

Abstract
Background
Actin, alpha, skeletal muscle 1 (ACTA1) is one of the causative genes of nemaline myopathy (NM) and congenital fiber‐type disproportion (CFTD). CFTD is characterized by type 1 fiber atrophy and distinguished from NM in the absence of rods. Eight patients with CFTD, including one patient with dilated cardiomyopathy (DCM), have previously been reported. Herein, we report the case of a 10‐year‐old boy presenting with CFTD and DCM.
Methods
We performed exome sequencing and analyzed the effect of Met327Lys mutations on cultured C2C12 muscle cells compared with that seen in the wild type (WT, ACTA1) and previously identified Asp294Val mutations associated with a severe phenotype of CFTD without cardiomyopathy.
Results
Exome sequencing revealed a de novo mutation, c.980 T > A, p.(Met327Lys), in ACTA1 (NM_001100.4). C2C12 cells transfected with the WT plasmid expressed ACTA1 in the nucleus and cytoplasm. Cells with the Asp294Val mutant showed needle‐like structures in the cytoplasm, whereas the expression of the Met327Lys mutant resulted in few aggregations but many apoptotic cells.
Conclusion
Apoptosis induced in Met327Lys‐transfected muscle cells supports the pathogenicity of the mutation and can be implicated as one of the histopathological features associated with CFTD, as in NM.
Keywords: actin, alpha, apoptosis, congenital fiber‐type disproportion, dilated cardiomyopathy, skeletal muscle 1
Actin, alpha, skeletal muscle 1 (ACTA1) is one of the causative genes of nemaline myopathy (NM) and congenital fiber‐type disproportion (CFTD). Herein, we report a 10‐year‐old boy presented with CFTD and dilated cardiomyopathy (DCM), who has a novel de novo mutation, c.980T>A, p.(Met327Lys), in ACTA1. Met327Lys‐transfected cells showed few aggregations and numerous apoptotic cells. This result supports the pathogenicity of the mutation and can be implicated as one of the histopathological features associated with CFTD, as in NM.
1. INTRODUCTION
There are six types of actins that play a role in cell adhesion, morphology, and motility. The α‐actin 1 encoded by the ACTA1 gene (OMIM: *102610) comprises the skeletal muscle and 20% of the adult myocardium (Feng & Marston, 2009). It is the principal actin filament and generates force by sliding with the myosin filament (Squire, 2019). Pathologically, ACTA1‐associated diseases can be divided into nemaline myopathy (NM), intranuclear rod myopathy (IRM), actin filament aggregate myopathy, myopathy with the core‐like area, and congenital fiber‐type disproportion (CFTD). The hotspot mutations for NM are between amino acid (AA) residues 144–165 of ACTA1 and overlap with the hotspot mutations for IRM and actin filament aggregate myopathy. Genotype–phenotype correlations have not been established for these diseases because only a few mutations—except for NM—have been identified to reach any inference (Feng & Marston, 2009; Laing et al., 2009). Moreover, single mutations exhibit different phenotypes and severity, and pathological findings sometimes coexist between the intranuclear rod and core‐like myopathies (Schröder et al., 2004). Therefore, an ACTA1‐associated disease can be regarded as a part of the pathological spectrum rather than a single disease (Laing et al., 2009). For NM, ACTA1 mutation accounts for 20%–30% of cases following nebulin (NEB) mutation (Jeannet et al., 2007). It ranges from a neonatal‐onset severe form to an adult‐onset mild form. NM is characterized by nemaline rods and derived from the Z lines. The Z lines are composed of α‐actinin, actin, tropomyosin, myotilin, γ‐filament, cofilin‐2, telethonin, and nebulin. Rod bodies are induced by the depletion of ATP and metabolic stress because actin needs ATP to form the filaments (Sewry et al., 2019). In CFTD, mutations are often located on the surface of the ACTA1 protein (Laing et al., 2004) to ensure that the association with specific proteins is predicted (Laing et al., 2009). It is an early onset, nonprogressive systemic weakness characterized by the atrophy of type 1 muscle fibers. This feature is shared by NM and many other neuromuscular diseases. CFTD is distinguished from NM in the absence of rods. However, analysis of cultured muscle cells reported that few CFTD mutations induced aggregations (Clarke et al., 2007). Eight different ACTA1 mutations have previously been reported in CFTD (Table S1). Cardiomyopathy is a rare finding accompanying CFTD with ACTA1 mutation.
