

ABSTRACT
VPS13 family proteins form conduits between the membranes of different organelles through which lipids are transferred. In humans, there are four VPS13 paralogs, and mutations in the genes encoding each of them are associated with different inherited disorders. VPS13 proteins contain multiple conserved domains. The Vps13 adaptor-binding (VAB) domain binds to adaptor proteins that recruit VPS13 to specific membrane contact sites. This work demonstrates the importance of a different domain in VPS13A function. The pleckstrin homology (PH) domain at the C-terminal region of VPS13A is required to form a complex with the XK scramblase and for the co-localization of VPS13A with XK within the cell. Alphafold modeling was used to predict an interaction surface between VPS13A and XK. Mutations in this region disrupt both complex formation and co-localization of the two proteins. Mutant VPS13A alleles found in patients with VPS13A disease truncate the PH domain. The phenotypic similarities between VPS13A disease and McLeod syndrome caused by mutations in VPS13A and XK, respectively, argue that loss of the VPS13A–XK complex is the basis of both diseases.
KEY WORDS: Neurodegeneration, Neuro-acanthocytosis syndromes, Lipid transport, PH domain, VPS13A, XK
Summary: Mutations in VPS13A and XK cause related neurodegenerative disorders. A pathological VPS13A mutation disrupts binding of the VPS13A and XK proteins, suggesting a common basis of both diseases.
INTRODUCTION
VPS13 family members belong to an important class of lipid-transfer proteins that localize to membrane contact sites created where membranes of different organelles come into close apposition (Dziurdzik and Conibear, 2021; Leonzino et al., 2021; McEwan and Ryan, 2021; Melia and Reinisch, 2022). In humans, there are four VPS13 paralogs, VPS13A, VPS13B, VPS13C and VPS13D. VPS13A is present at both endoplasmic reticulum (ER)–mitochondrial junctions and ER–lipid droplet junctions (Kumar et al., 2018; Muñoz-Braceras et al., 2019; Yeshaw et al., 2019). The rod-shaped VPS13 proteins span the gap between two organelles, and a hydrophobic groove running through the interior of the protein allows lipids to transit in bulk between the two membranes (Leonzino et al., 2021; Li et al., 2020).
In yeast, there is a single Vps13 protein that localizes at different membrane contact sites depending on the growth and differentiation state of the cell (Lang et al., 2015; Park et al., 2016). The distribution of Vps13 to these different sites is regulated by its interaction with organelle-specific adaptor proteins that compete for binding to a region of Vps13, termed the Vps13 adaptor-binding (VAB) domain (Fig. 1A) (Bean et al., 2018). Loss of a particular adaptor protein releases Vps13 from specific organelles, resulting in a subset of phenotypes observed in vps13 null mutants (Lang et al., 2015; Park et al., 2013).
Fig. 1.
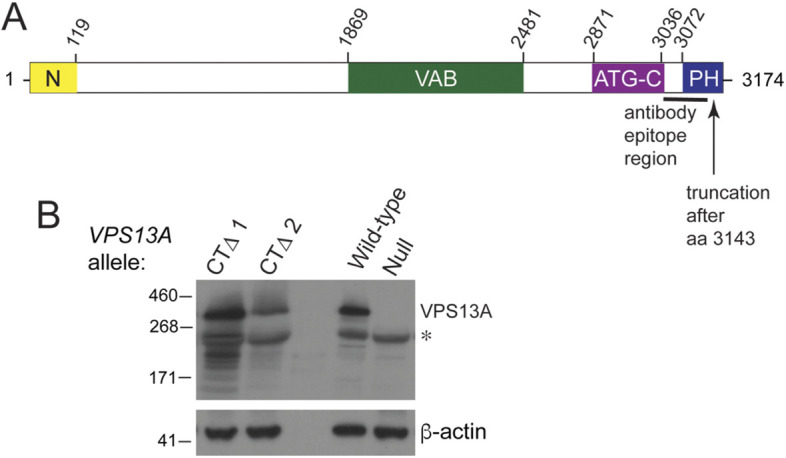
A truncation of the VPS13A C-terminus associated with VPS13A disease does not dramatically affect protein stability. (A) Schematic of the domains of the VPS13A protein. Yellow indicates the N-Chorein domain, green indicates the Vps13 adaptor-binding (VAB) domain, purple is the ATG2 C-terminal (ATG-C) domain and blue denotes the PH-like domain (Rzepnikowska et al., 2017). Numbers above indicate the amino acid positions within VPS13A. The black line indicates the position of the protein fragment used to produce the VPS13A antibody used in panel B, and the arrow indicates the end point of the VPS13A-CTΔ truncation. (B) Immunoblotting of VPS13A proteins from human blood samples. Samples from patients with VPS13A disease were probed with anti-VPS13A antibodies that recognize an epitope in the C-terminal region of the protein that does not overlap with the deleted region in VPS13A-CTΔ. CTΔ1 and CTΔ2 indicate samples from patients 1 and 2, respectively. WT is a control sample from a healthy individual, and ‘null’ is a sample from a patient diagnosed with VPS13A disease, which fails to exhibit the VPS13A protein (Niemela et al., 2020). The asterisk indicates a prominent cross-reacting species. β-actin was used as a loading control. The experiment was performed twice.
The yeast paradigm likely holds true for the VPS13 paralogs in human cells. Mutations in each VPS13 gene are associated with different inherited disorders (Gauthier et al., 2018; Kolehmainen et al., 2003; Lesage et al., 2016; Rampoldi et al., 2001; Seong et al., 2018; Ueno et al., 2001). For example, the adult-onset neurodegenerative disorder VPS13A disease (also known as chorea-acanthocytosis) is due to loss of VPS13A function (Rampoldi et al., 2001; Ueno et al., 2001; Walker and Danek, 2021). Some patients with VPS13A disease have missense mutations in the VPS13A VAB domain, indicating that VPS13A binding to adaptor proteins is likely important for its function (Dobson-Stone et al., 2002). One candidate for a VPS13 adaptor protein is XK. XK is a scramblase, which is able to flip lipids between the leaflets of a lipid bilayer (Adlakha et al., 2022; Ryoden et al., 2022). People with mutations in the XK gene exhibit a neurodegenerative disorder known as McLeod syndrome, which shares phenotypic similarities with VPS13A disease (Roulis et al., 2018). The XK protein co-localizes with VPS13A in cells by formation of a VPS13A–XK complex (Park and Neiman, 2020; Urata et al., 2019). However, a VAB-domain mutant was not found to disrupt formation of the VPS13A–XK complex in vivo (Park and Neiman, 2020). As VAB-domain mutants might disrupt binding to multiple different adaptor proteins, the question of whether VPS13A and XK function together remained open. This work shows that XK forms a complex with VPS13A, not by binding to the VAB domain, but instead by binding to a pleckstrin homology (PH) domain localized at the C-terminal end of VPS13A. Furthermore, we demonstrate that two unrelated patients with VPS13A disease carry an allele of VPS13A that encodes a truncated protein missing a portion of the PH domain. These results provide in vivo evidence that VPS13A disease and McLeod syndrome share the loss of functional VPS13A–XK complexes.
