potential clinical use in regenerative medicine to treat bone disorders.11,12 In addition to positive regulators that promote the transition from stem cell to differentiated tissues, negative regulators that suppress differentiation are also involved. This is demonstrated by evidence of ectopic bone formation in humans carrying inactivating mutations in the GNAS gene locus.13,14 Synthetic small interfering RNA (siRNA) libraries from stem cells have been used to identify the endogenous suppressors of osteogenic anabolism that, when silenced by the appropriate siRNA, could stimulate osteogenic differentiation of hMSCs and bone growth.15
siRNA oligonucleotides have been demonstrated to silence target mRNA transcripts in rodents, non-human primates,16,17 and humans.18 Silencing GNAS to stimulate bone growth is a logical approach given bone formation arising from inactivating mutations in GNAS.13,14,19 The use of siRNA to silence GNAS requires in vivo delivery of siRNA to the tissues where the target mesenchymal stem cells (MSCs) reside. The use of lipid nanoparticles (LNPs) for siRNA delivery is showing considerable promise to silence target genes in vivo. Potent LNP-siRNA systems have been developed that can silence hepatocytes genes at intravenous (i.v.) doses as low as 10 μg siRNA/kg body weight,17 and silence target genes in immune cells and tumors at somewhat higher doses.20,21
The aim of the study was to conduct proof-of-principle experiments to investigate whether lipid-based nanosystems can be designed to safely and effectively deliver gene-specific siRNAs to bone-residing MSCs that can modulate cell differentiation in favor of bone formation.
Given that accumulation of nanoparticles in bone requires particles of 50-nm diameter or less,22 we employ limited-size LNP-siRNA systems (50-nm diameter) to maximize “passive” accumulation of siRNA in bone tissue following i.v. administration. We demonstrate delivery of LNP-siRNAs to mouse bone MSCs, use these LNP-siRNA systems to silence GNAS, and characterize functional effects on key osteogenic markers of bone formation. We show that this results in accelerated differentiation of MSCs into osteoblasts, as assessed by the expression of specific markers.
Results
Culturing cells in osteogenic medium results in upregulation of bone osteogenic markers
MSC-like cells (mouse embryonic fibroblasts [MEFs] or C3H10T1/2 clone 8) are a good alternative source of primary bone-derived MSCs for investigating osteogenic differentiation in bone tissue engineering studies.23,24 MEFs or C3H10T1/2 clone 8 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) or in osteogenic medium (OM), and harvested at appropriate time points for the determination of GNAS and osteogenic markers using RT-qPCR. In cells treated with OM, a 3-fold increase in GNAS transcripts was observed after 3 days of treatment. However, GNAS transcripts decreased close to baseline levels by day 10 (Figure 1A), suggesting that cells had differentiated to a mature stage. Cells incubated with DMEM exhibited background levels of GNAS throughout the entire incubation period.
Figure 1.

Expression of GNAS transcripts and key osteogenic markers during induction of osteogenesis in vitro
(A–F) MSC-like cells were incubated in DMEM or OM and total RNA was extracted to measure (A) GNAS, (B) ALPL, (C) Sp7, (D) BGLAP, (E) IBSP, and (F) TBP mRNA at the indicated time points. Bar graphs represent expression of the target mRNA relative to the HPRT housekeeping gene with or without OM. mRNA values on day 0 are considered to be 100%. Each data point represents the arithmetic mean ± SD of three technical replicates. Data from one representative experiment of three independent experiments are shown. (G) Alkaline activity was visualized in MSC-like cells following incubation in DMEM or OM in triplicate on 12-well plates. Data are representative of three images captured with a 10× objective. Bar graphs depict quantification of images showing ALP activity of MSC-like cells following incubation with DMEM or OM medium. Data are normalized to DMEM incubation medium and each point represents the arithmetic mean ± SD of three technical replicates. Data from one representative experiment of three independent experiments are shown (∗p < 0.05). Images only from MEFs cells are shown. OM, osteogenic medium.
In order to confirm that MSC-like cells treated with OM demonstrate osteogenic characteristics, the expression of ALPL, a key marker of osteogenic differentiation, was examined. Osteogenic stimulation generated a significant increase in ALPL after 3 days of incubation in OM and increased up to 18-fold at 10 days’ incubation time, compared with cells treated with DMEM (Figure 1B). This increase was also confirmed using an enzymatic assay for alkaline phosphatase (ALP) protein expression in cells cultured in DMEM or OM over 3 weeks. After 2 weeks, the intensity of ALP staining in OM-treated cells was significantly increased relative to cells treated with DMEM, and after 3 weeks OM-treated cells exhibited the highest levels of ALP (Figure 1G). To further confirm the osteogenic identity of OM-treated cells, we also examined the expression of Sp7, BGLAP, and IBSP markers for osteogenic differentiation. It was observed that all three genes were significantly upregulated when cells were treated with OM, compared with those treated with DMEM (Figures 1C–1E). Finally, the housekeeping TATA-binding protein (TBP) gene did not change in the presence of OM, suggesting that the effect was specific for osteogenic markers. Taken together, the data indicate that MSC-like cells are a good in vitro expression system for GNAS, where expression occurs at significant levels following 3 days of incubation in OM. In addition, the increased expression of osteogenic markers associated with increased GNAS suggests that both MSC-like cells could be utilized effectively as an in vitro system to assess the effect of GNAS downregulation on bone osteogenic genes.
siRNA screening to identify the anti-GNAS duplex that mediates optimal knockdown of the GNAS gene
MSC-like cells (C3H10T1/2 cells and MEFs) were transfected with the fluorescently labeled transfection control duplex using RNAiMax and examined 24 h later. Microscopy revealed 70%–80 % transfection efficiency following incubation with 10 or 30 nM TYE 563-labeled siRNA respectively (data not shown). Cells pre-treated with OM for 48 h followed by incubation in DMEM were also transfected with six siRNA duplexes (A–F), including the HPRT positive control and a scrambled siRNA, at doses of 1, 3, and 10 nM (Table S1).
Relative mRNA levels were then determined using qRT-PCR at 4 days post transfection with a variety of siRNAs shown in Table S1. It was found that sequences C, D, and F were the most potent (Figures S1A and S1B). Further knockdown studies were conducted on MSC-like cells pre-incubated with OM, followed by treatment with sequences C, D, and F, and downregulation of the GNAS target gene was determined using qRT-PCR and droplet digital PCR (ddPCR). Since the ddPCR technique exhibits the best accuracy using a small number of cells, GNAS mRNA was determined in 10,000 plated cells receiving treatment with siRNA against GNAS (siGNAS). This study had a 2-fold aim, to confirm the most effective siRNA sequence, and to compare two different methods to be used in vivo to determine GNAS mRNA following systemic administration of LNP-siRNA. Data showed that sequence D was the most effective in silencing the GNAS target gene in vitro and that ddPCR data confirmed its robustness and accuracy in a small number of cells (Figure S1D), corroborating the qRT-PCR data (Figure S1C). Therefore, sequence D was selected to pursue further in vitro and in vivo experiments aiming at silencing the GNAS target gene.
As siRNA duplexes are susceptible to degradation in vivo, sequence D was chemically modified by adding 2′ O-methyl bases that block exonuclease activity and improve resistance to nucleases in vivo (i.e., stealth siRNA). To assess the efficacy and toxicity of the modified duplex, MSC-like cells were plated and treated with the modified (stealth) and unmodified (standard) siRNA using the same transfection method, and GNAS mRNA was determined using qRT-PCR. It was observed that stealth siGNAS showed similar efficacy to unmodified duplex, producing over 70% knockdown of GNAS without affecting cell viability assessed based on the RNA yield across the plates (Figure S2A). For consistency, all in vitro and in vivo experiments were done using the stealth siRNA. Data also demonstrated that GNAS silencing was sequence specific and gene specific, as TBP mRNA expression following siRNA treatment remained intact (Figure S2B). As well, a noticeable increase of osteogenic BGLAP gene transcripts was observed at relatively high siRNA doses, indicating the role of GNAS as a negative regulator (Figure S2C).
Transfection reagents such as Trifecta are not effective in vivo due to bioavailability and toxicity issues, requiring use of LNP systems such as those used for Onpattro. We therefore characterized the transfection potency of LNP-siGNAS systems, particularly the efficacy of siGNAS delivered with LNPs containing (R)-2,3-bis(octadecyloxy)propyl-1-(methoxy polyethylene glycol 2000) carbamate known as (PEG)-DMG or PEG-(R)-2,3-bis(stearyloxy)propyl-1-(methoxy poly(ethylene glycol) 2000 carbamate (DSG) known as PEG-DSG, at 1.5 mol %. LNP systems containing PEG-DSG exhibit longer circulation lifetimes in vivo,25 important for accessing compact bone. MSC-like cells were plated, pre-incubated with OM, and treated with LNP-siLuc control or LNP-siGNAS for 4 days. GNAS mRNA, two housekeeping gene transcripts, namely HPRT and TBP, were determined using qRT-PCR analysis. Cells treated with siGNAS delivered with LNPs containing PEG-DMG exhibited significant downregulation of the target gene relative to siCtrl-treated cells for all three doses tested, leading to up to 70%–80% knockdown after 2 days of treatment (Figure 2A). When siGNAS was delivered with PEG-DSG LNPs, however, significantly fewer transcripts of the target gene were measured after 2 days of treatment, reducing the GNAS expression by 50%–60% (Figure 2B), suggesting that LNPs-siRNAs containing PEG-DMG were more potent than those formulated using PEG-DSG. The expression TBP housekeeping used in addition to the HPRT remained constant across all doses tested, indicating that no off-target effects occurred during treatment (Figure 2C). Taken together, the data indicate that LNPs can efficiently transfect MSC-like cells with siRNA, resulting in specific and significant GNAS gene knockdown.
Figure 2.

The structure of PEG lipid in the LNP compositions reduces the potency of siRNA-mediated knockdown of GNAS in vitro
(A–C) MSC-like cells were seeded in multi-well plates and siGNAS or siCtrl encapsulated in LNPs containing PEG-DMG or PEG-DSG were administered in triplicate wells at indicated doses. Bar graphs represent expression of GNAS mRNA relative to HPRT determined 4 days after incubation with (A) PEG-DMG and (B) PEG-DSG LNPs as determined by qRT-PCR. The GNAS mRNA was normalized to the housekeeping genes HPRT and TBP. GNAS mRNA values following LNP-siCtrl treatments are considered 100%. Each data point represents arithmetic mean ± SD of three technical replicates. Data from one representative experiment of three independent experiments are shown. Bar graphs in (C) represent expression of TBP mRNA relative to HPRT. TBP mRNA following siCtrl is considered to be 100% (∗p < 0.05).
