

Abstract
Hydrolytic loss of nucleobases from the deoxyribose backbone of DNA is one of the most common unavoidable types of damage in synthetic and cellular DNA. The reaction generates abasic sites in DNA and it is important to understand the properties of these lesions. The acidic nature of the α-protons of the ring-opened abasic aldehyde residue facilitates β-elimination of the 3’-phosphoryl group. This reaction is expected to generate a DNA strand break with a phosphoryl group on the 5’-terminus and a trans-α,β-unsaturated aldehyde residue on the 3’-terminus; however, a handful of studies have identified noncanonical sugar remnants on the 3’-terminus, suggesting that the products arising from strand cleavage at AP sites in DNA may be more complex than commonly thought. We characterized the strand cleavage induced by treatment of an abasic site-containing DNA oligonucleotide with heat, NaOH, piperidine, spermine, and the base excision repair glycoslyases Fpg and Endo III. The results showed that, under multiple conditions, cleavage at an abasic site in a DNA oligomer generated noncanonical sugar remnants including cis-α,β-unsaturated aldehyde, 2-deoxyribose, and 3-thio-2,3-dideoxyribose products on the 3’-terminus of the strand break.
Graphical Abstract
INTRODUCTION
Apurinic/apyrimidinic (AP) sites are generated by spontaneous1-3 and enzyme-catalyzed4-7 hydrolysis of the glycosidic bonds connecting nucleobases to the deoxyribose backbone of DNA (Scheme 1). Chemical modification of the DNA bases also can accelerate hydrolysis of the glycosidic bonds to generate AP sites.3, 8, 9 These processes combine to make AP sites among the most common unavoidable lesions found in cellular and synthetic DNA.8, 10-14
Scheme 1.
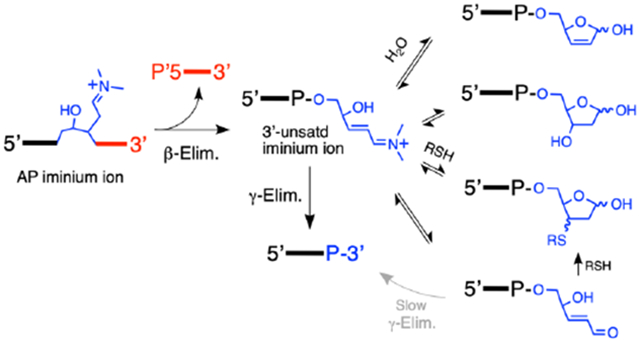
The canonical products arising from strand cleavage at an AP site in DNA are the 5’-phosphoryl terminus (5’P) and the trans-α,β-unsaturated aldehyde (3’PUA) sugar remnant generated by an initial β-elimination reaction, and the 3’-phosphoryl terminus (3’P) generated by subsequent γ,δ-elimination. The wavy lines annotated with 5’ and 3’ labels represent DNA strands. The P in this Scheme represents either a DNA phosphodiester linkage or a terminal phosphoryl group.
AP sites exist as an equilibrium mixture of the ring-closed hemiacetal alongside small amounts (~1%) of the ring-opened aldehyde (Scheme 1).15, 16 Much of the interesting chemistry associated with AP residues in DNA, as is the case for all aldose sugars, stems from equilibrium amounts of the reactive, ring-opened aldehyde. The electrophilic nature of the AP aldehyde residue enables generation of secondary lesions including DNA-DNA interstrand cross-links17-22 and DNA-protein cross-links.23-26 In addition, the acidic nature of the α-protons27 of the ring-opened AP aldehyde facilitates generation of DNA strand breaks via β-elimination of the 3’-phosphoryl group (Scheme 1).8, 28-33
In neutral aqueous buffers at 37 °C, AP sites in DNA are converted into strand breaks rather slowly, with half-times in the range of 200-2000 h.30, 34-36 However, the rate of strand cleavage at AP sites in DNA increases substantially with heating, under alkaline conditions, or in the presence of amines.3, 30, 37-43 Amine-catalyzed DNA strand cleavage may be biologically important because the cell nucleus is rich in low molecular weight polyamines such as spermine that efficiently catalyze β-elimination at AP sites.30, 31, 41, 44-48 In addition, amine residues on peptides, histones, and DNA repair proteins can catalyze strand cleavage at AP sites6, 32, 34, 40, 49-57 (in enzymology, this is classified as a β-lyase reaction58).
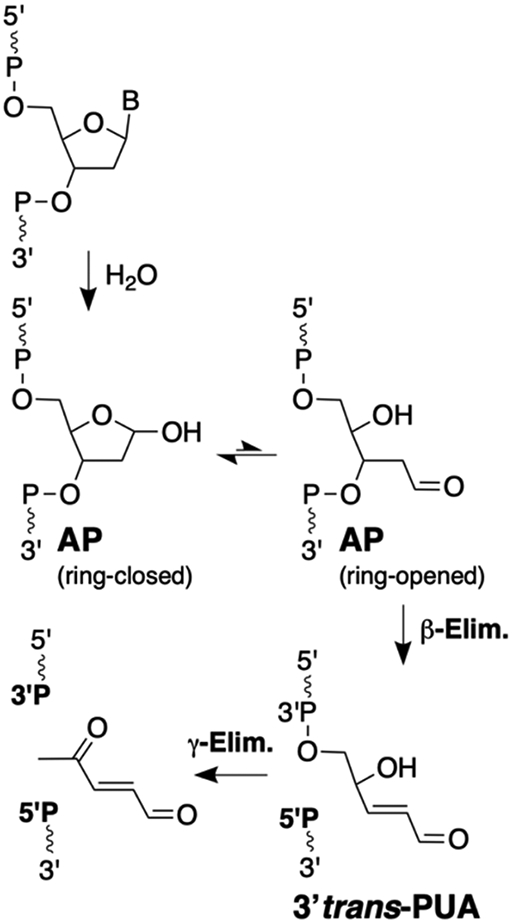
A series of early studies established that β-elimination of phosphate from an AP site in DNA induced by heat, NaOH, or amine catalysts generates a strand break with a phosphoryl group on the 5’-terminus (5’P) and a trans-α,β-unsaturated aldehyde residue on the 3’-terminus (Scheme 1).31, 32, 37, 40, 41, 53, 59 The 3’-trans-alkenal sugar remnant has been referred to by a variety of names including trans-4-hydroxy-2-pentenal 5-phosphate,60, 61, trans-2-hydroxy-5-oxopent-3-enyl,62 βE (β-elimination product),63 3’-Ald,64 3’dRP (3’deoxyribose phosphate),65, 66 3’ddR5P (2,3-didehydro-2,3-dideoxy-ribose-5-phosphate),48, 67 and 3’PUA (phospho-α,β-unsaturated aldehyde).68-70 Here we will use the 3’PUA nomenclature. Under more vigorous conditions, the 3’trans-PUA sugar remnant is removed from DNA altogether by a γ,δ-elimination reaction that leaves a single nucleotide gap flanked by 5’P and 3’-phosphoryl (3’P) groups (Scheme 1).8, 28-30, 41, 53 The 3’trans-PUA sugar remnant has been detected in the DNA of cultured human cells63 and there is evidence from studies involving dysregulated base excision repair that AP-derived strand breaks are more toxic to cells than the parent abasic site.71-74
Interestingly, a small number of studies have described product mixtures derived from the cleavage of AP sites in DNA that are different, and in some cases more complex, than the canonical 3’trans-PUA, 3’P, and 5’P cleavage products. These noncanonical 3’-sugar remnants include 3’cis-PUA,37, 75, 76 3’deoxyribose (3’dR),37, 75, 77-80 3’,4’-cyclized deoxyribose,28, 30, 41 and adducts arising from conjugate addition of nitrogen nucleophiles51, 81, 82 or thiols83-85 to the 3’trans-PUA group (Figure 1).
Figure 1.
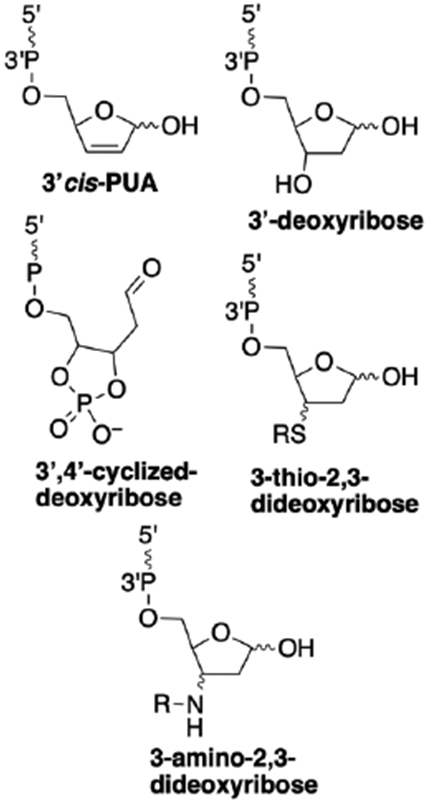
Possible noncanonical 3’end groups generated by cleavage at an AP site in DNA. The wavy lines annotated with 5’ and 3’ labels represent DNA strands.
It is important to define the secondary products arising from abundant lesions such as the AP site in synthetic and cellular DNA. With this in mind, we investigated the products arising from strand cleavage induced by treatment of an AP-containing DNA oligonucleotide with heat, NaOH, piperidine, spermine, and the base excision repair glycoslyases Fpg and Endo III. The results show that, under multiple conditions, noncanonical sugar remnants are generated on the 3’-terminus of AP-derived strand breaks in a DNA oligomer. These findings suggest that the products of strand cleavage at AP sites in DNA often may be more complex than expected.
EXPERIMENTAL PROCEDURES
Material.
Reagents were purchased from the following suppliers and were of the highest purity available: oligonucleotides were from Integrated DNA Technologies (IDT, Coralville, IA), uracil DNA glycosylase (UDG), endonuclease III (Nth, Endo III) and Fpg were from New England Biolabs (Ipswich, MA), BS Poly-prep columns were obtained from BioRad (Hercules, CA, USA) and other reagents, salts, and buffers were purchased from Sigma-Aldrich (St. Louis, MO). DMBAA (dimethylbutylammonium acetate) solutions used in the ESI-MS experiments was prepared as follows: a stock of N,N-dimethylbutyl amine (7.125 M) was diluted to 100 mM with water and adjusted to pH 7.1 with glacial acetic acid.
Enzymatic generation of AP site in DNA oligonucleotides.
The 2’-deoxyuridine-containing precursor oligonucleotide (5’TTTTTdUTTTTTTTTTT-3’, 5 μL of a stock solution containing 1 nmol/μL, 5 nmol total) was mixed with a stock solution of buffer (10 μL of 250 mM HEPES pH 7.4 containing 500 mM NaCl), followed by deionized water (28 μL) and the enzyme uracil DNA glycosylase (UDG, 7 μL of a 5000 U/mL solution, 35 U) to give a solution containing 100 μM of the dU-containing oligonucleotide in HEPES buffer (50 mM, pH 7.4, containing 100 mM NaCl), and 0.7 U/μL UDG, in a total volume of 50 μL. After incubating for 2 h at 37 °C, the enzyme was removed by phenol-chloroform extraction, the DNA ethanol precipitated, and the pellet washed with cold 80% ethanol in water. The resulting AP-containing DNA oligomer 5’TTTTTXTTTTTTTTTT (where X = AP) was dried in a Speed-vac concentrator and stored at −20 °C until use.
Non-enzymatic generation of a 3’-terminal AP site (3’-deoxyribose residue) in the DNA oligomer 5’-TTTTT-dR-3’ by acid-treatment of 5’-TTTTTA-3’.
The 2’-deoxyadenosine-containing precursor oligonucleotide 5’-TTTTTA-3’ (5 μL of a stock solution containing 1 nmol/μL, 5 nmol) was mixed with an aqueous solution of HCl (2 μL of a 100 mM stock solution), followed by dilution with deionized water (13 μL) to give a solution containing 250 μM of the 5’-TTTTTA oligodeoxynucleotide in 20 μL of 10 mM HCl. The microcentrifuge tube containing the mixture was heated at 65 °C for 1 h in a thermostat-controlled aluminum dry-block system to induce depurination of the adenine residue. The reaction mixture was then passed through a Sephadex spin-column prepared using G-25 beads swollen in 50 mM HEPES pH 7.4, 100 mM NaCl. The resulting solution of 5’-TTTTT-dR-3’ was frozen and stored at −20 °C until use.
Thermal cleavage of the AP-containing oligonucleotide.
The AP-containing oligodeoxynucleotide pellet from the UDG reaction described above was dissolved in 10 μL of 250 mM HEPES pH 7.4 and 500 mM NaCl and deionized water (40 μL) to give a solution containing approximately 100 μM of the AP-oligonucleotide and HEPES pH 7.4 (50 mM, containing 100 mM NaCl) in a total volume of 50 μL. The mixture was incubated in a thermostat-controlled aluminum dry-block system held at 85 °C. At selected times (0, 15, 45 min), aliquots of the reaction mixture (10 μL) were removed and subjected to HPLC analysis.
