

Abstract
Iron deficiency is the most common micronutrient deficiency worldwide. While iron deficiency is known to suppress embryonic organogenesis, its effect on the adult organ in the context of clinically relevant damage has not been considered. Here we report that iron deficiency is a risk factor for nephrotoxic intrinsic acute kidney injury of the nephron (iAKI). Iron deficiency exacerbated cisplatin-induced iAKI by markedly increasing non-heme catalytic iron and Nox4 protein which together catalyze production of hydroxyl radicals followed by protein and DNA oxidation, apoptosis and ferroptosis. Crosstalk between non-heme catalytic iron/Nox4 and downstream oxidative damage generated a mutual amplification cycle that facilitated rapid progression of cisplatin-induced iAKI. Iron deficiency also exacerbated a second model of iAKI, rhabdomyolysis, via increasing catalytic heme-iron. Heme-iron induced lipid peroxidation and DNA oxidation by interacting with Nox4-independent mechanisms, promoting p53/p21 activity and cellular senescence. Our data suggests that correcting iron deficiency and/or targeting specific catalytic iron species are strategies to mitigate iAKI in a wide range of patients with diverse forms of kidney injury.
Keywords: non-heme iron, heme, catalytic iron, Nox4, hydroxyl radicals, apoptosis, ferroptosis, cellular senescence
Introduction
Intrinsic acute kidney injury (iAKI) is induced by a myriad of stimuli that indirectly damage kidney epithelia, including endotoxins, cytotoxic medications, pancreatic enzymes, and metals including heme-iron containing proteins. These chemicals damage glomerular and tubular cells and as a result, disrupt kidney function producing life threatening ‘uremia’ [1, 2]. Yet, patients demonstrate a wide range of sensitivities to injurious stimuli, and variable duration of kidney dysfunction, which has not been adequately explained by specific molecular mechanisms. A number of risk factors have been found to increase susceptibility to intrinsic kidney injury including inflammation (TNFα) [3], metabolic disarray (hyperglycemia in diabetes) [4] , clinical characteristics (sex and aging) [5–8] and preexisting kidney disease (chronic kidney diseases) [9, 10], but it is likely that there are additional, potentially unifying risk-factors. Consequently, to understand the range of responses of the kidney to injurious stimuli, it is important to identify both genetic and non-genetic susceptibility factors.
Nephrotoxicity contributes to hospital-acquired iAKI but its frequency varies widely (8–60%) [11]. Cisplatin (dichlorodiamino platinum) is widely used in the treatment of a variety of solid malignant tumors but a subset of patients (~30%) develops nephrotoxicity [12] due to cisplatin-induced death of proximal tubular cells [13–16]. Rhabdomyolysis represents 7-10% of all cases of AKI in the United States [17]. This form of iAKI is incurred by the release of myoglobin from skeletal muscle under pathological conditions, and its capture and damage of the proximal tubule [18, 19]. Hence, cisplatin and rhabdomyolysis toxicities are two major causes of nephrotoxic iAKI but nonetheless, in both of these cases, the underlying factors that increase susceptibility to these toxins and modulate the duration of iAKI remains unknown.
The delivery of iron to the kidney and its appearance in the urine is thought to be an important mechanism that damages epithelia [20–23]. This notion is supported by the finding that excess iron can directly injure renal tubular cells in vitro and in vivo [24–29], and conversely iron chelators can provide functional and histologic protection in cisplatin-, rhabdomyolysis-, ischemia-reperfusion- and contrast-induced iAKI in animal models. It is thought that chelators remove “poorly bound catalytic iron” [20–22, 30]. Nonetheless, rather than accelerating iAKI, recent experiments suggest a protective role for physiologically replete cellular stores of “bound iron”. Prophylactic delivery of the Lipocalin-2:Siderophore:Iron complex [31], as well as increased baseline levels of serum iron and even systemic iron overload [32] were found to alleviate ischemic iAKI, perhaps by supplying iron for basic cellular functions such as mitochondrial respiration [33] and DNA synthesis [34, 35]. In sum, while an acute role of “catalytic iron” at the onset of iAKI has been explored, it still remains unknown whether different iron species play specific pathogenic roles. In addition, published data indicated that the set point of cellular iron stores may modulate the iAKI phenotype and provides new therapeutic options.
In order to understand the role of cellular iron in iAKI, we created models of nutritional iron deficiency, and determined how they impact nephrotoxicity. Prolonged iron deficiency markedly exacerbated both cisplatin- and rhabdomyolysis-induced iAKI while iron repletion mitigated iron deficiency stimulated iAKI. Given the global importance of iron deficiency in as many as two billion pregnancies and young children [36, 37], our data suggests that nutritional iron deficiency may be an important susceptibility factor for nephrotoxic iAKI worldwide.
Materials and Methods
Animal husbandry
Use of animals and corresponding protocols were approved by the Institutional Animal Care and Use Committee at Tongji University (IACUC No. TJLAC-014-021), Shanghai, China. C57BL/6 mice (Male, two months old, 20-22 g) were obtained from Shanghai Super–B & K Laboratory Animal Corp. Ltd, and a Nox4 KO mouse strain was obtained from Jackson Laboratories (Stock number: 022996). All mice were housed in the SPF Experimental Animal Center of Tongji University by following standard procedures. Iron deficient (FeD, ~27ppm), mild iron deficient (FeDM, ~56ppm) and iron sufficient (FeS, ~250ppm) mouse diets were constituted by supplementing basic mouse diet (D18756, Research Diets, NJ, USA) with ferric citrate, and the iron concentrations were quantitated by using an Optima 2100 atomic absorption spectrometer (Perkin-Elmer). The mice were fed either FeD, FeDM or FeS chows for three weeks in order to generate iron deficiency, mild iron deficiency and iron sufficiency, respectively, before iAKI were experimentally induced with cisplatin or rhabdomyolysis.
Animal models of Cisplatin- and Rhabdomyolysis-induced iAKI
Cisplatin-induced iAKI was established by intraperitoneally injecting mice a single dose of cisplatin (20mg/kg body weight, i.p.) or vehicle (Saline), as previously reported [117]. For the induction of rhabdomyolysis-iAKI, mice were deprived of water for 16 hours, anesthetized with isoflurane and intramuscularly injected 50% glycerol (glycerol:saline=1:1; 6ml/kg) or vehicle (saline) into each posterior thigh muscle with half the volume as previously reported [117]. Kidney injuries were analyzed three days after cisplatin injection and one-day post glycerol injection, respectively.
Intervention of cisplatin- and rhabdomyolysis-induced iAKI
For intervention with ferrostatin-1 (Fer-1), FeD, FeDM and FeS mice were inoculated with Fer-1 (2mg/kg in corn oil, i.p.) or vehicle (Corn oil) 30min before cisplatin or glycerol injection and then every 12 (for cisplatin model) or 6 (for rhabdomyolysis model) hours thereafter, respectively.
For intervention by restoring iron prior, FeD and FeS mice were supplemented iron sucrose (1mg/mouse for three consecutive days) or vehicle (saline) by i.p. injection 7 days before cisplatin or glycerol treatment.
For intervention with DFO or DMTU, FeD and FeS mice were administered DFO (50mg/kg) or DMTU (50mg/kg) or vehicle (saline) by i.p. injection 60min before cisplatin or glycerol treatment and then every 8 hours after cisplatin or glycerol injection.
For intervention by inhibiting Nox4 activity, FeD and FeS mice were administered GKT137831 (40mg/kg) or vehicle by gavage 30min before cisplatin injection and then every 12 hours after cisplatin injection.
Kidney injuries were analyzed three days after cisplatin injection and or one day post glycerol injection, respectively.
Quantitation of sCr and BUN
Serum sCr and BUN were measured by using the automatic biochemistry assay CHRMI-X-180 (Japan Syemex) and iSTAT-300G analyzer (Abbot) according to the manufacturer’s instructions.
Measurement of hematocrit, serum iron, transferrin saturation (TSAT), tissue non-heme iron and catalytic iron
Hematocrit was measured with hematocrit tubes [118]. Serum iron, transferrin saturation, non-transferrin-bound iron, tissue non-heme iron, and catalytic iron were quantitated by following protocols as previously described [22, 119].
Histology, immunohistochemistry and immunofluorescence
Paraffin sections of mouse kidneys were prepared and stained with hematoxylin and eosin (H-E) by following a standard protocol.
To immunodetect ferritin heavy chain (Fth1), Nox4 and 8-OHdG in the kidney, paraffin sections of the kidney were dewaxed and hydrated for antigen retrieval in a boiling buffer (0.01 mol/L sodium citrate, pH 6.0) for 20 min. Anti-Fth1 (1:500, Ab75973, Abcam), anti-Nox4 (1:500, A1528, Abclonal) or anti-8-OHdG (1:250, 4354-MC-050, Trevigen) and HRP-conjugated anti-rabbit antibody (Jackson Immunoresearch Labs) were sequentially applied, and the expression and localization of Fth1 and Nox4 proteins and 8-OHdG were colorized by using a DAB substrate kit (SK-4105, Vector Laboratories), and then visualized and imaged with a Nikon ECLIPSE 80i microscope.
To immunofluorescently detect DMT1 and Tfr1 proteins, duodenum was fixed in 4% paraformaldehyde, cryosectioned and immunostained by sequentially applying primary antibodies against DMT1 (1:500 dilution, ab123085, Abcam) and Tfr1 (1:500 dilution, 126800, Thermo) and secondary Alexa Fluor 488- and Alexa Fluor 594-conjugated donkey anti-mouse and anti-rat antibodies. Immunofluorescence was visualized and imaged for the expression and localization of DMT1 and Tfr1 proteins by using a Leica TCS SP8 confocal microscope. DAPI was used to fluorescently label nuclei.
Cytokine and chemokine measurement
Serum cytokines and chemokines, TNFα, IL6, MCP1 and KC, were quantitated using CBA kits (BD Biosciences) and a flow cytometer (BD, FACSVerse™) by following the manufacturer’s recommended protocols.
TUNEL staining
Tunel staining was performed to detect DNA fragmentation during apoptosis with an in situ Apoptosis Detection Kit (11684817910, Roche) by following the manufacturer’s protocols.
Western Blot
The kidney was homogenized in RIPA buffer (150mM NaCl, 50mM Tris-HCl, pH 7.4, 5mM EDTA, 1% Triton X-100, 0.5% deoxycholate, 0.1% SDS and 1X protease inhibitor cocktail from Sigma-Aldrich). The extracted proteins were fractionated by SDS-PAGE and immunoblotted for the immunodetection of specific proteins. Primary antibodies used in this study were anti-Nox1 (1:2000, ab131088, Abcam), anti-Nox2 (1:2000, ab129068, Abcam), anti-Nox4 (1:5000, A1528, Abclonal), anti-Alas1 (1:1000, ab154860, Abcam), anti-Alas2 (1:1000, ab184964, Abcam), anti-Hmox1 (1:2000, A1346, Abclonal), anti-Hmox2 (1:1000, A2745, Abclonal), anti-RIPK3 (1:2000, ab56164, Abcam), anti-p-RIPK3 (1:2000, phosphorylation at T231 and S232, ab201912, Abcam), anti-MLKL (1:2000, 28640, Cell Signaling), anti-p-MLKL (1:2000, phosphorylation at S345, 62233, Cell Signaling), anti-Bax (1:1000, 556467, BD Biosciences), anti-caspase 8 (1:1000, 8592, Cell Signaling), anti-cleaved caspase 3 (1:2000, 9664, Cell Signaling), anti-p16INK4A (1:2000, ab189034, Abcam), anti-p53 (1:2000, 10442-1-AP, ProteinTech), anti-p21 (1:2000, sc397, Santa Cruz), anti-γH2A.X (1:2000, ab26350, Abcam), anti-H2A.X (1:2000, ab11175, Abcam), anti-Lamin B1 (1:5000, ab16048, Abcam), anti-LC3A/B (1:1000, 21741S, Cell Signaling), anti-Fth1 (1:1000, ab75973, Abcam), anti-FPN (1:1000, NBP1-21502, Novus), anti-Tfr1 (1:1000, 12-6800, Thermo), anti-Ngal (1:5000, AF1857, R&D systems), and anti-β-actin (1:5000, HC201, Transgene). Urinary Ngal was immunodetected by using 5μl of urine for SDS-PAGE and immunoblotting. HRP-conjugated secondary antibodies and ECL kits were purchased from Jackson Immunoresearch Labs and Thermo Scientific, respectively. Chemiluminescence was detected and imaged by using an ImageQuant LAS 4000 mini system (GE Healthcare), and protein abundance was expressed relative to β-actin.
Analyses of protein, lipid and DNA oxidation
Protein oxidization in the kidney was analyzed by immunodetecting carbonyl proteins by using an Oxidized Protein Western Blot Detection kit (ab178020, Abcam). Lipid peroxidation in the kidney was analyzed by immunodetecting 4-HNE-protein adducts by following a standard Western blot protocol with an anti-4-HNE antibody (1:2000, ab46545, Abcam). DNA oxidation in the kidney was analyzed by quantitating 8-OHdG with an ELISA kit (SU-B20013, Nanjing Jiancheng Bioengineering institute) by following a manufacturer’s recommended protocol.
RNA isolation and real-time quantitative PCR.
Total RNA was isolated by using a RNAiso Plus kit (9109, Takara) and was reverse transcribed to cDNA with a PrimeScript™ RT kit with gDNA Eraser (RR047A, Takara). Quantitative PCR was performed in an ABI ViiA™ 7 Real time PCR system by using a SYBR Green kit (RR820A, Takara) and gene-specific primers (Table S1; Invitrogen). 18S rRNA was used an internal control for mRNA expression of genes.
Statistical analyses
All data are presented as mean ± SD, and statistically analyzed using one-way or two-way ANOVA followed by Tukey’s test. A p value of <0.05 was considered statistically significant.
Results
Iron deficiency worsened cisplatin and rhabdomyolysis iAKI
Nutritional iron deficiency was generated by following an established strategy [38, 39] i.e. feeding mice an iron deficient diet (FeD, ~27ppm; or FeDM, ~56 ppm) or an iron sufficient diet (FeS, ~250 ppm, similar to regular mouse chow) for three weeks. Iron deficiency was confirmed by reduction of serum iron and hepatic, splenic and renal non-heme iron (Supplemental Fig. 1A–D). Iron deficiency was also confirmed by a decrease in hepatic Hamp mRNA as well as an increase in duodenal Dcytb and DMT1 mRNA (IRE isoform) (Supplemental Fig. 1E–G). These classical responses varied according to the diet (FeD> FeDM), demonstrating a dose response to dietary iron.
Cisplatin:
Intrinsic AKI (iAKI) was induced by a single dose of cisplatin (Cis; 20mg/kg i.p.) to mice maintained on FeD, FeDM and FeS diets. By 72h after the dose of Cis, body weight decreased by ~18% (Supplemental Fig. 1H), indicating that cisplatin induced a systemic illness, regardless of diet [40]. However, histologic analyses demonstrated cisplatin-induced tubular cell damage in accordance with diet (FeD> FeDM >FeS kidneys) as evidenced by widespread structural disruption and cast accumulation in both proximal and distal tubules (Fig. 1A). The following markers confirmed that FeD and FeDM were more severely affected than FeS mice: Serum creatinine (sCr; Fig. 1B), blood urea nitrogen (BUN; Fig. 1C), kidney mRNA for tubular damage markers Kim-1 and Ngal (Lcn2) (Supplemental Fig. 1I and J), urinary Ngal (Supplemental Fig. 1K) , AKI-associated serum and kidney cytokines and chemokines, TNFα, IL6, MCP1 and KC [41–43], (Supplemental Fig. 1L–S) were more elevated in FeD and FeDM compared with FeS mice.
Fig. 1. Iron deficiency exacerbated cisplatin and rhabdomyolysis iAKI.