RESULTS
A small truncation of the VPS13A C-terminus results in VPS13A disease
Two unrelated patients with VPS13A disease both carried the VPS13A-c9431_9432delAG allele (hereafter VPS13A-CTΔ for C-terminal deletion) (Fig. 1A) (Dobson-Stone et al., 2002; Richard et al., 2019). This allele contains a frameshift mutation that removes the last 31 amino acids (aa) from the end of the 3174 aa-containing VPS13A protein and adds six additional amino acids to the C-terminus. The other alleles in patient 1 and patient 2 were VPS13A-c.6725delA and VPS13A-c.4956+1G>A, respectively. These alleles contain a nonsense and a splice-site mutation, respectively, which could produce only severely truncated proteins (Dobson-Stone et al., 2002). To determine whether the VPS13A-CTΔ protein was stably expressed, erythrocyte membrane samples from patients 1 and 2 were analyzed by immunoblotting using anti-VPS13A antibodies that recognize the C-terminal region of VPS13A (Fig. 1A). The VPS13A positive control came from a healthy individual, whereas the negative control used membrane samples from a patient with VPS13A disease in which the protein was known to be absent (Niemela et al., 2020). Samples from both patients carrying the VPS13A-CTΔ allele displayed mutant VPS13A proteins with a similar mobility to the wild-type (WT) protein as expected, given that the 3 kDa lost in the truncation makes up a small fraction of the full-length 349 kDa WT protein (Fig. 1B). The extreme C-terminal region of VPS13A is therefore critical for a function in humans that prevents development of VPS13A disease.
The VPS13A C-terminus is required for co-localization with XK at the ER in human cells
The VPS13A-CTΔ allele in humans affects the C-terminus in two ways: (1) removal of the last 31 amino acids and (2) addition of six amino acids to the end of the truncated protein. To determine whether it is the truncation that affects VPS13A function, a stop codon was introduced immediately after codon 3143 in an allele of VPS13A that carried an internal fusion of the gene encoding the mCherry fluorescent protein (designated VPS13A1–1343^mCherry). This mutant allele, as well as a VPS13A^mCherry control (Kumar et al., 2018), was transfected into the human embryonic kidney cell line HEK293T and the VPS13A proteins visualized by fluorescence microscopy.
Previous work showed that VPS13A localizes predominantly to short patches throughout the ER, representing ER–mitochondrial contacts, and on lipid droplets (Kumar et al., 2018; Muñoz-Braceras et al., 2019; Yeshaw et al., 2019). Consistent with these results, VPS13A^mCherry localization to lipid droplets was prominent (Fig. 2A). In contrast, the VPS13A1–3143^mCherry protein exhibited only a diffuse cytosolic fluorescence and co-localization with a lipid droplet marker was greatly reduced (Fig. 2A).
Fig. 2.

The VPS13A C-terminus is required for co-localization with XK in cells and for VPS13A–XK complex formation. (A) Cellular localization. HEK293T cells were transfected with plasmids expressing either VPS13A^mCherry (pVPS13A^mCherry) or VPS13A1–3143^mCherry (pJS160). Protein localization was examined by wide-field fluorescence microscopy. Lipid droplets were visualized using the blue fluorescent marker monodansylpentane (MDH) (Currie et al., 2014). Dotted lines indicate the outline of the cell. Arrows highlight a subset of examples of co-localization between VPS13A^mCherry and lipid droplets. For VPS13A^mCherry, 100% of lipid droplets displayed red fluorescence, whereas for VPS13A1–3143^mCherry, only 1.8% of lipid droplets displayed VPS13A^mCherry fluorescence (300 lipid droplets scored in each of three independent experiments). Scale bar: 5 µm. (B) HEK293T cells were co-transfected with plasmids expressing either VPS13^mCherry or VPS13A1–3143^mCherry together with a plasmid expressing GFP–XK. Arrowheads indicate co-localization of VPS13^mCherry with GFP–XK. Co-localization was scored in cells showing both fluorescent proteins. For VPS13A^mCherry, 100% co-localization with GFP–XK was seen, whereas for VPS13A1–3143^mCherry, 0% co-localization with GFP–XK was seen (25 cells examined in each of three independent experiments). Scale bar: 5 µm. (C) Co-immunoprecipitation. HEK293T cells were transfected with plasmids expressing either VPS13A^mCherry, VPS13A1–3143^mCherry or VPS13AW2460R^mCherry with or without a plasmid expressing GFP–XK [pcDNA3.1(+)-N-eGFP-XK]. Twenty-four hours after transfection, cells were lysed and GFP nanobodies coupled to agarose beads were used to immunoprecipitate GFP–XK. Bound proteins were probed on immunoblots with anti-GFP and anti-mCherry antibodies to detect XK and VPS13A, respectively. GFP–XK displayed a series of slower-mobility forms, likely due to incomplete denaturation of the multiple transmembrane regions of the XK protein. Right panels show total lysates prior to incubation with the GFP nanobodies. GAPDH was used as a loading control. The experiment was performed three times.
Localization of VPS13A^mCherry was altered when GFP–XK was simultaneously overexpressed, moving from lipid droplets to patches in the ER, where it co-localized with the XK protein (Fig. 2B) (Park and Neiman, 2020). However, when the plasmid expressing VPS13A1–3143^mCherry was co-transfected with the plasmid expressing GFP–XK, no co-localization with XK was observed (Fig. 2B). These results indicate that the last 31 amino acids of VPS13A are required for VPS13A–XK co-localization within the cell.
With the currently available XK antibodies, endogenous levels of the protein are cytologically undetectable, which is why over-expression of XK is required. It is possible that the high level of XK overexpression is responsible for the ER co-localization of VPS13^mCherry and XK. XK expression was therefore reduced by replacing the full CMV promoter with a smaller fragment that promotes weaker expression (Rodova et al., 2013). This promoter lowered the expression of GFP–XK such that it was not detected in total lysates, but immunoprecipitation of GFP–XK could still pull down endogenous VPS13A (Fig. S1A). When examined by fluorescence microscopy, the overall fluorescence signal from GFP–XK was lowered; however, GFP–XK still displayed co-localization with VPS13A^mCherry in ER patches (Fig. S1B). Although this co-localization within the ER might not represent the subcellular distribution of the endogenous complex formed when VPS13A and XK are expressed at their native levels, this cytological assay nonetheless provides a readout for the ability of the two proteins to interact in vivo. Because expression levels did not affect the results, the full CMV promoter constructs were used in all subsequent experiments so that GFP–XK could be assessed in total lysates.
VPS13A–XK complex formation requires the last 31 amino acids of VPS13A
In yeast, Vps13 is recruited to various locations through adaptor proteins localized to specific organelles that bind the VAB domain. This paradigm does not fit for VPS13A and XK, however, as a mutation in the VPS13A VAB domain (VPS13AW2460R) disrupts co-localization of VPS13A and XK, but does not disrupt the co-immunoprecipitation (co-IP) of the two proteins (Park and Neiman, 2020). The discovery that the VPS13A C-terminus is also required for XK localization in cells suggested that this might be the region responsible for the interaction between the two proteins.
The plasmid expressing GFP–XK was co-transfected into HEK293T cells with plasmids expressing either VPS13A^mCherry, VPS13A1–3143^mCherry or VPS13AW2460R^mCherry, and the GFP–XK protein was immunoprecipitated from cell lysates using anti-GFP nanobodies. These immunoprecipitates were then probed with anti-mCherry antibodies to detect VPS13A. As a specificity control, cells transfected with only the VPS13A^mCherry-encoding constructs were also tested. The VPS13A^mCherry protein was present in the co-IP with GFP–XK, but not when GFP–XK was absent (Fig. 2C). No co-IP was observed for GFP–XK and VPS13A1–3143^mCherry (Fig. 2C). Unlike the VPS13-CTΔ protein observed in the patient samples, (Fig. 1), the steady-state VPS13A1–3143^mCherry protein level was reduced in total lysates relative to the WT protein (whether this difference is due to the additional six amino acids added to the end of VPS13-CTΔ protein is not known). However, this is not the cause of the failure of VPS13A1–3143^mCherry to co-IP with GFP–XK, as the VPS13AW2460R protein level was similarly reduced in total lysates but still immunoprecipitated with GFP–XK (Fig. 2C). Quantification of the relative co-IP efficiency for each VPS13A protein, normalized for abundance in the total lysates, revealed that the level of VPS13AW2460R was reduced 30% relative to that of VPS13A, whereas the level of VPS13A1–3343 is reduced 20-fold. These results show that VPS13A–XK complex formation requires the C-terminal 31 aa of VPS13A.