Cleavage of the AP oligonucleotide with NaOH.
Four different conditions were examined. 1. 5 mM NaOH, 37 °C, 1 h. Approximately 1 nmol of the AP-containing oligonucleotide was dissolved in 18 μL of deionized water was mixed with 2 μL of a 50 mM stock solution of NaOH in water. The solution was heated at 37 °C for 1 h and then subjected to HPLC analysis. 2. 200 mM NaOH, 37 °C, 20 min. Approximately 1 nmol of the AP-containing oligonucleotide was dissolved in 16 μL of deionized water was mixed with 4 μL of a 1 M stock solution of NaOH in water. The solution was heated at 37 °C for 15 min and then subjected to HPLC analysis. 3. 200 mM NaOH, 37 °C, 5 h. Approximately 1 nmol of the AP-containing oligonucleotide was dissolved in 16 μL of deionized water was mixed with 4 μL of a 1 M stock solution of NaOH in water. The solution was heated at 37 °C for 5 h and then subjected to HPLC analysis. 4. Typical workup for monofunctional base excision repair glycosylase assay. Approximately 1 nmol of the AP-containing oligonucleotide in 10 μL of 20 mM HEPES buffer (pH 7.4 containing 100 mM NaCl) was mixed with 10 μL of a stock solution of 1 M NaOH in water. The solution was heated at 95 °C for 2 min and then subjected to HPLC analysis.
Cleavage of the AP oligonucleotide with piperidine.
Four different conditions were examined. 1. 5 mM piperidine, 37 °C, 1 h. Approximately 1 nmol of the AP-oligonucleotide in 18 μL of HEPES (50 mM, pH 7.4 containing 100 mM NaCl) was mixed with a 2 μL of a stock solution of 50 mM piperidine in water. The solution was heated at 37 °C for 1 h and then subjected to HPLC analysis. 2. 1 M piperidine, 50 °C, 20 min. Approximately 1 nmol of the AP-oligonucleotide in 10 μL of deionized water was mixed with a 40 μL of a stock solution of 1.25 M piperidine in water. The microcentrifuge tube containing the solution was heated in a 50 °C hot block for 20 min and then subjected to HPLC analysis. 3. 1 M piperidine, 95 °C, 30 min. Approximately 1 nmol of the AP-oligonucleotide in 10 μL of deionized water was mixed with a 40 μL of a stock solution of 1.25 M piperidine in water. The microcentrifuge tube containing the solution was heated in a 95 °C hot block for 30 min and then subjected to HPLC analysis. 4. A control reaction heating the AP-oligonucleotide at 95 °C for 30 min, in the absence of piperidine. A microcentrifuge tube containing approximately 1 nmol of the AP-oligonucleotide in 50 μL of deionized water was heated in a 95 °C hot block for 30 min and then subjected to HPLC analysis.
Cleavage of the AP oligonucleotide with spermine.
Approximately 10 nmol of the AP-containing DNA oligomer from the UDG reaction described above was dissolved in 20 μL of 250 mM HEPES pH 7.4 containing 500 mM NaCl and mixed with deionized water (70 μL), and spermine (10 μL of a 50 mM stock solution in water, with the pH adjusted to 7.4) to give a final mixture containing 100 μM of the AP-oligonucleotide and 5 mM spermine in HEPES buffer (50 mM, pH 7.4 containing 100 mM NaCl). The mixture was incubated at 37 °C and aliquots of the reaction mixture (10 μL) were removed at 5 min, 30 min, 1 h, 6 h, 12 h, 24 h and 48 h and subjected to HPLC analysis. The experiment examining cleavage of the AP oligonucleotide induced by spermine in the presence of 2-mercaptoethanol was carried out in a similar manner by dissolving approximately 5 nmol of the AP-containing DNA oligomer from the UDG reaction described above in 10 μL of HEPES buffer (250 mM, pH 7.4 containing 500 mM NaCl), deionized water (30 μL), spermine (5 μL of a 50 mM stock solution in water, with the pH adjusted to 7.4), and 2-mercaptoethanol (5 μL of a 50 mM stock solution in water).
Generation of a mixture containing 3’cis-PUA, 3’trans-PUA, and 3’dR cleavage products using the conditions of Kushida et al.75
The AP-containing oligodeoxynucleotide pellet from the UDG reaction described above was dissolved in 5 μL of Tris-borate buffer (200 mM, pH 7.5) and deionized water (45 μL) to give a final mixture containing approximately 100 μM of the AP-oligonucleotide in 50 μL of Tris-borate buffer (20 mM, pH 7.5). The mixture was incubated in a thermostat-controlled aluminum dry block system held at 90 °C, for 30 min and a 10 μL aliquot of the reaction mixture was removed and subjected to HPLC analysis.
Reactions of 2-mercaptoethanol with the products arising from cleavage of the AP oligonucleotide.
To an aliquot (10 μL) containing 1 nmol of DNA derived from cleavage of the AP-containing oligonucleotide was added 1.1 μL of a 50 mM stock solution of 2-mercaptoethanol in water. The resulting reaction mixtures, containing a 5 mM final concentration of 2-mercaptoethanol and 0.91x concentrations of the buffer and salt found in the original cleavage reaction mixtures, were incubated at 37 °C for 15 min and subjected to HPLC analysis.
Cleavage of an AP-containing duplex with Endonuclease III.
Cleavage of an AP-containing duplex by the base excision repair glycosylase Endo III was carried out by mixing 2 nmol of the AP-containing oligonucleotide 5’TTTTTXTTTTTTTTTT, (where X = AP) with 2 nmol of the complementary strand 5’dT2A16T2 in Tris-HCl buffer (20 mM, pH 8.4) containing EDTA (1 mM), DTT (1 mM), and the enzyme Endo III (20 units). The mixture was incubated at 37 °C. Aliquots (10 μL) were removed at 15 min and 2 h and subjected to HPLC analysis.
Cleavage of an AP-containing duplex by Fpg.
Cleavage of an AP-containing duplex by the base excision repair glycosylase Fpg was carried out by mixing 1 nmol of the AP-containing oligonucleotide 5’TTTTTXTTTTTTTTTT, (where X = AP) with 1 nmol of the complementary strand 5’dT2A16T2 in Bis-Tris-Propane-HCl (pH 7.0), MgCl2 (10 mM), DTT (1 mM), and the enzyme Fpg (16 units). The mixture was incubated at 37 °C for 6 h and subjected to HPLC analysis.
HPLC Analysis of the Products Arising from Cleavage of the AP-Containing Oligonucleotide.
HPLC analyses were conducted using a reverse phase column at 24 °C (Agilent AdvanceBio, C18, 4.6 x 50 mm, 2.7 μm) eluted with a linear gradient of 6-14% acetonitrile in aqueous 0.1 M triethylammonium acetate pH 7.0 over 20 min at a flow rate of 0.6 mL/min. The products were monitored by their absorbance at 260 nm.
ESI-QTOF-LC-MS analysis of AP-derived cleavage products.
Samples for mass spectrometric analysis were prepared using 5 nmol of the AP-containing oligonucleotide. LC-MS data were acquired on an Agilent Technologies 6520A Accurate Mass QTOF. Samples were analyzed according to the protocol of Studzinska and Buszewski,86 with slight modifications as outlined. Sample was injected onto a C8 trap column (Michrom Bioresources Captrap) at a flow rate of 5 μL/min of 10 mM DMBAA, pH 7.1 over 4 min. and separated by isocratic elution (either 80% or 42.5% methanol, 15 mM DMBAA, pH 7.1) at a flow rate of 0.4 μL/min on a 10 cm x 75 μm C8 analytical column (fused silica packed with Michrom Bioresources C8, 3.5 μm particles). Following the 4 min trap load, separation on the trap/analytical columns continued for 16 min, under isocratic elution conditions. Total run time was 20 min. Mass spectra were acquired using the following parameters: negative-ion mode; VCap 2500 V; mass range 290-3200 m/z; 0.63 spectra/second; fragmentor at 300 V (250 V for IDT oligo); internal MS recalibration was achieved using the K/Na adducted Hexakis 1221 Chip Cube High Mass Reference compound (m/z 1279.99). Samples were loaded in sequence as follows: blank (10 mM DMBAA), sample, and blank. Multiply-charged DNA peaks were deconvoluted using the maximum entropy algorithm in Qualitative Analysis software (version B.07.00 Agilent Technologies) with the following parameters: adduct = proton-loss; m/z range = 600-1500 m/z; mass range = expected mass ±2 kDa; peak height to calculate mass = 25%. The m/z values reported are neutral deconvoluted masses.
RESULTS AND DISCUSSION
Generation of the AP-containing 2’-oligodeoxynudeotide.
We examined the products arising from strand cleavage at an AP site embedded in a polythymidine oligodeoxynucleotide (Scheme 2). The AP site was installed 5 nucleotides from the 5’-end and 10 nucleotides from the 3’-end by treatment of a 2’-deoxyuridine-containing precursor with the enzyme uracil DNA glycosylase (UDG, Scheme 2).19, 87-89 The cleavage products generated under various conditions were characterized by reverse-phase HPLC, nanospray ESI-QTOF mass spectrometry, comparision to authentic synthetic standards, and by diagnostic chemical reactions (e.g. conjugate addition of 2-mercaptoethanol). The exact retention times varied slightly from run-to-run, but the products consistently eluted in the relative order: 3’P, 3’dR, 3’trans-PUA, 3’cis-PUA, 3’PUA-thiol adducts, 5’P, AP-oligo, and dU-oligo (early to late, Figure S1).
Scheme 2.
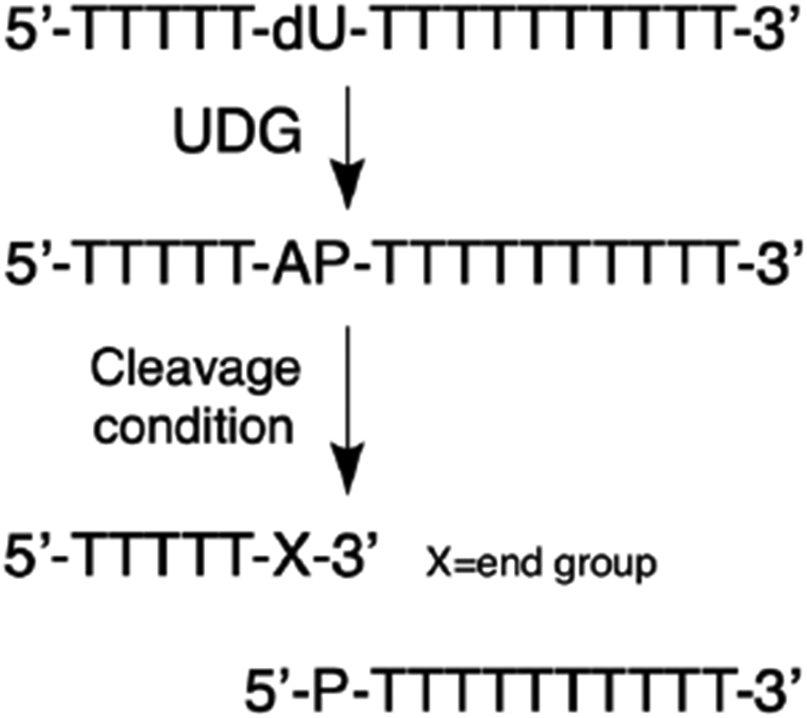
The AP-containing oligodeoxynucleotide was generated by treatment of a 2’-deoxyuridine (dU) containing precursor oligodeoxynucleotide with the enzyme uracil DNA glycosylase (UDG). In this Scheme, X corresponds to the 3’-end groups generated under various cleavage conditions and P corresponds to a terminal phosphoryl group.
Thermal cleavage of the AP-containing DNA oligomer generates 3’PUA and 3’P.
Sugiyama showed that thermolysis of an AP-containing trinucleotide at 90 °C in pH 7 sodium cacodylate buffer generated 3’trans-PUA as the major early product (20 min).37 At longer reaction times (1 h), 10-20% yields of the 3’cis-PUA and 3’dR products were observed alongside the 3’trans-PUA product while, after extended heating (>1.5 h), the 3’P product dominated (see Scheme 1 and Figure 1 for structures of these products).
We found that heating our AP-containing oligonucleotide at 85 °C in HEPES buffer (50 mM, pH 7.4, containing 100 mM NaCl) for 15 min generated two major cleavage products eluting at 12.4 and 14.4 min in the reverse-phase HPLC analysis (Figure 2A, HPLC trace c). The 14.4 min peak corresponded to the 5’P(T)10 product derived from β-elimination at the AP site. Based on Sugiyama’s work, we suspected that the 12.4 min peak corresponded to the 3’trans-PUA cleavage product.37 Evidence for this assignment was provided by investigating the reaction of this material with 2-mercaptoethanol. Thiols readily undergo conjugate addition to α,β-unsaturated aldehydes in neutral aqueous solution.90-94 More specifically, our recent work along with earlier precedents show that thiols readily add to the 3’trans-PUA group to generate diastereomeric 3-thio-2,3-dideoxyribose (3-thio-ddR) adducts.83-85, 95 Indeed, we found that addition of 2-mercaptoethanol (5 mM) to the thermolysis reaction mixture, followed by incubation for 15 min at 37 °C, led to disappearance of the putative 3’trans-PUA product at 12.4 min, with concomitant appearance of two new peaks eluting at approximately 13.0 and 13.5 min (Figure 3A). We ascribed these new peaks to the expected diastereomeric 3-thio-ddR products on the 3’-terminus of the strand break (Figure 3B). Similar results were observed using other thiols (Figure S2).