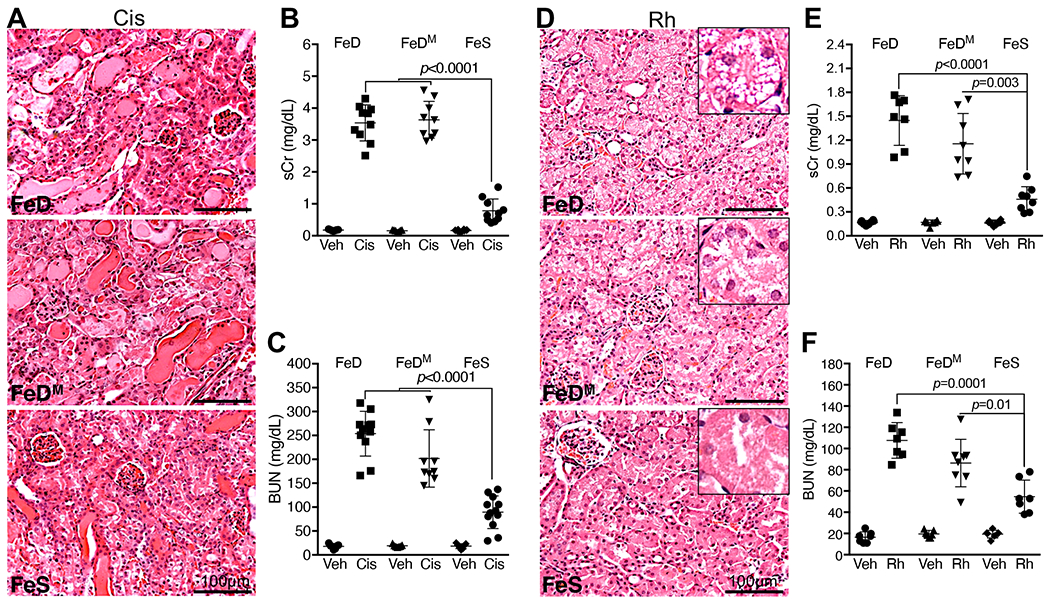
(A) H.E. staining showed more severe tubular damages in FeD and FeDM than in FeS kidneys 72h after cisplatin treatment (Cis), scale bar: 100μm. (B, C) Higher levels of serum creatinine (sCr; B) and blood urea nitrogen (BUN; C) in FeD and FeDM than in FeS mice 72h after cisplatin treatment. n=9-10. (D) H.E. staining showed more severe vacuolization of proximal tubular cells in FeD and FeDM than in FeS kidneys 24h after rhabdomyolysis (Rh) initiation, scale bars: 100μm. (E, F) Higher levels of sCr and BUN in FeD and FeDM than in FeS mice 24h after Rh initiation. n=7-8. Data are means ± SD, and are representatives of three independent experiments. (B, C, E and F) Two-way ANOVA–Tukey’s post hoc test.
Rhabdomyolysis:
A similar pattern emerged when we examined iron deficiency in a second type of iAKI incurred by glycerol-induced rhabdomyolysis (Rh; 6ml/kg, 50% glycerol). Consistent with renal tubular vacuolization observed in human Rh-iAKI patients [44], histologic analysis 24h after Rh demonstrated vacuolated renal tubular cells particularly in FeD and FeDM proximal tubules (Fig. 1D). Markers sCr (Fig. 1E), BUN (Fig. 1F) and kidney Ngal protein (Supplemental Fig. 1T) were markedly elevated in FeD and FeDM compared with FeS Rh mice.
In sum, preexisting iron deficiency worsened iAKI induced by either cisplatin or rhabdomyolysis.
Iron deficiency increases catalytic iron in cisplatin and Rh kidneys
Catalytic iron abruptly emerges during tissue injury and is thought to play important roles in the pathogenesis of cisplatin- and rhabdomyolysis- induced iAKI [21, 22, 27, 30, 45].
Cisplatin:
We found that cisplatin increased catalytic iron in all kidneys at 72h (Cis vs Veh: FeD, ~6.4 folds; FeS, ~3.8 folds) particularly in FeD kidneys (Cis: FeD vs FeS, ~1.6 folds; Fig. 2A). While proximal tubular cytochrome P450 might be the source [22], catalytic iron in FeD cisplatin-treated kidney may derive from systemic rather than kidney iron, given the much lower baseline levels of non-heme iron in FeD kidneys (p=0.004; Fig. 2B) and because cisplatin is a systematic toxicant [40]. To understand the origin of catalytic iron in FeD kidneys (Fig. 2A), we analyzed the impact of cisplatin on systemic iron homeostasis. Cisplatin markedly increased serum iron in both FeD and FeS mice particularly in FeD mice (FeD/Cis vs FeD/Veh: ~1.84; FeS/Cis vs FeS/Veh: ~1.5; Fig. 2C), which resulted in elevated transferrin-bound iron as demonstrated by increased transferrin saturation (TSAT; Fig. 2D) and non-transferrin bound iron (NTBI; Fig. 2E). Cisplatin also increased non-heme iron in the liver (FeD/Cis vs FeD/Veh: ~1.76; FeS/Cis vs FeS/Veh: ~1.35; Fig. 2F), spleen (FeD/Cis vs FeD/Veh: ~7.36; FeS/Cis vs FeS/Veh: ~2.63; Fig. 2G) and kidney (FeD/Cis vs FeD/Veh: ~1.58; FeS/Cis vs FeS/Veh: ~1.35;Fig. 2B). A number of these parameters were enhanced when the mouse was subject to FeD diets.
Fig. 2. Iron deficiency increased catalytic iron in cisplatin and Rh kidneys.
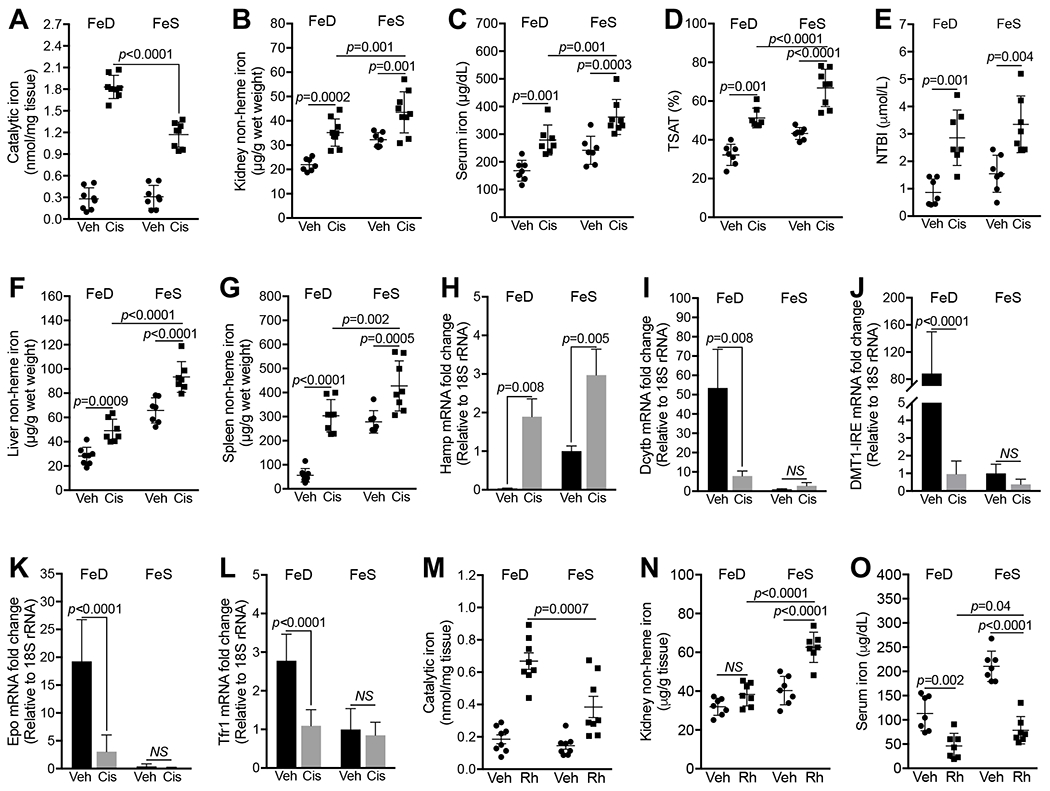
(A) Markedly higher catalytic iron levels in FeD than in FeS kidneys 72h after cisplatin treatment. n=7. (B-F) Levels of renal non-heme iron (B), serum iron (C), transferrin saturation (TSAT; D), non-transferrin-bound iron (NTBI; E), hepatic (F) and splenic (G) non-heme iron were increased in FeD and FeS mice 72h after cisplatin treatment. n=7-8. (H-L) Q-PCR showed the upregulation of hepatic Hamp mRNA (H) and the downregulation of duodenal Dcytb (I) and DMT1 (IRE isoform; J) and kidney Epo (K) and Tfr1 (L) mRNAs in FeD mice 72h after cisplatin treatment. n=7-8. (M) Higher catalytic iron levels in FeD than in FeS kidneys 24h after Rh initiation. n=8. (N) Renal non-heme iron levels were increased in FeS but not in FeD Rh mice. n=7. (O) Serum iron was reduced in FeD and FeS mice 24h after Rh initiation. n=7. Data are means ± SD. (A-O) Two-way ANOVA–Tukey’s post hoc test.
Consistent with elevated systemic iron, cisplatin induced changes in the iron regulatory system, that were particularly prominent in FeD mice. Cisplatin caused much higher fold increase of hepatic Hamp mRNA (FeD/Cis vs FeD/Veh: ~53; FeS/Cis vs FeS/Veh: ~3; Fig. 2H), and larger fold decrease of duodenal Dcytb (FeD/Cis vs FeD/Veh: ~0.15; FeS/Cis vs FeS/Veh: NS; Fig. 2I) and DMT1 (IRE isoform; FeD/Cis vs FeD/Veh: ~0.011; FeS/Cis vs FeS/Veh: NS; Fig. 2J), and kidney Epo (FeD/Cis vs FeD/Veh: ~0.16; FeS/Cis vs FeS/Veh: NS; Fig. 2K), DMT1 (IRE isoform; FeD/Cis vs FeD/Veh: ~0.24; FeS/Cis vs FeS/Veh: NS; Supplemental Fig. 2A) and Tfr1 (FeD/Cis vs FeD/Veh: ~0.4; FeS/Cis vs FeS/Veh: NS; Fig. 2L) in FeD than in FeS mice, reflecting in part different set points in steady state iron balance.
Taken together, cisplatin dramatically increased non-heme iron in injured tissues particularly in FeD mice. We suggest that as a result, systemic iron homeostasis is disrupted, reflected in an increase in serum TSAT (Fig. 2D) and NTBI (Fig. 2E). In addition, because FeD results in much lower basal levels of non-heme iron (Fig. 2B, Supplemental Fig. 1D) resulting in much lower levels of iron binding protein ferritin (Fth1; Supplemental Fig. 2B) prior to injury, cells have less capacity to metabolize cellular iron. Together with endogenous mechanisms such as a marked increase of heme oxygenase 1 (Homx1; Supplemental Fig. 2C) which degrades heme and releases ferrous iron in the proximal tubule [46], acutely increased serum iron likely made a major contribution to non-heme iron and in the setting of limited cellular ferritin produced higher levels catalytic iron in FeD than in FeS cisplatin kidneys.
Rhabdomyolysis:
Rh also markedly increased catalytic iron particularly in FeD kidneys (FeD/Rh vs FeD/Veh: 3.6 folds; and FeS/Rh vs FeS/Veh: 2.65 folds; FeD/Rh vs FeS/Rh: 1.74 folds; Fig. 2M) in the setting of lower baseline non-heme iron levels in FeD kidneys (Fig. 2N). However, Rh differed from cisplatin since Rh caused a marked reduction of serum iron in FeD and FeS mice (Fig. 2O), and did not change non-heme iron content in either FeD or FeS livers (Supplemental Fig. 2D) and spleens (Supplemental Fig. 2E). Hence, while Rh diverted a large quantity of muscular myoglobin-bound heme to the kidney [18, 19, 47], it did not promote the redistribution of systemic non-heme iron via serum to the kidney as did Cis. Interestingly, the conversion of myoglobin heme into non-heme iron was reduced in FeD compared with FeS Rh kidneys (Fig. 2N) despite the upregulation of Hmox1 protein in injured kidney in response to iron deficiency (Supplemental Fig. 2F), suggesting heme accumulation in FeD Rh kidney and efficient heme degradation to release ferrous iron that could be subsequently neutralized by ferritin and exported into circulation by ferroportin. The different heme metabolism in FeD and FeS Rh kidneys may lead to higher catalytic iron levels in FeD than FeS Rh kidneys. Hence, while increased catalytic iron in the kidney characterizes the two forms of iAKI, basic components of iron metabolism appear to differ.
Iron deficiency aggravated protein and DNA oxidation and activated apoptosis and ferroptosis in cisplatin-treated kidneys
Catalytic iron produces highly toxic hydroxyl radicals that attack cell components in close proximity to generate oxidized proteins [48], lipids [49, 50] and 8-OHdG [51, 52]. Lipophilic catalytic iron can also directly catalyze the oxidative reaction of polyunsaturated lipids by removing hydrogen atoms from polyunsaturated fatty acids in the lipid bilayers of organelle membranes [53, 54]. We found markedly higher levels of oxidized carbonyl proteins [55], but not products of lipid peroxidation such as 4-Hydroxynonenal (4-HNE) protein adducts [49], in FeD and FeDM than in FeS kidneys 72h after cisplatin treatment (Fig. 3A). In addition, 8-hydroxydeoxyguanosine (8-OHdG), a product of DNA oxidation and a marker of hydroxyl radicals [51] was largely increased particularly in proximal tubules (Supplemental Fig. 3A) of FeD and FeDM cisplatin-treated kidneys (Cis vs Veh: FeD, ~3 folds; FeDM, ~3 folds; FeS, ~2.3 folds; p<0.0001), compared to FeS kidneys (FeD/Cis or FeDM/Cis vs FeS/Cis, ~1.4 folds; p<0.0001; Fig. 3B). Provided that carbonyl proteins and 8-OHdG were major oxidized products of proteins and DNA by hydroxyl radicals [51, 54, 56], these data indicated that iron deficiency markedly enhanced the production of reactive oxidative species (ROS) particularly hydroxyl radicals in cisplatin-treated kidneys via FeD enhanced production of catalytic iron.
Fig. 3. Iron deficiency stimulated distinct oxidative damage pathways in cisplatin and Rh kidneys.
