

Abstract
Atherosclerotic cardiovascular disease (ASCVD) is epidemic throughout the world and is etiologic for such acute cardiovascular events as myocardial infarction, ischemic stroke, unstable angina, and death. ASCVD also impacts risk for dementia, chronic kidney disease peripheral arterial disease and mobility, impaired sexual response, and a host of other visceral impairments that adversely impact the quality and rate of progression of aging. The relationship between low-density lipoprotein cholesterol (LDL-C) and risk for ASCVD is one of the most highly established and investigated issues in the entirety of modern medicine. Elevated LDL-C is a necessary condition for atherogenesis induction. Basic scientific investigation, prospective longitudinal cohorts, and randomized clinical trials have all validated this association. Yet despite the enormous number of clinical trials which support the need for reducing the burden of atherogenic lipoprotein in blood, the percentage of high and very high-risk patients who achieve risk stratified LDL-C target reductions is low and has remained low for the last thirty years. Atherosclerosis is a preventable disease. As clinicians, the time has come for us to take primordial and primary prevention more serously. Despite a plethora of therapeutic approaches, the large majority of patients at risk for ASCVD are poorly or inadequately treated, leaving them vulnerable to disease progression, acute cardiovascular events, and poor aging due to loss of function in multiple visceral organs. Herein we discuss the need to greatly intensify efforts to reduce risk, decrease disease burden, and provide more comprehensive and earlier risk assessment to optimally prevent ASCVD and its complications. Evidence is presented to support that treatment should aim for far lower goals in cholesterol management, should take into account many more factors than commonly employed today and should begin significantly earlier in life.
Keywords: Atherosclerosis, Cholesterol, Coronary artery disease, Dementia, Lipoproteins, Myocardial infarction, Prevention, Stroke
Abbreviations: ASCVD, Atherosclerotic cardiovascular disease; LDL-C, low-density lipoprotein cholesterol; MI, myocardial infarction; CAD, coronary artery disease; HMG CoA, 3-hydroxymethyl-3-methylglutaryl coenzyme A; RCT, Randomized controlled trial; CHD, Coronary Heart Disease; HDL-C, High-density lipoprotein cholesterol; NMR, Nuclear Magnetic Resonance; PCSK9, Proprotein convertase subtilisin:kexin type 9; apoB, apolipoprotein B; VLDL, Very low-density lipoprotein; IDL, Intermediate-density lipoprotein; LLT, Lipid-lowering therapy; NFT, Neurofibrillary tangle; MCI, Mild cognitive impairment; PAV, Percent Atheroma Volume; FCT, Fibrous Cap Thickness; LCBI, Lipid core burden index; CAC, Coronary artery calcium; CCTA, Coronary computed tomographic angiography; PAD, Peripheral Arterial Disease; NPV, Negative predictive value; FH, Familial hypercholesterolemia
Graphical abstract
1. Introduction
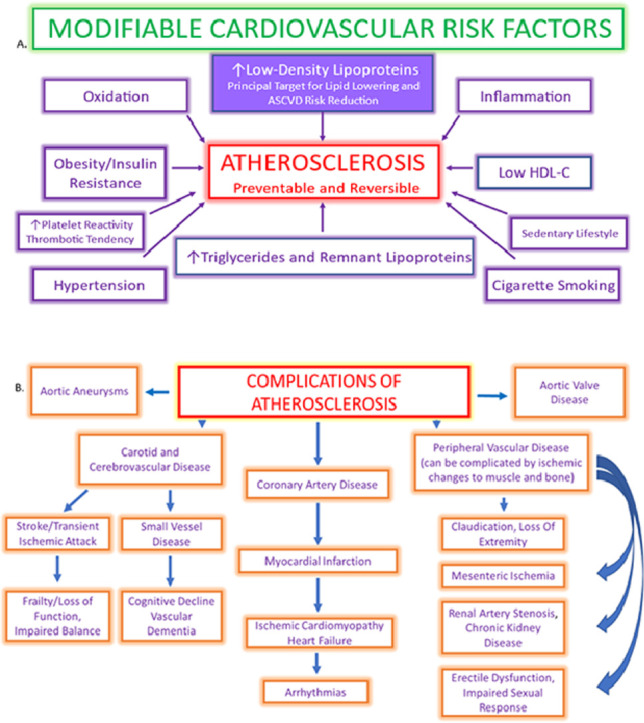
Atherosclerosis is the leading cause of disease, disability, and death in the United States and globally [1,2]. Current medical practice has made progress, but agonizingly slowly considering the millions of people still adversely afflicted by atherosclerotic complications despite use of current treatments. This review examines how new approaches can significantly reduce the human cost of atherosclerosis. In light of the continued high rate of atherosclerotic disease, what seems needed is what Martin Luther King, Jr. called “the fierce urgency of now” [3]. An entire paradigm shift is required such that preventive efforts are embraced much earlier in life, as discussed later in the paper. We propose that preventing and controlling atherosclerosis, the greatest killer of both men and women, be the top priority of medical care in the United States.
While there has been a significant reduction in heart attack and stroke [4,5], large numbers of Americans still sustain myocardial and cerebral infarctions and other complications of atherosclerotic cardiovascular disease (ASCVD) [6,7] Despite the wealth of evidence and the availability of effective preventive interventions, declines in ASCVD hit a nadir, and in fact, cardiovascular mortality has been on the rise over the last decade in both men and women in the US [8]. and throughout the world [2] Even though modern technology has helped more victims of acute cardiovascular events survive, significant numbers of patients who survive due to stents and other interventions in the immediate acute phase nevertheless often experience long-term disability, reinfarction, and death secondary to inadequate treatment [9,10].
Atherosclerosis causes or contributes to many other diseases besides coronary artery disease. Success cannot be claimed until they are equally addressed and reduced.
Current practices are certainly not eliminating atherosclerotic disease. Atherosclerotic disease is preventable since its drivers of risk are largely modifiable (e.g., hyperlipidemia, hypertension, diabetes, cigarette smoking, sedentary lifestyle, obesity). A more intensive, more precise approach applied earlier than is current practice is delineated in this paper, which will also explain why doing so has a higher likelihood of significantly reducing the total burden of atherosclerotic disease. Delay and inadequate care leave patients at heightened risk for ASCVD-related events and complications and all of the many other manifestations of atherosclerosis. Guidelines and risk assessment tools used to prevent events and other complications of atherosclerosis need to have high treatment and prediction success rates. Unfortunately, that has not been the case in many instances.
Multiple studies have shown that the guidelines would not have recommended treatment for at least half of patients who subsequently suffered proven myocardial infarctions, including those with MI's under age 50, those from a high-risk population (India) and those over age 65[[11], [12], [13], [14]]. A better approach is required and justified.
2. Not just coronary artery disease and stroke
Current practice focuses essentially on preventing acute events from coronary artery disease (CAD), but atherosclerosis affects many other arterial beds [15], [16], [17] in ways that develop slowly over many years. Atherosclerosis causes disability and death from its contributions to:
-
•
Disabling consequences of cerebral vascular accidents and cerebral ischemia
-
•
Dementia
-
•
Peripheral arterial disease
-
•
Heart failure
-
•
Renal artery stenosis
-
•
Carotid artery stenosis and embolization
-
•
Kidney failure
-
•
Hypertension
-
•
Aortic disease
-
•
Mesenteric artery disease
-
•
Erectile dysfunction
-
•
Frailty
- •
These can take so long to manifest that they are ignored in randomized controlled trials (RCTs). Assuming that just reducing acute events will also prevent long-term consequences of atherosclerosis is unwarranted by current evidence and has not been adequately studied. These slower to develop manifestations of atherosclerotic disease should also be prioritized and equal efforts should be made to prevent them.
2.1. Low-density lipoprotein cholesterol and ASCVD
The most important atherosclerosis treatment breakthrough occurred in 1987 when the FDA approved lovastatin, the first 3-hydroxymethyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitor [22]. One year later the National Cholesterol Education Program published the first guidelines for clinicians to prevent myocardial infarctions by reducing cholesterol levels [23,24]. While many countries have developed their own national guidelines, most follow a similar premise – intensity of preventive efforts should match the individual's estimated global ASCVD risk [25], [26], [27], [28]. While various national guideline recommendations are largely overlapping, important differences exist. Sources of variation are primarily related to two issues:
-
•
Risk and principal drivers of ASCVD differ in different racial and ethnic groups.
-
•
Different guideline committees around the globe evaluate the same evidence and yet reach very different conclusions as to what the evidence means and what recommendations it supports.
2.2. Limits of guidelines
Cardioprevention guidelines are intended to provide physicians with a single consensus point of view, providing algorithms, calculators and tables based on pooled cohorts for quick reference, and establishing evidence-based standards. However, most guidelines have important limitations, among which are inadequate personalization of care; slow incorporation of new knowledge; and relatively conservative treatment strategies. Moreover, many are lengthy and complex, making them inaccessible to many practitioners, who have multiple guidelines with which they need to be facile [29].
2.3. Slow adoption of optimal medical treatment
Physicians in clinical practice tend to be slow in adopting new approaches and in changing how they care for patients (i.e., “clinical inertia”) [30], [31], [32]. Partly that is due to the time pressures in modern medicine and partly due to the conservative nature of physicians to avoid changes until the evidence demonstrates that a newer strategy is clearly superior. Most cholesterol and prevention guidelines prioritize the evaluation and treatment of hypercholesterolemia. The evidence supporting this approach is incontrovertible, yet the majority of patients with dyslipidemia are under-treated [33,34]. Even among treated high-risk patients, 50% of such individuals discontinue their statin therapy within 6 months and by 5 years only 20% remain adherent to it [35]. There are likely multiple reasons: socioeconomic factors; media attacks on statins [36]; shortened visits in modern medical practice; lack of understanding of what specific drugs do to prevent events and preserve health on the part of patients; and tolerability issues, among others. It might also be that many practicing physicians have yet to recognize that preventing atherosclerosis is the most impactful action they can take. If so, that lack of urgency can be communicated to patients. Whatever the reason, premature discontinuation of lipid-lowering therapy is associated with a rapid rise in risk for ASCVD events [37,38]. Among high-risk patients, statin titration occurs infrequently in patients not meeting their risk-stratified Low-Density Lipoprotein Cholesterol (LDL-C) goals [39]. Despite widespread availability of adjuvant therapies that can dramatically increase LDL-C goal attainment rates, these are vastly and conspicuously underutilized [40].
Acute events occurring despite what appears to be optimal medical treatment are attributed to ‘residual cardiovascular risk’ [41], [42], [43]. The clinical goal in patient management should be to lower the remaining total burden of disease to an extremely low level. Moreover, the concept of residual risk, as it is typically formulated, does not consider the risk of non-acute events. This review will explore how treating earlier in the course of atherosclerosis, treating more intensively and more precisely, and individualizing care can help accomplish that goal.
Interest in the relationship between hypercholesterolemia and atherosclerosis first took root in 1913 when Anichkow first fed rabbits cholesterol and saw atherosclerosis develop in a mammal that never develops it in the wild [44,45]. Scientific advances now provide a more granular and extensive understanding of atherogenesis, though there is still much more to learn. There has been exponential growth in scientific tools and methods that have accelerated our understanding of the complex mechanisms that result in atherosclerosis and its consequences [46], [47], [48], [49], [50], [51], [52], [53], [54], [55], [56], [57]. Atherosclerosis begins as lipid deposition in the intima of arteries, widening the intimal space, then progresses to a plaque, then to an unstable, vulnerable plaque, and then (subsequent to loss of plaque integrity) to thrombosis inside the artery. The earlier that sequence can be halted, the fewer occlusive thrombi there will be, and the fewer complications of atherosclerosis that can result. There is a highly sophisticated toolbox to recognize atherosclerosis early [15], to measure it and its causes precisely, and to use that information coupled with evidence-based preventive interventions (therapeutic lifestyle changes and medications) to arrest its progression. If LDL-C can be kept very low from birth, atherosclerosis will not occur. Where that early prevention is not possible to institute, even larger plaques can potentially be controlled in nearly every case if treated intensively enough – doing so would likely prevent most of the thrombi that lead to acute cardiovascular events or more or larger plaques, as long as treatment is not delayed for too long nor inadequately applied.
2.4. Atherosclerosis represents a clinical paradox: it is potentially the most preventable or treatable chronic disease, yet it remains the greatest cause of disability and death throughout the world. This does not have to be the case
There has been compelling and convincing justification for some time that an approach that includes keeping plasma atherogenic lipoproteins low from early in life will greatly reduce risk for ASCVD. As detailed by Ference et al. “initiating lipid-lowering therapy after a person has already been exposed to a cumulative burden of 6250 mg-years of LDL by age 50 years means that person has very likely already developed a large atherosclerotic plaque burden … lowering LDL after this cumulative exposure to LDL should reduce the risk of cardiovascular events, but this person will remain at relatively high “residual” risk of experiencing an acute cardiovascular event because one of the underlying plaques can still disrupt to cause an acute coronary syndrome … [that] may explain much of the high residual risk of cardiovascular events observed among people enrolled in lipid-lowering randomized trials” [58].
2.5. Normal LDL-C is 20–40 mg/dL
Humans were never meant to harbor the low-density lipoprotein cholesterol (LDL-C) levels that are now commonplace. In one series of 147 full-term neonates, the average LDL-C was 20 ± 10 mg/dL [59] Despite the extraordinary rate of development and need for myelination, even neonates need very little LDL-C [60], [61], [62], [63]. The fact that animals, non-human primates, and humans who maintain low cholesterol levels from early in life have very little atherosclerosis all suggest that a ‘normal’ non-atherogenic LDL-C level is 20–40 mg/dl. That is of course difficult to achieve in a modern society and, as described herein, is not necessary for most people.
Based on the log-linear relationship of LDL-C to the hazard ratio for an acute ASCVD event, the LDL-C level where no excess risk occurs is approximately 38 mg/dL or 1 mmol/L [64] (Fig. 1). This value is consistent with the LDL-C levels observed among hunter-gatherer populations [65,66]. In the Framingham Study, the average LDL-C of a man presenting with an Acute Coronary Syndrome (ACS) is approximately 150 mg/dL [67]. In the Cooper Center Longitudinal Study, even when LDL-C at baseline was < 100 mg/dL, there was a continuous rise in risk for Coronary Heart Disease (CHD) mortality over a mean follow-up time of 26.5 yrs [68,69] Hence, it is crucial that exposure to atherogenic lipoproteins be dramatically reduced early and over the long-term.
Fig. 1.

Log-linear relationship between LDL-C levels and relative risk for CHD. Reproduced with permission from Grundy et al. [64].
3. Despite many recommendations, early treatment has not become common practice
There are practical tools available to recognize atherosclerosis very early, to assess lipids more accurately, and to uncover and treat ancillary risk factors. These concepts and tools allow precision management of atherosclerosis, but they are employed too little and too late in many cases. Current approaches using algorithms and calculators are based on generalized data rather than precisely individualized to each patient [29,[70], [71], [72]].
3.1. The art of medicine
As important and vital as the science is, the practice of medicine is the art of medicine, which means to apply the scientific evidence to each individual tailored to unique needs [72]. Confounding factors are rigorously excluded from RCTs, but humans contain them in multitudes unique to each person [72]. RCTs test hypotheses about specific interventions, but they do not tell physicians how to treat a person [73]. As Virgil Brown noted, “Just as evidence is not the law, evidence is not the art of medicine. Considering evidence provides for inductive reasoning, but this requires deductive considerations to actually apply evidence in the most effective ways” [74].
3.2. The rationale for keeping LDL-C at very low levels
Our understanding of atherosclerosis, its molecular pathways [18,75], genetic influences [76], [77], [78], inflammatory mechanisms [79], [80], [81], [82], interaction of comorbidities [83], and the role of lifestyle [84,85] and the environment [86,87], have expanded rapidly [46,88]. There is now a powerful therapeutic armamentarium to lower cholesterol, stabilize the arterial wall, and prevent the plaques and thrombi that incur considerable acute, progressive and chronic injury [89,90]. Anti-inflammatory treatments have proven beneficial and are being refined, and new medications are likely coming to the clinical setting [91], [92], [93]. Vaccines against targets etiologic for atherosclerosis are being explored [94]. Current medicines are already remarkably effective and it will be ever more likely to completely halt progression of atherosclerosis before harm is done in most cases, and sooner than current common practice patterns accomplish. Even before these new treatments come to fruition, we propose that there are enough tools to nearly eliminate “residual risk.”
3.3. A childhood disease
Atherosclerosis begins in earliest childhood, sometimes even during gestation, presenting as yellow streaks in arterial walls [95], [96], [97], [98]. It is a chronic disease: absent intervention, it slowly progresses throughout life, unevenly, sometimes rapidly [16], but inevitably worsening over time [18,[99], [100], [101], [102], [103], [104]]. It has been shown that the progression can be halted, and even reversed to some degree with depletion of the lipid core, if plaque is not extensively fibrotic or calcified [18,105,106]. Previously believed to just be part of normal aging, atherosclerosis is actually a pediatric disease that progresses into adulthood [107], [108], [109], [110], [111]. Advanced disease recognized in very young people was observed in young men killed in Korea and Vietnam, as well as victims of trauma as young as 20–25 years of age [112]. Those fatty streak lesions in early childhood are usually the sites of more advanced lesions later in life [113]. Mothers who are not overweight, diabetic, hypertensive or hypercholesterolemic in pregnancy and have otherwise healthy lifestyles are less likely to have children with hyperlipidemia [82]. Childhood risk factors have been shown to predict future clinical atherosclerotic disease by midlife [114]. Atherosclerosis would likely be far less common if from birth everyone maintained a healthy weight and diet and avoided toxic habits and environmental exposures. Unfortunately, such goals have proven very difficult to achieve in modern society and the success rates of lifestyle improvement are low at any age [85,115,116].
3.4. Atherosclerosis is not inevitable
Mammals, primates, those living indigenous lives away from ‘modern civilization’ , and those with mutations that cause extremely low LDL-C from birth [117] develop little or no significant atherosclerosis [118]. The fact that animals, non-human primates, and humans who maintain low cholesterol levels from early in life have very little atherosclerosis all support the conclusion that a ‘normal’ non-atherogenic LDL-C level is below 38 mg/dl, as noted previously. Other than those with genetically low LDL, what those with little or no atherosclerosis have in common from birth are: [1] low intake of saturated fats, salt, and sugars and other refined carbohydrates, [2] primarily plant-based diets, [3] absence of harmful substance abuse and less polluted environments, and [4] physically active, non-sedentary lives. The Tsimane tribe of Bolivia, for example, live unexposed to ‘developed’ life and are essentially free of atherosclerotic disease [119]. The mean LDL-C and HDL-C in the Tsimane people are at 90 mg/dL and 39.5 mg/dL, respectively [120]. People with the least risk factors fare much better [121], but unfortunately the vast majority have one or many major risk factors. In 2010, 47% of Americans had one or more of uncontrolled high blood pressure, uncontrolled high levels of LDL-C, or were current smokers [122,123]. When you add in those with other risks, as noted in Table 3, the percentage would be much higher (Table 4).
Table 3.