The C-terminal PH domain of VPS13A is sufficient for XK binding
The final 31 aa of VPS13A are part of a PH domain, a common structural motif known to be involved in both protein–protein and protein–lipid interactions (Scheffzek and Welti, 2012). To determine whether the intact PH domain is required for XK interaction or just the extreme C-terminus, GFP fusions to different parts of the VPS13A C-terminus were constructed. GFP–VPS13A2751–3174 included the ATG-related C-terminal (ATG-C) and PH domains (Fig. 1A), GFP–VPS13A3927–3174 contained only the intact PH domain, GFP–VPS13A3027–3143 contained the PH domain with the 31 aa truncation and GFP–VPS13A3144–3174 contained only the last 31 aa. Plasmids encoding these alleles were transfected into HEK293T cells with and without the plasmid encoding XK and steady-state VPS13A protein levels examined by immunoblotting using anti-GFP antibodies. The three larger fusion proteins exhibited similar protein levels regardless of the presence or absence of XK (Fig. 3A). Levels of the smallest fusion protein were also similar in both conditions, although because GFP–VPS133144–3174 would not be resolved from GFP alone in this gel system, proteolytic cleavage of the 31 aa could not be ruled out.
Fig. 3.

The VPS13A PH domain is sufficient for VPS13A–XK complex formation. (A) Co-immunoprecipitation of XK with different fragments of VPS13A fused to GFP. HEK293T cells were transfected with plasmids expressing GFP–VPS13A2751–3174, which contains the ATG-C (purple box) and PH (blue box) domains (pJS164); GFP–VPS13A3027–3174, which contains only the PH domain (pJS166); GFP–VPS13A3027–3143, which contains the 31 aa truncation of the PH domain (pJS171); or GFP–VPS13A3144–3174, which contains only the last 31 aa of VPS13A (pJS167); with or without co-transfections with a vector expressing an untagged XK gene (pJS169). Twenty-four hours after transfection, cells were lysed and total lysates were probed with anti-XK antibodies or anti-GFP antibodies to detect different GFP–VPS13A fusions. Anti-GAPDH antibody was used as a loading control. Nanobodies coupled to agarose beads were used to isolate the GFP–VPS13A fusion proteins and the immunoprecipitates were then probed for the total lysates. Four biological replicates were performed. (B) Localization. HEK293T cells were transfected with the same GFP–VPS13A vectors as in (A) in combination with the vector expressing mRFP–XK (pJS142-D3). Then, protein localization was examined by confocal fluorescence microscopy. Arrowheads in the upper left panel indicate examples of enrichment of GFP fluorescence on the lipid droplet surface. When expressed alone, only GFP–VPS13A2751–3174 displayed a clear signal on the lipid droplet surface (69% of cells versus 0% for all other constructs, >100 cells scored in two independent experiments). For GFP–VPS13A2751–3174 and GFP–VPS13A3027–3174, the changes in distribution seen with co-expression of mRFP–XK were seen in 100% of co-transfected cells (n>50 in each of two independent experiments), whereas no change was seen in 100% of co-transfected cells for GFP–VPS13A3027–3243 and GFP–VPS13A3144–3174 (n>100 over two independent experiments). Residue numbers and representation of the domains is shown on the left. Scale bar: 5 µm.
Fluorescence microscopy was used to examine the localization patterns of the different fusion proteins in the presence or absence of mRFP–XK. In the absence of exogenous mRFP–XK, GFP–VPS13A2751–3174 localized diffusely in the cell with some localization on the surface of lipid droplets (arrowheads in Fig. 3B), as described previously (Kumar et al., 2018). This localization was lost when mRFP–XK was overexpressed, with GFP–VPS13A2751–3174 instead mirroring the distribution of mRFP–XK throughout the endomembrane system (Fig. 3B). This is similar to the behavior of full-length VPS13A^mCherry, in that overexpression of XK recruits VPS13A away from lipid droplets to the ER (Park and Neiman, 2020). Thus, the C-terminal 424 aa of VPS13A contain the region essential for XK association, but other regions are necessary to limit their co-localization to the ER.
The GFP–VPS133027–3174 C-terminal fusion protein containing only the PH domain localized diffusely throughout both the cytoplasm and nucleoplasm without any clear localization on lipid droplets (Fig. 3B). Thus, the ATG-C domain is necessary for lipid droplet localization, consistent with the results of Kumar et al. (2018). When co-expressed with mRFP–XK, the distribution of GFP–VPS133027–3174 within the cell changed to mirror that of XK, similar to what was observed for GFP–VPS13A2751–3174 (Fig. 3B). Therefore, the PH domain of VPS13A is sufficient for association with XK. Removal of the last 31 aa from the PH domain abolished co-localization with XK, as did the fusion with just the 31 aa alone (Fig. 3B). These results show that the C-terminal 31 aa of VPS13A are necessary but not sufficient for co-localization with XK.
To determine whether these co-localization defects reflect changes in the ability of the VPS13A fragments to bind to XK, the same constructs were transfected into HEK293T cells with or without a vector expressing untagged XK. The GFP fusion proteins were immunoprecipitated and the co-immunoprecipitation of XK assessed using anti-XK antibodies (Fig. 3C). Note that endogenous XK protein levels were too low to be detected under these conditions either in total lysates or in the GFP–VPS13A immunoprecipitates (Fig. 3A,C, lanes 1 and 2). Consistent with the localization results, overexpressed XK protein co-immunoprecipitated with both GFP–VPS13A2751–3174 and GFP–VPS133027–3174 but with neither of the two shorter fragments. Taken together with the localization data, these results demonstrate that the PH domain at the C-terminus of VPS13A is the region responsible for binding to the XK protein.
Mutations in the second intracellular loop of XK disrupt binding to the VPS13A PH domain
XK is an integral membrane protein with ten predicted hydrophobic regions. An Alphafold structural prediction of XK suggested that two of these hydrophobic stretches are intramembrane hairpins rather than transmembrane domains (Fig. 4A), as was previously proposed (Jumper et al., 2021; Varadi et al., 2022). A recent cryo-electron microscopy structure of the related protein XKR8 supports this topological arrangement for XK (Ryoden et al., 2022). The consequence of this change is that three loops previously predicted to be extracellular loops are actually cytoplasmic. XKR2 (also known as XKRX) encodes the most closely related paralog of XK in human cells and co-immunoprecipitated with VPS13A in a proteomic study (Huttlin et al., 2015; Suzuki et al., 2014). On the assumption that the VPS13A binding site is conserved between XKR2 and XK, the cytoplasmic loops of these two proteins were aligned, revealing an identical 6 aa sequence in the second intracellular loop of both proteins (Fig. 4B). Based on this alignment, an allele of XK was constructed in which these six conserved residues (EEPYVS at aa 97–102 of XK) were mutated to alanine (XK6A).
Fig. 4.

Amino acids in the second intracellular loop of XK are required for interaction with VPS13A. (A) Predicted topology of the XK protein based on Alphafold structural prediction (Jumper et al., 2021; Varadi et al., 2022). The second intracellular loop is highlighted in red. (B) Alignment of sequences from the second intracellular loops of XK and XKR2. The invariant residues highlighted in red were mutated to alanines in the XK6A allele. (C) Localization. HEK293T cells were transfected with constructs expressing either GFP–XK or GFP–XK6A with or without VPS13A^mCherry. Arrowheads highlight co-localization of GFP–XK and VPS13A^mCherry. Scale bar: 5 µm. (D) Co-immunoprecipitation of endogenous VPS13A with GFP–XK or GFP–XK6A. HEK293T cells were transfected with constructs expressing either GFP–XK [pcDNA3.1(+)-N-eGFP-XK] or GFP–XK6A (pJS165). GFP–XK was immunoprecipitated with GFP nanobodies as in Fig. 2C. Precipitates and total lysates were probed on immunoblots using anti-VPS13A and anti-GFP antibodies (left panels). GAPDH was used as a loading control. (E) Co-immunoprecipitation of overexpressed untagged XK or XK6A with the VPS13A PH domain. HEK293T cells were transfected with a construct expressing the GFP–VPS13A3027–3174 fusion protein alone (pJS166) or with untagged XK (pJS169) or XK6A (pJS173) and processed as described in Fig. 3A. The experiments in D and E were each performed twice.
