

Abstract
The highly homologous protein lysine methyltransferases G9a and GLP, which catalyze mono- and dimethylation of histone H3 lysine 9 (H3K9), have been implicated in various human diseases. To investigate functions of G9a and GLP in human diseases, we and others reported several noncovalent reversible small-molecule inhibitors of G9a and GLP. Here, we report the discovery of the first-in-class G9a/GLP covalent irreversible inhibitors, 1 and 8 (MS8511), by targeting a cysteine residue at the substrate binding site. We characterized these covalent inhibitors in enzymatic, mass spectrometry based and cellular assays and using X-ray crystallography. Compared to the noncovalent G9a/GLP inhibitor UNC0642, covalent inhibitor 8 displayed improved potency in enzymatic and cellular assays. Interestingly, compound 8 also displayed potential kinetic preference for covalently modifying G9a over GLP. Collectively, compound 8 could be a useful chemical tool for studying the functional roles of G9a and GLP by covalently modifying and inhibiting these methyltransferases.
Graphical Abstract

INTRODUCTION
Lysine methylation on histone tails has been well-established as important for regulating gene expression through its chromatin remodeling capabilities.1–3 Recent reports suggest that, in addition to histone methylation, lysine methylation on non-histone proteins also plays a critical role in some signaling pathways.4–6 Protein lysine methyltransferases (PKMTs) catalyze the transfer of a methyl group from the cofactor S-adenosyl-L-methionine (SAM) to the ε-amino group of a lysine residue of their substrates.7–9 The resulting mono-, di-, or trimethylation on histones controls gene activation1 and repression, whereas lysine methylation on non-histone proteins affects protein stability,10,11 protein translocalization,12–14 and protein–protein interactions.15,16 Recent studies highlight the strong positive correlation between aberrant PKMT expression and disease progression,17–22 including various cancers23–25 and developmental disorders.26–28
G9a (also known as euchromatic histone lysine methyltransferase 2 (EHMT2)) and its closely related paralogue GLP (G9a-like protein, also known as euchromatic histone lysine methyltransferase 1 (EHMT1)) can catalyze the methylation of both histone and non-histone substrates. The catalytic function of these two PKMTs in the mono- and dimethylation of histone 3 lysine 9 (H3K9) has been extensively studied.29,30 G9a and GLP share approximately 80% sequence identity in their conserved catalytic SET domains, posing a challenge for developing paralogue-selective inhibitors.30 It has been reported that G9a overexpression is associated with proliferation and metastasis in several types of cancer including brain,31 breast,32 ovarian,33 lung,34 bladder,35,36 melanoma,37 and colorectal cancer.38 Moreover, it has been shown that G9a is involved in embryonic stem cell maintenance29,30 and T-cell differentiation39 and is implicated in other diseases such as Alzheimer’s disease (AD),40,41 sickle cell disease42 and Prader–Willi syndrome (PWS).43
To date, a number of G9a/GLP dual inhibitors have been developed by targeting either the SAM binding site or the substrate binding pocket.44–56 Among them, our substrate-competitive G9a/GLP dual inhibitors, UNC0638 and UNC0642, have been widely used by the research community as cellular and in vivo chemical probes, respectively.45,47 We also developed a highly selective GLP inhibitor, MS012.57,58 The cocrystal structures of UNC0638 and MS012 in complex with G9a or GLP revealed that both G9a and GLP have a cysteine residue (Cys1098 in G9a, Cys1186 in GLP) at the inhibitor binding site (Figure 1 and Figure S1).45,57 We hypothesized that this unexploited cysteine residue could be targeted to generate covalent inhibitors that may offer potential advantages over noncovalent inhibitors. While covalent inhibitors of G9a and/or GLP have not been reported, several epigenetic regulators have been successfully targeted by covalent inhibitors.59–61
Figure 1.

Design of G9a/GLP covalent inhibitors. (A) Chemical structures of UNC0638 and UNC0642. The cyclohexyl group of UNC0638 (highlighted in blue) was replaced by various covalent warheads. (B) Cocrystal structure of G9a in complex with UNC0638 (PDB ID 3RJW).45
Here, we report the discovery of the first covalent inhibitors of G9a and GLP, 1 and 8 (MS8511). A battery of mass spectrometry (MS) based assays and kinetic studies revealed the covalent inhibition mechanism of these compounds. The cocrystal structures of compound 1 in complex with G9a and GLP further confirmed that this compound covalently modified Cys1098 in G9a or the equivalent Cys1186 in GLP. In addition, compound 8 displayed excellent selectivity for G9a and GLP over 21 other PKMTs and protein arginine methyltransferases (PRMTs). Compared to the reversible inhibitor UNC0642, compound 8 displayed better binding affinity in a biophysical assay, was more potent in inhibiting the G9a methyltransferase activity in a biochemical assay, and exhibited improved cellular activities in reducing H3K9 dimethylation and inhibiting the proliferation of cancer cells. Overall, compound 8 is a potent and selective G9a/GLP covalent inhibitor, which could be useful for exploring biological functions of G9a and GLP.
RESULTS AND DISCUSSION
Design and Initial Evaluation of Covalent Inhibitors.
We analyzed the cocrystal structure of UNC0638 in complex with G9a (PDB ID 3RJW)45 and found that the distance between the thiol group of Cys1098 and the extended carbon atom at the C-2 position of UNC0638 was 4.1 Å, which makes it feasible to design covalent inhibitors (Figure 1). By installing a few known covalent warheads at the C-2 position of the quinazoline scaffold, we designed and synthesized several Cys1098-targeting covalent inhibitors (compounds 1–7) (Table 1). Interestingly, only compound 1, which contains an acrylamide group, showed covalent modification of G9a (40%) using MS-based analysis after 1 h of incubation (Table 1 and Figure 2). Moreover, compound 6, which carries a N-methyl substituted acrylamide group, completely lost its ability to covalently modify G9a (Figure 2). Previously, we and others reported that the quinoline scaffold could lead to better inhibitory potency than the quinazoline scaffold by increasing the basicity of N-1, which is crucial for binding.58,62 Therefore, we design and synthesized compound 8, a quinoline analogue of compound 1 (Table 1). Indeed, compound 8 exhibited enhanced covalent modification of G9a (50%; Figure 2 and Table 1).
Table 1.
SARs of Compounds with Various Covalent Warheads

| |||
|---|---|---|---|
|
| |||
| Compound | X | R | G9a adduct (%)a |
|
| |||
| 1 | N |

|
40% |
| 2 | N |

|
ND |
| 3 | N |

|
ND |
| 4 | N |

|
ND |
| 5 | N |

|
ND |
| 6 | N |

|
ND |
| 7 | N |

|
ND |
| 8 (MS8511) | C |
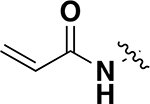
|
50% |
Formation of G9a adduct was monitored by mass spectrometry. G9a adduct (%) = (G9a adduct/(G9a + G9a adduct)) × 100.
ND: G9a adduct was not detected.
Figure 2.

Representative results of mass spectrometry based analysis of G9a incubated with compound 1, 6, or 8. The G9a–inhibitor adduct was quantified after 1 h incubation of G9a (5 μM) with DMSO or the indicated compound (50 μM) at room temperature. Molecular weight of G9a, 34 314 Da; molecular weight of G9a + compound 1, 34 811 Da; molecular weight of G9a + compound 8, 34 810 Da.
Characterization of Compounds 1 and 8 in Enzymatic and Biophysical Assays.
We next characterized compounds 1 and 8 in G9a and GLP biochemical and biophysical assays. In these experiments, we used UNC0642 rather than UNC0638 as a control because UNC0642, 1 and 8 have a nitrogen atom that directly attaches to the C-2 position of the quinazoline/quinoline core, while UNC0638 does not have this nitrogen atom. First, we evaluated the inhibitory effect of these compounds on G9a and GLP enzymatic activities using a S-adenosyl-L-homocysteine hydrolase (SAHH) coupled enzyme assay (Figure 3).63 Compared to UNC0642, compound 1 was 14-fold less potent against G9a and 10-fold less potent against GLP. Compound 8, however, displayed 2-fold improved potency for G9a and a similar potency for GLP. It is worth noting that most published quinazoline-based inhibitors displayed better potency for GLP over G9a.47,57 While compound 1 and UNC0642 exhibited a slight preference for GLP over G9a, compound 8 displayed a slight preference for G9a over GLP (IC50 = 100 ± 6 nM for G9a, IC50 = 140 ± 5 nM for GLP; Figure 3 and Table 2).
Figure 3.

Concentration-dependent inhibition of G9a and GLP by compounds 1 and 8 in SAHH-coupled enzymatic assays. The assays were performed after 10 min of preincubation with G9a (A) and GLP (B). Data are the means ± SD from four independent experiments.
Table 2.
IC50 Values of Compounds 1 and 8 in G9a and GLP SAHH-Coupled Enzymatic Assaysa
| compd 1 | compd 8 | UNC0642 | |
|---|---|---|---|
| IC50 for G9a (nM) | 3100 ± 260 | 100 ± 6 | 220 ± 14 |
| IC50 for GLP (nM) | 1800 ± 240 | 140 ± 5 | 180 ± 21 |
IC50 values are reported as the mean ± SD from four independent experiments.
We next assessed the binding affinities of these covalent inhibitors to G9a and GLP using isothermal titration calorimetry (ITC). Compared to UNC0642, compound 1 displayed reduced binding affinities to G9a (3.4-fold) and GLP (13-fold), while compound 8 exhibited significantly improved binding affinity to G9a (5-fold) and slightly improved binding affinity to GLP (1.3-fold). While UNC0642 showed a better binding affinity for GLP (Kd = 62 ± 16 nM) than G9a (Kd = 230 ± 17 nM) (Figure 4A), compound 1 (Kd = 790 ± 9 nM for G9a, Kd = 790 ± 22 nM for GLP) and compound 8 (Kd = 44 ± 1 nM for G9a, Kd = 46 ± 15 nM for GLP) exhibited similar binding affinities for G9a and GLP (Figure 4B,C).
Figure 4.

