

Abstract
Introduction:
Metformin is currently first line therapy for type 2 diabetes (T2D). The mechanism of action of metformin involves activation of AMP-activated protein kinase (AMPK) to enhance mitochondrial function (for example, biogenesis, refurbishment and dynamics) and autophagy. Many neurodegenerative diseases of the central and peripheral nervous systems arise from metabolic failure and toxic protein aggregation where activated AMPK could prove protective.
Areas covered:
The authors review literature on metformin treatment in Parkinson’s disease, Huntington’s disease and other neurological diseases of the CNS along with neuroprotective effects of AMPK activation and suppression of the mammalian target of rapamycin (mTOR) pathway on peripheral neuropathy and neuropathic pain. The authors compare the efficacy of metformin with the actions of resveratrol.
Expert opinion:
Metformin, through activation of AMPK and autophagy, can enhance neuronal bioenergetics, promote nerve repair and reduce toxic protein aggregates in neurological diseases. A long history of safe use in humans should encourage development of metformin and other AMPK activators in preclinical and clinical research. Future studies in animal models of neurological disease should strive to further dissect in a mechanistic manner the pathways downstream from metformin-dependent AMPK activation, and to further investigate mTOR dependent and independent signaling pathways driving neuroprotection.
Keywords: AMP-activated protein kinase (AMPK), autophagy, axon regeneration, diabetes, mitochondria, mTOR, neurodegenerative disease, neuropathic pain, neuropathy, PGC-1α
1. Metformin and management of type 2 diabetes mellitus
Metformin has become one of the most frequently prescribed drugs worldwide [1] due to the high prevalence of type 2 diabetes (T2D), an expanding scope of use and favorable safety profile. Although medical management of T2D has progressed rapidly in recent years, metformin remains a cornerstone of anti-hyperglycemic regimens. First marketed for T2D in 1957, metformin has commanded significant market share in the ensuing decades and is a mainstay of treatment, with current prescription rates of 77% in T2D patients [2]. With the prevalence of diabetes quadrupling over the past 3 decades, use of metformin has experienced similar growth [3].
Depending on presenting glycated hemoglobin (HbA1c) and fasting plasma glucose levels, metformin may or may not be prescribed at the time of diagnosis of T2D. Lifestyle modifications, generally consisting of improved low-calorie diet and regular physical activity, are also routinely recommended by Diabetes Canada [4] and have been adopted by major diabetes associations worldwide [5,6]. Metformin is inexpensive and usually well tolerated with few contraindications, negligible weight gain, and rarely induces hypoglycemia. The side effect profile is generally favorable, with short lived diarrhea, nausea, and abdominal pain upon initiation of metformin treatment often mitigated by slowing dose titration. Chronic use has been associated with vitamin B12 deficiency [7]. One serious but rare side effect of metformin use is precipitation of a metabolic lactic acidosis, a high mortality acid-base disturbance related to lactic acid overproduction and reduced excretion [8]. Risk factors for lactic acidosis are advanced age, renal failure, and liver failure. Renal failure is of particular concern as metformin is cleared via the kidneys [9]. Poor renal function is therefore a potential contraindication to metformin use and must be monitored at regular intervals to evaluate for development and progression of diabetic nephropathy. Determination of estimated glomerular filtration rate (eGFR) has become the standard of practice for approximating renal function in patients on metformin [10,11]. Prescriber caution is advised at an eGFR between 30–59 mL/min/1.73 m2 (CKD Stage 3); an eGFR below 30 mL/min/1.73 m2 (CKD Stage 4–5) is an absolute contraindication to metformin use due to increased likelihood of inducing lactic acidosis.
Metformin has not been approved for prevention of T2D by the FDA. However, several randomized control trials (RCTs) have investigated its efficacy in preventing both the development of T2D in high risk groups and the development of micro- and macrovascular complications of T2D [12]. In the Diabetes Prevention Program Research Group RCT, a standard dose of metformin (850 mg BID) was paired with basic lifestyle modifications in those at high risk for developing T2D [13]. After 3 years, the metformin treatment arm had a 31% lower incidence of T2D than the placebo group but was not as effective as the intensive lifestyle modification treatment arm which saw a 58% lower incidence than placebo. The efficacy of metformin was greatest in those with higher BMI and fasting plasma glucose values. Reductions in mortality and vascular complications of T2D have also been observed with metformin use [14]. Compared to lifestyle modifications and insulin and sulfonylurea therapy, metformin achieved a 32% risk reduction in all diabetes-related micro- and macrovascular complications. Similarly, a significant reduction in all-cause mortality was observed in the metformin treatment group compared to the insulin and sulfonylurea cohort. Long-term continuation of benefit was observed after 10 years of treatment [15].
Use of metformin in special populations varies by jurisdiction [16]. In one study, metformin was shown to be an effective means of glycemic control in pediatric patients aged 8–16 with reductions in HbA1c of 1.4% [17]. Fasting plasma glucose values also saw significant reduction over placebo. Adverse events were similar to those experienced in adult populations. In older adults, aggressive glycemic control increased mortality with little effect on cardiovascular events [18]. While no RCT has determined the efficacy of metformin in older adults, it is routinely used in clinical practice for glycemic control and cancer risk reduction [19].