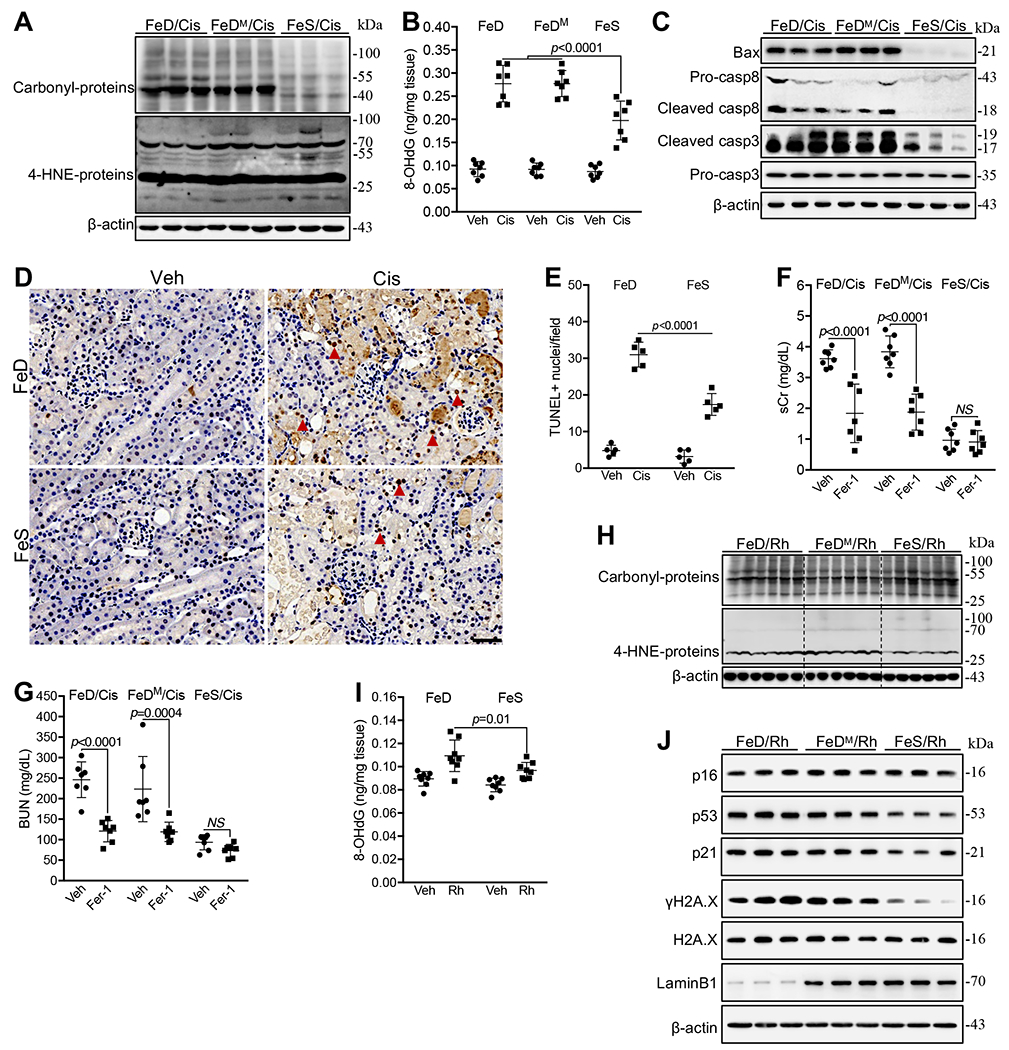
(A) Western blot showed the marked increase of oxidized proteins (carbonyl-proteins) but not oxidized lipids (4-HNE-protein adducts) in FeD and FeDM compared with FeS kidneys 72h after cisplatin (Cis) treatment. (B) Higher levels of 8-OHdG in FeD and FeDM than in FeS kidneys 72h after cisplatin treatment. n=7. (C) Western blot showed a marked increase of Bax, cleaved forms of caspase 8 (Casp8) and 3 (casp3) proteins, in FeD and FeDM compared with FeS kidneys 72h after cisplatin treatment. (D, E) Tunel staining showed the enhanced apoptosis in FeD and FeDM compared with FeS kidneys 72h after cisplatin treatment (D), which was quantified as TUNEL+ cells (Arrow head) per field (E). (F, G) Fer-1 markedly reversed sCr (F) and BUN (G) levels in FeD and FeDM but not in FeS mice 72h after cisplatin treatment. n=7. (H) Western blot showed a marked increase of 4-HNE-proteins but not carbonyl-proteins in FeD and FeDM compared with FeS kidneys 24h after Rh initiation. (I) Slightly higher 8-OHdG levels in FeD and FeDM than in FeS kidneys 24h after Rh initiation. n=8. (J) Western blot showed an increase of p53, p21 and γH2A.X and a decrease of Lamin B1 in FeD and FeDM compared with FeS kidneys 24h after Rh initiation. Data are means ± SD. (B, E, F, G and I) Two-way ANOVA–Tukey’s post hoc test.
ROS activates apoptosis [57], necroptosis[58–61], ferroptosis[62–65] and cellular senescence [66, 67]. We found that key proapoptotic regulators, Bax and cleaved forms of caspase 8/3 [14, 68], were markedly increased in FeD and FeDM compared with FeS kidneys 72h after cisplatin treatment (Fig. 3C), further confirmed by Tunel staining (Fig. 3D, E). Next we probed the ferroptosis pathway by applying a lipid-ROS scavenger and specific ferroptosis inhibitor, Ferrostatin-1 (Fer-1; 2mg/kg; i.p.) [62, 64], which markedly reduced sCr (Fig. 3F), BUN (Fig. 3G) and serum inflammatory factors, TNFα, IL6, MCP1 and KC (Supplemental Fig. 3B to E) in FeD and FeDM but not in FeS cisplatin-treated mice. Fer-1 did not significantly regulate cleaved caspase 3 and the phosphorylation of key regulators of necroptosis, receptor interacting serine/threonine kinase 3 (p-RIPK3) and mixed lineage kinase domain-like (p-MLKL) [13, 16, 69] (fig. S3F). Indeed, iron deficiency did not regulate p-MLKL (Supplemental Fig. 3G), ruling out the necroptosis pathway. In addition, key regulators of cellular senescence, p16, p53 and p21, and a marker of DNA damage and cellular senescence, gamma H2A.X (γH2A.X) [70–75] were not modulated in cisplatin-treated kidneys by dietary iron restriction (Supplemental Fig. 3H). These data indicated that iron deficiency enhanced apoptosis and activated Fer-1-inhibitable ferroptosis in cisplatin-treated kidneys, but it had negligible impact on cisplatin-induced necroptosis and cellular senescence.
Iron deficiency aggravated lipid peroxidation and cellular senescence in Rh kidneys
Distinct from Cis-iAKI, iron deficiency in Rh-iAKI increased lipid peroxidation as demonstrated by more 4-HNE-protein adducts (Fig. 3H) as well as moderately increased 8-OHdG (Rh vs Veh: FeD, ~1.2 folds; FeS, ~1.15 folds; Fig. 3H), but the effect on 8-OHdG were not pronounced (FeD/Rh vs FeS/Rh, ~1.13 folds, p=0.01; Fig. 3I), compared with cisplatin-treated counterparts (Cis vs Veh: FeD, ~3 folds; FeS, ~2.3 folds; FeD/Cis vs FeS/Cis, ~1.4 folds; Fig. 3B). Hence, iron deficiency likely increased the production of ROS such as hydroxyl radicals in Rh kidneys but less drastically than in cisplatin-treated counterparts.
Iron deficiency in Rh-iAKI increased p53 protein and its target gene product p21 but not p16INK4A, and also increased γH2A.X and reduced Lamin B1 proteins, markers of cellular senescence [76] in dose response to iron level in diet (FeD> FeDM > FeS Rh mice; Fig. 3J). In contrast, iron deficiency did not significantly regulate p-RIPK3, p-MLKL, cleaved caspase 3, and LC3-I/II, a marker of autophagy [77], in kidneys of Rh mice (Supplemental Fig. 3I), and Fer-1 failed to regulate sCr and BUN in all Rh mice (Fig. Supplemental Figure 3J and K) although ferroptosis was reported to facilitate the pathogenesis of Rh-iAKI [78].
Taken together, iron deficiency promoted lipid peroxidation and oxidative damage and promoted cellular senescence by upregulating the p53/p21 pathway in the Rh kidney, distinguishing Rh from cisplatin forms of iAKI.
Hydroxyl radical was a key oxidant worsening cisplatin- and Rh-iAKI in FeD mice
We next examined whether iron-catalyzed hydroxyl radical was a key downstream oxidant worsening these two types of iAKI in FeD mice. We found that a hydroxyl radical scavenger, N,N’-dimethylthiourea (DMTU; 50mg/kg, i.p.) [79], markedly reduced 8-OHdG (Fig. 4A), p-RIPK3, p-MLKL, Bax, cleaved caspase 8 and 3 proteins (Fig. 4B) to similar levels in FeD and FeS cisplatin-treated kidneys. As a result, DMTU nearly reversed sCr (DMTU/FeD/Cis: 0.26 ± 0.06 mg/dL; DMTU/FeS/Cis: 0.25 ± 0.12 mg/dL) and BUN (DMTU/FeD/Cis: 27.5 ± 4.8 mg/dL; DMTU/FeS/Cis: 34.6 ± 10.8 mg/dL) levels in FeD and FeS cisplatin-treated mice to the normal range (sCr: ≤0.2 mg/dL) (Fig. 4C and D). Likewise, Ngal protein was suppressed by DMTU treatment (Supplemental Fig. 4A). Similarly, DMTU also suppressed the levels of 8-OHdG (Fig. 4E), p53, p21 and γH2A.X proteins (Fig. 4F) in kidneys of Rh mice and reduced sCr and BUN levels but less effectively in FeD than FeS Rh mice (Fig. 4G and H).
Fig. 4. DMTU diminished cisplatin- and Rh-induced iAKI.
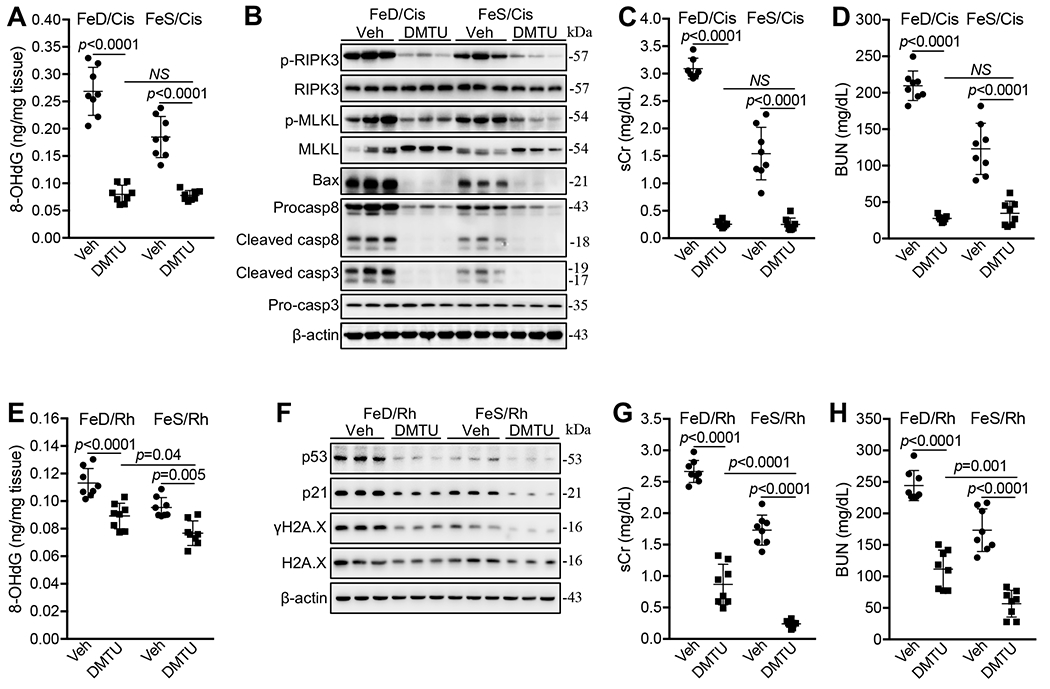
(A) DMTU markedly reduced 8-OHdG levels in FeD and FeS kidneys 72h after cisplatin treatment (Cis). n=7-8. (B) Western blot showed that DMTU markedly reduced p-RIPK3 and p-MLKL proteins and diminished Bax, caspase 8 and cleaved caspase 3 proteins in FeD and FeS kidneys 72h after cisplatin treatment. (C, D) DMTU nearly reversed sCr (C) and BUN (D) levels in FeD and FeS mice 72h after cisplatin treatment. n=8. (E) DMTU reduced 8-OHdG levels in FeD and FeS kidneys 24h after Rh initiation. n=8. (F) Western blot showed that DMTU markedly reduced p53, p21 and γH2A.X proteins in FeD and FeS kidneys 24h after Rh initiation. (G, H) DMTU reduced sCr (G) and BUN (H) levels in all mice 24h after Rh initiation but less efficiently in FeD Rh mice. n=8. Data are means ± SD. (A, C-E, G and H) Two-way ANOVA–Tukey’s post hoc test.
Interestingly, DMTU markedly reduced catalytic iron levels (Supplemental Fig. 4B and C), downregulated ferroportin (FPN; Slc40a1), Fth1, Hmox1 and Hmox2, and upregulated Alas1 and Alas2 in both FeD and FeS kidneys subject to either cisplatin (Supplemental Fig. 4D) or Rh (Supplemental Fig. 4E), suggesting that DMTU mitigated the progression of cisplatin- or Rh- induced oxidative kidney damage and consequently may prevent further generation of catalytic iron in injured kidneys.
In sum, DMTU nearly reversed cisplatin-induced iAKI in FeD and FeS mice and markedly reduced Rh-induced iAKI but less efficiently in FeD mice, despite the fact that these stimuli induced different pathways downstream of catalytic iron. These data indicate that the increased production of hydroxyl radical is a key pathogenic mechanism for worsening these two types of iAKI in FeD mice.
Non-heme catalytic iron played a major role in worsening cisplatin-induced iAKI in FeD mice
Catalytic iron includes both non-heme iron (hydrophilic) and heme-iron (lipophilic) species that are mutually interconvertible through the regulation of heme synthesis and degradation, respectively [46]. To define the pathogenic role of non-heme catalytic iron, we administered mice with an efficient non-heme iron chelator, deferoxamine (DFO) [30], and examined whether DFO could remove renal catalytic iron and suppress cisplatin-induced iAKI in FeD mice. DFO reduced the levels of both non-heme iron (Fig. 5A) and catalytic iron (Fig. 5B) in kidneys of all cisplatin-treated mice, which was consistent with the downregulation of FPN and ferritin and the upregulation of Tfr1 especially in FeD cisplatin-treated kidneys (Supplemental Fig. 5A). It was notable however, that even after DFO treatment, catalytic iron levels remained higher in FeD than FeS cisplatin-treated kidneys (p=0.01; Fig. 5B) despite lower non-heme iron levels (p<0.0001; Fig. 5A), indicating the presence of higher levels of DFO-unchelatable catalytic iron species. Additionally, DFO markedly downregulated key enzymes of heme degradation, Hmox1 and Hmox2 proteins and upregulated key enzymes of heme synthesis, Alas1 and Alas2 proteins, in both FeD and FeS cisplatin-treated kidneys (Supplemental Fig. 5A) perhaps by activating hypoxia signaling pathways [80–84]. Taken together, DFO reduced non-heme catalytic iron but also probably inhibited heme degradation particularly in FeD cisplatin-treated kidneys suggesting the possibility that residual catalytic iron species probably included heme.
Fig. 5. DFO reduced catalytic iron and suppressed cisplatin-iAKI but not Rh-iAKI in iron deficient mice.
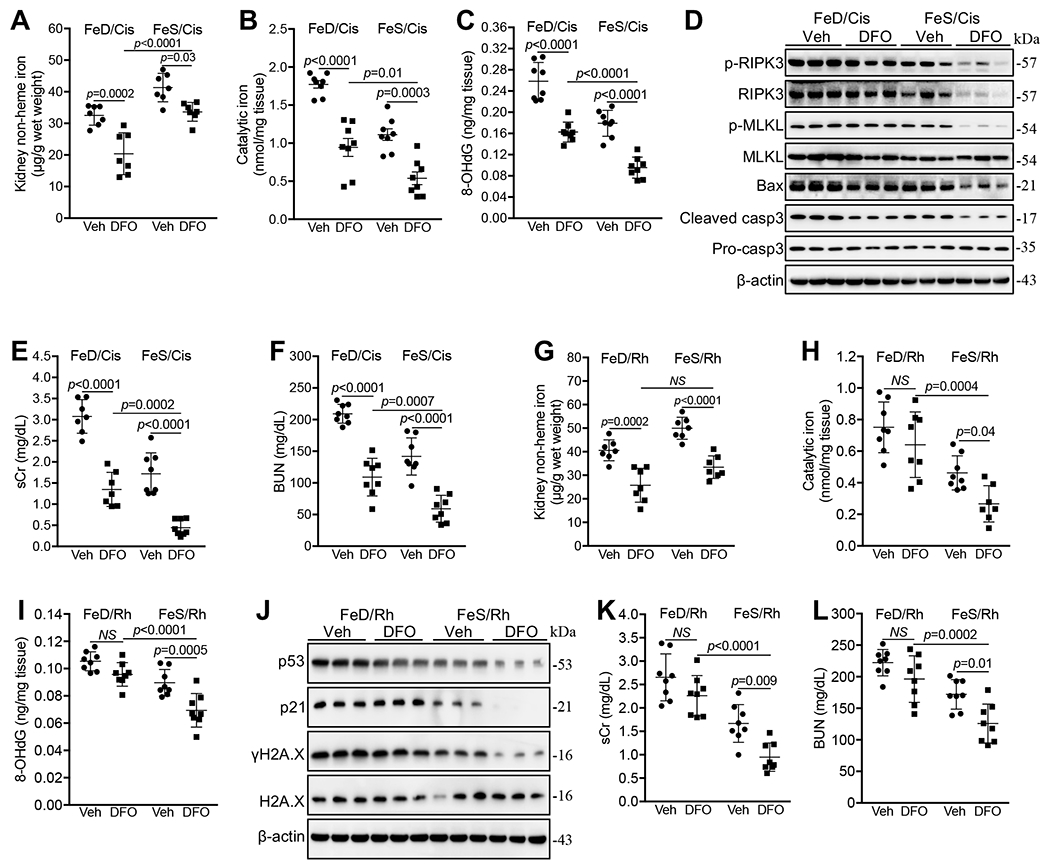
(A-C) DFO reduced non-heme iron (A), catalytic iron (B), and 8-OHdG (C) levels in FeD and FeS kidneys 72h after cisplatin treatment. n=7-8. (D) Western blot showed that DFO reduced Bax and cleaved caspase 3 proteins in FeD kidneys, and p-RIPK3, p-MLKL, Bax and cleaved caspase 3 proteins in FeS kidneys 72h after cisplatin treatment. (E, F) DFO reduced sCr (E) and BUN (F) levels in FeD and FeS mice 72h after cisplatin treatment. n=7-8. (G) DFO reduced non-heme iron levels in FeD and FeS kidneys 24h after Rh initiation. n=7. (H, I) DFO reduced catalytic iron (H) and 8-OHdG (I) levels in FeS but not in FeD kidneys 24h after Rh initiation. n=7-8. (J) Western blot showed that DFO reduced p53, p21 and γH2A.X proteins in FeS but not in FeD kidneys 24h after Rh initiation. (K, L) DFO reduced sCr (K) and BUN (L) levels in FeS but not in FeD mice 24h after Rh initiation. n=8. Data are means ± SD. (A-C, E-I, K and L) Two-way ANOVA–Tukey’s post hoc test.