ITC titrations of UNC0642, compound 1, and compound 8 into G9a and GLP. Representative ITC titrations are shown for UNC0642 (A), compound 1 (B), and compound 8 (C) with G9a (left) and GLP (right). The calculated values represent the means ± SD from two independent experiments.
It is worth noting that the SAHH-coupled biochemical assays were performed with 10 min of preincubation time while the biophysical ITC experiments were carried out over a relatively long experimental time (50 min). It is likely that a only small fraction of G9a/GLP was covalently modified in the SAHH-coupled biochemical assays due to the short incubation time. Under the ITC assay conditions, however, a relatively larger portion of G9a/GLP could be covalently modified due to the longer experimental time.
Time-Dependent Covalent Modification of G9a and GLP by Compound 8.
To explore the kinetics of the G9a and GLP covalent modification by compound 8, we performed time-dependent experiments using two complementary assays: a radioactivity-based scintillation proximity biochemical assay and mass spectrometry based analysis. First, a radioactivity-based scintillation proximity assay was performed using different preincubation times (5, 15, 45 min). Interestingly, compound 8 not only displayed improved inhibitory activity against G9a and GLP with increased preincubation time; it also more effectively inhibited G9a than GLP at all three time points (Figure 5A). Next, we utilized mass spectrometry to monitor the formation of protein adduct at different time points (0, 15, 30, 45, 60, 75, 90, 105, 120 min; Figure 5B). The formation of the G9a–compound 8 adduct was faster than that of the GLP–compound 8 adduct. In particular, the results at early time points (such as 30 and 60 min) suggest that compound 8 might prefer covalent modification for G9a over GLP kinetically (Figure 5C).
Figure 5.

Time-dependent inhibition and covalent modification of G9a and GLP by compound 8. (A) Inhibitory activity of compound 8 in G9a and GLP radioactivity-based scintillation proximity biochemical assays following different preincubation times (5, 15, 45 min). Data are the means ± SD from four replicates. (B) Quantification of time-dependent protein–inhibitor adduct formation using mass spectrometry based analysis. Data are the means ± SD from two independent experiments. (C) Representative mass spectrometric analysis results of G9a (left) and GLP (right) treated with compound 8 for 30 min (upper) or 60 min (lower). Molecular weight of G9a, 34 314 Da; molecular weight of G9a + compound 8, 34 810 Da; molecular weight of GLP, 34 687 Da; molecular weight of GLP + compound 8, 35 183 Da. In (B) and (C) 5 μM G9a or GLP and 50 μM compound 8 were used.
Design and Characterization of a Noncovalent Inhibitor Control of Compound 8.
To determine whether covalent modification plays a role in the appeared slight kinetic preference of compound 8 for G9a over GLP, we designed and synthesized compound 9 as a noncovalent inhibitor control (Figure 6). Compound 9 contains a structurally similar propionamide group, which is incapable of covalently modifying the cysteine residue of G9a and GLP. Using MS-based analysis, we confirmed that compound 9 did not covalently modify G9a after 1 h of incubation (Figure 7A). As a noncovalent inhibitor, compound 9 displayed a preference similar to that of UNC0642 for GLP over G9a in biophysical and biochemical assays. In ITC experiments, compound 9 showed a higher binding affinity for GLP than for G9a (Kd = 48 ± 4 nM for G9a, Kd = 16 ± 1 nM for GLP; Figure 7B). In the SAHH-coupled biochemical assays, compound 9 exhibited slightly better potency for GLP than for G9a (IC50 = 211 ± 25 nM for G9a, IC50 = 151 ± 12 nM for GLP; Figure 7C). These results (Figure 7) together with the results of compound 8 in Figures 3–5 suggest that covalent modification may play a role in the potential kinetic preference of compound 8 for G9a over GLP.
Figure 6.

Design of noncovalent inhibitor control of compound 8.
Figure 7.

(A) Representative mass spectrometric analysis results of G9a incubated with compound 9 or compound 8 (as a positive control). After treatment with the indicated compound (or DMSO) for 1 h, the formation of the protein–inhibitor adduct was quantified by using mass spectrometry. Data are shown as G9a + DMSO (top), G9a + compound 8 (middle), and G9a + compound 9 (bottom). (B) ITC titrations of compound 9 into G9a and GLP. The calculated values represent the means ± SD from two independent experiments. (C) Concentration-dependent inhibition of G9a and GLP by compound 9 in SAHH-coupled biochemical assays. The calculated values represent the means ± SD from four independent experiments.
Cocrystal Structures of G9a and GLP in Complex with Compound 1.
We solved cocrystal structures of compound 1 in complex with either G9a (PDB ID 7T7L) or GLP (PDB ID 7T7M), which confirmed the covalent modifications of Cys1098 in G9a and Cys1186 in GLP by compound 1 (Figure 8A,B). Notably, we observed the covalent modification by compound 1 in chains A, B, and D of G9a and chain D of GLP in the cocrystal structures. Surprisingly, both the G9a and GLP cocrystal structures displayed very similar binding modes for compound 1. From the overlaid cocrystal structures using noncovalent modified chains of G9a and GLP, which capture the ligand binding modes before covalent modification, we found that the α helix of G9a indicated by the dashed arrow in Figure 8C moved downward slightly compared to the same α helix of GLP, leading to more space in the G9a binding site. This difference in G9a and GLP binding sites was also confirmed by structural alignment of previously published G9a (PDB ID 5TUY)43 and GLP (PDB ID 5TUZ)43 cocrystal structures (Figure S1). This increased space in the binding pocket of G9a compared to that of GLP could also potentially explain why noncovalent inhibitors such as compound 9 have lower binding affinities to G9a than to GLP: noncovalent inhibitors such as compound 9 fit more loosely in the G9a binding pocket than in the GLP binding pocket. On the other hand, the formation of a covalent bond might be able to limit the movement of the covalent binder, which could potentially compensate the effect of G9a’s slightly enlarged binding pocket, leading to compound 8’s similar binding affinities for G9a and GLP. We also attempted to obtain a cocrystal structure of G9a or GLP in complex with compound 8, but we were unsuccessful.
Figure 8.

X-ray cocrystal structures of compound 1 in complex with G9a and GLP. (A) Cocrystal structure of compound 1 bound to G9a (PDB ID 7T7L). Cys1098 was covalently modified by compound 1. (B) Cocrystal structure of compound 1 bound to GLP (PDB ID 7T7M). Cys1186 was covalently modified by compound 1. (C) Overlay of the crystal structures of G9a in complex with compound 1 (in orange) and GLP in complex with compound 1 (in cyan). G9a and GLP structures that were not covalently modified were used for overlay. The dashed arrow indicates the slight downward movement of the α helix of G9a (in orange) compared to that of GLP (in cyan).
Selectivity and Cellular Activities of Compound 8.
We further characterized compound 8 by determining its selectivity against 21 other methyltransferases. Compound 8 at 1 μM did not display a significant inhibition effect (>50%) against any of these 21 PKMTs and PRMTs (Figure 9).
Figure 9.

Selectivity of compound 8 against 21 other methyltransferases. Data are the means ± SD from two independent experiments.
Lastly, we assessed the cellular activities of compounds 1 and 8 in MDA-MB-231 and K562 cells, using UNC0642 as a control. First, we assessed the reduction of the H3K9 dimethylation (H3K9me2) level in MDA-MB-231 cells treated with 1, 8, 9, or UNC0642 at 0.2, 1, or 5 μM for 24, 48, or 72 h (Figure 10A). As expected, compound 8 effectively reduced the H3K9me2 mark in a concentration- and time-dependent manner. Consistent with the results in the enzymatic inhibition assays (Figure 3 and Table 2), compound 8 displayed improved potency compared to UNC0642 and compound 9, while compound 1 exhibited slightly lower potency than UNC0642 and compound 9, especially after treatment for 72 h (Figure 10A). It is worth noting that compound 9 (noncovalent negative control) displayed some cellular potency as a reversible inhibitor but was not as potent as compound 8. Next, we assessed the effects of 1, 8, and UNC0642 on reducing the H3K9me2 level in K562 cells treated with the inhibitors for 48 h. Similar to the results in MDA-MB-231 cells, both covalent inhibitors (1 and 8) reduced the H3K9me2 level in K562 cells (Figure 10B), and compound 8 showed much better potency compared to compound 1 and slightly better potency compared to UNC0642 (Figure 10C).
Figure 10.

Effective reduction of H3K9me2 by compound 8 in cells in a concentration- and time-dependent manner. (A) Effects of compound 1, compound 8, compound 9, and UNC0642 on reducing the H3K9me2 level in MDA-MB-231 cells. MDA-MB-231 cells were treated with DMSO or the indicated compound at the indicated concentration for 24, 48, or 72 h. Western blot results are representative from at least two independent experiments. (B) Effects of compound 1, compound 8, and UNC0642 on reducing the H3K9me2 level in K562 cells. K562 cells were treated with DMSO or the indicated compound at the indicated concentration for 48 h. Western blot results are representative from at least two independent experiments. (C) Quantification of Western blot results shown in (B) and biological repeats. Blots were quantified by Image Studio and analyzed by using unpaired student t test. ns, P > 0.05; **, P < 0.01; ***, P < 0.001.
We also assessed the effects of compound 1, compound 8, and UNC0642 on inhibiting the cell growth in MDA-MB-231 cells and found that compound 8 was more effective than compound 1 and UNC0642 in suppressing the growth of MDA-MB-231 cells (Figure S2). This result is consistent with the result that compound 8 was more effective in reducing the H3K9me2 level than compound 1 and UNC0642 in MDA-MB-231 cells (Figure 10A).
Chemical Synthesis.
The synthetic route for preparing compounds 1 and 2 is shown in Scheme 1. Intermediate 1047 was reacted with sodium azide, with copper iodide as a catalysis, under microwave conditions to yield related amine 11.64 Coupling of compound 11 with the corresponding acid chlorides yielded compounds 1 and 2, respectively. Compounds 3 and 4 were synthesized with the use of a seven-step reaction sequence shown in Scheme 2. Intermediate 1245 was first reacted with N-Boc glycine to yield intermediate 13, which was cyclized under heating conditions to afford compound 14. Substitution of compound 14’s chloro group with pyrrolidine yielded compound 15, which was subsequently reacted with POCl3 to afford compound 16. Substitution of compound 16’s chloro group with (1-isopropylpiperidin-4-yl)amine under basic conditions yielded compound 17. The Boc protecting group removal of compound 17, followed by amide coupling with the corresponding acid chlorides, afforded compounds 3 and 4, respectively. Compound 5 was prepared by the reaction of amine 21, which was synthesized in two steps, with intermediate 10 under microwave conditions (Scheme 3). It is worth noting that we tried to remove both Boc protecting groups in a single step, but we were unsuccessful. Compound 6 was synthesized similarly to the synthesis of compound 1 (Scheme 4). Intermediate 10 was first reacted with methylamine to afford intermediate 22, which was further reacted with acryloyl chloride to yield compound 6. Compound 7 was synthesized by reacting intermediate 10 with compound 23 using a palladium catalyst (Scheme 5). Lastly, compounds 8 and 9 were prepared according to the synthetic route shown in Scheme 6. Previously reported dichloroquinoline intermediate 2456 was first reacted with 4-methoxylbenzylamine to afford intermediate 25, which was further reacted with (1-isopropylpiperidin-4-yl)amine to yield intermediate 26. The PMB protection group was removed to afford intermediate 27, which was coupled with acryloyl chloride and propionyl chloride to yield compounds 8 and 9, respectively.
Scheme 1. Synthesis of Compounds 1 and 2a.