2. Mechanism of action of metformin
The glucose-lowering effect of metformin is believed to be due to its ability to inhibit hepatic gluconeogenesis while also improving glucose uptake in peripheral tissues [20–23]. Metformin reduces glycated hemoglobin and fasting plasma glucose while inducing mild weight loss [22]. The specific biochemical mechanism of action of metformin has eluded researchers for decades as there continues to be disagreement and an absence of definitive proof. As evidenced by its large volume of distribution, metformin penetrates multiple tissue types throughout the body [24]. Particular significance is attributed to the action of metformin in hepatocytes, adipocytes, enterocytes, and neurons. Regardless of cell type, metformin activates the heterotrimer AMP-activated protein kinase (AMPK), a key regulator of cellular metabolism activated by rising intracellular AMP:ATP ratios [25]. This energy sensing enzyme controls activation of a number of pathways designed to enhance mitochondrial function, including the transcriptional coactivator peroxisome proliferator-activated receptor-λ coactivator 1-α (PGC-1α) and fatty acid oxidation (β-oxidation). AMPK is considered a master regulator of cellular bioenergetics and the numerous signaling cascades that control mitochondrial biogenesis, refurbishment and function have been reviewed in detail elsewhere [25–28]. Inhibition of Complex I of the respiratory chain is also believed to be a key step in metformin action [29] and putative effects on mitochondrial function stemming from this are currently under investigation [30]. See Box 1 and Figure 1 for an overview of the AMPK pathway and impact of metformin.
Box 1. The AMPK/PGC-1α signaling axis as a sentinel modulating the metabolic needs of the cell.
AMPK, through its ability to sense the AMP:ATP ratio, adjusts cellular metabolic output toward a more catabolic phenotype. Under conditions of low ATP, as triggered by metformin, the enzyme is activated via phosphorylation at T172 and mediates phosphorylation of PGC-1α on residues T177 and S538 [31]. This pathway is supplemented through the sirtuin-dependent sensing of redox state where, under conditions of high NAD+/NADH, there is elevated deacetylation and further activation of PGC-1α. Within the nucleus, PGC-1α interacts with a variety of transcription factors to modulate key metabolic pathways. Under low ATP a primary role of AMPK/PGC-1α activation is to drive mitochondrial biogenesis, dynamics (mainly augmented mitochondrial fusion via mitofusin 2 [32]) and refurbishment via raising mitophagy to enhance electron transport and oxidative phosphorylation. In parallel, fatty acid oxidation is elevated as an alternative and high capacity supply of ATP. Metformin also inhibits gluconeogenesis through activation of AMPK and the HNF4α and FOXO1 axis, but this effect is further supported by lowered ATP directly inhibiting enzymes of the gluconeogenic pathway [29]. Finally, PGC-1α acts as a negative regulator of lipogenesis. In the context of diabetes, especially in the liver and skeletal muscle, the blockade of gluconeogenesis and suppression of lipogenesis are key aspects of metformin’s ability to lower systemic hyperglycemia and free fatty acid levels. AMPK can also directly and indirectly modulate autophagy (discussed later in the context of mammalian target of rapamycin (mTOR) signaling).
Figure 1. Metformin-dependent activation of AMPK and downstream targets.
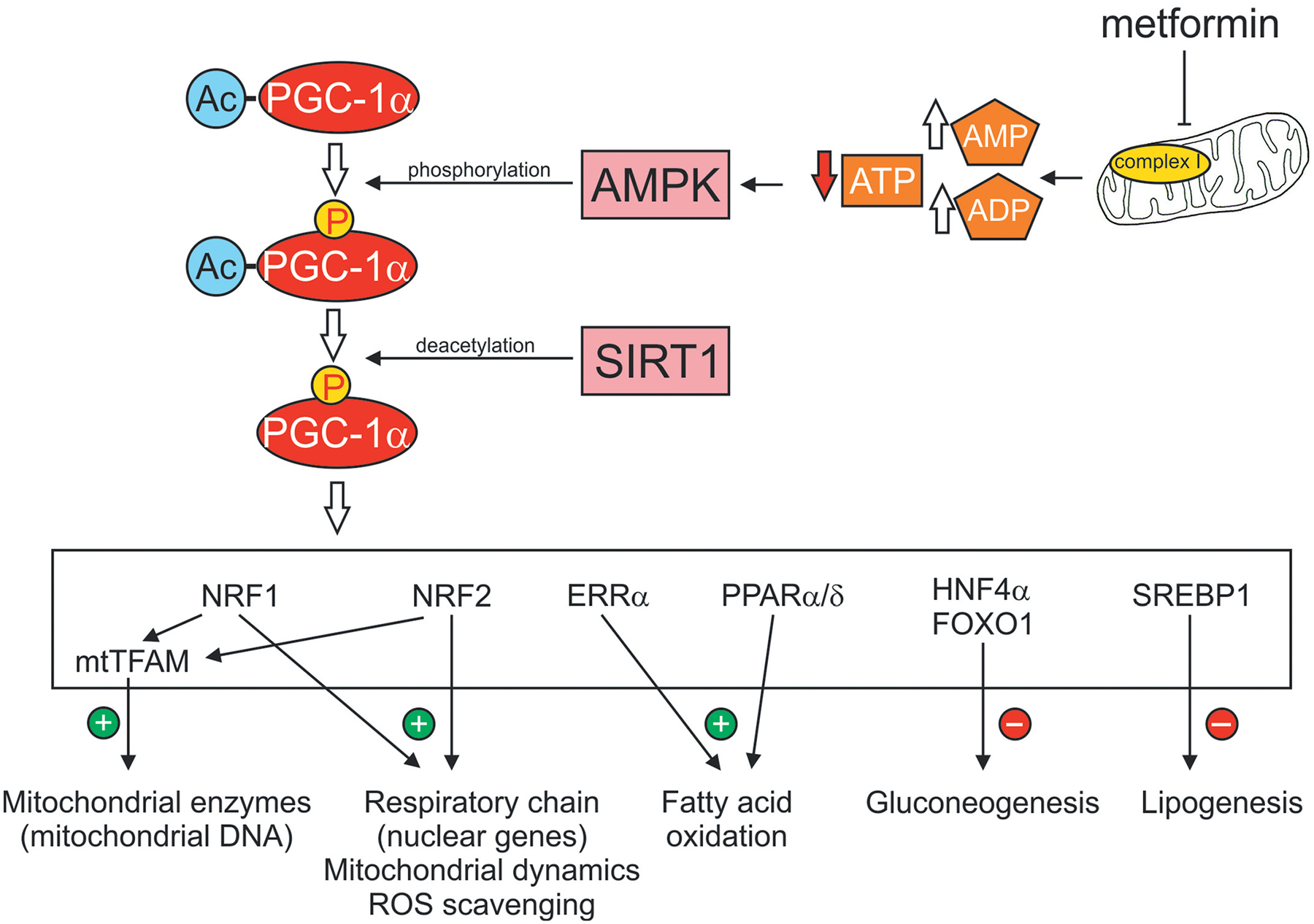