Consistent with the reduction of catalytic iron (Fig. 5B), DFO suppressed the levels of 8-OHdG (Fig. 5C), Bax and cleaved caspase 3 (Fig. 5D) in FeD and FeS cisplatin-treated kidneys. Interestingly, DFO markedly decreased the levels of p-RIPK3 and p-MLKL in FeS but not in FeD cisplatin-treated kidneys (Fig. 5D). In addition, DFO did not regulate the levels of p53, p21, p16, and γH2A.X proteins in all cisplatin-treated kidneys (Supplemental Fig. 5B). As a result, DFO treatment suppressed sCr and BUN levels but less efficiently in cisplatin-treated FeD mice (Fig. 5E and F).
Heme catalytic iron played a major role in worsening Rh-iAKI in FeD mice
DFO treatment reduced renal non-heme iron in both FeD and FeS Rh mice (Fig. 5G), and consistently downregulated FPN and Fth1 protein and upregulated Tfr1 protein (Supplemental Fig. 5C). However, while DFO suppressed catalytic iron in the kidney of FeS Rh mice [21, 30], suggesting that non-heme catalytic iron species was present in FeS kidney, DFO failed to reduce catalytic iron in FeD counterparts (Fig. 5H), suggesting that heme might be its major catalytic iron species. Moreover, DFO markedly downregulated key enzymes of heme degradation, Hmoxl and Hmox2 proteins, in kidneys of both FeD and FeS Rh mice (Supplemental Fig. 5C), indicating that DFO likely inhibited heme degradation particularly in the kidneys of FeD Rh mice.
Consistent with its impact on catalytic iron, DFO reduced the levels of 8-OHdG (Fig. 5I), p53, p21 and γH2A.X (Fig. 5J) in kidneys of FeS but not in FeD Rh mice. As a result, DFO markedly reduced sCr and BUN levels in FeS but not in FeD Rh mice (Fig. 5K and L).
Nox4 was upregulated to interact with non-heme catalytic iron to exacerbate cisplatin- but not Rh- iAKI in FeD mice
Nox enzymes are major source of ROS in biologic systems by catalyzing the production of superoxide which can be converted to highly reactive hydroxyl radicals by catalytic iron [45]. Nox1, Nox2 and Nox4 were expressed in the kidney [85–88] and while their expression was not changed by iron deficiency alone (Supplemental Fig. 6A) there was massive upregulation of Nox4 protein (but not Nox1 or Nox2) in proximal tubules, and to a lesser extent, in the loop of Henle by FeD and FeDM but not by FeS after cisplatin-treatment (Fig. 6A and B). Conversely, Nox4 protein was markedly downregulated by Fer-1 in FeD and FeDM cisplatin-treated kidney (Fig. 6C) and by DFO (Fig. 6D) and DMTU (Fig. 6E) in both FeD and FeS cisplatin-treated kidneys. Taken together, these data suggested that Nox4 may play important roles in worsening cisplatin-induced iAKI in FeD mice.
Fig. 6. NOX4 was upregulated to interact with non-heme catalytic iron to exacerbate cisplatin-iAKI but not Rh-iAKI in iron deficient mice.
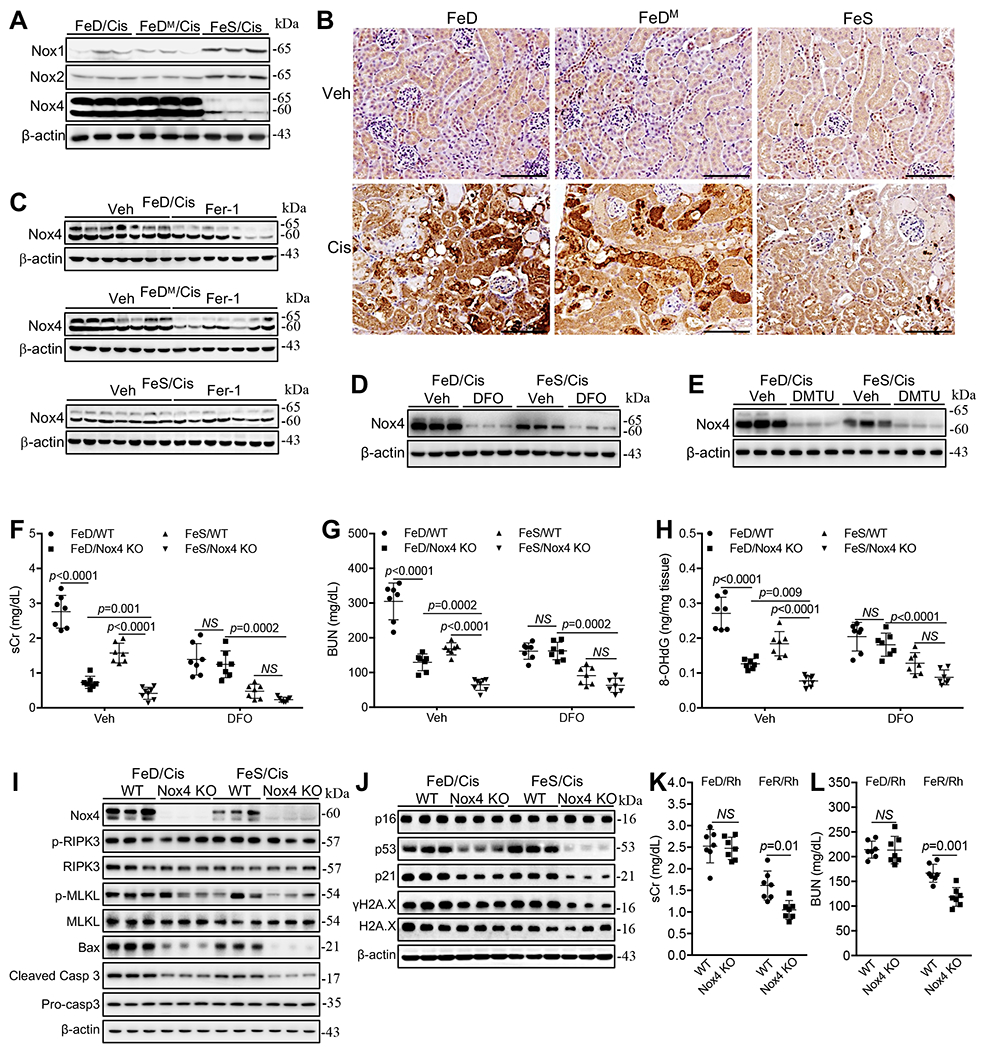
(A) Western bot showed the marked upregulation of Nox4 protein in FeD and FeDM compared with FeS kidneys 72h after cisplatin treatment. (B) Immunohistochemical staining showed that Nox4 protein was upregulated mainly in the proximal tubules of FeD and FeDM compared with FeS kidneys 72h after cisplatin treatment, scale: 100μm. (C-E) Fer-1 reduced Nox4 protein in FeD kidneys (C), while DFO (D) and DMTU (E) markedly downregulated Nox4 protein in FeD and FeS kidneys 72h after cisplatin treatment. (F, G) Nox4 KO markedly reduced sCr (G) and BUN (H) levels in all mice but less efficiently in FeD mice 72h after cisplatin treatment. n=7-8, but it failed to regulate these molecules in FeD and FeS cisplatin kidneys when treated with DFO. (H) Nox4 KO markedly reduced 8-OHdG levels in all kidneys but less efficiently in FeD mice 72h after cisplatin treatment, but it failed to regulate this molecule in FeD and FeS cisplatin kidneys when treated with DFO. n=7-8. (I) Western blot showed that Nox4 KO markedly reduced Bax and cleaved caspase 3 but not p-RIPK3 and p-MLKL proteins in FeD and FeS kidneys 72h after cisplatin treatment. (J) Western blot showed that Nox4 KO reduced p53, p21 and γH2A.X proteins in all kidneys but less efficiently in FeD kidneys 72h after cisplatin treatment. (K, L) Nox4 KO reduced sCr (K) and BUN (L) levels in FeS but not FeD mice 24h after Rh initiation. n=7. Data are means ± SD from two independent experiments. (F-H, K and L) Two-way ANOVA–Tukey’s post hoc test.
We examined the roles of Nox4 in cisplatin-induced iAKI by using both a specific and potent inhibitor of Nox1/Nox4 [89], GKT137831 (40mg/kg; i.p.), and a Nox4 knockout (KO) murine model. GKT137831 markedly reduced sCr and BUN levels in cisplatin-treated FeD mice (Supplemental Fig. 6B and C) and genetic deletion of Nox4 caused a marked reduction of sCr and BUN levels in both FeD and FeS cisplatin-treated mice albeit less efficiently in FeD mice (Fig. 6F and G). Likewise, Nox4 KO reduced the levels of 8-OHdG (Fig. 6H) and Bax and cleaved caspase 3 but not p-RIPK3 and p-MLKL proteins (Fig. 6I) in cisplatin-treated kidneys but less efficiently in FeD mice. Surprisingly, while the levels of p53, p21 and γH2A.X proteins were comparable in cisplatin-treated kidneys of FeD and FeS WT mice, these molecules were reduced in kidneys of all Nox4 KO mice but less efficiently in FeD Nox4 KO mice, which resulted in higher levels of these proteins in FeD than in FeS Nox4 KO kidneys (Fig. 6J). Nox4 KO also markedly reduced renal catalytic iron levels but less efficiently in FeD than in FeS cisplatin-treated kidneys (Supplemental Fig. 6D). Interestingly, Nox4 KO markedly increased renal non-heme iron (Supplemental Fig. 6E), upregulated FPN, Fth1, Alas1 and Alas2 proteins particularly in FeD cisplatin-treated kidney and diminished the expression of Hmox1 and Hmox2 in both FeD and FeS cisplatin-treated kidneys (Supplemental Fig. 6F), indicating that Nox4 KO reduced catalytic iron also possibly through increasing the expression of ferritin and FPN proteins.
When DFO was administered and reduced catalytic iron to similar levels in cisplatin-treated kidneys of WT and Nox4 KO mice under either FeD or FeS conditions (Supplemental Fig. 6G), Nox4 KO failed to significantly reduced renal 8-OHdG production (Fig. 6H) and sCr (Fig. 6F) and BUN (Fig. 6G) levels in either FeD or FeS mice when compared to their WT counterparts, indicating that non-heme catalytic iron was required for the pathogenic role of Nox4 in cisplatin-iAKI. Taken together, Nox4 was a major pathogenic contributor to cisplatin-induced iAKI and its upregulation was a major mechanism exacerbating cisplatin-induced iAKI in FeD mice likely by interacting with non-heme catalytic iron and producing hydroxyl radicals.
In Rh mice, the expression of Nox4 protein was slightly upregulated in FeD compared with FeS kidneys (Supplemental Fig. 6H). However, Nox4 KO failed to regulate sCr and BUN levels in FeD Rh mice while it reduced their levels in FeS Rh mice (Fig. 6K and L), indicating that Nox4 may not play significant roles in the pathogenesis of Rh-induced iAKI under iron deficient conditions, but it was likely involved in the regulation of this process under iron sufficient conditions.
Prior iron correction prevented cisplatin- and Rh- induced iAKI in FeD mice
To examine whether iron correction prior to injury can prevent the worsening effects of iron deficiency on cisplatin- and Rh- induced iAKI, we supplemented mice with iron sucrose (1mg/mouse/day for three consecutive days) by i.p. injection 7 days prior to iAKI induction. Iron sucrose increased non-heme iron content in kidneys of both cisplatin-treated and Rh mice (Fig. 7A and B), and upregulated Fth1 and FPN proteins particularly in kidneys of FeD cisplatin or Rh mice (Supplemental Fig. 7A and B). Indeed, iron supplementation markedly reduced the levels of catalytic iron (Fig. 7C), 8-OHdG (Fig. 7D), and p-RIPK3, p-MLKL, Bax and cleaved caspase 3 proteins (Fig. 7E), and consequently suppressed sCr and BUN in FeD cisplatin-treated mice to similar levels in vehicle-injected FeS counterparts (Fig. 7F and G). Remarkably, iron supplementation also markedly reduced renal catalytic iron, 8-OHdG, and p-RIPK3, p-MLKL, Bax and cleaved caspase 3 proteins, and sCr and BUN levels in FeS cisplatin-treated mice (Fig. 7C–G).
Fig. 7. Prior iron correction protected from cisplatin- and Rh-iAKI.
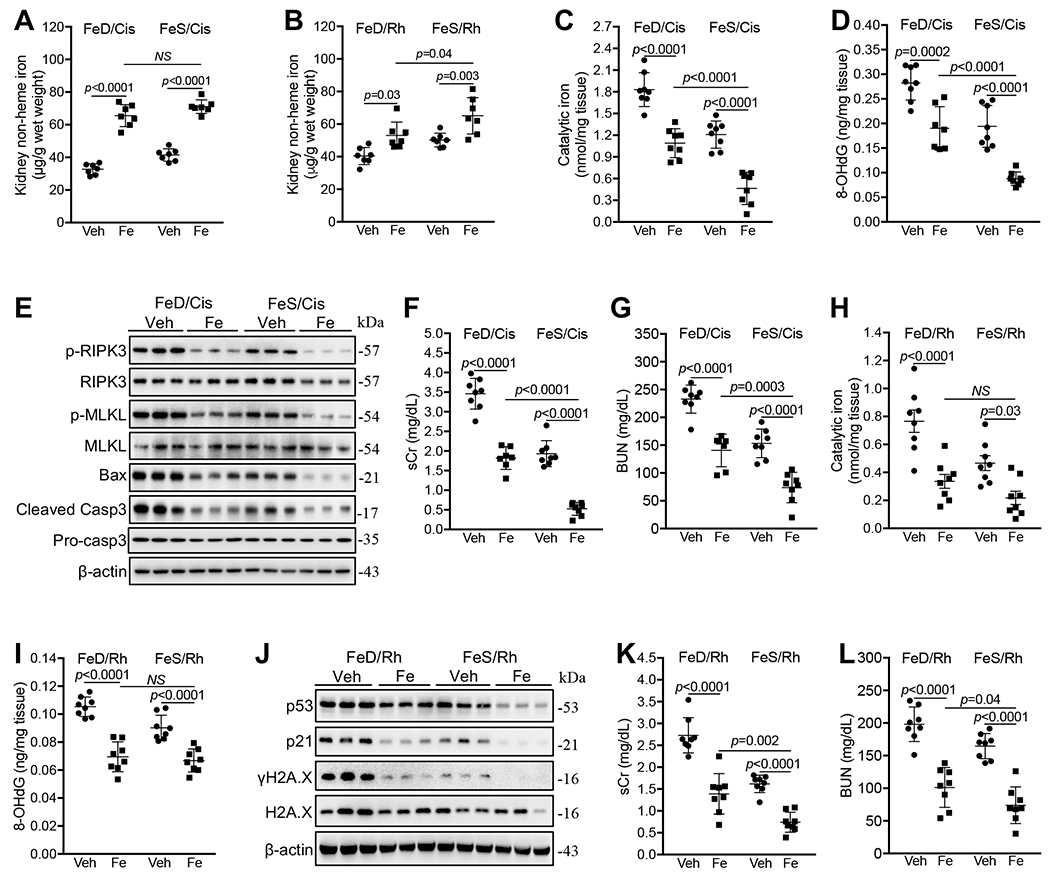
(A, B) Prior iron supplementation (Fe) increased renal non-heme iron content 72h after cisplatin treatment (A) and 24h after Rh induction (B). n=7. (C, D) Prior iron supplementation reduced catalytic iron (C) and 8-OHdG (D) levels in FeD and FeS cisplatin-treated kidneys. n=8. (E) Western blot showed that prior iron supplementation reduced p-RIPK3, p-MLKL, Bax and cleaved caspase 3 proteins in FeD and FeS cisplatin-treated kidneys. (F, G) Prior iron supplementation reduced sCr (C) and BUN (D) levels in FeD and FeS cisplatin-treated mice. n=7-8. (H, I) Prior iron supplementation reduced catalytic iron (H) and 8-OHdG (I) levels in FeD and FeS kidneys 24h after Rh initiation. n=8. J. Western blot showed that prior iron supplementation reduced p53, p21 and γH2A.X proteins in FeD and FeS kidneys 24h after Rh initiation. (K, L) Prior iron supplementation reduced sCr (K) and BUN (L) levels in FeD and FeS Rh mice. n=8. Data are means ± SD. (A-D, F to I, K and L) Two-way ANOVA–Tukey’s post hoc test. (A-L) A representative of two independent experiments.