Factors to consider in assessing risk of atherosclerosis and individualizing treatment.
| Factor | Details of increased risk |
|---|---|
| Presence of plaque in any vascular bed | Either non-calcified as seen on ultrasound or other modalities and/or calcified plaque seen in aorta, peripheral arteries or by CAC. Plaque is a sign of advanced atherosclerosis. Calcified plaque is an even later finding. Thickening Intimal-media is also of concern and is the earliest sign. |
| Insulin resistance, diabetes, metabolic syndrome* | Adiposopathy [261), insulin resistance, prediabetes, diabetes, metabolic syndrome [253], excess visceral fat. Hemoglobin A1C predicts subclinical atherosclerosis [254]. |
| Hypertension* | Essential, secondary, or primary aldosteronism [154] Blood pressure is safest at or below 120/70 mm Hg at any age [255], [256], [257], [258] |
| Elevated Lipoprotein (a) [259], [260], [261], [262], [263], [264]* | Levels > 75 Nmol/L [265], [266], [267], [268], [269] Elevated levels are likely as common as 20% of the population. A major contributor to ASCVD and calcific aortic stenosis. Also increases risk of stroke in children [263,270]. Measurement in nmol/L is preferable to mg/dl. |
| Familial Hypercholesterolemia [271]* | Heterozygous FH is the most common monogenic condition, affecting between 1 in 200–300 Americans, and as frequent as 1 in 24 of those with ASCVD [272]. |
| Elevated hsCRP [273] or GlycA [274], [275], [276]* | Indicators of active inflammation. However, hsCRP can be lower at times despite even advanced atherosclerosis with plaque (probably due to temporary inactivity of inflammation). A normal hsCRP does not negate the risks of other important factors, as inflammation activity can wax and wane. High Sensitivity CRP > 1.0 mg/dl denotes even higher risk. |
| LDL-C, non-HDL-C, ApoB* |
LDL-C of 20–40 mg/dl seems to be the healthy level for humans from birth on, but impractical to achieve in developed societies. Apolipoproteins are the primary cause of atherosclerosis [277]. ApoB and non-HDL-C can help refine atherogenic particle levels. |
| Triglycerides* | Shaik and Rosenson [278], [279], [280], [281] Risk begins to increase above 100 mg/dl [282] Post-prandial surge important as well. |
| Remnant Cholesterol | Remnant cholesterol (approximation) = Total cholesterol – HDL-C – LDL-C. Levels above 10 mg/dl indicate risk [283]. Apolipoprotein B levels and non-HDL Cholesterol also include atherogenic remnants and are likely more predictive than LDL-C alone. |
| Age⁎⁎ | Risk increases with age (age is the most determinative factor in risk calculators). Even over age 75 years, treatment is effective, safe and appropriate [284], [285], [286], [287], [288], [289] |
| Family history* | Early ASCVD, diabetes , hypertension [290] all increase risk for descendants [291], [292], [293], [294], [295], [296] |
| Obesity, visceral fat | Major cause of metabolic syndrome and atherosclerosis, even when at first ‘metabolically healthy’ [252,[297], [298], [299], [300], [301]]. |
| Chronic kidney disease* | CKD and atherosclerosis each increases risk and pathology of the other [302], [303], [304], [305] |
| Non-alcoholic fatty liver disease | Closely related to atherosclerosis and contributory to it [169]. |
| Other co-morbidities | Hypo- or hyperthyroidism [306], gout [307], sleep apnea [308], gut microbiome (theoretical) [309], Cushing's syndrome [310], many others. The microbiome is not currently actionable. |
| Some medications | Some increase LDL (Corticosteroids [311], Androgenic steroids [312], Progestogens, Thiazide diuretics, Beta-blockers, Retinoic acid derivatives, Oral estrogens [313]). Chronic corticosteroids increase risks even at low doses. Many others [314]. |
| Substance use* | Tobacco [315,316], marijuana [317], [318], [319], alcohol [320], cocaine [321] |
| Autoimmune disease* | Rheumatoid arthritis [322,323], Systemic Lupus Erythematosus [324], psoriasis and psoriatic arthritis [325,326], ankylosing spondylitis [327], scleroderma [328], inflammatory bowel disease [329], probably others |
| immunological disease and inflammation elsewhere in the body [82,159]; clonal hematopoiesis of indeterminate potential (CHIP) [160], [161], [162], [163]; neutrophil extracellular traps [164], [165], [166], [167] | These are not yet readily actionable and further research is needed. |
| Genetic factors and social determinants of health | Knowledge and applicability are developing rapidly, already useful for FH and some other genetic variants [330], [331], [332], [333], [334] |
| Race/ethnicity (All people are complex genetic mixtures, but some genetic factors are alerted by ethnicity in some cases. While of course not universal or definitive, Race/ethnicity can signal risk requiring deeper evaluation)* | South Asian (much higher risk of atherosclerosis, high Lipoprotein (a) and diabetes and at early ages) [280,[335], [336], [337], [338], [339], [340], [341]], African American (hypertension, renal disease); Non-white Hispanic (diabetes, obesity, CAD); many others. Some of Asian heritage have lower risk. Ethnicity affects incidence of biomarkers [342] |
| Other lab parameters | Elevated Microalbumin/creatine ratio [343,344], even in children [104]. High uric acid [345,346], low vitamin D [347], periodontitis [383], elevated ceramides [348] and others [384] are associated with increased risk of atherosclerosis, though causation remains to be fully determined and thus treatment not yet justified just to reduce risk of atherosclerosis. Their presence implies increased risk even if causation not proven. Thus, a high uric acid would raise concern but lowering uric acid just for that reason is not indicated. |
| Testosterone deficiency and treatment | Reasonable evidence that hypogonadism increases risk of atherosclerosis, less certain if treatment affects risk. Excess testosterone treatment probably increases risk. Must use replacement therapy carefully [349]. |
| Female reproductive* | Premature menopause, high cholesterol in pregnancy (cholesterol usually increases in pregnancy and in menopause), preeclampsia, eclampsia, gestational diabetes mellitus and polycystic ovary syndrome all increase risk [350], [351], [352] |
| Social factors* | Socioeconomic status [353]; discrimination [238,354] and financial barriers to access to healthcare [355]. Culture, beliefs, life views, etc. that affect use of medical science. Lack of belief in science. Desire for ‘natural’ approach. Poor compliance and long-term adherence [356,357]. Unjustified fear of LLT medications [36] |
| Mental health [358] | Depression (associated and possibly causal [359]), stress [360], anxiety or anger syndromes [361]. Some anti-psychotic medications increase atherosclerotic risk [314]. |
| Lifestyle [362]* | Atherogenic diet (highly processed food, high salt and simple carbohydrates, poorly balanced nutrition) [363], [364], [365], [366]; saturated fat [367], [368], [369], [370]; trimethylamine-N-oxide (TMAO) [371,372]. Inadequate aerobic and resistance exercise [373,374]. Excess sedentary time independent of exercise [375], [376], [377]. Mediterranean diet [378], vegan diet [379,380], DASH Diet [102] are proven much healthier and less atherogenic. Overweight, obesity and (most important) excess visceral fat. |
| Environment | Air pollution [86,381] contributes to atherosclerosis risk, as do excess noise [381], [382], [383], [384] and chemical pollution [87,168] |
| Acute respiratory syndrome-coronavirus-2 (SARS-CoV-2) | This virus causes significant endothelial changes in many arteries. This causes immediate cardiac pathology in some patients, even in mild cases. Whether it will have long-term consequences remains to be seen, but all COVID patients should be carefully monitored for the development of cardiac problems over time and for accelerated atherosclerosis. |
(Some of these, designated with *, are partly or wholly addressed in the 2019 AHA guidelines, as contributors or risk enhancers, but often partially or at higher thresholds than recommended in this paper. Age (⁎⁎) is a major determinant of the AHA risk calculator).
Table 4.
Factors not included in risk calculators.
| Patient A | Patient B | |
|---|---|---|
| Required by calculator | ||
| Age | 40 | 40 |
| Gender | Male | Male |
| Race | Non-African American | Non-African American |
| Smoker | No | No |
| Treated for diabetes | No | No |
| Treated for Hypertension | No | No |
| Total Cholesterol (mg/dl) | 175 | 220 |
| HDL cholesterol (mg/dl) | 55 | 55 |
| Systolic blood pressure (mmHg) | 110 | 110 |
| Diastolic blood pressure (mmHg) | 70 | 70 |
| Not included in calculator | Patient A | Patient B |
| Fasting Triglycerides (mg/dl) | 80 | 210 |
| LDL-C (mg/dl) | 75 | 150 |
| LDL-P (Nmol/L) | 800 | 2300 |
| Strong family history of ASCVD | No | Yes |
| Hemoglobin A1C (%) | 5.4 | 6.3 |
| Waist circumference (inches) | 35 | 42 |
| Lipoprotein (a) (mg/dl) | 70 | 250 |
| AHA/ACC Risk Score by calculator | ||
| (% ten-year risk) | 0.6% | 0.9% |
Consider two hypothetical male non-African-American patients, each 40 years old and their AHA Risk Calculator scores as follows (Patient B has a common profile) (Table 4). The latest guidelines recommend taking ancillary factors into account, as for Patient B, but with a risk score below 1%, most calculator users would be unlikely to recommend treatment for Patient B, yet he appears at very high risk of an acute event in the relatively near future, as well as slow-developing manifestations of atherosclerosis. Changing only the age for Patient B to 60 years in the AHA Risk Calculator means a Risk Score of only 6.8%, still below the treatment threshold of 7.5%.
Every decade of delay in treatment could mean risk of an acute event, damage to other organs, poor aging and ever greater physiological resistance to treatment once finally begun.
Treating Patient B at age 40 or younger would likely prevent premature morbidity and mortality and would be significantly easier, safer and more effective.
Interventions to improve lifestyle in Americans usually have low success rates or are often inadequate to fully control risk even with excellent adherence. Medications are important adjuncts and can significantly attenuate the impact of poor lifestyle, environmental pollution and genetics if begun early and intensively. Even under conditions of adverse genetics, toxic environmental exposures, and comorbidities, most atherosclerosis can be slowed significantly or completely halted if also treated early and intensively.
3.5. Cells do not need LDL-C
Cholesterol is essential for modulating cell membrane fluidity, cell transporters, and intracellular signaling systems, and is a precursor to myelin, bile salts, Vitamin D, steroid hormones (corticosteroids, sex hormones, mineralocorticoids), and establishes impermeability of the skin. All somatic cells, including astrocytes and oligodendrocytes in the brain, make cholesterol through the same pathway that the liver utilizes, and can obtain some from High-Density Lipoprotein (HDL) [57,124,125]. Even when LDL-C is extremely low, there is no impairment of cellular cholesterol production and utilization within the brain because the brain produces its own pool of cholesterol [126], as do all cells in the body. No tissues depend on cholesterol transfer from LDL-C (the ovaries, testes, and adrenals produce cholesterol de novo or import it via SR-B1 receptors from HDL particles). Currently, common practice considers an LDL-C of 100 mg/dl as acceptable, but atherosclerosis exists even below an LDL-C of 55 mg/dl and even lower [127].
3.6. The primary role of lipoproteins is excretion of excess cholesterol
While apolipoproteins play the dual role of distributing triglyceride and cholesterol to systemic tissues, their primary role is to facilitate excretion of cholesterol from the bloodstream and the body [128]. Atherosclerosis occurs when those mechanisms are inadequate and lead to excess circulating cholesterol that is deposited in the intimal space of medium to large arteries by transcytosis of LDL particles and atherogenic apo B remnants [18,129]. Atherogenic apolipoprotein B (ApoB) lipoproteins include LDL, Very Low-Density Lipoprotein (VLDL), and Intermediate-Density Lipoprotein (IDL). These lipoproteins are toxic because they deliver sterols, oxysterols, oxidized phospholipids, and toxic lipids (e.g., oxidized fatty acids) into the arterial vasculature and potentiate inflammation, a primary driving force of atherogenesis [130].
Thus the initiating event for atherosclerosis is the deposition of lipids into the intimal space beyond what that space can hold, as noted by Tabas et al. [131], Williams et al. [132] and Boren and Williams [133].
-
•
If there is no such lipid deposition, then there will be little or no inflammation in the endothelium and intimal space. Keeping LDL-C very low will accomplish that.
-
•
If there is no inflammation, then there will be no atherogenesis.
-
•
If no atherosclerosis, there are no complications and much better vigor and longer aging.
As Sniderman et al. said, “After all, if disease in the wall is prevented, there will be no events to predict” [134].
Brown and Goldstein demonstrated in 1974 that there is a receptor feedback-controlled limit to cholesterol deposition of about 25 mg/dL and that the LDL receptor is critical to understanding atherosclerosis [46,75,135]. Any cholesterol in excess of that generates an inflammatory response in the intima and endothelium, mediated by the immune system, in which monocytes migrate into the intimal space and transform into resident macrophages [136]. A cascade of immunological reactions follows thereafter, mediated by interleukins, cytokines, oxygen free radicals, and growth factors produced by T helper cells, mast cells, neutrophils, and platelets [,[137]. This causes or contributes to arterial consequences in the entire body, not just the coronary arteries:
-
•
Brain (dementia, including Alzheimer's disease) [19,[138], [139], [140], [141], [142], [143], [144], [145], [146], [147], [148]].
-
•
Heart (adverse forms of structural remodeling, heart failure, fibrosis, arrhythmias including atrial fibrillation and malignant ventricular arrhythmias).
-
•
Kidney (renal artery stenosis, chronic kidney disease).
-
•
Arteries in the lower extremities (peripheral arterial disease).
-
•
Pudendal artery (erectile dysfunction).
-
•
Mesenteric arteries (mesenteric ischemia).
-
•
Aorta (aneurysms) and aortic valve (calcific aortic stenosis).
The accumulated effect can be any combination of reduced cognition or dementia, weakness and fatigue, dyspnea, frailty, vital organ failure, poor aging and premature mortality. These peripheral but critical manifestations of atherosclerosis are devastating and common, yet difficult to capture in RCT's. As Kuller has noted, “The incubation period for the development of brain pathology, i.e., amyloid plaques and neurofibrillary tangles (NFTs), for example, to cognitive decline, mild cognitive impairment (MCI) and dementia is very long, perhaps as much as 20 years or more. Both the longitudinal studies and especially the clinical trials may not have been followed long enough to see a beneficial effect” [149].
LIMITS OF RCTs
The relative brevity of RCTs is likely one of the reasons that current practice is directed essentially only against limited acute events, mainly in people who already have advanced disease that would manifest within the time frame and the number of subjects. In addition, few primary prevention studies are powered enough to detect effects on mortality, even from infarctions. Preventing heart attacks and strokes seems extremely likely to also reduce mortality over the longer term. As Kostapanos and Elisaf note, “no long-term placebo-controlled primary prevention statin trials are available, nor is there a current ethical basis for designing one” [150]. Statin trials are not powered to detect reductions in mortality but reducing acute events and long-term consequences seems essential to reducing mortality as well.
3.7. Low-enough LDL-C prevents atherosclerosis
If LDL-C in blood is kept very low routinely – under 85 mg/dL for life, or correspondingly low by other measures discussed below, including LDL particle number by nuclear magnetic resonance (NMR) spectroscopy, ApoB or non-HDL-C (total cholesterol-HDL cholesterol), atherosclerosis seems unlikely to occur to any clinically meaningful degree. Hypertension, diabetes, and some inflammatory conditions cause inflammation and damage to the endothelium, but if ApoB containing lipoproteins are kept low relatively early in life, as low as at birth, they will likely have far less pathophysiologic impact. Peter Libby [18] noted that “If the entire population maintained LDL concentrations akin to those of a neonate (or to those of adults of most other animal species), atherosclerosis might well be an orphan disease” [18]. Based on the preponderance of evidence, it seems best to set LDL-C goals below 40 mg/dl (1 mmol/dl), or even lower for even higher risk. This is also consistent with current recommendations from the European Society of Cardiology/European Atherosclerosis Society guidelines for the management of dyslipidemia [151]. However, that would likely be challenging to achieve widely, but as will be shown in this review, in those with no enhancing risk factors, keeping LDL-C below at most 85 mg/dl from birth throughout life would likely delay onset of complications until age 100. This has been derived from experience with those with a heterozygous deficiency in PCSK9. For those with additional risk factors or more advanced atherosclerosis, keeping LDL-C below 38 mg/dl (depending on severity) would also likely maintain good health until very late in life.
3.8. Co-factors are also extremely important
There are many factors that damage the endothelium, contribute to atherosclerosis in other ways and activate the immune system, such as insulin resistance [152,153], hypertension [154], [155], [156], smoking [157,158], immunological disease and inflammation elsewhere in the body [82,159], clonal hematopoiesis of indeterminate potential (CHIP) [160], [161], [162], [163], neutrophil extracellular traps [164,[165], [166], [167]], environmental pollutants [86,87,168], Non-alcoholic Fatty Liver Disease (NAFLD) [169], and many others (Table 3). Good control of those should help preserve healthy arteries. This is a ripe area for research, as it seems likely that controlling atherosclerosis very early and intensively will minimize the impact of these factors. Because such early studies might not be feasible due to needed length and power, extrapolating from known information would strongly suggest that early and intensive treatment would preclude the adverse consequences of many co-factors participating in the progression of atherosclerosis. If LDL-C is kept out of the intimal space, there will be no atherosclerosis to be a cofactor for.
Triglycerides have long been of concern, but there is now growing recognition of their importance in atherogenesis [170], [171], [172], [173]. Higher triglyceride levels displace cholesterol in lipoprotein particles leading to smaller, more atherogenic particles [172], [173], [174], [175], [176]. As noted, the greater the number of ApoB containing lipoprotein particles there are in circulation, the higher the likelihood they will penetrate the intimal space, be oxidized and initiate a chronic maladaptive inflammatory response.
3.9. The lower the better
Many studies have confirmed that the lower the LDL-C, the lower the risk and the fewer complications of atherosclerosis, with no evidence of any clinically significant harm no matter how low the LDL-C level [177,178]. Logarithmic scales including many historical trials of lipid-lowering show a direct relationship of disease level with lower LDL-C level achieved [179], [180], [181], [182]. The Cholesterol Treatment Trialists [177], the Justification for the Use of Statin in Prevention: An Intervention Trial Evaluating Rosuvastatin (JUPITER) study [183,184], and the Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk (FOURIER) [185] and ODYSSEY Outcomes: Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab [186] demonstrated that even for patients with LDL-C below 70–100 mg/dl, further reduction in LDL-C improved outcomes in several clinical scenarios. It seems logical that the same would be even more effective before advanced atherosclerosis has developed. These trials showed that for every 1 mmol/L (38.67 mg/dl) reduction in LDL-C, the rate of adverse events is reduced by about 22% [187].
FOURIER and ODYSSEY made clear that no matter how low LDL-C, even below 20 mg/dl, there was no greater incidence of adverse events than from placebo. As cognitive effects of very low LDL-C have been of theoretical concern, the Evaluating PCSK9 Binding antiBody Influence oN coGnitive HeAlth in High cardiovascUlar Risk Subjects (EBBINGHAUS) Trial examined neurocognitive function using the Cambridge Neuropsychological Test Automated Battery (CANTAB) battery of tests and found no difference between baseline and end of study. Even when ultra-low LDL-C was achieved (< 10 mg/dL), no between-group differences could be discerned. In a subsequent analysis of questionnaires filled out by Ebbinghaus participants, the study found no evidence of neurocognitive harm between the start and end of the study [188].
3.10. Mendelian randomization also very convincingly shows that the lower the LDL-C, the less atherosclerosis and the fewer the resulting ASCVD-related events [189]
Some recommend that percent reduction in LDL-C is most important, particularly at low levels of LDL-C [190,191]. Other studies, such as FOURIER and the Mendelian randomization studies previously referenced, show that the level of LDL-C is highly determinative, not just the percentage reduction. That is consistent with our understanding of the pathophysiology of atherosclerosis, in which the fewer LDL particles that enter the intimal space, the less inflammation and its consequences that will occur and the lower the loading with cholesterol that macrophages will undergo. Were it true that the degree of reduction is all that matters, then it would imply that a patient with Familial Hypercholesterolemia [192] with an LDL-C of 500 mg/dl would no longer be at risk if treatment reduced the LDL-C fifty-percent to 250 mg/dL. Those patients would certainly still be at greater risk than if their LDL could be reduced to very low levels with treatment and, if needed, LDL apheresis [193].
3.11. LDL-C is a vascular toxin
Fortunately, mainstream lipid-lowering therapies (LLT) are remarkably safe [126,194]. The low incidence of side effects is dwarfed by the events protected and the lives saved, for a very positive benefit/risk ratio [195]. Consistent with this, in the world of cardiovascular disease prevention, “it is vital that we rid the system of its most potent toxin: LDL-C, a metabolite responsible for the death and disability of more people than any other known product of human physiology” [31].
Is it reasonable to view LDL-C as a vascular toxin? Yes. LDL particles represent the end-product of lipoprotein metabolism. LDL particles have two routes for removal: (1) clearance by hepatic LDL receptors, or (2) uptake into the intimal space and scavenged by macrophages [196]. LDL particles induce endothelial dysfunction, and promote the development of a pro-oxidative, pro-inflammatory, prothrombotic phenotype along the arterial wall. Mendelian randomization studies are quite consistent when it comes to LDL: the higher the serum level, irrespective of genetic polymorphism, the higher the risk for ASCVD [197]. The opposite of this is also true: the lower the level of LDL-C, the lower the risk. This is consistent with the principles of toxicology.