GNAS knockdown in vitro, mediated by LNP-siRNA, modulates differentiation of MSC-like cells
To determine the anabolic effect of RNAi-mediated GNAS knockdown, we measured the expression of early and late markers of osteogenic differentiation following treatment with LNP-siGNAS. Cells were incubated with LNP-siGNAS or LNP-siCtrl, and RNA transcripts of bone osteogenic markers were determined at 7 days using qRT-PCR analysis. Data showed that cells treated with LNP-siGNAS generated modest increases in ALPL and COL1A1 transcripts compared with cells treated with siCtrl at doses of 5 and 10 μg/mL (Figures 3A and 3B). To confirm the osteogenic identity of the cell transformation, we also examined the expression of two additional late-stage osteogenic markers (Table S2). BGLAP and IBSP were significantly upregulated following treatment with siGNAS compared with siCtrl, and this upregulation was largely dose dependent (Figures 3C and 3D). It is well established that enhanced ALP is a marker for osteoblast differentiation in vitro and for bone formation. To further confirm the osteogenic differentiation of MSC-like cells following siRNA-mediated GNAS knockdown, cells were briefly (48 h) incubated with OM and replenished with DMEM followed by treatment with LNP-siCtrl and LNP-siGNAS for 3 weeks. Red staining associated with production of ALP was significantly enhanced in cells treated with LNP-siGNAS compared with LNP-siCtrl, suggesting a significant increase in ALP activity compared with siCtrl control, presumably caused by GNAS knockdown (Figures 3E and 3F). The ALP staining intensity began to increase 2 weeks after LNP-siGNAS treatment and remained significantly higher than that for cells treated with LNP-siCtrl. These results indicate that siRNA-mediated knockdown of GNAS gene exhibits an osteogenic effect as demonstrated by increased ALP activity and differentiation to osteoblasts.
Figure 3.

siRNA-mediated GNAS knockdown increases expression of bone osteogenic markers and enhances ALP activity in vitro
(A–D) MSC-like cells were treated with PBS control, LNP-siGNAS, or LNP-siCtrl (comprising PEG-DMG) at indicated doses, and mRNA expression of bone osteogenic markers was analyzed using qRT-PCR at 7 days following treatment. Expression of target genes, relative to HPRT, was determined and normalized to siCtrl levels, which were considered to be 100%. Graphs depict increased mRNA transcripts of bone osteogenic markers (A) ALPL, (B) COL1A1, (C) BGLAP, and (D) IBSP following treatment with siGNAS relative to siCtrl at 7 days after treatment. Each data point represents arithmetic mean ± SD of three technical replicates. Data from one representative experiment of three independent experiments are shown (∗p < 0.05). MSC-like cells were seeded in plates at 60 × 104 cells per cm2 and incubated with OM for 48 h. Next, they were incubated with LNP-siGNAS or LNP-siCtrl (PEG-DMG) for 1 week at doses of 10 mg/mL, including PBS controls. ALP activity was assessed using photomicroscopy 2 and 3 weeks later based on red staining of ALP. (E) Data from one representative image of a minimum of three images per treatment are shown. Images were analyzed using the OpenLab software and quantified as described in section “materials and methods.” (F) Bar graphs were obtained from the quantification of images and depict fold increase (mean ± SD, n = 3) of ALP activity over time, relative to cells treated with PBS or siCtrl. Data from one representative experiment of two independent experiments are shown (∗p < 0.05).
LNP-siRNA induced extended GNAS silencing in vitro without affecting cell viability
Since phenotypic changes and cell differentiation may require silencing of GNAS for an extended time period, it was important to examine the duration of RNAi-induced silencing in vitro after a single treatment. Following a brief period of GNAS induction, MSC-like cells were treated with LNP-siGNAS or LNP-siCtrl at a dose of 5 μg/mL. GNAS stimulation was performed to slightly boost gene expression levels to achieve a reliable siGNAS dose-response curve in vitro. Treatment was stopped after 2 days by replacing the medium with DMEM, and GNAS mRNA was determined every 3 days for 2 weeks using real-time qRT-PCR. Data showed that GNAS transcripts were reduced by 80% at 3 days after single treatment with siGNAS, compared with siCtrl (Figure 4A). GNAS expression started to increase after 6 days and continued to increase during the 2-week observation period. However, GNAS expression remained significantly reduced compared with LNP-siCtrl-treated MSC-like cells even at 2 weeks. Microscopic monitoring of cells did not reveal evidence of toxicity over the 15-day incubation period. Importantly, despite significant downregulation of GNAS, the TBP housekeeping gene remained unchanged, confirming target specificity of RNA interference (data not shown). To determine the osteogenic effects on MSCs following single treatments, mRNA transcripts of two key osteogenic genes, IBSP and BGLAP, were also determined. A moderate upregulation of the mRNA of both biochemical markers was observed early during the observation period, which tended to increase significantly over time (Figures 4B and 4C). Hence, a single-dose treatment with LNP-siGNAS at 5 μg/mL produced a silencing effect on the GNAS gene for several days without compromising cell viability assessed by the RNA yield, and elicited a moderate but consistent osteogenic effect on MSC-like cells.
Figure 4.

LNP encapsulation induces prolonged siRNA-mediated GNAS knockdown in vitro and stimulates expression of osteogenic markers
(A–C) MSC-like cells were treated with LNP-siGNAS or LNP-siCtrl (PEG-DMG) at 5 μg/mL following brief induction of GNAS. After 3 days, plates were replenished with DMEM and total RNA was extracted every 3 days for a period of 2 weeks. Total RNA was extracted and mRNA of (A) GNAS, (B) IBSP, and (C) BGLAP was determined using qRT-PCR. Bar graphs represent mRNA expression of GNAS and selected bone osteogenic markers over time relative to an HPRT housekeeping gene. GNAS mRNA transcripts, following siCtrl treatment, were considered to be 100%. Each data point represents arithmetic mean ± SD of three technical replicates. Data from one representative experiment of two independent experiments are shown (∗p < 0.05).
LNP-siRNA silences the reporter GFP gene in primary bone-derived MSCs in vitro
To demonstrate that LNP-siRNAs were effective in silencing genes in primary MSCs, it was important to isolate and confirm the identity of primary MSCs. Freshly prepared cells, derived from compact bone of green fluorescence protein (GFP) transgenic mice, were stained with a cocktail of antibodies to PDGFRα, Sca-1, CD45, CD31, and TER119, and analyzed by flow cytometry. Viable cells were detected as propidium iodide (PI) negative and Hoechst positive and hematopoietic (CD45+, CD31+, and TER119+) cells were removed from the analysis by gating out these marker signals. Sca-1 and PDGFRα receptors was considered to be markers for the target MSCs (Figure 5A). PDGFRα+/Sca-1+ cells were isolated and cultured in the presence of mouse mesenchymal stem cell stimulatory supplements (MesenCult MSC Basal Medium, STEMCELL Technologies, Vancouver, BC, Canada) in a hypoxic environment (5% O2, 10% CO2, 85% N, 25% CO2), known to trigger upregulation of genes involved in metabolism, cell proliferation, and survival. Small and medium colonies developed into spindle-shaped fibroblast-like cells, features that became more evident on day 14 and 21, indicating a MSCs-like phenotype (Figure 5B) that could be used for future in vitro characterization and silencing investigations. Cells were further expanded and their potential to differentiate into osteoblasts was tested following incubation with MesenCult Osteogenic Stimulatory Kit (STEMCELL Technologies, Vancouver, BC, Canada) or OM followed by staining for ALP, a well-defined cytochemical marker of osteogenesis. While incubation with DMEM caused only minor changes in ALP, incubation of cells with OM significantly increased ALP activity (red staining) in primary MSCs after 21 days, reflecting their differentiation into osteoblasts (Figure 5C). This was supported by von Kossa staining, which allows indirect visualization of calcium deposits (black spots, Figure 5C). This confirmed the osteogenic identity of MSCs cultured in vitro, which differentiated into osteoblasts following 3 weeks of incubation with OM.
Figure 5.

Silencing of GFP mediated by LNP-siRNA in well-defined primary MSCs in vitro is specific and dose dependent
Enrichment of PDGFRa/Sca1 MSCs population of GFP-expressing mice.
(A) Representative flow cytometric profiles of compact bone-derived mononuclear cells (top left FSC/SSC) and gated nucleated live cells (Hoechst positive/propidium iodide negative, top right) stained with CD45, TER119, CD31, biotinylated CD140 (PDGFRα, APA5, bottom left), SA-PE, and Sca-1 antibodies (bottom right). (B) Phase-contrast micrographs showing the morphology of a colony of MSC-derived cells sorted from 5,000 PDGFRα+/Sca-1+ cells at 7, 14, and 21 days after plating and culturing in MesenCult medium. Adipogenesis (bottom) as indicated by lipid vacuoles present intracellularly. (C) Osteogenesis of MSCs cultured for 14 days in DMEM or OM as detected by ALP (top) or von Kossa staining (bottom). (D–F) Data depict the presence of Tye 563-labeled siRNA (red) intracellularly in bone MSCs visualized using a Leica confocal microscope. Bone MSCs derived from GFP transgenic mice were treated with LNP-siGFP or LNP-siCtrl (PEG-DSG) at indicated doses. Bar graphs depict relative GFP expression of MSCs determined by flow cytometry at 4 (E) and 7 (F) days after treatment, following detection of MSCs using the appropriate antibodies. Data from one representative experiment of two independent experiments are shown (∗p < 0.05).