aReagents and conditions: (a) NaN3, CuI, VC, N1,N2-dimethylethane-1,2-diamine, EtOH/H2O, microwave, 130 °C, 30 min, 70%; (b) acid chloride, DMF, 60 °C, 2 h, 40–45%.
Scheme 2. Synthesis of Compounds 3 and 4a.

aReagents and conditions: (a) TBTU, TEA, DCM, rt, overnight, 49%; (b) H2O2, NaOH, MeOH/H2O, reflux, 30 min, 98%; (c) pyrrolidine, DIEA, DMSO, microwave, 100 °C 1 h, 32%; (d) POCl3, PhNMe2, CHCl3, 80 °C, 2 h; (e) (1-isopropylpiperidin-4-yl)amine, K2CO3, DMF, 80 °C, 1 h, 30% over two steps; (f) HCl, 1,4-dioxane, rt, 30 min; (g) acid chloride, DIEA, DCM, rt, 2 h, 60–70% over two steps.
Scheme 3. Synthesis of Compound 5a.

aReagents and conditions: (a) Sc(OTf)3, DCM, rt, 2 h, 90%; (b) TFA, DCM, rt, 30 min, 80%; (c) intermediate 10, DIEA, IPA, microwave, 130 °C, 30 min, 53%.
Scheme 4. Synthesis of Compound 6a.

aReagents and conditions: (a) methylamine, HCl, IPA, microwave, 160 °C, 15 min, 82%; (b) acryloyl chloride, DMF, 60 °C, 2 h, 40%.
Scheme 5. Synthesis of Compound 7a.

aReagents and conditions: (a) Pd2(dba)3, K3PO4, BINAP, 1,4-dioxane, microwave, 160 °C, 30 min, 27%.
Scheme 6. Synthesis of Compounds 8 and 9a.