Herein, we report a 10‐year‐old boy presented with CFTD and dilated cardiomyopathy (DCM), who has a novel de novo mutation, c.980 T > A, p.(Met327Lys), in ACTA1 (NM_001100.4). We have analyzed the effect of mutation on cultured muscle cells.
2. METHODS
2.1. Exome sequencing and direct sequencing
Genomic DNA was extracted from the peripheral blood lymphocytes of the patient and his parents using the standard method. Exome sequencing was performed using the TruSeq Exome platform (Illumina, San Diego, USA). The pair‐end reads were aligned to the GRCh37 reference by the Burrows‐Wheeler Aligner. Variant annotations were performed by the Variant studio (Illumina, San Diego, USA). The interpretation was in accordance with the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) guidelines (Richards et al., 2015). Direct sequencing was performed by FASMAC (Atsugi, Japan).
2.2. Expression of ACTA1–GFP fusion protein in C2C12 cells
Wild‐type (WT) ACTA1 cDNA (Otsuki et al., 2005) was subcloned from a plasmid vector (RIKEN, Tsukuba, Japan) into the pAcGFP‐N1 vector (Clontech; Takara, Kusatsu, Japan), with GFP fused to its C‐terminal. Plasmids with mutations Met327Lys and Asp294Val were constructed by PCR‐based site‐directed mutagenesis on WT ACTA1 cDNA. The mutation p.(Asp294Val) was previously reported to be responsible for CFTD without cardiomyopathy (Clarke et al., 2007) (Table S1), which was taken as a positive control for our experiment.
To confirm protein expression, the three vectors (WT‐GFP, Met327LysK‐GFP, and Asp294Val‐GFP) were transfected into HEK293 cells. The GFP signal was detected by fluorescence microscopy (BZ‐X800; Keyence, Osaka, Japan), and protein expression was determined by western blotting using an ACTA1‐specific antibody (data not shown). Next, C2C12 cells (subcloned from a myoblast line established from normal adult mouse muscle) obtained from RIKEN (Tsukuba, Japan) were transfected with these vectors using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) following the product’s protocol. The transfected C2C12 cells were cultured in eight‐strip chamber slides, fixed, and subjected to actin and nuclear staining.
3. RESULTS
3.1. Clinical phenotype
The patient was a 10‐year‐old boy born by cesarean section at 38 weeks after infertility to nonconsanguineous healthy parents. At birth, his height was 43.5 cm [−2.13 standard deviations (SD)], his body weight (BW) was 1905 g (−2.74 SD), and his occipitofrontal circumference was 31.2 cm (−1.32 SD). The Apgar scores were 6 and 8 at 1 and 5 min, respectively. After birth, the patient had respiratory distress and was given nasal continuous positive airway pressure for 2 days. He also had hypotonia and muscle weakness. At 2 months of age, he presented stridor and sometimes aspirated milk. At 7 months of age, he was admitted to our hospital for examination. At the time of admission, his height and BW were 66.6 cm (−1.07 SD) and 5.9 kg (−2.83 SD), respectively, and he showed a high‐arched palate. Neurological evaluation revealed hypotonia and loss of deep tendon reflex. His joints showed hyperextension. As for laboratory data, creatinine phosphokinase was 65 U/L, aspartate aminotransferase 40 U/L, and lactate 1.4 mmol/L. Nerve conduction velocity was within the normal range and electromyography showed low amplitude. His left ventricular ejection fraction (LVEF) was 76.5% on echocardiography. Muscle biopsy of his left quadriceps femoris (Figure S1a,b) showed a bimodal distribution of fiber size, ranging from 10 to 30 μm in diameter, upon hematoxylin and eosin staining. Rimmed vacuoles, nemaline bodies, and ragged‐red fibers were not detected upon modified Gomori Trichrome staining. Of the total, type 1, 2A, and 2B fibers comprised 42%, 22%, and 36%, respectively. Type 1 fiber atrophy was seen, and fiber size disproportion was 58% (Figure S1a). In some fascicles, type 1 fibers predominated (Figure S1b). These findings were consistent with CFTD.