Metformin interacts with components of the mitochondria, including the respiratory chain at Complex 1, to inhibit electron flow and oxidative phosphorylation. The increase in ratio of AMP:ATP activates AMPK and leads to elevated phosphorylation of the co-transcriptional activator, PGC-1α. SIRT1, a sensor of the NAD+/NADH redox state, further enhances PGC-1α activity via deacetylation. Activated PGC-1α then can interact with a variety of transcription factors (shown in the white box) to modulate a variety of signal transduction pathways associated with mitochondrial function and cellular metabolism. Abbreviations: Ac, acetylation; AMPK, AMP-activated protein kinase; FOXO1, forkhead box O 1; ERRα, estrogen receptor-relatedα; HNF4α, hepatocyte nuclear factor 4α PGC-1α, peroxisome proliferator-activated receptor λ coactivator 1-α; PPAR α/δ, peroxisome proliferator-activated receptor α/δ; NRF1, nuclear respiratory factor 1; NRF2, nuclear factor erythroid 2-related factor; SIRT1, sirtuin1 or silent mating type information regulation 2 homolog; SREBP1, sterol regulatory element-binding protein 1; TFAM, mitochondrial transcription factor A.
2.1. Metformin signaling in hepatocytes
AMPK phosphorylation and activation by metformin was first demonstrated in rat hepatocytes [33]. Metformin induces the phosphorylation and activation of AMPK at T172 in both isoforms of the catalytic α-subunit. Inhibition of acetyl-CoA carboxylase (ACC) and increased fatty acid oxidation were identified as downstream effects. Liver kinase B1 (LKB1), the upstream kinase of AMPK, was implicated as LKB1 knockout mice were resistant to AMPK activation and did not exhibit reduced serum glucose levels following metformin treatment [34]. However, the LKB1-AMPK pathway may not be absolutely required to lower hepatic glucose production [35]. Using mice with defective α1/2 AMPK subunits, metformin decreased intracellular ATP concentration in primary hepatocytes as well as causing an associated drop in hepatic glucose production. Increased levels of AMP, an allosteric activator of AMPK, were also detected.
Other effects of metformin on mitochondrial respiratory function have also been observed. Mitochondrial respiratory chain complex 1 (NADH:ubiquinone oxidoreductase) was inhibited in primary rat hepatocytes by metformin treatment [36]. Metformin also prevented glutamate and malate oxidation in a dose and time dependent manner, further suggesting inhibition of complex 1. This effect required intact cells and did not occur in isolated mitochondria, suggesting that metformin did not directly inhibit complex 1 but operated indirectly [37]. More recent research has shown that isolated mitochondrial complexes can be weakly and reversibly inhibited by metformin, most likely by acting on conserved core regions of the enzyme [38]. Complex 3 of the respiratory chain and ATP synthase were also inhibited by metformin. The redox state of mitochondria, as determined by NADH:NAD+ ratios in hepatocytes, responded to metformin treatment in a manner similar to that of rotenone, a known inhibitor of complex 1 [39].
2.2. Metformin signaling in other non-neuronal cell types
In enterocytes, metformin increased glucose uptake and utilization, potentially through the activation of AMPK [40,41] which then induced translocation of glucose transporters (GLUT2) to the apical membrane [42]. In skeletal muscle, AMPK activation by metformin led to an increase in glucose uptake, a process that was prevented with AMPK siRNA knockdown [33,43]. Both brown and white adipose tissue responded to metformin treatment by activating AMPK in a dose-dependent manner without affecting the total amount of AMPK protein [23]. Inactivation of ACC also occurred in adipose tissue following metformin treatment. AMPK activation was observed in both human and mouse adipose tissue [44]. Decreases in lipogenic gene expression and lipid droplet formation were also observed [45]. Metformin exhibited mitochondrial effects in brown adipose tissue, inducing mitochondrial biogenesis and thermogenesis [46]. Similarly, metformin accumulated in brown adipose tissue and reduced oxygen consumption rate, suggesting that metformin acted negatively on mitochondrial function [47].