aReagents and conditions: (a) 4-methoxylbenzylamine, Pd(OAc)2, BINAP, t-BuONa, THF, microwave, 80 °C, 30 min, 48%; (b) (1-isopropylpiperidin-4-yl)amine, Brettphos-G1-Pd, Brettphos, LHMDS, 1,4-dioxane, microwave, 160 °C, 30 min, 70%; (c), TFA, 50 °C, 1 h, 90%; (d) acid chloride, DIEA, CHCl3, rt, overnight, 30–40%.
CONCLUSIONS
In this study, we discovered two first-in-class potent and selective G9a/GLP covalent inhibitors, compounds 1 and 8, by targeting a cysteine residue at the substrate binding site of G9a (Cys1098) or GLP (Cys1186). Compared to the noncovalent G9a/GLP inhibitor UNC0642, compound 8 displayed 2-fold improved binding affinity to G9a in an ITC biophysical assay and 5-fold improved potency in inhibiting the G9a methyltransferase activity in an SAHH-coupled biochemical assay while exhibiting comparable binding affinity and potency for GLP. Kinetic studies using an MS-based protein covalent modification assay and a radioactivity-based scintillation proximity assay revealed that compound 8 covalently modified G9a faster than it modified GLP, which may contribute to its slight inhibition preference for G9a over GLP, compared to the noncovalent control, compound 9. We also obtained two cocrystal structures of 1 in complex with G9a and GLP, respectively, which confirmed the covalent inhibition mechanism of this compound at the atomic level. In cellular assays, compared to UNC0642, compound 8 displayed improved potency in reducing the H3K9me2 level and enhanced antiproliferation activity. Therefore, compound 8 is a highly potent, selective, and cell-active covalent inhibitor of G9a and GLP, which could be a useful chemical tool for investigating the physiological and pathophysiological functions of G9a and GLP.
EXPERIMENTAL SECTION
Chemistry General Procedures.
All commercial chemical reagents and solvents were used for the reactions without further purification. Microwave-heated reactions were conducted with a Discover SP microwave system with an Explorer 12 Hybrid Autosampler by CEM (Buckingham, U.K.). Flash column chromatography was performed on a Teledyne ISCO CombiFlash Rf+ instrument equipped with a 220/254/280 nm wavelength UV detector and a fraction collector. Reverse phase column chromatography was conducted on HP C18 RediSep Rf columns to purify some polar compounds. All final compounds were purified with preparative high-performance liquid chromatography (HPLC) on an Agilent Prep 1200 series with the UV detector set to 220/254 nm at a flow rate of 40 mL/min. Samples were injected onto a Phenomenex Luna 750 × 30 mm, 5 μm C18 column, and the gradient was set to 10% acetonitrile in H2O containing 0.1% TFA progressing to 100% acetonitrile. All compounds assessed for biological activity have purity of >95% as determined by an Agilent 1200 series system with a DAD detector and a 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column for chromatography and high-resolution mass spectra (HRMS) that were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Samples (2 μL) were injected onto a C18 column at room temperature, and the flow rate was set to 0.4 mL/min with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B. Nuclear magnetic resonance (NMR) spectra were acquired on either Bruker DRX 600 MHz, 500, MHz, or 400 MHz for proton (1H NMR) and 151 or 101 MHz for carbon (13C NMR). Chemical shifts for all compounds are reported in parts per million (ppm, δ). The format of chemical shift was reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant (J values in hertz), and integration. All final compounds had >95% purity using the HPLC methods described above and existed as trifluoroacetate salt forms.
N-(4-((1-Isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)-quinazolin-2-yl)acrylamide (1).
To a microwave reaction tube, compound 1047 (93 mg, 0.2 mmol), sodium azide (27 mg, 0.40 mmol), CuI (76 mg, 0.40 mmol), sodium ascorbate (4 mg, 0.02 mmol)m and N1,N2-dimethylethane-1,2-diamine (2 μL, 0.02 mmol) were mixed in EtOH/H2O (2 mL, 4:1). The mixture was heated under microwave irradiation conditions at 130 °C for 30 min. After cooling to rt, the mixture was diluted with ammonia solution (2 mL) and extracted with CHCl3 (3 × 5 mL). The organic layer was collected. After drying with Na2SO4, the solvent was removed to yield intermediate 11 as a pale yellow solid (62 mg, 70%). 1H NMR (600 MHz, methanol-d4) δ 7.75 (s, 1H), 7.00 (s, 1H), 4.61–4.53 (m, 1H), 4.31 (t, J = 5.5 Hz, 2H), 3.97 (s, 3H), 3.87–3.79 (m, 2H), 3.66–3.56 (m, 3H), 3.48 (t, J = 7.3 Hz, 2H), 3.27–3.19 (m, 2H), 3.19–3.12 (m, 2H), 2.47–2.40 (m, 2H), 2.40–2.31 (m, 2H), 2.28–2.17 (m, 2H), 2.13–2.02 (m, 4H), 1.43 (d, J = 6.0 Hz, 6H).
Compound 11 (44 mg, 0.10 mmol) was dissolved in DMF (1 mL), and acryloyl chloride (0.1 mL, 1.14 mmol) was added. The mixture was heated at 60 °C for 4 h. After cooling to room temperature, MeOH (0.1 mL) was added to quench the reaction. The crude mixture was purified by prep-HPLC to yield compound 1 (20 mg, 40%) as a white solid. 1H NMR (600 MHz, methanol-d4) δ 7.88 (s, 1H), 7.53 (s, 1H), 6.71 (dd, J = 17.0, 1.6 Hz, 1H), 6.64 (dd, J = 16.9, 10.0 Hz, 1H), 6.10 (dd, J = 10.0, 1.6 Hz, 1H), 4.62 (tt, J = 11.7, 4.1 Hz, 1H), 4.37 (t, J = 5.5 Hz, 2H), 4.02 (s, 3H), 3.87–3.80 (m, 2H), 3.70–3.56 (m, 3H), 3.49 (t, J = 7.3 Hz, 2H), 3.28–3.11 (m, 4H), 2.50–2.33 (m, 4H), 2.28–2.03 (m, 6H), 1.44 (d, J = 6.7 Hz, 6H). 13C NMR (151 MHz, methanol-d4) δ 167.46, 159.91, 155.46, 150.11, 149.74, 133.56, 131.73, 128.62, 105.40, 103.31, 100.88, 66.94, 58.34, 55.78, 54.04, 52.78, 48.16, 28.12, 25.18, 22.59, 15.62. HRMS (TOF): calcd for C27H41N6O3+ [M + H]+ 497.3235, found 497.3223.
2-Chloro-N-(4-((1-isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinazolin-2-yl)acetamide (2).
Compound 2 was prepared by using similar procedures for synthesizing compound 1 as a white solid, with 45% yield. 1H NMR (600 MHz, methanol-d4) δ 7.89 (s, 1H), 7.55 (s, 1H), 4.66–4.57 (m, 1H), 4.46 (s, 2H), 4.37 (t, J = 5.5 Hz, 2H), 4.02 (s, 3H), 3.87–3.78 (m, 2H), 3.70–3.59 (m, 3H), 3.49 (t, J = 7.3 Hz, 2H), 3.26–3.13 (m, 4H), 2.48–2.34 (m, 4H), 2.26–2.02 (m, 6H), 1.44 (d, J = 6.7 Hz, 6H). HRMS (TOF): calcd for C26H40ClN6O3+ [M + H]+ 519.2845, found 519.2817.
N-((4-((1-Isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinazolin-2-yl)methyl)acrylamide (3).
Compound 1265 (480 mg, 2.00 mmol), N-Boc glycine (1.75 g, 10.00 mmol), TBTU (1.6 g, 5.00 mmol), and TEA (1.4 mL, 10.00 mmol) were dissolved in DCM (20 mL). The solution was stirred at room temperature for 24 h. Then, the mixture was poured into water (20 mL); the organic layer was separated and collected. After drying with Na2SO4, the solvent was removed; the residue was purified by ISCO to yield compound 13 (390 mg, 49%) as a yellow solid. 1H NMR (600 MHz, chloroform-d) δ 7.29 (s, 1H), 6.97 (s, 1H), 5.30–5.18 (m, 1H), 4.26 (t, J = 6.1 Hz, 2H), 4.00 (d, J = 6.2 Hz, 2H), 3.87 (s, 3H), 3.77 (t, J = 6.3 Hz, 2H), 2.36–2.31 (m, 2H), 1.52 (s, 9H).
Compound 13 (390 mg, 0.98 mmol) was dissolved in MeOH/H2O (5 mL, 4:1). NaOH (80 mg, 2.00 mmol) and H2O2 (one drop) were added subsequently. The mixture was heated at 100 °C for 30 min. After cooling to room temperature, the pH value of the mixture was adjusted to pH 7.0 by concentrated HCl. The solid precipitated and was collected; it was dried to yield compound 14 as a brown solid. The crude product was dissolved in DMSO (5 mL), and pyrrolidine (0.17 mL, 2.08 mmol) was added followed by DIEA (0.35 mL, 2.00 mmol). The mixture was heated under microwave irradiation at 100 °C for 1 h. After cooling to room temperature, the mixture was purified by reverse-ISCO to yield compound 15 (174 mg, 32%, two steps) as a brown oil. 1H NMR (600 MHz, methanol-d4) δ 7.56 (s, 1H), 7.23 (s, 1H), 4.36 (s, 2H), 4.31 (t, J = 5.6 Hz, 2H), 3.97 (s, 3H), 3.90–3.79 (m, 2H), 3.50 (t, J = 7.2 Hz, 2H), 3.22–3.10 (m, 2H), 2.41–2.32 (m, 2H), 2.29–2.17 (m, 2H), 2.10 (s, 2H), 1.49 (s, 9H).
Compound 15 (174 mg, 0.40 mmol) was mixed with CHCl3 (2 mL) added with POCl3 (0.14 mL, 1.50 mmol) followed by PhNMe2 (0.57 mL, 4.52 mmol). The mixture was heated at 80 °C for 2 h. After cooling to room temperature, the mixture was poured into ice–water and neutralized with NaHCO3. Then, DCM (3 × 5 mL) added for extraction. The organic layer was collected and dried. After the solvent was removed, the residue was used for the next step without further purification. The obtained crude intermediate 16 was dissolved in DMF (1 mL), and (1-isopropylpiperidin-4-yl)amine (0.1 mL, 0.60 mmol) was added followed by K2CO3 (70 mg, 0.51 mmol). The mixture was heated at 80 °C for 1 h. After cooling to room temperature, the mixture was filtered and the filtrate was collected and purified by reverse-ISCO to yield compound 17 (71 mg, 30%, two steps). 1H NMR (600 MHz, methanol-d4) δ 7.91 (s, 1H), 7.29 (s, 1H), 4.74 (s, 2H), 4.67–4.58 (m, 1H), 4.38 (t, J = 5.5 Hz, 2H), 4.04 (s, 3H), 3.87–3.79 (m, 2H), 3.70–3.57 (m, 3H), 3.50 (t, J = 7.3 Hz, 2H), 3.22–3.11 (m, 4H), 2.46–2.36 (m, 4H), 2.28–2.19 (m, 2H), 2.19–2.05 (m, 4H), 1.43 (d, J = 6.7 Hz, 6H).
Compound 17 (71 mg, 0.13 mmol) was added with HCl/dioxane solution (4 M, 1 mL). The mixture was stirred at room temperature for 30 min, and then the solvent was removed. The obtained white solid intermediate 18 was used for the next step without further purification. Intermediate 18 (30 mg, 0.05 mmol) was dissolved in DMF (0.5 mL), followed by acryloyl chloride (5 μL, 0.06 mmol) and DIEA (0.1 mL, 0.60 mmol). The solution was stirred at room temperature for 2 h. After quenching with MeOH (0.1 mL), the mixture was purified by prep-HPLC to yield compound 3 (22 mg, 60%, two steps) as a pale yellow solid. 1H NMR (600 MHz, methanol-d4) δ 7.90 (s, 1H), 7.30 (d, J = 1.1 Hz, 1H), 6.46 (dd, J = 17.1, 10.3 Hz, 1H), 6.34 (d, J = 17.1 Hz, 1H), 5.84 (d, J = 10.3 Hz, 1H), 4.74–4.63 (m, 3H), 4.36 (t, J = 5.5 Hz, 2H), 4.04 (s, 3H), 3.88–3.78 (m, 2H), 3.66–3.57 (m, 3H), 3.50 (t, J = 7.2 Hz, 2H), 3.26–3.12 (m, 4H), 2.47–2.36 (m, 4H), 2.23 (d, J = 7.7 Hz, 2H), 2.16–2.04 (m, 4H), 1.43 (d, J = 7.0 Hz, 6H). HRMS (TOF): calcd for C28H43N6O3+ [M + H]+ 511.3391, found 511.3402.