The patient’s developmental milestones were delayed, as he could control his head at 12 months and walk at 3 years of age. At 9 years of age, LVEF was decreased to 50% in echocardiography without any symptoms. Therefore, he was diagnosed with DCM. At 10 years of age, he was again admitted to our hospital because of cardiac failure. Chest X‐ray showed cardiac enlargement, and the cardiothoracic rate was 60% (Figure S1c); LVEF declined to 20% (Figure S1d). Despite intensive treatment, he died at 10 years of age.
3.2. Exome sequencing and direct sequencing
Exome sequencing revealed a novel de novo mutation, c.980 T > A, p.(Met327Lys), in the ACTA1 gene (NM_001100.4), which was confirmed by Sanger sequencing (Figure S2a). The parents of the patient did not have the same mutation (Figure S2a). Maternity and paternity were confirmed by exome sequencing. In addition, this mutation has not been reported in gnomAD, sbSNP, or jMorp. The site of the p.(Met327Lys) mutation was highly conserved from Homo sapiens to Danio rerio (Figure S2b). In terms of clinical variation, the mutation c.981G > C, p.(Met327Ile) was also registered in nemaline myopathy 3 in ClinVar, although the variant was considered of uncertain significance. Our mutation was predicted to be pathogenic by three out of four online programs (Figure S2c) and was predicted to be likely pathogenic according to the ACMG/AMP guidelines (Richards et al., 2015).
3.3. Expression of ACTA1–GFP fusion protein in C2C12 cells
The transfected C2C12 cells with WT cDNA expressed ACTA1 in the nucleus and cytoplasm with few aggregations (Figure 1a,b). Asp294Val‐transfected cells showed needle‐like structures in the cytoplasm (Figure 1c,d). Met327Lys‐transfected cells showed few aggregations in the cytoplasm and numerous apoptotic cells (Figure 1e–h).
FIGURE 1.
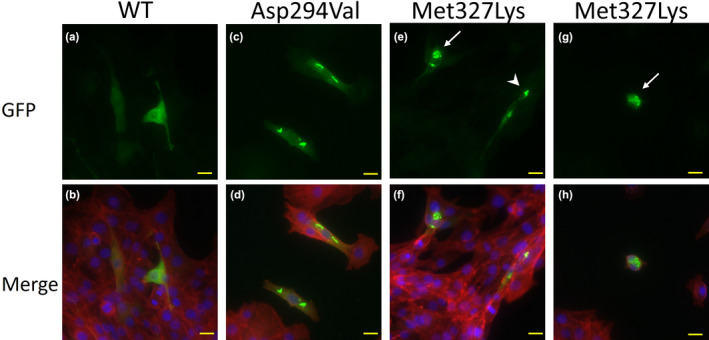
WT and mutant ACTA1 proteins were expressed in C2C12 cells. The nucleus is stained by Fluoro‐KEEPER Antifade Reagent Nonhardening Type with DAPI (Nacalai Tesque), and F‐actin is stained by ActinRed™555 (Invitrogen). pAcGFP‐N1 plasmids with ACTA1‐WT, ACTA1‐Asp294Val, and ACTA1‐Met327Lys are transfected into C2C12 cells. The yellow bar indicates 20 μm. WT ACTA1 is expressed in the nucleus and cytoplasm with few aggregations (a, b). Asp294Val mutation (previously reported), positive control, shows needle‐like structures in the cytoplasm (c, d). Met327Lys mutation, as found in our case here, shows few aggregations in the cytoplasm and numerous apoptotic cells (e–h). White arrows indicate apoptotic cells (e, g), and the arrowhead indicates aggregations (e).