The transfection efficiency of siRNA in 80% confluent primary MSCs, incubated with 10 nM TYE 563-labeled siRNA transfection control duplexes, at 24 h was approximately 70%–80% based on number of cells expressing the fluorescent red dye (red label, Figure 5D). To determine if transfection efficiency translates to a knockdown of the GFP reporter gene in primary MSCs when siRNA is delivered with LNPs, cells were treated with increasing doses of LNP-siGFP and LNP-siCtrl as well as phosphate-buffered saline (PBS). Four and 7 days later, GFP expression was determined in MSCs (Sca-1+/PDGFRα+) using flow cytometry. After 4 days of treatment, a moderate but dose-dependent downregulation of GFP signal was observed, relative to siCtrl at 5 and 10 μg/mL (Figure 5E). Following incubation with LNP-siGFP, GFP transcripts were also significantly reduced at 7 days post treatment for all doses compared with siCtrl (Figure 5F). Hence, primary MSCs can be successfully transfected with siRNA using LNPs, and LNP-siRNA nanosystems can silence the GFP reporter gene in a sequence- and dose-dependent manner.
siRNA-LNPs exhibit significant uptake by MSCs in vivo
Having characterized the osteogenic impact of GNAS silencing in vitro, we next moved to in vivo studies. First, the uptake of LNP-siRNAs by bone residing MSCs following an i.v. injection was investigated. PDGFRα-GFP transgenic mice that express the GFP reporter gene only in MSCs were given intravenous administrations of LNP-siRNA labeled with DiI (red) or PBS controls. Forty-eight hours later, bone sections devoid of bone marrow were viewed by confocal microscopy. In mice treated with PBS, MSCs were visualized as compact bone cells expressing nuclear GFP, predominantly in the outer layers of bone, as indicated by the green MSC and blue nuclear stain (Figure 6A). In those receiving LNP-siRNA, a proportion of MSCs and nuclei overlapped with DiI-red-labeled LNPs, indicating that a considerable number of MSCs had taken up the LNP-siRNAs (Figure 6B). To quantitatively examine the uptake of LNP-siRNA by MSCs, either 10 mg/kg LNP-siRNA labeled with DiO (green) or PBS (controls) was administered i.v. into C57Bl6 mice via tail-vein injection. The presence of LNP-siRNAs was then detected by flow cytometry in MSCs identified as PDGFRα+/Sca-1+ cells at 48 and 72 h following injection (Figure 6C). Fluorescence-activated cell sorting (FACS) analysis showed that a significant proportion of MSCs (approximately 70%) contained the DiO-labeled LNP-siRNAs (Figure 6D). Taken together, the data demonstrate that, following systemic administration, a substantial quantity of LNP-siRNAs was also taken up by a large proportion of bone-derived MSCs, demonstrated by a 15-fold increase of the mean fluorescence intensity (MFI). Aside from the compact bone and bone marrow, the expression of GNAS is identified in a number of tissues, including adipose tissues, dental tissues, and human placenta. However, due to the difficulty in characterization of these cells in other tissues, assessment of LNP-siRNA uptake by MSCs in those tissues was not pursued.
Figure 6.

LNP-siRNA are delivered successfully to mouse MSCs following systemic administration
(A and B) Images depict representative areas of 5-μm bone sections captured 2 days following systemic administration of (A) PBS and (B) DiI-labeled LNP-siRNAs (red), comprising PEG-DSG analyzed by a confocal microscopy. Images demonstrate a degree of overlap between MSCs (green) and LNP-siRNAs (red), indicating the intracellular enrichment of bone MSCs with LNP-siRNAs. Scale bar, 5 μm. The LNP-siRNA delivery to MSCs was also quantified using flow cytometry. (C) The contour plots (top) represent MSCs, defined as PDGFRα+/Sca-1+ cells, at the indicated time points. The contour plots in the bottom panel represent the percentage of DiI-positive MSCs defined as PDGFRα+/Sca-1+ cells of the upper panel, comprising high levels of labeled LNP-siRNAs following their systemic delivery, compared with PBS controls. Plots are representative of two experiments performed in duplicate. (D) Bar graphs demonstrate the percentage of the MSCs (top) containing LNP-siRNAs relative to MSCs of PDGFRα-GFP-expressing mice treated with PBS, and the fold increase of DiI MFI signal in MSCs (bottom) relative to PBS treatments, at the indicated time points. Data from one representative experiment of two independent experiments are shown. Each data point represents arithmetic mean ± SD of three mice per treatment (∗p < 0.001). MFI, mean fluorescence intensity.
LNP-siRNA mediates silencing in GFP reporter genes in vivo following systemic administration
Having achieved knockdown of GNAS in vitro, we determined whether or not LNP-siRNAs could mediate silencing of a reporter gene in MSCs in vivo. Transgenic mice expressing C57Bl6-GFP received increasing doses of LNP-siGFP i.v., including PBS and siCtrl as controls. Since MSCs constitute a very rare population representing only 0.001% in the bone marrow and less in the compact bone, after removing the marrow, bones of three mice were harvested, pooled, and MSC isolates were identified as PDGFRα+/Sca-1+ cells. Approximately 5,000 cells were sorted for further testing. Referring to color intensity in the heatmap, gene expression analysis demonstrated reductions in numbers of GFP mRNA transcripts with increasing doses of LNP-siRNA targeted against GFP (Figure S3A). Gene silencing, determined by qRT-PCR, occurred 1 week following systemic administration, particularly at high doses. SiCtrl or siGFP administration did not alter the expression HPRT or TBP housekeeping genes, demonstrating that GFP knockdown was gene specific, which ruled out any siRNA-mediated off-target effects. No signs of toxicity or weight loss were observed following systemic administration of all doses of LNP-siGFP, as indicated by clinical assessments and weight measurements throughout the observation time (Figure S3B). In addition, FACS analysis demonstrated downregulation of GFP in MSCs derived from compact bone, reaching up to 60% knockdown compared with LNP-siCtrl treatment at doses of 10 mg/kg LNP-siGFP (Figure S3C). GFP signals were also significantly lower in MSCs originated from bone marrow (p < 0.05) following treatment with 10 mg/kg LNP-siRNA, overall correlating with GFP mRNA transcripts (Figure S3D). Taken together, these data demonstrate that substantial silencing of the GFP reporter gene in MSCs in vivo was obtained with LNP-siRNA.
GNAS knockdown mediated by LNP-siRNA stimulates osteogenic differentiation of MSCs in vivo
The functional effect of silencing of the negative regulator GNAS gene in MSCs was assessed by determining the molecular expression of key osteogenic markers, following systemic administration of LNP-siRNA targeting GNAS. Meanwhile, it was important to identify molecular changes in MSCs in vivo that would allow us to monitor any phenotypic differentiation toward osteoblasts. Cell isolates from C57Bl6 and PDGFRα-GFP transgenic mice were stained with a cocktail of antibodies, including the MSC-specific markers PDGFRα and Sca-1. C57Bl6-derived cells had a distinct cell population expressing high levels of the MSC-specific receptors PDGFRa and Sca-1 (Figure S4A, left). However, cells derived from the compact bone of PDGFRα GFP transgenic mice (Figure S4A, middle) had (1) a cell population that co-expressed GFP associated with PDGFRα receptor and Sca-1, as well as (2) a cell population expressing PDGFRα-GFP alone. Staining these cells with the anti-PDGFRα antibody confirmed the presence of two sub-populations, one that co-expressed PDGFRα and the Sca-1 receptor, and one that expressed PDGFRα but lacked expression of Sca-1. This became evident by overlaying the contour plots of MSCs from transgenic mice expressing PDGFRα-GFP, as detected by the PDGFRα antibody, and the plots of the same cell population as detected by GFP expression alone (Figure S4A, right).
To carry out the molecular characterization of MSCs, 3,000–5,000 cells co-expressing PDGFRα and Sca-1 were sorted from C57Bl6 mice and three subsets of cells were obtained from transgenic mice expressing PDGFRα-GFP, namely (1) cells co-expressing PDGFRα and Sca-1, (2) cells expressing PDGFRα alone, and (3) cells expressing neither of these receptors (negative control). RNA was extracted and expression of six key osteogenic markers, including HPRT and GNAS, was determined quantitatively using ddPCR. Data showed that PDGFRα+/Sca-1+ MSCs, isolated from C57Bl6 mice, expressed low (Sp7 and RUNX2), intermediate (ALPL and IBSP), and relatively high (COLlA1 and BGLAP) levels of osteogenic transcripts involved in bone differentiation and maturation (Figure S4B). However, there was a noticeable difference for cell populations sorted from mice expressing PDGFRα-GFP; cells identified as PDGFRα+/Sca-1+ exhibited somewhat lower expression of key osteogenic transcripts relative to the same cell population derived from C57Bl6. Further data analysis demonstrated that a second cell population, identified as PDGFRα+/Sca-1−, exhibited higher levels of osteogenic transcripts than PDGFRα+/Sca-1+ cells for most of the osteogenic markers. Expression of osteogenic markers in the PDGFRα−/Sca-1− was barely detectable (Figure S4C). Collectively, these observations suggest that transgenic mice expressing PDGFRα-GFP are a good model to detect MSCs and examine the response of osteogenic markers following silencing of a negative regulator such as GNAS, provided both PDGFRα+/Sca-1+ and PDGFRα+/Sca-1− cell populations are investigated.
We examined the efficacy of LNP-siRNA targeting GNAS, and the response of osteogenic markers, in both MSC populations. Transgenic mice expressing PDGFRα-GFP were treated i.v. with LNP-siGNAS and LNP-siCtrl, and, 7 days after treatment, 3,000–5,000 PDGFRα+/Sca-1+ MSCs and PDGFRα+/Sca-1- MSCs were sorted (Figure 7A). RNA was extracted and expression of GNAS transcripts along with osteogenic markers were determined using ddPCR. It was observed that GNAS transcripts were moderately downregulated relative to LNP-siCtrl treatments in both MSC cell subsets at 7 days following LNP-siGNAS administration (Figure 7B). Importantly, ddPCR data analysis also revealed that intermediate and late osteogenic marker transcripts were moderately upregulated in the PDGFRα+/Sca-1+ MSCs subset, with COL1A1 and BGLAP demonstrating a considerable increase relative to LNP-siCtrl 7 days following systemic administration of LNP-siRNAs (Figure 7C). Furthermore, when the PDGFRα+/Sca-1− MSCs subset was analyzed, mRNA transcripts of intermediate and late osteogenic markers were further upregulated, suggesting that moderate decrease of the GNAS target gene was sufficient to trigger a significant increase of some of the key osteogenic markers involved in differentiation (Figure 7C).
Figure 7.

Downregulation of GNAS mediated by LNP-siRNA is associated with upregulation of osteogenic markers
LNP-siGNAS or LNP-siCtrl (PEG-DSG) at doses of 10 mg/kg, or PBS, were administered i.v. into PDGFRα+ transgenic mice. GNAS and osteogenic marker transcripts of cells expressing either PDGFRα+/Sca-1+ (gate 1) or PDGFRα+/Sca-1+- (gate 2), defined as MSCs, were determined in bones using ddPCR. (A) Dot plots displaying two populations of MSCs (1 and 2) following administration of siCtrl (left) and siGNAS (right). Bar graphs depicting mRNA GNAS transcripts relative to siCtrl treatments for populations of PDGFRα+/Sca-1+ cells (gate 1) or PDGFRα+/Sca-1+- cells (gate 2). mRNA GNAS transcripts following siCtrl treatments were considered to be 100%. (B and C) Heatmap depicting GNAS mRNA transcript expression of PDGFRα+/Sca-1+ cell populations following treatment of PDGFRα transgenic mice with siCtrl or siGNAS-LNPs. Each square represents the signal obtained from analysis of MSCs of three pooled mice. (C) Heatmap showing transcript expression of osteogenic markers in MSC populations defined in gate 1 and 2 of PDGFRα+ transgenic mice following treatment with siCtrl or LNP-siGNAS. Each square represents the signal obtained from MSCs of three pooled mice. Data from one representative experiment of two independent experiments are shown.