HEK293T cells expressing GFP–XK6A in the presence of endogenous VPS13A displayed similar levels of fluorescence and subcellular distribution as the WT GFP–XK protein (Fig. 4C; 140 cells scored over three independent experiments). When VPS13A^mCherry was co-expressed with GFP–XK, 100% of the cells exhibited co-localization of the two proteins (Fig. 4C). The fraction of cells displaying co-localization was reduced tenfold when GFP–XK6A was co-expressed with VPS13A^mCherry (350 cells scored over five independent experiments).
The conserved residues in the second intracellular loop not only promote VPS13A^mCherry and XK co-localization, they are also required for VPS13A–XK complex formation. Endogenous VPS13A co-immunoprecipitated with GFP–XK, but not GFP–XK6A, even though more of the mutant protein was present in the immunoprecipitate (Fig. 4D). Furthermore, this region of XK specifically interacts with the VPS13A PH domain, as untagged XK6A failed to co-immunoprecipitate with the GFP–VPS133027–3174 fragment (Fig. 4E).
Alphafold prediction of the VPS13A–XK binding interface
To define more precisely the binding interface between VPS13A and XK, the multimer mode of Alphafold was used to predict the structure of the PH domain of VPS13A in complex with the second intracellular loop of XK (Fig. 5A) (Evans et al., 2022 preprint; Jumper et al., 2021). The XK sequence in this region is largely unstructured except for two short β-strands that align with three β-strands of the PH domain in VPS13A to create a short β-sheet. Thus, the predicted interface between the two proteins is the contact between one β-strand from XK and one from VPS13A. The structured portions of this model are predicted with high confidence based on the predicted local distance difference test and predicted aligned error scores (Fig. S2).
Fig. 5.

Identification of an XK–VPS13A binding interface. (A) Alphafold multimer assembly of the VPS13A PH domain (blue) with the second intracellular loop of XK (red) showing antiparallel alignment of a predicted β-strand in XK (aa 100–106) with one in VPS13A (aa 3144–3150). Green shows the position of I3148 of VPS13A. (B) Cellular localization. The localization of GFP–VPS13A3027–3174 or GFP–VPS13A3027–3174-I3148P (pJS172) was analyzed in HEK293T cells with and without co-expression of mRFP–XK. Scale bar: 5 µm. Images shown are representative of >100 cells examined for each transfection. (C) HEK293T cells were transfected with constructs expressing GFP–VPS13A3027–3174 (indicated by the blue box) or GFP–VPS13A3027–3174-I3148P (indicated by the blue box with an asterisk) with or without untagged XK and processed as in Fig. 3A. Three biological replicates were performed.
Our results above are consistent with this model as the 31 aa truncation of VPS13A that disrupts the XK interaction removes this critical β-strand in VPS13A. Similarly, the mutations present in the XK6A allele mutate three residues in the β-strand of XK that aligns with VPS13A. To test the prediction more directly, the I3148P mutation was introduced into GFP–VPS133027–3174. This mutation is predicted to introduce a twist in the VPS13A β-strand, which could disrupt interaction with XK. The WT and I3148P versions of GFP–VPS133127–3174 were both expressed in HEK293T cells and examined for co-localization with mRFP–XK. Although both VPS13A proteins localized throughout the cell when expressed alone, the subcellular localization of GFP–VPS13A3217–3174-I3148P did not change in the presence of mRFP–XK, unlike GFP–VPS13A3217–3174 (Fig. 5B) (100% of co-transfected cells examined, n>50 in each for two independent experiments). Similarly, introduction of the I3148P mutation in the GFP–VPS133027–3174 protein abolished the co-IP with XK without affecting the stability of GFP–VPS133027–3174 (Fig. 5C). Thus, this point mutation was sufficient to disrupt the interaction between the VPS13A and XK, providing additional support for the Alphafold model of the binding interface.
DISCUSSION
The paradigm developed in yeast is that Vps13 is recruited to specific membrane contact sites via interaction of the VAB domain with organelle-specific adaptor proteins (Bean et al., 2018; John Peter et al., 2017; Park et al., 2013). Point mutations in the VAB domain have been identified in patients with VPS13A-, VPS13B- and VPS13D-related diseases, indicating that this domain is critical for VPS13 function in human cells as well (Dobson-Stone et al., 2002; Gauthier et al., 2018; Kolehmainen et al., 2003). However, it has become clear that Vps13 recruitment and function is more complex than this simple paradigm. For example, a recent report in yeast showed that the Arf1 protein binds to the PH domain of Vps13 to recruit Vps13 to the Golgi complex (Kolakowski et al., 2021).
This work has revealed that the PH domain of VPS13A plays a critical function in human cells by providing an interaction interface with XK. A mutation in the VPS13A VAB domain disrupts VPS13–XK co-localization without disrupting VPS13–XK complex formation (Park and Neiman, 2020) (Fig. 2C). By contrast, mutations that truncate the PH domain in VPS13A, change a conserved cytoplasmic loop in XK, or disrupt a predicted interaction surface between the VPS13A PH domain and XK eliminate both complex formation and co-localization. These results argue that the interaction between VPS13A and XK is direct and provide strong support for the idea that the phenotypic similarities between VPS13A disease and McLeod syndrome are due to disruption of this complex.
We propose that the presence of multiple partner-binding sites reflects regulation of VPS13 protein localization via combinatorial recruitment. For example, there are at least three domains in VPS13A that are necessary for lipid droplet localization: (1) the VAB domain, (2) the PH domain and (3) the ATG-C domain (Kumar et al., 2018; Park and Neiman, 2020) (Fig. 3B). VPS13A disease mutations in either the VAB or PH domain alone disrupt localization to lipid droplets (Park and Neiman, 2020) (Fig. 2A). As these domains likely bind discrete partners, this suggests that interaction between VPS13A and at least two other proteins is necessary for recruitment to lipid droplets.
The VPS13A PH domain is required both for lipid droplet localization and for binding to XK. However, XK is unlikely to be directly responsible for recruitment of VPS13A to lipid droplets. XK itself is not localized at lipid droplets and, in fact, XK overexpression recruits VPS13A away from lipid droplets (Fig. 2A; Park and Neiman, 2020). Thus, analogous to the situation in yeast in which multiple Vps13 adaptor proteins compete for binding to the VAB domain, our results suggest that XK competes with an unknown lipid-droplet-localized VPS13A partner for binding to the VPS13A PH domain.
The idea that multiple interactions regulate VPS13A association with different membranes is also supported by an Alphafold structural prediction of the full-length protein (Fig. 6). Alphafold was used to generate a full-length model of the VPS13A–XK complex by dividing VPS13A into four slightly overlapping sections, predicting the structure of each section independently, and then aligning the overlapping regions of the structures (Fig. 6). The XK protein was then docked onto VPS13A using the predicted structure of the interface shown in Fig. 5A. The position of the transmembrane domains of XK indicate where the organelle membrane would be located and allow the orientation of the C-terminal end of VPS13A with respect to the membrane. Interestingly, the VAB, ATG-C and PH domains in the C-terminal half of the protein were all in position to link the VPS13A to the membrane (Fig. 6). The VAB and PH domains likely do this by binding to membrane-associated proteins, e.g. by binding to adaptor proteins and XK, respectively. Additionally, this structure predicts that the ATG-C domain consists of four amphipathic helices that expose a hydrophobic surface on the end of the protein, potentially constituting a direct membrane-binding site. Consistent with this idea, the ATG-C domain in the yeast protein binds directly to lipids (De et al., 2017). Combinatorial binding through these three different motifs regulates the association of the C-terminal end of VPS13A with different membrane organelles, for example the mitochondrion, lipid droplets or the plasma membrane, whereas the N-terminal end of VPS13A is anchored to the ER through binding to VAP family proteins (Kumar et al., 2018; Yeshaw et al., 2019).