2-Chloro-N-((4-((1-isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinazolin-2-yl)methyl)acetamide (4).
Compound 4 was synthesized following similar procedures for preparing compound 3 (70%, two steps). 1H NMR (600 MHz, methanol-d4) δ 7.91 (s, 1H), 7.29 (s, 1H), 4.75–4.68 (m, 1H), 4.66 (s, 2H), 4.37 (t, J = 5.5 Hz, 2H), 4.29 (s, 2H), 4.05 (s, 3H), 3.87–3.80 (m, 2H), 3.69–3.59 (m, 3H), 3.50 (t, J = 7.3 Hz, 2H), 3.29–3.23 (m, 2H), 3.22–3.14 (m, 2H), 2.51–2.36 (m, 4H), 2.29–2.18 (m, 2H), 2.18–2.02 (m, 4H), 1.43 (d, J = 6.7 Hz, 6H). HRMS (TOF): calcd for C27H42ClN6O3+ [M + H]+ 533.3001, found 533.2983.
Methyl 2-(((4-((1-Isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)-quinazolin-2-yl)amino)methyl)acrylate (5).
Compound 1966 (320 mg, 1.02 mmol) was dissolved in DCM (5 mL) and Sc(OTf)3 (49 mg, 0.10 mmol). After stirring for 2 h, the mixture was diluted with EtOAc (10 mL) and washed with water. The organic phase was dried with Na2SO4 and evaporated to give compound 20 (190 mg, 90%) as a colorless oil. 1H NMR (600 MHz, chloroform-d) δ 6.25 (s, 1H), 5.79 (s, 1H), 5.04–4.90 (m, 1H), 3.95 (s, 2H), 3.77 (s, 3H), 1.44 (s, 9H).
Compound 20 (190 mg, 0.88 mmol) was dissolved in DCM (5 mL), and TFA (2.5 mL) was added. After stirring at room temperature for 30 min, the solvent was removed to yield to intermediate 21. 1H NMR (600 MHz, methanol-d4) δ 6.54 (s, 1H), 6.10 (s, 1H), 3.85 (s, 2H), 3.83 (s, 3H).
Intermediate 21 (46 mg, 0.20 mmol), intermediate 10 (46 mg, 0.10 mmol), DIEA (0.17 mL, 0.10 mmol), and IPA (1 mL) were mixed together. The mixture was heated under microwave irradiation at 130 °C for 30 min. After cooling to room temperature, the mixture was purified by prep-HPLC to yield compound 5 (44 mg, 53%) as a yellow solid. 1H NMR (600 MHz, methanol-d4) δ 7.75 (s, 1H), 7.03 (s, 1H), 6.33 (s, 1H), 5.89 (s, 1H), 4.57–4.49 (m, 1H), 4.45 (s, 2H), 4.31 (t, J = 5.4 Hz, 2H), 3.97 (s, 3H), 3.87–3.80 (m, 5H), 3.66–3.57 (m, 3H), 3.48 (t, J = 7.2 Hz, 2H), 3.28–3.21 (m, 2H), 3.21–3.12 (m, 2H), 2.47–2.39 (m, 2H), 2.39–2.33 (m, 2H), 2.27–2.19 (m, 2H), 2.12–2.01 (m, 4H), 1.43 (d, J = 6.7 Hz, 6H). HRMS (TOF): calcd for C29H45N6O4+ [M + H]+ 541.3497, found 541.3469.
N-(4-((1-Isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)-quinazolin-2-yl)-N-methylacrylamide (6).
Intermediate 10 (92 mg, 0.20 mmol), methylamine (1 M in THF, 0.4 mL, 0.40 mmol), and HCl (4 M in dioxane, 0.2 mL, 0.80 mmol) and IPA (1 mL) were mixed. The mixture was heated under microwave irradiation at 160 °C for 15 min. After cooling to room temperature, the mixture was purified by prep-HPLC to yield compound 22 (114 mg, 82%) as a yellow solid. 1H NMR (600 MHz, methanol-d4) δ 7.74 (s, 1H), 7.00 (s, 1H), 4.69–4.48 (m, 1H), 4.30 (t, J = 5.5 Hz, 2H), 3.96 (s, 3H), 3.87–3.79 (m, 2H), 3.68–3.54 (m, 3H), 3.48 (t, J = 7.2 Hz, 2H), 3.30–3.22 (m, 2H), 3.20–3.13 (m, 2H), 3.10 (s, 3H), 2.52–2.41 (m, 2H), 2.41–2.29 (m, 2H), 2.27–2.17 (m, 2H), 2.17–2.03 (m, 4H), 1.42 (d, J = 6.7 Hz, 6H).
Compound 6 was prepared similarly to the synthesis of compound 1 by reaction of intermediate 22 with acryloyl chloride with 40% yield. 1H NMR (500 MHz, methanol-d4) δ 7.90 (s, 1H), 7.43 (s, 1H), 7.01 (dd, J = 16.6, 10.5 Hz, 1H), 6.62 (dd, J = 16.7, 1.6 Hz, 1H), 6.09 (dd, J = 10.5, 1.6 Hz, 1H), 4.70–4.62 (m, 1H), 4.37 (t, J = 5.5 Hz, 2H), 4.03 (s, 3H), 3.87–3.80 (m, 2H), 3.76 (s, 3H), 3.67–3.59 (m, 3H), 3.50 (t, J = 7.1 Hz, 2H), 3.31–3.25 (m, 2H), 3.20–3.13 (m, 2H), 2.50–2.42 (m, 2H), 2.39 (s, 2H), 2.27–2.14 (m, 4H), 2.09 (s, 2H), 1.43 (d, J = 6.7 Hz, 6H). HRMS (TOF): calcd for C28H43N6O3+ [M + H]+ 511.3391, found 511.3409.
1-(4-((1-Isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)-quinazolin-2-yl)-3-methylenepiperidin-2-one (7).
Intermediate 2367 (22 mg, 0.20 mmol), intermediate 9 (46 mg, 0.10 mmol), Pd2(dba)3 (18 mg, 0.02 mmol), BINAP (27 mg, 0.04 mmol), and K3PO4 (64 mg, 0.30 mmol) were mixed in dioxane (1 mL). The mixture was heated under microwave irradiation at 160 °C for 30 min. After cooling to room temperature, the mixture was filtered; the filtrate was collected and purified by prep-HPLC to yield compound 7 (21 mg, 27%) as a brown solid. 1H NMR (600 MHz, methanol-d4) δ 7.91 (s, 1H), 7.48 (s, 1H), 6.57 (s, 1H), 5.79 (s, 1H), 4.72–4.62 (m, 1H), 4.38 (t, J = 5.5 Hz, 2H), 4.30 (t, J = 6.0 Hz, 2H), 4.03 (s, 3H), 3.84 (t, J = 9.9 Hz, 2H), 3.68–3.58 (m, 3H), 3.50 (t, J = 7.4 Hz, 2H), 3.37–3.29 (m, 2H), 3.23–3.12 (m, 2H), 2.82–2.76 (m, 2H), 2.50–2.43 (m, 2H), 2.42–2.35 (m, 2H), 2.28–2.20 (m, 2H), 2.14 (d, J = 24.9, 12.1, 4.4 Hz, 6H), 1.44 (d, J = 6.6 Hz, 6H). HRMS (TOF): calcd for C30H45N6O3+ [M + H]+ 537.3548, found 537.3519.
N-(4-((1-Isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinolin-2-yl)acrylamide (8).
Intermediate 2468 (141 mg, 0.40 mmol), 4-methoxylbenzylamine (69 mg, 0.50 mmol), Pd(OAc)2 (9 mg, 0.04 mmol), BINAP (50 mg, 0.08 mmol), and t-BuONa (96 mg, 1.00 mmol) were mixed with THF (5 mL). The mixture was heated under microwave irradiation at 80 °C for 30 min. After cooling to room temperature, the mixture was filtered; the filtrate was collected and purified by reverse-ISCO to yield compound 25 (88 mg, 48%) as a yellow solid. 1H NMR (500 MHz, chloroform-d) δ 7.31 (d, J = 8.6 Hz, 2H), 7.10 (s, 1H), 6.88 (d, J = 8.7 Hz, 2H), 6.62 (s, 1H), 4.83 (s, 1H), 4.57 (d, J = 5.4 Hz, 2H), 4.22 (t, J = 6.3 Hz, 2H), 3.96 (s, 3H), 3.80 (s, 3H), 3.01–2.74 (m, 6H), 2.35–2.21 (m, 2H), 1.98–1.88 (m, 4H).
Compound 25 (88 mg, 0.19 mmol), (1-isopropylpiperidin-4-yl)amine (0.05 mL, 0.30 mmol), Brettphos Pd G1 (16 mg, 0.02 mmol), Brettphos (11 mg, 0.02 mmol), and LHMDS (1 M in THF, 0.3 mL, 0.30 mmol) were mixed together with dioxane (2 mL). The mixture was heated under microwave irradiation at 160 °C for 30 min. After cooling to room temperature, the mixture was filtered; the filtrate was collected and purified by prep-HPLC to yield compound 26 (79 mg, 70%) as a pale yellow solid. 1H NMR (600 MHz, methanol-d4) δ 7.61 (s, 1H), 7.01 (s, 1H), 5.98 (s, 1H), 4.27 (t, J = 5.5 Hz, 2H), 3.96 (s, 3H), 3.94–3.87 (m, 1H), 3.87–3.80 (m, 2H), 3.69–3.56 (m, 3H), 3.48 (t, J = 7.2 Hz, 2H), 3.31–3.23 (m, 2H), 3.20–3.13 (m, 2H), 2.42 (ddd, J = 13.5, 5.2, 2.9 Hz, 2H), 2.39–2.27 (m, 2H), 2.26–2.17 (m, 2H), 2.15–2.04 (m, 4H), 1.42 (d, J = 6.7 Hz, 6H).
Compound 26 (56 mg, 0.10 mmol) was mixed with TFA (1 mL) and heated at 50 °C for 1 h. After cooling to room temperature, the mixture was purified by prep-HPLC to yield compound 27 (70 mg, 90%) as a brown solid. 1H NMR (600 MHz, methanol-d4) δ 7.61 (s, 1H), 7.01 (s, 1H), 5.98 (s, 1H), 4.27 (t, J = 5.5 Hz, 2H), 3.96 (s, 3H), 3.94–3.87 (m, 1H), 3.87–3.80 (m, 2H), 3.69–3.56 (m, 3H), 3.48 (t, J = 7.2 Hz, 2H), 3.31–3.23 (m, 2H), 3.20–3.13 (m, 2H), 2.42 (ddd, J = 13.5, 5.2, 2.9 Hz, 2H), 2.39–2.27 (m, 2H), 2.26–2.17 (m, 2H), 2.15–2.04 (m, 4H), 1.42 (d, J = 6.7 Hz, 6H).
Compound 27 (39 mg, 0.09 mmol), acryloyl chloride (0.01 mL, 0.11 mmol), and DIEA (0.17 mL, 0.97 mmol) were mixed together with CHCl3 (1 mL). After stirring at room temperature overnight, MeOH (0.1 mL) was added to quench the reaction. The mixture was purified by prep-HPLC to yield compound 8 (13 mg, 30%) as a white solid. 1H NMR (600 MHz, methanol-d4) δ 7.83 (s, 1H), 7.47 (s, 1H), 6.81 (s, 1H), 6.65 (dd, J = 17.0, 2.0 Hz, 1H), 6.60 (dd, J = 16.9, 9.6 Hz, 1H), 6.06 (dd, J = 9.6, 2.0 Hz, 1H), 4.36 (t, J = 5.5 Hz, 2H), 4.10–4.00 (m, 4H), 3.88–3.81 (m, 2H), 3.69–3.60 (m, 3H), 3.50 (t, J = 7.3 Hz, 2H), 3.36–3.30 (m, 2H), 3.21–3.13 (m, 2H), 2.48–2.42 (m, 2H), 2.42–2.35 (m, 2H), 2.27–2.12 (m, 4H), 2.12–2.05 (m, 2H), 1.44 (d, J = 6.6 Hz, 6H). 13C NMR (101 MHz, methanol-d4) δ 166.91, 154.33, 153.62, 149.29, 147.27, 132.22, 130.77, 129.15, 109.75, 102.05, 101.19, 86.57, 66.75, 58.39, 55.77, 54.08, 52.91, 48.68, 48.30, 28.23, 25.23, 22.59, 15.62. HRMS (TOF): calcd for C28H42N5O3+ [M + H]+ 496.3282, found 496.3285.
N-(4-((1-Isopropylpiperidin-4-yl)amino)-6-methoxy-7-(3-(pyrrolidin-1-yl)propoxy)quinolin-2-yl)propionamide (9).
Compound 9 was synthesized similarly to the synthesis of compound 8 by reaction of compound 27 with propionyl chloride to give a white solid with 40% yield. 1H NMR (600 MHz, methanol-d4) δ 7.82 (s, 1H), 7.46 (s, 1H), 6.63 (s, 1H), 4.35 (t, J = 5.5 Hz, 2H), 4.07–4.00 (m, 4H), 3.87–3.81 (m, 2H), 3.69–3.59 (m, 3H), 3.50 (t, J = 7.2 Hz, 3H), 3.32–3.29 (m, 2H), 3.21–3.15 (m, 2H), 2.64 (q, J = 7.5 Hz, 2H), 2.48–2.41 (m, 2H), 2.41–2.34 (m, 2H), 2.23 (q, J = 7.5 Hz, 2H), 2.19–2.04 (m, 4H), 1.44 (d, J = 6.7 Hz, 6H), 1.27 (t, J = 7.5 Hz, 3H). 13C NMR (151 MHz, methanol-d4) δ 176.85, 154.32, 153.52, 149.18, 147.33, 132.07, 109.62, 102.09, 101.17, 85.99, 66.73, 58.35, 55.76, 54.06, 52.91, 48.63, 48.16, 29.64, 28.20, 25.22, 22.59, 15.61, 7.62. HRMS (TOF): calcd for C28H44N5O3+ [M + H]+ 498.3439, found 498.3420.
SAHH-Coupled Enzymatic Assay.
The assay was performed with the use of the previously published protocol.63 Briefly, tested compounds were predissolved in assay buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 0.01% Triton X-100, 3 mM MgCl2, 0.1 mg/mL BSA) with 5 μM purified S-adenosylhomocysteine hydrolase (SAHH), 1 unit of adenosine deaminase (ADA; Sigma-Aldrich, USA), 10 μM H3 peptide (1–25) (Anaspec, USA) and 5 nM G9a (or GLP) as 30 μL in a black 96-well plate. After 5 min, 20 μL of SAM (25 μM) (NEB, USA) in assay buffer was added. After incubation for 10 min at room temperature, thiol fluorescent probe IV (20 μM, 50 μL) (Sigma-Aldrich, USA) was added. After 10 min, the fluorescence of the probe was measured at excitation = 400/emission = 465 by an Infinite M Plex (Tecan, USA).