To further validate these observations, we investigated GNAS downregulation and any effects on osteogenic markers during an 8-day observation time. C57Bl6 mice were administered i.v. with LNP-siCtrl, LNP-siGNAS, or a PBS control at a dose of 10 mg/kg. After 4 and 8 days following treatment, mice of each group were divided in four subgroups of two mice each. Following dissection, femurs and tibia of two mice were pooled to enable a sufficient MSC yield, hence producing a biological replicate for each treatment and time point. In wild-type mice, as expected, a single population of PDGFRα+/Sca-1+ MSCs was identified and sorted at day 4 and 8 following systemic administration. The data demonstrated a statistically significant downregulation of GNAS transcripts at 4 days post treatment with LNP-siGNAS, relative to LNP-siCtrl (p < 0.05), and it remained low as long as 8 days post treatment when a marginal increase of GNAS was detected (Figure 8A). After 4 days post treatment, no change of osteogenic markers was observed (Figure 8B). However, after 8 days, ddPCR analysis showed that most of the osteogenic transcripts were increased relative to controls, with both COL1A1 and BGLAP reaching statistical significance (p < 0.05, Figure 8C) suggesting a time lag in the response and molecular cell differentiation. It is important to note that no treatment-related toxic effects were observed, as determined by monitoring the mice weight loss (Figures S6A and S6B). In addition, clinical chemistry testing did not reveal alteration of the markers, indicating that treatment and the dose employed were well tolerated (Table S3). Finally, to investigate a possible downregulation of GNAS in other tissues, GNAS expression was first determined in various organs of three C57Bl6 mice. Next, 6 days following the systemic administration of LNP-siGNAS (n = 3) or LNPsi-Ctrl (n = 3), tissues were harvested and subjected to RNA extraction. Residual GNAS mRNA was determined using RT-qPCR and normalized to the respective LNP-siCtrl, which was considered 100%. Tissues exhibited various patterns of GNAS expression (Figure S6A). Furthermore, it was observed that GNAS was significantly downregulated in the liver, moderately in the spleen, as well as in the bone marrow. However, GNAS transcripts remained unchanged in a broad range of other tissues tested where isolation yielded RNA of reliable quality (Figure S6B). In fact, this pattern correlates with biodistribution of nucleic acid-loaded LNPs used in this study, whereby liver takes up the majority of these LNPs, followed by spleen and bone marrow.25 Nonetheless, it is difficult to assess GNAS silencing in MSCs of these organs comprising the highest expression of GNAS, since isolation of MSCs from tissues apart from compact bone and bone marrow has not yet been optimized. Importantly, these organs particularly liver, did not show any functional impairment as demonstrated by the blood chemistry (Table S3).
Figure 8.

Silencing of GNAS by LNP-siRNA in C57Bl6 wild-type mice leads to osteogenic differentiation of MSCs in vivo
Osteogenic marker transcripts of PDGFRα+/Sca-1+ MSCs were determined in pooled bones (femur and tibiae) 4 and 8 days following treatment with LNP-siGNAS or LNP-siCtrl (PEG-DSG), using ddPCR. (A) Dot plots (left) display PDGFRα+/Sca-1+ MSCs following administration of siCtrl and dot plots (right) display PDGFRα+/Sca-1+ MSCs following administration of siGNAS. Bar graphs (middle) depict mRNA GNAS transcripts of PDGFRα+/Sca-1+ cell populations relative to siCtrl at indicated time points. mRNA GNAS transcripts following siCtrl treatment were considered to be 100%. (B and C) Bar graphs depict GNAS mRNA transcripts of osteogenic markers relative to siCtrl of PDGFRα+/Sca-1+ MSC populations at (B) 4 days and (C) 8 days following systemic administration of LNP-siRNA. mRNA GNAS transcripts following siCtrl treatment were considered to be 100%. Each data point represents arithmetic mean ± SD of two mice. Data from one representative experiment of two independent experiments are shown (∗p < 0.05).
Taken together, these data demonstrate that, overall, the systemic administration of LNP-siGNAS produced downregulation of GNAS in mouse bone-derived MSCs at a level that affected the expression of key osteogenic markers. This indicated that MSCs could undergo transcription to form cells with the molecular characteristics of osteoblasts.
Discussion
The results of this investigation show that siRNA-induced silencing of the suppressor gene GNAS in vitro in primary MSCs and MSC-like cells moves MSCs toward osteoblast formation. Brief pre-incubation of MSC-like cells with OM was done to enhance the GNAS mRNA, as its expression was lower than in primary MSCs. This optimized the siRNA knockdown conditions in vitro, whereby higher levels of the target gene allowed detection of a dose-dependent silencing. However, enhancement of GNAS was halted well before eliciting any effect on osteogenic markers, and cells were subjected to treatment with LNP-siGNAS when these osteogenic markers were at the lowest levels. Further, we show that in vivo delivery of optimized siRNA to silence GNAS packaged in LNP results in osteogenic differentiation of MSCs in compact bone. There are three topics that require further discussion. The first concerns the need for agents that boost osteogenesis and the potential of GNAS as a gene silencing target for enhancing osteogenesis in MSCs. The second topic concerns the LNP-siRNA delivery system employed here and the ways this approach needs improvement. The final topic concerns the animal model employed and its relevance to bone disease.
There is a clear clinical need for new bone anabolic agents, and recently efforts to promote osteogenic marker expression for osteoporosis treatment have been made.26,27 Targeting MSCs to undergo osteogenic differentiation in the bone, followed by mobilization of the osteoblastic progenitors to the bone surface, is a physiologically relevant approach to treat bone diseases associated with bone loss. MSCs expand rapidly,28,29 thus enhancing osteoblast differentiation of MSCs is a promising approach15,30,31 and a better alternative to procedures for MSC transplantation in vivo that are facing major challenges due to their inability to home to the bone surfaces and promote osteogenic responses.32,33
Regarding the targeting of LNP-siRNAs to bone MSCs, although LNP delivery to the skeleton has been a challenge, the capillary supply of bone multicellular unit (BMU) provides the vascular supply for multipotent stem and lining cells residing under the canopy.26 As such, the marrow capillary of approximately 3-μm diameter, enables targeting of LNPs to MSCs. With regard to the GNAS as a relevant target with predictable outcome, herein we provide validation of GNAS as a silencing target for enhancing osteoblast differentiation in vitro. However, the GNAS gene is a critical regulator of osteoblast differentiation in mesenchymal progenitor cells, and modulation of GNAS can have a wide variety of potential effects. For example, extreme Wnt or Hedgehog signaling following complete knockout of GNAS could inhibit bone formation, whereas an abundance of GNAS can produce ectopic bone formation in soft tissues.34 However, other work has shown that the GNAS gene is an osteogenic-specific negative regulator of bone formation that can cause skeletal abnormalities when mutated in mice and humans.13,14,35 This is consistent with observations indicating that GNAS (or its Gsα subunit) is a negative regulator of osteogenic commitment and that decreased GNAS expression enhances an osteogenic cell fate in MSCs.15,36 The in vitro work performed here is consistent with these latter studies, showing that GNAS is present in MSC-like cells and that GNAS silencing leads to characteristics of osteoblasts in target cells. This is corroborated by the in vivo studies that clearly showed that a moderate downregulation of GNAS was sufficient to produce an appreciable osteogenic differentiation of MSCs following treatment. However, due to the potential disparate effects of GNAS modulation, including its effect in other tissues, it is necessary to identify alternative target genes for safer and more specific control of bone formation before clinical potential can be exploited.
With regard to the LNP delivery system employed here, previous investigators have used lipid-based formulations containing cationic lipids 1,2-Dioleoyl-3-trimethylammonium propane (DOTAP) to show that siRNA-induced silencing of genes that inhibit bone formation in osteogenic-lineage cells has promise.19,37 However, the clinical utility of delivery systems containing permanently positively charged lipids such as DOTAP is limited due to the toxicity of these agents.38,39 LNP systems containing ionizable cationic lipids have been shown to be effective agents for gene silencing in the liver in vivo with therapeutic indices as high as 1,00040,41 resulting in the US Food and Drug Administration (FDA)-approved drug Onpattro.18 Nonetheless, the efficacy of these systems for gene silencing in non-hepatic tissues is limited due to rapid accumulation in liver tissue following i.v. administration. In the present study, we attempted to improve distribution to non-hepatic tissue by incorporating 1.5 mol % PEG-DSG rather than PEG-DMG in the LNP-siRNA system as the longer residence time of PEG-DSG in the LNP particle would be expected to lead to longer circulation lifetimes25 and thus improved distribution to bone. However, these systems still exhibited rapid clearance (t1/2 < 15 min), resulting in the need to administer LNP-siRNA at the dose levels of 10 mg/kg siRNA to achieve appreciable distribution to bone. This dose is higher than the dose level needed to induce gene silencing in hepatocytes,17,42 but below the maximum tolerated dose. As a result, the LNP systems employed here may have limited clinical potential. While the circulation lifetimes could be improved by incorporating higher levels of PEG-DSG, such levels render the LNP-siRNA systems increasingly less transfection competent.25,43 Alternative methods of producing LNP-siRNA systems with long circulation lifetimes that are potent transfection agents are urgently required. Although this approach demonstrated efficacy with regard to delivery to the targeted cells with a functional effect as assessed by increased expression of relevant bone formation genes, the delivery system may indeed require improvement. In this regard, screening of other formulations with longer circulation life times are being investigated.
A final point is that the efficacy of the LNP-siRNA nanosystems was tested only in healthy animals, free of bone abnormalities. It is possible that a functional response to gene silencing resulting in stimulated bone formation could be reduced in these animals. Further experiments are needed on disease models with bone anomalies resulting from gene mutations to bring the RNAi technology and gene delivery closer to clinical applications.