Fig. 6.
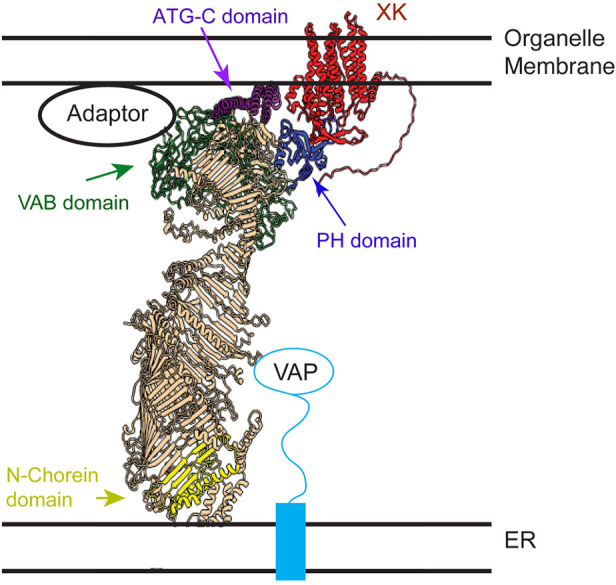
Model of VPS13A arrangement at a membrane contact site. A model structure of the VPS13A–XK complex based on assembly of Alphafold predictions is shown relative to the predicted locations of the ER membrane and a second organellar membrane (e.g. lipid droplet, mitochondria or plasma membrane). The XK protein is in red and the different domains of VPS13A are highlighted: yellow for the N-Chorein domain, green for the Vps13 adaptor binding (VAB) domain, purple for the ATG-C domain and blue for the PH-like domain. Binding to the VAP protein anchors the C-terminal end of VPS13A to the ER (Yeshaw et al., 2019).
Our results are consistent with the idea that VPS13A disease and McLeod Syndrome both result from loss of an XK–VPS13A complex. An important question is where that complex is localized. We note that the co-localization of VPS13A and XK in ER subdomains in transfected, cultured cells, albeit a useful tool for examining protein association, likely represents a consequence of overexpression of both proteins rather than the localization of the complex at native expression levels. In red blood cells, XK localizes to the plasma membrane as a component of the Kell antigen (Russo et al., 1999). Moreover, both XK and VPS13A are present in the membrane fraction of red blood cells and levels of VPS13A in this fraction are reduced in cells from patients lacking XK, suggesting the two proteins might interact at the plasma membrane of red blood cells (Urata, et al., 2019).
Red blood cells lack intracellular membranes and, thus, it is still unclear where VPS13A or XK localize in nucleated cells. While this work was in preparation, Guillen-Samander et al. (2022) preprint reported mapping the interaction of VPS13A and XK to the PH domain and the second intracellular loop, respectively, consistent with the results presented here. Additionally, they demonstrated in cultured cells that VPS13A and XK can co-localize at ER–plasma membrane junctions (Guillen-Samander et al., 2022). Thus, ER–plasma membrane junctions are a likely site of VPS13A–XK activity.
XK encodes a lipid scramblase, capable of flipping individual phospholipids between the leaflets of a membrane bilayer (Adlakha et al., 2022; Ryoden et al., 2022). XK is required for extracellular exposure of phosphatidylserine in response to extracellular ATP in immune cells and VPS13A is necessary for XK activity in this context (Ryoden et al., 2022). These results suggest that mediating exposure of phosphatidylserine on certain neurons might be the critical function of VPS13A–XK, although XK scramblase activity is not specific to phosphatidylserine in vitro (Adlakha et al., 2022).
Physical interaction with lipid scramblases has emerged as a common property of the VPS13 protein family and related lipid-transport proteins (Adlakha et al., 2022; Ryoden et al., 2022). This linking of intermembrane and intra-bilayer transport might be important for promoting transport through the VPS13 protein channel by maintaining a concentration gradient in lipids between the donor and acceptor leaflets. Knockdown of VPS13A in PC12 cells has been shown to lower phosphatidylinositol-4-phosphate levels at the plasma membrane (Park et al., 2015). Interestingly, maintenance of phosphatidylinositol-4-phosphate levels in the plasma membrane requires continual phosphatidylinositol synthesis in the ER, suggesting that continual delivery of phosphatidylinositol from the ER to the plasma membrane is required (Pemberton et al., 2020). Thus, an alternative possibility is that movement of lipids from the inner leaflet of the plasma membrane to the outer leaflet via XK helps maintain a directional flow of phosphatidylinositol from the ER to the plasma membrane via VPS13A. If true, then defects in phosphatidylinositol levels rather than phosphatidylserine transport could be the basis for the neurological effects of VPS13A and XK mutations. Further studies will be required to define the critical cellular role of the VPS13A–XK complex, which, when abolished, results in disease.
MATERIALS AND METHODS
Cell culture and transfections
HEK293T cells were a gift from Kevin Czaplinski (Stony Brook University). The cells were tested for contamination prior to freezing in aliquots. Cells were thawed and passaged no more than 25 times before fresh aliquots were thawed. Cells were maintained at 37°C in a humidified atmosphere at 5% CO2 in Dullbecco's Modified Eagle Medium (DMEM, Gibco 11995-065) supplemented with 10% fetal bovine serum (Gibco 16000-044). Transfection for HEK293T cells was done using Lipofectamine 2000 (Thermo Fisher Scientific, 11668-019) following the manufacturer's instructions. Briefly, for microscopy, 2 µg plasmid DNA and 3 µl Lipofectamine 2000 were separately added to 200 µl OptiMEM (Gibco, 31985-062). After 5 min incubation at room temperature, the Lipofectamine 2000 mixed with OptiMEM was added to the plasmid DNA with OptiMEM, and this mixture was incubated at room temperature for 20 min. The plasmid/Lipofectamine 2000 mixture in OptiMEM medium was then added dropwise to HEK293T cells. For live-cell microscopy, HEK293T cells were grown in gelatin-coated 35-mm glass-bottomed culture dishes (Millipore, ES-006-B). For immunoprecipitation and western blot analysis, HEK293T cells were grown in gelatin-coated 10-cm culture dishes and 10 µg plasmid DNA and 20 µl of Lipofectamine 2000 were used for transfection.
Plasmid construction
Plasmids and primers used in this study are listed in Tables S1 and S2, respectively. Plasmid JS142-D3 expressing mRFP–XK was constructed by Gibson assembly using NEBuilder HiFi DNA Assembly (New England BioLabs, E2611L) with an mRFP-coding fragment amplified with oligos JSO620 and JSO628 and linearized pcDNA3.1(+)-N-XK as the template. To construct pJS160, which expresses VPS13A1–3143^mCherry, two fragments were amplified using the oligo pairs JSO731/JSO732 and JSO733/JSO734 with pVPS13A^mCherry as template. These primers amplify two overlapping halves of the template, creating a deletion of the 31 codons at the 3′ end of the VPS13A coding region. To construct plasmids expressing various VPS13A fragments fused to GFP, each fragment was amplified with a pair of oligos by PCR; VPS13A2751–3174 with JSO756/JSO757, VPS13A3027–3174 with JSO775/JSO757, VPS13A3027–3143 with JSO775/JSO781 and VPS13A3144–3474 with JSO776/JSO757, resulting in pJS164, pJS166, pJS171 and pJS167, respectively. The vector pEGFP-C2 was linearized by digestion with XhoI and XmaI, and fragments were incorporated into the linearized vector by Gibson assembly. To create the point mutation I3148P within VPS13A3027–3174, the coding region for this VPS13A region was amplified in two overlapping fragments from pJS166 using the pairs of oligos JSO775/JSO783 and JSO784/JSO757. The mutation I3148P (‘ATA’ to ‘CCA’) was incorporated into the overlap between these two fragments. The two fragments then were introduced into the linearized vector pEGFP-C2 by Gibson assembly, leading to plasmid JS172.