Figure 2.
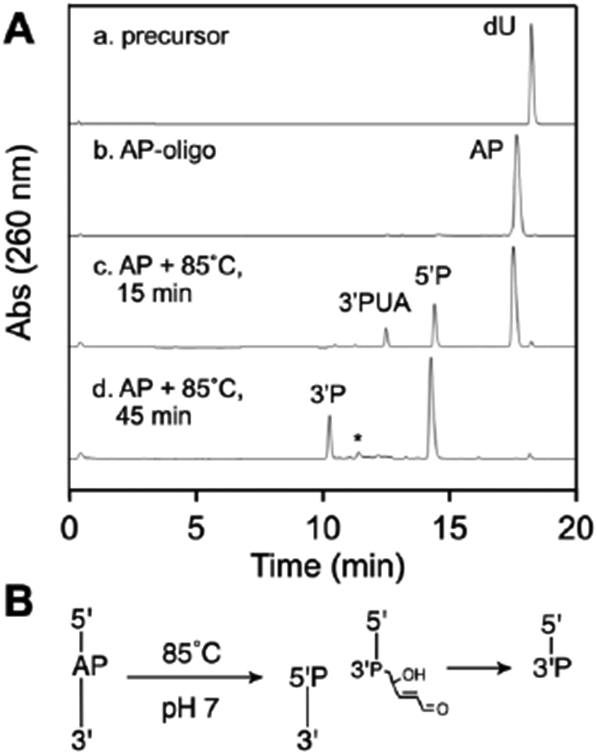
HPLC analysis of thermal cleavage of an AP site in DNA (panel A) and a schematic depiction of the product structures, where P represents a terminal phosphoryl group or a phosphodiester linkage (panel B).
Figure 3.

HPLC analysis of the products arising from reaction of 2-mercaptoethanol with the 3’trans-PUA thermal cleavage product (panel A) to generate a diastereomeric mixture of 3-alkylthio-2,3-dideoxyribose (3-thio-ddR) products on the 3’-terminus of the strand break with structures depicted in panel B, where P represents a phosphodiester linkage.
The 3’trans-PUA and 3-thio-ddR end products were stable in pH 7.4 buffer at 37 °C. Specifically, incubation of the 3’trans-PUA product for 6-12 h led to production of only small amounts of the 3’P product (Figure S3). The diastereomeric 3-thio-2,3-ddR products generated by addition of 2-mercaptoethanol to the 3’-trans-PUA cleavage product showed no decomposition over the course of 24 h (Figure S4).
Extended heating of the AP-containing oligonucleotide for 45 min at 85 °C led to complete strand cleavage, with generation of the 5’P and 3’P products, eluting at 10.4 and 14.4 min respectively (Figure 2A, HPLC trace d). A very small amount of the 3’dR product may be seen eluting near 11 min (marked by an asterisk in Figure 2A, HPLC trace d).
Nanospray ESI-TOF-MS analysis provided support for the product assignments described above and shown Figures 1 and 2. The reaction mixture at 15 min revealed strong signals consistent with the starting AP oligodeoxynucleotide, the 5’P product, the 3’trans-PUA product, the 3’P product, and a weak, but distinct, signal corresponding to the 3’dR product (Figure S5). The observed m/z values and relative signal intensities in the isotope clusters closely matched those calculated for the proposed product structures (Figure S5). The reaction mixture generated by heating the AP-oligodeoxynucleotide at 85 °C for 45 min showed signals for the 5’P product and the 3’P product consistent with the HPLC data (Figure S6). Mass spectrometric analysis of the mixture generated by thermolysis of the AP-oligonucleotide followed by reaction with 2-mercaptoethanol showed a strong signal consistent with the 3-thio-ddR products on the 3’-terminus of the strand break (Figure S7).
Overall, the data indicate that heating the AP-containing oligonucleotide at 85 °C in pH 7.4 buffer induces a β-elimination reaction that generates the 3’trans-PUA cleavage product, as expected.37 Extended heating generated the 3’P product via γ-elimination of the sugar remnant. Under our reaction conditions, there was no evidence that the 3’cis-PUA product was formed (the properties of this product are discussed further below). We observed only a trace of the 3’dR product.
Cleavage of the AP-oligonucleotide with NaOH generates 3’trans-PUA, 3’P, and the noncanonical 3’dR product.
Sodium hydroxide is often used to cleave DNA AP sites in biochemical assays. For example, assays designed to measure the activity of monofunctional DNA glycosylases often feature a NaOH workup42, 43 The general expectation is that mild NaOH treatment generates a cleavage product bearing the 3’trans-PUA group by β-elimination at the AP sites.8 For example, Mazumdar et al. reported (based on HPLC retention time) that the 3’trans-PUA group was generated by mild NaOH cleavage of a single-stranded, AP-containing, undecameric oligodeoxynucleotide (100 mM NaOH, pH 13, 4 °C).40 More vigorous NaOH treatments were reported to generate the 5’P and 3’P DNA-cleavage products via sequential α,β-and γ,δ-elimination reactions (Scheme 1).38-40
We characterized the products generated by NaOH-mediated cleavage of the AP-containing oligodeoxynucleotide under several different conditions. We found that a very mild treatment of the AP-containing oligonucleotide with NaOH (5 mM, 37 °C, 1 h) generated low yields of strand cleavage with a mixture of the 3’trans-PUA (12.4 min) and 5’P (14.4 min) termini at the strand break (Figure 4A, HPLC trace d). Interestingly, a somewhat more vigorous NaOH treatment (200 mM, 37 °C, 20 min) generated a product eluting at 11.2 min, clearly distinct from the 3’trans-PUA product (Figure 4A, HPLC trace c). We assigned this material as the 3’dR product, based on its co-elution with an authentic standard of 5’-TTTTT-dR-3’ prepared by acid depurination of a 5’-TTTTTA-3’ precursor according to the method of Bailly and Verly (10 mM HCl, 1 h, 65 °C).83
Figure 4.
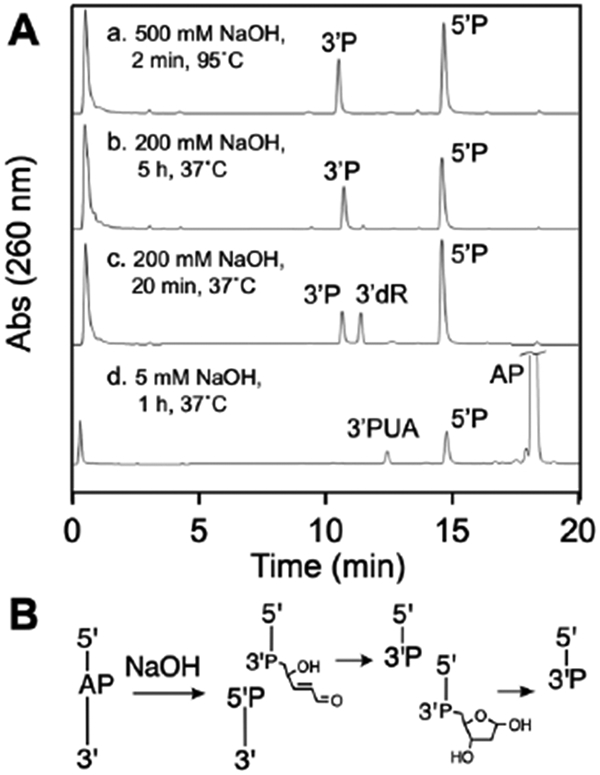
HPLC analysis of the products generated by NaOH-mediated strand cleavage of an AP site in DNA (panel A) and a schematic depiction of the product structures, where P represents a terminal phosphoryl group or a phosphodiester linkage (panel B).
To the best of our knowledge, generation of the 3’dR cleavage product from NaOH-mediated cleavage of an AP site in DNA has not been reported previously. However, a general precedent explaining this reaction may be found in the conjugate addition of water to the low molecular weight α,β-unsaturated aldehyde, acrolein, in aqueous solution.96 Thus, the 3’dR product generated by treatment of the AP oligonucleotide with 5 mM NaOH may arise via addition of water to the initial 3’trans-PUA cleavage product. Consistent with this idea, a control experiment showed that an authentic sample of the 3’trans-PUA product generated by thermolysis of the AP-oligonucleotide was, indeed, converted to a mixture of 3’P and 3’dR upon treatment with 5 mM NaOH, 37 °C for 6 h (Figure S8).
Treatment of the AP-containing DNA with 200 mM NaOH at 37 °C for 5 h cleanly generated the 5’P and 3’P products resulting from sequential α,β-and γ,δ-elimination reactions (Figure 4A, HPLC trace b). We also examined an NaOH workup commonly used in the study of monofunctional base excision repair DNA glycosylases,42 involving addition of an equal volume of 500 mM NaOH to the AP-oligonucleotide in 20 mM HEPES pH 7.4 containing 100 mM NaCl, followed by heating at 95 °C for 2 min. As expected, this procedure gave the 5’P and 3’P cleavage products (Figure 4A, HPLC trace a).
Mass spectrometric analyses supported the structural assignments described above. The mixture generated by mild NaOH treatment (5 mM, 1 h, 37 °C) showed strong signals for the starting AP-oligonucleotide, the 5’P product, and the 3’trans-PUA product (Figure S9). Analysis of the AP-oligodeoxynucleotide treated with 200 mM NaOH for 20 min at 37 °C revealed strong signals consistent with 5’P, 3’dR, and 3’P (Figure S10). The product mixture generated by addition of 500 mM NaOH to the AP-oligonucleotide in 20 mM HEPES pH 7.4 containing 100 mM NaCl, followed by heating at 95 °C for 2 min gave strong signals for the 5’P and 3’P cleavage products (Figure S11).
Overall, the results show that treatment of an AP-containing oligodeoxynucleotide with very mild NaOH (5 mM NaOH, 1 h, 37 °C) generates low yields of strand cleavage with 5’P and 3’trans-PUA at the termini of the break. Slightly more vigorous conditions (200 mM NaOH, 20 min, 37 °C) gave complete cleavage of the AP site and, unexpectedly, generated a mixture of the 3’dR and 3’P end products. An NaOH workup commonly used to detect AP sites in assays of monofunctional DNA glycosylase cleanly generated the 3’P cleavage product.
Cleavage of the AP oligonucleotide with piperidine generates 3’trans-PUA, 3’P, and a noncanonical 3’-piperidinyl adduct.
Treatment with hot piperidine (0.1-1 M, 90-95 °C, 15-30 min) is a standard method to convert various DNA modifications into strand cleavages that can easily be detected using gel electrophoresis.3, 97, 98 For example, piperidine workup is employed in classical Maxam-Gilbert sequencing reactions.99 Piperidine workup of a DNA oligonucleotide containing an AP site is expected to generate a strand break with 5’P and 3’P termini via sequential α,β-and γ,δ-elimination reactions.3, 41, 97-99
We examined the products generated by treatment of our AP-containing oligodeoxynucleotide with piperidine under several different conditions. We found that heating the AP-oligonucleotide under standard Maxam-Gilbert conditions99 involving piperidine (1 M) at 95 °C for 30 min in either HEPES (50 mM, pH 7.4 containing 100 mM NaCl) or water cleanly generated the expected 3’P and 5’P cleavage products eluting at 10.4 and 14.4 min, respectively (Figure 5A, HPLC trace a). Electrospray mass spectrometric analysis of the mixtures generated under these conditions showed the expected signals for the 5’P and 3’P products (Figure S12). For comparison, treatment of the AP-oligonucleotide under the same conditions (95 °C for 30 min) without piperidine generated a cleavage mixture composed of the 5’P, 3’trans-PUA, 3’dR, and 3’P products (Figure S13).
Figure 5.