2.3. Metformin signaling in neurons
Activation of AMPK occurs in a manner similar to that of other cell types [48,49]. Phosphorylation and inactivation of the downstream effector ACC has also been observed in human neural stem cells and rat dorsal root ganglia (DRG) after metformin treatment [50,51]. Interestingly, the intracellular action of metformin may differ between different types of neurons as a decrease in AMPK activation has been reported in rat hypothalamic neurons following metformin treatment [52]. Selective inhibition of respiratory chain complex 1 also occurs in cultured embryonic cortical neurons after metformin exposure [53].
2.4. Does resveratrol mimic metformin action?
Resveratrol is a naturally occurring polyphenol found in high concentrations in peanuts, pomegranate and the skin of grapes [54]. Resveratrol has been widely used as a natural medicine and dietary supplement and is attributed with a variety of biochemical and metabolic properties. Resveratrol significantly improved the longevity and morbidity of mice fed a high calorie diet along with improving insulin sensitivity and decreasing hepatic lipid accumulation [55]. Decreased severity of cardiovascular disease [56] and improved sensory neuron function in diabetic mice [57] have also been reported.
Like metformin, the mechanism of resveratrol involves the phosphorylation and activation of AMPK [58]. Downstream activity on metabolic effectors such as ACC also occurs [59]. Interestingly, changes to cellular ATP levels are not always involved in AMPK activation as AMP:ATP ratios remain largely unchanged after resveratrol treatment [58,60]. AMPK activity was dependent on the presence and activity of SIRT1, a prominent deacetylase with genetic, metabolic, and glucose homeostatic functions [61,62]. SIRT1 knockout prevented the resveratrol-induced increase of mitochondrial membrane potential and ATP levels in primary myoblasts. Similarly, SIRT1 inhibition with nicotinamide prevented glucose uptake and AMPK activation after resveratrol treatment.
3. Rationale for using metformin to treat neurological disorders
There is growing interest in the potential therapeutic use of metformin in diseases of the central nervous system. Most neurodegenerative diseases are heterogeneous in nature and it is becoming recognized that many share pathological mechanisms of neuronal and support cell damage that parallel stress pathways in other diseases, for example in type 1 diabetes (T1D) and T2D [63].
One example of a pathogenic mechanism common to multiple disorders is the mTOR signaling pathway. mTOR signaling contributes to a collective of brain functions including maintenance of neural stem cells and behaviors such as sleeping and eating [64]. Disruption of mTOR signaling, a characteristic of T2D, also occurs in neurodegenerative and neuropsychiatric disorders such as tuberous sclerosis complex, hamartoma tumor syndrome, neurofibromatosis, autism spectrum disorder (ASD), Fragile-X syndrome, epilepsy, Parkinson’s disease, Huntington’s disease, major depressive disorder and schizophrenia [63]. Notably, metformin inhibited the mechanistic target of mTOR signaling, rapamycin complex 1 (mTORC1), via AMPK-dependent as well as AMPK-independent pathways. This supports the idea that metformin has the potential to influence a variety of neuropsychiatric and neurodegenerative diseases due to common mechanistic defects shared with T2D [64]. See Box 2 and Figure 2 for a summary of the interactions between AMPK and the mTOR pathway and regulation of cell growth and metabolism.
Box 2. AMPK and the mTOR pathway interact to provide antagonistic and exquisite regulation of cellular growth and metabolism.
Key degenerative processes that contribute to neurological disease include accumulation of toxic proteins and mitochondrial dysfunction with attendant energy loss. For example, the accumulation of toxic protein aggregates in Parkinson’s disease (Lewy bodies, synuclein) and Huntington’s disease (mutant huntingtin-induced protein accumulation) have generated great interest as drivers of catabolic processes to clear cells of these damaging intracellular entities. Activators of autophagy such as AMPK, via novel compounds or via resveratrol or metformin, are a major focus. AMPK also drives synthesis of components of the respiratory complexes to elevate mitochondrial function and thus overcome energy deficits seen in many neurodegenerative diseases. Distal fiber loss in PNS disease and synaptic pruning in CNS disease are early features of nerve damage and require therapeutic approaches that drive axonal/dendrite regeneration and building of new pre- and post-synaptic densities. The key role of mTOR pathway in driving neuronal growth was first demonstrated through studies ablating the upstream negative regulator of mTOR, phosphatase and tensin homolog (PTEN). In both adult retinal ganglion and sensory neurons, PTEN knockout resulted in enhanced axonal survival and regeneration [65,66]. However, to achieve neuronal protection in the CNS and PNS a fine balance is required between activation of AMPK and generation of its catabolic effects that might act antagonistically to the known beneficial effects of the anabolic processes linked to mTOR activation. An important component of this multifaceted interaction is the metformin induced AMPK-dependent optimization of mitochondrial function that can enhance efficiency of ATP production in the neuron and support cells to maintain or regenerate nerve structure and oppose metabolic failure. Thus, complex regulatory pathways must balance growth factor or hormone signaling via Akt and activation of the mTOR pathway with the need to maintain sufficient activation of AMPK to support mitochondrial function. An example is the ability of IGF-1 to activate both Akt and AMPK to drive neurite outgrowth and mitochondrial function [67]. Metformin therapy allows endogenous pathways, such as neurotrophin signaling, to continue activating PI 3-K/Akt and the mTOR pathway while stimulating AMPK to augment mitochondrial function and autophagic processes. The nature and maintenance of this complex and inter-dependent signaling repertoire will vary dependent on cell and disease context and is discussed by Koley et al [68].