3.12. Treating early is far more effective than starting treatment after disease develops
No reasonable clinician would wait for kidney damage or a cerebrovascular event before treating hypertension, delay managing hyperglycemia until kidney failure or retinal hemorrhage, hold off on an antibiotic for pneumonia or cellulitis or let joints deteriorate before treating rheumatoid arthritis. In contrast, addressing hypercholesterolemia is frequently delayed until after a cardiovascular event occurs.
Brown and Goldstein noted that the discovery of proprotein convertase subtilisin: kexin type 9 (PCSK9) made clear a vital lesson that many leading lipidologists had been proposing for a long time: the earlier LDL-C is kept very low, the lower the burden of atherosclerosis that will develop. They compared the reduction in risk in those born with PCSK9 deficiency to those who were treated in later life with a statin in a five-year trial [117]. They calculated that in both cases LDL-C was 40 mg/dl lower than if either no treatment or no PCSK9 mutation. Table 1 shows a comparison based on their paper [198].
Table 1.
Effect of low lifelong LDL-C (from birth due to genetic causes) versus from five years of treatment.
| LDL level reduction | By | Duration of reduction | Reduction in events |
|---|---|---|---|
| 40 mg/dl reduction | Statin treatment in a trial | 5 years | 23% |
| 40 mg/dl reduction | Loss of function mutation in PCSK9 | From birth | 88% |
They concluded: “Early intervention may well put an end to the epidemic of coronary heart disease that ravaged Western populations in the 20th century.”
As early as 1996 they had said, “If we wait for susceptible individuals to develop symptoms before deciding to treat, the earliest symptom is often sudden death” [199]. Others in the forefront of atherosclerosis research and conceptualization have said the same [113,134].
3.13. The pathogenesis of atherosclerosis explains why early and lower are better
This corresponds mechanistically to the rapidly developing understanding of the pathogenesis of atherosclerosis. It is an inflammatory disease. The more it progresses, the more intense the immunological reaction becomes, which sensitizes circulating immune cells and causes significant changes in the endothelium. As an atheroma develops, it is likely that the process will eventually become at least somewhat independent of additional LDL-C infiltration of the intima, resulting in resistance to treatment. If initially the immunological atherosclerotic process can be prevented or halted by keeping the influx of LDL-C very low, then it seems unlikely that a self-propagating immunologic reaction would become irreversible. Inadequately controlled inflammation is posited as part of the reason for residual risk, and it certainly plays a defining role in advanced atherosclerosis. However, if atherosclerosis is halted very early on, it is unlikely that inflammation would continue and therefore would not frustrate efforts to prevent atherosclerotic complications. The evidence of the protective effect of maintaining very low LDL-C from very early on seems to support this concept. In the unfortunately much more common case, where opportunity for early life intervention does not exist, then counteracting inflammation and dramatically lowering the circulation of ApoB lipoproteins should stop the process and even allow regression, which has been shown where there is adequate intensity of treatment [132,[200], [201], [202], [203], [204]].
Many things, such as systemic or localized inflammation (autoimmune diseases, periodontal disease, others), hypertension, diabetes and more, can damage the endothelium in ways that might induce an immune reaction [205,206]. Managing the immune component of atherosclerosis is vital, though currently specific tools to do so are limited [207]. In any case, if LDL-C is kept very low very early on, that would mute the effect of inflammation on the arterial lining because there would not be the cholesterol to initiate the immune response and induce foam cell formation and an atheroma to develop and then progress to plaques and formation of overlying thrombi. When Reverse Cholesterol Transport (RCT) is intact, as in most people, then foam cells and atheromas already in place should be reduced and stabilized by achieving very low cholesterol levels.
3.14. The trajectory of disease can be altered if treated early
Studies show that the trajectory of developing atherosclerotic plaque to acute events can be altered (Fig. 2). The lower and the earlier LDL-C is reduced, the larger the rightward shift along the clinical event horizon, and the more delayed the onset of clinically apparent disease will be [208]. Horton et al. calculated that the degree of atherosclerosis progression relates to level of apo B lipoproteins times the duration of the exposure of the intima to LDL-C. In their seminal paper on PCSK9 they noted that a useful measure would be the total accumulation of LDL-C over time (g/dl LDL-C x years of exposure) [209]. They drew Kaplan-Meier curves estimating that for each level of LDL-C over time there was a predictable age range at which the curve would cross the threshold where acute coronary events became more likely. The threshold for homozygous FH was under age 15 years (some need coronary bypass graft surgery by age 6); early thirties for heterozygous FH; mid-sixties for average Americans (calculated from National Health and Nutrition Education Survey III); above ninety years for PCSK9 heterozygous for loss of function. Thresholds were judged fifteen years lower for males and those with cofactors like hypertension, diabetes, and smoking and ten years higher for females with no cofactors. Early treatment extends the time before events become more likely. Subsequent studies strongly substantiated this finding [58,[210], [211], [212]].
Fig. 2.

Examples of area under the LDL-C versus age curves.
Each color represents a different patient population plotting cumulative low-density lipoprotein cholesterol (LDL-C) years versus age and the average onset of atherosclerotic cardiovascular disease (ASCVD)(black dashed horizontal line). Individuals with genetically determined severe hypercholesterolemia from birth (e.g., familial hypercholesterolemia [FH]) have the largest area under the curve at any given age (red dashed vertical line) and steepest slope of LDL-C versus age. Thus, they experience the earliest onset of ASCVD. Individuals with moderate hypercholesterolemia starting in the teenage years secondary to genetics and/or suboptimal lifestyle habits are at risk for relatively early ASCVD due to a lengthy cumulative exposure to LDL-C. Those with modest hypercholesterolemia from adulthood, often due to suboptimal lifestyle habits, generally develop ASCVD later than the other 2 groups. Individuals genetically endowed with low LDL-C from birth have a markedly reduced risk of developing ASCVD. (Figure and legend reproduced from Shapiro and Bhatt with permission [208]).
Steinberg and Grundy concluded that current guidelines are too conservative. The evidence for the value of early intervention is so strong, even in the thirty-year age group, they said, that it misses the opportunity to reduce the toll of CHD. They identified the core principles of LDL-C reduction as “the lower the better,” and “the earlier the better” [213]. They also argued that an RCT would be prohibitively prolonged and would require unaffordable numbers of subjects [213]. In addition, they noted that, because the evidence is so compelling, an RCT is unnecessary, just as it is obvious smoking is harmful even though that has never been ‘proven’ by an RCT (nor have parachutes been shown by an RCT to be beneficial after jumping out of an airplane [214]). Ference and colleagues state, “such a trial may not be logistically feasible because it would take several decades to complete and because adherence to the allocated treatment over such a prolonged follow-up period would be difficult to maintain. As a result, such a trial is unlikely ever to be conducted” [58].
Log linear plots of results of lipid-lowering trials all show that the lower the LDL-C, the lower the risk down to the lowest LDL-C levels [64],]. In the analysis by Beokholdt et al., the relationship between CV events and attained LDL-C on a statin is linear between 25 and 200 mg/dL [215]. Similarly, in the FOURIER trial, investigators observed a monotonic relationship between attained LDL-C on lipid-lowering therapy with a statin and evolocumab and CV risk all the way down to < 10 mg/dL without an increase in adverse events [216].
3.15. Lifestyle modification is beneficial but seldom successful
If LDL-C can be kept very low early by lifestyle alone, it would likely produce great benefit, but further significant lifestyle change is unlikely for the vast majority of Americans (of whom 73.6% percent are overweight or obese [217] and over 34 million still smoke [218]), beyond what has been achieved to this point. Society can do both – work toward a healthier national lifestyle and treat those at risk.
The accumulating evidence that it is not just level of cholesterol, but duration of elevation above healthy levels, would suggest that now is the time to reconsider current approaches in this regard. Further delay in early management of atherosclerosis would continue to expose a large population of younger people to later risk that could have been prevented and would burden society with the cost, loss of productivity, and the human tragedy of unnecessary disease. Yet despite strong evidence and repeated calls by leaders in the field to treat much earlier, early intervention has not been adopted and likely few practitioners are aware of the rationale for this approach.
3.16. Adequate treatment can significantly reverse atherosclerosis
The Vascular Effects of Alirocumab in Acute MI-Patients (PACMAN-AMI) trial [204] showed conclusively that atheroma can regress. Changes in Percent Atheroma Volume (PAV), Lipid Core Burden Index (LCBI) and Fibrous Cap Thickness (FCT) were biphasic and improved with larger beneficial changes observed as LDL-C fell below 50 mg/dl when participants were treated with either statin monotherapy or with a statin and alirocumab.
3.17. The rationale for assessing risk more precisely
Controlling atherosclerosis as early and effectively as possible, preferably far earlier than in current practice, requires early identification of intimal changes, precise measurement of lipid levels, and recognition of all the comorbidities and risk factors listed in Table 3. As noted, atherosclerosis begins as lipid streaks in the intima, but eventually widens the intimal space to a degree that is measurable by a variety of techniques.
3.18. Advantages of coronary artery calcium scoring
Coronary Artery Calcium Scoring (CAC) is now widely recommended as a means of detecting early atherosclerosis [191,219].
-
•
It is not operator-dependent.
-
•
Is inexpensive.
-
•
Is easy to do and interpret.
-
•
Has a rich body of studies supporting it.
-
•
A positive CAC score (>0) is highly predictive of increased ASCVD risk.
-
•
CAC 0 has a Negative Predictive Value (NPV) of 99% for acute events over the following ten years.
However, for those with no CAC but a high-risk profile (particularly those with diabetes, history of smoking, or family history of premature ASCVD), who often have considerable non-calcified plaque, coronary CT angiography (CCTA) may be indicated [220,221] and treatment begun.
3.18.1. Limits of CAC
As with all tests, CAC has its limitations:
-
•
Plaque calcification is a late event and thus does not accomplish early, pre-plaque detection (the goal being to prevent any plaque from forming in the first place)
-
•
Non-calcified plaque is just as likely to cause intraarterial thrombi and is shown to be significantly present in patients with CAC scores of 0[222], [223], [224], [225], [226].
-
•
Calcification usually continues to deposit even when atherosclerosis stabilizes, thus making serial CAC of less value.
-
•
It is calibrated for age 40 years and above.
-
•
As a screening tool, it would entail exposing large numbers of people to radiation
-
•
Preventive cardiology has begun looking ahead to lifetime risk rather than the ten years for CAC and most risk calculators [28,29].
3.18.2. Risk is lifetime, not just a decade and people are living longer with better life expectancy
While it is good that a calcium score of zero makes an acute event less likely in the subsequent decade, looking beyond ten years and considering that atherosclerosis can have effects on other organ systems besides the coronary arteries, it seems worthwhile instituting earlier prevention in even those with CAC 0 who have a high-risk profile, as outlined in this review.
3.18.3. There is compelling evidence and significant agreement, as reviewed above, that
-
•
Atherosclerosis begins early in childhood as LDL-C exceeds 20–40 mg/dl, and then progresses unless therapeutic intervention is instituted.
-
•
Atherosclerosis is essentially universal in the United States and developed countries.
-
•
Powerful evidence shows that the earlier treatment is begun, the more successful it will be.
-
•
Atherosclerosis burdens the young [227], [228], [229], middle-aged and old.
-
•
LDL-C is the initial and primary driver of atherosclerosis. The lower the LDL-C that is achieved, and the earlier, the lower the likelihood of atherosclerosis progression and the greater the chance of stabilization or regression.
-
•
There is no apparent clinically significant harm (no signal for neurocognitive impairment/dementia, hemorrhagic stroke, increase in neoplasms, risk for demyelination, etc.) from lowering LDL to even the very lowest levels (< 10 mg/dL) [230,231].
-
•
Statins and other LLT are remarkably safe and the few adverse effects that occur affect far fewer people than the many lives saved, an extremely favorable benefit/risk ratio [195,[232], [233], [234]].
-
•
Other manifestations of atherosclerosis are as dangerous as coronary artery disease and stroke but are mostly unaddressed in prevention. They are not reflected in current risk assessments. The new paradigm would incorporate preserving all vital functions that atherosclerosis can degrade, which early, intensive treatment would likely accomplish.
-
•
Detailed individualization of treatment is preferable to generic tools.
-
•
As valuable as randomized controlled trials are, they are not the only form of valid evidence and they have important limitations, as discussed above. When RCTs are not feasible or adequate to answer important needs, such as diseases that take very long times to develop, that should not prevent establishing important goals and approaches when there is ample other evidence demonstrating their value.
-
•
Considering the considerable ‘residual risk’ and the high potential of reducing it with precise, early, intensive treatment, it seems urgent that new approaches as described herein be adopted to reduce that toll. As numerous references noted above have said, if treatment were to begin early and intensively, atherosclerosis could become so rare as to be an ‘orphan disease’ in all who followed the advice.
3.19. Early and intensive precision prevention can be cost effective
The downside of early detection and management of atherosclerosis is that so many more Americans would require treatment. Can we afford to take care of them all? A better question would be: can we afford not to? The American Heart Association estimated current direct and indirect costs of cardiovascular disease in 2021 and projected to 2035 if trends continue [235], listed in Table 2. Indirect costs are major contributors to cost of atherosclerosis, even with only ASCVD events being assessed [236].
Table 2.
Direct and indirect costs of specific atherosclerotic disease current and projected to 2035, adapted from AHA report [235].
| Condition | Current total of direct and indirect costs (in billions of dollars) | Projected 2035 total costs (in billions of dollars) |
|---|---|---|
| Hypertension | 110 | 221 |
| CHD | 188 | 366 |
| Congestive Heart Failure | 29 | 64 |
| Stroke | 67 | 143 |
| Totals | 394 | 794 |
Dementia currently costs the United States $305 billion [237], also likely to at least double by 2035 if the trends noted above apply. Since the US population is aging rapidly, an even faster increase in costs for dementia seem likely. Not included in these numbers are the costs of all the other manifestations of atherosclerosis and the effect on vigor of aging. Correcting disparities in healthcare would also bring considerable savings [238]. If the projections are fulfilled, within thirteen years US expense for these diseases will well exceed $1 trillion. The cost of the early, precise, and intensive treatment of atherosclerosis as reviewed above would likely cost a fraction of that, especially when early detection means beginning management at a stage that would be much more effective and less costly. There would be huge savings from prevented acute events, procedures and long-term complications [239]. Increased productivity and reduced presenteeism from chronic atherosclerotic disease would also bring considerable cost savings.
The cost effectiveness of generic statins has been reported . The study of screening and treating FH in the very young also showed cost-effectiveness [240], [241], [242]. At current costs (The Medical Letter, September 23, 2019), rosuvastatin costs the pharmacy $144 per year. Ezetimibe $444 per year. Giving both would cost $588 per year. There were about 100 million Americans ages 20–44 in the 2020 census. If 20% qualified for treatment under the recommendations in this paper and half of those agreed to treatment, that would cost about $6 billion per year for the cost of the two drugs, which would be about 0.14% of US healthcare expenditures of around $4.3 trillion in 2021. Even counting the costs of laboratory testing and doctors’ visits it would still be a tiny percentage. While a more detailed accounting is beyond the scope of this paper, early treatment appears to be cost effective, as has been judged by multiple analyses [243], [244], [245].
3.20. Lack of informed consent
Guidelines recommend physicians engage in Shared Decision Making with patients, which we unequivocally endorse, as it respects patient autonomy and dignity. Currently, patients are only informed of one approach to manage their atherosclerosis and prevent complications. True informed consent and Shared Decision Making ethically requires that patients be informed about all well-founded approaches, such as the differences from current guidelines reviewed here and urged by many of the leading figures in preventive cardiology and lipidology for decades.
3.21. Summary of specific recommendations
Based on all the above, we make the following recommendations:
-
•Principles:
-
○Atherosclerosis is by far the greatest cause of disease, disability, death and cost. It should be the number one priority of the healthcare system.
-
○Risk is proportional to the level of LDL-C and the duration of the exposure and increased by enhancing factors.
-
■Thus, the earlier elevated LDL-C is lowered, the better.
-
■
-
○That means screening at the earliest age possible.
-
○Each patient should be evaluated thoroughly and individually for all factors that contribute to risk (Table 3).
-
○The optimal LDL-C appears to be the level present at birth (20–40 mg/dl).
-
■That is probably not necessary for everyone nor likely practical to attain widely.
-
■
-
○We recommend:
-
•Progression of atherosclerosis seems unlikely when all these conditions are met:
-
•LDL-C in all previous years never exceeds 85 mg/dl
-
•Non-HDL-C is below 100 mg/dl
-
•There are no significant enhancing factors
-
•The patient is fit and follows a healthy lifestyle (such as the AHA Seven Healthy Habits)
-
•
-
•Medications would not be indicated for these patients unless they develop plaque, but of course continued healthy lifestyle is vital.
-
•These patients should be evaluated yearly to estimate risk if and when it develops.
-
•
-
•Treatment to reduce or at least halt progression of intimal changes is indicated when one or a combination of the following exists:
-
•Patients have LDL-C consistently greater than 100 mg/dl or non-HDL-C > 110 mg/dl.
-
•Plaque
-
•Significant enhancing factors
-
•Established ASCVD.
-
•
-
•Studies have shown that reversal of plaque begins when LDL-C falls below approximately 60–80 mg/dl and treatment is begun before significant scarring and calcification occurs [246], [247], [248].
-
•That should also make new intimal thickening, plaque and intraarterial thrombi much less likely, if blood pressure and all other factors are well controlled.
-
•
-
•Thus, getting LDL-C below 40 mg/dl seems the most effective goal for patients in the category of more advanced atherosclerosis.
-
•Very advanced disease or very high risk would benefit from lowering LDL-C < 20 mg/dl.
-
•There are now excellent, safe treatments to achieve these goals in most patients.
-
•As duration of arterial exposure to excess cholesterol is the other primary determinant, the earlier and faster those lower levels are achieved, the better.
-
•It requires only six weeks to see the effect of most medication changes, so physicians can act quickly to ensure getting patients to goal as soon as possible to reduce continued exposure of the vascular bed to excess cholesterol.
-
•It is very important to identify all enhancing factors and to do all possible to correct as many as are amenable to improvement.
-
•The presence of plaque on imaging should spur even more careful management.
-
•If atherosclerosis can be recognized and controlled from an early age, residual risk could be virtually eliminated, and the “epidemic of vascular disease” ended.
-
○
-
•Most patients are encountered later in life.
-
•If they have had lifelong low LDL-C, no plaque and no risk factors, as noted, observe only.
-
•If they have more advanced signs, as noted above (LDL-C consistently greater than 100 mg/dl or non-HDL-C > 110 mg/dl, plaque, significant enhancing factors, and/or established ASCVD), then treatment to goal < 40 mg/dl.
-
•If they have more severe disease – any one or more of many significant risk factors, major plaque, high CAC, history of ASCVD events or other complications of atherosclerosis (Mild Cognitive Impairment, PAD, etc.), then LDL-C goal should be < 20 mg/dl.
-
•In all cases, it is critical to manage all controllable enhancing factors.
-
•
-
•Screening should begin as early as possible
-
•Whenever a patient is first encountered
-
•Screening from birth would have even better results, as follows:
-
•In the first year: test Lipoprotein (a) and LDL-C (high Lipoprotein (a) increases the risk of stroke in children and FH needs to be treated very early, depending on severity).
-
•Within eight years, do a full lipid panel.
-
•If it indicates Familial Hypercholesterolemia not previously detected, treat immediately in cooperation with the pediatrician and follow closely. (Note discovery of FH should always be accompanied by cascade screening of blood relatives)
-
•Other than FH, if there are no significant enhancing factors and LDL-C is below 85 mg/dl, improved lifestyle is likely all that is needed. Regular follow-up is important to detect changes.
-
•If there are significant enhancing factors (Table 3), consider lifestyle management for six months. If no improvement in lifestyle, weight, lipids and other factors, statin and ezetimibe might be needed from age eight on.