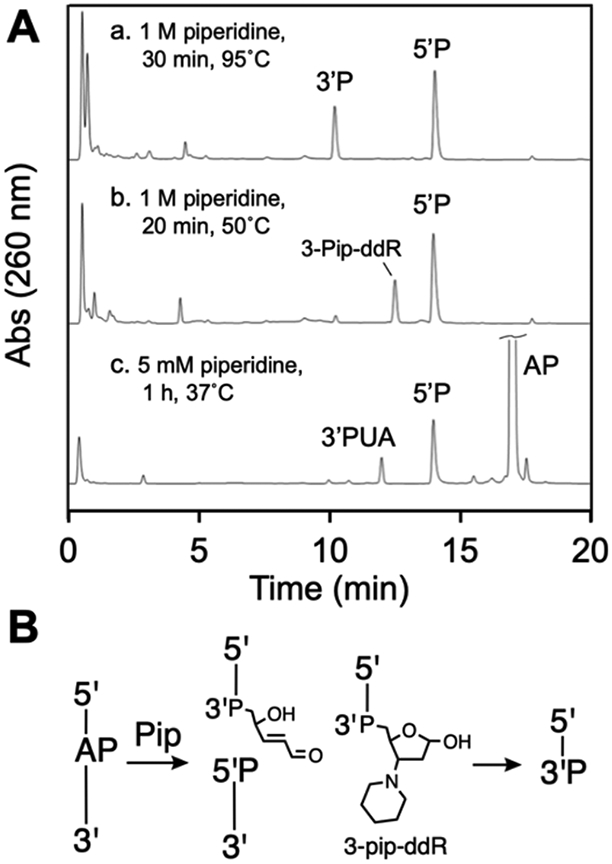
HPLC analysis of the products generated by piperidine-mediated strand cleavage of an AP in DNA (panel A) and schematic depiction of the product structures, where P represents a terminal phosphoryl group or a phosphodiester linkage (panel B).
A milder condition involving treatment of the AP-containing oligonucleotide with 1 M piperidine in HEPES (50 mM, pH 7.4 containing 100 mM NaCl) at 50 °C for 20 min gave complete cleavage of the AP-oligonucleotide but generated an unexpected product alongside the typical 5’P elimination product (Figure 5A, HPLC trace b). The retention time of the unknown product was clearly distinct from that of either the 3’P or 3’trans-PUA products, eluting at about 12.9 min, consistently later than 3’trans-PUA. The retention time was not altered by addition of 2-mercaptoethanol (5 mM, 15 min), indicating the product was not an α,β-unsaturated aldehyde or iminium ion. Nanospray ESI-TOF mass spectrometric analysis of the reaction products revealed a strong signal consistent with a 3-piperidinyl-2,3-dideoxyribose adduct on the 3’-terminus of the strand break (3-pip-ddR, Figure 5B, Figure S14). We characterized the analogous product generated by a nucleoside model system that mimics amine-catalyzed strand cleavage.95 In addition, similar 1,4-addition products previously have been proposed to arise from putrescine- and 9-aminoellipticine-mediated cleavage of AP-containing oligonucleotides.81, 82
A very mild piperidine workup (5 mM piperidine, 1 h, 37 °C) produced low yields of cleavage at the AP site, with 5’P and 3’trans-PUA groups at the termini of the strand break (Figure 5A, HPLC trace c).
Overall, the results show that typical Maxam-Gilbert workup of the AP-containing oligodeoxynucleotide (1 M, 95 °C, 30 min) generates a strand break with the expected 3’P and 5’P termini at the gap. Less vigorous conditions (1 M, 50 °C, 20 min), induced cleavage at the AP site, with generation of an unexpected 3-piperidino-2,3-dideoxyribose adduct (3-pip-ddR) on the 3’-terminus of the strand break.
Spermine-mediated cleavage of the AP-oligonucleotide generates a dynamic mixture of 3’trans-PUA, 3’cis-PUA, and 3’dR end products that ultimately converge on 3’P as the final cleavage product.
Spermine is a biogenic amine that is present in the cell at millimolar concentrations.44 This polyamine efficiently catalyzes strand cleavage at AP sites in DNA via conversion of the AP aldehyde to the corresponding iminium ion.38, 45, 48, 83, 100 Elimination of the 3’-phosphoryl group is facilitated by the dramatic increase in the acidity of the α-protons of the iminium ion compared to those of the corresponding aldehyde (Scheme 3).101, 102 The β-elimination reaction generates an α,β-unsaturated iminium ion intermediate that is substantially more reactive than the corresponding α,β-unsaturated aldehyde, with respect to both the conjugate addition of nucleophiles and the acidity of the γ-proton (Scheme 3).103-105
Scheme 3.

Conversion of the AP aldehyde residue to the corresponding iminium ion facilitates elimination via increased acidity of the α-protons. The resulting α,β-unsaturated iminium has the potential to undergo conjugate addition of water (or other nucleophiles, red arrow) or γ,δ-elimination (blue arrow) to generate the 3’P end group. For brevity, the amine catalyst is shown as a simple dialkylamine (the actual structure of spermine is shown in Figure 6). The wavy lines annotated with 5’ and 3’ labels represent DNA strands. The P in this Scheme represents either a DNA phosphodiester linkage or a terminal phosphoryl group.
The products generated by spermine-catalyzed cleavage at an AP in DNA have not previously been characterized by any means other than their gel electrophoretic mobility.38, 41, 48, 83, 100 Early work provided evidence that the amine-containing tripeptide, Lys-Trp-Lys, generates the 3’trans-PUA and 3’P products.40 However, our recent results obtained using a nucleoside system to model amine-catalyzed strand cleavage at an AP site suggested that the reaction has the potential to generate complex mixtures including the 3’trans-PUA, 3’cis-PUA, and 3’dR products (Scheme 4).95
Scheme 4.

Proposed equilibria involved in the evolution of 3’-end groups following spermine-catalyzed cleavage of an AP site in DNA. All of the reactions and equilibria shown here are subject to catalysis by amines. For brevity, the amine catalyst is shown as a simple dialkylamine (the actual structure of spermine is shown in Figure 6).
Indeed, we found that spermine-induced cleavage of the AP-containing oligodeoxynucleotide under physiologically-relevant conditions (5 mM spermine, pH 7.4 buffer, 37 °C) generated a complex mixture of products that evolves over time. At early times (15 min), we observed the intact AP-containing oligodeoxynucleotide, the 5’P elimination product, and two closely-spaced peaks eluting near 12.5 min in the HPLC chromatogram (Figure 6A, HPLC trace a). Based upon our recent work,95 we suspected that these two closely-spaced peaks might correspond to the 3’trans-PUA and 3’cis-PUA isomers.
Figure 6.
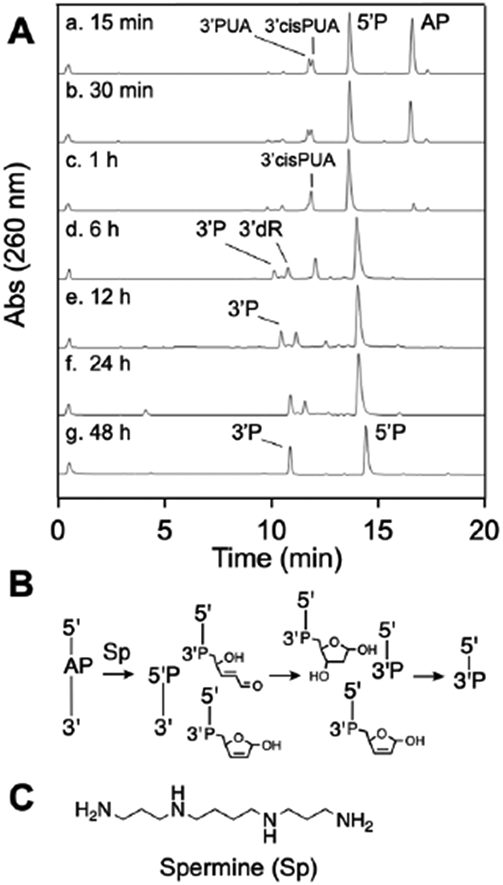
HPLC analysis of products generated from spermine-mediated strand cleavage at an AP site in DNA (panel A) and a schematic depiction of the product structures, where P represents a terminal phosphoryl group or a phosphodiester linkage (panel B). The chemical structure of spermine is shown in panel C.
Addition of 2-mercaptoethanol to this early product mixture caused disappearance of the earlier peak in the doublet, with concomitant generation of two new peaks corresponding to the diastereomeric mixture of 3-thio-2,3-ddR isomers arising from conjugate addition of the 2-mercaptoethanol to the 3’trans-PUA group (Figure 7). This suggested that the first peak in the doublet was the 3’trans-PUA product. The later-eluting peak in the doublet was unaffected by addition of 2-mercaptoethanol, consistent with the less reactive nature of the 3’cis-PUA group.95 Support for the idea that 3’cis-PUA arises from amine-catalyzed isomerization of the initially-formed 3’trans-PUA was provided by a separate experiment showing that the authentic 3’trans-PUA product generated by thermolysis of the AP-oligonucleotide was converted to the cis-trans doublet of HPLC peaks upon treatment with spermine (Figure S15).
Figure 7.
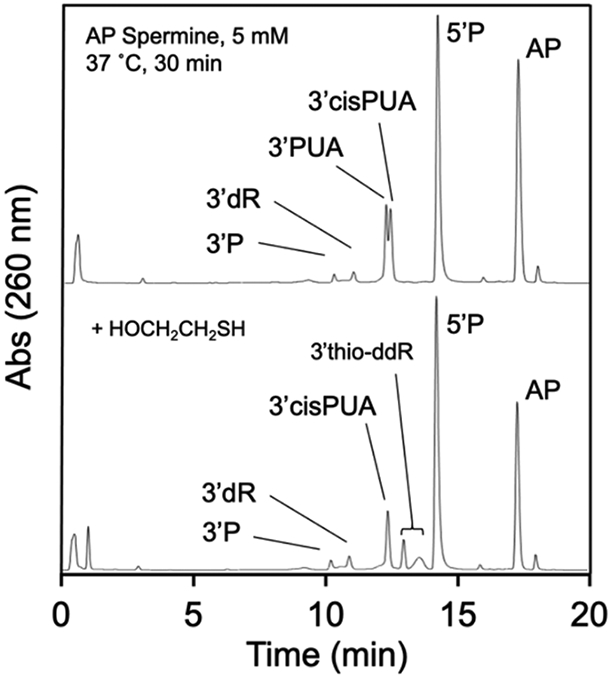
Addition of 2-mercaptoethanol to the product mixture generated by spermine-catalyzed cleavage of an AP-containing oligodeoxynucleotide leads to disappearance of one component of the doublet eluting at approximately 12.5 min, with concomitant generation of the 3-thio-ddR adduct on the 3’-terminus.
ESI-MS analysis of the reaction mixture generated by spermine-catalyzed cleavage of the AP oligonucleotide at early times supported these product assignments. Specifically, when the reaction was allowed to proceed for 30 min, followed by workup with 2-mercaptoethanol (5 mM, 15 min), we observed strong signals consistent with the intact AP oligonucleotide, 5’P, the 3-thio-ddR isomers, and the 3’cis-PUA product. Note that the 3’cis-PUA product is isobaric with 3’trans-PUA, but is resistant to reaction with the added thiol under these conditions (Figure S16).
To further support our assignment that the closely-spaced peaks eluting near 12.5 minutes in our HPLC analyses were the cis- and trans-PUA cleavage products we used a literature protocol to generate an authentic sample of these products. We employed the conditions of Kushida et al. showed that heating an AP-containing oligodeoxynucleotide in Tris-borate buffer (pH 7.5) generated a 4:1 mixture of the cis- and trans-PUA cleavage products along with the 3’dR cleavage product.75 Indeed, HPLC analysis of the product mixture generated by heating our AP-containing oligonucleotide using the Kushida conditions revealed a closely-spaced doublet of peaks eluting near 12.5 min in which the later-eluting product (3’cis-PUA) dominated. The 3’dR cleavage product, eluting near 11.5 min, also was evident in this reaction mixture, as reported by Kushida et al. Addition of 2-mercaptoethanol to this product mixture caused disappearance of the early-eluting (3’trans-PUA) component of the doublet along with the appearance of characteristic peaks corresponding to the mixture of 3-thio-ddR isomers arising from conjugate addition of the thiol to the 3’trans-PUA group (Figure 8). As noted above, the later-eluting 3’cis-PUA component of the doublet was resistant to reaction with 2-mercaptoethanol, because the aldehyde residue in this isomer exists primarily as the ring-closed hemiacetal.95 Overall, the results support our conclusion that spermine-mediated cleavage of the AP-oligonucleotide generates a mixture of 3’cis- and 3’trans-PUA end groups at early reaction times.
Figure 8.