4. DISCUSSION
To date, ten patients presenting cardiomyopathy have been reported with ACTA1 mutation, including our patient (Figure 2a). DCM was reported in five patients, and hypertrophic cardiomyopathy (HCM) in the other five patients. ACTA1 mutations associated with HCM are related to Ca2+ hypersensitivity, whereas, with DCM, Ca2+ hyposensitivity is observed (Feng & Marston, 2009). The type of cardiomyopathy might not depend on the myopathy itself. For example, four cases with ACTA1 gene mutations were associated with cardiomyopathy (both HCM and DCM) and NM (D'Amico et al., 2006; Gatayama et al., 2013; Kim et al., 2011; Yokoyama et al., 2016); two cases were associated with CFTD and DCM (Tadokoro et al., 2018). One HCM case was associated with congenital myopathy with the core, whereas for another, the myopathy was unclassified (Kaindl et al., 2004). Likewise, two cases were associated with DCM alone (Carnevale et al., 2020; Reza et al., 2018). Genotype–phenotype correlation has not been elucidated; however, 327–328 AA and 336–338 AA residues in the ACTA1 sequence might accumulate mutations related to cardiomyopathy (Figure 2b,c).
FIGURE 2.
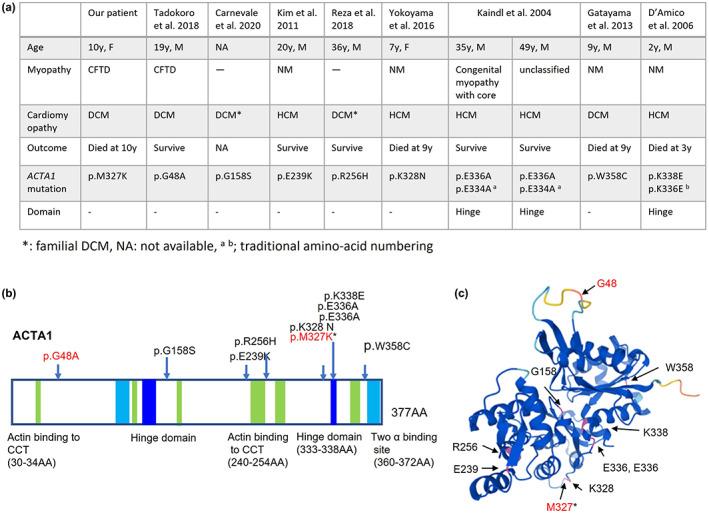
Clinical features of cardiomyopathy with ACTA1 mutations (a), schematic representation of the ACTA1 gene (b), and three‐dimensional (3D) model of the ACTA1 monomer (c). (b)The hinge domains are at 137–150 amino acids (AA) and 333–338 AA. Two actin‐binding sites are at 112–125 AA and 360–372 AA. Sites of actin‐binding to cytosolic chaperonin containing TCP‐1 (CCT) are at 30–34 AA, 135–139 AA, 170–174 AA, 240–254 AA, 265–274 AA, and 340–349 AA. (b, c) Arrows indicate the mutations in Figure 2a. The mutations written in red are associated with congenital fiber‐type disproportion (CFTD). Asterisk indicates our patient. The ACTA1 monomer has been illustrated by AlphaFold Protein Structure Database (https://alphafold.ebi.ac.uk/).