Iron supplementation also markedly suppressed renal catalytic iron (Fig. 7H), 8-OHdG (Fig. 7I), and p53, p21 and γH2A.X proteins (Fig. 7J), and resultantly sCr (Fig. 7K) and BUN (Fig. 7L) in FeD Rh mice to similar levels in vehicle-injected FeS counterparts. Again, iron supplementation suppressed renal catalytic iron, 8-OHdG, and p53, p21 and γH2A.X proteins and sCr and BUN levels in FeS Rh mice (Fig. 7H–L).
Taken together, iron restoration prior to iAKI induction prevented the worsening effects of iron deficiency on cisplatin- and Rh- induced iAKI likely by upregulating ferritin and ferroportin proteins which could contribute to neutralizing catalytic iron [23] ameliorating of iAKIs in FeD mice.
Conclusively, iron deficiency is a susceptibility factor that worsens cisplatin- and Rh- iAKI by increasing non-heme/heme and heme catalytic iron species to interact with Nox4 and Nox4-independent mechanisms, respectively, and resultantly promoting stimuli-specific downstream oxidative kidney injury pathways, and prior iron supplementation restores metabolic iron balance and upregulates ferritin and ferroportin (FPN), which protects from iAKI (Figure 8).
Fig. 8. Mechanistic pathways by which iron deficiency worsened cisplatin- and Rh-iAKI.
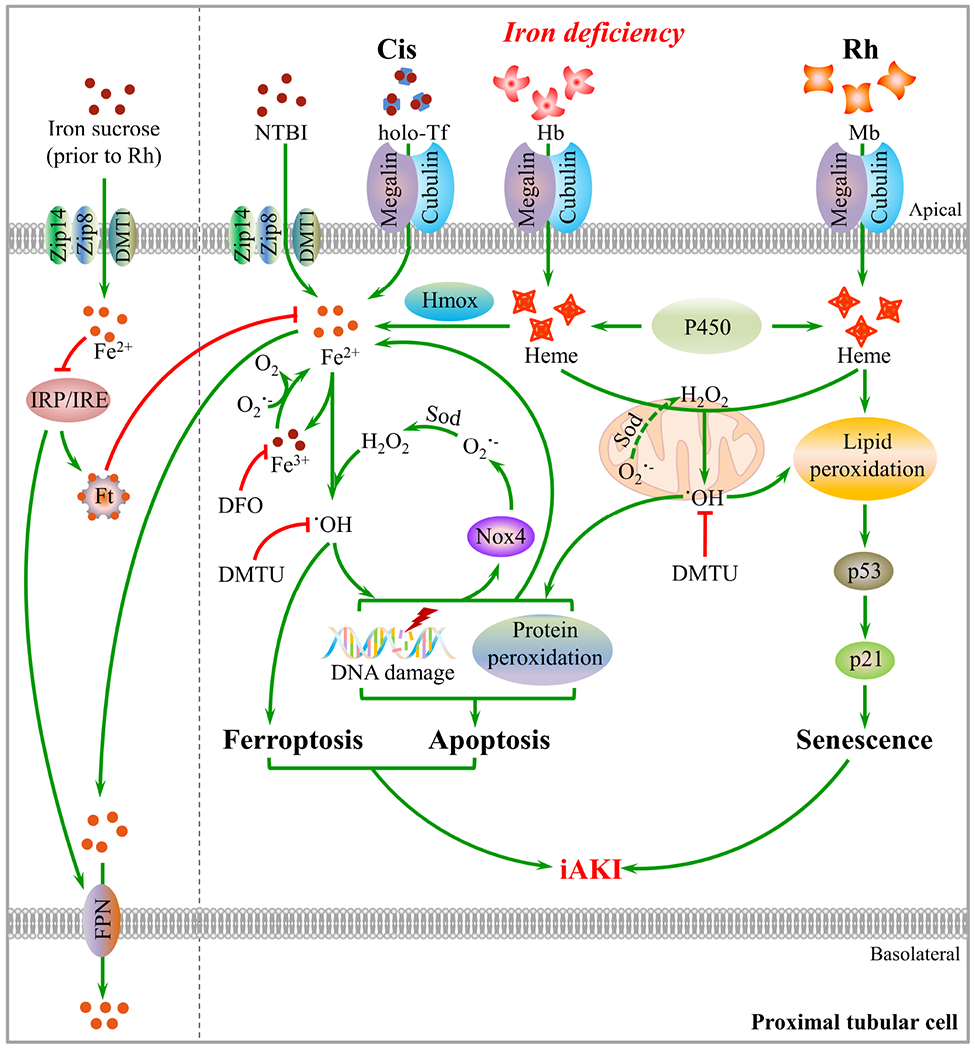
Iron deficiency exacerbates cisplatin (Cis)-iAKI by markedly increasing non-heme and heme catalytic iron which interact with upregulated Nox4 and probably mitochondrion-derived peroxide, respectively, to produce massive hydroxyl radicals, and resultantly aggravating protein and DNA oxidation, apoptosis and ferroptosis. Crosstalk between non-heme catalytic iron/Nox4 and oxidative damage generates a mutual amplification cycle that facilitates rapid progression of cisplatin-iAKI. Iron deficiency aggravated Rh-iAKI by increasing catalytic heme-iron that induced lipid peroxidation and DNA oxidation by interacting with Nox4-independent mechanisms, promoting p53/p21 activity and cellular senescence. Prior iron supplementation restored metabolic iron balance and upregulates ferritin (Ft) and ferroportin (FPN), which protects from iAKI.
Discussion
In this study, we demonstrated that iron deficiency is a risk factor for two different types of nephrotoxic iAKIs, cisplatin and rhabdomyolysis. Both types of iAKI increase specific non-heme and/or heme catalytic iron species, which subsequently activate different downstream pathophysiologic mechanisms. Consistent with this finding, recent data from Vaugier et al demonstrated that elderly patients with increased baseline levels of serum iron and ferritin were predicted to have better renal allograft survival [32]. Moreover, iron overload in HFE knockout mice markedly reduced the severity of post-ischemic AKI [32], and conversely, patients with lower hematocrits had more severe colistin induced iAKI [90]. Because iron deficiency is the most common micronutrient deficiency in the world, these findings demonstrate a new clinical strategy to prevent iAKI by correcting iron deficiency.
Cisplatin-mediated disruption of systemic iron homeostasis and acute redistribution of systemic iron particularly in FeD mice likely caused higher levels of catalytic iron in FeD kidneys. Given that cisplatin damages most tissues [91, 92], newly transported iron might derive from damaged iron-enriched tissues such as liver and spleen where non-heme iron levels markedly increased after cisplatin treatment and could then be released via upregulating iron exporter ferroportin [93–96] and transported to damaged kidneys through repetitive renal filtration and reabsorption. In addition, newly transported iron might also derive from cisplatin-induced hemolysis [97]. A red blood cell source of iron would be particularly damaging because it includes hemoglobin-iron, heme-iron, and non-transferrin-bound iron which are known to be transported to the kidney [98, 99] and are catalytically active [24–27]. A similar process involving cytochrome P-450 is known to occur within the kidney subsequent to cisplatin treatment [22]. Among these iron species, heme could be degraded to release non-heme catalytic iron by heme oxygenases, Hmox1 and Hmox2, that were markedly upregulated in cisplatin-injured kidneys particularly in FeD mice [100, 101]. However, heme might be less efficiently degraded in the presence of DFO likely because DFO-induced iron deficiency (particularly in FeD mice) may limit the supply of NAPDH required for Hmox1/2 activities (from NADPH cytochrome P450 reductase and the pentose phosphate pathway) [102] as well as by downregulating Hmox1 and Hmox2 proteins likely through the activation of hypoxia signaling pathways [82–84]. Hence, clinical use of DFO needs to be carefully evaluated particularly in iron deficient patients.
Iron deficiency induced the overproduction of hydroxyl radical in cisplatin-injured kidneys by increasing non-heme catalytic iron species. We found several lines of evidence that non-heme catalytic iron species played a major pathogenic role in cisplatin-induced iAKI, including (1) the marked increase of both non-heme iron and catalytic iron in FeD cisplatin-treated kidney; (2) the marked reduction of both non-heme iron and catalytic iron and oxidative kidney damage by a non-heme iron chelator deferoxamine; (3) the reversal of oxidative kidney damage in cisplatin-treated kidneys by a water soluble hydroxyl radical scavenger DMTU; and, (4) the markedly worsened protein and DNA oxidation in FeD compared with FeS cisplatin-treated kidneys, consistent with the fact that non-heme catalytic iron reacts with superoxide and peroxide to produce hydroxyl radicals in a broad hydrophilic cellular environment and cause widespread oxidative damage [51, 54, 56].
Iron deficiency induced the overproduction of hydroxyl radical also by dysregulating Nox4 in cisplatin-injured kidneys. Nox4 is a major form of NADPH oxidases in the kidney and an important source of ROS for normal renal physiological functions and in the setting of kidney injury [103, 104]. Its abrupt upregulation in FeD cisplatin-injured kidneys that could be reversed by Fer-1[62, 64], DFO and DMTU and the reversal of a number of metrics of cisplatin-induced iAKI by its inhibition and genetic deletion conclusively demonstrated that Nox4 played a key role in the pathogenesis of cisplatin-induced iAKI exacerbating the kidney injury in FeD mice. Moreover, findings that DFO and preventive iron supplement reduced non-heme catalytic iron and Nox4 protein and DFO abolished the pathogenic roles of Nox4 in cisplatin-induced iAKI indicated that Nox4-mediated cell damage was downstream of non-heme catalytic iron accumulation in both FeD and FeS cisplatin-injured kidneys. Provided that non-heme catalytic iron reacts with hydrogen peroxide, an end product of Nox4, to generate hydroxyl radicals, these data indicate that non-heme catalytic iron mainly interacts with Nox4 to facilitate cisplatin-induced iAKI particularly in FeD mice. Further, because the increase of catalytic iron and Nox4 in cisplatin-treated kidneys worsened oxidative damage and conversely the inhibition of oxidative damage by DMTU and Nox4 KO reduced catalytic iron/Nox4 and catalytic iron, respectively, non-heme catalytic iron/Nox4 and downstream oxidative damage likely formed a mutual amplification cycle that facilitated the rapid progression of cisplatin-induced iAKI. Therefore, strategies that target this cycle particularly by removing the upstream non-heme catalytic iron or inhibiting downstream Nox4 or eliminating downstream hydroxyl radicals are predicted to be efficacious for the intervention of cisplatin-induced iAKI.
Iron deficiency increased apoptosis a major cell death mechanism in the pathophysiology of cisplatin-induced iAKI [13–16]. Iron deficiency specifically increased pro-apoptotic proteins, Bax, active caspase 8 and active caspase 3, which could be reversed by treatment with iron supplement, DFO, DMTU and Nox4 KO, indicating that apoptosis-mediated cell damage is downstream of catalytic iron/Nox4 accumulation and hydroxyl radical-mediated oxidation in FeD cisplatin-injured kidneys. In contrast, key regulators of necroptosis, p-RIPK3 and p-MLKL, were not induced by iron deficiency, nor ameliorated by DFO treatment and Nox4 KO in FeD cisplatin-treated kidneys. Hence, non-heme catalytic iron/NOX4-mediated hydroxyl radical accumulation is a distinct pathway which enhanced the activity of apoptotic pathways.
Iron deficiency also induced the accumulation of lipid-ROS that activate ferroptosis in cisplatin-injured kidneys. Ferroptosis is thought to be an iron dependent cell death pathway [105] induced by lipid-ROS and plays important roles in the pathophysiology of several types of kidney injury, including folic acid-induced iAKI, ischemia-reperfusion iAKI, rhabdomyolysis iAKI, and oxalate nephropathy [65, 78, 106]. Fer-1 is a lipid-ROS scavenger and it partially reversed the exacerbation of cisplatin-iAKI in FeD mice apparently by quenching lipid-ROS and regulating Nox4. Since Nox4 needed non-heme catalytic iron to promote oxidative damage, we suggest that crosstalk between non-heme catalytic iron/NOX4 pathway and ferroptosis might also be a contributor to cisplatin-induced iAKI under FeD conditions. In fact, the interaction of Nox and ferroptosis pathways was previously suggested by experiments showing that Nox enzymes provide a source of ROS inducing ferroptosis in Calu-1 and HT-1080 cells treated with a ferroptosis inducer, erastin [62].
Nox4-mediated hydroxyl radical accumulation represented an important pathway that induced cellular senescence particularly in FeS cisplatin-injured kidney. This notion was supported by the findings that Nox4 KO reduced p53, p21 and γH2A.X proteins in all cisplatin-treated kidneys particularly in FeS mice, which resulted in higher levels of these proteins in FeD than FeS cisplatin-injured kidneys. Since Nox4 interacted with the upstream non-heme catalytic iron to fulfill its pathogenic activity, non-heme catalytic iron/Nox4 may be an important mechanism for the activation of cellular senescence. Surprisingly, DFO failed to regulate p53, p21 and γH2A.X proteins in kidneys of either FeD or FeS cisplatin-treated mice. It is known that iron deficiency or iron deficiency followed by excessive iron resupply causes mitochondrial damage with reduced respiratory efficiency and increased levels of oxidant, mtDNA oxidation and lipid peroxidation in rat liver and kidney [33, 107]. Hence, it is possible that DFO-induced iron deficiency worsened damage of organelles [108] that may consequently exacerbate cellular senescence in FeD and FeS cisplatin-injured kidneys despite simultaneously removing non-heme catalytic iron to alleviate cellular senescence.
Non-heme catalytic iron may also interact with Nox4-independent pathways and consequently worsened cisplatin-induced iAKI in FeD mice because of more severe DNA oxidation, apoptosis and cellular senescence in FeD than FeS cisplatin-injured kidneys of Nox4 KO mice. Since iron deficiency increases oxidative damage of organelles particularly mitochondria in the kidney [33, 107], it was possible that subsequent cisplatin treatment could exacerbate mitochondrial damage to generate higher levels of superoxide and peroxide that interact with elevated non-heme catalytic iron to produce massive hydroxyl radicals and consequently worsen oxidative damage in FeD kidney. This possibility was supported by that finding that the removal of hydroxyl radicals by DMTU markedly reduced DNA oxidation, necroptosis, apoptosis and finally sCr and BUN to similar levels in FeD and FeS cisplatin-treated mice, respectively.