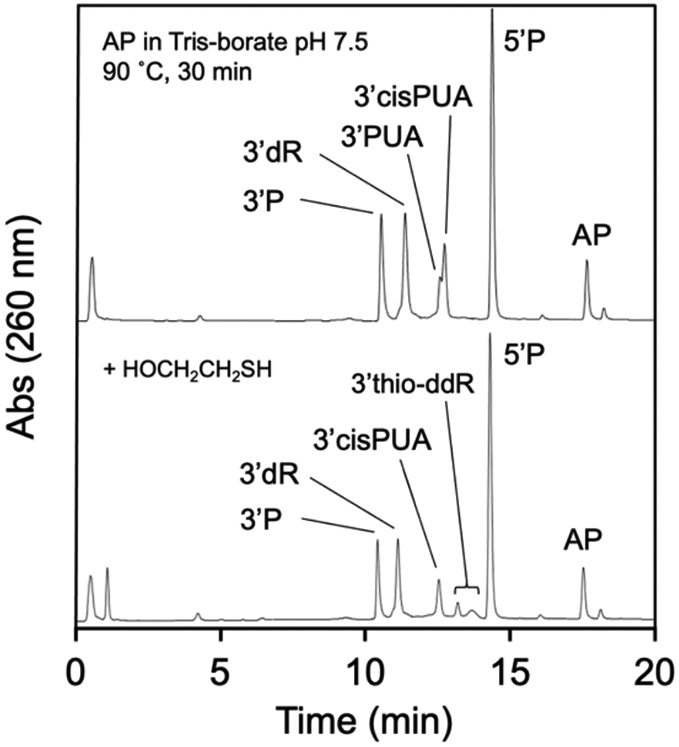
HPLC analysis of authentic cis- and trans-PUA. Upper HPLC trace shows an authentic mixture of 3’P, 3’dR, 3’PUA, and 3’cis-PUA generated by incubation of the AP-containing oligonucleotide in Tris-borate buffer according to the method of Kushida et al.75 The lower HPLC trace shows that 2-mercaptoethanol reacts with the later-eluting component 3’trans-PUA to generate a characteristic 3-thio-2,3-ddR residue on the 3’-terminus, while the early-eluting 3’cis-PUA is resistant to reaction with the thiol.
The products generated by spermine-mediated strand cleavage of the AP-containing DNA oligomer evolved into more complex mixtures at intermediate reaction times. At 1 h, the amount of the 3’trans-PUA product decreased, while the 3’cis-PUA product persisted. At reaction times of 6-12 h, the product mixture consisted of 3’cis-PUA, 3’dR and 3’P (Figure 6A, HPLC traces d and e). The identity of the 3’dR product eluting at 11.2 min was confirmed based on co-elution with an authentic standard of 5’TTTTT-dR-3’ prepared by acid treatment of a 5’-TTTTTA-3’ precursor (10 mM HCl, 1 h, 65 °C).83 Similarly, the identity of the 3’P product was confirmed based on co-elution with an authentic sample of this material.
From 12-24 h, 3’dR and 3’P were the major products (Figure 6A, HPLC traces e and f) and, by 48 h, the 3’P and 5’P were the only products remaining from the spermine-mediated strand cleavage process (Figure 6A, HPLC trace g). In a separate experiment designed to model the final stages of this product evolution, we showed that an authentic sample of 3’dR is stable in pH 7.4 buffer (in the absence of spermine) but is, indeed, converted to the 3’P product by incubation with spermine (5 mM) for 24 h at 37 ° C (Figure S17). ESI-MS analysis of the reaction at 24 h supported the product assignments shown in Figure 6 (Figure S18).
Scheme 4 shows the putative equilibria involved in the evolution of the product mixture generated by spermine-catalyzed cleavage of the AP-containing DNA oligomer. Iminium ion catalysis is central to the formation, interconversion, and ultimate disappearance of the various 3’-sugar remnants observed in this experiment (illustrated for the reversible formation of 3’dR and its ultimate conversion to 3’P, in Scheme 5).
Scheme 5.
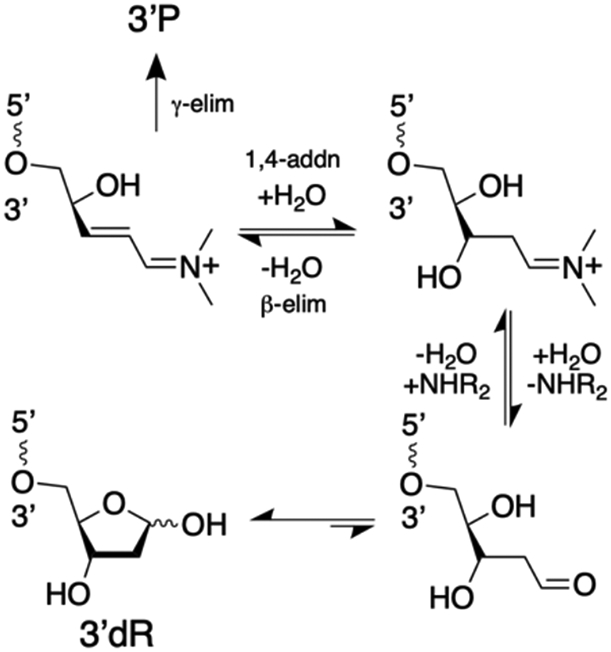
Iminium ion intermediates are central to the formation, decomposition, and interconversion of various 3’-sugar remnants generated in amine-catalyzed strand cleavage. Reactions involved in the amine-catalyzed reversible formation of various 3’-sugar remnants are shown here in the context of the 3’dR end group. For brevity, the amine catalyst is shown as a simple dialkylamine. The wavy lines annotated with 5’ and 3’ labels represent DNA strands.
Modeling cleavage of an AP site in DNA under cellular conditions: amine-catalyzed strand cleavage in the presence of thiol.
Cells contain millimolar concentrations of thiols including protein thiols and the low molecular weight thiol, glutathione.106-109 Accordingly, we felt it would be interesting to examine spermine-mediated cleavage of the AP oligonucleotide in the presence of thiol. These reactions may model one of the possible chemical fates of DNA AP sites in the cellular environment.
We treated the AP-containing oligodeoxynucleotide with spermine (5 mM) in the presence of 2-mercaptoethanol (5 mM) in pH 7.4 HEPES buffer (50 mM, containing 100 mM NaCl). At an early reaction time (1 h), the only products observed were 5’P and the characteristic adducts resulting from addition of 2-mercaptoethanol to the 3’-α,β-unsaturated iminium ion (Scheme 6, Figure 9).
Scheme 6.

Proposed equilibria involved in the evolution of various 3’-end groups following spermine-mediated cleavage of an AP site in the presence of thiol. All of the reactions and equilibria shown are subject to amine catalysis. For brevity the amine catalyst is shown as a simple dialkylamine (the actual structure of spermine is shown in Figure 6).
Figure 9.
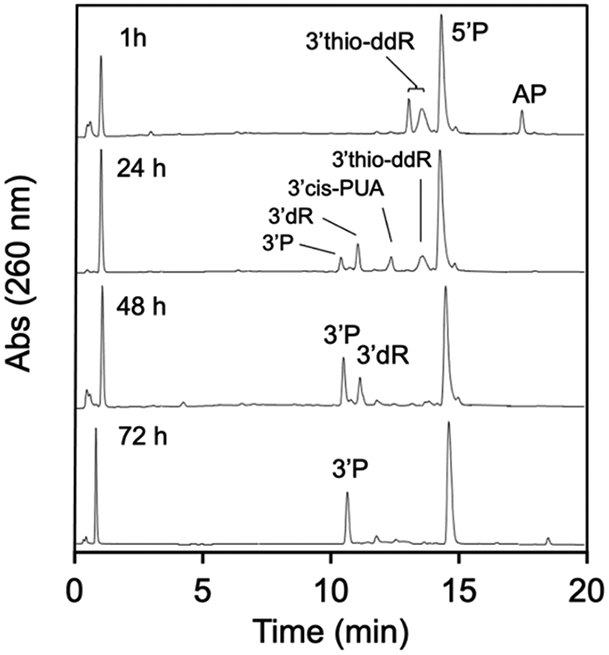
HPLC analysis of the time-course for the evolution of products resulting from cleavage of the AP-containing oligodeoxynucleotide in pH 7.4 buffer induced by spermine (5 mM) in the presence of 2-mercaptoethanol (5 mM).
By 24 h, the product mixture contained a subset of the diastereomeric thiol adducts, along with 3’cis-PUA, 3’dR, and 3’P. We infer that the species eluting near 12.5 min must be the 3’cis-PUA cleavage product because the 3’trans-PUA does not persist in the presence of thiols. It is uncertain why a subset of the diastereomeric thiol adducts is more persistent than the others (only one of the two HPLC peaks corresponding to the diastereomeric thiol adducts remains at 24 h).
At 48 h, the only significant products remaining were 3’dR and 3’P end products. By 72 h, the mixture evolved to 3’P and 5’P as the final, stable end products. In a control experiment designed to model the final stages of product evolution under these conditions, we showed that an authentic sample of the 3’dR cleavage product was stable in pH 7.4 buffer containing 2-mercaptoethanol but does, in fact, generate the 3’P end group as the major product when incubated in the presence of spermine (5 mM) and 2-mercaptoethanol (5 mM) for 24 h (Figure S17, Scheme 5).
It is noteworthy that the presence of thiol in the reaction mixture substantially slows generation of the final 3’P product relative to the same conditions without thiol (compare Figures 6 and 9). Scheme 6 shows the putative equilibria involved in the evolution of products generated by spermine-catalyzed cleavage of the DNA AP site in the presence of thiol.
The AP lyase activity of the DNA glycosylase Endo III generates 3’dR (not 3’PUA) as the major strand cleavage product.
The AP-lyase activity of bifunctional DNA glycosylase enzymes induces strand cleavage at AP sites and may be important in cellular DNA repair.54, 110, 111 The typical expectation is that these lyase reactions generate either the 3’trans-PUA product via β-elimination or the 3’P product by sequential α,β- and γ,δ-elimination reactions.4, 6, 40, 53, 55, 59, 112 Here we examined the AP lyase activity of two well-studied glycosylases, Fpg and Endo III.
The lyase activity of Fpg on AP-containing DNA was expected to generate a one nucleotide gap flanked by 3’P and 5’P groups via sequential α,β- and γ,δ-elimination reactions.4, 113 On the other hand, the expectations surrounding the lyase activity of Endo III were somewhat less clear. It is widely believed that Endo III generates the 3’trans-PUA product. For example, the New England Biolabs catalog notes that Endo III “cleaves 3’ to the AP site leaving a 5’-phosphate and a 3’-phospho-α,β-unsaturated aldehyde” (p. 109, 2019-20 catalog).114 This expectation was born in the early reports that characterized this enzyme31, 32, 40, 115, 116 and is now well entrenched in the literature of base excision repair. Interestingly, however, the Sowers and Cadet groups independently presented MALDI-MS evidence indicating that the combined glycosylase-AP lyase action of E. coli Endo III generates the 3’dR cleavage product – not 3’trans-PUA – following removal of 5,6-dihydrothymine, 5-hydroxycytosine, or 5-fluorouracil from oligomeric DNA duplexes.77, 78 In addition, more recent studies showed that the lyase action of Endo III on AP-containing duplexes generates the 3’dR cleavage product.79, 80
The amplified reactivity of the 3’PUA-lysine-120 iminium ion intermediate generated in the catalytic cycle of Endo III may enable conjugate addition of water that ultimately produces the 3’dR cleavage product (Scheme 7). The would make formation of the 3’dR product by Endo III mechanistically analogous to the spermine-catalyzed generation of 3’dR described above. The exact reasons for the apparently discordant results in the literature regarding the nature of the 3’-end group generated by the lyase action of E. coli Endo III are not completely clear, but may arise from difficulties in separating the 3’dR and 3’PUA cleavage products by gel electrophoretic and chromatographic methods.79, 80 For example, when analyzed using denaturing 20% polyacrylamide gels, the 3’dR and 3’PUA products on a 5’-Cy3-labeled 11 mer displayed only slightly different mobilities80 while, in a 5’-32P-phosphorylated 17 mer, no clear resolution of these products was observed.79 In addition, the ability of the 3’dR and 3’PUA end groups to interconvert under some conditions (as seen in Figures S8 and S17) may present a confounding factor in some analyses.116
Scheme 7.
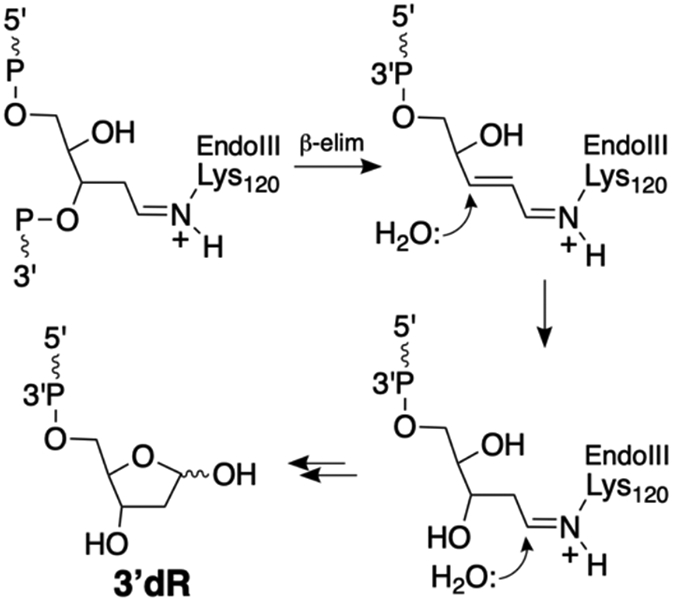
The AP-lyase action of the DNA glycosylase Endo III generates 3’dR – not 3’PUA – as the major product. The wavy lines annotated with 5’ and 3’ labels represent DNA strands and P represents a phosphodiester linkage.