To construct the plasmid JS163, the full-length CMV promoter was deleted from pcDNA3.1(+)-N-eGFP-XK by digestion with MluI and NheI, and the vector backbone was purified using the QIAGEN Gel Purification Kit (169021320). A smaller CMV promoter fragment (−125 bp) was amplified from pcDNA3.1(+)-N-eGFP-XK with the oligo pair JSO741/JSO742 and introduced into the gel-purified pcDNA3.1(+)-N-eGFP-XK backbone by Gibson assembly. To generate the plasmid JS165, which expresses GFP–XK6A, overlapping fragments were amplified from pcDNA3.1(+)-N-eGFP-XK with oligo pairs JSO758/JSO759 and JSO760/JSO76. These amplifications generate the 5′ and 3′ ends of the XK coding region with an overlap in the region of codons 90 to 110. In this overlap, the oligos alter the coding region for the amino acid sequence EEPYVS to AAAAAA (5′-GAAGAGCCTTATGTCAGT-3′ to 5′-GCAGCGGCTGCTGCCGCT-3′). pcDNA3(+)-N-eGFP-XK was digested with EcoRI and XbaI to remove the XK gene, and the remaining backbone of the plasmid was assembled with the two PCR fragments by Gibson assembly. To remove the GFP tag from GFP–XK and GFP–XK6A, pcDNA3.1(+)-N-eGFP-XK and pJS165 were first digested with HindIII and EcoRI to delete the GFP coding region. The 5′ and 3′ ends were filled in using DNA polymerase I Large (Klenow) fragment (New England Biolabs, M0210S), and the blunt ends were ligated using T4 DNA ligase (New England Biolabs, M0202S), resulting in the plasmids pJS169 and pJS173.
Coding sequences from all constructed plasmids were confirmed by sequencing performed by the Stony Brook University DNA sequencing facility.
Immunoprecipitation and western blot analysis
Transfected cells were washed with ice-cold PBS (Life Technologies, 14190-144), collected using a cell scraper into a 1.5 ml microcentrifuge tube, and stored at −80°C. GFP pull-downs were performed as described in Park and Neiman (2020) with minor changes. Cell pellets were resuspended in 300 µl lysis buffer [10 mM Tris/HCl, pH 7.5; 150 mM NaCl; 0.5 mM EDTA; 0.5% NP-40; 1 mM phenylmethylsulfonyl fluoride (PMSF); and protease inhibitor cocktail (Thermo Fisher Scientific, A32955; one tablet per 10 ml)] and lysed by sonication with a microprobe (Qsonica Sonicators) at amplitude 40 for 15 s, three times per sample. Cell lysates were centrifuged at 14,000 g for 10 min at 4°C and supernatants were transferred into new 1.5 ml microcentrifuge tubes. 30 µl lysate per each supernatant was saved and mixed with 2× SDS sample buffer (100 mM Tris/HCl, pH 6.8; 4% SDS; 20% glycerol; 0.2 mg/ml Bromophenol Blue; and 0.72 M 2-mercaptoethanol). To immunoprecipitate GFP fusion proteins from the supernatants, 500 µl of washing buffer (10 mM Tris/HCl, pH 7.5; 150 mM NaCl; and 0.5 mM EDTA) containing 1 mM PMSF and protease inhibitor cocktail was added to the remaining supernatant to dilute the NP-40 to 0.2%, and then 25 µl of GFP-trap agarose beads (Chromotek, GTA020), pre-washed twice with 500 µl washing buffer, were added. The supernatant and GFP-trap agarose bead mixtures were incubated for 1.5 h at 4°C on a rotator. Agarose beads were pelleted by centrifugation at 2500 g for 2 min at 4°C. Beads were washed twice with 500 µl washing buffer. Finally, 2× SDS sample buffer was added to the beads and samples were heated at 50°C for 10 min.
Protein samples were fractionated on 7.5% or 10% SDS-polyacrylamide gels (Bio-Rad, 1610171 or 1610173). For the transfer of VPS13A full-length protein tagged with or without mCherry, transfer of the 7.5% gel to a polyvinylidene difluoride (PVDF) membrane was done overnight at 4°C as described in Park et al. (2013). The primary antibodies used for immunoblotting analysis were as follows: anti-VPS13A (Sigma-Aldrich, HPA021652, 1:700), anti-mCherry (Rockland, 600-401-P16, 1:1000), anti-GFP (Clontech, 632381, 1:1000), anti-XK (Sigma-Aldrich, HPA019036, 1:2500) and anti-GAPDH (ProteinTech, 60004, 1:5000). The secondary antibodies used were ECL anti-mouse IgG (GE Healthcare, NA931V, 1:5000 and ECL anti-rabbit IgG (GE Healthcare, NA934V, 1:5000).
Preparation of and western blotting of human blood samples
Patient samples were provided by Paul Goldsmith (Newcastle, UK) and Naomi Lubarr (New York, USA) with informed consent. The study protocol was approved the Ethics committee at Ludwig Maximilian University in Munich. All clinical investigation was conducted according to the principles expressed in the Declaration of Helsinki. 2.5 ml aliquot of EDTA/citrate blood was defrosted, filled up to 12 ml with red blood cell (RBC) wash buffer (5 mM Na2HPO4 and 154 mM NaCl), mixed carefully and centrifuged for 10 min at 2500 g. Most of the supernatant was discarded, and 1.5–2 ml was left at the bottom with the pellet. Washing steps were repeated for pellet, and the supernatant was removed and discarded. The pellet was distributed to two 1.5 ml standard lab reaction tubes, and 500 µl RBC lysis buffer (5 mM Na2HPO4) was added to each reaction tube. Samples were centrifuged for 5 min at 16,000 g, and the supernatant was carefully removed and discarded. Samples was washed with RBC lysis buffer at least twice. After that, samples were distributed in 5 µl aliquots and stored at −20°C.
For immunoblotting of these samples, 15 µl 2× Laemmli buffer was added to 5 µl aliquots of blood samples. Samples were incubated at 70°C for 10 min. A total volume of 10 µl per lane was loaded on a NuPAGE 3–8% Tris-Acetate gel (Invitrogen, EA0375BOX). After electrophoresis, probes were transferred to PVDF membranes. Membranes were blocked for 1 h in PBS with 0.1% Tween 20 (PBST) with 0.2% I-Block (Invitrogen, T2015). The membranes were incubated overnight with primary antibody in blocking solution at 4°C. After washing four times for 15 min with PBST, the membranes were incubated with secondary antibodies for 1 h in PBST with 0.2% I-Block. Development of the membrane after four 15 min PBST washes was performed with ECL plus (Pierce, 32134). The anti-VPS13A antibody (Sigma-Aldrich, HPA021662) was used at 1:10,000 dilution and detected with HRP-conjugated anti-rabbit antibodies (Promega, W4011) used at a 1:10,000 dilution. β-actin was detected using primary antibodies (Sigma, A5316) at 1:50,000 dilution and HRP-conjugated anti-mouse antibodies (Promega, W4021) at 1:5000 dilution.