In summary, the studies presented here provide proof-of-principle confirmation that LNP delivery of siRNA to silence genes causing downregulation of osteogenesis in MSCs in compact bone can lead to osteogenic effects both in vitro and in vivo. While the gene targets and the delivery systems to achieve these effects require further improvement, the approach to treating genetic diseases in bones by correcting underlying gene dysfunctions in stem cells clearly has significant clinical potential. We note that gene silencing in MSCs in vivo facilitates a natural mechanism to treat disease and has led to success stories in other areas of medicine.44
Materials and methods
Preparation of LNPs and siRNA encapsulation
LNPs were prepared as described previously.45 Briefly, one volume each of dilinoleylmethyl-4-dimethylaminobutyrate (DLin-MC3-DMA; courtesy of Dr. Marco Ciufolini, Department of Chemistry, Faculty of Science, University of British Columbia), distearoylphosphatidylcholine (DSPC; from Avanti Polar Lipids, Alabaster AL, USA), cholesterol (Sigma-Aldrich, ON, Canada), and (PEG-DSG (from Avanti Polar Lipids, Alabaster AL, USA), at 50:10:38.5:1.5 mole ratio was dissolved in ethanol and mixed with three volumes of siRNA (1:10 w/w siRNA to lipid) suspended in acetate buffer. The cationic lipid was synthesized by Biofine International (Vancouver, BC, Canada). A detailed description of the cationic lipid and its activities has been given previously.42 Formulation was performed in a microfluidic mixer Chip provided by Precision Nanosystems (Vancouver, BC, Canada), by pumping both solutions through the micromixer at a combined flow rate of 4 mL/min (1 mL/min for ethanol and 3 mL/min for aqueous buffer). The resultant mixture was dialyzed against PBS, pH 7.4, for 16 h to remove ethanol, and then filtered. A more detailed description of the method has been given previously.46 To prepare fluorescently labeled LNPs, 0.2 mol % 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindodicarbocyanine-5,5′-disulfonic acid (DiI-C18), red) or 3,3′-dioctadecyloxacarbocyanine perchlorate (DiO-C18, green) (Invitrogen), was added to the lipid mix. Dynamic light scattering was used to determine LNP size (number weighting) using the Malvern Zetasizer NanoZS (Worcestershire, UK). Size measurements were performed in PBS (pH 7.4). Encapsulation was measured using the Quant-iT Ribogreen RNA assay (Life Technologies, Burlington, ON). Particles were incubated at 37°C in the presence or absence of 1% Triton X-100 (Sigma-Aldrich, St. Louis, MO) for 10 min. Following incubation, Quant-iT Ribogreen RNA reagent was added and fluorescence intensity was measured (Ex/Em 480/520nm). Total siRNA was measured following treatment with 1% Triton X-100 (Sigma-Aldrich, ON, Canada) while non-treated samples represented un-encapsulated siRNA. Particle size was 52 nm polydispersity index (PDI) = 0.03) and 52.5 nm (PDI = 0.05) for siGNAS and siLuciferase particles, respectively. Encapsulation efficiency measured using Quant-iT Ribogreen (Thermo Fisher Scientific MA, USA) RNA assay was greater than 90% for both types of LNPs (91.4% ± 0.5% for siGNAS and 94.5% ± 1.4% for siLuciferase particles). The pKa of the ionizable cationic lipid component of the particles, DLin-MC3-DMA, is 6.4, so the particles have a near-neutral surface charge at a physiological pH of 7.4.
Cell culture, gene expression, siRNA transfection, and sequence identification
The murine mesenchymal cell line MEFs and C3H10T1/2 clone 8 murine cells have many features of MSCs and therefore were both used as MSC-like cells to investigate osteogenic differentiation in vitro.23,24 They were purchased from the American Type Culture Collection (ATCC; Rockville, MD, USA) and monolayer cultures of cells were routinely maintained in standard 100-cm2 cell culture dishes in DMEM (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% (v/v) fetal calf serum (FCS; Gibco, Gaithersburg, MD, USA), 1% L-glutamine and 1% penicillin and streptomycin (P/S) at 37°C in a 5% carbon dioxide atmosphere. MEFs or C3H10T1/2 cells were seeded in 12-well plates in triplicate wells at a density of 6 × 104 cells/cm2 and incubated with DMEM, 10% FBS, and 1% P/S. At 80% confluence, cells were incubated in DMEM or in MesenCult Osteogenic Stimulatory Kit (STEMCELL Technologies, Vancouver, BC, Canada), known as OM, which is formulated to stimulate in vitro differentiation of mouse mesenchymal progenitor stem cells from compact bone and bone marrow into osteoblasts. Media were changed every second day for 15 days. On days 0, 3, 7, and 10, cells from triplicate wells were lysed using 350 μL of Lysis Buffer (Invitrogen, Carlsbad, CA, USA) and stored at −80°C. At the end of the experiment, cell lysates were thawed, mixed with equal amounts of 70% ethanol, and RNA was extracted according to the manufacturer’s directions (PureLink RNA extraction Kit, Life Technologies). Extricated total RNA was used to determine mRNA expression of GNAS, Sp7 (an early marker of osteoblast differentiation), ALP (ALPL), an intermediate bone osteogenic marker, and two other late osteogenic markers, BSP, a component of mineralized tissues, and osteocalcin (BGLAP), a noncollagenous protein hormone found in bone. The mRNA expression levels of GNAS or bone osteogenic genes were quantified in cells cultured in DMEM or OM at various times by real-time quantitative RT-PCR. To identify the best siRNA sequence and test the efficacy of gene-specific siRNA, MSC-like cells (MEFs or C3H10T1/2 cells) were cultured in OM for 48 h to induce expression of GNAS. Next, wells were replenished with DMEM and treated with six predesigned duplexes (TriFECTa, Integrated DNA Technologies), namely A, B, C, D, E, and F, all from the RefSeq database in GenBank (Table S1). Included in the treatment were a “universal” negative control duplex (NC1) that targets a site that is absent from human, mouse, and rat genomes, and a positive control duplex (HPRT-S1 DS positive control) that targets a site in the hypoxanthine phosphoribosyntransferase (HPRT) 1 gene common between human, mouse, and rat. Cells were seeded in 24-well plates and forward transfection was performed in triplicate according to the manufacturer’s directions, by mixing the RNAiMax (Invitrogen) transfection reagent with siRNA to obtain final concentrations of 10 and 20 nM, including positive and negative controls. Transfection progressed for 2 h before the first medium renewal. After 24 h in culture, cells were lysed with 350 μL of Lysis Buffer (Life Technologies) and stored at −80°C. Transfection efficiency was also visualized and monitored using Tye 563-labeled siRNA imaged in a Zeiss Axiovert 200 microscope with a QImaging Retiga EX mono 12-bit camera. The mRNA expression levels of GNAS or bone osteogenic markers were quantified by qRT-PCR as described.45 To confirm the efficacy of the anti-GNAS sequence, cells were plated in 24-well plates in triplicates and treated with three of the best-performing siRNA duplexes at 10 and 20 nM siRNA doses, including negative and positive controls, and GNAS mRNA was determined using qRT-PCR. To validate the findings, GNAS mRNA levels were also determined in 5,000 cells following incubation with selected siRNA duplexes using droplet digital PCR (ddPCR), a method based on sample compartmentalization in single oil droplets that represent independent PCR reactions. This permits the absolute quantification of target DNA and greater precision relative to qRT-PCR.47 Cells were lysed and RNA was extracted using RNeasy Plus Micro Kit (Qiagen) as per manufacturer’s directions, followed by cDNA synthesis using High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City CA, USA). GNAS transcribed cDNA was determined using the QX100 Droplet Digital PCR system (Bio-Rad Laboratories, Hercules, CA). A sample premixture was produced by diluting the cDNA 2.5 times in TE-Buffer pH-8 (Invitrogen, Thermo Fisher Scientific) in Eppendorf tubes, and 9 μL of this premixture was loaded in eight wells of a 96-well plate. Next, 1 μL of each primer assay (times the number of total samples) was mixed with 10 μL (times the number of total samples) of ddPCR Supermix for Probes (Bio-Rad) and loaded onto the wells of the 96-well plate containing 9 μL of the mixture. After a quick vortex, the plate was spun down at 1,000 rpm for 1 min. Next, droplets were generated by a QX200 Droplet Generator (Bio-Rad Laboratories) and the endpoint PCR was performed on a T100 Thermal Cycler (Bio-Rad Laboratories) following the manufacturer’s recommendations. PCR products were loaded into the QX100 Droplet Reader and analyzed by QuantaSoft version 1.2 (Bio-Rad Laboratories). For the ddPCR experiments, the same primers and probes were used as in qRT-PCR, with the identical nucleotide sequence and dye/quenchers 6-FAM/ZEN/IBFQ (Integrated DNA Technologies, Iowa, USA).
In vitro knockdown efficiency and quantitative real-time PCR
To investigate the siRNA-mediated knockdown, MSC-like cells were seeded in 12-well plates as described. The sequence of the mouse GNAS gene (GenBank: NM_019690) was extracted from the NCBI Entrez nucleotide database. The anti-GNAS siRNA (siGNAS) was composed of two complementary RNA strands: sense strand (GNAS-S) 5′-5′-mGmCrAmCrCmArUrUrGrUrGmArAmGrCmArGmArUrGrArGrGmAT-3′ and antisense strand (GNAS-AS): 5′-rArUmCrCrUrCrArUmCrUmGrCmUrUrCrArCrArArUrGrGmUrGmCmUmU-3′-3′. The two strands of the GNAS siRNA are modified oligoribonucleotides that include multiple 2′-OMe modifications (denoted by the letter m) introduced into the siRNA to improve nuclease stability and to reduce the risk of pure RNA triggering an innate immune response in mammalian cells.48 In addition, the 2′ O-methyl bases are a modification of RNA that increases stability of interactions, making them resistant to attack by single-stranded ribonucleases and less susceptible to DNases, thereby increasing stability and binding affinity to the target. Cells were incubated in OM to induce expression of GNAS for 48 h in culture. Next, the selected siRNA duplexes targeting GNAS (Table S1), or negative control siRNA (siLuciferase S, cuuAcGcuGAGuAcuucGAdTsdT; AS, UCGAAGuACUcAGCGuAAGdTsdT) encapsulated in DLinMC3-DMA LNPs, were added at 2.5, 5, and 10-μg/mL final concentration and incubated at 37°C (5% CO2 and 5% O2). The control siRNA against luciferase contained phosphorothioate linkages (indicated as the letter s) between the 3′-deoxythymidine (dT) overhangs and includes multiple 2′-OMe modifications (indicated by lower-case letters). To assess the efficacy of the modified duplex and rule out toxicity, cells were plated and treated with the modified (stealth) and non-modified (standard) siRNA using the same transfection method, and GNAS mRNA transcripts were measured using qRT-PCR. Each siRNA dose was administered in triplicate. One triplicate of wells was treated with PBS and served as a negative control. The medium was changed every 3 days, while the siRNA was maintained at the required concentrations throughout the entire incubation time. The incubation time varied between 4 days, when testing the in vitro efficacy, and 14 days when assessing the osteogenic stimulation.