To the best of our knowledge, among all cardiomyopathy cases associated with ACTA1 mutation, one patient with HCM died at 3 years of age; two patients, one with HCM and the other with DCM, died at 9 years of age; four patients over 19 years of age were alive at the time of writing (Figure 2a). Our patient was also diagnosed with DCM at 9 years of age. The skeletal α‐actin is less expressed in a neonatal heart compared with that in an adult heart and is reported to be more abundant in failing hearts (Feng & Marston, 2009). Patients with severe cases may die by 10 years of age, whereas patients with mild cases can survive into adulthood (Figure 2a). Nine years of age might be the critical age for developing severe cardiomyopathy, and it might be important to prevent cardiac failure at an early stage.
Although muscle biopsy revealed a lack of nemaline bodies, C2C12 cells transfected with Asp294Val mutant showed needle‐like structures in the cytoplasm. The formation of similar cytoplasmic structures was reported previously with the mutation p.(Asp294Val) responsible for CFTD without cardiomyopathy. The affected patient died at 3 years of age due to respiratory complications (Clarke et al., 2007).
In addition, Met327Lys‐transfected cells showed few aggregations in the cytoplasm and many of the cells were apoptotic. Apoptosis, rather than aggregation, usually contributes to NM; 13 out of 15 ACTA1 mutations in NM cases have been reported to induce apoptosis in myoblast cells and myotubes with various degrees of aggregation. The cell death is suspected of being related to muscular atrophy with a lack of type 2 fibers (Vandamme et al., 2009). In contrast, CFTD has been reported to cause disruptions in actin functions such as in vitro motility, actin‐myosin force generation, and bindings (Feng & Marston, 2009). However, our findings of apoptotic cells associated with Met327Lys mutation suggest the involvement of muscle cell death in this condition. These findings are consistent with the microscopic findings of type 2 fiber decrease (Figure S1b). Hence, these results suggest that muscle cell death is one of the mechanisms of CFTD, as in NM.
In conclusion, Met327Lys is identified to be a severe mutation of ACTA1 associated with CFTD and DCM. Our results of Met327Lys‐transfected cells, showing few aggregations and many apoptotic cells, support the pathogenicity of the mutation. Muscle cell death is suggested to be a histopathological feature associated with CFTD, as observed in NM. The critical age for developing severe cardiomyopathy might be 9 years and perhaps crucial for preventing cardiac failure at an early stage.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest associated with this manuscript.
ETHICS STATEMENT
Written informed consent for genetic analyses of the patient was obtained from the parents. This research was approved by the bioethics committee for human gene analysis at Jichi Medical University.
AUTHORS’ CONTRIBUTION
Ayumi Matsumoto, Hidetoshi Tsuda, Kazuhisa Watanabe, Sadahiko Iwamoto, and Takanori Yamagata conceived and collected the data and wrote the manuscript. Sadahiro Furui, Masako Kawada‐Nagashima, Mitsuru Seki, Kazuhiro Muramatsu, and Hitoshi Osaka performed the clinical evaluation of the patient. Ichizo Nishino made a pathological diagnosis. Tatsuya Anzai, Kazuhiro Muramatsu, and Hitoshi Osaka recruited the patient. All authors reviewed and approved the manuscript.
Supporting information
Figure S1
Figure S2
{kind=link}
Table S1
ACKNOWLEDGMENTS
This work was supported by the JSPS KAKENHI (grant number 20 K08265 for AM). We thank the Research Association for Biotechnology, Dr. Yoshihide Hayashizaki of RIKEN, and Dr. Sumio Sugano of Tokyo University.