We found that treatment of an AP-containing duplex with Fpg cleanly generated the expected 3’P and 5’P cleavage products (Figure 11, HPLC trace c). Treatment of an AP-containing duplex for 15 min with Endo III generated primarily the 3’dR product alongside a small amount of a 3’PUA product. Mass spectrometric analysis of the mixture supported these structural assignments (Figure S19). When the AP-oligonucleotide was incubated for 2 h with Endo III in the presence of 2-mercaptoethanol (5 mM), 3’dR was still the major product (Figure S20). This control reaction provided evidence that 3’dR is produced directly by the AP-lyase activity “on the enzyme”, rather than by conversion of an initially-generated 3’trans-PUA product to 3’dR. At 2 h, 3’dR is the only product observed. It may be interesting to note that the products generated by the AP-lyase-hydration reactions catalyzed by spermine and Endo III are formally equivalent to those resulting from hydrolysis of the phosphoryl group on the 3’side of the AP site.
Figure 11.
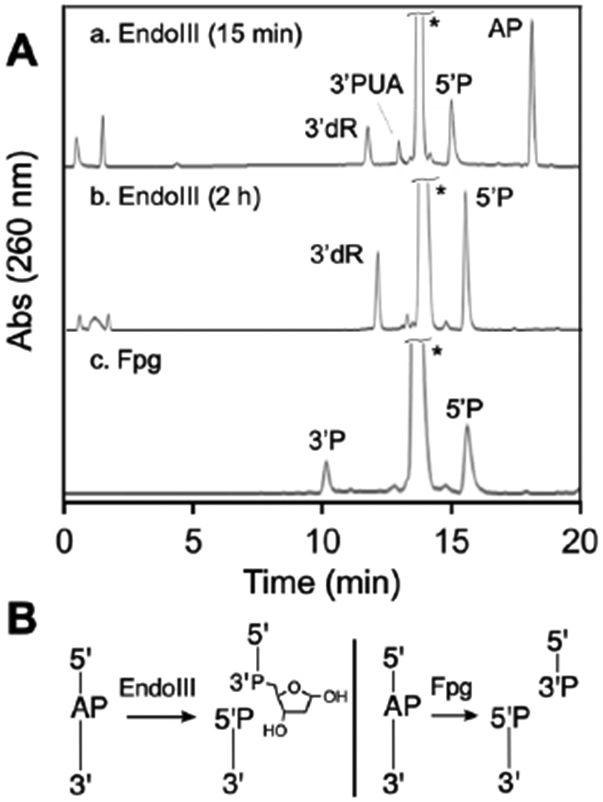
HPLC analysis of products generated by Fpg- and Endo III-mediated strand cleavage of an AP site in duplex DNA (panel A). The AP-containing strand is 5’TTTTTXTTTTTTTTTT3’, where X=AP. The large peak labeled with (*) corresponds to the complementary strand 5’TTA16TT. Panel B provides a schematic depiction of the product structures, where P represents a terminal phosphoryl group or a phosphodiester linkage.
CONCLUSIONS
In the work described here, we characterized the strand cleavage products arising from treatment of an AP-containing DNA oligonucleotide with heat, NaOH, hot piperidine, spermine, and the base excision repair glycoslyases Fpg and Endo III (Table 1). Our findings mesh well with existing literature, but also identified unexpected products generated under many of the strand cleavage conditions examined.
Table 1.
Products Generated by Cleavage of an AP-containing Oligodeoxynucleotide Under Various Conditions (HPLC data is shown in the Figures and exact conditions are discussed in the text).
| Condition |
cis- PUA |
trans- PUA |
3’dR | 3’P |
|---|---|---|---|---|
| 85C, 15 min | √ | |||
| 85C, 45 min | √ | √ | ||
| NaOH, 500 mM 2 min, 95C |
√ | |||
| NaOH, 200 mM 5 h, 37°C |
√ | |||
| NaOH, 200 mM 20 min, 37°C |
√ | √ | ||
| NaOH, 5 mM 1 h, 37°C |
√ | √ | ||
| Piperidine, 1 M 30 min, 95°C |
√ | |||
| Piperidine, 1 M 1 h, 37°C |
√ | √ | ||
| Spermine, 5 mM 37°C, 30 min |
√ | √ | ||
| 1 h | √ | |||
| 12 h | √ | √ | ||
| 48 h | √ | |||
| Fpg | √ | |||
| Endo III | √ |
Thermal treatment of the AP-containing DNA oligomer (85 °C, 15 min) in pH 7.4 buffer produced the 3’trans-PUA product as expected.37 The 3’trans-PUA product was rather stable in pH 7.4 buffer at 37 °C (in the absence of an amine catalyst) decomposing via a slow γ-elimination process to produce small amounts of the 3’P product over 12 h. In the presence of thiol, however, the 3’trans-PUA sugar remnant was rapidly converted to a diastereomeric mixture of 3-thio-2,3-ddR adducts on the 3’-end of the strand break. The thiol adducts were stable in the absence of an amine catalyst. Our reactions, carried out at 85 °C, did not produce significant amounts of the 3’dR or 3’cis-PUA end groups that were observed previously when an AP-containing trinucleotide was heated at 90 °C in pH 7 buffer.37
We found that a mild NaOH treatment (5 mM, 1 h, 37 °C) of the AP-oligonucleotide gave the expected 3’trans-PUA cleavage product. A more vigorous NaOH treatment (500 mM, 2 min, 95 °C) cleanly generated the expected 5’P and 3’P products. Interestingly, a NaOH treatment of intermediate intensity (200 mM, 20 min, 37 °C) generated significant amounts of the 3’dR product. To the best of our knowledge, the 3’dR cleavage product has not previously been observed to arise from NaOH treatment of an AP site in DNA.
We found that a standard piperidine workup (1 M, 30 min, 95 °C)99 completely cleaved the AP-containing oligonucleotide, with generation of the expected 5’P and 3’P termini at the strand break. Very mild conditions (5 mM, 1 h, 37 °C) gave a mixture of 3’P and 3’trans-PUA groups. When the AP-oligonucleotide was subjected to a piperidine workup of intermediate intensity (1 M, 20 min, 50 °C) we observed an unprecedented piperidinyl adduct on the 3’-terminus of the strand break, arising from conjugate addition of the amine to the α,β-unsaturated iminium ion intermediate.
Perhaps the most interesting reactions reported here, both from the perspective of biological significance and unexpected complexity, involved spermine-catalyzed cleavage of the AP-containing oligodeoxynucleotide. The cell nucleus is rich in spermine and other polyamines,45 making this process of likely significance in cellular DNA. The results show that amine-catalyzed cleavage of an AP site in DNA under physiologically-relevant generates complex, interconverting mixtures that include several noncanonical sugar remnants on the 3’-terminus of the strand break. Iminium ion catalysis underlies the formation, interconversion, and ultimate removal of various sugar remnants on the 3’-terminus of a AP-derived strand breaks (illustrated in Schemes 3 and 5).
We found that spermine-catalyzed strand cleavage initially generated a mixture of the 3’cis- and 3’trans-PUA isomers on the 3’-terminus of the strand break. The 3’trans-PUA isomer gave way to 3’dR and 3’P products, while the 3’cis-PUA isomer persisted. The 3’cis-PUA is relatively unreactive because the aldehyde group of this isomer exists predominantly in the masked, hemiacetal form. After 1 h, the AP-containing oligodeoxynucleotide was almost completely consumed and the 3’cis-PUA product was the major cleavage product. At 12-24 h, 3’dR was the only remaining 3’-sugar remnant and, finally, 3’P is generated as the ultimate, chemically stable end product. It is striking that two noncanonical cleavage products, the 3’cis-PUA and 3’dR, are the major sugar remnants on the 3’-terminus of the AP-derived strand break during the central portion of the reaction time-course. It may be useful to point out that, in gel electrophoretic experiments commonly used to characterize DNA-cleavage reactions, the noncanonical cleavage 3’cis-PUA and 3’dR could remain unnoticed because they may not be clearly distinguished from the expected 3’trans-PUA cleavage product.
Cells contain millimolar concentrations of thiols including the tripeptide glutathione and protein thiols.106-109 Thus, our experiments examining spermine-catalyzed cleavage of the AP-containing oligonucleotide in the presence of thiol may model the chemical fate of an AP site in cellular DNA. At early reaction times the only products observed under these conditions were the 5’P elimination product and the diastereomeric 3-thio-ddR adducts on the 3’-terminus arising from conjugate addition of thiol to the α,β-unsaturated iminium ion intermediate (Scheme 6). The thiol adducts gave way to a mixture of the 3’dR and 3’cis-PUA at 48 h and, finally, to the 3’P product by 72 h. Notably, the canonical 3’trans-PUA product was not observed in the presence of thiol.
These results suggests that, in the glutathione-rich environment of the cell,106-109 a diastereomeric mixture 3-glutathionyl-2,3-dideoxyribose end groups will be the major products arising from cleavage at AP sites in DNA. Furthermore, our results support the early report of and Bailly and Verly who noted that the formation of thiol-adducts generated by amine-catalyzed cleavage of an AP site in the presence of thiol significantly slowed the subsequent amine-catalyzed γ,δ-elimination reaction that generates the final 3’P product.38, 83
Finally, our findings reinforce earlier reports,77-80 indicating that the AP-lyase activity E. coli Endo III generates 3’dR – and not 3’PUA – as the major product. This enzymatic process is mechanistically analogous to the generation of 3’dR by the low molecular weight amine spermine, likely involving conjugate addition of water to an electrophilic α,β-unsaturated iminium ion intermediate attached to the active site lysine (Scheme 7).57 Generation of 3’dR by spermine-and Endo III-catalyzed cleavage of AP sites in DNA may have important functional consequences, as these processes evades production of the more electrophilic (and potentially genotoxic74 or cytotoxic48, 79) α,β-unsaturated aldehyde residue on the 3’-terminus of an AP-derived strand break. This could be especially important to the extent that this enzyme and its homologs function in APE-independent base excision repair pathways.64-66, 70, 110 It may be interesting to determine whether the lyase action of the human and S. cerevisiae Endo III homologs, hNth and Ntg1/2p similarly generates 3’dR.
Overall, our results reveal that the products arising from strand cleavage at an AP site in DNA can be more complex than commonly expected. Our results offer a deeper understanding of the possible degradation products arising from synthetic oligodeoxynucleotides used in molecular biology, materials science, and medicine. In addition, the results expand our understanding of 3’-blocked ends that must be chemically or enzymatically removed as part of cellular single-strand break repair and base excision repair pathways.117, 118 Finally, it is significant that some of these 3’-sugar remnants have the potential to generate interstrand cross-links at AP-derived strand breaks in duplex DNA.48, 79
Supplementary Material
Funding
We are grateful to the National Institutes of Health (ES021007) and the National Science Foundation (NSF-CHE 1808672) for support of this work.
Footnotes
SUPPORTING INFORMATION
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org.
HPLC and MS analysis of the products generated by cleavage of AP-containing DNA oligomers.