Radioactivity-Based Scintillation Proximity Biochemical Assay.
The transfer of the 3H-labeled methyl group from SAM to the substrate was monitored to assess the inhibitory activities of compound 8 against G9a (5 nM) and GLP (5 nM). The assay was performed for different incubation times (5, 15, 45 min) with H3 peptide (0.05 μM) as the substrate and SAM (1 μM) as the cofactor. Reaction Biology Corp. (USA) performed the assay as four replicates.
Expression and Purification of G9a, GLP, and SAHH.
Catalytic domains of human G9a (913–1193) and GLP (982–1266) were cloned, expressed, and purified according to the previously described protocol.58 The full length of Sulfolobus solfataricus SAHH was cloned, expressed, and purified according to a published protocol.63
Isothermal Titration Calorimetry (ITC).
Binding of compound 1, compound 8, compound 9, and UNC0642 was analyzed with a MicroCal iTC200 (Malvern) in 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% DMSO at 25 °C. After an initial 0.4 μL injection, 13 injections (19 injections for MS8511N) from the syringe solution (500 μM of compounds) were titrated into 300 μL of the protein solution (50 μM G9a or GLP) in the cell, which was stirred at 750 rpm. The data were fitted by the single binding site model using Microcal Origin 7.0 (Malvern). The reported values represent the mean ± SD from two independent measurements.
Western Blot Analysis for H3K9me2.
K562 and MDA-MB-231 cells were seeded in six-well plates (Thermo Fisher Scientific, USA) at 5 × 105 cells per well and treated with the indicated compound for 48 h. Cells were then lysed for 30 min on ice with RIPA buffer containing 1× proteasome/phosphatase inhibitor (Thermo Fisher Scientific, USA). Following lysis, samples were centrifuged for 10 min at 15 000 rpm and 4 °C. Supernatant was collected and mixed with 1× Laemmli buffer (Bio-Rad, USA) and then heated at 100 °C for 10 min. The protein concentration was quantified with the Pierce Rapid Gold BCA kit (Thermo Fisher Scientific, USA). For each sample 10 μg was loaded into a 4–20% Tris-glycine gel (Bio-Rad, USA). SDS-PAGE was run at 90 V for 30 min and at then 120 V for 50 min. Following separation, proteins were transferred onto a PVDF membrane with the use of a Trans-Blot Turbo Transfer system (Bio-Rad, USA). Membranes were blocked with TBS Odyssey Blocking Buffer (LI-COR, USA) for 1 h at room temperature and then incubated in primary antibody (1:1000) overnight at 4 °C. The next day, membranes were washed with 0.1% TBST and TBS and then incubated with secondary antibody (1:10 000) for 1 h at room temperature. After washing, blots were imaged with the use of the Odyssey system (LI-COR, USA) and quantified with Image Studio (LI-COR, USA).
Crystallography.
G9a or GLP in complex with compound 1 was crystallized according to a previously published protocol.43 Briefly, purified GLP and G9a were mixed with S-adenosyl-L-methionine (SAM; Sigma-Aldrich, USA) and compound 1 (protein/SAM/compound 1 molar ratio of 1:2:4). The mixture was coconcentrated to about 20 mg/mL and crystallized with the use of the sitting drop method at 17 °C. The GLP–SAM–compound 1 and G9a–SAM–compound 1 complexes were crystallized in 2% v/v 1,4-dioxane and 10% w/v polyethylene glycol 20 000 for G9a and 0.2 magnesium formate and 15% w/v polyethylene glycol 3350 for GLP. X-ray diffraction data were collected at 100 K at the NE-CAT beamline 24-ID-E of the Advanced Photon Source at Argonne National Laboratory. The data were processed with MOSFLM, SCALA, and other programs from the CCP4 suite. The structures of the G9a and GLP complexes were solved by molecular replacement using the PHASER. Phenix was used for structure refinement. The graphic program COOT was used for model building and visualization.
Selectivity Assays.
Selectivity assays against other methyltransferases were performed by Reaction Biology Corp. (USA) using the same 3H-labeled SAM based assay format as described above. All the experiments were performed using 1 μM compound in duplicates. The compound enzyme inhibition effect was calculated as a percentage of inhibition against control enzyme activity.
Mass Spectrometric Analysis.
Mass spectrometric analysis was performed by an Agilent LC/MSD time-of-flight (TOF) mass spectrometer (Agilent Technologies, USA) equipped with an electrospray ion source. To investigate the covalent modification of G9a and GLP by synthesized compounds, 5 μM purified G9a (or GLP) in a buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl) was incubated with 50 μM indicated compound for the indicated time at room temperature.
Cytotoxicity Assay.
MDA-MB-231 cells were seeded in 96-well plates (Thermo Fisher Scientific, USA) at 1 × 104 cells per well and treated with the indicated compounds for 3 days at various concentrations. The viability was assessed by CCK-8 (Dojindo, USA) following its protocol. All values were plotted from triplicate experiments.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported in part by Grant R01HD088626 (to J.J.) from the National Institutes of Health (NIH), Levo Therapeutics, and an endowed professorship by the Icahn School of Medicine at Mount Sinai (to J.J.). This work utilized the NMR Spectrometer Systems at Mount Sinai acquired with funding from National Institutes of Health SIG Grants 1S10OD025132 and 1S10OD028504. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on the 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We also thank the staff at the 24-ID-E beamline of the Advanced Photon Source at Argonne National Laboratory for facilitating X-ray data collection. We thank Dr. Jithesh Kottur for helping with crystallographic refinement.
ABBREVIATIONS USED
- BINAP
2,2′-bis(diphenylphosphino)-1,1′-binaphthyl
- Boc
tert-butyloxycarbonyl
- DCM
dichloromethane
- DIEA
N,N-diisopropylethylamine
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- EHMT1
euchromatic histone lysine methyltransferase 1
- EHMT2
euchromatic histone lysine methyltransferase 2
- H3K9
histone H3 lysine 9
- H3K9me2
dimethylation of lysine 9 on histone H3
- HMT
histone methyl transferase
- IC50
half-maximal inhibitory concentration
- ITC
isothermal titration calorimetry
- IPA
isopropyl alcohol
- PKMTs
protein lysine methyltransferases
- PRMTs
protein arginine methyltransferases
- LHMDS
lithium bis-(trimethylsilyl)amide
- MS
mass spectrometry
- PMB
p-methoxybenzyl
- SAM
S-adenosyl-L-methionine
- SAHH
S-adenosyl-L-homocysteine hydrolase
- SET
suppressor of variegation 3–9, enhancer of zeste, and trithorax
- TBTU
2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethylaminium tetrafluoroborate
- TEA
triethylamine
- TFA
trifluoroacetic acid
- VC
vanadium carbide
Footnotes
The authors declare the following competing financial interest(s): J.J. is a cofounder and equity shareholder in Cullgen Inc. and a consultant for Cullgen Inc., EpiCypher Inc., and Accent Therapeutics Inc. The Jin laboratory received research funds from Celgene Corporation, Levo Therapeutics, Cullgen, Inc. and Cullinan Oncology
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.2c00652.
Overlay of cocrystal structures of G9a and GLP in complex with MS0124; crystallography data and refinement statistics for G9a and GLP cocrystal structures in complex with compound 1; 1H NMR, 13C NMR, and/or LC–MS spectra of compounds 1–9 (PDF)
Molecular formula strings for all compounds (CSV)
PDB validation report for 7T7L (PDF)
PDB validation report for 7T7M (PDF)
Accession Codes
The structure of G9a in complex with 1 has been deposited under PDB ID 7T7L. The structure of GLP in complex with 1 has been deposited under PDB ID 7T7M. Authors will release the atomic coordinates and experimental data upon article publication.
Contributor Information
Kwang-Su Park, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Yan Xiong, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Hyerin Yim, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Julia Velez, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Nicolas Babault, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Prashasti Kumar, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Jing Liu, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
Jian Jin, Mount Sinai Center for Therapeutics Discovery, Departments of Pharmacological Sciences, Oncological Sciences and Neuroscience, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, New York, New York 10029, United States.
REFERENCES
- (1).Husmann D; Gozani O Histone lysine methyltransferases in biology and disease. Nat. Struct Mol. Biol. 2019, 26 (10), 880–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Black JC; Van Rechem C; Whetstine JR Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol. Cell 2012, 48 (4), 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Bhat KP; Umit Kaniskan H; Jin J; Gozani O Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat. Rev. Drug Discovery 2021, 20 (4), 265–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hamamoto R; Saloura V; Nakamura Y Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat. Rev. Cancer 2015, 15 (2), 110–124. [DOI] [PubMed] [Google Scholar]