Matsumoto, A. , Tsuda, H. , Furui, S. , Kawada‐Nagashima, M. , Anzai, T. , Seki, M. , Watanabe, K. , Muramatsu, K. , Osaka, H. , Iwamoto, S. , Nishino, I. , & Yamagata, T. (2022). A case of congenital fiber‐type disproportion syndrome presenting dilated cardiomyopathy with ACTA1 mutation. Molecular Genetics & Genomic Medicine, 10, e2008. 10.1002/mgg3.2008
REFERENCES
- Carnevale, A. , Rosas‐Madrigal, S. , Rosendo‐Gutiérrez, R. , López‐Mora, E. , Romero‐Hidalgo, S. , Avila‐Vazzini, N. , Jacobo‐Albavera, L. , Domínguez‐Pérez, M. , Vargas‐Alarcón, G. , Pérez‐Villatoro, F. , Navarrete‐Martínez, J. I. , & Villarreal‐Molina, M. T. (2020). Genomic study of dilated cardiomyopathy in a group of Mexican patients using site‐directed next generation sequencing. Molecular Genetics & Genomic Medicine, 8, e1504. 10.1002/mgg3.1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, N. F. , Ilkovski, B. , Cooper, S. , Valova, V. A. , Robinson, P. J. , Nonaka, I. , Feng, J. J. , Marston, S. , & North, K. (2007). The pathogenesis of ACTA1‐related congenital fiber type disproportion. Annals of Neurology, 61, 552–561. 10.1002/ana.21112 [DOI] [PubMed] [Google Scholar]
- D'Amico, A. , Graziano, C. , Pacileo, G. , Petrini, S. , Nowak, K. J. , Boldrini, R. , Jacques, A. , Feng, J. J. , Porfirio, B. , Sewry, C. A. , Santorelli, F. M. , Limongelli, G. , Bertini, E. , Laing, N. , & Marston, S. B. (2006). Fatal hypertrophic cardiomyopathy and nemaline myopathy associated with ACTA1 K336E mutation. Neuromuscular Disorders: NMD, 16, 548–552. 10.1016/j.nmd.2006.07.005 [DOI] [PubMed] [Google Scholar]
- Feng, J. J. , & Marston, S. (2009). Genotype‐phenotype correlations in ACTA1 mutations that cause congenital myopathies. Neuromuscular Disorders: NMD, 19, 6–16. 10.1016/j.nmd.2008.09.005 [DOI] [PubMed] [Google Scholar]
- Gatayama, R. , Ueno, K. , Nakamura, H. , Yanagi, S. , Ueda, H. , Yamagishi, H. , & Yasui, S. (2013). Nemaline myopathy with dilated cardiomyopathy in childhood. Pediatrics, 131, e1986‐1990. 10.1542/peds.2012-1139 [DOI] [PubMed] [Google Scholar]
- Jeannet, P. Y. , Mittaz, L. , Dunand, M. , Lobrinus, J. A. , Bonafe, L. , & Kuntzer, T. (2007). Autosomal dominant nemaline myopathy: a new phenotype unlinked to previously known genetic loci. Neuromuscular Disorders: NMD, 17, 6–12. 10.1016/j.nmd.2006.10.005 [DOI] [PubMed] [Google Scholar]
- Kaindl, A. M. , Rüschendorf, F. , Krause, S. , Goebel, H. H. , Koehler, K. , Becker, C. , Pongratz, D. , Müller‐Höcker, J. , Nürnberg, P. , Stoltenburg‐Didinger, G. , Lochmüller, H. , & Huebner, A. (2004). Missense mutations of ACTA1 cause dominant congenital myopathy with cores. Journal of Medical Genetics, 41, 842–848. 10.1136/jmg.2004.020271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. Y. , Park, Y. E. , Kim, H. S. , Lee, C. H. , Yang, D. H. , & Kim, D. S. (2011). Nemaline myopathy and non‐fatal hypertrophic cardiomyopathy caused by a novel ACTA1 E239K mutation. Journal of the Neurological Sciences, 307, 171–173. 10.1016/j.jns.2011.04.022 [DOI] [PubMed] [Google Scholar]
- Laing, N. G. , Clarke, N. F. , Dye, D. E. , Liyanage, K. , Walker, K. R. , Kobayashi, Y. , Shimakawa, S. , Hagiwara, T. , Ouvrier, R. , Sparrow, J. C. , Nishino, I. , North, K. N. , & Nonaka, I. (2004). Actin mutations are one cause of congenital fibre type disproportion. Annals of Neurology, 56, 689–694. 10.1002/ana.20260 [DOI] [PubMed] [Google Scholar]