Figure 2. AMPK regulation of the mTOR pathway and control of cell growth and autophagy.
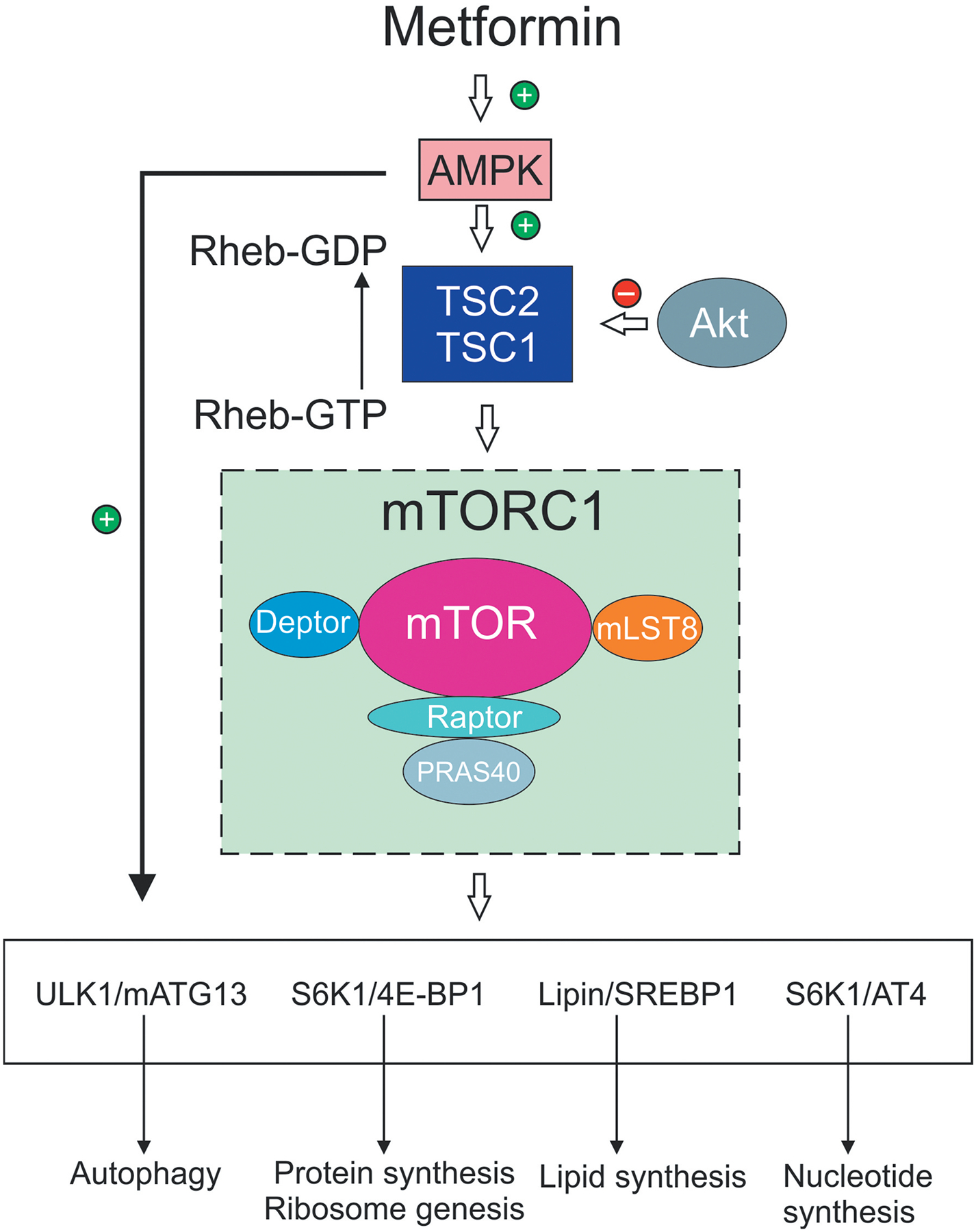
AMPK and signaling via PI 3-K/Akt, for example by growth factors, act as antagonistic modulators of the mTOR pathway to control cell phenotype. AMPK activates the TSC1/TSC2 complex to drive conversion of Rheb-GTP, an activator of the mTORC1 complex, to Rheb-GDF. This conversion effectively inhibits mTORC1 complex activity (which includes mTOR and associated proteins) and thus suppresses mTOR pathway function. Akt is inhibitory to TSC1/TSC2 thus suppressing formation of Rheb-GDP and enhancing mTORC1 activation. The mTOR pathway when active drives protein synthesis (including ribosome biogenesis), nucleotide and lipid synthesis. mTOR activation is a negative regulator of autophagy and acts antagonistically to AMPK, which also acts as a direct activator of autophagy. The mobilization of these anabolic pathways augmented cell growth and proliferation under conditions of sufficient energy and metabolite supply. AMPK activation, via detection of lowered energy and metabolite supply, suppresses these activities toward a more catabolic phenotype. Abbreviations: 4E-BP1, translation initiation factor 4E-binding protein 1; Akt, protein kinase B; AT4, activating transcription factor 4; ATG13, autophagy-related protein 13; mTOR, mammalian target of rapamycin; mTORC1, mTOR complex 1; PI 3-K, phosphoinositide 3-kinase; Rheb, Ras homolog enriched in brain; S6K1, ribosomal protein S6 kinase 1; TSC1 and 2, tuberous sclerosis 1 and 2; ULK1, Unc-51-like autophagy activating kinase 1.