Iron deficiency caused drastic increase of heme catalytic iron species in Rh-injured kidney. Rh diverts a large quantity of myoglobin-bound heme to the kidney for degradation by Hmox1 that was responsively upregulated [47, 109]. Indeed, Rh increased non-heme catalytic iron levels in FeS kidney that could be removed by DFO, indicating that non-heme catalytic iron species were involved in the pathogenesis of Rh-induced iAKI in FeS mice. However, Rh largely increased DFO-unchelatable catalytic iron in FeD kidneys, indicating that heme was not efficiently degraded despite the marked upregulation of Hmox1 and Hmox2 proteins perhaps attributable to limited NADPH supply caused by iron deficiency [102]. Therefore, heme was likely major catalytic iron species in the kidney of FeD Rh mice. Further supporting evidence came from the findings that iron deficiency worsened lipid peroxidation but not protein oxidation in the kidney, which is consistent with the lipophilic propensity of heme that confers heme the capability to intercalate into organellar membranes to feasibly catalyze lipid peroxidation [110]. Moreover, Rh moderately increased 8-OHdG, a quantitative marker of hydroxyl radicals [51, 54, 56], and DMTU reduced oxidative kidney damage, indicating that heme catalytic iron could also catalyze the production of hydroxyl radicals through Fenton-like mechanism [110]. However, the mild increase of 8-OHdG particularly in FeD Rh kidney compared with its massive increase in FeD cisplatin-treated kidney suggested that Rh-induced oxidative damage might be confined to specific regions likely in close proximity to organellar membranes consistent with the lipophilicity of heme catalytic iron.
Iron deficiency exacerbated rhabdomyolysis-induced iAKI through heme catalytic iron-initiated cellular senescence. This notion was supported by (1) markedly increased vacuolization in proximal tubular cells, a characteristic of cellular senescence [71], (2) markedly increased heme catalytic iron species, (3) increased lipid peroxidation that can induce cellular senescence [111–114], (4) upregulation of p53 and p21 but not p16INK4A expression, regulators of cellular senescence [73, 75, 115], (5) increased DNA oxidative damages and their specific responsive protein γH2A.X, a reliable quantitative indicator of cellular senescence [70]; and (6) decrease of LaminB1 expression, a reliable marker of cellular senescence [76]. The p53/p21 pathway inhibits cyclin-dependent kinases and cell cycle, and commonly mediate the activation of the senescence program [72–75, 115]. p53 is thought to mediate pro-senescence signals emerging from unscheduled oncogene activation, telomere dysfunction, DNA damage, and ROS [116], and its downstream target p21 can drive the transition of cells into senescence[49, 52].
Provided with that 4-HNE-protein adducts derived from lipid peroxidation has been found to induce cellular senescence through activating critical cell cycle sentinels such as p53 [111–114], iron deficiency likely exacerbated cellular senescence by markedly increasing 4-HNE-protein adducts and subsequently activated the p53/p21 pathway in the kidney subject to Rh. In addition, since the activation of the DNA damage response including formation of DNA damage foci containing activated γ-H2A.X at sites of persistent DNA strand breaks is a major trigger of cell senescence [70], elevated γ-H2A.X protein in response to increased oxidative DNA damages in the kidney of FeD Rh mice indicated the activation of a process leading to cellular senescence. Hence, heme catalytic iron may be identified to be a primary upstream inducer of cellular senescence particularly in FeD Rh-injured kidney.
Collaborative interactions between Nox4 and non-heme catalytic iron likely also exist in Rh-induced iAKI under FeS conditions because DFO, Nox4 KO and DMTU could markedly reduce oxidative damage in FeS Rh mice. However, neither DFO nor Nox4 KO significantly impacted Rh-induced kidney damage in FeD mice despite the fact that Nox4 was moderately upregulated in the kidney, indicating that Nox4 could not interact with heme catalytic iron species in the kidney of FeD Rh mice.
Taken together, nutritional iron deficiency is a “risk factor” for iAKI that synergizes with cisplatin or rhabdomyolysis to severely exacerbate kidney injury. The crosstalk between iron levels and cell damage suggest that clinical studies designed to normalize iron levels will improve the outcome of tissue damaging therapies. Conversely, medications or procedures that induce cell damage are inappropriate during periods of iron deficiency. While speculative, the impact of iron correction before medical therapy could be quite large, since iron deficiency is the most common micronutrient deficiency, involving large populations on all continents, particularly women of childbearing age.
Supplementary Material
Fig. S1. Verification of nutritional iron deficiency and its worsening effects on cisplatin- and Rh- induced AKI. (A to D) Reduction of serum iron (A), and hepatic (B), splenic (C) and renal (D) non-heme iron in mice which were fed FeD and FeDM compared with FeS diets for 21 days. n=7-8. (E to G) Q-PCR showed the upregulation of duodenal Dcytb and DMT1 mRNA and the downregulation of hepatic Hamp mRNA in FeD and FeDM compared with FeS mice. n=6. (H) Weight reduction of FeD, FeDM and FeS mice 72h after cisplatin treatment. n=7. (I and J) Q-PCR showed the upregulation of Kim-1 and Ngal mRNA in FeD and FeDM compared with FeS kidneys 72h after cisplatin treatment. n=7. (K) Western blot showed a marked increase of urinary Ngal protein in FeD and FeDM compared with FeS mice 72h after cisplatin treatment. (L to S) Higher levels of serum TNFα (L), IL-6 (M), MCP-1 (N) and KC (O) and their kidney mRNA (P-S) in FeD and FeDM than FeS mice 72h after cisplatin treatment. n=7. (T) Western blot showed an increase of Ngal protein in FeD and FeDM compared with FeS kidneys 24h after Rh initiation. Data are means ± SD. (A to G and K) One-way ANOVA–Tukey’s post hoc test; (H to J and L to S) Two-way ANOVA–Tukey’s post hoc test.
Fig. S2. Systemic iron regulation in FeD cisplatin and Rh mice. (A) Q-PCR showed the downregulation of kidney DMT1 (IRE isoform) mRNA in FeD mice 72h after cisplatin treatment. n=7. (B) Immunohistochemical staining showed much lower levels of ferritin (Fth1) in FeD than FeS kidneys prior to injury. (C) Western blot showed the marked upregulation of heme oxygenase 1 (Hmox1) in FeD compared FeS cisplatin-treated kidneys. (D and E) Rh had negligible effect on hepatic (D) and splenic (E) non-heme iron levels in either FeD or FeS mice. n=7. (F) Western blot showed the marked upregulation of heme oxygenase 1 (Hmox1) in FeD compared FeS kidneys subject to Rh. Data are means ± SD. (A, D and E) Two-way ANOVA–Tukey’s post hoc test.
Fig. S3. Iron deficiency enhanced specific tubular cell damage pathways in cisplatin and Rh kidneys. (A) Immunohistochemical staining showed that 8-OHdG was increased mainly in the proximal tubules of FeD compared with FeS kidneys 72h after cisplatin treatment. (B to E) Fer-1 treatment reduced serum TNFα (B), IL-6 (C), MCP-1 (D) and KC (E) levels in FeD and FeDM but not in FeS mice 72h after cisplatin treatment. n=7. (F) Western blot showed negligible changes of p-RIPK3, p-MLKL and cleaved caspase 3 proteins in FeD, FeDM and FeS cisplatin-treated kidneys 72h after Fer-1 treatment. (G and H) Western blot showed comparable levels of p-RIPK3 and p-MLKL (G), p16, p53, p21, γH2A.X and H2A.X proteins (H) in FeD, FeDM and FeS kidneys 72h after cisplatin treatment. (I) Western blot showed comparable levels of p-RIPK3 and p-MLKL, cleaved caspase 3 and LC3-I/II in FeD, FeDM and FeS kidneys 24h after Rh induction. n=7-8. (J and K) Fer-1 did not regulate sCr (J) and BUN (K) levels in FeD, FeDM and FeS mice 24h after Rh induction. n=7-8. Data are means ± SD. (B to E, J and K) Two-way ANOVA–Tukey’s post hoc test.
Fig. S4. DMTU reduced cisplatin- and Rh- induced iAKI. (A) DMTU diminished the expression of Ngal protein in FeD and FeS kidneys 72h after cisplatin treatment. (B and C) DMTU markedly reduced catalytic iron levels in FeD and FeS kidneys subject to cisplatin (Cis; B) and Rh (C). n=8. (D and E) DMTU downregulated ferroportin (FPN), ferritin (Fth1), Hmox1 and Hmox2 proteins while it upregulated transferrin receptor 1 (Tfr1), Alas1 and Alas2 proteins particularly in FeD kidneys subject to cisplatin (D) and Rh (E). Data are means ± SD. (B and C) Two-way ANOVA–Tukey’s post hoc test.
Fig. S5. Regulation of heme and non-heme iron metabolism and cellular senescence in Cis and Rh kidneys by DFO. (A) DFO downregulated ferroportin (FPN), ferritin (Fth1), Hmoxl and Hmox2 proteins, while upregulated transferrin receptor 1 (Tfr1), Alas1 and Alas2 proteins in both FeD and FeS cisplatin kidneys. (B) DFO had negligible effects on cellular senescence in FeD and FeS cisplatin-treated kidneys. (C) DFO downregulated ferroportin (FPN), ferritin (Fth1), Hmox1 and Hmox2 proteins, while upregulated transferrin receptor 1 (Tfr1), Alas1 and Alas2 proteins in both FeD and FeS kidneys subject to Rh.
Fig. S6. Nox4 was upregulated to promote cisplatin- but not Rh-iAKI in FeD mice. (A) Western blot showed comparable levels of Nox1, Nox2 and Nox4 proteins in FeD, FeDM and FeS mice prior to injury. (B and C) GKT137831 reduced sCr (B) and BUN (C) levels in FeD mice 72h after cisplatin treatment. n=5-7. (D) Nox4 KO reduced catalytic iron levels in FeD and FeS kidneys 72h after cisplatin treatment. (E) Nox4 KO increased non-heme iron levels in FeD and FeS kidneys 72h after cisplatin treatment. n=7. (F) Nox4 KO upregulated ferroportin (FPN), ferritin (Fth1), Alas1 and Alas2 and downregulated Hmox1 and Hmox2 proteins in both FeD and FeS cisplatin-treated kidneys. (G) Nox4 KO did not significantly regulate catalytic iron levels in FeD and FeS kidneys 72h after cisplatin treatment when treated with DFO. n=7. (H) Western blot showed moderate increase of Nox4 but not Nox1 and Nox2 proteins in kidneys of FeD compared with FeS Rh mice. Data are means ± SD.
Fig. S7. Iron regulation in cisplatin and Rh kidneys by prior iron supplementation. (A and B) Iron supplementation increased the expression of ferroportin (FPN) and ferritin (Fth1) proteins and suppressed the expression of transferrin receptor 1 (Tfr1) protein in FeD and FeS kidneys subject to cisplatin (Cis; A) and Rh (B).
Table S1. Gene-specific primers for Q-PCR.
Acknowledgements
AQ is supported by grants from China National Science Foundation 31271551 and 81970593. JB is supported by National Institute of Health grants 1U54DK104309-01, 2R01DK073462 and RFA-DK-16-026, and Kidney Precision Medicine Project.
Abbreviation
- 4-HNE
4-Hydroxynonenal
- 8-OHdG
8-hydroxydeoxyguanosine
- AKI
Acute kidney injury
- Alas
5’-Aminolevulinate Synthase
- BUN
Blood urea nitrogen
- Cis
Cisplatin
- DFO
Deferoxamine
- DMT1
Divalent metal transporter
- DMTU
N,N’-dimethylthiourea
- FeD
Iron deficient diet
- Fer-1
Ferrostatin-1
- Ft
Ferritin
- Fth1
Ferritin heavy chain
- FeS
Iron sufficient diet
- FPN
Ferroportin; Slc40a1
- γH2A.X
Gamma H2A.X
- Hmox
Heme oxygenase
- IL-1
Interleukin-1
- IL-6
Interleukin-6
- KC
Kupffer cells
- KO
Knockout
- Ngal
Neutrophil gelatinase-associated lipocalin
- Nox
NADPH oxidase
- NTBI
Non-transferrin bound iron
- MCP1
Monocyte chemotactic protein 1
- p-MLKL
Phospho-mixed lineage kinase domain-like
- p-RIPK3
Phospho-serine/threonine kinase 3
- Rh
Rhabdomyolysis
- ROS
Reactive oxidative species
- sCr
Serum creatinine
- Tfr1
Transferrin receptor 1
- TNF-α
Tumor Necrosis Factor alpha
- TSAT
Transferrin saturation
Footnotes
Competing interests
There are no conflicts of interest to disclose, and the data contained in this article have not been previously published.
Data and materials availability
All data are available in the main text or the supplementary materials. All materials used in the analysis are available subject to materials transfer agreements (MTAs).