Immunostaining and microscopy
To visualize lipid droplets in live cells, ∼70–80% confluent cells in gelatin-coated 35 mm glass-bottomed culture dishes were washed once with Hank's balanced salt solution (HBSS) (Life Technologies, 14025092) and incubated with 1 ml HBSS containing 5 µM monodansylpentane (MDH) (Abcepta, SM1000a) for 15 min in a 37°C humidified incubator. To determine fluorescent protein localization in live cells, cells at 24 h after transfection were washed once and incubated with HBSS buffer. Live cells in the HBSS buffer were observed using a wide-field Zeiss Observer A1 microscope with an attached Orca II ERG camera (Hamamatsu) or a Zeiss LSM 510 META NLO two-photon laser scanning confocal microscope coupled with a Coherent Chameleon laser system. ZEN 2012 Blue edition software was used to acquire and process images from wide-field microscopy. Zeiss LSM version 4.0 software and ZEN Black edition software were used to acquire and process images from the confocal microscope, respectively.
Alphafold predictions
Alphafold predictions were done using Alphafold 2.1 on the Google Colaboratory site (https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb). Because of constraints on the size of the protein sequence that the program could use for predictions, the VPS13A model was constructed by first generating Alphafold predictions of four overlapping segments of VPS13A: aa 1–1000, 900–1850, 1750–2600 and 2500–3174. The full model was constructed by using ChimeraX software to align the overlapping regions. For the prediction of the VPS13A PH domain in complex with the second intracellular loop of XK, the multimer mode of Alphafold was used. ChimeraX was used to align this structure with the VPS13A model and a prediction of XK structure to generate the model of the complex in Fig. 6.
Supplementary Material
Acknowledgements
We acknowledge the work of Prof. Adrian Danek, Dr Benedikt Bader (Department of Neurology, Ludwig-Maximilians-Universität München, Germany) and Mrs G. Kwiatkowski (Department of Neuropathology, Ludwig-Maximilians-Universität München) in establishing the repository for the patient samples used in this study, as well as the support of the Advocacy for Neuroacanthocytosis Patients, the ERA-net E-Rare consortium EMINA (European Multidisciplinary Initiative on Neuroacanthocytosis; BMBF01GM1003) and all the collaborating clinicians who sent samples for analysis, particularly Paul Goldsmith, Naomi Lubarr and Ruth Walker. We are grateful to Bettina Schmid and Adrian Danek for comments on the manuscript. We also thank the Stony Brook Central Microscopy Facility for access to the confocal microscope and members of the Neiman Lab for helpful discussion.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: J.-S.P.; Methodology: J.-S.P.; Formal analysis: J.-S.P., Y.H., N.M.H., A.M.N.; Investigation: J.-S.P., Y.H., G.M.-M., A.M.N.; Resources: G.M.-M., A.M.N.; Writing - original draft: J.-S.P., A.M.N.; Writing - review & editing: J.-S.P., Y.H., N.M.H., G.M.-M.; Supervision: A.M.N.; Funding acquisition: A.M.N.
Funding
This work was supported by National Institutes of Health grant R01 GM720540 to A.M.N. Deposited in PMC for release after 12 months.
Peer review history
The peer review history is available online at https://journals.biologists.com/jcs/lookup/doi/10.1242/jcs.260227.reviewer-comments.pdf.
References
- Adlakha, J., Hong, Z., Li, P. Q. and Reinisch, K. M. (2022). Structural and biochemical insights into lipid transport by VPS13 proteins. J. Cell Biol. 221, e202202030. 10.1083/jcb.202202030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, B. D. M., Dziurdzik, S. K., Kolehmainen, K. L., Fowler, C. M. S., Kwong, W. K., Grad, L. I., Davey, M., Schluter, C. and Conibear, E. (2018). Competitive organelle-specific adaptors recruit Vps13 to membrane contact sites. J. Cell Biol. 217, 3593-3607. 10.1083/jcb.201804111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie, E., Guo, X., Christiano, R., Chitraju, C., Kory, N., Harrison, K., Haas, J., Walther, T. C. and Farese, R. V., Jr. (2014). High confidence proteomic analysis of yeast LDs identifies additional droplet proteins and reveals connections to dolichol synthesis and sterol acetylation. J. Lipid Res. 55, 1465-1477. 10.1194/jlr.M050229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De, M., Oleskie, A. N., Ayyash, M., Dutta, S., Mancour, L., Abazeed, M. E., Brace, E. J., Skiniotis, G. and Fuller, R. S. (2017). The Vps13p-Cdc31p complex is directly required for TGN late endosome transport and TGN homotypic fusion. J. Cell Biol. 216, 425-439. 10.1083/jcb.201606078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson-Stone, C., Danek, A., Rampoldi, L., Hardie, R. J., Chalmers, R. M., Wood, N. W., Bohlega, S., Dotti, M. T., Federico, A., Shizuka, M.et al. (2002). Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis. Eur. J. Hum. Genet. 10, 773-781. 10.1038/sj.ejhg.5200866 [DOI] [PubMed] [Google Scholar]
- Dziurdzik, S. K. and Conibear, E. (2021). The Vps13 family of lipid transporters and its role at membrane contact sites. Int. J. Mol. Sci. 22, 2905. 10.3390/ijms22062905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, R., O'Neill, M., Pritzel, A., Antropova, N., Senior, A. W., Green, T., Zidek, A., Bates, R., Blackwell, S., Yim, J.et al. (2022). Protein complex prediction with AlphaFold-Multimer. bioRxiv. 10.1101/2021.10.04.463034 [DOI] [Google Scholar]
- Gauthier, J., Meijer, I. A., Lessel, D., Mencacci, N. E., Krainc, D., Hempel, M., Tsiakas, K., Prokisch, H., Rossignol, E., Helm, M. H.et al. (2018). Recessive mutations in VPS13D cause childhood onset movement disorders. Ann. Neurol. 83, 1089-1095. 10.1002/ana.25204 [DOI] [PubMed] [Google Scholar]
- Guillen-Samander, A., Wu, Y., Pineda, S. S., Garcia, F. J., Eisen, J. N., Leonzino, M., Ugur, B., Kellis, M., Heiman, M. and De Camilli, P. (2022). A partnership of the lipid scramblase XK and of the lipid transfer protein VPS13A at the plasma membrane. bioRxiv. 10.1101/2022.03.30.486314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin, E. L., Ting, L., Bruckner, R. J., Gebreab, F., Gygi, M. P., Szpyt, J., Tam, S., Zarraga, G., Colby, G., Baltier, K.et al. (2015). The BioPlex network: a systematic exploration of the human interactome. Cell 162, 425-440. 10.1016/j.cell.2015.06.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- John Peter, A. T., Herrmann, B., Antunes, D., Rapaport, D., Dimmer, K. S. and Kornmann, B. (2017). Vps13-Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J. Cell Biol. 216, 3219-3229. 10.1083/jcb.201610055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Zidek, A., Potapenko, A.et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596, 583-589. 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolakowski, D., Rzepnikowska, W., Kaniak-Golik, A., Zoladek, T. and Kaminska, J. (2021). The GTPase Arf1 is a determinant of yeast Vps13 localization to the Golgi Apparatus. Int. J. Mol. Sci. 22, 12274. 10.3390/ijms222212274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolehmainen, J., Black, G. C., Saarinen, A., Chandler, K., Clayton-Smith, J., Traskelin, A. L., Perveen, R., Kivitie-Kallio, S., Norio, R., Warburg, M.et al. (2003). Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am. J. Hum. Genet. 72, 1359-1369. 10.1086/375454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, N., Leonzino, M., Hancock-Cerutti, W., Horenkamp, F. A., Li, P., Lees, J. A., Wheeler, H., Reinisch, K. M. and De Camilli, P. (2018). VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 217, 3625-3639. 