Other disorders common to both T2D and many neurodegenerative processes that may respond to metformin include insulin resistance and mitochondrial dysfunction [69]. Metformin improves glucose metabolism and hence may be able to alleviate insulin resistance. This could be of particular value given that the CNS is subject to an energetically impoverished environment in many neurodegenerative diseases. One factor driving this energy loss in the CNS is mitochondrial dysfunction. The brain has a high energy demand to mass ratio and neurons are exquisitely sensitive to changing energy demands. A common feature of mitochondrial dysfunction in neurological disease is loss of AMPK signaling. Therefore, metformin-dependent activation of the AMPK pathway may provide the rationale for repurposing the drug to treat neurodegenerative diseases [69].
Our main focus is to provide a synthesis of the literature with regards to effects of metformin in the CNS compared to the PNS, highlighting metformin-induced activation of the AMPK signal transduction pathway and its protective properties against neurodegenerative disease. Below we briefly discuss the influence of metformin on a specific set of CNS neurological disorders, since this area has recently been covered extensively elsewhere [63,69–71]. Our focus on AMPK leads to exclusion of Alzheimer’s disease as only a limited number of metformin studies have been performed in AD and the literature is sparse on pathways activated by metformin. We refer the reader to [72] for an overview of metformin and AD.
3.1. Parkinson’s disease
Parkinson’s Disease (PD) is the second most prevalent neurodegenerative disease following Alzheimer’s disease [73]. The main features of PD are accumulation of Lewy Bodies composed primarily of α-synuclein (α-syn), loss of dopaminergic (DA) neurons in the substantia nigra pars compacta and various motor symptoms [74]. Meta-analysis of human genome-wide association studies (GWAS) in PD patients identified down-regulated expression of genes downstream of PGC-1α that are associated with mitochondrial electron transport, glucose utilization and glucose sensing [75]. Furthermore, in complementary animal studies, death of DA neurons induced by over-expression of α-syn was prevented by up-regulation of PGC-1α [75]. Subsequent preclinical studies have reported that treatment with metformin had a beneficial effect on PD-like symptoms, reduced degeneration of substantia nigra pars compacta DA neurons, ameliorated loss of cell viability, increased dopamine levels and improved motor functions in neurotoxin, 1-methyl-4-phenyl-1,2,3,6-tetrehydropyridine (MPTP) rodent models of PD [76–79].
Progression of PD has been attributed to high levels of reactive oxygen species (ROS) and increased oxidative stress driving dopaminergic neuronal loss [80]. Metformin treatment lowered oxidative stress and increased expression of the antioxidant enzymes superoxide dismutase and catalase in MPTP-induced mice [78]. Interestingly, in transgenic mice with conditional knockout of AMPK α1 and α2 subunits in DA cells, metformin treatment for 3 weeks continued to show neuroprotective effects, implying targets other than AMPK for its site of action [79]. There have been numerous documentations of metformin exhibiting positive effects on patients with PD. The underlying mechanisms through which this occurs remain elusive and contributions from mechanistic pathways independent of AMPK, such as brain-derived neurotrophic factor (BDNF) signaling, should not be overlooked. For example, blocking trkB, the tyrosine kinase receptor for BDNF, resulted in the loss of neuroprotective effects of metformin in MPTP-induced mice. Furthermore, BDNF signaling was increased in MPTP PD-induced mice treated with metformin [77,78].
PD has also been linked to nonfunctional HTRA2 and PINK1 in humans and animals [81]. It is suggested that TRAP1 may be a downstream effector of HTRA2 and PINK1, and TRAP1 over-expression could remedy HTRA2 and PINK1 induced mitochondrial dysfunction in human cells [81]. Metformin recovered the impaired mitochondrial membrane potential induced by the Hsp90 family/TRAP1 inhibitor 17-AAG [81]. Moreover, metformin suppressed downstream events of the mitochondrial unfolded protein response such as elevated turnover of mitochondria [81]. Finally, metformin has proven beneficial in minimizing the long-term side effects of L-DOPA treatment, such as dyskinesias, in animal models of PD, without interfering with the positive therapeutic effect of L-DOPA [82].
3.2. Huntington’s disease
Huntington’s disease (HD) is a hereditary disorder that results in progressive cognitive decline, psychiatric phenotypes and chorea [83]. This neurodegenerative disease involves autosomal dominant inheritance of the defective huntingtin (htt) gene [69]. When the mutated htt gene has more than 36 CAG repeats it translates with an extended polyglutamine tract, rendering the protein unstable [83]. Instability of the extended homopolymer leads to misfolding of the mutant huntingtin protein (mHtt) that triggers and overloads the ubiquitin-proteasomal degradation system [84]. mHtt forms toxic aggregates with itself and other proteins, leading to depletion of several molecules and neuronal degeneration, primarily in the striatum [85,86].