References
- [1].Coca SG, King JT Jr., Rosenthal RA, Perkal MF, Parikh CR, The duration of postoperative acute kidney injury is an additional parameter predicting long-term survival in diabetic veterans, Kidney international 78(9) (2010) 926–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Xu K, Rosenstiel P, Paragas N, Hinze C, Gao X, Huai Shen T, Werth M, Forster C, Deng R, Bruck E, Boles RW, Tornato A, Gopal T, Jones M, Konig J, Stauber J, D’Agati V, Erdjument-Bromage H, Saggi S, Wagener G, Schmidt-Ott KM, Tatonetti N, Tempst P, Oliver JA, Guarnieri P, Barasch J, Unique Transcriptional Programs Identify Subtypes of AKI, Journal of the American Society of Nephrology : JASN 28(6) (2017) 1729–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gao G, Zhang B, Ramesh G, Betterly D, Tadagavadi RK, Wang W, Reeves WB, TNF-alpha mediates increased susceptibility to ischemic AKI in diabetes, American journal of physiology. Renal physiology 304(5) (2013) F515–F521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Peng J, Li X, Zhang D, Chen JK, Su Y, Smith SB, Dong Z, Hyperglycemia, p53, and mitochondrial pathway of apoptosis are involved in the susceptibility of diabetic models to ischemic acute kidney injury, Kidney international 87(1) (2015) 137–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Boddu R, Fan C, Rangarajan S, Sunil B, Bolisetty S, Curtis LM, Unique sex- and age-dependent effects in protective pathways in acute kidney injury, American journal of physiology. Renal physiology 313(3) (2017) F740–F755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Anderson S, Eldadah B, Halter JB, Hazzard WR, Himmelfarb J, Horne FM, Kimmel PL, Molitoris BA, Murthy M, O’Hare AM, Schmader KE, High KP, Acute kidney injury in older adults, Journal of the American Society of Nephrology : JASN 22(1) (2011) 28–38. [DOI] [PubMed] [Google Scholar]
- [7].Nath KA, Grande JP, Farrugia G, Croatt AJ, Belcher JD, Hebbel RP, Vercellotti GM, Katusic ZS, Age sensitizes the kidney to heme protein-induced acute kidney injury, American journal of physiology. Renal physiology 304(3) (2013) F317–F325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Xue JL, Daniels F, Star RA, Kimmel PL, Eggers PW, Molitoris BA, Himmelfarb J, Collins AJ, Incidence and mortality of acute renal failure in Medicare beneficiaries, 1992 to 2001, Journal of the American Society of Nephrology : JASN 17(4) (2006) 1135–42. [DOI] [PubMed] [Google Scholar]
- [9].Leelahavanichkul A, Huang Y, Hu X, Zhou H, Tsuji T, Chen R, Kopp JB, Schnermann J, Yuen PS, Star RA, Chronic kidney disease worsens sepsis and sepsis-induced acute kidney injury by releasing High Mobility Group Box Protein-1, Kidney international 80(11) (2011) 1198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chawla LS, Eggers PW, Star RA, Kimmel PL, Acute kidney injury and chronic kidney disease as interconnected syndromes, N Engl J Med 371(1) (2014) 58–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Schetz M, Dasta J, Goldstein S, Golper T, Drug-induced acute kidney injury, Curr Opin Crit Care 11(6) (2005) 555–65. [DOI] [PubMed] [Google Scholar]
- [12].Lebwohl D, Canetta R, Clinical development of platinum complexes in cancer therapy: an historical perspective and an update, Eur J Cancer 34(10) (1998) 1522–34. [DOI] [PubMed] [Google Scholar]
- [13].Tristao VR, Goncalves PF, Dalboni MA, Batista MC, Durao Mde S Jr., Monte JC, Nec-1 protects against nonapoptotic cell death in cisplatin-induced kidney injury, Renal failure 34(3) (2012) 373–7. [DOI] [PubMed] [Google Scholar]
- [14].Wei Q, Dong G, Franklin J, Dong Z, The pathological role of Bax in cisplatin nephrotoxicity, Kidney international 72(1) (2007) 53–62. [DOI] [PubMed] [Google Scholar]
- [15].Wei Q, Dong G, Yang T, Megyesi J, Price PM, Dong Z, Activation and involvement of p53 in cisplatin-induced nephrotoxicity, American journal of physiology. Renal physiology 293(4) (2007) F1282–F1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, Wang Y, Huang Z, Ren J, Liu S, Chen X, Han J, A Role for Tubular Necroptosis in Cisplatin-Induced AKI, Journal of the American Society of Nephrology : JASN 26(11) (2015) 2647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bosch X, Poch E, Grau JM, Rhabdomyolysis and acute kidney injury, N Engl J Med 361(1) (2009) 62–72. [DOI] [PubMed] [Google Scholar]
- [18].Zager RA, Studies of mechanisms and protective maneuvers in myoglobinuric acute renal injury, Laboratory investigation; a journal of technical methods and pathology 60(5) (1989) 619–29. [PubMed] [Google Scholar]
- [19].Zager RA, Foerder CA, Effects of inorganic iron and myoglobin on in vitro proximal tubular lipid peroxidation and cytotoxicity, The Journal of clinical investigation 89(3) (1992) 989–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Baliga R, Ueda N, Shah SV, Increase in bleomycin-detectable iron in ischaemia/reperfusion injury to rat kidneys, Biochem J 291 ( Pt 3) (1993) 901–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Baliga R, Zhang Z, Baliga M, Shah SV, Evidence for cytochrome P-450 as a source of catalytic iron in myoglobinuric acute renal failure, Kidney international 49(2) (1996) 362–9. [DOI] [PubMed] [Google Scholar]
- [22].Baliga R, Zhang Z, Baliga M, Ueda N, Shah SV, Role of cytochrome P-450 as a source of catalytic iron in cisplatin-induced nephrotoxicity, Kidney international 54(5) (1998) 1562–9. [DOI] [PubMed] [Google Scholar]
- [23].Zarjou A, Bolisetty S, Joseph R, Traylor A, Apostolov EO, Arosio P, Balla J, Verlander J, Darshan D, Kuhn LC, Agarwal A, Proximal tubule H-ferritin mediates iron trafficking in acute kidney injury, The Journal of clinical investigation 123(10) (2013) 4423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sponsel HT, Alfrey AC, Hammond WS, Durr JA, Ray C, Anderson RJ, Effect of iron on renal tubular epithelial cells, Kidney international 50(2) (1996) 436–44. [DOI] [PubMed] [Google Scholar]
- [25].Sheerin NS, Sacks SH, Fogazzi GB, In vitro erythrophagocytosis by renal tubular cells and tubular toxicity by haemoglobin and iron, Nephrol Dial Transplant 14(6) (1999) 1391–7. [DOI] [PubMed] [Google Scholar]
- [26].Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA, Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution, Blood 116(26) (2010) 6054–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Martines AM, Masereeuw R, Tjalsma H, Hoenderop JG, Wetzels JF, Swinkels DW, Iron metabolism in the pathogenesis of iron-induced kidney injury, Nat Rev Nephrol 9(7) (2013) 385–98. [DOI] [PubMed] [Google Scholar]
- [28].Moulouel B, Houamel D, Delaby C, Tchernitchko D, Vaulont S, Letteron P, Thibaudeau O, Puy H, Gouya L, Beaumont C, Karim Z, Hepcidin regulates intrarenal iron handling at the distal nephron, Kidney international 84(4) (2013) 756–66. [DOI] [PubMed] [Google Scholar]
- [29].Scindia Y, Dey P, Thirunagari A, Liping H, Rosin DL, Floris M, Okusa MD, Swaminathan S, Hepcidin Mitigates Renal Ischemia-Reperfusion Injury by Modulating Systemic Iron Homeostasis, Journal of the American Society of Nephrology : JASN 26(11) (2015) 2800–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sharma S, Leaf DE, Iron Chelation as a Potential Therapeutic Strategy for AKI Prevention, Journal of the American Society of Nephrology : JASN 30(11) (2019) 2060–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mori K, Lee HT, Rapoport D, Drexler IR, Foster K, Yang J, Schmidt-Ott KM, Chen X, Li JY, Weiss S, Mishra J, Cheema FH, Markowitz G, Suganami T, Sawai K, Mukoyama M, Kunis C, D’Agati V, Devarajan P, Barasch J, Endocytic delivery of lipocalin-siderophore-iron complex rescues the kidney from ischemia-reperfusion injury, The Journal of clinical investigation 115(3) (2005) 610–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vaugier C, Amano MT, Chemouny JM, Dussiot M, Berrou C, Matignon M, Ben Mkaddem S, Wang PHM, Fricot A, Maciel TT, Grapton D, Mathieu JRR, Beaumont C, Peraldi MN, Peyssonnaux C, Mesnard L, Daugas E, Vrtovsnik F, Monteiro RC, Hermine O, Ginzburg YZ, Benhamou M, Camara NOS, Flamant M, Moura IC, Serum Iron Protects from Renal Postischemic Injury, Journal of the American Society of Nephrology : JASN 28(12) (2017) 3605–3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Walter PB, Knutson MD, Paler-Martinez A, Lee S, Xu Y, Viteri FE, Ames BN, Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats, Proceedings of the National Academy of Sciences of the United States of America 99(4) (2002) 2264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].O’Brien E, Holt ME, Thompson MK, Salay LE, Ehlinger AC, Chazin WJ, Barton JK, The [4Fe4S] cluster of human DNA primase functions as a redox switch using DNA charge transport, Science 355(6327) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Netz DJ, Stith CM, Stumpfig M, Kopf G, Vogel D, Genau HM, Stodola JL, Lill R, Burgers PM, Pierik AJ, Eukaryotic DNA polymerases require an iron-sulfur cluster for the formation of active complexes, Nat Chem Biol 8(1) (2011) 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kassebaum NJ, Jasrasaria R, Naghavi M, Wulf SK, Johns N, Lozano R, Regan M, Weatherall D, Chou DP, Eisele TP, Flaxman SR, Pullan RL, Brooker SJ, Murray CJ, A systematic analysis of global anemia burden from 1990 to 2010, Blood 123(5) (2014) 615–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Stevens GA, Finucane MM, De-Regil LM, Paciorek CJ, Flaxman SR, Branca F, Pena-Rosas JP, Bhutta ZA, Ezzati M, Nutrition G Impact Model Study, Global, regional, and national trends in haemoglobin concentration and prevalence of total and severe anaemia in children and pregnant and non-pregnant women for 1995-2011: a systematic analysis of population-representative data, Lancet Glob Health 1(1) (2013) e16–e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sangkhae V, Fisher AL, Wong S, Koenig MD, Tussing-Humphreys L, Chu A, Lelic M, Ganz T, Nemeth E, Effects of maternal iron status on placental and fetal iron homeostasis, The Journal of clinical investigation 130(2) (2020) 625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lisle SJ, Lewis RM, Petry CJ, Ozanne SE, Hales CN, Forhead AJ, Effect of maternal iron restriction during pregnancy on renal morphology in the adult rat offspring, The British journal of nutrition 90(1) (2003) 33–9. [DOI] [PubMed] [Google Scholar]
- [40].Hesketh PJ, Chemotherapy-induced nausea and vomiting, N Engl J Med 358(23) (2008) 2482–94. [DOI] [PubMed] [Google Scholar]
- [41].Faubel S, Lewis EC, Reznikov L, Ljubanovic D, Hoke TS, Somerset H, Oh DJ, Lu L, Klein CL, Dinarello CA, Edelstein CL, Cisplatin-induced acute renal failure is associated with an increase in the cytokines interleukin (IL)-1beta, IL-18, IL-6, and neutrophil infiltration in the kidney, J Pharmacol Exp Ther 322(1) (2007) 8–15. [DOI] [PubMed] [Google Scholar]
- [42].Ramesh G, Reeves WB, TNF-alpha mediates chemokine and cytokine expression and renal injury in cisplatin nephrotoxicity, The Journal of clinical investigation 110(6) (2002) 835–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Liu M, Chien CC, Burne-Taney M, Molls RR, Racusen LC, Colvin RB, Rabb H, A pathophysiologic role for T lymphocytes in murine acute cisplatin nephrotoxicity, Journal of the American Society of Nephrology : JASN 17(3) (2006) 765–74. [DOI] [PubMed] [Google Scholar]
- [44].Liapis H, Boils C, Hennigar R, Silva F, Myoglobin casts in renal biopsies: immunohistochemistry and morphologic spectrum, Hum Pathol 54 (2016) 25–30. [DOI] [PubMed] [Google Scholar]
- [45].Shah SV, Rajapurkar MM, Baliga R, The role of catalytic iron in acute kidney injury, Clinical journal of the American Society of Nephrology : CJASN 6(10) (2011) 2329–31. [DOI] [PubMed] [Google Scholar]
- [46].N.V.B.a.C.-E. Ha, Metabolism of Iron and Heme, Essentials of Medical Biochemistry Second edition (2015) Chapter 27.
- [47].Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME, Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat, The Journal of clinical investigation 90(1) (1992) 267–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Xu G, Chance MR, Hydroxyl radical-mediated modification of proteins as probes for structural proteomics, Chemical reviews 107(8) (2007) 3514–43. [DOI] [PubMed] [Google Scholar]
- [49].Castro JP, Jung T, Grune T, Siems W, 4-Hydroxynonenal (HNE) modified proteins in metabolic diseases, Free radical biology & medicine 111 (2017) 309–315. [DOI] [PubMed] [Google Scholar]
- [50].Yin H, Xu L, Porter NA, Free radical lipid peroxidation: mechanisms and analysis, Chemical reviews 111(10) (2011) 5944–72. [DOI] [PubMed] [Google Scholar]
- [51].Valavanidis A, Vlachogianni T, Fiotakis C, 8-hydroxy-2’ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis, Journal of environmental science and health. Part C, Environmental carcinogenesis & ecotoxicology reviews 27(2) (2009) 120–39. [DOI] [PubMed] [Google Scholar]
- [52].Kaori Eguchi TA , Kiriu Takahiro Minamiyama Yukiko, Shigeru Okada Apoptosis and 8-hydroxydeoxyguanosine levels in renal tissue after an injection of ferric nitrilotriacetate, a renal carcinogen, in male Wistar rats maintained on vitamin E-deficient, -normal or- supplemented diets, Pathophysiology 6 (1999) 149–156. [Google Scholar]
- [53].Halliwell B, Gutteridge JM, Role of free radicals and catalytic metal ions in human disease: an overview, Methods in enzymology 186 (1990) 1–85. [DOI] [PubMed] [Google Scholar]
- [54].Shah SV, Baliga R, Rajapurkar M, Fonseca VA, Oxidants in chronic kidney disease, Journal of the American Society of Nephrology : JASN 18(1) (2007) 16–28. [DOI] [PubMed] [Google Scholar]
- [55].Suzuki YJ, Carini M, Butterfield DA, Protein carbonylation, Antioxidants & redox signaling 12(3) (2010) 323–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Ha H, Kim C, Son Y, Chung MH, Kim KH, DNA damage in the kidneys of diabetic rats exhibiting microalbuminuria, Free radical biology & medicine 16(2) (1994) 271–4. [DOI] [PubMed] [Google Scholar]
- [57].Redza-Dutordoir M, Averill-Bates DA, Activation of apoptosis signalling pathways by reactive oxygen species, Biochimica et biophysica acta 1863(12) (2016) 2977–2992. [DOI] [PubMed] [Google Scholar]
- [58].Schulze-Osthoff K, Bakker AC, Vanhaesebroeck B, Beyaert R, Jacob WA, Fiers W, Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation, The Journal of biological chemistry 267(8) (1992) 5317–23. [PubMed] [Google Scholar]
- [59].Goossens V, Grooten J, De Vos K, Fiers W, Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity, Proceedings of the National Academy of Sciences of the United States of America 92(18) (1995) 8115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Schenk B, Fulda S, Reactive oxygen species regulate Smac mimetic/TNFalpha-induced necroptotic signaling and cell death, Oncogene 34(47) (2015) 5796–806. [DOI] [PubMed] [Google Scholar]
- [61].Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, Cai Q, Yang ZH, Huang D, Wu R, Han J, RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome, Nature communications 8 (2017) 14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR, Ferroptosis: an iron-dependent form of nonapoptotic cell death, Cell 149(5) (2012) 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch A, Eggenhofer E, Basavarajappa D, Radmark O, Kobayashi S, Seibt T, Beck H, Neff F, Esposito I, Wanke R, Forster H, Yefremova O, Heinrichmeyer M, Bornkamm GW, Geissler EK, Thomas SB, Stockwell BR, O’Donnell VB, Kagan VE, Schick JA, Conrad M, Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice, Nature cell biology 16(12) (2014) 1180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, Stockwell BR, Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models, Journal of the American Chemical Society 136(12) (2014) 4551–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Linkermann A, Skouta R, Himmerkus N, Mulay SR, Dewitz C, De Zen F, Prokai A, Zuchtriegel G, Krombach F, Welz PS, Weinlich R, Vanden Berghe T, Vandenabeele P, Pasparakis M, Bleich M, Weinberg JM, Reichel CA, Brasen JH, Kunzendorf U, Anders HJ, Stockwell BR, Green DR, Krautwald S, Synchronized renal tubular cell death involves ferroptosis, Proceedings of the National Academy of Sciences of the United States of America 111(47) (2014) 16836–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Colavitti R, Finkel T, Reactive oxygen species as mediators of cellular senescence, IUBMB life 57(4–5) (2005) 277–81. [DOI] [PubMed] [Google Scholar]
- [67].Sell TNT, Mitochondrial Reactive Oxygen Species in Cellular Senescence, Cellular Ageing and Replicative Senescence (2016) 169–185. [Google Scholar]
- [68].Linkermann A, Chen G, Dong G, Kunzendorf U, Krautwald S, Dong Z, Regulated cell death in AKI, Journal of the American Society of Nephrology : JASN 25(12) (2014) 2689–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Xu Y, Han J, The Necrosome in Acute Kidney Injury, Seminars in nephrology 36(3) (2016) 199–207. [DOI] [PubMed] [Google Scholar]