- Laing, N. G. , Dye, D. E. , Wallgren‐Pettersson, C. , Richard, G. , Monnier, N. , Lillis, S. , Winder, T. L. , Lochmüller, H. , Graziano, C. , Mitrani‐Rosenbaum, S. , Twomey, D. , Sparrow, J. C. , Beggs, A. H. , & Nowak, K. J. (2009). Mutations and polymorphisms of the skeletal muscle alpha‐actin gene (ACTA1). Human Mutation, 30, 1267–1277. 10.1002/humu.21059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuki, T. , Ota, T. , Nishikawa, T. , Hayashi, K. , Suzuki, Y. , Yamamoto, J. , Wakamatsu, A. , Kimura, K. , Sakamoto, K. , Hatano, N. , Kawai, Y. , Ishii, S. , Saito, K. , Kojima, S. , Sugiyama, T. , Ono, T. , Okano, K. , Yoshikawa, Y. , Aotsuka, S. , … Isogai, T. (2005). Signal sequence and keyword trap in silico for selection of full‐length human cDNAs encoding secretion or membrane proteins from oligo‐capped cDNA libraries. DNA Research, 12, 117–126. 10.1093/dnares/12.2.117 [DOI] [PubMed] [Google Scholar]
- Reza, N. , Garg, A. , Merrill, S. L. , Chowns, J. L. , Rao, S. , & Owens, A. T. (2018). ACTA1 novel likely pathogenic variant in a family with dilated cardiomyopathy. Circulation: Genomic and Precision Medicine, 11, e002243. 10.1161/circgen.118.002243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , & Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder, J. M. , Durling, H. , & Laing, N. (2004). Actin myopathy with nemaline bodies, intranuclear rods, and a heterozygous mutation in ACTA1 (Asp154Asn). Acta Neuropathologica, 108, 250–256. 10.1007/s00401-004-0888-1 [DOI] [PubMed] [Google Scholar]
- Sewry, C. A. , Laitila, J. M. , & Wallgren‐Pettersson, C. (2019). Nemaline myopathies: A current view. Journal of Muscle Research and Cell Motility, 40, 111–126. 10.1007/s10974-019-09519-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squire, J. (2019). Special issue: The actin‐myosin interaction in muscle: Background and overview. International Journal of Molecular Sciences, 20, 5715. 10.3390/ijms20225715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro, K. , Ohta, Y. , Sasaki, R. , Takahashi, Y. , Sato, K. , Shang, J. , Takemoto, M. , Hishikawa, N. , Yamashita, T. , Nakamura, K. , Nishino, I. , & Abe, K. (2018). Congenital myopathy with fiber‐type disproportion accompanied by dilated cardiomyopathy in a patient with a novel p.G48A ACTA1 mutation. Journal of the Neurological Sciences, 393, 142–144. 10.1016/j.jns.2018.08.015 [DOI] [PubMed] [Google Scholar]
- Vandamme, D. , Lambert, E. , Waterschoot, D. , Cognard, C. , Vandekerckhove, J. , Ampe, C. , Constantin, B. , & Rommelaere, H. (2009). alpha‐Skeletal muscle actin nemaline myopathy mutants cause cell death in cultured muscle cells. Biochimica et Biophysica Acta, 1793, 1259–1271. 10.1016/j.bbamcr.2009.04.004 [DOI] [PubMed] [Google Scholar]
- Yokoyama, S. , Koide, A. , Nishino, I. , Hayashi, Y. , Ohki, H. , Miura, M. , & Shibuya, K. (2016). Hypertrophic cardiomyopathy associated with nemaline myopathy due to ACTA1 mutation. Pediatric Cardiology and Cardiac Surgery, 32, 181–186. 10.9794/jspccs.32.181 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Table S1