Studies have been performed in transgenic Caenorhabditis elegans models of HD with an 88 or 128 polyglutamine expansion at the N-terminal of Htt [87]. Following systemic metformin treatment, the nematodes demonstrated improvements in neuronal function, including improved sensitivity to touch [88]. Furthermore, inclusion of aak-2 (ok524), a loss of function allele that is the worm homolog of AMPKα1, into the 128Q C. elegans model blocked the positive effects of metformin [88].
Male mice carrying the 150 CAG extended mHtt protein exhibit early stages of HD and oral metformin treatment corrected abnormal neuronal function and prevented anxiety behaviors detected prior to onset of overt disease. This was associated with increased neuronal hyperactivity in layers 2 and 3 of the visual cortex in heterozygous Hdh150 mice [89]. Consequently, metformin modulated the MIDI/PP2A/mTOR protein complex to decrease translation of mRNA coding for mHtt protein and depress mHtt load [89]. AMPK activation seems to exert more extensive neuroprotective effects when triggered in early phases of HD progression. Detrimental effects of overactivated AMPK leading to neurodegeneration were investigated in R6/2 mice with 212 ±9 CAG repeats, a model of more advanced HD [90]. The AMPK activator aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) increased mHtt aggregates, worsened motor coordination and increased apoptotic neuronal death in the striatum [90]. Activation of AMPK by metformin may therefore be a treatment option only in early stages of HD when levels of AMPK are low [86,88].
Metformin has shown positive outcomes in human trials. In a cohort study consisting of 4325 HD patients, 121 of them were using metformin for treatment of T2D. Patients using metformin performed significantly better in cognitive and executive functioning tasks compared to HD patients not receiving metformin [86].
3.3. Multiple sclerosis
Multiple sclerosis (MS) is a chronic autoimmune disease in which CD4 T cells, astrocytes and microglia invade the CNS and cause damage via neuroinflammation, demyelination and destruction of neuronal support cells [91,92]. Recent therapeutic approaches for MS include controlling regulatory immune responses and preservation of oligodendrocyte function.
Metformin improved pathological features of MS in a commonly used mouse model of MS induced by the toxin cuprizone [93]. Metformin alleviated signs of oxidative stress in the corpus callosum and mitochondrial dysfunction [94]. PGC-1α expression was downregulated in this MS mouse model and this was also prevented by metformin treatment [93]. The increased expression of PGC-1α gene elicited by metformin treatment was associated with elevated expression of the mitofusin-2 (MFN2) gene, which promotes mitochondrial fusion [93]. Treatment with metformin maintained a significantly higher number of oligodendrocytes and its precursor cells, improved myelination and lowered activated astrocytes and microglia, thus reducing gliosis [93]. Metformin also significantly (p < 0.05) increased the expression of other genes that are critical for proper mitochondrial and metabolic function such as NRF-1 (controls mitochondrial DNA expression), TFAM (activator of mitochondrial transcription) and GAPDH (responsible for regulating bioenergetics) [93]. Metformin activated the AMPK pathway in pertussis toxin (PTX)-induced experimental autoimmune encephalomyelitis (EAE), a mouse model of MS and this was associated with a reduced number of inflammatory lesions and diminished severity of demyelination [95]. In supporting work, metformin increased mRNA transcripts for neuroprotective CNTF, interferon-λ and PDGF in mixed glial cell cultures and this was associated with AMPK activation [95].
3.4. Stroke
Stroke is the 2nd leading cause of death globally [96,97]. In addition to stroke, cardiac dysfunctions commonly lead to cerebral ischemia and reperfusion injuries [98]. Ischemic injuries drive neuron and support cell damage and can involve mitochondrial deficits, oxidative stress, disruption of the blood-brain barrier, elevated neuronal apoptosis and neuroinflammation [98]. Not only has metformin been accredited with the ability to reduce the probability of future stroke occurrences, but studies have also suggested that acute and chronic metformin treatment are both effective in repairing cerebral damage post-stroke.
M2-activated microglia promote clearance of cellular debris and heighten the expression of growth factors and anti-inflammatory molecules such as interleukin-10, interleukin-4 and TGF-β [95,98,99]. Activated microglia are polarized into the neuroprotective M2 phenotype in response to ischemic injury but quickly revert back to M1 phenotype [99]. An in vitro study utilized lipopolysaccharide to simulate ischemic injury in microglia and then evaluated the potential of metformin to promote protection [100]. Metformin triggered the lipopolysaccharide injured microglia to up-regulate AMPK and release elevated levels of interleukin-10, an anti-inflammatory cytokine [100]. Metformin induced an M2 polarization state of BV2 microglia through an AMPK-dependent mechanism [100]. In addition, metformin administered for 30 days following middle cerebral artery occlusion to replicate damage and neural recovery signaling succedent to a stroke, improved angiogenesis and neurogenesis [100].
In rats receiving metformin for 2 weeks prior to stroke induction by sudden cardiac arrest, there was a 30% increase in survival after 7 days [101], with significantly higher neurological function in assessments done 24, 48 and 72 hours after return of regular blood flow compared to the vehicle group [101]. Immunohistochemical assessment 7 days after the return of regular blood flow showed a significantly reduced quantity of viable neurons in the vehicle group compared to the metformin pre-treatment group [101]. The increase in neuroprotective effects seen in the pre-treated metformin group were abolished with intracerebroventricular administration of AMPK-inhibitor compound C [101]. AMPK activation in the hippocampus of the metformin treated group was accompanied by an enhanced autophagic response, possibly linked to augmented removal of aberrant cellular protein accumulated due to stress, and may have contributed to functional recovery [101].