To investigate duration of silencing of the GNAS in vitro, cells were cultured in 12-well plates in DMEM or OM and, after 48 h, medium was removed and cells were replenished with DMEM plus additives. Next, they were treated with LNP-siRNA Ctrl or LNP-siRNA targeting GNAS at 5 μg/mL dose. After 4 days, LNP-siRNA treatment was stopped by removing the medium and replenishing the cells with fresh DMEM. Cells were harvested every 3 days for a period of 15 days. At different time points, cells were lysed with 350 μL of RLT RNeasy Plus lysis buffer containing 3.5 μL of 2-mercaptoethanol, vortexed, spun, and stored in a −80°C freezer. The mRNA expression levels of GNAS or bone osteogenic genes were quantified by qRT-PCR and RNA was reverse transcribed to cDNA from 1 μg of total RNA. Real-time PCR was performed on a StepONEPlus Real-Time PCR system, which included forward and reverse primers and the FAM-labeled probe for the target gene respectively (Table S2). Relative expression between samples was quantified using the comparative ΔΔCT algorithm, and the amount of mRNA of each gene was normalized to two housekeeping genes: mouse hypoxanthine phosphoribosyltransferase (HPRT) and TBP, a general transcription factor that binds specifically to a DNA sequence called the TATA box. When indicated, the mRNA extracted from siRNA-treated cells was normalized to mRNA of cells treated with control siRNA (siCtrl), which was considered as 100% (.i.e., no efficacy).
MSC isolation, characterization, culture, and siRNA silencing of a reporter gene in vitro and in vivo
Transgenic mice carrying GFP C57BLKa-Thy1.1 (GFP, CMV-IE, chicken β-actin promoter49) were a kind donation of Dr. Irvin Weissman, Stanford University School of Medicine. Bone MSCs were isolated from mice as described, with minor modifications.50 Briefly, femurs and tibias were dissected and minced into very small pieces with scissors on a mortar placed on ice containing 5 mL of cold PBS. The minced bones were gently washed once with PBS containing 2% FBS and 10 mM HEPES to wash marrow from bone tissue. The pellet of bone fragments was collected and incubated for 1 h in a shaker at 37°C in 15 mL of RPMI medium containing 3 mg/mL collagenase/dispase (Roche, Mississauga, ON, Canada) and 2% FBS. Next, the cell mix was resuspended in 15 mL of ice-cold (4°C) PBS and filtered through a cell strainer (Falcon) to remove debris and bone fragments. The remaining bones were transferred to a mortar containing 5 mL of ice-cold PBS and tapped gently with a pestle. The mix was passed through the cell strainer into the same collecting tube and the procedure was repeated three times. The filtered cell pellet was then collected by centrifugation at 300 × g for 10 min at 4°C, washed twice in ice-cold PBS/FBS 2%, and stained for 30 min on ice with the following monoclonal antibodies (mAbs): biotinylated CD140 (PDGFRα, APA5) that was visualized with a second step staining with Streptavidin-PE or Alexa 488, Sca-1 conjugated to PE-Cy7 (Ly6A/E), CD45 (30-F11), TER119 (TER-119), and CD31 all conjugated to allophycocyanin, PI, and Hoechst for labeling the nucleated cells. All mAbs were purchased from eBioscience. PI fluorescence was measured and a live cell gate was defined that excluded the cells positive for PI and included Hoechst-positive cells. Gates were drawn to exclude cells that were stained positive for CD45, TER119, and CD31 (lineage positive) and cells stained positive for PDGFRα and Sca-1, identified following staining with the isotype controls, were defined as the target cells. Flow cytometry analysis and sorting were performed on a BD FACS Aria III or BD Influx cell sorter. Sorted cells (between 3,000 and 5,000 per groups of three or four mice) were seeded in 12-well plates and cultured in MesenCult Expansion Kit (Mouse), STEMCELL Technologies, 1% L-glutamine, 1% penicillin, and streptomycin. Bone MSCs were expanded in 12-well plates and, to demonstrate their osteogenic potential, they were cultured for 21 days in MesenCult medium (MM) or MesenCult Osteogenic (OM) Stimulatory Kit (Mouse) purchased from STEMCELL Technologies. Osteogenic differentiation was detected by ALP and silver nitrate (von Kossa) staining as described below. To demonstrate pluripotency, MSCs were cultured in adipocyte differentiation medium containing DMEM, 10% FBS, 1 μM dexamethasone, 1 μM troglitazone, and 5 μg/mL insulin (Sigma-Aldrich, ON, Canada) and the medium was changed every 2 days. Transfection efficiency was assessed following visualization of Tye 563-labeled siRNA in bone MSCs using a Zeiss Axiovert 200 microscope with a QImaging Retiga EX mono 12-bit camera. Fluorescence and phase-contrast images were overlaid to visualize the intracellular siRNA. Next, bone MSCs were cultured in 12-well plates and treated with siRNA targeting GFP gene (siGFP) at doses of 1, 5, and 10 μg/mL. Four and 7 days after treatment, cells were harvested and MSCs were identified using antibodies as described above, and GFP expression was determined using a BD LSRII flow cytometer following detection of Sca1+/PDGFRα+ cells.
To demonstrate the efficacy of LNP-siRNA against GFP reporter gene in vivo, LNP-siGFP was administered i.v. to GFP transgenic mice (n = 3) at doses of 5 and 10 mg/kg, including siLuciferase (siCtrl) at 10 mg/kg. One week later, bones were harvested, pooled, processed as described, treated with collagenase/dispase, and stained with antibodies to detect bone MSCs. After gating out lineage-positive cells and gating in PI-negative and Hoechst-positive cells, GFP expression was measured in PDGFRa+/Sca1+-expressing cells following acquisition of approximately 1 million cells. In another set of experiments, aimed at determining GFP mRNA levels, following i.v. administration of LNP-siGFP or siCtrl, bone MSCs from pooled murine bones (n = 3) were sorted in 350 μL of RLT RNeasy Plus lysis buffer containing 3.5 μL of β2-mercaptoethanol and stored at −80°C. GFP mRNA expression was determined using ddPCR as described. To determine the expected range of gene expression, MSCs were isolated from 10-week-old PDGFRα H-2Kb-GFP transgenic mice (Rossi Lab, Biomedical Research Center, University of British Columbia [UBC]51 and C57Bl6 mice). From 3,000 to 5,000 cells from three different cell populations were sorted; i.e., PDGFRα+-GFP/Sca-1+ (double positive), PDGFRα+-GFP/Sca-1− (single positive) and PDGFRα−-GFP−/Sca-1− (double negative). cDNA was extracted and ddPCR was conducted using primer assays to determine expression of HPRT and TBP housekeeping genes, GNAS, and key early, intermediate, and late osteogenic markers of osteoblast differentiation (Sp7, RunX2, ALPL, COL1A1, IBSP, and BGLAP). The same procedure was followed for the C57Bl6 mice except that double-positive (PDGFRα+/Sca-1+) populations only were sorted and the PDGFRα receptor was detected with a two-step procedure; i.e., biotinylated CD140 (PDGFRα), followed by streptavidin-PE. Following acquisition using FACSDiva software, contour plots obtained from data analysis of cells derived from transgenic and wild-type mice were overlaid to confirm the identification of targeted MSC populations.
ALP and von Kossa staining
MSCs were cultured for 21 days in MM or OM and incubated at 37°C and 5% CO2 and O2. At the end of observation period, osteogenic differentiation was detected by ALP activity and von Kossa staining. This procedure was also applied to evaluate the functional effect of silencing of GNAS, following 7 days of treatment with LNP-siGNAS, as assessed by the expression of ALP. After 2 weeks in culture, plates were washed with PBS and fixed for 5 min with 5% formaldehyde at room temperature. The fixative was rinsed thoroughly and the cells were incubated with the ALP staining solution containing Napthol AS-MX phosphate (Sigma-Aldrich) 0.1mg/mL and Fast Red Violet LB Salt (Sigma-Aldrich) at 0.65 mg/mL for 1 h, protected from light. Alkaline-dye mixture was then discarded, the cells were rinsed thoroughly for 2 minutes with deionized water, and plates were allowed to dry. In the last few minutes of the incubation, von Kossa solution (2.5% silver nitrate in dH2O) was added to the cells and they were incubated for 1 h in the presence of strong light. Cells were then washed with tap water, allowed to dry, covered with coverslips, and viewed under a Zeiss Axiovert 200 microscope. The data were analyzed using the OpenLab software. Quantification of ALP staining was done in at least three acquired images and in multiple areas of each image using the NIH ImageJ win-32 software. The resulting Red, Green and Blue (RGB) color images were analyzed as previously described using the Color Deconvolution plugin 1.7 and, for each region of interest (ROI), the FastRed and FastBlue option was selected to quantify the staining.52 The numerical intensities were converted to optical density (OD) values with the following formula: OD = log (maximum intensity/mean intensity), where maximum intensity = 255 for 8-bit images. A minimum of three images were examined for each treatment and time point. The data were analyzed using the OpenLab software.