- (5).Zhang X; Huang YL; Shi XB Emerging roles of lysine methylation on non-histone proteins. Cell. Mol. Life Sci. 2015, 72 (22), 4257–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Cornett EM; Ferry L; Defossez PA; Rothbart SB Lysine methylation regulators moonlighting outside the epigenome. Mol. Cell 2019, 75 (6), 1092–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Luo M Chemical and biochemical perspectives of protein lysine methylation. Chem. Rev. 2018, 118 (14), 6656–6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kaniskan HU; Martini ML; Jin J Inhibitors of protein methyltransferases and demethylases. Chem. Rev. 2018, 118 (3), 989–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kaniskan HU; Konze KD; Jin J Selective inhibitors of protein methyltransferases. J. Med. Chem. 2015, 58 (4), 1596–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Subramanian K; Jia D; Kapoor-Vazirani P; Powell DR; Collins RE; Sharma D; Peng JM; Cheng XD; Vertino PM Regulation of estrogen receptor alpha by the SET7 lysine methyltransferase. Mol. Cell 2008, 30 (3), 336–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chuikov S; Kurash JK; Wilson JR; Xiao B; Justin N; Ivanov GS; McKinney K; Tempst P; Prives C; Gamblin SJ; Barlev NA; Reinberg D Regulation of p53 activity through lysine methylation. Nature 2004, 432 (7015), 353–360. [DOI] [PubMed] [Google Scholar]
- (12).Deng XL; Hamamoto R; Vougiouklakis T; Wang R; Yoshioka Y; Suzuki T; Dohmae N; Matsuo Y; Park JH; Nakamura Y Critical roles of SMYD2-mediated beta-catenin methylation for nuclear translocation and activation of Wnt signaling. Oncotarget 2017, 8 (34), 55837–55847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Cho HS; Shimazu T; Toyokawa G; Daigo Y; Maehara Y; Hayami S; Ito A; Masuda K; Ikawa N; Field HI; Tsuchiya E; Ohnuma S; Ponder BAJ; Yoshida M; Nakamura Y; Hamamoto R Enhanced HSP70 lysine methylation promotes proliferation of cancer cells through activation of aurora kinase B. Nat. Commun. 2012, 3, 1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Fang L; Teng HQ; Wang YL; Liao GH; Weng LJ; Li YX; Wang XB; Jin JL; Jiao CC; Chen L; Peng XP; Chen JY; Yang YZ; Fang HQ; Han DY; Li C; Jin XL; Zhang SL; Liu ZC; Liu M; Wei Q; Liao LJ; Ge X; Zhao B; Zhou DW; Qin HL; Zhou J; Wang P SET1a-mediated monomethylation at K342 regulates YAP activation by blocking its nuclear export and promotes tumorigenesis. Cancer Cell 2018, 34 (1), 103–118. [DOI] [PubMed] [Google Scholar]
- (15).Tsusaka T; Kikuchi M; Shimazu T; Suzuki T; Sohtome Y; Akakabe M; Sodeoka M; Dohmae N; Umehara T; Shinkai Y Trimethylation of ATF7IP by G9a/GLP recruits the chromodomain protein MPP8. Epigenet. Chromatin 2018, 11, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kori S; Ferry L; Matano S; Jimenji T; Kodera N; Tsusaka T; Matsumura R; Oda T; Sato M; Dohmae N; Ando T; Shinkai Y; Defossez PA; Arita K Structure of the UHRF1 tandem tudor domain bound to a methylated non-histone protein, LIG1, reveals rules for binding and regulation. Structure 2019, 27 (3), 485–496. [DOI] [PubMed] [Google Scholar]
- (17).Osipovich AB; Gangula R; Vianna PG; Magnuson MA SETD5 is essential for mammalian development and the cotranscriptional regulation of histone acetylation. Development 2016, 143 (24), 4595–4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wen H; Li YY; Xi YX; Jiang SM; Stratton S; Peng DN; Tanaka K; Ren YF; Xia Z; Wu J; Li B; Barton MC; Li W; Li HT; Shi XB ZMYND11 links histone H3.3K36me3 to transcription elongation and tumour suppression. Nature 2014, 508 (7495), 263–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Viaene A; Santi M; Rosenbaum J; Li M; Surrey L; Nasrallah M SETD2 mutations in primary central nervous system tumors. Acta Neuropathol. Commun. 2018, 6, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bennett RL; Swaroop A; Troche C; Licht JD The role of nuclear receptor-binding SET domain family histone lysine methyltransferases in cancer. Cold Spring Harbor Perspect. Med. 2017, 7 (6), a026708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhu L; Li Q; Wong SHK; Huang M; Klein BJ; Shen JF; Ikenouye L; Onishi M; Schneidawind D; Buechele C; Hansen L; Duque-Afonso J; Zhu FF; Martin GM; Gozani O; Majeti R; Kutateladze TG; Cleary ML ASH1l links histone H3 lysine 36 dimethylation to MLL leukemia. Cancer Discov 2016, 6 (7), 770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Metzger E; Wang S; Urban S; Willmann D; Schmidt A; Offermann A; Allen A; Sum M; Obier N; Cottard F; Ulferts S; Preca BT; Hermann B; Maurer J; Greschik H; Hornung V; Einsle O; Perner S; Imhof A; Jung M; Schule R KMT9 monomethylates histone H4 lysine 12 and controls proliferation of prostate cancer cells. Nat. Struct Mol. Biol. 2019, 26 (5), 361–376. [DOI] [PubMed] [Google Scholar]
- (23).Carlson SM; Gozani O Nonhistone lysine methylation in the regulation of cancer pathways. Cold Spring Harbor Perspect. Med. 2016, 6 (11), a026435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Levy D Lysine methylation signaling of non-histone proteins in the nucleus. Cell. Mol. Life Sci. 2019, 76 (15), 2873–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Chen Y; Ren B; Yang JS; Wang HY; Yang G; Xu RY; You L; Zhao YP The role of histone methylation in the development of digestive cancers: a potential direction for cancer management. Signal Transduction Targeted Ther. 2020, 5 (1), 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Faundes V; Newman WG; Bernardini L; Canham N; Clayton-Smith J; Dallapiccola B; Davies SJ; Demos MK; Goldman A; Gill H; Horton R; Kerr B; Kumar D; Lehman A; McKee S; Morton J; Parker MJ; Rankin J; Robertson L; Temple IK; Banka S; et al. Histone lysine methylases and demethylases in the landscape of human developmental disorders. Am. J. Hum. Genet. 2018, 102 (1), 175–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Butcher DT; Cytrynbaum C; Turinsky AL; Siu MT; Inbar-Feigenberg M; Mendoza-Londono R; Chitayat D; Walker S; Machado J; Caluseriu O; Dupuis L; Grafodatskaya D; Reardon W; Gilbert-Dussardier B; Verloes A; Bilan F; Milunsky JM; Basran R; Papsin B; Stockley TL; Scherer SW; Choufani S; Brudno M; Weksberg R Charge and kabuki syndromes: gene-specific dna methylation signatures identify epigenetic mechanisms linking these clinically overlapping conditions. Am. J. Hum. Genet. 2017, 100 (5), 773–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Majumder S; Thieme K; Batchu SN; Alghamdi TA; Bowskill BB; Kabir MG; Liu YA; Advani SL; White KE; Geldenhuys L; Tennankore KK; Poyah P; Siddiqi FS; Advani A Shifts in podocyte histone H3K27me3 regulate mouse and human glomerular disease. J. Clin Invest 2018, 128 (1), 483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tachibana M; Sugimoto K; Nozaki M; Ueda J; Ohta T; Ohki M; Fukuda M; Takeda N; Niida H; Kato H; Shinkai Y G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Gene Dev 2002, 16 (14), 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Link PA; Gangisetty O; James SR; Woloszynska-Read A; Tachibana M; Shinkai Y; Karpf AR Fistinct roles for histone methyltransferases G9a and GLP in cancer germ-line antigen gene regulation in human cancer cells and murine embryonic stem cells. Mol. Cancer Res. 2009, 7 (6), 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Guo AS; Huang YQ; Ma XD; Lin RS Mechanism of G9a inhibitor BIX-01294 acting on U251 glioma cells. Mol. Med. Rep 2016, 14 (5), 4613–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tu WB; Shiah YJ; Lourenco C; Mullen PJ; Dingar D; Redel C; Tamachi A; Ba-Alawi W; Aman A; Al-Awar R; Cescon DW; Haibe-Kains B; Arrowsmith CH; Raught B; Boutros PC; Penn LZ MYC Interacts with the G9a histone methyltransferase to drive transcriptional repression and tumorigenesis. Cancer Cell 2018, 34 (4), 579–595.e8. [DOI] [PubMed] [Google Scholar]
- (33).Hua KT; Wang MY; Chen MW; Wei LH; Chen CK; Ko CH; Jeng YM; Sung PL; Jan YH; Hsiao M; Kuo ML; Yen ML The H3K9 methyltransferase G9a is a marker of aggressive ovarian cancer that promotes peritoneal metastasis. Mol. Cancer 2014, 13, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Sun T; Zhang KQ; Pangeni RP; Wu J; Li WD; Du Y; Guo YM; Chaurasiya S; Arvanitis L; Raz DJ G9a promotes invasion and metastasis of non-small cell lung cancer through enhancing focal adhesion kinase activation via NF-kappa B signaling pathway. Mol. Cancer Res. 2021, 19 (3), 429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cao YP; Sun JY; Li MQ; Dong Y; Zhang YH; Yan J; Huang RM; Yan X Inhibition of G9a by a small molecule inhibitor, UNC0642, induces apoptosis of human bladder cancer cells. Acta Pharmacol Sin 2019, 40 (8), 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Segovia C; San José-Enériz E; Munera-Maravilla E; Martínez-Fernández M; Garate L; Miranda E; Vilas-Zornoza A; Lodewijk I; Rubio C; Segrelles C; Valcárcel LV; Rabal O; Casares N; Bernardini A; Suarez-Cabrera C; López-Calderón FF; Fortes P; Casado JA; Dueñas M; Villacampa F; Lasarte JJ; Guerrero-Ramos F; de Velasco G; Oyarzabal J; Castellano D; Agirre X; Prósper F; Paramio JM Inhibition of a G9a/DNMT network triggers immune-mediated bladder cancer regression. Nat. Med. 2019, 25 (7), 1073–1081. [DOI] [PubMed] [Google Scholar]