- •
-
•Children deemed at very high risk can be considered for treatment even at an early age depending on individual cases and clinical judgment.
-
•
-
•
-
•When a person enters care by age 13 on:
-
•If there is evidence that LDL-C has not exceeded 85–100 mg/dl previously and there are no significant enhancing factors, continue to observe yearly and encourage healthy lifestyle.
-
•When there is intimal thickening or plaque or enhancing factors that cannot first be reversed by lifestyle change, then more aggressive efforts can be considered.
-
•Plaque can be surprisingly present in even young adults, sometimes without obvious causes.
-
•Few between 20–50 years old would be rated eligible for treatment by current guidelines but many would benefit from early intervention.
-
•Screening for plaque can be useful to detect those cases at an early stage.
-
•Carotid Intimal-medial Thickness (CIMT) by 2D B-mode ultrasound, if it can be done reliably, detects atherosclerosis at the earliest stage. However, it is very operator dependent and is useful only where dedicated technique can be carefully controlled.
-
•3D carotid plaque burden by ultrasound is more reliable and can follow plaque development serially.
-
•Any presence of arterial plaque in the aorta, femoral artery, etc., signifies plaque and advanced atherosclerosis.
-
•If risk is much higher, then CT angiography and, if necessary, MRA (Magnetic Resonance Angiography) can be considered.
-
•CAC is calibrated only for those over 40 years of age.
-
•
-
•The goal for LDL-C when plaque is present is at least < 38 mg/dl (plus control of all enhancing factors).
-
•Those at higher risk (longstanding high cholesterol, major plaque, prior events and/or major enhancing factors), will in most cases require lowering LDL-C to below 20 mg/dl, as in the PCSK9 trials.
-
•
-
•Considerations in female patients
-
•Pregnancy, complications of pregnancy, menopause, Polycystic Ovary Disease and other such factors unique to women tend to elevate cholesterol to high levels and increase risk later in life. While the cholesterol elevations might be of shorter duration, the risk is still considerable even in women with no other risk factors.
-
•Treatment would be indicated immediately after pregnancy, breastfeeding and future pregnancy are no longer issues.
-
•
-
•
3.22. Summary
Concern for costs tend to dominate discussions of public policy, but the human suffering from disease, disability, and death caused by atherosclerosis is overwhelming and far more important. As Brown and Goldstein said, it is time to end the epidemic and, as Peter Libby said, demote atherosclerosis from the leading killer to a rare disease. We should do the same for all the other atherosclerotic-driven diseases.
Assessing precisely, beginning very early, and achieving intensive goals has been shown to be efficacious, safe and cost effective. With the urging of so many leaders in the field for so long and the compelling evidence laid out in this review, the question remains: what is the profession waiting for? There is that fierce urgency of now: every day of delay means more people losing arterial health, with all the tragic consequences that result. We have the means; do we have the will?
CRediT authorship contribution statement
Michael E. Makover: Conceptualization, Writing – original draft, Writing – review & editing. Michael D. Shapiro: Conceptualization, Writing – review & editing. Peter P. Toth: Conceptualization, Writing – review & editing.
Declaration of Competing Interest
Michael Makover: None. Michael Shapiro: Dr Shapiro has participated in the Scientific Advisory Boards for Amgen, Esperion, Novartis, and Novo Nordisk. Peter P. Toth: Dr. Toth is a member of the speakers bureau for Amarin, Amgen, Esperion, and Novo-Nordisk; he is a consultant to Amarin, Kowa, Merck, Novartis, and Resverlogix.
Acknowledgments
Makover wishes to dedicate this paper to the late Michael Schloss, of New York University, a colleague who was a pioneer of many of the concepts discussed.
References
- 1.Song P., Fang Z., Wang H., et al. Global and regional prevalence, burden, and risk factors for carotid atherosclerosis: a systematic review, meta-analysis, and modelling study. Lancet Glob Health. 2020;8:e721–e729. doi: 10.1016/S2214-109X(20)30117-0. [DOI] [PubMed] [Google Scholar]
- 2.Roth G.A., Mensah G.A., Johnson C.O., et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 study. J Am Coll Cardiol. 2020;76:2982–3021. doi: 10.1016/j.jacc.2020.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jr. MLk. Beyond Vietnam: A Time to Break the Silence. Addressing a crowd of 3,000 at Riverside Church in New York City. 1967.
- 4.Mensah G.A., Wei G.S., Sorlie P.D., et al. Decline in cardiovascular mortality: possible causes and Implications. Circ Res. 2017;120:366–380. doi: 10.1161/CIRCRESAHA.116.309115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ford E.S., Capewell S. Proportion of the decline in cardiovascular mortality disease due to prevention versus treatment: public health versus clinical care. Annu Rev Public Health. 2011;32:5–22. doi: 10.1146/annurev-publhealth-031210-101211. [DOI] [PubMed] [Google Scholar]
- 6.Dalen J.E., Alpert J.S., Goldberg R.J., Weinstein R.S. The epidemic of the 20(th) century: coronary heart disease. Am J Med. 2014;127:807–812. doi: 10.1016/j.amjmed.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 7.Virani S.S., Alonso A., Aparicio H.J., et al. Heart disease and stroke statistics-2021 update: a report from the American heart association. Circulation. 2021;143:e254–e743. doi: 10.1161/CIR.0000000000000950. [DOI] [PubMed] [Google Scholar]
- 8.Mehta N.K., Abrams L.R., Myrskylä M. US life expectancy stalls due to cardiovascular disease, not drug deaths. Proc Natl Acad Sci. 2020;117:6998–7000. doi: 10.1073/pnas.1920391117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doll J.A. Quality of life after myocardial infarction: more progress needed. Heart. 2020;106(8) doi: 10.1136/heartjnl-2019-315871. [DOI] [PubMed] [Google Scholar]
- 10.Kulik A. Quality of life after coronary artery bypass graft surgery versus percutaneous coronary intervention: what do the trials tell us? Curr Opin Cardiol. 2017;32:707–714. doi: 10.1097/HCO.0000000000000458. [DOI] [PubMed] [Google Scholar]
- 11.Dalton J.E., Rothberg M.B., Dawson N.V., Krieger N.I., Zidar D.A., Perzynski A.T. Failure of traditional risk factors to adequately predict cardiovascular events in older populations. J Am Geriatr Soc. 2020;68:754–761. doi: 10.1111/jgs.16329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arora S., Qamar A., Gupta P., et al. Guideline based eligibility for primary prevention statin therapy - Insights from the North India ST-elevation myocardial infarction registry (NORIN-STEMI) J Clin Lipidol. 2022;16:227–236. doi: 10.1016/j.jacl.2021.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Singh A., Collins B.L., Gupta A., et al. Cardiovascular risk and statin eligibility of young adults after an MI: partners YOUNG-MI registry. J Am Coll Cardiol. 2018;71:292–302. doi: 10.1016/j.jacc.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miedema M.D., Garberich R.F., Schnaidt L.J., et al. Statin eligibility and outpatient care prior to ST-segment elevation myocardial infarction. J Am Heart Assoc. 2017;6 doi: 10.1161/JAHA.116.005333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fernández-Friera L., Peñalvo J.L., Fernández-Ortiz A., et al. Prevalence, vascular distribution, and multiterritorial extent of subclinical atherosclerosis in a middle-aged cohort: the PESA (progression of early subclinical atherosclerosis) study. Circulation. 2015;131:2104–2113. doi: 10.1161/CIRCULATIONAHA.114.014310. [DOI] [PubMed] [Google Scholar]
- 16.López-Melgar B., Fernández-Friera L., Oliva B., et al. Short-term progression of multiterritorial subclinical atherosclerosis. J Am Coll Cardiol. 2020;75:1617–1627. doi: 10.1016/j.jacc.2020.02.026. [DOI] [PubMed] [Google Scholar]
- 17.Bos D., Rijk M.J.M.V.D., Geeraedts T.E.A., et al. Intracranial carotid artery atherosclerosis. Stroke. 2012;43:1878–1884. doi: 10.1161/STROKEAHA.111.648667. [DOI] [PubMed] [Google Scholar]
- 18.Libby P. The changing landscape of atherosclerosis. Nature. 2021;592:524–533. doi: 10.1038/s41586-021-03392-8. [DOI] [PubMed] [Google Scholar]
- 19.Snowdon D.A. Aging and Alzheimer's disease: lessons from the nun study. Gerontologist. 1997;37:150–156. doi: 10.1093/geront/37.2.150. [DOI] [PubMed] [Google Scholar]
- 20.Wang Q., Wang Y., Lehto K., Pedersen N.L., Williams D.M., Hägg S. Genetically-predicted life-long lowering of low-density lipoprotein cholesterol is associated with decreased frailty: a Mendelian randomization study in UK biobank. EBioMedicine. 2019;45:487–494. doi: 10.1016/j.ebiom.2019.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh M., Stewart R., White H. Importance of frailty in patients with cardiovascular disease. Eur Heart J. 2014;35:1726–1731. doi: 10.1093/eurheartj/ehu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tobert J.A. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nat Rev Drug Discov. 2003;2:517–526. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- 23.Hulley S.B. The US national cholesterol education program. Adult treatment guidelines. Drugs. 1988;36(3):100–104. doi: 10.2165/00003495-198800363-00021. Suppl. [DOI] [PubMed] [Google Scholar]
- 24.Report of the national cholesterol education program expert panel on detection, evaluation, and treatment of high blood cholesterol in adults. The expert panel. Arch Intern Med. 1988;148:36–69. [PubMed] [Google Scholar]
- 25.Stone N.J., Blumenthal R.S., Lloyd-Jones D., Grundy S.M. Comparing primary prevention recommendations. Circulation. 2020;141:1117–1120. doi: 10.1161/CIRCULATIONAHA.119.044562. [DOI] [PubMed] [Google Scholar]
- 26.Yebyo H.G., Zappacosta S., Aschmann H.E., Haile S.R., Puhan M.A. Global variation of risk thresholds for initiating statins for primary prevention of cardiovascular disease: a benefit-harm balance modelling study. BMC Cardiovasc Disord. 2020;20:418. doi: 10.1186/s12872-020-01697-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartlomiejczyk M.A., Penson P., Banach M. Worldwide dyslipidemia guidelines. Curr Cardiovasc Risk Rep. 2019;13(2) [Google Scholar]
- 28.Tokgözoğlu L., Casula M., Pirillo A., Catapano A.L. Similarities and differences between European and American guidelines on the management of blood lipids to reduce cardiovascular risk. Atheroscler Suppl. 2020;42:e1–e5. doi: 10.1016/j.atherosclerosissup.2021.01.001. [DOI] [PubMed] [Google Scholar]
- 29.Allan S., Pencina M., Thanassoulis G. Clinical reasoning and prevention of cardiovascular disease. J Clin Lipidol. 2021;15:394–398. doi: 10.1016/j.jacl.2021.04.001. [DOI] [PubMed] [Google Scholar]
- 30.Tufekci Z. Why did it take so long to accept the facts about COVID? New York Times 7 May 2021.
- 31.Toth P.P. Low-density lipoprotein cholesterol treatment rates in high risk patients: more disappointment despite ever more refined evidence-based guidelines. Am J Prev Cardiol. 2021;6 doi: 10.1016/j.ajpc.2021.100186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramsaran E., Preusse P., Sundaresan D., et al. Adherence to blood cholesterol treatment guidelines among physicians managing patients with atherosclerotic cardiovascular disease. Am J Cardiol. 2019;124:169–175. doi: 10.1016/j.amjcard.2019.04.017. [DOI] [PubMed] [Google Scholar]
- 33.Yang Y.S., Lee S.Y., Kim J.S., et al. Achievement of LDL-C targets defined by ESC/EAS (2011) guidelines in risk-stratified Korean patients with dyslipidemia receiving lipid-modifying treatments. Endocrinol Metab. 2020;35:367–376. doi: 10.3803/EnM.2020.35.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blom D.J., Almahmeed W., Al-Rasadi K., et al. Low-density lipoprotein cholesterol goal achievement in patients with familial hypercholesterolemia in countries outside Western Europe: the international cholesterol management practice study. J Clin Lipidol. 2019;13:594–600. doi: 10.1016/j.jacl.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 35.Toth P.P., Granowitz C., Hull M., Anderson A., Philip S. Long-term statin persistence is poor among high-risk patients with dyslipidemia: a real-world administrative claims analysis. Lipids Health Dis. 2019;18:175. doi: 10.1186/s12944-019-1099-z. 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Statin Denial: an internet-driven cult with deadly consequences. Ann Intern Med. 2017;167:281–282. doi: 10.7326/M17-1566. [DOI] [PubMed] [Google Scholar]
- 37.Rea F., Biffi A., Ronco R., et al. Cardiovascular outcomes and mortality associated with discontinuing statins in older patients receiving polypharmacy. JAMA Netw Open. 2021;4 doi: 10.1001/jamanetworkopen.2021.13186. e2113186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Serban M.-.C., Colantonio L.D., Manthripragada A.D., et al. Statin intolerance and risk of coronary heart events and all-cause mortality following myocardial infarction. J Am Coll Cardiol. 2017;69:1386–1395. doi: 10.1016/j.jacc.2016.12.036. [DOI] [PubMed] [Google Scholar]
- 39.Adusumalli S., Westover J.E., Jacoby D.S., et al. Effect of passive choice and active choice interventions in the electronic health record to cardiologists on statin prescribing: a cluster randomized clinical trial. JAMA Cardiol. 2021;6:40–48. doi: 10.1001/jamacardio.2020.4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baum S.J., Rane P.B., Nunna S., et al. Geographic variations in lipid-lowering therapy utilization, LDL-C levels, and proportion retrospectively meeting the ACC/AHA very high-risk criteria in a real-world population of patients with major atherosclerotic cardiovascular disease events in the United States. Am J Prev Cardiol. 2021;6 doi: 10.1016/j.ajpc.2021.100177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Makover M.E., Schloss M. The very high residual degree of death and disease from atherosclerosis needs new approaches. J Clin Lipidol. 2016;10:466–468. doi: 10.1016/j.jacl.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 42.Kones R. Molecular sources of residual cardiovascular risk, clinical signals, and innovative solutions: relationship with subclinical disease, undertreatment, and poor adherence: implications of new evidence upon optimizing cardiovascular patient outcomes. Vasc Health Risk Manag. 2013;9:617–670. doi: 10.2147/VHRM.S37119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho K.I., Yu J., Hayashi T., Han S.H., Koh K.K. Strategies to overcome residual risk during statins era. Circ J. 2019;83:1973–1979. doi: 10.1253/circj.CJ-19-0624. [DOI] [PubMed] [Google Scholar]
- 44.Finking G., Hanke H. Nikolaj Nikolajewitsch Anitschkow (1885-1964) established the cholesterol-fed rabbit as a model for atherosclerosis research. Atherosclerosis. 1997;135:1–7. doi: 10.1016/s0021-9150(97)00161-5. [DOI] [PubMed] [Google Scholar]
- 45.Steinberg D. Thematic review series: the pathogenesis of atherosclerosis. An interpretive history of the cholesterol controversy: part I. J Lipid Res. 2004;45:1583–1593. doi: 10.1194/jlr.R400003-JLR200. [DOI] [PubMed] [Google Scholar]
- 46.Brown M.S., Goldstein J.L. Cholesterol feedback: from Schoenheimer's bottle to Scap's MELADL. J Lipid Res. 2009;50:S15–S27. doi: 10.1194/jlr.R800054-JLR200. Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sorci-Thomas M.G., Thomas M.J. Microdomains, inflammation, and atherosclerosis. Circ Res. 2016;118:679–691. doi: 10.1161/CIRCRESAHA.115.306246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Basatemur G.L., Jørgensen H.F., Clarke M.C.H., Bennett M.R., Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. 2019;16:727–744. doi: 10.1038/s41569-019-0227-9. [DOI] [PubMed] [Google Scholar]
- 49.Moreau P.R., Tomas Bosch V., Bouvy-Liivrand M., et al. Profiling of primary and mature miRNA expression in atherosclerosis associated cell types. Arterioscler Thromb Vasc Biol. 2021 doi: 10.1161/ATVBAHA.121.315579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thomas D.G., Wei Y., Tall A.R. Lipid and metabolic syndrome traits in coronary artery disease: a Mendelian randomization study. J Lipid Res. 2021;62 doi: 10.1194/jlr.P120001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Allayee H. Genetic evidence for independent causal relationships between metabolic biomarkers and risk of coronary artery diseases. J Lipid Res. 2021;62 doi: 10.1016/j.jlr.2021.100064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pedersen T.R. The success story of LDL cholesterol lowering. Circ Res. 2016;118:721–731. doi: 10.1161/CIRCRESAHA.115.306297. [DOI] [PubMed] [Google Scholar]
- 53.Ren J., Bi Y., Sowers J.R., Hetz C., Zhang Y. Endoplasmic reticulum stress and unfolded protein response in cardiovascular diseases. Nat Rev Cardiol. 2021;18:499–521. doi: 10.1038/s41569-021-00511-w. [DOI] [PubMed] [Google Scholar]
- 54.Hevonoja T., Pentikäinen M.O., Hyvönen M.T., Kovanen P.T., Ala-Korpela M. Structure of low density lipoprotein (LDL) particles: basis for understanding molecular changes in modified LDL. Biochim Biophys Acta BBA. 2000;1488:189–210. doi: 10.1016/s1388-1981(00)00123-2. Molecular and Cell Biology of Lipids. [DOI] [PubMed] [Google Scholar]
- 55.Torres N., Guevara-Cruz M., Velázquez-Villegas L.A., Tovar A.R. Nutrition and atherosclerosis. Arch Med Res. 2015;46:408–426. doi: 10.1016/j.arcmed.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 56.Ference B.A., Kastelein J.J.P., Catapano A.L. Lipids and lipoproteins in 2020. JAMA. 2020;324:595–596. doi: 10.1001/jama.2020.5685. [DOI] [PubMed] [Google Scholar]
- 57.Varghese D.S., Ali B.R. Pathological crosstalk between oxidized LDL and ER stress in human diseases: a comprehensive review. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.674103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ference B.A., Graham I., Tokgozoglu L., Catapano A.L. Impact of lipids on cardiovascular health: JACC health promotion series. J Am Coll Cardiol. 2018;72:1141–1156. doi: 10.1016/j.jacc.2018.06.046. [DOI] [PubMed] [Google Scholar]
- 59.Donegá S., Oba J., Maranhão R.C. Concentration of serum lipids and apolipoprotein B in newborns. Arq Bras Cardiol. 2006;86:419–424. doi: 10.1590/s0066-782x2006000600003. [DOI] [PubMed] [Google Scholar]
- 60.Nayak C.D., Agarwal V., Nayak D.M. Correlation of cord blood lipid heterogeneity in neonates with their anthropometry at birth. Indian J Clin Biochem. 2013;28:152–157. doi: 10.1007/s12291-012-0252-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsang R.C., Fallat R.W., Glueck C.J. Cholesterol at Birth and Age 1: comparison of normal and hypercholesterolemic neonates. Pediatrics. 1974;53:458. [PubMed] [Google Scholar]
- 62.Bansal N., Cruickshank J.K., McElduff P., Durrington P.N. Cord blood lipoproteins and prenatal influences. Curr Opin Lipidol. 2005;16:400–408. doi: 10.1097/01.mol.0000174154.61307.16. [DOI] [PubMed] [Google Scholar]