10.1083/jcb.201807019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang, A. B., John Peter, A. T., Walter, P. and Kornmann, B. (2015). ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J. Cell Biol. 210, 883-890. 10.1083/jcb.201502105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonzino, M., Reinisch, K. M. and De Camilli, P. (2021). Insights into VPS13 properties and function reveal a new mechanism of eukaryotic lipid transport. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1866, 159003. 10.1016/j.bbalip.2021.159003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage, S., Drouet, V., Majounie, E., Deramecourt, V., Jacoupy, M., Nicolas, A., Cormier-Dequaire, F., Hassoun, S. M., Pujol, C., Ciura, S.et al. (2016). Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am. J. Hum. Genet. 98, 500-513. 10.1016/j.ajhg.2016.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P., Lees, J. A., Lusk, C. P. and Reinisch, K. M. (2020). Cryo-EM reconstruction of a VPS13 fragment reveals a long groove to channel lipids between membranes. J. Cell Biol. 219, e202001161. 10.1083/jcb.202001161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan, D. G. and Ryan, K. M. (2021). ATG2 and VPS13 proteins: molecular highways transporting lipids to drive membrane expansion and organelle communication. FEBS J. 10.1111/febs.16280 [DOI] [PubMed] [Google Scholar]
- Melia, T. J. and Reinisch, K. M. (2022). A possible role for VPS13-family proteins in bulk lipid transfer, membrane expansion and organelle biogenesis. J. Cell Sci. 135, jcs259357. 10.1242/jcs.259357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Braceras, S., Tornero-Ecija, A. R., Vincent, O. and Escalante, R. (2019). VPS13A is closely associated with mitochondria and is required for efficient lysosomal degradation. Dis. Models Mech. 12, dmm036681. 10.1242/dmm.036681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemela, V., Salih, A., Solea, D., Lindvall, B., Weinberg, J., Miltenberger, G., Granberg, T., Tzovla, A., Nordin, L., Danfors, T.et al. (2020). Phenotypic variability in chorea-acanthocytosis associated with novel VPS13A mutations. Neurol. Genet. 6, e426. 10.1212/NXG.0000000000000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. S. and Neiman, A. M. (2020). XK is a partner for VPS13A: a moleular link between Chorea-Acanthocytosis and McLeod syndrome. Mol. Biol. Cell 31, 2425-2436. 10.1091/mbc.E19-08-0439-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. S., Okumura, Y., Tachikawa, H. and Neiman, A. M. (2013). SPO71 encodes a developmental stage-specific partner for VPS13 in Saccharomcyes cerevisiae. Eukaryot. Cell 12, 1530-1537. 10.1128/EC.00239-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. S., Halegoua, S., Kishida, S. and Neiman, A. M. (2015). A conserved function in phosphatidylinositol metabolism for mammalian Vps13 family proteins. PLoS One 10, e0124836. 10.1371/journal.pone.0124836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, J. S., Thorsness, M. K., Policastro, R., McGoldrick, L., Hollingsworth, N. M., Thorsness, P. E. and Neiman, A. M. (2016). Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol. Biol. Cell 27, 2435-2449. 10.1091/mbc.e16-02-0112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton, J. G., Kim, Y. J., Humpolickova, J., Eisenreichova, A., Sengupta, N., Toth, D. J., Boura, E. and Balla, T. (2020). Defining the subcellular distribution and metabolic channeling of phosphatidylinositol. J. Cell Biol. 219, e201906130. 10.1083/jcb.201906130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampoldi, L., Dobson-Stone, C., Rubio, J. P., Danek, A., Chalmers, R. M., Wood, N. W., Verellen, C., Ferrer, X., Malandrini, A., Fabrizi, G. M.et al. (2001). A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat. Genet. 28, 119-120. 10.1038/88821 [DOI] [PubMed] [Google Scholar]
- Richard, A., Hsu, J., Baum, P., Alterman, R. and Simon, D. K. (2019). Efficacy of deep brain stimulation in a patient with genetically confirmed Chorea-Acanthocytosis. Case Rep. Neurol. 11, 199-204. 10.1159/000500951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodova, M., Jayini, R., Singasani, R., Chipps, E. and Islam, M. R. (2013). CMV promoter is repressed by p53 and activated by JNK pathway. Plasmid 69, 223-230. 10.1016/j.plasmid.2013.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulis, E., Hyland, C., Flower, R., Gassner, C., Jung, H. H. and Frey, B. M. (2018). Molecular basis and clinical overview of McLeod syndrome compared with other neuroacanthocytosis syndromes: a review. JAMA Neurol. 75, 1554-1562. 10.1001/jamaneurol.2018.2166 [DOI] [PubMed] [Google Scholar]
- Russo, D., Lee, S. and Redman, C. (1999). Intracellular assembly of Kell and XK blood group proteins. Biochim. Biophys. Acta 1461, 10-18. 10.1016/S0005-2736(99)00148-0 [DOI] [PubMed] [Google Scholar]
- Ryoden, Y., Segawa, K. and Nagata, S. (2022). Requirement of Xk and Vps13a for the P2X7-mediated phospholipid scrambling and cell lysis in mouse T cells. Proc. Natl. Acad. Sci. USA 119, e2119286119. 10.1073/pnas.2119286119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzepnikowska, W., Flis, K., Munoz-Braceras, S., Menezes, R., Escalante, R. and Zoladek, T. (2017). Yeast and other lower eukaryotic organisms for studies of Vps13 proteins in health and disease. Traffic 18, 711-719. 10.1111/tra.12523 [DOI] [PubMed] [Google Scholar]
- Scheffzek, K. and Welti, S. (2012). Pleckstrin homology (PH) like domains - versatile modules in protein-protein interaction platforms. FEBS Lett. 586, 2662-2673. 10.1016/j.febslet.2012.06.006 [DOI] [PubMed] [Google Scholar]
- Seong, E., Insolera, R., Dulovic, M., Kamsteeg, E. J., Trinh, J., Bruggemann, N., Sandford, E., Li, S., Ozel, A. B., Li, J. Z.et al. (2018). Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann. Neurol. 83, 1075-1088. 10.1002/ana.25220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, J., Imanishi, E. and Nagata, S. (2014). Exposure of phosphatidylserine by Xk-related protein family members during apoptosis. J. Biol. Chem. 289, 30257-30267. 10.1074/jbc.M114.583419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno, S., Maruki, Y., Nakamura, M., Tomemori, Y., Kamae, K., Tanabe, H., Yamashita, Y., Matsuda, S., Kaneko, S. and Sano, A. (2001). The gene encoding a newly discovered protein, chorein, is mutated in chorea-acanthocytosis. Nat. Genet. 28, 121-122. 10.1038/88825 [DOI] [PubMed] [Google Scholar]
- Urata, Y., Nakamura, M., Sasaki, N., Shiokawa, N., Nishida, Y., Arai, K., Hiwatashi, H., Yokoyama, I., Narumi, S., Terayama, Y.et al. (2019). Novel pathogenic XK mutations in McLeod syndrome and interaction between XK protein and chorein. Neurol. Genet 5, e328. 10.1212/NXG.0000000000000328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., Yuan, D., Stroe, O., Wood, G., Laydon, A.et al. (2022). AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 50, D439-D444. 10.1093/nar/gkab1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, R. H. and Danek, A. (2021). “Neuroacanthocytosis” - Overdue for a taxonomic update. Tremor. Other Hyperkinet. Mov. 11, 1. 10.5334/tohm.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeshaw, W. M., van der Zwaag, M., Pinto, F., Lahaye, L. L., Faber, A. I. E., Gomez-Sanchez, R., Dolga, A. M., Poland, C., Monaco, A. P., van IJzendoorn, S. C. D.et al. (2019). Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. Elife 8, e43561. 10.7554/eLife.43561 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.