The authors declare no conflicts of interest
REFERENCES
- (1).Lindahl T, and Karlstrom O (1973) Heat-induced depyrimidination of deoxyribonucleic acid in solution. Biochemistry 12, 5151–5154. [DOI] [PubMed] [Google Scholar]
- (2).Lindahl T, and Nyberg B (1972) Rate of depurination of native deoxyribonucleic acid. Biochemistry 11, 3610–3618. [DOI] [PubMed] [Google Scholar]
- (3).Gates KS, Nooner T, and Dutta S (2004) Biologically relevant chemical reactions of N7-alkyl-2'-deoxyguanosine adducts in DNA. Chem. Res. Toxicol 17, 839–856. [DOI] [PubMed] [Google Scholar]
- (4).David SS, and Williams SD (1998) Chemistry of glycosylases and endonucleases involved in base-excision repair. Chem. Rev 98, 1221–1261. [DOI] [PubMed] [Google Scholar]
- (5).Stivers JT, and Jiang YJ (2003) A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev 103, 2729–2759. [DOI] [PubMed] [Google Scholar]
- (6).Brooks SC, Adhikary S, Rubinson EH, and Eichman BF (2013) Recent advances in the structural mechanisms of DNA glycosylases. Biochim. Biophys. Acta 1834, 247–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Imai K, Slupphaug G, Lee W, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, Ochs HD, Fischer A, and Durandy A (2003) Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol 4, 1023–1028. [DOI] [PubMed] [Google Scholar]
- (8).Gates KS (2009) An overview of chemical processes that damage cellular DNA: spontaneous hydrolysis, alkylation, and reactions with radicals. Chem. Res. Toxicol 22, 1747–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Nooner T, Dutta S, and Gates KS (2004) Chemical properties of the leinamycin 2'-deoxyguanosine adduct. Chem. Res. Toxicol 17, 942–949. [DOI] [PubMed] [Google Scholar]
- (10).Nakamura J, and Swenberg JA (1999) Endogenous apurinic/apyrimidinic sites in genomic DNA of mammalian tissues. Cancer Res. 59, 2522–2526. [PubMed] [Google Scholar]
- (11).De Bont R, and van Larebeke N (2004) Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19, 169–185. [DOI] [PubMed] [Google Scholar]
- (12).Swenberg JA, Moeller BC, Gao L, Upton PB, Nakamura J, and Starr T (2011) Endogenous versus exogenous DNA adducts: their role in carginogenesis, epidemiology, and risk assessment. Toxicol. Sci 120, S130–S145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Septak M (1996) Kinetic studies on depurination and detritylation of CPG-bound intermediates during oligonucleotide synthesis. Nucleic Acids Res. 24, 3053–3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chen H, Yao L, Brown C, Rizzo CJ, and Turesky RJ (2019) Quantitation of Apurinic/Apyrimidinic Sites in Isolated DNA and in Mammalian Tissue with a Reduced Level of Artifacts. Anal. Chem , 7403–7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wilde JA, Bolton PH, Mazumdar A, Manoharan M, and Gerlt JA (1989) Characterization of the equilibrating forms of the abasic site in duplex DNA using 17O-NMR. J. Am. Chem. Soc 111, 1894–1896. [Google Scholar]
- (16).Overend WG (1950) Deoxy-sugars. Part XIII. Some observations on the Feulgen nucleal reaction. J. Chem. Soc, 2769–2774. [Google Scholar]
- (17).Imani-Nejad M, Price NE, Haldar T, Lewis C, Wang Y, and Gates KS (2019) Interstrand DNA cross-links derived from reaction of a 2-aminopurine residue with an abasic site. ACS Chem. Biol 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gamboa Varela J, and Gates KS (2015) A Simple, High-Yield Synthesis of DNA Duplexes Containing a Covalent, Thermally-Reversible Interstrand Cross-link At a Defined Location Angew. Chem. Int. Ed. Eng 54, 7666–7669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Johnson KM, Price NE, Wang J, Fekry MI, Dutta S, Seiner DR, Wang Y, and Gates KS (2013) On the Formation and Properties of Interstrand DNA-DNA Cross-links Forged by Reaction of an Abasic Site With the Opposing Guanine Residue of 5’-CAp Sequences in Duplex DNA. J. Am. Chem. Soc 135, 1015–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Price NE, Johnson KM, Wang J, Fekry MI, Wang Y, and Gates KS (2014) Interstrand DNA–DNA Cross-Link Formation Between Adenine Residues and Abasic Sites in Duplex DNA. J. Am. Chem. Soc 136, 3483–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Catalano MJ, Price NE, and Gates KS (2016) Effective molarity in a nucleic acid controlled reaction. Bioorg. Med. Chem. Lett 26, 2627–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gamboa Varela J, Pierce LE, Guo X, Price NE, Johnson KM, Yang Z, Wang Y, and Gates KS (2021) Interstrand Cross-Link Formation Involving Reaction of a Mispaired Cytosine Residue with an Abasic Site in Duplex DNA. Chem. Res. Toxicol 34, 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM, and Cortez D (2019) HMCES maintains genome integrity by shielding abasic sites in single-strand DNA. Cell 176, 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Thompson PS, Amidon KM, Mohni KN, Cortez D, and Eichman BF (2019) Protection of abasic sites during DNA replication by a stable thiazolidine protein-DNA cross-link. Nature Struct. Mol. Biol 26, 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Halabelian L, Ravichandran M, Li Y, Zeng H, Rao A, Aravind L, and Arrowsmith CH (2019) Strutural basis of HMCES interactions with abasic DNA and multivalent substrate recognition. Nature Struct. Mol. Biol 26, 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Chan W, Ham Y-H, Jin L, Chan HW, Wong Y-L, Chan C-K, and Chung P-Y (2019) Quantification of a novel DNA-protein cross-link product formed by reacting apurinc/apyrimidinic sites in DNA with cysteine residues in protein by liquid chromotography-tandem mass spectrometry coupled with the stable isotope-dilution method. Anal. Chem 91, 4987–4994. [DOI] [PubMed] [Google Scholar]
- (27).Guthrie JP, Cossar J, and Klym A (1984) Halogenation of acetone. A method for determining pKas of ketones in aqueous solution, with an examination of the thermodynamics and kinetics of alkaline halogenation and a discussion of the best value for the rate constant for a diffusion-controlled reaction. Energetic requirements for a diffusion-controlled reaction involving heavy-atom bond formation. J. Am. Chem. Soc 106, 1351–1360. [Google Scholar]
- (28).Bayley CR, Brammer KW, and Jones AS (1961) The nucleotide sequence in deoxyribonucleic acids. Part V. The alkaline degradation of apurinic sites. J. Chem. Soc, 1903–1907. [Google Scholar]
- (29).Crine P, and Verly WG (1976) A study of DNA spontaneous degradation. Biochim. Biophys. Acta 442, 50–57. [DOI] [PubMed] [Google Scholar]
- (30).Lindahl T, and Andersson A (1972) Rate of chain breakage at apurinic sites in double-stranded deoxyribonucleic acid. Biochemistry 11, 3618–3623. [DOI] [PubMed] [Google Scholar]
- (31).Bailly V, and Verly WG (1987) Escherichia coli endonuclease III is not an endonuclease but a beta-elimination catalyst. Biochem. J 242, 565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kim J, and Linn S (1988) The mechanism of action of E. coli endonuclease III and T7 UV endonuclease (endonuclease V) on AP sites. Nucleic Acids Res. 16, 1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Brown DM, and Todd AR (1955) Nucleic acids. Ann. Rev. Biochem 24, 311–338. [DOI] [PubMed] [Google Scholar]
- (34).Zhou C, Sczepanski JT, and Greenberg MM (2012) Mechanistic studies on histone catalyzed cleavage of apyrimidinic/apurinic sites in nucleosome core particles. J. Am. Chem. Soc 134, 16734–16741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lawley PD, Lethbridge J, Edwards PA, and Shooter K (1969) Inactivation of bacteriophage T7 by mono- and difunctional sulphur mustards in relation to cross-linking and depurination of bacteriophage DNA. J. Mol. Biol 39. [DOI] [PubMed] [Google Scholar]
- (36).Laurence DJR (1963) Chain breakage of deoxyribonucleic acid following treatment with low doses of sulphur mustard. Proc. Royal. Soc. A 271, 520–530. [Google Scholar]
- (37).Sugiyama H, Fujiwara T, Ura A, Tashiro T, Yamamoto K, Kawanishi S, and Saito I (1994) Chemistry of thermal degradation of abasic sites in DNA. mechanistic investigation on thermal DNA strand cleavage of alkylated DNA. Chem. Res. Toxicol 7, 673–683. [DOI] [PubMed] [Google Scholar]
- (38).Bailly V, Derydt M, and Verly WG (1989) δ-Elimination in the repair of AP (apurinic/apyrimidinic) sites in DNA. Biochem. J 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bailly V, and Verly WG (1988) Possible roles of β-elimination and δ-elimination reactions in the repair of DNA containing AP (apurinic/apyrimidinic) sites in mammalian cells. Biochem. J 253, 553–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Mazumder A, Gerlt JA, Absalon MJ, Stubbe J, Cunningham RP, Withka J, and Bolton PH (1991) Stereochemical studies of the b-elimination reactions at aldehydic abasic sites in DNA: endonuclease III from Escherichia coli, sodium hydroxide, and Lys-Trp-Lys. Biochemistry 30, 1119–1126. [DOI] [PubMed] [Google Scholar]
- (41).McHugh PJ, and Knowland J (1995) Novel reagents for chemical cleavage at abasic sites and UV photoproducts in DNA. Nucleic Acids Res. 23, 1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Liu M, Bandaru V, Bond JP, Jaruga P, Zhao XS, Christov PP, Burrows CJ, Rizzo CJ, Dizdaroglu M, and Wallace SS (2010) The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Nat. Acad. Sci. USA 107, 4925–4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Imani-Nejad M, Housh K, Rodriguez AA, Haldar T, Kathe S, Wallace SS, Eichman BF, and Gates KS (2020) Unhooking of an interstrand cross-link at DNA fork structures by the DNA glycosylase NEIL3. DNA Repair 86, 102752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sarhan S, and Seiler N (1989) On the subcellular localization of the polyamines. Biol. Chem. Hoppe Seyler 370, 1279–1284. [DOI] [PubMed] [Google Scholar]
- (45).Male R, Fosse VM, and Kleppe K (1982) Polyamine-induced hydrolysis of apurinic sites in DNA and nucleosomes. Nucleic Acid Res. 10, 6305–6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Raspaud E, Chaperon I, Leforestier A, and Livolant F (1999) Spermine-induced aggregation of DNA, nucleosome, and chromatin. Biophys. J 77, 1547–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wallace HM, Fraser AV, and Hughes A (2003) A perspective of polyamine metabolism. Biochem. J 376, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yang Z, Price NE, Johnson KM, Wang Y, and Gates KS (2017) Interstrand cross-links arising from strand breaks at true abasic sites in duplex DNA. Nucleic Acids Res. 45, 6275–6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Sczepanski JT, Wong RS, McKnight JN, Bowman GD, and Greenberg MM (2010) Rapid DNA-protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proc. Nat. Acad. Sci. USA 107, 22475–22480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sczepanski JT, Zhou C, and Greenberg MM (2013) Nucleosome core particle-catalyzed strand scission at abasic sites. Biochemistry 52, 2157–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Pruss D, Gravin IM, Melnik S, and Bavykin SG (1999) DNA-protein cross-linking applications for chromatin studies in vitro and in vivo. Methods Enzymol. 304, 516–533. [DOI] [PubMed] [Google Scholar]
- (52).Stande NT, Carvajal J, Hallett RA, Waters CA, Roberts SA, Strom C, Kuhlman B, and Ramsden DA (2014) Requirements for 5'-dRP/AP lyase activity in Ku. Nucleic Acids Res. 42, 11136–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Mazumdar A, Gerlt JA, Rabow L, Absalon MJ, Stubbe J, and Bolton PH (1989) UV endonuclease V from bacteriophage T4 catalyzes DNA strand cleavage at aldehydic abasic sites by a syn beta-elimination reaction. J. Am. Chem. Soc 111, 8029–8030. [Google Scholar]
- (54).Maher RL, Wallace SS, and Pederson DS (2019) The lyase activity of bifunctional DNA glycosylases and the 3'-phosphodiesterase activity of APE1 contribute to the repair of oxidized bases in nucleosomes. Nucleic Acids Res. 47, 2922–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Krokan HE, and Bjørås M (2013) Base Excision Repair. Cold Spring Harb. Perspect. Biol 5, a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).McCullough AK, Sanchez A, Dodson ML, Marapaka P, Taylor JS, and Lloyd RS (2001) The reaction mechanism of DNA glycosylase/AP lyases at abasic sites. Biochemistry 40, 561–568. [DOI] [PubMed] [Google Scholar]
- (57).Fromme JC, and Verdine GL (2003) Structure of a trapped endonuclease III-DNA covalent intermediate. EMBO J. 22, 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Cornish-Bowden A (2014) Current IUBMB recommendations on enzyme nomenclature and kinetics. Perspect. Sci 1, 74–87. [Google Scholar]
- (59).Mazumdar A, Gerlt JA, Rabow L, Absalon MJ, Stubbe J, and Bolton PH (1991) UV Endonuclease V from Bacteriophage T4 catalyzes DNA strand cleavage at aldehydic sites by a syn beta-elimination reaction. J. Am. Chem. Soc 111, 8029–8030. [Google Scholar]
- (60).Piersen CE, McCullough AK, and Lloyd RS (2000) AP lyases and dRPases: commonality of mechanism. Mutation Res. 459, 43–53. [DOI] [PubMed] [Google Scholar]