- [70].Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T, DNA damage response and cellular senescence in tissues of aging mice, Aging cell 8(3) (2009) 311–23. [DOI] [PubMed] [Google Scholar]
- [71].Cho S, Hwang ES, Fluorescence-based detection and quantification of features of cellular senescence, Methods in cell biology 103 (2011) 149–88. [DOI] [PubMed] [Google Scholar]
- [72].Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW, A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy, Cell 109(3) (2002) 335–46. [DOI] [PubMed] [Google Scholar]
- [73].Chan HM, Narita M, Lowe SW, Livingston DM, The p400 E1A-associated protein is a novel component of the p53 --> p21 senescence pathway, Genes & development 19(2) (2005) 196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E, Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence, Nature cell biology 8(11) (2006) 1291–7. [DOI] [PubMed] [Google Scholar]
- [75].Fujita K, Mondal AM, Horikawa I, Nguyen GH, Kumamoto K, Sohn JJ, Bowman ED, Mathe EA, Schetter AJ, Pine SR, Ji H, Vojtesek B, Bourdon JC, Lane DP, Harris CC, p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence, Nature cell biology 11(9) (2009) 1135–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Freund A, Laberge RM, Demaria M, Campisi J, Lamin B1 loss is a senescence-associated biomarker, Mol Biol Cell 23(11) (2012) 2066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Tanida I, Ueno T, Kominami E, LC3 and Autophagy, Methods in molecular biology 445 (2008) 77–88. [DOI] [PubMed] [Google Scholar]
- [78].Guerrero-Hue M, Garcia-Caballero C, Palomino-Antolin A, Rubio-Navarro A, Vazquez-Carballo C, Herencia C, Martin-Sanchez D, Farre-Alins V, Egea J, Cannata P, Praga M, Ortiz A, Egido J, Sanz AB, Moreno JA, Curcumin reduces renal damage associated with rhabdomyolysis by decreasing ferroptosis-mediated cell death, FASEB J 33(8) (2019) 8961–8975. [DOI] [PubMed] [Google Scholar]
- [79].Novogrodsky A, Ravid A, Rubin AL, Stenzel KH, Hydroxyl radical scavengers inhibit lymphocyte mitogenesis, Proceedings of the National Academy of Sciences of the United States of America 79(4) (1982) 1171–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hofer T, Wenger RH, Kramer MF, Ferreira GC, Gassmann M, Hypoxic up-regulation of erythroid 5-aminolevulinate synthase, Blood 101(1) (2003) 348–50. [DOI] [PubMed] [Google Scholar]
- [81].Grek CL, Newton DA, Spyropoulos DD, Baatz JE, Hypoxia up-regulates expression of hemoglobin in alveolar epithelial cells, American journal of respiratory cell and molecular biology 44(4) (2011) 439–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Zhang Y, Furuyama K, Kaneko K, Ding Y, Ogawa K, Yoshizawa M, Kawamura M, Takeda K, Yoshida T, Shibahara S, Hypoxia reduces the expression of heme oxygenase-2 in various types of human cell lines. A possible strategy for the maintenance of intracellular heme level, The FEBS journal 273(14) (2006) 3136–47. [DOI] [PubMed] [Google Scholar]
- [83].Shibahara S, Han F, Li B, Takeda K, Hypoxia and heme oxygenases: oxygen sensing and regulation of expression, Antioxidants & redox signaling 9(12) (2007) 2209–25. [DOI] [PubMed] [Google Scholar]
- [84].Kyung Jin Woo T-JL, Park Jong-Wook, Kwon Taeg Kyu., Desferrioxamine, an iron chelator, enhances HIF-1α accumulation via cyclooxygenase-2 signaling pathway., Biochemical and Biophysical Research Communications. 343(1) (2006) 8–14. [DOI] [PubMed] [Google Scholar]
- [85].Geiszt M, Kopp JB, Varnai P, Leto TL, Identification of renox, an NAD(P)H oxidase in kidney, Proceedings of the National Academy of Sciences of the United States of America 97(14) (2000) 8010–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Li N, Yi FX, Spurrier JL, Bobrowitz CA, Zou AP, Production of superoxide through NADH oxidase in thick ascending limb of Henle’s loop in rat kidney, American journal of physiology. Renal physiology 282(6) (2002) F1111–9. [DOI] [PubMed] [Google Scholar]
- [87].Etoh T, Inoguchi T, Kakimoto M, Sonoda N, Kobayashi K, Kuroda J, Sumimoto H, Nawata H, Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment, Diabetologia 46(10) (2003) 1428–37. [DOI] [PubMed] [Google Scholar]
- [88].Asaba K, Tojo A, Onozato ML, Goto A, Quinn MT, Fujita T, Wilcox CS, Effects of NADPH oxidase inhibitor in diabetic nephropathy, Kidney international 67(5) (2005) 1890–8. [DOI] [PubMed] [Google Scholar]
- [89].Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, Molango S, Heitz F, Merlot C, Szyndralewiez C, Page P, Brenner DA, Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent, Hepatology 56(6) (2012) 2316–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Miano TA, Lautenbach E, Wilson FP, Guo W, Borovskiy Y, Hennessy S, Attributable Risk and Time Course of Colistin-Associated Acute Kidney Injury, Clinical journal of the American Society of Nephrology : CJASN 13(4) (2018) 542–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Paken J, Govender CD, Pillay M, Sewram V, Cisplatin-Associated Ototoxicity: A Review for the Health Professional, Journal of toxicology 2016 (2016) 1809394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Aldossary SA, Review on Pharmacology of Cisplatin: Clinical Use, Toxicity and Mechanism of Resistance of Cisplatin, Biomed Pharmacol J 12(1) (2019). [Google Scholar]
- [93].Abboud S, Haile DJ, A novel mammalian iron-regulated protein involved in intracellular iron metabolism, The Journal of biological chemistry 275(26) (2000) 19906–12. [DOI] [PubMed] [Google Scholar]
- [94].Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI, Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter, Nature 403(6771) (2000) 776–81. [DOI] [PubMed] [Google Scholar]
- [95].McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ, A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation, Molecular cell 5(2) (2000) 299–309. [DOI] [PubMed] [Google Scholar]
- [96].Zhang DL, Hughes RM, Ollivierre-Wilson H, Ghosh MC, Rouault TA, A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression, Cell metabolism 9(5) (2009) 461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Getaz EP, Beckley S, Fitzpatrick J, Dozier A, Cisplatin-induced hemolysis, N Engl J Med 302(6) (1980) 334–5. [DOI] [PubMed] [Google Scholar]
- [98].Paller MS, Hemoglobin- and myoglobin-induced acute renal failure in rats: role of iron in nephrotoxicity, Am J Physiol 255(3 Pt 2) (1988) F539–F544. [DOI] [PubMed] [Google Scholar]
- [99].Tracz MJ, Alam J, Nath KA, Physiology and pathophysiology of heme: implications for kidney disease, Journal of the American Society of Nephrology : JASN 18(2) (2007) 414–20. [DOI] [PubMed] [Google Scholar]
- [100].Agarwal A, Balla J, Alam J, Croatt AJ, Nath KA, Induction of heme oxygenase in toxic renal injury: a protective role in cisplatin nephrotoxicity in the rat, Kidney international 48(4) (1995) 1298–307. [DOI] [PubMed] [Google Scholar]
- [101].Shiraishi F, Curtis LM, Truong L, Poss K, Visner GA, Madsen K, Nick HS, Agarwal A, Heme oxygenase-1 gene ablation or expression modulates cisplatin-induced renal tubular apoptosis, American journal of physiology. Renal physiology 278(5) (2000) F726–36. [DOI] [PubMed] [Google Scholar]
- [102].Dhur A, Galan P, Hercberg S, Effects of different degrees of iron deficiency on cytochrome P450 complex and pentose phosphate pathway dehydrogenases in the rat, J Nutr 119(1) (1989) 40–7. [DOI] [PubMed] [Google Scholar]
- [103].Meng XM, Ren GL, Gao L, Yang Q, Li HD, Wu WF, Huang C, Zhang L, Lv XW, Li J, NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation, Laboratory investigation; a journal of technical methods and pathology 98(1) (2018) 63–78. [DOI] [PubMed] [Google Scholar]
- [104].You YH, Quach T, Saito R, Pham J, Sharma K, Metabolomics Reveals a Key Role for Fumarate in Mediating the Effects of NADPH Oxidase 4 in Diabetic Kidney Disease, Journal of the American Society of Nephrology : JASN 27(2) (2016) 466–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, Mikulska-Ruminska K, Shrivastava IH, Kenny EM, Yang Q, Rosenbaum JC, Sparvero LJ, Emlet DR, Wen X, Minami Y, Qu F, Watkins SC, Holman TR, VanDemark AP, Kellum JA, Bahar I, Bayir H, Kagan VE, PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals, Cell 171(3) (2017) 628–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Martin-Sanchez D, Ruiz-Andres O, Poveda J, Carrasco S, Cannata-Ortiz P, Sanchez-Nino MD, Ruiz Ortega M, Egido J, Linkermann A, Ortiz A, Sanz AB, Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI, Journal of the American Society of Nephrology : JASN 28(1) (2017) 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].M D Knutson PBW , B N Ames F E Viteri, Both iron deficiency and daily iron supplements increase lipid peroxidation in rats, J Nutr 130(3) (2000) 621–628. [DOI] [PubMed] [Google Scholar]
- [108].Ziegler DV, Wiley CD, Velarde MC, Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging, Aging cell 14(1) (2015) 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Wei Q, Hill WD, Su Y, Huang S, Dong Z, Heme oxygenase-1 induction contributes to renoprotection by G-CSF during rhabdomyolysis-associated acute kidney injury, American journal of physiology. Renal physiology 301(1) (2011) F162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Skibsted CUCKSMH., Heme-iron in lipid oxidation., Coordination Chemistry Reviews. 249(3–4) (2005) 485–498. [Google Scholar]
- [111].Rufini A, Tucci P, Celardo I, Melino G, Senescence and aging: the critical roles of p53, Oncogene 32(43) (2013) 5129–43. [DOI] [PubMed] [Google Scholar]
- [112].Qian Y, Chen X, Senescence regulation by the p53 protein family, Methods in molecular biology 965 (2013) 37–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Sahin E, DePinho RA, Axis of ageing: telomeres, p53 and mitochondria, Nature reviews. Molecular cell biology 13(6) (2012) 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Liu D, Xu Y, p53, oxidative stress, and aging, Antioxidants & redox signaling 15(6) (2011) 1669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Yosef R, Pilpel N, Papismadov N, Gal H, Ovadya Y, Vadai E, Miller S, Porat Z, Ben-Dor S, Krizhanovsky V, p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling, EMBO J 36(15) (2017) 2280–2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Kuilman T, Michaloglou C, Mooi WJ, Peeper DS, The essence of senescence, Genes & development 24(22) (2010) 2463–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Singh AP, Junemann A, Muthuraman A, Jaggi AS, Singh N, Grover K, Dhawan R, Animal models of acute renal failure, Pharmacol Rep 64(1) (2012) 31–44. [DOI] [PubMed] [Google Scholar]
- [118].Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL, A heme export protein is required for red blood cell differentiation and iron homeostasis, Science 319(5864) (2008) 825–8. [DOI] [PubMed] [Google Scholar]
- [119].Zhang D, Okada S, Kawabata T, Yasuda T, An improved simple colorimetric method for quantitation of non-transferrin-bound iron in serum, Biochem Mol Biol Int 35(3) (1995) 635–41. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Verification of nutritional iron deficiency and its worsening effects on cisplatin- and Rh- induced AKI. (A to D) Reduction of serum iron (A), and hepatic (B), splenic (C) and renal (D) non-heme iron in mice which were fed FeD and FeDM compared with FeS diets for 21 days. n=7-8. (E to G) Q-PCR showed the upregulation of duodenal Dcytb and DMT1 mRNA and the downregulation of hepatic Hamp mRNA in FeD and FeDM compared with FeS mice. n=6. (H) Weight reduction of FeD, FeDM and FeS mice 72h after cisplatin treatment. n=7. (I and J) Q-PCR showed the upregulation of Kim-1 and Ngal mRNA in FeD and FeDM compared with FeS kidneys 72h after cisplatin treatment. n=7. (K) Western blot showed a marked increase of urinary Ngal protein in FeD and FeDM compared with FeS mice 72h after cisplatin treatment. (L to S) Higher levels of serum TNFα (L), IL-6 (M), MCP-1 (N) and KC (O) and their kidney mRNA (P-S) in FeD and FeDM than FeS mice 72h after cisplatin treatment. n=7. (T) Western blot showed an increase of Ngal protein in FeD and FeDM compared with FeS kidneys 24h after Rh initiation. Data are means ± SD. (A to G and K) One-way ANOVA–Tukey’s post hoc test; (H to J and L to S) Two-way ANOVA–Tukey’s post hoc test.
Fig. S2. Systemic iron regulation in FeD cisplatin and Rh mice. (A) Q-PCR showed the downregulation of kidney DMT1 (IRE isoform) mRNA in FeD mice 72h after cisplatin treatment. n=7. (B) Immunohistochemical staining showed much lower levels of ferritin (Fth1) in FeD than FeS kidneys prior to injury. (C) Western blot showed the marked upregulation of heme oxygenase 1 (Hmox1) in FeD compared FeS cisplatin-treated kidneys. (D and E) Rh had negligible effect on hepatic (D) and splenic (E) non-heme iron levels in either FeD or FeS mice. n=7. (F) Western blot showed the marked upregulation of heme oxygenase 1 (Hmox1) in FeD compared FeS kidneys subject to Rh. Data are means ± SD. (A, D and E) Two-way ANOVA–Tukey’s post hoc test.
Fig. S3. Iron deficiency enhanced specific tubular cell damage pathways in cisplatin and Rh kidneys. (A) Immunohistochemical staining showed that 8-OHdG was increased mainly in the proximal tubules of FeD compared with FeS kidneys 72h after cisplatin treatment. (B to E) Fer-1 treatment reduced serum TNFα (B), IL-6 (C), MCP-1 (D) and KC (E) levels in FeD and FeDM but not in FeS mice 72h after cisplatin treatment. n=7. (F) Western blot showed negligible changes of p-RIPK3, p-MLKL and cleaved caspase 3 proteins in FeD, FeDM and FeS cisplatin-treated kidneys 72h after Fer-1 treatment. (G and H) Western blot showed comparable levels of p-RIPK3 and p-MLKL (G), p16, p53, p21, γH2A.X and H2A.X proteins (H) in FeD, FeDM and FeS kidneys 72h after cisplatin treatment. (I) Western blot showed comparable levels of p-RIPK3 and p-MLKL, cleaved caspase 3 and LC3-I/II in FeD, FeDM and FeS kidneys 24h after Rh induction. n=7-8. (J and K) Fer-1 did not regulate sCr (J) and BUN (K) levels in FeD, FeDM and FeS mice 24h after Rh induction. n=7-8. Data are means ± SD. (B to E, J and K) Two-way ANOVA–Tukey’s post hoc test.
Fig. S4. DMTU reduced cisplatin- and Rh- induced iAKI. (A) DMTU diminished the expression of Ngal protein in FeD and FeS kidneys 72h after cisplatin treatment. (B and C) DMTU markedly reduced catalytic iron levels in FeD and FeS kidneys subject to cisplatin (Cis; B) and Rh (C). n=8. (D and E) DMTU downregulated ferroportin (FPN), ferritin (Fth1), Hmox1 and Hmox2 proteins while it upregulated transferrin receptor 1 (Tfr1), Alas1 and Alas2 proteins particularly in FeD kidneys subject to cisplatin (D) and Rh (E). Data are means ± SD. (B and C) Two-way ANOVA–Tukey’s post hoc test.
Fig. S5. Regulation of heme and non-heme iron metabolism and cellular senescence in Cis and Rh kidneys by DFO. (A) DFO downregulated ferroportin (FPN), ferritin (Fth1), Hmoxl and Hmox2 proteins, while upregulated transferrin receptor 1 (Tfr1), Alas1 and Alas2 proteins in both FeD and FeS cisplatin kidneys. (B) DFO had negligible effects on cellular senescence in FeD and FeS cisplatin-treated kidneys. (C) DFO downregulated ferroportin (FPN), ferritin (Fth1), Hmox1 and Hmox2 proteins, while upregulated transferrin receptor 1 (Tfr1), Alas1 and Alas2 proteins in both FeD and FeS kidneys subject to Rh.
Fig. S6. Nox4 was upregulated to promote cisplatin- but not Rh-iAKI in FeD mice. (A) Western blot showed comparable levels of Nox1, Nox2 and Nox4 proteins in FeD, FeDM and FeS mice prior to injury. (B and C) GKT137831 reduced sCr (B) and BUN (C) levels in FeD mice 72h after cisplatin treatment. n=5-7. (D) Nox4 KO reduced catalytic iron levels in FeD and FeS kidneys 72h after cisplatin treatment. (E) Nox4 KO increased non-heme iron levels in FeD and FeS kidneys 72h after cisplatin treatment. n=7. (F) Nox4 KO upregulated ferroportin (FPN), ferritin (Fth1), Alas1 and Alas2 and downregulated Hmox1 and Hmox2 proteins in both FeD and FeS cisplatin-treated kidneys. (G) Nox4 KO did not significantly regulate catalytic iron levels in FeD and FeS kidneys 72h after cisplatin treatment when treated with DFO. n=7. (H) Western blot showed moderate increase of Nox4 but not Nox1 and Nox2 proteins in kidneys of FeD compared with FeS Rh mice. Data are means ± SD.
Fig. S7. Iron regulation in cisplatin and Rh kidneys by prior iron supplementation. (A and B) Iron supplementation increased the expression of ferroportin (FPN) and ferritin (Fth1) proteins and suppressed the expression of transferrin receptor 1 (Tfr1) protein in FeD and FeS kidneys subject to cisplatin (Cis; A) and Rh (B).
Table S1. Gene-specific primers for Q-PCR.
Data Availability Statement
All data are available in the main text or the supplementary materials. All materials used in the analysis are available subject to materials transfer agreements (MTAs).