- 63.Dietschy J.M., Turley S.D. Thematic review series: brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res. 2004;45:1375–1397. doi: 10.1194/jlr.R400004-JLR200. [DOI] [PubMed] [Google Scholar]
- 64.Grundy S.M., Cleeman J.I., Merz C.N.B., et al. Implications of recent clinical trials for the national cholesterol education program adult treatment panel III guidelines. Circulation. 2004;110:227–239. doi: 10.1161/01.CIR.0000133317.49796.0E. [DOI] [PubMed] [Google Scholar]
- 65.Pontzer H., Wood B.M., Raichlen D.A. Hunter-gatherers as models in public health. Obes Rev. 2018;19:24–35. doi: 10.1111/obr.12785. [DOI] [PubMed] [Google Scholar]
- 66.O'Keefe J.H., Cordain L., Harris W.H., Moe R.M., Vogel R. Optimal low-density lipoprotein is 50 to 70mg/dl: lower is better and physiologically normal. J Am Coll Cardiol. 2004;43:2142–2146. doi: 10.1016/j.jacc.2004.03.046. [DOI] [PubMed] [Google Scholar]
- 67.Kannel W.B. Range of serum cholesterol values in the population developing coronary artery disease. Am J Cardiol. 1995;76 doi: 10.1016/s0002-9149(99)80474-3. 69C–77C. [DOI] [PubMed] [Google Scholar]
- 68.Abdullah S.M., Defina L.F., Leonard D., et al. Long-term association of low-density lipoprotein cholesterol with cardiovascular mortality in individuals at low 10-year risk of atherosclerotic cardiovascular disease. Circulation. 2018;138:2315–2325. doi: 10.1161/CIRCULATIONAHA.118.034273. [DOI] [PubMed] [Google Scholar]
- 69.Shapiro M.D., Bhatt D.L. Cholesterol-years” for ASCVD risk prediction and treatment∗. J Am Coll Cardiol. 2020;76:1517–1520. doi: 10.1016/j.jacc.2020.08.004. [DOI] [PubMed] [Google Scholar]
- 70.Kavousi M., Leening M.J., Nanchen D., et al. Comparison of application of the ACC/AHA guidelines, adult treatment panel III guidelines, and European society of cardiology guidelines for cardiovascular disease prevention in a European cohort. JAMA. 2014;311:1416–1423. doi: 10.1001/jama.2014.2632. [DOI] [PubMed] [Google Scholar]
- 71.Sniderman A.D., LaChapelle K.J., Rachon N.A., Furberg C.D. The necessity for clinical reasoning in the era of evidence-based medicine. Mayo Clin Proc. 2013;88:1108–1114. doi: 10.1016/j.mayocp.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 72.Sniderman A.D., D'Agostino Sr R.B., Pencina M.J. The role of physicians in the era of predictive analytics. JAMA. 2015;314:25–26. doi: 10.1001/jama.2015.6177. [DOI] [PubMed] [Google Scholar]
- 73.Hampton J.R. Evidence-based medicine, opinion-based medicine, and real-world medicine. Perspect Biol Med. 2002;45:549–568. doi: 10.1353/pbm.2002.0070. [DOI] [PubMed] [Google Scholar]
- 74.Brown W.V. From the editor: new guidelines are coming. J Clin Lipidol. 2017;11:1–2. doi: 10.1016/j.jacl.2017.02.011. [DOI] [PubMed] [Google Scholar]
- 75.Brown M.S., Goldstein J.L. Receptor-mediated control of cholesterol metabolism. Science. 1976;191:150. doi: 10.1126/science.174194. [DOI] [PubMed] [Google Scholar]
- 76.Biros E., Karan M., Golledge J. Genetic variation and atherosclerosis. Curr Genomics. 2008;9:29–42. doi: 10.2174/138920208783884856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nurnberg S.T., Zhang H., Hand N.J., et al. From loci to biology: functional genomics of genome-wide association for coronary disease. Circ Res. 2016;118:586–606. doi: 10.1161/CIRCRESAHA.115.306464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Saigusa R., Winkels H., Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. 2020;17:387–401. doi: 10.1038/s41569-020-0352-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alfaddagh A., Martin S.S., Leucker T.M., et al. Inflammation and cardiovascular disease: from mechanisms to therapeutics. Am J Prev Cardiol. 2020;4 doi: 10.1016/j.ajpc.2020.100130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Matsuura E., Atzeni F., Sarzi-Puttini P., Turiel M., Lopez L.R., Nurmohamed M.T. Is atherosclerosis an autoimmune disease? BMC Med. 2014;12:47. doi: 10.1186/1741-7015-12-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wolf D., Ley K. Immunity and inflammation in atherosclerosis. Circ Res. 2019;124:315–327. doi: 10.1161/CIRCRESAHA.118.313591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Libby P., Loscalzo J., Ridker P.M., et al. Inflammation, immunity, and infection in atherothrombosis: JACC review topic of the week. J Am Coll Cardiol. 2018;72:2071–2081. doi: 10.1016/j.jacc.2018.08.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Escárcega R.O., Lipinski M.J., García-Carrasco M., Mendoza-Pinto C., Galvez-Romero J.L., Cervera R. Inflammation and atherosclerosis: cardiovascular evaluation in patients with autoimmune diseases. Autoimmun Rev. 2018;17:703–708. doi: 10.1016/j.autrev.2018.01.021. [DOI] [PubMed] [Google Scholar]
- 84.Lechner K., von Schacky C., McKenzie A.L., et al. Lifestyle factors and high-risk atherosclerosis: pathways and mechanisms beyond traditional risk factors. Eur J Prev Cardiol. 2020;27:394–406. doi: 10.1177/2047487319869400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rippe J.M. Lifestyle strategies for risk factor reduction, prevention, and treatment of cardiovascular disease. Am J Lifestyle Med. 2019;13:204–212. doi: 10.1177/1559827618812395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bevan G.H., Al-Kindi S.G., Brook R.D., Münzel T., Rajagopalan S. Ambient air pollution and atherosclerosis: insights into dose, time, and mechanisms. Arterioscler Thromb Vasc Biol. 2021;41:628–637. doi: 10.1161/ATVBAHA.120.315219. [DOI] [PubMed] [Google Scholar]
- 87.Lind P.M., Lind L. Are persistent organic pollutants linked to lipid abnormalities, atherosclerosis and cardiovascular disease? A review. J Lipid Atheroscler. 2020;9:334–348. doi: 10.12997/jla.2020.9.3.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ramsey S.A., Gold E.S., Aderem A. A systems biology approach to understanding atherosclerosis. EMBO Mol Med. 2010;2:79–89. doi: 10.1002/emmm.201000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miedema M.D., Nauffal V.D., Singh A., Blankstein R. Statin therapy for young adults: a long-term investment worth considering. Trends Cardiovasc Med. 2020;30:48–53. doi: 10.1016/j.tcm.2019.02.004. [DOI] [PubMed] [Google Scholar]
- 90.Ahmad Z., Banerjee P., Hamon S., et al. Inhibition of angiopoietin-like protein 3 with a monoclonal antibody reduces triglycerides in hypertriglyceridemia. Circulation. 2019;140:470–486. doi: 10.1161/CIRCULATIONAHA.118.039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Libby P. Inflammation in atherosclerosis-no longer a theory. Clin Chem. 2021;67:131–142. doi: 10.1093/clinchem/hvaa275. [DOI] [PubMed] [Google Scholar]
- 92.Libby P., Everett B.M. Novel antiatherosclerotic therapies. Arterioscler Thromb Vasc Biol. 2019;39:538–545. doi: 10.1161/ATVBAHA.118.310958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bäck M., Yurdagul A., Tabas I., Öörni K., Kovanen P.T. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16:389–406. doi: 10.1038/s41569-019-0169-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.García-González V., Delgado-Coello B., Pérez-Torres A., Mas-Oliva J. Reality of a vaccine in the prevention and treatment of atherosclerosis. Arch Med Res. 2015;46:427–437. doi: 10.1016/j.arcmed.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 95.Newman W.P., Freedman D.S., Voors A.W., et al. Relation of serum lipoprotein levels and systolic blood pressure to early atherosclerosis. The Bogalusa heart study. N Engl J Med. 1986;314:138–144. doi: 10.1056/NEJM198601163140302. [DOI] [PubMed] [Google Scholar]
- 96.Holman R.L., Mc G.H., Strong J.P., Geer J.C. The natural history of atherosclerosis: the early aortic lesions as seen in New Orleans in the middle of the of the 20th century. Am J Pathol. 1958;34:209–235. [PMC free article] [PubMed] [Google Scholar]
- 97.Stary H.C. Macrophages, macrophage foam cells, and eccentric intimal thickening in the coronary arteries of young children. Atherosclerosis. 1987;64:91–108. doi: 10.1016/0021-9150(87)90234-6. [DOI] [PubMed] [Google Scholar]
- 98.Laitinen T.T., Nuotio J., Rovio S.P., et al. Dietary fats and atherosclerosis from childhood to adulthood. Pediatrics. 2020;145 doi: 10.1542/peds.2019-2786. [DOI] [PubMed] [Google Scholar]
- 99.Strong J.P., Malcom G.T., McMahan C.A., et al. Prevalence and extent of atherosclerosis in adolescents and young adults: implications for prevention from the pathobiological determinants of atherosclerosis in youth study. JAMA. 1999;281:727–735. doi: 10.1001/jama.281.8.727. [DOI] [PubMed] [Google Scholar]
- 100.Gooding H.C., Gidding S.S., Moran A.E., et al. Challenges and opportunities for the prevention and treatment of cardiovascular disease among young adults: report from a national heart, lung, and blood institute working group. J Am Heart Assoc. 2020;9 doi: 10.1161/JAHA.120.016115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gidding S.S., Rana J.S., Prendergast C., et al. Pathobiological determinants of atherosclerosis in youth (PDAY) risk score in young adults predicts coronary artery and abdominal aorta calcium in middle age. Circulation. 2016;133:139–146. doi: 10.1161/CIRCULATIONAHA.115.018042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chiavaroli L., Viguiliouk E., Nishi S.K., et al. DASH dietary pattern and cardiometabolic outcomes: an umbrella review of systematic reviews and meta-analyses. Nutrients. 2019:11. doi: 10.3390/nu11020338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Navar-Boggan A.M., Peterson E.D., D'Agostino R.B., Neely B., Sniderman A.D., Pencina M.J. Hyperlipidemia in early adulthood increases long-term risk of coronary heart disease. Circulation. 2015;131:451–458. doi: 10.1161/CIRCULATIONAHA.114.012477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kosmeri C., Milionis H., Vlahos A.P., et al. The impact of dyslipidemia on early markers of endothelial and renal dysfunction in children. J Clin Lipidol. 2021;15:292–300. doi: 10.1016/j.jacl.2020.12.003. [DOI] [PubMed] [Google Scholar]
- 105.Daida H., Dohi T., Fukushima Y., Ohmura H., Miyauchi K. The goal of achieving atherosclerotic plaque regression with lipid-lowering therapy: insights from IVUS trials. J Atheroscler Thromb. 2019;26:592–600. doi: 10.5551/jat.48603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fisher E.A. Regression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:226–235. doi: 10.1161/ATVBAHA.115.301926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Murray R., Godfrey K.M., Lillycrop K.A. The early life origins of cardiovascular disease. Curr Cardiovasc Risk Rep. 2015;9:15. [Google Scholar]
- 108.Raitakari O.T., Juonala M., Kähönen M., et al. Cardiovascular risk factors in childhood and carotid artery intima-media thickness in adulthood: the cardiovascular risk in young finns study. JAMA. 2003;290:2277–2283. doi: 10.1001/jama.290.17.2277. [DOI] [PubMed] [Google Scholar]
- 109.Shea S., Stein J.H., Jorgensen N.W., et al. Cholesterol mass efflux capacity, incident cardiovascular disease, and progression of carotid plaque. Arterioscler Thromb Vasc Biol. 2019;39:89–96. doi: 10.1161/ATVBAHA.118.311366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Françoso L.A., Coates V. Anatomicopathological evidence of the beginning of atherosclerosis in infancy and adolescence. Arq Bras Cardiol. 2002;78:131–142. [PubMed] [Google Scholar]
- 111.Joseph A., Ackerman D., Talley J.D., Johnstone J., Kupersmith J. Manifestations of coronary atherosclerosis in young trauma victims – an autopsy study. J Am Coll Cardiol. 1993;22:459–467. doi: 10.1016/0735-1097(93)90050-b. [DOI] [PubMed] [Google Scholar]
- 112.Strong J.P. Coronary Atherosclerosis in soldiers: a clue to the natural history of atherosclerosis in the young. JAMA. 1986;256:2863–2866. doi: 10.1001/jama.256.20.2863. [DOI] [PubMed] [Google Scholar]
- 113.Steinberg D. The rationale for initiating treatment of hypercholesterolemia in young adulthood. Curr Atheroscler Rep. 2013;15:296. doi: 10.1007/s11883-012-0296-2. [DOI] [PubMed] [Google Scholar]
- 114.Jacobs D.R., Woo J.G., Sinaiko A.R., et al. Childhood cardiovascular risk factors and adult cardiovascular events. N Engl J Med. 2022;386:1877–1888. doi: 10.1056/NEJMoa2109191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Srivastava G., Browne N., Kyle T., et al. Caring for US children: barriers to effective treatment in children with the disease of obesity. Obes. 2021;29 doi: 10.1002/oby.22987. [DOI] [PubMed] [Google Scholar]
- 116.Srivastava G., Browne N., Kyle T.K., et al. Caring for US children: barriers to effective treatment in children with the disease of obesity. Obesity. 2021;29:46–55. doi: 10.1002/oby.22987. [DOI] [PubMed] [Google Scholar]
- 117.Cohen J., Pertsemlidis A., Kotowski I.K., Graham R., Garcia C.K., Hobbs H.H. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–165. doi: 10.1038/ng1509. [DOI] [PubMed] [Google Scholar]
- 118.Zhao Z., Tuakli-Wosornu Y., Lagace T.A., et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79:514–523. doi: 10.1086/507488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kaplan H., Thompson R.C., Trumble B.C., et al. Coronary atherosclerosis in indigenous South American Tsimane: a cross-sectional cohort study. Lancet. 2017;389:1730–1739. doi: 10.1016/S0140-6736(17)30752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kaplan H., Thompson R.C., Trumble B.C., et al. Coronary atherosclerosis in indigenous South American Tsimane: a cross-sectional cohort study. Lancet North Am Ed. 2017;389:1730–1739. doi: 10.1016/S0140-6736(17)30752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Greenland P., Lloyd-Jones D. Time to end the mixed—and often incorrect—messages about prevention and treatment of atherosclerotic cardiovascular disease. J Am Coll Cardiol. 2007;50:2133–2135. doi: 10.1016/j.jacc.2007.05.055. ⁎⁎editorials published in the journal of the American College of cardiologyreflect the views of the authors and do not necessarily represent the views of JACCor the American college of cardiology. [DOI] [PubMed] [Google Scholar]
- 122.Fryar C.D., Chen T.C., Li X. Prevalence of uncontrolled risk factors for cardiovascular disease: united States, 1999-2010. NCHS Data Brief. 2012:1–8. [PubMed] [Google Scholar]
- 123.Rodriguez B.L., Fujimoto W.Y., Mayer-Davis E.J., et al. Prevalence of cardiovascular disease risk factors in U.S. children and adolescents with diabetes. Diabetes Care. 2006;29:1891. doi: 10.2337/dc06-0310. [DOI] [PubMed] [Google Scholar]
- 124.Shen W.J., Azhar S., Kraemer F.B. SR-B1: a unique multifunctional receptor for cholesterol influx and efflux. Annu Rev Physiol. 2018;80:95–116. doi: 10.1146/annurev-physiol-021317-121550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hu J., Zhang Z., Shen W.J., Azhar S. Cellular cholesterol delivery, intracellular processing and utilization for biosynthesis of steroid hormones. Nutr Metab. 2010;7:47. doi: 10.1186/1743-7075-7-47. (Lond) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Masana L., Girona J., Ibarretxe D., et al. Clinical and pathophysiological evidence supporting the safety of extremely low LDL levels – the zero-LDL hypothesis. J Clin Lipidol. 2018;12:292–299. doi: 10.1016/j.jacl.2017.12.018. e3. [DOI] [PubMed] [Google Scholar]
- 127.Al Rifai M., Martin S.S., McEvoy J.W., et al. The prevalence and correlates of subclinical atherosclerosis among adults with low-density lipoprotein cholesterol <70 mg/dL: the multi-ethnic study of atherosclerosis (MESA) and Brazilian longitudinal study of adult health (ELSA-Brasil) Atherosclerosis. 2018;274:61–66. doi: 10.1016/j.atherosclerosis.2018.04.021. [DOI] [PubMed] [Google Scholar]
- 128.Mehta A., Shapiro M.D. Apolipoproteins in vascular biology and atherosclerotic disease. Nat Rev Cardiol. 2021 doi: 10.1038/s41569-021-00613-5. [DOI] [PubMed] [Google Scholar]
- 129.Nordestgaard B.G. Triglyceride-rich lipoproteins and atherosclerotic cardiovascular disease: new insights from epidemiology, genetics, and biology. Circ Res. 2016;118:547–563. doi: 10.1161/CIRCRESAHA.115.306249. [DOI] [PubMed] [Google Scholar]
- 130.Geovanini G.R., Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci. 2018;132:1243–1252. doi: 10.1042/CS20180306. Lond. [DOI] [PubMed] [Google Scholar]
- 131.Tabas I., Williams K.J., Borén J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 132.Williams K.J., Feig J.E., Fisher E.A. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat Clin Pract Cardiovasc Med. 2008;5:91–102. doi: 10.1038/ncpcardio1086. [DOI] [PubMed] [Google Scholar]
- 133.Borén J., Williams K.J. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27:473–483. doi: 10.1097/MOL.0000000000000330. [DOI] [PubMed] [Google Scholar]
- 134.Sniderman A.D., Lawler P.R., Williams K., Thanassoulis G., de Graaf J., Furberg C.D. The causal exposure model of vascular disease. Clin Sci. 2012;122:369–373. doi: 10.1042/CS20110449. Lond. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Goldstein J.L., Brown M.S. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–438. doi: 10.1161/ATVBAHA.108.179564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Linton M.F., Fazio S. Macrophages, inflammation, and atherosclerosis. Int J Obes Relat Metab Disord. 2003;27(3):S35–S40. doi: 10.1038/sj.ijo.0802498. Suppl. [DOI] [PubMed] [Google Scholar]
- 137.Linton M.F., Yancey P.G., Davies S.S., et al. In: Endotext. Feingold KR, Anawalt B, Boyce A, et al., editors. South Dartmouth; (MA): 2000. The role of lipids and lipoproteins in atherosclerosis.https://www.ncbi.nlm.nih.gov/books/NBK343489/ editors. [Google Scholar]
- 138.Shi J., Perry G., Smith M.A., Friedland R.P. Vascular abnormalities: the insidious pathogenesis of Alzheimer's disease. Neurobiol Aging. 2000;21:357–361. doi: 10.1016/s0197-4580(00)00119-6. [DOI] [PubMed] [Google Scholar]
- 139.Lathe R., Sapronova A., Kotelevtsev Y. Atherosclerosis and Alzheimer-diseases with a common cause? Inflammation, oxysterols, vasculature. BMC Geriatr. 2014;14:36. doi: 10.1186/1471-2318-14-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dolan H., Crain B., Troncoso J., Resnick S.M., Zonderman A.B., Obrien R.J. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore longitudinal study of aging cohort. Ann Neurol. 2010;68:231–240. doi: 10.1002/ana.22055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kalaria R.N. The role of cerebral ischemia in Alzheimer's disease. Neurobiol Aging. 2000;21:321–330. doi: 10.1016/s0197-4580(00)00125-1. [DOI] [PubMed] [Google Scholar]
- 142.Cortes-Canteli M., Gispert J.D., Salvadó G., et al. Subclinical atherosclerosis and brain metabolism in middle-aged individuals. J Am Coll Cardiol. 2021;77:888–898. doi: 10.1016/j.jacc.2020.12.027. [DOI] [PubMed] [Google Scholar]