- (37).Kato S; Weng QY; Insco ML; Chen KY; Muralidhar S; Pozniak J; Diaz JMS; Drier Y; Nguyen N; Lo JA; van Rooijen E; Kemeny LV; Zhan Y; Feng Y; Silkworth W; Powell CT; Liau BB; Xiong Y; Jin J; Newton-Bishop J; Zon LI; Bernstein BE; Fisher DE Gain-of-function genetic alterations of G9a drive oncogenesis. Cancer Discov 2020, 10 (7), 980–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bergin CJ; Zouggar A; Haebe JR; Masibag AN; Desrochers FM; Reilley SY; Agrawal G; Benoit YD G9a controls pluripotent-like identity and tumor-initiating function in human colorectal cancer. Oncogene 2021, 40 (6), 1191–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Antignano F; Burrows K; Hughes MR; Han JM; Kron KJ; Penrod NM; Oudhoff MJ; Wang SKH; Min PH; Gold MJ; Chenery AL; Braam MJS; Fung TC; Rossi FMV; McNagny KM; Arrowsmith CH; Lupien M; Levings MK; Zaph C Methyltransferase G9a regulates T cell differentiation during murine intestinal inflammation. J. Clin Invest 2014, 124 (5), 1945–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Grinan-Ferre C; Marsal-Garcia L; Bellver-Sanchis A; Kondengaden SM; Turga RC; Vazquez S; Pallas M Pharmacological inhibition of G9a/GLP restores cognition and reduces oxidative stress, neuroinflammation and beta-amyloid plaques in an early-onset Alzheimer’s disease mouse model. Aging-Us 2019, 11 (23), 11591–11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zheng Y; Liu A; Wang ZJ; Cao Q; Wang W; Lin L; Ma K; Zhang F; Wei J; Matas E; Cheng J; Chen GJ; Wang X; Yan Z Inhibition of EHMT1/2 rescues synaptic and cognitive functions for Alzheimer’s disease. Brain 2019, 142 (3), 787–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Krivega I; Byrnes C; de Vasconcellos JF; Lee YT; Kaushal M; Dean A; Miller JL Inhibition of G9a methyltransferase stimulates fetal hemoglobin production by facilitating LCR/γ-globin looping. Blood 2015, 126 (5), 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kim Y; Lee HM; Xiong Y; Sciaky N; Hulbert SW; Cao XY; Everitt JI; Jin J; Roth BL; Jiang YH Targeting the histone methyltransferase G9a activates imprinted genes and improves survival of a mouse model of Prader-Willi syndrome. Nat. Med. 2017, 23 (2), 213–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Chang YQ; Ganesh T; Horton JR; Spannhoff A; Liu J; Sun AM; Zhang X; Bedford MT; Shinkai YC; Snyder JP; Cheng XD Adding a lysine mimic in the design of potent inhibitors of histone lysine methyltransferases. J. Mol. Biol. 2010, 400 (1), 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Vedadi M; Barsyte-Lovejoy D; Liu F; Rival-Gervier S; Allali-Hassani A; Labrie V; Wigle TJ; DiMaggio PA; Wasney GA; Siarheyeva A; Dong AP; Tempel W; Wang SC; Chen X; Chau I; Mangano TJ; Huang XP; Simpson CD; Pattenden SG; Norris JL; Kireev DB; Tripathy A; Edwards A; Roth BL; Janzen WP; Garcia BA; Petronis A; Ellis J; Brown PJ; Frye SV; Arrowsmith CH; Jin J A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol. 2011, 7 (8), 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Liu F; Barsyte-Lovejoy D; Allali-Hassani A; He YL; Herold JM; Chen X; Yates CM; Frye SV; Brown PJ; Huang J; Vedadi M; Arrowsmith CH; Jin J Optimization of cellular activity of G9a inhibitors 7-aminoalkoxy-quinazolines. J. Med. Chem. 2011, 54 (17), 6139–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Liu F; Barsyte-Lovejoy D; Li FL; Xiong Y; Korboukh V; Huang XP; Allali-Hassani A; Janzen WP; Roth BL; Frye SV; Arrowsmith CH; Brown PJ; Vedadi M; Jin J Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J. Med. Chem. 2013, 56 (21), 8931–8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sweis RF; Pliushchev M; Brown PJ; Guo J; Li FL; Maag D; Petros AM; Soni NB; Tse C; Vedadi M; Michaelides MR; Chiang GG; Pappano WN Discovery and development of potent and selective inhibitors of histone methyltransferase G9a. ACS Med. Chem. Lett. 2014, 5 (2), 205–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Milite C; Feoli A; Horton JR; Rescigno D; Cipriano A; Pisapia V; Viviano M; Pepe G; Amendola G; Novellino E; Cosconati S; Cheng XD; Castellano S; Sbardella G Discovery of a novel chemotype of histone lysine methyltransferase EHMT1/2 (GLP/G9a) inhibitors: rational design, synthesis, biological evaluation, and co-crystal structure. J. Med. Chem. 2019, 62 (5), 2666–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Liu F; Chen X; Allali-Hassani A; Quinn AM; Wasney GA; Dong A; Barsyte D; Kozieradzki I; Senisterra G; Chau I; Siarheyeva A; Kireev DB; Jadhav A; Herold JM; Frye SV; Arrowsmith CH; Brown PJ; Simeonov A; Vedadi M; Jin J Discovery of a 2,4-Diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase G9a. J. Med. Chem. 2009, 52 (24), 7950–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Liu F; Chen X; Allali-Hassani A; Quinn AM; Wigle TJ; Wasney GA; Dong AP; Senisterra G; Chau I; Siarheyeva A; Norris JL; Kireev DB; Jadhav A; Herold JM; Janzen WP; Arrowsmith CH; Frye SV; Brown PJ; Simeonov A; Vedadi M; Jin JA Protein lysine methyltransferase G9a inhibitors: design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J. Med. Chem. 2010, 53 (15), 5844–5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Kubicek S; O’Sullivan RJ; August EM; Hickey ER; Zhang Q; Teodoro ML; Rea S; Mechtler K; Kowalski JA; Homon CA; Kelly TA; Jenuwein T Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol. Cell 2007, 25 (3), 473–481. [DOI] [PubMed] [Google Scholar]
- (53).Katayama K; Ishii K; Tsuda E; Yotsumoto K; Hiramoto K; Suzuki M; Yasumatsu I; Igarashi W; Torihata M; Ishiyama T; Katagiri T Discovery of novel histone lysine methyltransferase G9a/GLP (EHMT2/1) inhibitors: design, synthesis, and structure-activity relationships of 2,4-diamino-6-methylpyrimidines. Bioorg. Med. Chem. Lett. 2020, 30 (20), 127475. [DOI] [PubMed] [Google Scholar]
- (54).Katayama K; Ishii K; Terashima H; Tsuda E; Suzuki M; Yotsumoto K; Hiramoto K; Yasumatsu I; Torihata M; Ishiyama T; Muto T; Katagiri T Discovery of DS79932728: a potent, orally available G9a/GLP inhibitor for treating beta-thalassemia and sickle cell disease. ACS Med. Chem. Lett. 2021, 12 (1), 121–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Leenders R; Zijlmans R; van Bree B; van de Sande M; Trivarelli F; Damen E; Wegert A; Muller D; Ehlert JE; Feger D; Heidemann-Dinger C; Kubbutat M; Schachtele C; Lenstra DC; Mecinovic J; Muller G Novel SAR for quinazoline inhibitors of EHMT1 and EHMT2. Bioorg. Med. Chem. Lett. 2019, 29 (17), 2516–2524. [DOI] [PubMed] [Google Scholar]
- (56).San Jose-Eneriz ES; Agirre X; Rabal O; Vilas-Zornoza A; Sanchez-Arias JA; Miranda E; Ugarte A; Roa S; Paiva B; de Mendoza AEH; Alvarez RM; Casares N; Segura V; Martin-Subero JI; Ogi FX; Soule P; Santiveri CM; Campos-Olivas R; Castellano G; de Barrena MGF; Rodriguez-Madoz JR; Garcia-Barchino MJ; Lasarte JJ; Avila MA; Martinez-Climent JA; Oyarzabal J; Prosper F Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies. Nat. Commun. 2017, 8, 15424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Xiong Y; Li FL; Babault N; Dong AP; Zeng H; Wu H; Chen X; Arrowsmith CH; Brown PJ; Liu J; Vedadi M; Jin J Discovery of potent and selective inhibitors for G9a-like protein (GLP) lysine methyltransferase. J. Med. Chem. 2017, 60 (5), 1876–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Xiong Y; Li FL; Babault N; Wu H; Dong AP; Zeng H; Chen X; Arrowsmith CH; Brown PJ; Liu J; Vedadi M; Jin J Structure-activity relationship studies of G9a-like protein (GLP) inhibitors. Bioorgan Med. Chem. 2017, 25 (16), 4414–4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Zhang Y; Rong DQ; Li BB; Wang YX Targeting epigenetic regulators with covalent small-molecule inhibitors. J. Med. Chem. 2021, 64 (12), 7900–7925. [DOI] [PubMed] [Google Scholar]
- (60).Shen YD; Li FL; Szewczyk MM; Halabelian L; Park KS; Chau I; Dong AP; Zeng H; Chen H; Meng FY; Barsyte-Lovejoy D; Arrowsmith CH; Brown PJ; Liu J; Vedadi M; Jin J Discovery of a first-in-class protein arginine methyltransferase 6 (PRMT6) covalent inhibitor. J. Med. Chem. 2020, 63 (10), 5477–5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Butler KV; Ma AQ; Yu WY; Li FL; Tempel W; Babault N; Pittella-Silva F; Shao J; Wang JY; Luo MK; Vedadi M; Brown PJ; Arrowsmith CH; Jin J Structure-based design of a covalent inhibitor of the set domain-containing protein 8 (SETD8) lysine methyltransferase. J. Med. Chem. 2016, 59 (21), 9881–9889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Srimongkolpithak N; Sundriyal S; Li FL; Vedadi M; Fuchter MJ Identification of 2,4-diamino-6,7-dimethoxyquinoline derivatives as G9a inhibitors. Medchemcomm 2014, 5 (12), 1821–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Collazo E; Couture JF; Bulfer S; Trievel RC A coupled fluorescent assay for histone methyltransferases. Anal. Biochem. 2005, 342 (1), 86–92. [DOI] [PubMed] [Google Scholar]
- (64).Andersen J; Madsen U; Bjorkling F; Liang X Rapid synthesis of aryl azides from aryl halides under mild conditions. Synlett 2005, 2005 (14), 2209–2213. [Google Scholar]
- (65).Zhang YL; Gao HL; Liu RJ; Liu J; Chen L; Li XB; Zhao LJ; Wang W; Li BL Quinazoline-1-deoxynojirimycin hybrids as high active dual inhibitors of EGFR and alpha-glucosidase. Bioorg. Med. Chem. Lett. 2017, 27 (18), 4309–4313. [DOI] [PubMed] [Google Scholar]
- (66).Luhr S; Holz J; Zayas O; Seidelmann O; Domke L; Borner A Synthesis of enantiopure beta(2)-homoalanine derivatives via rhodium catalyzed asymmetric hydrogenation. Tetrahedron-Asymmetr 2013, 24 (7), 395–401. [Google Scholar]
- (67).Liu XG; Wang YY; Zhang X; Gao ZY; Zhang SP; Shi PZ; Zhang X; Song L; Hendrickson H; Zhou DH; Zheng GR Senolytic activity of piperlongumine analogues: Synthesis and biological evaluation. Bioorgan Med. Chem. 2018, 26 (14), 3925–3938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Rabal O; San Jose-Eneriz E; Agirre X; Sanchez-Arias JA; Vilas-Zornoza A; Ugarte A; de Miguel I; Miranda E; Garate L; Fraga M; Santamarina P; Fernandez Perez R; Ordonez R; Saez E; Roa S; Garcia-Barchino MJ; Martinez-Climent JA; Liu YY; Wu W; Xu MS; Prosper F; Oyarzabal J Discovery of reversible DNA methyltransferase and lysine methyltransferase G9a inhibitors with antitumoral in vivo efficacy. J. Med. Chem. 2018, 61 (15), 6518–6545. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.