- (61).Sandigursky M, Yacoub A, Kelley MR, Xu Y, Franklin WA, and Deutsch WA (1997) The yeast 8-oxoguanine DNA glycosylase (Ogg1) contains a DNA deoxyribophosphodiesterase (dRpase) activity. Nucleic Acids Res 25, 4557–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Golan G, Zharkov DO, Grollman AP, Dodson ML, McCullough AK, Lloyd RS, and Shoham G (2006) Structure of T4 pyrimidine dimer glycosylase in a reduced imine covalent complex with abasic site-containing DNA. J. Mol. Biol 362, 241–258. [DOI] [PubMed] [Google Scholar]
- (63).Rahimoff R, Kosmatchev O, Kirchner A, Pfaffeneder j., Spada F, Brantl V, Müller M, and Carell T (2017) 5-Formyl- and 5-carboxydeoxycytidines do not cause accumulation of harmful repair intermediates in stem cells. J. Am. Chem. Soc 139, 10359–10364. [DOI] [PubMed] [Google Scholar]
- (64).Silhan J, Nagorska K, Zhao Q, Jensen K, Freemont PS, Tang CM, and Baldwin GS (2012) Specialization of an Exonuclease III family enzyme in the repair of 3' DNA lesions during base excision repair in the human pathogen Neisseria meningitidis. Nucleic Acids Res 40, 2065–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Wiederhold L, Leppard JB, Kedar P, Karimi-Busheri F, Rasouli-Nia A, Weinfeld M, Tomkinson AE, Izumi T, Prasad R, Wilson SH, Mitra S, and Hazra TK (2004) AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 15, 209–220. [DOI] [PubMed] [Google Scholar]
- (66).Nilsen L, Forstrom RJ, Bjoras M, and Alseth I (2012) AP endonuclease independent repair of abasic sites in Schizosaccharomyces pombe. Nucleic Acids Res 40, 2000–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Levin JD, Johnson AW, and Demple B (1988) Homogeneous Escherichia coli endonuclease IV. Characterization of an enzyme that recognizes oxidative damage in DNA. J. Biol. Chem 263, 8066–8071. [PubMed] [Google Scholar]
- (68).Ren M, Shang M, Wang H, Xi Z, and Zhou C (2021) Histones participate in base excision repair of 8-oxodGuo by transiently cross-linking with active repair intermediates in nucleosome core particles. Nucleic Acids Res. 49, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Wilson DM III (2007) Processing of nonconventional DNA strand break ends. Env. Mol. Mutagenesis 48, 772–782. [DOI] [PubMed] [Google Scholar]
- (70).Barbado C, Cordoba-Canero D, Ariza RR, and Roldan-Arjona T (2018) Nonenzymatic release of N7-methylguanine channels repair of abasic sites into an AP endonuclease-independent pathway in Arabidopsis. Proc Natl Acad Sci U S A 115, E916–E924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Ma W, Westmoreland JW, Gordenin DA, and Resnick MA (2011) Alkylation base damage is converted into repairable double-strand breaks and complex intermediates in G2 cells lacking AP endonuclease. PLoS Genetics 7, e1002059;1002051-1002015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Guillet M, and Bioteux S (2002) Endogenous DNA abasic sites cause cell death in the absence of Apn1, Apn2, and Rad1/Rad10 in Saccaromyces cerevisae. EMBO J. 21, 2833–2841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Boiteux S, and Guillet M (2004) Abasic sites in DNA: repair and biological consequences in Saccaromyces cerevisiae. DNA Repair 3, 1–12. [DOI] [PubMed] [Google Scholar]
- (74).Simonelli V, Narciso L, Dogliotti E, and Fortini P (2005) Base excision repair intermediates are mutagenic in mammalian cells. Nucleic Acids Res 33, 4404–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Kushida T, Uesugi M, Sugiura Y, Kigoshi H, Tanaka H, Hirokawa J, Ojika M, and Yamada K (1994) DNA damage by ptaquiloside, a potent bracken carcinogen: detection of selective strand breaks and identification of DNA-cleavage products. J. Am. Chem. Soc 116, 479–486. [Google Scholar]
- (76).Chung SJ, and Verdine GL (2004) Structures of end products resulting from lesion processing by a DNA glycosylase/lyase. Chem. Biol 11, 1643–1649. [DOI] [PubMed] [Google Scholar]
- (77).Darwanto A, Farrel A, Rogstad DK, and Sowers LC (2009) Characterization of DNA glycosylase activity by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Biochem 394, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).D'Ham C, Romieu A, Jaquinod M, Gaparutto D, and Cadet J (1999) Excision of 5,6-dihydroxy-5,6-dihydrothymine, 5,6-dihydrothymine, and 5-hydroxycytosine from define sequence oligonucleotides by Escherichia coli endonuclease III and Fpg proteins: Kinetic and mechanistic aspects. Biochemistry 38, 3335–3344. [DOI] [PubMed] [Google Scholar]
- (79).Housh K, Jha JS, Yang Z, Haldar T, Johnson KM, Yin J, Wang Y, and Gates KS (2021) Formation and Repair of an Interstrand DNA Cross-Link Arising from a Common Endogenous Lesion. J. Am. Chem. Soc 143, 15344–15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Alexeeva M, Moen MN, Grosvik K, Tesfahun AN, Xu XM, Muruzabal-Lecumberri I, Olsen KM, Rasmussen A, Ruoff P, Kirpekar F, Klungland A, and Bjelland S (2019) Excision of uracil from DNA by hSMUG1 includes strand incision and processing. Nucleic Acids Res 47, 779–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Shishkina IG, and Johnson F (2000) A new method for the postsynthetic generation of abasic sites in oligomeric DNA. Chem. Res. Toxicol 13, 907–912. [DOI] [PubMed] [Google Scholar]
- (82).Vasseur J-J, Rayner B, Imbach J-L, Verma S, McCloskey JA, Lee M, Chang D-K, and Lown JW (1987) Structure of the adduct formed between 3-aminocarbazole and the apurinic site oligonucleotide model d[Tp(Ap)pT]. J. Org. Chem 52, 4994–4998. [Google Scholar]
- (83).Bailly V, and Verly WG (1988) Importance of thiols in the repair mechanisms of DNA containing AP (apurinic/apyrimidinic) sites. Nucleic Acids Res. 16, 9489–9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Bricteux-Grégoire S, and Verly WG (1989) The use of thioglycolate to distinguish between 3' AP (apurinic/apyrimidinic) endonucleases and AP lyases. Nucleic Acids Res. 17, 6269–6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Manoharan M, Mazumdar A, Ransom SC, Gerlt JA, and Bolton PH (1988) Mechanism of UV endonuclease V cleavage of abasic sites in DNA determined by 13C labeling. J. Am. Chem. Soc 110, 2690–2691. [Google Scholar]
- (86).Studzińska S, and Buszewski B (2014) Evaluation of ultra high-performance [corrected] liquid chromatography columns for the analysis of unmodified and antisense oligonucleotides. Anal. Bioanal. Chem 406, 7127–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Lindahl T, Ljunquist S, Siegert W, Nyberg B, and Sperens B (1977) DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. . J. Biol. Chem 252, 3286–3294. [PubMed] [Google Scholar]
- (88).Varshney U, and van de Sande JH (1991) Specificities and kinetics of uracil excision from uracil-containing DNA oligomers by Escherichia coli uracil DNA glycosylase. Biochemistry 30, 4055–4061. [DOI] [PubMed] [Google Scholar]
- (89).Stuart GR, and Chambers RW (1987) Synthesis and properties of oligodeoxynucleotides with an AP site at a preselected position. Nucleic Acids Res. 15, 7451–7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Seiner DR, and Gates KS (2007) Kinetics and mechanism of protein tyrosine phosphatase 1B inactivation by acrolein. Chem. Res. Toxicol 20, 1315–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Esterbauer H, Ertl A, and Scholz N (1976) The reaction of cysteine with α,β-unsaturated aldehydes. Tetrahedron 32, 285–289. [Google Scholar]
- (92).Esterbauer H, Zollner H, and Scholz N (1975) Reaction of glutathione with conjugated carbonyls. Z. Naturforsh 30, 466–473. [DOI] [PubMed] [Google Scholar]
- (93).Wakita C, Maeshima T, Yamazaki A, T. S, Ito S, Akagawa M, Ojika M, Yodoi J, and Uchida K (2009) Stereochemical configuration of 4-hydroxy-2-nonenal-cysteine adducts and their stereoselective formation in a redox-regulated protein. J. Biol. Chem 284, 28810–28822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Fekry MI, Price N, Zang H, Huang C, Harmata M, Brown P, Daniels JS, and Gates KS (2011) Thiol-Activated DNA Damage By α-Bromo-2-cyclopentenone. Chem. Res. Toxicol 24, 217–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Jha JS, Nel C, Haldar T, Peters D, Housh K, and Gates KS (2021) Products generated by amine-catalyzed strand cleavage at apurinic/apyrimidinic sites in DNA: new insights from a biomimetic nucleoside model system. Chem. Res. Toxicol , Manuscript under review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Engels C, Schwab C, Zhang J, Stevens MJA, Bieri C, Ebert M-O, McNeill K, Sturla SJ, and Lacroix C (2016) Acrolein contributes strongly to antimicrobial and heterocyclic amine transformation activities of reuterin. Sci. Rep 6, doi: 10.1038/srep36246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Mattes WB, Hartley JA, and Kohn KW (1986) Mechanism of DNA strand breakage by piperidine at sites of N7 alkylation. Biochim. Biophys. Acta 868, 71–76. [DOI] [PubMed] [Google Scholar]
- (98).Zang H, and Gates KS (2003) Sequence specificity of DNA alkylation by the antitumor natural product leinamycin. Chem. Res. Toxicol 16, 1539–1546. [DOI] [PubMed] [Google Scholar]
- (99).Maxam AM, and Gilbert W (1980) Sequencing end-labeled DNA with base-specific chemical cleavages. Methods Enzymol. 65, 499–560. [DOI] [PubMed] [Google Scholar]
- (100).Minko IG, Jacobs AC, de Leon AR, Gruppi F, Donley N, Harris TM, Rizzo CJ, McCullough AK, and Lloyd RS (2016) Catalysts of DNA strand cleavage at apurinic/apyrmidinic sites. Sci. Rep 6, DOI: 10.1038/srep28894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Bender ML, and Williams A (1966) Ketimine intermediates in amine-catalyzed enolization of acetone. J. Am. Chem. Soc 88, 2502–2508. [Google Scholar]
- (102).Gallopo AR, and Cleland WW (1979) Properties of 3-hydroxypropionaldehyde 3-phosphate. Arch. Biochem. Biophys 195, 152–154. [DOI] [PubMed] [Google Scholar]
- (103).Langenbeck W, and Sauerbier R (1937) Organic catalysts. XVII. The hydration of crotonaldehyde to aldol. Chem. Ber 70, 1540, 1540–1541. [Google Scholar]
- (104).Paras NA, and MacMillan DW (2002) The enantioselective organocatalytic 1,4-addition of electron-rich benzenes to alpha,beta-unsaturated aldehydes. J Am Chem Soc 124, 7894–7895. [DOI] [PubMed] [Google Scholar]
- (105).Enders D, Wang C, and Liebich JX (2009) Organocatalytic Asymmetric Aza - Michael Additions. Chem. Eur. J 15, 11058–11076. [DOI] [PubMed] [Google Scholar]
- (106).Meister A, and Anderson ME (1983) Glutathione. Ann. Rev. Biochem 52, 711–760. [DOI] [PubMed] [Google Scholar]
- (107).Szapacs ME, Riggins JN, Zimmerman LJ, and Liebler DC (2007) Covalent adduction of human serum albumin by 4-hydroxy-2-nonenal: kinetic analysis of competing alkylation reactions. Biochemistry 45, 10521–10528. [DOI] [PubMed] [Google Scholar]
- (108).Chen Y, Cong Y, Quan B, Lan T, Chu X, Ye Z, Hou X, and Wang C (2017) Chemoproteomic profiling of targets of lipid-derived electrophiles by bioorthogonal aminooxy probe. Redox Biol 12, 712–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Hansen RE, Roth D, and Winther JR (2009) Quantifying the global cellular thiol-disulfide status. Proc. Nat. Acad. Sci. USA 106, 422–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Meadows KL, Song B, and Doetsch PW (2003) Characterization of AP lyase activities of Saccharomyces cerevisiae Ntg1p and Ntg2p: implications for biological function. Nucleic Acids Res 31, 5560–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Alseth I, Korvald H, Osman F, Seeberg E, and Bjoras M (2004) A general role of the DNA glycosylase Nth1 in the abasic sites cleavage step of base excision repair in Schizosaccharomyces pombe. Nucleic Acids Res 32, 5119–5125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Mullins EA, Rodriguez AA, Bradley NP, and Eichman BF (2019) Emerging Roles of DNA Glycosylases and the Base Excision Repair Pathway. Trends Biochem. Sci 44, 765–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Bhagwat M, and Gerlt JA (1996) 3’- and 5’-Strand Cleavage Reactions Catalyzed by the Fpg Protein from Escherichia coli Occur via Successive β- and δ-Elimination Mechanisms, Respectively. Biochemistry 35, 659–665. [DOI] [PubMed] [Google Scholar]
- (114).New England Biolabs Catalog 2019–20. https://www.neb.com/products/m0268-endonuclease-iii-nth#Product%20Information (accessed 2021-08-30), pp 107 and 109.
- (115).Kow YW, and Wallace SS (1987) Mechanism of action of Escherichia coli Endonuclease III. Biochemistry 26, 8200–8206. [DOI] [PubMed] [Google Scholar]
- (116).Sandigursky M, Lalezari I, and Franklin WA (1992) Excision of sugar-phosphate products at apurinic/apyrimidinic sites by DNA deoxyribophosphodiesterase of Escherichia coli. Radiat. Res 131, 332–337. [PubMed] [Google Scholar]
- (117).Caldecott KW (2008) Single-strand break repair and genetic disease. Nat. Rev. Genetics 9, 619–631. [DOI] [PubMed] [Google Scholar]
- (118).Hegde ML, Hazra TK, and Mitra S (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res 18, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.