- 143.Iadecola C. Revisiting atherosclerosis and dementia. Nat Neurosci. 2020;23:691–692. doi: 10.1038/s41593-020-0626-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Iadecola C., Gottesman R.F. Cerebrovascular alterations in Alzheimer Disease. Circ Res. 2018;123:406–408. doi: 10.1161/CIRCRESAHA.118.313400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gottesman R.F., Albert M.S., Alonso A., et al. Associations between midlife vascular risk factors and 25-year incident dementia in the atherosclerosis risk in communities (ARIC) cohort. JAMA Neurol. 2017;74:1246–1254. doi: 10.1001/jamaneurol.2017.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Nichol A.D., Bailey M., Cooper D.J. Challenging issues in randomised controlled trials. Injury. 2010;41(1):S20–S23. doi: 10.1016/j.injury.2010.03.033. Suppl. [DOI] [PubMed] [Google Scholar]
- 147.Shepardson N.E., Shankar G.M., Selkoe D.J. Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch Neurol. 2011;68:1239–1244. doi: 10.1001/archneurol.2011.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Wingo T.S., Cutler D.J., Wingo A.P., et al. Association of Early-onset Alzheimer disease with elevated low-density lipoprotein cholesterol levels and rare genetic Coding variants of APOB. JAMA Neurol. 2019;76:809–817. doi: 10.1001/jamaneurol.2019.0648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kuller L.H. Statins, lipids and dementia? J Clin Lipidol. 2021;15:18–21. doi: 10.1016/j.jacl.2020.12.011. [DOI] [PubMed] [Google Scholar]
- 150.Kostapanos M.S., Elisaf M.S. Statins and mortality: the untold story. Br J Clin Pharmacol. 2017;83:938–941. doi: 10.1111/bcp.13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mach F., Baigent C., Catapano A.L., et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. 2020;41:111–188. doi: 10.1093/eurheartj/ehz455. [DOI] [PubMed] [Google Scholar]
- 152.Di Pino A., DeFronzo R.A. Insulin resistance and atherosclerosis: implications for insulin-sensitizing agents. Endocr Rev. 2019;40:1447–1467. doi: 10.1210/er.2018-00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ormazabal V., Nair S., Elfeky O., Aguayo C., Salomon C., Zuñiga F.A. Association between insulin resistance and the development of cardiovascular disease. Cardiovasc Diabetol. 2018;17:122. doi: 10.1186/s12933-018-0762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nosalski R., McGinnigle E., Siedlinski M., Guzik T.J. Novel immune mechanisms in hypertension and cardiovascular risk. Curr Cardiovasc Risk Rep. 2017;11:12. doi: 10.1007/s12170-017-0537-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hollander W. Role of hypertension in atherosclerosis and cardiovascular disease. Am J Cardiol. 1976;38:786–800. doi: 10.1016/0002-9149(76)90357-x. [DOI] [PubMed] [Google Scholar]
- 156.Hurtubise J., McLellan K., Durr K., Onasanya O., Nwabuko D., Ndisang J.F. The different facets of dyslipidemia and hypertension in atherosclerosis. Curr Atheroscler Rep. 2016;18:82. doi: 10.1007/s11883-016-0632-z. [DOI] [PubMed] [Google Scholar]
- 157.Siasos G., Tsigkou V., Kokkou E., et al. Smoking and atherosclerosis: mechanisms of disease and new therapeutic approaches. Curr Med Chem. 2014;21:3936–3948. doi: 10.2174/092986732134141015161539. [DOI] [PubMed] [Google Scholar]
- 158.McEvoy J.W., Nasir K., DeFilippis A.P., et al. Relationship of cigarette smoking with inflammation and subclinical vascular disease: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:1002–1010. doi: 10.1161/ATVBAHA.114.304960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Dragoljevic D., Kraakman M.J., Nagareddy P.R., et al. Defective cholesterol metabolism in haematopoietic stem cells promotes monocyte-driven atherosclerosis in rheumatoid arthritis. Eur Heart J. 2018;39:2158–2167. doi: 10.1093/eurheartj/ehy119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jaiswal S., Natarajan P., Silver A.J., et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121. doi: 10.1056/NEJMoa1701719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lee M.K.S., Dragoljevic D., Bertuzzo Veiga C., Wang N., Yvan-Charvet L., Murphy A.J. Interplay between clonal hematopoiesis of indeterminate potential and metabolism. Trends Endocrinol Metab. 2020;31:525–535. doi: 10.1016/j.tem.2020.02.005. [DOI] [PubMed] [Google Scholar]
- 162.Hoermann G., Greiner G., Griesmacher A., Valent P. Clonal hematopoiesis of indeterminate potential: a multidisciplinary challenge in personalized hematology. J Pers Med. 2020;10 doi: 10.3390/jpm10030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sánchez-Cabo F., Fuster J.J. Clonal haematopoiesis and atherosclerosis: a chicken or egg question? Nat Rev Cardiol. 2021;18:463–464. doi: 10.1038/s41569-021-00554-z. [DOI] [PubMed] [Google Scholar]
- 164.Reiner R.C., Wiens K.E., Deshpande A., et al. Mapping geographical inequalities in childhood diarrhoeal morbidity and mortality in low-income and middle-income countries, 2000–17: analysis for the global burden of disease study 2017. Lancet North Am Ed. 2020;395:1779–1801. doi: 10.1016/S0140-6736(20)30114-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Thålin C., Hisada Y., Lundström S., Mackman N., Wallén H. Neutrophil Extracellular Traps. Arterioscler Thromb Vasc Biol. 2019;39:1724–1738. doi: 10.1161/ATVBAHA.119.312463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Josefs T., Barrett T.J., Brown E.J., et al. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight. 2020;5 doi: 10.1172/jci.insight.134796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Moschonas I.C., Tselepis A.D. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis. 2019;288:9–16. doi: 10.1016/j.atherosclerosis.2019.06.919. [DOI] [PubMed] [Google Scholar]
- 168.Lind P.M., van Bavel B., Salihovic S., Lind L. Circulating levels of persistent organic pollutants (POPs) and carotid atherosclerosis in the elderly. Environ Health Perspect. 2012;120:38–43. doi: 10.1289/ehp.1103563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Stols-Gonçalves D., Hovingh G.K., Nieuwdorp M., Holleboom A.G. NAFLD and atherosclerosis: two sides of the same dysmetabolic coin? Trends Endocrinol Metab. 2019;30:891–902. doi: 10.1016/j.tem.2019.08.008. [DOI] [PubMed] [Google Scholar]
- 170.Nichols G.A., Philip S., Reynolds K., Granowitz C.B., Fazio S. Increased residual cardiovascular risk in patients with diabetes and high versus normal triglycerides despite statin-controlled LDL cholesterol. Diabetes Obes Metab. 2019;21:366–371. doi: 10.1111/dom.13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Jorgensen A.B., Frikke-Schmidt R., Nordestgaard B.G., Tybjaerg-Hansen A. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41. doi: 10.1056/NEJMoa1308027. [DOI] [PubMed] [Google Scholar]
- 172.Ivanova E.A., Myasoedova V.A., Melnichenko A.A., Grechko A.V., Orekhov A.N. Small dense low-density lipoprotein as biomarker for atherosclerotic diseases. Oxid Med Cell Longev. 2017;2017 doi: 10.1155/2017/1273042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ference B.A., Kastelein J.J.P., Ray K.K., et al. Association of triglyceride-lowering LPL variants and LDL-C–lowering LDLR variants with risk of coronary heart disease. JAMA. 2019;321:364–373. doi: 10.1001/jama.2018.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kathiresan S., Otvos J.D., Sullivan L.M., et al. Increased small low-density lipoprotein particle number. Circulation. 2006;113:20–29. doi: 10.1161/CIRCULATIONAHA.105.567107. [DOI] [PubMed] [Google Scholar]
- 175.Woodman R.J., Watts G.F., Playford D.A., Best J.D., Chan D.C. Oxidized LDL and small LDL particle size are independently predictive of a selective defect in microcirculatory endothelial function in type 2 diabetes. Diabetes Obes Metab. 2005;7:612–617. doi: 10.1111/j.1463-1326.2005.00478.x. [DOI] [PubMed] [Google Scholar]
- 176.Chan D.C., Watts G.F. Apolipoproteins as markers and managers of coronary risk. QJM Int J Med. 2006;99:277–287. doi: 10.1093/qjmed/hcl027. [DOI] [PubMed] [Google Scholar]
- 177.Fulcher J., O'Connell R., Voysey M., et al. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet. 2015;385:1397–1405. doi: 10.1016/S0140-6736(14)61368-4. [DOI] [PubMed] [Google Scholar]
- 178.Olsson A.G., Angelin B., Assmann G., et al. Can LDL cholesterol be too low? Possible risks of extremely low levels. J Intern Med. 2017;281:534–553. doi: 10.1111/joim.12614. [DOI] [PubMed] [Google Scholar]
- 179.Karagiannis A.D., Mehta A., Dhindsa D.S., et al. How low is safe? The frontier of very low (<30mg/dL) LDL cholesterol. Eur Heart J. 2021 doi: 10.1093/eurheartj/ehaa1080. [DOI] [PubMed] [Google Scholar]
- 180.Ference B.A., Ginsberg H.N., Graham I., et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European atherosclerosis society consensus panel. Eur Heart J. 2017;38:2459–2472. doi: 10.1093/eurheartj/ehx144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Silverman M.G., Ference B.A., Im K., et al. Association between lowering LDL-C and cardiovascular risk reduction among different therapeutic interventions: a systematic review and meta-analysis. JAMA. 2016;316:1289–1297. doi: 10.1001/jama.2016.13985. [DOI] [PubMed] [Google Scholar]
- 182.Sabatine M.S., Wiviott S.D., Im K., Murphy S.A., Giugliano R.P. Efficacy and safety of further lowering of low-density lipoprotein cholesterol in patients starting with very low levels: a meta-analysis. JAMA Cardiol. 2018;3:823–828. doi: 10.1001/jamacardio.2018.2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mora S., Ridker P.M. Justification for the use of statins in primary prevention: an intervention trial evaluating rosuvastatin (JUPITER)—can C-reactive protein be used to target statin therapy in primary prevention? Am J Cardiol. 2006;97:33–41. doi: 10.1016/j.amjcard.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 184.Ridker P.M., Danielson E., Fonseca F.A., et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 185.Sabatine M.S., Giugliano R.P., Keech A.C., et al. Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. doi: 10.1056/NEJMoa1615664. [DOI] [PubMed] [Google Scholar]
- 186.Schwartz G.G., Steg P.G., Szarek M., et al. Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097–2107. doi: 10.1056/NEJMoa1801174. [DOI] [PubMed] [Google Scholar]
- 187.Mihaylova B., Emberson J., Blackwell L., et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Gencer B., Mach F., Guo J., et al. Cognition after lowering LDL-cholesterol with evolocumab. J Am Coll Cardiol. 2020;75:2283–2293. doi: 10.1016/j.jacc.2020.03.039. [DOI] [PubMed] [Google Scholar]
- 189.Ference B.A., Yoo W., Alesh I., et al. Effect of long-term exposure to lower low-density lipoprotein cholesterol beginning early in life on the risk of coronary heart disease. J Am Coll Cardiol. 2012;60:2631–2639. doi: 10.1016/j.jacc.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 190.Leibowitz M., Cohen-Stavi C., Basu S., Balicer R.D. Targeting LDL cholesterol: beyond absolute goals toward personalized risk. Curr Cardiol Rep. 2017;19:52. doi: 10.1007/s11886-017-0858-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Grundy S.M., Stone N.J., Bailey A.L., et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: executive summary: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. J Am Coll Cardiol. 2019;73:3168–3209. doi: 10.1016/j.jacc.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 192.Näslund U., Ng N., Lundgren A., et al. Visualization of asymptomatic atherosclerotic disease for optimum cardiovascular prevention (VIPVIZA): a pragmatic, open-label, randomised controlled trial. Lancet. 2019;393:133–142. doi: 10.1016/S0140-6736(18)32818-6. [DOI] [PubMed] [Google Scholar]
- 193.Sachais B.S., Shaz B.H. Apheresis to mitigate atherosclerotic vascular disease. Am J Hypertens. 2018;31:945–949. doi: 10.1093/ajh/hpy068. [DOI] [PubMed] [Google Scholar]
- 194.Zhao Z., Du S., Shen S., et al. Comparative efficacy and safety of lipid-lowering agents in patients with hypercholesterolemia: a frequentist network meta-analysis. Medicine. 2019;98:e14400. doi: 10.1097/MD.0000000000014400. (Baltimore) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Jacobson T.A. NLA task force on statin safety-2014 update. J Clin Lipidol. 2014;8:S1–S4. doi: 10.1016/j.jacl.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 196.May P., Woldt E., Matz R.L., Boucher P. The LDL receptor-related protein (LRP) family: an old family of proteins with new physiological functions. Ann Med. 2007;39:219–228. doi: 10.1080/07853890701214881. [DOI] [PubMed] [Google Scholar]
- 197.Lotta L.A., Sharp S.J., Burgess S., et al. Association between low-density lipoprotein cholesterol–lowering genetic variants and risk of type 2 diabetes: a meta-analysis. JAMA. 2016;316:1383–1391. doi: 10.1001/jama.2016.14568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Brown M.S., Goldstein J.L. Lowering LDL-not only how low, but how long? Science. 2006;311:1721. doi: 10.1126/science.1125884. [DOI] [PubMed] [Google Scholar]
- 199.Brown M.S., Goldstein J.L. Heart attacks: gone with the century? Science. 1996;272:629. doi: 10.1126/science.272.5262.629. [DOI] [PubMed] [Google Scholar]
- 200.Fisher E.A. Regression of atherosclerosis: the journey from the liver to the plaque and back. Arterioscler Thromb Vasc Biol. 2016;36:226–235. doi: 10.1161/ATVBAHA.115.301926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Nicholls S.J., Tuzcu E.M., Sipahi I., et al. Statins, high-density lipoprotein cholesterol, and regression of coronary atherosclerosis. JAMA. 2007;297:499–508. doi: 10.1001/jama.297.5.499. [DOI] [PubMed] [Google Scholar]
- 202.Nissen S.E., Tuzcu E.M., Schoenhagen P., et al. Effect of intensive compared with moderate lipid-lowering therapy on progression of coronary atherosclerosisa randomized controlled trial. JAMA. 2004;291:1071–1080. doi: 10.1001/jama.291.9.1071. [DOI] [PubMed] [Google Scholar]
- 203.Nicholls S.J., Puri R., Anderson T., et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316:2373–2384. doi: 10.1001/jama.2016.16951. [DOI] [PubMed] [Google Scholar]
- 204.Räber L., Ueki Y., Otsuka T., et al. Effect of alirocumab added to high-intensity statin therapy on coronary atherosclerosis in patients with acute myocardial infarction: the PACMAN-AMI randomized clinical trial. JAMA. 2022;327:1771–1781. doi: 10.1001/jama.2022.5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gimbrone M.A., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–636. doi: 10.1161/CIRCRESAHA.115.306301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Alexander Y., Osto E., Schmidt-Trucksäss A., et al. Endothelial function in cardiovascular medicine: a consensus paper of the European society of cardiology working groups on atherosclerosis and vascular biology, aorta and peripheral vascular diseases, coronary pathophysiology and microcirculation, and thrombosis. Cardiovasc Res. 2021;117:29–42. doi: 10.1093/cvr/cvaa085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Fredman G., Tabas I. Boosting inflammation resolution in atherosclerosis: the next frontier for therapy. Am J Pathol. 2017;187:1211–1221. doi: 10.1016/j.ajpath.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Shapiro Michael D., Bhatt Deepak L. Cholesterol-years” for ASCVD risk prediction and treatment∗. J Am Coll Cardiol. 2020;76:1517–1520. doi: 10.1016/j.jacc.2020.08.004. [DOI] [PubMed] [Google Scholar]
- 209.Horton J.D., Cohen J.C., Hobbs H.H. PCSK9: a convertase that coordinates LDL catabolism. J Lipid Res. 2009;50:S172–S177. doi: 10.1194/jlr.R800091-JLR200. Suppl. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Domanski M.J., Tian X., Wu C.O., et al. Time course of LDL cholesterol exposure and cardiovascular disease event risk. J Am Coll Cardiol. 2020;76:1507–1516. doi: 10.1016/j.jacc.2020.07.059. [DOI] [PubMed] [Google Scholar]
- 211.Borén J., Chapman M.J., Krauss R.M., et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European atherosclerosis society consensus panel. Eur Heart J. 2020;41:2313–2330. doi: 10.1093/eurheartj/ehz962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Packard C., Chapman M.J., Sibartie M., Laufs U., Masana L. Intensive low-density lipoprotein cholesterol lowering in cardiovascular disease prevention: opportunities and challenges. Heart. 2021;107:1369–1375. doi: 10.1136/heartjnl-2020-318760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Steinberg D., Grundy S.M. The case for treating hypercholesterolemia at an earlier age. J Am Coll Cardiol. 2012;60:2640–2642. doi: 10.1016/j.jacc.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 214.Smith G.C., Pell J.P. Parachute use to prevent death and major trauma related to gravitational challenge: systematic review of randomised controlled trials. BMJ. 2003;327:1459–1461. doi: 10.1136/bmj.327.7429.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Boekholdt S.M., Hovingh G.K., Mora S., et al. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta-analysis of statin trials. J Am Coll Cardiol. 2014;64:485–494. doi: 10.1016/j.jacc.2014.02.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Giugliano R.P., Pedersen T.R., Park J.-.G., et al. Clinical efficacy and safety of achieving very low LDL-cholesterol concentrations with the PCSK9 inhibitor evolocumab: a prespecified secondary analysis of the Fourier trial. Lancet North Am Ed. 2017;390:1962–1971. doi: 10.1016/S0140-6736(17)32290-0. [DOI] [PubMed] [Google Scholar]
- 217.Fryar C.D., Carroll M.D., Afful J. Prevalence of overweight, obesity, and severe obesity among adults aged 20 and over: United States, 1960–1962 through 2017–2018. NCHS Health E-Stats. 2020 [Google Scholar]
- 218.Creamer M.R., Wang T.W., Babb S., et al. Tobacco product use and cessation indicators among adults - United States, 2018. MMWR Morb Mortal Wkly Rep. 2019;68:1013–1019. doi: 10.15585/mmwr.mm6845a2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Orringer C.E., Blaha M.J., Blankstein R., et al. The national lipid association scientific statement on coronary artery calcium scoring to guide preventive strategies for ASCVD risk reduction. J Clin Lipidol. 2021;15:33–60. doi: 10.1016/j.jacl.2020.12.005. [DOI] [PubMed] [Google Scholar]
- 220.Gabriel F.S., Gonçalves L.F.G., Melo E.V., et al. Atherosclerotic plaque in patients with zero calcium score at coronary computed tomography angiography. Arq Bras Cardiol. 2018;110:420–427. doi: 10.5935/abc.20180063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang X., Le E.P.V., Rajani N.K., et al. A zero coronary artery calcium score in patients with stable chest pain is associated with a good prognosis, despite risk of non-calcified plaques. Open Heart. 2019;6 doi: 10.1136/openhrt-2018-000945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Goldstein J.A., Demetriou D., Grines C.L., Pica M., Shoukfeh M., O'Neill W.W. Multiple complex coronary plaques in patients with acute myocardial infarction. N Engl J Med. 2000;343:915–922. doi: 10.1056/NEJM200009283431303. [DOI] [PubMed] [Google Scholar]
- 223.Strauss H.W., Nakahara T., Narula N., Narula J. Vascular calcification: the evolving relationship of vascular calcification to major acute coronary events. J Nucl Med. 2019;60:1207. doi: 10.2967/jnumed.119.230276. [DOI] [PubMed] [Google Scholar]
- 224.Bauer R.W., Thilo C., Chiaramida S.A., Vogl T.J., Costello P., Schoepf U.J. Noncalcified atherosclerotic plaque burden at coronary CT angiography: a better predictor of ischemia at stress myocardial perfusion imaging than calcium score and stenosis severity. Am J Roentgenol. 2009;193:410–418. doi: 10.2214/AJR.08.1277. [DOI] [PubMed] [Google Scholar]