Assessment of LNP-siRNA delivery in MSCs in vivo
Ten-week-old PDGFRα-GFP transgenic mice and C57Bl/6 female mice (from Charles River) were used throughout. All animal experiments in this study were performed according to approved protocols and in compliance with the guidelines and requirements of Canadian Council and UBC Committee on Animal Care. DiI-labeled LNP-siRNA (DiIC18, Invitrogen) formulations were administered i.v. to mice via tail-vein injections at a dose of 10 mg/kg. Mice were weighed prior to and post systemic administration of PBS and siLuc (siCtrl) or siGFP encapsulated in LNPs. Two days following injections, mice were euthanized, bones were dissected, the marrow was flushed, and bones were fixed in 10% phosphate-buffered formalin. Bones were then decalcified in a 0.5 M ethylenediaminetetraacetic acid (EDTA; Sigma-Aldrich, ON, Canada) solution containing 0.2% glycerol and 0.6% sodium hydroxide, pH 7.1, for several days then processed overnight in two changes of 95% ethanol, four changes of 100% ethanol, three changes of xylene, and three changes of wax for a total of 15.5 h. Bones were then embedded in wax and sectioned at 5-μm thickness using a Leica microtome. Once dry, bone tissues were placed on slides and transferred to an oven at 60° for about 2 h to dewax, stained for 30 s with TOTO-3 (Invitrogen), washed, and covered with coverslips. All z stack images were acquired using a Leica TCS SP8 laser scanning confocal microscope using 20× or 60× objective (Leica, Germany) and images were analyzed using LAS AF 3 software. The GFP was excited with a 488-nm argon laser and DiI was excited with a 561-nm laser. The presence of DiI-labeled LNP-siRNA in GFP-expressing PDGFRa+ cells was determined based on the overlap of nuclei-blue, MSCs-green, and LPN-siRNA red. To confirm and quantify LNP-siRNA uptake by MSCs in vivo, C57Bl6 mice were injected i.v. with DiO-labeled LNP-siRNA at doses of 10 mg/kg or PBS for controls. Forty-eight and 72 h following injections, femurs and tibiae were dissected and digested with collagenase/dispase and bone MSCs were identified as described. Fluorescence intensity produced by DiO-labeled LNP uptake by bone MSCs was measured in 1 million events, acquired using BD LSRII flow cytometer.
GNAS silencing in vivo and expression of osteogenic markers
The ultimate goal was to silence the negative regulator GNAS and investigate the response of key osteogenic markers that would point to a shift of MSCs from a progenitor to a more differentiated, osteoblast-like stage. For this, three mouse models were used. In the first model, 10-week-old GFP transgenic mice were divided into groups of four with three mice per group and received LNP-siGFP or LNP-siCtrl i.v. at doses of 5, 10, and 15 mg/kg. In the second model, 10-week-old PDGFRa-GFP mice divided into two groups of three mice each were subjected to systemic administration of 10 mg/kg LNP-siGNAS or siCtrl. One week later, animals were euthanized and, in order to obtain a workable number of MSCs (3,000–5,000), femurs and tibiae of each group (n = 3) were harvested, pooled, and processed as described. For the third model, wild-type C57Bl/6 female mice (Charles River) were divided into four groups with four animals per group and subjected to systemic administration of LNP-siCtrl (n = 8) and LNP-siGNAS (n = 8). An additional group of three mice was added and received PBS. After 4 and 8 days following treatment, mice of each group were divided into four subgroups of two mice each. Following dissection, femurs and tibia of two mice were pooled to enable a sufficient MSC yield, hence producing a biological replicate for each treatment and time point. Mice were weighed prior to and post systemic administration LNP-siRNAs and PBS to assess any weight loss during the follow-up. For all the three models, the MSCs were identified using the same staining strategy and approximately 5,000 cells were sorted directly in 350 μL of RLT RNeasy Plus lysis buffer containing 3.5 μL of β2-mercaptoethanol and stored in a −80°C freezer. Of note, during analysis of PDGFRα-GFP transgenic mice, somewhat unexpectedly, two subpopulation of MSCs were identified (PDGFRα+-GFP/Sca-1+ and PDGFRα+-GFP/Sca-1−) and both were sorted for further molecular characterization. In the C57Bl6, however, only one cell population was identified co-expressing PDGFRa and Sca1. Following cDNA synthesis, GNAS mRNA transcripts and the osteogenic markers were determined by ddPCR using the respective predesigned primer assays. Finally, to investigate downregulation of GNAS in other tissues, two groups of three mice each were subjected to LNP-siGNAS or LNP-siCtrl via tail-vein injection at a dose of 10 mg/kg. Six days following systemic administration, tissues were harvested form a broad range of organs, RNA was extracted as described, and residual mRNA expression was determined by RT-qPCR. Relative expression of GNAS was also determined in all tissues tested. In a last experiment, six C57Bl6 mice divided into two groups of three mice each received PBS or LNP-siLuc systemically at a dose of 10 mg/kg. At day 4 following treatment, blood was withdrawn and transferred to BD Microtainer blood collection tubes (BD, NJ, USA) allowed to clot at room temperature for 1 h, and centrifuged at 2,000 rpm. Serum was then transferred to Eppendorf tubes and kept at 4°C. Custom clinical chemistry tests were performed by IDEXX BioAnalytics, Sacramento, CA, USA.
Statistical analysis
Experimental values are presented as means ± SD of three values and expressed as percentage of the control baselines. The n value represents the number of experiments conducted for analysis. Statistical analyses were performed using a two-tailed Student’s t test, where two groups were compared at each treatment or time point indicated (i.e., siCtrl versus siGNAS or PBS versus OM-treated samples). The analysis (paired or two-sample equal variance homoscedastic), was determined based on the variation of the standard deviation of two populations. p < 0.05 was accepted as statistically significant (∗p < 0.05).
Acknowledgments
The authors would like to acknowledge support from the Canadian Institutes for Health Research (CIHR) under UOP grant 122069 and the NanoMedicines Innovation Network (NMIN), a Canadian Networks of Centers of Excellence (NCE) in nanomedicine. D.W. is supported by the Swiss National Science Foundation (#183923).
Author contributions
G.B., R.N.Y., F.M.V.R., and P.R.C. conceptualized the study. A.G.C. and K.Y.T.C. prepared the formulations. G.B., A.G.C., and T.P. conducted all in vitro and in vivo experiments. G.B. wrote the original draft. D.W. analyzed the images and revised the manuscript. P.R.C. did the final editing of the manuscript. R.N.Y. and P.R.C. acquired the funding.
Declaration of interests
P.R.C. has financial interests in Precision NanoSystems, Acuitas Therapeutics, and Mesentech.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.06.012.
Supplemental information
References
- 1.Rogers M.J., Crockett J.C., Coxon F.P., Mönkkönen J. Biochemical and molecular mechanisms of action of bisphosphonates. Bone. 2011;49:34–41. doi: 10.1016/j.bone.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 2.Davey R.A., Findlay D.M. Calcitonin: physiology or fantasy? J. Bone Miner. Res. 2013;28:973–979. doi: 10.1002/jbmr.1869. [DOI] [PubMed] [Google Scholar]
- 3.Cummings S.R., Martin J.S., McClung M.R., Siris E.S., Eastell R., Reid I.R., Delmas P., Zoog H.B., Austin M., Wang A., et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 2009;361:756–765. doi: 10.1056/NEJMoa0809493. [DOI] [PubMed] [Google Scholar]
- 4.Finkelstein J.S., Wyland J.J., Lee H., Neer R.M. Effects of teriparatide, alendronate, or both in women with postmenopausal osteoporosis. J. Clin. Endocrinol. Metab. 2010;95:1838–1845. doi: 10.1210/jc.2009-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delmas P.D., Bjarnason N.H., Mitlak B.H., Bruce R., Shah A.S., Anne-Catherine S., Aartis S., Huster W.J., Draper M., Christiansen C. Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N. Engl. J. Med. 1997;337:1641–1647. doi: 10.1056/NEJM199712043372301. [DOI] [PubMed] [Google Scholar]
- 6.Das S., Crockett J.C. Osteoporosis – a current view of pharmacological prevention and treatment Physiological control of bone remodeling. Drug Des. Devel. Ther. 2013;7:435–448. doi: 10.2147/DDDT.S31504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vahle J.L., Sato M., Long G.G., Young J.K., Francis P.C., Engelhardt J.A., Westmore M.S., Ma Y.L., Nold J.B. Skeletal changes in rats given daily subcutaneous injections of recombinant human parathyroid hormone (1-34) for 2 years and relevance to human safety. Toxicol. Pathol. 2002;30:312–321. doi: 10.1080/01926230252929882. [DOI] [PubMed] [Google Scholar]
- 8.Baron R., Hesse E. Update on bone anabolics in osteoporosis treatment: rationale, current status, and perspectives. J. Clin. Endocrinol. Metab. 2012;97:311–325. doi: 10.1210/jc.2011-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McClung M.R., Grauer A., Boonen S., Bolognese M.A., Brown J.P., Diez-Perez A., Langdahl B.L., Reginster J.Y., Zanchetta J.R., Wasserman S.M., et al. Romosozumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2014;370:412–420. doi: 10.1056/NEJMoa1305224. [DOI] [PubMed] [Google Scholar]
- 10.Pittenger M.F., Martin B.J. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ. Res. 2004;95:9–20. doi: 10.1161/01.RES.0000135902.99383.6f. [DOI] [PubMed] [Google Scholar]
- 11.Dezawa M., Kanno H., Hoshino M., Cho H., Matsumoto N., Itokazu Y., Tajima N., Yamada H., Sawada H., Ishikawa H., et al. Specific induction of neuronal cells from bone marrow stromal cells and application for autologous transplantation. J. Clin. Invest. 2004;113:1701–1710. doi: 10.1172/jci200420935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horwitz E.M., Gordon P.L., Koo W.K.K., Marx J.C., Neel M.D., McNall R.Y., Muul L., Hofmann T. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc. Natl. Acad. Sci. USA. 2002;99:8932–8937. doi: 10.1073/pnas.132252399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eddy M.C., Jan de beur S.M., Yandow S.M., McAlister W.H., Shore E.M., Kaplan F.S., Whyte M.P., Levine M.A. Deficiency of the α-subunit of the stimulatory G protein and severe extraskeletal ossification. J. Bone Miner. Res. 2000;15:2074–2083. doi: 10.1359/jbmr.2000.15.11.2074. [DOI] [PubMed] [Google Scholar]