- 225.Naghavi M., Libby P., Falk E., et al. From vulnerable plaque to vulnerable patient. Circulation. 2003;108:1664–1672. doi: 10.1161/01.CIR.0000087480.94275.97. [DOI] [PubMed] [Google Scholar]
- 226.Mandrola J., Foy A. The case against coronary artery calcium scoring for cardiovascular disease risk assessment. Am Fam Physician. 2019;100:734–735. [PubMed] [Google Scholar]
- 227.Gulati R., Behfar A., Narula J., et al. Acute myocardial infarction in young individuals. Mayo Clin Proc. 2020;95:136–156. doi: 10.1016/j.mayocp.2019.05.001. [DOI] [PubMed] [Google Scholar]
- 228.Egred M., Viswanathan G., Davis G.K. Myocardial infarction in young adults. Postgrad Med J. 2005;81:741–745. doi: 10.1136/pgmj.2004.027532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Wu W.Y., Berman A.N., Biery D.W., Blankstein R. Recent trends in acute myocardial infarction among the young. Curr Opin Cardiol. 2020;35:524–530. doi: 10.1097/HCO.0000000000000781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Giugliano R.P., Mach F., Zavitz K., et al. Cognitive function in a randomized trial of evolocumab. N Engl J Med. 2017;377:633–643. doi: 10.1056/NEJMoa1701131. [DOI] [PubMed] [Google Scholar]
- 231.Jukema J.W., Zijlstra L.E., Bhatt D.L., et al. Effect of alirocumab on stroke in odyssey outcomes. Circulation. 2019;140:2054–2062. doi: 10.1161/CIRCULATIONAHA.119.043826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Florido R., Elander A., Blumenthal R.S., Martin S.S. Statins and incident diabetes: can risk outweigh benefit? Curr Cardiovasc Risk Rep. 2015;9:14. [Google Scholar]
- 233.Strilchuk L., Fogacci F., Cicero A.F. Safety and tolerability of injectable lipid-lowering drugs: an update of clinical data. Expert Opin Drug Saf. 2019;18:611–621. doi: 10.1080/14740338.2019.1620730. [DOI] [PubMed] [Google Scholar]
- 234.Wood F.A., Howard J.P., Finegold J.A., et al. N-of-1 trial of a statin, placebo, or no treatment to assess side effects. N Engl J Med. 2020;383:2182–2184. doi: 10.1056/NEJMc2031173. [DOI] [PubMed] [Google Scholar]
- 235.Association American Heart Cardiovascular disease: a costly burden for America. Projections through 2035. https://www.heart.org/-/media/files/get-involved/advocacy/burden-report-consumer-report.pdf
- 236.Grover S.A., Ho V., Lavoie F., Coupal L., Zowall H., Pilote L. The importance of indirect costs in primary cardiovascular disease prevention: can we save lives and money with statins? Arch Intern Med. 2003;163:333–339. doi: 10.1001/archinte.163.3.333. [DOI] [PubMed] [Google Scholar]
- 237.Wong W. Economic burden of Alzheimer disease and managed care considerations. Am J Manag Care. 2020;26:S177–s183. doi: 10.37765/ajmc.2020.88482. [DOI] [PubMed] [Google Scholar]
- 238.Graham G. Disparities in cardiovascular disease risk in the United States. Curr Cardiol Rev. 2015;11:238–245. doi: 10.2174/1573403X11666141122220003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Shah P., Glueck C.J., Jetty V., et al. Pharmacoeconomics of PCSK9 inhibitors in 103 hypercholesterolemic patients referred for diagnosis and treatment to a cholesterol treatment center. Lipids Health Dis. 2016;15:132. doi: 10.1186/s12944-016-0302-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Ademi Z., Norman R., Pang J., et al. Health economic evaluation of screening and treating children with familial hypercholesterolemia early in life: many happy returns on investment? Atherosclerosis. 2020;304:1–8. doi: 10.1016/j.atherosclerosis.2020.05.007. [DOI] [PubMed] [Google Scholar]
- 241.Kohli-Lynch C.N., Bellows B.K., Thanassoulis G., et al. Cost-effectiveness of low-density lipoprotein cholesterol level–guided statin treatment in patients with borderline cardiovascular risk. JAMA Cardiol. 2019;4:969–977. doi: 10.1001/jamacardio.2019.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Heidenreich P.A., Clarke S.L., Maron D.J. Time to Relax the 40-year age threshold for pharmacologic cholesterol lowering*. J Am Coll Cardiol. 2021;78:1965–1967. doi: 10.1016/j.jacc.2021.08.072. [DOI] [PubMed] [Google Scholar]
- 243.Pandya A., Sy S., Cho S., Weinstein M.C., Gaziano T.A. Cost-effectiveness of 10-year risk thresholds for initiation of statin therapy for primary prevention of cardiovascular disease. JAMA. 2015;314:142–150. doi: 10.1001/jama.2015.6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lazar L.D., Pletcher M.J., Coxson P.G., Bibbins-Domingo K., Goldman L. Cost-effectiveness of statin therapy for primary prevention in a low-cost statin era. Circulation. 2011;124:146–153. doi: 10.1161/CIRCULATIONAHA.110.986349. [DOI] [PubMed] [Google Scholar]
- 245.Heller D.J., Coxson P.G., Penko J., et al. Evaluating the impact and cost-effectiveness of statin use guidelines for primary prevention of coronary heart disease and stroke. Circulation. 2017;136:1087–1098. doi: 10.1161/CIRCULATIONAHA.117.027067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Li Y., Deng S., Liu B., et al. The effects of lipid-lowering therapy on coronary plaque regression: a systematic review and meta-analysis. Sci Rep. 2021;11:7999. doi: 10.1038/s41598-021-87528-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Nicholls S.J., Ballantyne C.M., Barter P.J., et al. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365:2078–2087. doi: 10.1056/NEJMoa1110874. [DOI] [PubMed] [Google Scholar]
- 248.Di Giovanni G., Nicholls S.J. Intensive lipid lowering agents and coronary atherosclerosis: insights from intravascular imaging. Am J Prev Cardiol. 2022;11 doi: 10.1016/j.ajpc.2022.100366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Juliani F.C., Miname M.H., Castelo M.H.C.G., et al. Efficacy and safety of early lipid lowering treatment in children with familial hypercholesterolemia. J Am Coll Cardiol. 2022;79:1472. 1472. [Google Scholar]
- 250.Luirink I.K., Wiegman A., Kusters D.M., et al. 20-Year follow-up of statins in children with familial hypercholesterolemia. N Engl J Med. 2019;381:1547–1556. doi: 10.1056/NEJMoa1816454. [DOI] [PubMed] [Google Scholar]
- 251.Juliani Fabiana C., Miname Marcio H., Castelo Maria Helane Costa G., et al. Efficacy and safety of early lipid lowering treatment in children with familial hypercholesterolemia. J Am Coll Cardiol. 2022;79:1472. 1472. [Google Scholar]
- 252.Adiposopathy Bays H. "sick fat," Ockham's razor, and resolution of the obesity paradox. Curr Atheroscler Rep. 2014;16:409. doi: 10.1007/s11883-014-0409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Vega G.L., Wang J., Grundy S.M. Utility of metabolic syndrome as a risk enhancing factor in decision of statin use. J Clin Lipidol. 2021;15:255–265. doi: 10.1016/j.jacl.2021.01.012. [DOI] [PubMed] [Google Scholar]
- 254.Rossello X., Raposeiras-Roubin S., Oliva B., et al. Glycated hemoglobin and subclinical atherosclerosis in people without diabetes. J Am Coll Cardiol. 2021;77:2777–2791. doi: 10.1016/j.jacc.2021.03.335. [DOI] [PubMed] [Google Scholar]
- 255.Whelton P.K., Carey R.M., Aronow W.S., et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Hypertension. 2018;71:e13–e115. doi: 10.1161/HYP.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 256.Del Pinto R., Ferri C. Hypertension management at older age: an update. High Blood Press Cardiovasc Prev. 2019;26:27–36. doi: 10.1007/s40292-018-0290-z. [DOI] [PubMed] [Google Scholar]
- 257.Wald N., Wald D., Kellermann A.L. When guidelines cause hypertension. Am J Med. 2018;131:1402–1404. doi: 10.1016/j.amjmed.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 258.Yamada M.H., Fujihara K., Kodama S., et al. Associations of systolic blood pressure and diastolic blood pressure with the incidence of coronary artery disease or cerebrovascular disease according to glucose status. Diabetes Care. 2021 doi: 10.2337/dc20-2252. [DOI] [PubMed] [Google Scholar]
- 259.Tsimikas S. A test in context: lipoprotein(a) J Am Coll Cardiol. 2017;69:692–711. doi: 10.1016/j.jacc.2016.11.042. [DOI] [PubMed] [Google Scholar]
- 260.Liu T., Yoon W.S., Lee S.R. Recent updates of lipoprotein(a) and cardiovascular disease. Chonnam Med J. 2021;57:36–43. doi: 10.4068/cmj.2021.57.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Littmann K., Wodaje T., Alvarsson M., et al. The association of lipozprotein(a) plasma levels with prevalence of cardiovascular disease and metabolic control status in patients with type 1 diabetes. Diabetes Care. 2020;43:1851. doi: 10.2337/dc19-1398. [DOI] [PubMed] [Google Scholar]
- 262.Nordestgaard B.G., Chapman M.J., Ray K., et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.McNeal C.J. Lipoprotein(a): its relevance to the pediatric population. J Clin Lipidol. 2015;9:S57–S66. doi: 10.1016/j.jacl.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 264.Momiyama Y., Ohmori R., Fayad Z.A., et al. Associations between serum lipoprotein(a) levels and the severity of coronary and aortic atherosclerosis. Atherosclerosis. 2012;222:241–244. doi: 10.1016/j.atherosclerosis.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 265.Prendergast C.J., Kelley J.C., Linton E.F., Linton M.F. Lp(a) in childhood. Curr Cardiovasc Risk Rep. 2017;11:26. [Google Scholar]
- 266.Schmidt K., Noureen A., Kronenberg F., Utermann G. Structure, function, and genetics of lipoprotein (a) J Lipid Res. 2016;57:1339–1359. doi: 10.1194/jlr.R067314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Page M.M., Watts G.F. Contemporary perspectives on the genetics and clinical use of lipoprotein(a) in preventive cardiology. Curr Opin Cardiol. 2021;36:272–280. doi: 10.1097/HCO.0000000000000842. [DOI] [PubMed] [Google Scholar]
- 268.Borrelli M.J., Youssef A., Boffa M.B., Koschinsky M.L. New frontiers in Lp(a)-targeted therapies. Trends Pharmacol Sci. 2019;40:212–225. doi: 10.1016/j.tips.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 269.Lüscher T.F. The next chapter of prevention: from LDL-cholesterol to lipoprotein(a) and triglycerides. Eur Heart J. 2020;41:2227–2230. doi: 10.1093/eurheartj/ehaa552. [DOI] [PubMed] [Google Scholar]
- 270.Teber S., Deda G., Akar N., Soylu K. Lipoprotein (a) levels in childhood arterial ischemic stroke. Clin Appl Thromb Hemost. 2010;16:214–217. doi: 10.1177/1076029609334124. [DOI] [PubMed] [Google Scholar]
- 271.Lui D.T.W., Lee A.C.H., Tan K.C.B. Management of familial hypercholesterolemia: current status and future perspectives. J Endocr Soc. 2020;5 doi: 10.1210/jendso/bvaa122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Hu P., Dharmayat K.I., Stevens C.A.T., et al. Prevalence of familial hypercholesterolemia among the general population and patients with atherosclerotic cardiovascular disease. Circulation. 2020;141:1742–1759. doi: 10.1161/CIRCULATIONAHA.119.044795. [DOI] [PubMed] [Google Scholar]
- 273.Lawler P.R., Bhatt D.L., Godoy L.C., et al. Targeting cardiovascular inflammation: next steps in clinical translation. Eur Heart J. 2021;42:113–131. doi: 10.1093/eurheartj/ehaa099. [DOI] [PubMed] [Google Scholar]
- 274.Connelly M.A., Otvos J.D., Shalaurova I., Playford M.P., Mehta N.N. GlycA, a novel biomarker of systemic inflammation and cardiovascular disease risk. J Transl Med. 2017;15:219. doi: 10.1186/s12967-017-1321-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Ballout R.A., Remaley A.T. GlycA: a new biomarker for systemic inflammation and cardiovascular disease (CVD) risk assessment. J Lab Precis Med. 2020;5 doi: 10.21037/jlpm.2020.03.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Fashanu O.E., Oyenuga A.O., Zhao D., et al. GlycA, a novel inflammatory marker and its association with peripheral arterial disease and carotid plaque: the multi-ethnic study of atherosclerosis. Angiology. 2019;70:737–746. doi: 10.1177/0003319719845185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Richardson T.G., Sanderson E., Palmer T.M., et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable Mendelian randomisation analysis. PLoS Med. 2020;17 doi: 10.1371/journal.pmed.1003062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Shaik A., Rosenson R.S. Genetics of triglyceride-rich lipoproteins guide identification of pharmacotherapy for cardiovascular risk reduction. Cardiovasc Drugs Ther. 2021;35:677–690. doi: 10.1007/s10557-021-07168-0. [DOI] [PubMed] [Google Scholar]
- 279.Toth P.P. Triglyceride-rich lipoproteins as a causal factor for cardiovascular disease. Vasc Health Risk Manag. 2016;12:171–183. doi: 10.2147/VHRM.S104369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Kalra D., Vijayaraghavan K., Sikand G., et al. Prevention of atherosclerotic cardiovascular disease in South Asians in the US: a clinical perspective from the national lipid association. J Clin Lipidol. 2021 doi: 10.1016/j.jacl.2021.03.007. [DOI] [PubMed] [Google Scholar]
- 281.Takaeko Y., Maruhashi T., Kajikawa M., et al. Lower triglyceride levels are associated with better endothelial function. J Clin Lipidol. 2021 doi: 10.1016/j.jacl.2021.04.004. [DOI] [PubMed] [Google Scholar]
- 282.Wang N., Fulcher J., Abeysuriya N., et al. Intensive LDL cholesterol-lowering treatment beyond current recommendations for the prevention of major vascular events: a systematic review and meta-analysis of randomised trials including 327 037 participants. Lancet Diabetes Endocrinol. 2020;8:36–49. doi: 10.1016/S2213-8587(19)30388-2. [DOI] [PubMed] [Google Scholar]
- 283.Davidson M.H. Triglyceride-rich lipoprotein cholesterol (TRL-C): the ugly stepsister of LDL-C. Eur Heart J. 2018;39:620–622. doi: 10.1093/eurheartj/ehx741. [DOI] [PubMed] [Google Scholar]
- 284.Sinnaeve P.R., Schwartz G.G., Wojdyla D.M., et al. Effect of alirocumab on cardiovascular outcomes after acute coronary syndromes according to age: an odyssey outcomes trial analysis. Eur Heart J. 2019;41:2248–2258. doi: 10.1093/eurheartj/ehz809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Ridker P.M., Lonn E., Paynter N.P., Glynn R., Yusuf S. Primary prevention with statin therapy in the elderly: new meta-analyses from the contemporary Jupiter and Hope-3 randomized trials. Circulation. 2017;135:1979–1981. doi: 10.1161/CIRCULATIONAHA.117.028271. [DOI] [PubMed] [Google Scholar]
- 286.Raal F.J., Mohamed F. Never too old to benefit from lipid-lowering treatment. Lancet North Am Ed. 2020;396:1608–1609. doi: 10.1016/S0140-6736(20)32333-3. [DOI] [PubMed] [Google Scholar]
- 287.Gencer B., Marston N.A., Im K., et al. Efficacy and safety of lowering LDL cholesterol in older patients: a systematic review and meta-analysis of randomised controlled trials. Lancet North Am Ed. 2020;396:1637–1643. doi: 10.1016/S0140-6736(20)32332-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Mortensen M.B., Nordestgaard B.G. Elevated LDL cholesterol and increased risk of myocardial infarction and atherosclerotic cardiovascular disease in individuals aged 70–100 years: a contemporary primary prevention cohort. Lancet North Am Ed. 2020;396:1644–1652. doi: 10.1016/S0140-6736(20)32233-9. [DOI] [PubMed] [Google Scholar]
- 289.Cho Y., Jeong Y., Seo D.H., et al. Use of statin for the primary prevention of cardiovascular outcomes in elderly patients: a propensity-matched cohort study. Atherosclerosis. 2021;328:92–99. doi: 10.1016/j.atherosclerosis.2021.05.022. [DOI] [PubMed] [Google Scholar]
- 290.Amaral J.F., Borsato D.M.A., Freitas I.M.G., Toschi-Dias E., Martinez D.G., Laterza M.C. Autonomic and Vascular control in prehypertensive subjects with a family history of arterial hypertension. Arq Bras Cardiol. 2018;110:166–174. doi: 10.5935/abc.20180006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Rissanen A.M. Familial occurrence of coronary heart disease: effect of age at diagnosis. Am J Cardiol. 1979;44:60–66. doi: 10.1016/0002-9149(79)90251-0. [DOI] [PubMed] [Google Scholar]
- 292.Leander K., Hallqvist J., Reuterwall C., Ahlbom A., de Faire U. Family history of coronary heart disease, a strong risk factor for myocardial infarction interacting with other cardiovascular risk factors: results from the Stockholm heart epidemiology program (SHEEP) Epidemiology. 2001;12:215–221. doi: 10.1097/00001648-200103000-00014. [DOI] [PubMed] [Google Scholar]
- 293.Panagiotakos D.B. Family history of coronary heart disease as a predictor of the incidence and progression of coronary artery calcification. Atherosclerosis. 2014;233:30–31. doi: 10.1016/j.atherosclerosis.2013.07.059. [DOI] [PubMed] [Google Scholar]
- 294.Pandey A.K., Blaha M.J., Sharma K., et al. Family history of coronary heart disease and the incidence and progression of coronary artery calcification: multi-ethnic study of atherosclerosis (MESA) Atherosclerosis. 2014;232:369–376. doi: 10.1016/j.atherosclerosis.2013.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Park G.M., Cho Y.R., Lee S.W., et al. Family history of diabetes and the risk of subclinical atherosclerosis. Diabetes Metab. 2016;42:170–177. doi: 10.1016/j.diabet.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 296.Solini A., Santini E., Passaro A., Madec S., Ferrannini E. Family history of hypertension, anthropometric parameters and markers of early atherosclerosis in young healthy individuals. J Hum Hypertens. 2009;23:801–807. doi: 10.1038/jhh.2009.26. [DOI] [PubMed] [Google Scholar]
- 297.Jung C.H., Lee M.J., Hwang J.Y., et al. Association of metabolically healthy obesity with subclinical coronary atherosclerosis in a Korean population. Obesity. 2014;22:2613–2620. doi: 10.1002/oby.20883. [DOI] [PubMed] [Google Scholar]
- 298.Umashanker D., Shukla A.P., Saunders K.H., Aronne L.J. Is obesity the new hypertension? Parallels in the evolution of obesity and hypertension as recognized disease states. Curr Atheroscler Rep. 2017;19:35. doi: 10.1007/s11883-017-0671-0. [DOI] [PubMed] [Google Scholar]
- 299.Bays H.E., Toth P.P., Kris-Etherton P.M., et al. Obesity, adiposity, and dyslipidemia: a consensus statement from the national lipid association. J Clin Lipidol. 2013;7:304–383. doi: 10.1016/j.jacl.2013.04.001. [DOI] [PubMed] [Google Scholar]
- 300.Powell-Wiley T.M., Poirier P., Burke L.E., et al. Obesity and cardiovascular disease: a scientific statement from the American heart association. Circulation. 2021;143:e984–e1010. doi: 10.1161/CIR.0000000000000973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Choi H.M., Doss H.M., Kim K.S. Multifaceted physiological roles of adiponectin in inflammation and diseases. Int J Mol Sci. 2020;21 doi: 10.3390/ijms21041219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Ohtake T., Kobayashi S. Chronic kidney disease and atherosclerosis: an important implication of carotid intima-media thickness. J Atheroscler Thromb. 2021;28:471–473. doi: 10.5551/jat.ED146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Pang Y., Sang Y., Ballew S.H., et al. Carotid intima-media thickness and incident ESRD: the atherosclerosis risk in communities (ARIC) study. Clin J Am Soc Nephrol. 2016;11:1197–1205. doi: 10.2215/CJN.11951115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Düsing P., Zietzer A., Goody P.R., et al. Vascular pathologies in chronic kidney disease: pathophysiological mechanisms and novel therapeutic approaches. J Mol Med (Berl) 2021;99:335–348. doi: 10.1007/s00109-021-02037-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Kon V., Yang H., Fazio S. Residual cardiovascular risk in chronic kidney disease: role of high-density lipoprotein. Arch Med Res. 2015;46:379–391. doi: 10.1016/j.arcmed.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Bano A., Chaker L., Mattace-Raso F.U.S., et al. Thyroid function and the risk of atherosclerotic cardiovascular morbidity and mortality. Circ Res. 2017;121:1392–1400. doi: 10.1161/CIRCRESAHA.117.311603. [DOI] [PubMed] [Google Scholar]
- 307.Kurup R., Galougahi K.K., Figtree G., Misra A., Patel S. The role of colchicine in atherosclerotic cardiovascular disease. Heart Lung Circ. 2021;30:795–806. doi: 10.1016/j.hlc.2020.11.010. [DOI] [PubMed] [Google Scholar]
- 308.Lévy P., Pépin J.L., Arnaud C., Baguet J.P., Dematteis M., Mach F. Obstructive sleep apnea and atherosclerosis. Prog Cardiovasc Dis. 2009;51:400–410. doi: 10.1016/j.pcad.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 309.Almendros I., Farré N. Obstructive sleep apnea and atherosclerosis: both the gut microbiome and hypercapnia matter. Am J Respir Cell Mol Biol. 2017;57:501–503. doi: 10.1165/rcmb.2017-0253ED. [DOI] [PubMed] [Google Scholar]
- 310.Neary N.M., Booker O.J., Abel B.S., et al. Hypercortisolism is associated with increased coronary arterial atherosclerosis: analysis of noninvasive coronary angiography using multidetector computerized tomography. J Clin Endocrinol Metab. 2013;98:2045–2052. doi: 10.1210/jc.2012-3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Nashel D.J. Is atherosclerosis a complication of long-term corticosteroid treatment? Am J Med. 1986;80:925–929. doi: 10.1016/0002-9343(86)90639-x. [DOI] [PubMed] [Google Scholar]
- 312.Achar S., Rostamian A., Narayan S.M. Cardiac and metabolic effects of anabolic-androgenic steroid abuse on lipids, blood pressure, left ventricular dimensions, and rhythm. Am J Cardiol. 2010;106:893–901. doi: 10.1016/j.amjcard.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Kopin L., Lowenstein C. Dyslipidemia. Ann Intern Med. 2017;167:ITC81–ITC96. doi: 10.7326/AITC201712050. [DOI] [PubMed] [Google Scholar]
- 314.Herink M., Ito M.K., et al. In: Endotext. Feingold K.R., Anawalt B., Boyce A., et al., editors. MDText.com, Inc; South Dartmouth (MA): 2000. Medication induced changes in lipid and lipoproteins. editors. Copyright © 2000-2021, MDText.com, Inc. [PubMed] [Google Scholar]
- 315.Messner B., Bernhard D. Smoking and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2014;34:509–515. doi: 10.1161/ATVBAHA.113.300156. [DOI] [PubMed] [Google Scholar]
- 316.Ambrose J.A., Barua R.S. The pathophysiology of cigarette smoking and cardiovascular disease: an update. J Am Coll Cardiol. 2004;43:1731–1737. doi: 10.1016/j.jacc.2003.12.047. [DOI] [PubMed] [Google Scholar]
- 317.Mehta J.L. Marijuana and coronary heart disease. https://www.acc.org/latest-in-cardiology/articles/2016/09/22/08/58/marijuana-and-coronary-heart-disease
- 318.Pacher P., Steffens S., Haskó G., Schindler T.H., Kunos G. Cardiovascular effects of marijuana and synthetic cannabinoids: the good, the bad, and the ugly. Nat Rev Cardiol. 2018;15:151–166. doi: 10.1038/nrcardio.2017.130. [DOI] [PubMed] [Google Scholar]
- 319.Singla S., Sachdeva R., Mehta J.L. Cannabinoids and atherosclerotic coronary heart disease. Clin Cardiol. 2012;35:329–335. doi: 10.1002/clc.21962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Piano M.R. Alcohol's effects on the cardiovascular system. Alcohol Res. 2017;38:219–241. [PMC free article] [PubMed] [Google Scholar]
- 321.Kim S.T., Park T. Acute and chronic effects of cocaine on cardiovascular health. Int J Mol Sci. 2019;20 doi: 10.3390/ijms20030584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Skeoch S., Bruce I.N. Atherosclerosis in rheumatoid arthritis: is it all about inflammation? Nat Rev Rheumatol. 2015;11:390–400. doi: 10.1038/nrrheum.2015.40. [DOI] [PubMed] [Google Scholar]
- 323.Reiss A.B., Silverman A., Khalfan M., et al. Accelerated atherosclerosis in rheumatoid arthritis: mechanisms and treatment. Curr Pharm Des. 2019;25:969–986. doi: 10.2174/1381612825666190430113212. [DOI] [PubMed] [Google Scholar]
- 324.Liu Y., Kaplan M.J. Cardiovascular disease in systemic lupus erythematosus: an update. Curr Opin Rheumatol. 2018;30:441–448. doi: 10.1097/BOR.0000000000000528. [DOI] [PubMed] [Google Scholar]
- 325.Masson W., Lobo M., Molinero G. Psoriasis and cardiovascular risk: a comprehensive review. Adv Ther. 2020;37:2017–2033. doi: 10.1007/s12325-020-01346-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Sanda G.E., Belur A.D., Teague H.L., Mehta N.N. Emerging associations between neutrophils, atherosclerosis, and psoriasis. Curr Atheroscler Rep. 2017;19:53. doi: 10.1007/s11883-017-0692-8. [DOI] [PubMed] [Google Scholar]
- 327.Łosińska K., Korkosz M., Kwaśny-Krochin B. Endothelial dysfunction in patients with ankylosing spondylitis. Reumatologia. 2019;57:100–105. doi: 10.5114/reum.2019.84815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Dimitroulas T., Baniotopoulos P., Pagkopoulou E., et al. Subclinical atherosclerosis in systemic sclerosis and rheumatoid arthritis: a comparative matched-cohort study. Rheumatol Int. 2020;40:1997–2004. doi: 10.1007/s00296-020-04677-3. [DOI] [PubMed] [Google Scholar]
- 329.Bigeh A., Sanchez A., Maestas C., Gulati M. Inflammatory bowel disease and the risk for cardiovascular disease: does all inflammation lead to heart disease? Trends Cardiovasc Med. 2020;30:463–469. doi: 10.1016/j.tcm.2019.10.001. [DOI] [PubMed] [Google Scholar]
- 330.Groenen A.G., Westerterp M. A new small molecule increases cholesterol efflux. Arterioscler Thromb Vasc Biol. 2021;41:1851–1853. doi: 10.1161/ATVBAHA.121.315930. [DOI] [PubMed] [Google Scholar]
- 331.Lusis A.J. Genetics of atherosclerosis. Trends Genet. 2012;28:267–275. doi: 10.1016/j.tig.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Forgo B., Medda E., Hernyes A., Szalontai L., Tarnoki D.L., Tarnoki A.D. Carotid artery atherosclerosis: a review on heritability and genetics. Twin Res Hum Genet. 2018;21:333–346. doi: 10.1017/thg.2018.45. [DOI] [PubMed] [Google Scholar]
- 333.McPherson R., Tybjaerg-Hansen A. Genetics of coronary artery disease. Circ Res. 2016;118:564–578. doi: 10.1161/CIRCRESAHA.115.306566. [DOI] [PubMed] [Google Scholar]
- 334.Roy H., Bhardwaj S., Yla-Herttuala S. Molecular genetics of atherosclerosis. Hum Genet. 2009;125:467–491. doi: 10.1007/s00439-009-0654-5. [DOI] [PubMed] [Google Scholar]
- 335.Makshood M., Post W.S., Kanaya A.M. Lipids in South Asians: epidemiology and management. Curr Cardiovasc Risk Rep. 2019;13:24. doi: 10.1007/s12170-019-0618-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Talegawkar S.A., Jin Y., Kandula N.R., Kanaya A.M. Cardiovascular health metrics among South Asian adults in the United States: prevalence and associations with subclinical atherosclerosis. Prev Med. 2017;96:79–84. doi: 10.1016/j.ypmed.2016.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Parikh N.I., Aurora M.S., Dash R., Shin J.J., Palaniappan L. Assessment of obesity and cardiovascular risk in South Asians. Curr Cardiovasc Risk Rep. 2014;9:425. [Google Scholar]
- 338.Volgman A.S., Palaniappan L.S., Aggarwal N.T., et al. Atherosclerotic cardiovascular disease in South Asians in the United States: epidemiology, risk factors, and treatments: a scientific statement from the American heart association. Circulation. 2018;138:e1–e34. doi: 10.1161/CIR.0000000000000580. [DOI] [PubMed] [Google Scholar]
- 339.Hussain S.M., Oldenburg B., Wang Y., Zoungas S., Tonkin A.M. Assessment of cardiovascular disease risk in South Asian populations. Int J Vasc Med. 2013;2013 doi: 10.1155/2013/786801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Misra R., Patel T., Kotha P., et al. Prevalence of diabetes, metabolic syndrome, and cardiovascular risk factors in US Asian Indians: results from a national study. J Diabetes Complicat. 2010;24:145–153. doi: 10.1016/j.jdiacomp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 341.Gupta M., Brister S. Is South Asian ethnicity an independent cardiovascular risk factor? Can J Cardiol. 2006;22:193–197. doi: 10.1016/s0828-282x(06)70895-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Veeranna V., Zalawadiya S.K., Niraj A., Kumar A., Ference B., Afonso L. Association of novel biomarkers with future cardiovascular events is influenced by ethnicity: results from a multi-ethnic cohort. Int J Cardiol. 2013;166:487–493. doi: 10.1016/j.ijcard.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 343.Kong X., Jia X., Wei Y., et al. Association between microalbuminuria and subclinical atherosclerosis evaluated by carotid artery intima-media in elderly patients with normal renal function. BMC Nephrol. 2012;13:37. doi: 10.1186/1471-2369-13-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Park H.E., Heo N.J., Kim M., Choi S.Y. Significance of microalbuminuria in relation to subclinical coronary atherosclerosis in asymptomatic nonhypertensive, nondiabetic subjects. J Korean Med Sci. 2013;28:409–414. doi: 10.3346/jkms.2013.28.3.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Chen Y., Xu B., Sun W., et al. Impact of the serum uric acid level on subclinical atherosclerosis in middle-aged and elderly Chinese. J Atheroscler Thromb. 2015;22:823–832. doi: 10.5551/jat.26260. [DOI] [PubMed] [Google Scholar]
- 346.Maruhashi T., Hisatome I., Kihara Y., Higashi Y. Hyperuricemia and endothelial function: from molecular background to clinical perspectives. Atherosclerosis. 2018;278:226–231. doi: 10.1016/j.atherosclerosis.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 347.Rimondi E., Marcuzzi A., Casciano F., et al. Role of vitamin D in the pathogenesis of atheromatosis. Nutr Metab Cardiovasc Dis. 2021;31:344–353. doi: 10.1016/j.numecd.2020.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.McGurk K.A., Keavney B.D., Nicolaou A. Circulating ceramides as biomarkers of cardiovascular disease: evidence from phenotypic and genomic studies. Atherosclerosis. 2021;327:18–30. doi: 10.1016/j.atherosclerosis.2021.04.021. [DOI] [PubMed] [Google Scholar]
- 349.Gencer B., Bonomi M., Adorni M.P., Sirtori C.R., Mach F., Ruscica M. Cardiovascular risk and testosterone – from subclinical atherosclerosis to lipoprotein function to heart failure. Rev Endocr Metab Disord. 2021;22:257–274. doi: 10.1007/s11154-021-09628-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Abouzeid C., Bhatt D., Amin N. The top five women's health issues in preventive cardiology. Curr Cardiovasc Risk Rep. 2018;12:6. [Google Scholar]
- 351.McKibben R.A., Al Rifai M., Mathews L.M., Michos E.D. Primary prevention of atherosclerotic cardiovascular disease in women. Curr Cardiovasc Risk Rep. 2015;10:1. doi: 10.1007/s12170-015-0480-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Gianos E., Karalis D.G., Gaballa D., et al. Managing cardiometabolic risk factors across a woman's lifespan: a lipidologist's perspective. J Clin Lipidol. 2021 doi: 10.1016/j.jacl.2021.03.005. [DOI] [PubMed] [Google Scholar]
- 353.Thurston R.C., Khoudary S.R.E., Derby C.A., et al. Low socioeconomic status over 12 years and subclinical cardiovascular disease. Stroke. 2014;45:954–960. doi: 10.1161/STROKEAHA.113.004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Hailu E.M., Needham B.L., Lewis T.T., et al. Discrimination, social support, and telomere length: the multi-ethnic study of atherosclerosis (MESA) Ann Epidemiol. 2020;42:58–63. doi: 10.1016/j.annepidem.2019.12.009. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Lynch J., Kaplan G.A., Salonen R., Cohen R.D., Salonen J.T. Socioeconomic status and carotid atherosclerosis. Circulation. 1995;92:1786–1792. doi: 10.1161/01.cir.92.7.1786. [DOI] [PubMed] [Google Scholar]
- 356.Dearborn-Tomazos J.L., Hu X., Bravata D.M., et al. Deintensification or no statin treatment is associated with higher mortality in patients with ischemic stroke or transient ischemic attack. Stroke. 2022 doi: 10.1161/STROKEAHA.120.030089. 0:STROKEAHA.120.030089. [DOI] [PubMed] [Google Scholar]
- 357.Mann D.M., Woodward M., Muntner P., Falzon L., Kronish I. Predictors of nonadherence to statins: a systematic review and meta-analysis. Ann Pharmacother. 2010;44:1410–1421. doi: 10.1345/aph.1P150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Hakulinen C., Pulkki-Råback L., Elovainio M., et al. Childhood psychosocial cumulative risks and carotid intima-media thickness in adulthood: the cardiovascular risk in young finns study. Psychosom Med. 2016;78:171–181. doi: 10.1097/PSY.0000000000000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Khan S.A., Shahzad U., Zarak M.S., Channa J., Khan I., Ghani M.O.A. Association of depression with subclinical coronary atherosclerosis: a systematic review. J Cardiovasc Transl Res. 2020 doi: 10.1007/s12265-020-09985-4. [DOI] [PubMed] [Google Scholar]
- 360.Fioranelli M., Bottaccioli A.G., Bottaccioli F., Bianchi M., Rovesti M., Roccia M.G. Stress and inflammation in coronary artery disease: a review psychoneuroendocrineimmunology-based. Front Immunol. 2018;9:2031. doi: 10.3389/fimmu.2018.02031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Julkunen J., Salonen R., Kaplan G.A., Chesney M.A., Salonen J.T. Hostility and the progression of carotid atherosclerosis. Psychosom Med. 1994;56:519–525. doi: 10.1097/00006842-199411000-00007. [DOI] [PubMed] [Google Scholar]
- 362.Arnett D.K., Blumenthal R.S., Albert M.A., et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American college of cardiology/American heart association task force on clinical practice guidelines. Circulation. 2019;140:e596–e646. doi: 10.1161/CIR.0000000000000678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Juul F., Vaidean G., Lin Y., Deierlein A.L., Parekh N. Ultra-processed foods and incident cardiovascular disease in the framingham offspring study. J Am Coll Cardiol. 2021;77:1520–1531. doi: 10.1016/j.jacc.2021.01.047. [DOI] [PubMed] [Google Scholar]
- 364.Widmer R.J., Flammer A.J., Lerman L.O., Lerman A. The Mediterranean diet, its components, and cardiovascular disease. Am J Med. 2015;128:229–238. doi: 10.1016/j.amjmed.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Spence J.D., Srichaikul K., Jenkins D.J.A. Cardiovascular harm from egg yolk and meat: more than just cholesterol and saturated fat. J Am Heart Assoc. 2021;10 doi: 10.1161/JAHA.120.017066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Chen X., Zhang Z., Yang H., et al. Consumption of ultra-processed foods and health outcomes: a systematic review of epidemiological studies. Nutr J. 2020;19:86. doi: 10.1186/s12937-020-00604-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Zhuang P., Zhang Y., He W., et al. Dietary fats in relation to total and cause-specific mortality in a prospective cohort of 521 120 individuals with 16 years of follow-up. Circ Res. 2019;124:757–768. doi: 10.1161/CIRCRESAHA.118.314038. [DOI] [PubMed] [Google Scholar]
- 368.Guasch-Ferré M., Babio N., Martínez-González M.A., et al. Dietary fat intake and risk of cardiovascular disease and all-cause mortality in a population at high risk of cardiovascular disease. Am J Clin Nutr. 2015;102:1563–1573. doi: 10.3945/ajcn.115.116046. [DOI] [PubMed] [Google Scholar]
- 369.Rocha D.M., Caldas A.P., Oliveira L.L., Bressan J., Hermsdorff H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis. 2016;244:211–215. doi: 10.1016/j.atherosclerosis.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 370.Schaffer A.E., D'Alessio D.A., Guyton J.R. Extreme elevations of low-density lipoprotein cholesterol with very low carbohydrate, high fat diets. J Clin Lipidol. 2021 doi: 10.1016/j.jacl.2021.04.010. [DOI] [PubMed] [Google Scholar]
- 371.Janeiro M.H., Ramírez M.J., Milagro F.I., Martínez J.A., Solas M. Implication of trimethylamine N-Oxide (TMAO) in disease: potential biomarker or new therapeutic target. Nutrients. 2018;10 doi: 10.3390/nu10101398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Tang W.H., Wang Z., Levison B.S., et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Aengevaeren V.L., Mosterd A., Sharma S., et al. Exercise and coronary atherosclerosis. Circulation. 2020;141:1338–1350. doi: 10.1161/CIRCULATIONAHA.119.044467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Fletcher G.F., Balady G., Blair S.N., et al. Statement on exercise: benefits and recommendations for physical activity programs for all Americans. Circulation. 1996;94:857–862. doi: 10.1161/01.cir.94.4.857. [DOI] [PubMed] [Google Scholar]
- 375.Schmidt-Trucksäss A. Does sedentary lifestyle touch arterial health? Atherosclerosis. 2016;244:222–223. doi: 10.1016/j.atherosclerosis.2015.10.107. [DOI] [PubMed] [Google Scholar]
- 376.Schmid D., Ricci C., Leitzmann M.F. Associations of objectively assessed physical activity and sedentary time with all-cause mortality in US adults: the NHANES study. PLoS One. 2015;10 doi: 10.1371/journal.pone.0119591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Cappola A.R., Ladenson P.W. Hypothyroidism and atherosclerosis. J Clin Endocrinol Metab. 2003;88:2438–2444. doi: 10.1210/jc.2003-030398. [DOI] [PubMed] [Google Scholar]
- 378.Estruch R., Ros E., Salas-Salvadó J., et al. Primary prevention of cardiovascular disease with a mediterranean diet. N Engl J Med. 2013;368:1279–1290. doi: 10.1056/NEJMoa1200303. [DOI] [PubMed] [Google Scholar]
- 379.Orlich M.J., Singh P.N., Sabaté J., et al. Vegetarian dietary patterns and mortality in Adventist health study 2. JAMA Intern. Med. 2013;173:1230–1238. doi: 10.1001/jamainternmed.2013.6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Marrone G., Guerriero C., Palazzetti D., et al. Vegan diet health benefits in metabolic syndrome. Nutrients. 2021;13 doi: 10.3390/nu13030817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 381.Kälsch H., Hennig F., Moebus S., et al. Are air pollution and traffic noise independently associated with atherosclerosis: the Heinz Nixdorf recall study. Eur Heart J. 2014;35:853–860. doi: 10.1093/eurheartj/eht426. [DOI] [PubMed] [Google Scholar]
- 382.Münzel T., Schmidt F.P., Steven S., Herzog J., Daiber A., Sørensen M. Environmental noise and the cardiovascular system. J Am Coll Cardiol. 2018;71:688–697. doi: 10.1016/j.jacc.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 383.Münzel T., Gori T., Babisch W., Basner M. Cardiovascular effects of environmental noise exposure. Eur Heart J. 2014;35:829–836. doi: 10.1093/eurheartj/ehu030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Hahad O., Kröller-Schön S., Daiber A., Münzel T. The cardiovascular effects of noise. Dtsch Arztebl Int. 2019;116:245–250. doi: 10.3238/arztebl.2019.0245. [DOI] [PMC free article] [PubMed] [Google Scholar]