- 14.Shore E.M., Ahn J., De Beur S.J., Li M., Xu M., Gardner R.M., Zasloff M.A., Whyte M.P., Levine M.A., Kaplan F.S. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N. Engl. J. Med. 2002;346:99–106. doi: 10.1056/NEJMoa011262. [DOI] [PubMed] [Google Scholar]
- 15.Zhao Y., Ding S. A high-throughput siRNA library screen identifies osteogenic suppressors in human mesenchymal stem cells. Proc. Natl. Acad. Sci. USA. 2007;104:9673–9678. doi: 10.1073/pnas.0703407104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Fougerolles A., Vornlocher H.P., Maraganore J., Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat. Rev. Drug Discov. 2007;6:443–453. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Semple S.C., Akinc A., Chen J., Sandhu A.P., Mui B.L., Cho C.K., Sah D.W.Y., Stebbing D., Crosley E.J., Yaworski E., et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010;28:172–176. doi: 10.1038/nbt.1602. [DOI] [PubMed] [Google Scholar]
- 18.Adams D., Gonzalez-Duarte A., O’Riordan W.D., Yang C.C., Ueda M., Kristen A.V., Tournev I., Schmidt H.H., Coelho T., Berk J.L., et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018;379:11–21. doi: 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- 19.Zhang G., Guo B., Wu H., Tang T., Zhang B.T., Zheng L., He Y., Yang Z., Pan X., Chow H., et al. A delivery system targeting bone formation surfaces to facilitate RNAi-based anabolic therapy. Nat. Med. 2012;18:307–314. doi: 10.1038/nm.2617. [DOI] [PubMed] [Google Scholar]
- 20.Basha G., Novobrantseva T.I., Rosin N., Tam Y.Y.C., Hafez I.M., Wong M.K., Sugo T., Ruda V.M., Qin J., Klebanov B., et al. Influence of cationic lipid composition on gene silencing properties of lipid nanoparticle formulations of siRNA in antigen-presenting cells. Mol. Ther. 2011;19:2186–2200. doi: 10.1038/mt.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee J.B., Zhang K., Tam Y.Y.C., Tam Y.K., Belliveau N.M., Sung V.Y.C., Lin P.J.C., LeBlanc E., Ciufolini M.A., Rennie P.S., Cullis P.R. Lipid nanoparticle siRNA systems for silencing the androgen receptor in human prostate cancer in vivo. Int. J. Cancer. 2012;131:781–790. doi: 10.1002/ijc.27361. [DOI] [PubMed] [Google Scholar]
- 22.Sarin H. Physiologic upper limits of pore size of different blood capillary types and another perspective on the dual pore theory of microvascular permeability. J. Angiogenes. Res. 2010;2:14. doi: 10.1186/2040-2384-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garreta E., Genové E., Borrós S., Borrós S., Semino C.E. Osteogenic differentiation of mouse embryonic stem cells and mouse embryonic fibroblasts in a three-dimensional self-assembling peptide scaffold. Tissue Eng. 2006;12:2215–2227. doi: 10.1089/ten.2006.12.2215. [DOI] [PubMed] [Google Scholar]
- 24.Zhao L., Li G., Chan K.M., Wang Y., Tang P.F. Comparison of multipotent differentiation potentials of murine primary bone marrow stromal cells and mesenchymal stem cell line C3H10T1/2. Calcif. Tissue Int. 2009;84:56–64. doi: 10.1007/s00223-008-9189-3. [DOI] [PubMed] [Google Scholar]
- 25.Chen S., Tam Y.Y.C., Lin P.J.C., Sung M.M.H., Tam Y.K., Cullis P.R. Influence of particle size on the in vivo potency of lipid nanoparticle formulations of siRNA. J. Control Release. 2016;235:236–244. doi: 10.1016/j.jconrel.2016.05.059. [DOI] [PubMed] [Google Scholar]
- 26.Khosla S., Westendorf J.J., Oursler M.J. J. Clin. Invest. 2008;118:421–428. doi: 10.1172/JCI33612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mora-Raimundo P., Lozano D., Manzano M., Vallet-Regí M. Nanoparticles to knockdown osteoporosis-related gene and promote osteogenic marker expression for osteoporosis treatment. ACS Nano. 2019;13:5451–5464. doi: 10.1021/acsnano.9b00241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Prockop D.J. Adult stem cells gradually come of age. Nat. Biotechnol. 2002;20:791–792. doi: 10.1038/nbt0802-791. [DOI] [PubMed] [Google Scholar]
- 29.Dezawa M., Ishikawa H., Itokazu Y., Yoshihara T., Hoshino M., Takeda S.I., Ide C., Nabeshima Y.I. Developmental biology: bone marrow stromal cells generate muscle cells and repair muscle degeneration. Science. 2005;309:314–317. doi: 10.1126/science.1110364. [DOI] [PubMed] [Google Scholar]
- 30.Slater B.J., Kwan M.D., Gupta D.M., Panetta N.J., Longaker M.T. Mesenchymal cells for skeletal tissue engineering. Expert Opin. Biol. Ther. 2008;8:885–893. doi: 10.1517/14712598.8.7.885. [DOI] [PubMed] [Google Scholar]
- 31.Seong J.M., Kim B.C., Park J.H., Kwon I.K., Mantalaris A., Hwang Y.S. Stem cells in bone tissue engineering. Biomed. Mater. 2010;5:062001. doi: 10.1088/1748-6041/5/6/062001. [DOI] [PubMed] [Google Scholar]
- 32.Gao J., Dennis J.E., Muzic R.F., Lundberg M., Caplan A.I. The dynamic in vivo distribution of bone marrow-derived mesenchymal stem cells after infusion. Cells Tissues Organs. 2001;169:12–20. doi: 10.1159/000047856. [DOI] [PubMed] [Google Scholar]
- 33.Meyerrose T.E., De Ugarte D.A., Hofling A.A., Herrbrich P.E., Cordonnier T.D., Shultz L.D., Eagon J.C., Wirthlin L., Sands M.S., Hedrick M.A., Nolta J.A. In vivo distribution of human adipose-derived mesenchymal stem cells in novel xenotransplantation models. Stem Cells. 2007;25:220–227. doi: 10.1634/stemcells.2006-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Regard J.B., Malhotra D., Gvozdenovic-Jeremic J., Josey M., Chen M., Weinstein L.S., Lu J., Shore E.M., Kaplan F.S., Yang Y. Activation of hedgehog signaling by loss of GNAS causes heterotopic ossification. Nat. Med. 2013;18:25–38. doi: 10.1016/j.devcel.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinstein L.S., Shenker A., Friedman E., Spiegel A.M., Gejman P.V., Merino M.J. Activating mutations of the stimulatory g protein in the mcCune–albright syndrome. N. Engl. J. Med. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 36.Lietman S.A., Ding C., Cooke D.W., Levine M.A. Reduction in Gsalpha induces osteogenic differentiation in human mesenchymal stem cells. Clin. Orthop. Relat. Res. 2005;434:231–238. doi: 10.1097/01.blo.0000153279.90512.38. [DOI] [PubMed] [Google Scholar]
- 37.Liang C., Guo B., Wu H., Shao N., Li D., Liu J., Dang L., Wang C., Li H., Li S., et al. Aptamer-functionalized lipid nanoparticles targeting osteoblasts as a novel RNA interference-based bone anabolic strategy. Nat. Med. 2015;21:288–294. doi: 10.1038/nm.3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chonn A., Cullis P.R., Devine D.V., Chonn A. The role of surface charge in the activation of the classical and alternative pathways of complement by liposomes. J. Immunol. 1991;146:4234–4241. [PubMed] [Google Scholar]
- 39.Wei X., Shao B., He Z., Ye T., Luo M., Sang Y., Liang X., Wang W., Luo S., Yang S., et al. Cationic nanocarriers induce cell necrosis through impairment of Na+/K+-ATPase and cause subsequent inflammatory response. Cell Res. 2015;25:237–253. doi: 10.1038/cr.2015.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coelho T., Adams D., Silva A., Lozeron P., Hawkins P.N., Mant T., Perez J., Chiesa J., Warrington S., Tranter E., et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N. Engl. J. Med. 2013;369:819–829. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 41.Fitzgerald K., Frank-Kamenetsky M., Shulga-Morskaya S., Liebow A., Bettencourt B.R., Sutherland J.E., Hutabarat R.M., Clausen V.A., Karsten V., Cehelsky J., et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60–68. doi: 10.1016/S0140-6736(13)61914-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jayaraman M., Ansell S.M., Mui B.L., Tam Y.K., Chen J., Du X., Butler D., Eltepu L., Matsuda S., Narayanannair J.K., et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. - Int. Ed. 2012;51:8529–8533. doi: 10.1002/anie.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mui B.L., Tam Y.K., Jayaraman M., Ansell S.M., Du X., Tam Y.Y.C., Lin P.J.C., Chen S., Narayanannair J.K., Rajeev K.G., et al. Influence of polyethylene glycol lipid desorption rates on pharmacokinetics and pharmacodynamics of siRNA lipid nanoparticles. Mol. Ther. - Nucleic Acids. 2013;2:e139. doi: 10.1038/mtna.2013.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Genovese P., Schiroli G., Escobar G., Di Tomaso T., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C., et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Basha G., Ordobadi M., Scott W.R., Cottle A., Liu Y., Wang H., Cullis P.R. Lipid nanoparticle delivery of siRNA to osteocytes leads to effective silencing of SOST and inhibition of sclerostin in vivo. Mol. Ther. - Nucleic Acids. 2016;5:e363. doi: 10.1038/mtna.2016.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen S., Tam Y.Y.C., Lin P.J.C., Leung A.K.K., Tam Y.K., Cullis P.R. Development of lipid nanoparticle formulations of siRNA for hepatocyte gene silencing following subcutaneous administration. J. Control Release. 2014;196:106–112. doi: 10.1016/j.jconrel.2014.09.025. [DOI] [PubMed] [Google Scholar]
- 47.Hindson C.M., Chevillet J.R., Briggs H.A., Gallichotte E.N., Ruf I.K., Hindson B.J., Vessella R.L., Tewari M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods. 2013;10:1003–1005. doi: 10.1038/nmeth.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rettig G.R., Behlke M.A. Progress toward in vivo use of siRNAs-II. Mol. Ther. 2012;20:483–512. doi: 10.1038/mt.2011.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wright D.E., Cheshier S.H., Wagers A.J., Randall T.D., Christensen J.L., Weissman I.L. Cyclophosphamide/granulocyte colony-stimulating factor causes selective mobilization of bone marrow hematopoietic stem cells into the blood after M phase of the cell cycle. Blood. 2001;97:2278–2285. doi: 10.1182/blood.v97.8.2278. [DOI] [PubMed] [Google Scholar]
- 50.Houlihan D.D., Mabuchi Y., Morikawa S., Niibe K., Araki D., Suzuki S., Okano H., Matsuzaki Y. Isolation of mouse mesenchymal stem cells on the basis of expression of Sca-1 and PDGFR-α. Nat. Protoc. 2012;7:2103–2111. doi: 10.1038/nprot.2012.125. [DOI] [PubMed] [Google Scholar]
- 51.Hamilton T.G., Klinghoffer R.A., Corrin P.D., Soriano P. Evolutionary divergence of platelet-derived growth factor Alpha receptor signaling mechanisms. Mol. Cell. Biol. 2003;23:4013–4025. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Varghese F., Bukhari A.B., Malhotra R., De A. IHC profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One. 2014;9:e96801. doi: 10.1371/journal.pone.0096801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.