3.5. Metformin and epilepsy
Currently there are several anti-epileptic medications available but none address disease progression and all have various side effects [102]. Epilepsy has been repeatedly linked to upregulated mTOR signaling [103]. AMPK protein levels were significantly lower in patients with temporal lobe epilepsy and in animal models with acute and chronic seizures [104]. Activated AMPK is able to block mTOR signaling downstream by phosphorylating mTOR regulatory proteins such as regulatory associated protein of mTOR known as raptor and tuberous sclerosis complex 2 (TSC2), a tumor suppressor also known as tuberin [105]. The concurrent activation of AMPK alongside restriction of mTOR provides promising therapeutic attributes to support seizure termination by metformin.
Treating rats with pilocarpine hydrochloride-induced epilepsy for 5 days with metformin decreased levels of p-mTOR protein and elevated levels of p-AMPK protein along with having anti-epileptic and anti-convulsant effects [106]. It has recently been suggested that mTOR regulation of autophagy may be responsible for the anti-convulsant effects [107]. Metformin treatment also suppressed expression of BDNF and its receptor, trkB, which are hyper-expressed in the epileptic brain and represent a pathological signaling cascade in mesial temporal lobe epilepsy [106,108]. Chronic metformin treatment also reduced the duration and mortality from generalized tonic-clonic seizures in a pentylenetetrazol acute seizure model [104]. Interestingly, metformin contributed to acute seizure termination but did not influence their initiation or severity [104].
4. Diseases of the peripheral nervous system
Distal dying-back or degeneration of nerve fibers is observed in many peripheral neuropathies including diabetic neuropathy, chemotherapy-induced peripheral neuropathy (CIPN), Friedreich’s ataxia, Charcot-Marie-Tooth disease type 2 and human immunodeficiency virus (HIV)-associated distal-symmetric neuropathy. It is becoming increasingly recognized that all of these neuropathies have some degree of mitochondrial dysfunction [109–112]. This is pertinent, as the growth cone motility required to maintain fields of innervation in the constantly changing environment of the epidermis consumes 50% of ATP supplies in neurons due to high rates of actin treadmilling [113]. Maintenance of plastic innervation therefore requires high consumption of ATP [114,115]. Unmyelinated axons are also more energetically demanding than myelinated axons, consuming 2.5–10-fold more energy per action potential [116]. Mitochondria are known to concentrate in regions of high metabolic demand [117] and sensory terminal boutons are packed with mitochondria [118]. Consequently, there is an ongoing focus on the AMPK pathway both as a lesion site and as a target for therapeutic intervention given its key role in modulating cellular bioenergetics. Studies with resveratrol and other AMPK activators indicate a capacity to drive nerve repair in a number of peripheral neuropathies. The efficacy of metformin is less well developed and, in the context of diabetes, is accompanied by concerns that it may exacerbate nerve damage.
4.1. Peripheral nerve regeneration and repair
Peripheral nerves possess endogenous repair systems but with age and concomitant disease it becomes increasingly difficult to recover from injury [119]. There are recent reports that metformin may improve the rate of axon regeneration in the PNS following nerve injury.
4.1.1. Metformin and nerve fiber regeneration
Axon regeneration is an energetically expensive process that requires coordinated mitochondrial function. Antioxidants increase neurite outgrowth and stabilize microtubules [120], which are highly vulnerable to oxidation induced structural instability that fosters axonal degeneration [120,121]. In a recent study, metformin protected the spinal cord from damage by activating an antioxidant pathway involving nuclear factor erythroid 2-related factor (Nrf2) that binds to the antioxidant response element (ARE) to improve mitochondrial function and reduce oxidative stress [122]. This pathway was activated via the PI 3-K/Akt pathway and blocked by inhibitors of PI-3 kinase, with heightened expression of acetylated tubulin and microtubule associated protein providing stabilization of microtubules in axons and integrity of dendrites, respectively [122].
Autophagy clears cellular debris that accumulates at the site of peripheral nerve damage and provides optimal conditions for nerve repair [123]. Metformin induced inhibition of the mTORC1 pathway has been linked to stimulating autophagy [124]. LC3-II is a commonly used marker to assess activity of lysosomal mediated autophagy [119] and expression was upregulated in rats with sciatic nerve injury and autophagy following metformin treatment [119]. Increased autophagy also correlated with reduced cell mortality, enhanced myelination and improved motor function recovery while addition of 3-methyl adenine, an autophagy inhibitor, blocked the positive effects of metformin [119]. Finally, metformin protected cultured Schwann cells from hypoxia-induced stress and up-regulated AMPK and an array of neurotrophic molecules to enhance Schwann cell survival, adhesion and migration [125].
Our own recent studies show that metformin can directly enhance axon regeneration in vitro, independent of putative effects on Schwann cell biology and myelination. Adult DRG neurons isolated from control rats and exposed to 0.3 mM metformin for 24 hours showed a significant elevation in total neurite outgrowth (Figure 3A–E). These structural effects of metformin also corresponded with a significant activation of AMPK over a 1 hour treatment period Figure 3F, G. Analysis of mitochondrial function confirmed that metformin was significantly suppressing electron transport and generating a reduced maximal and spare respiratory capacity Figure 4. The latter parameter is important because it is indicative of a reduced capacity for the cultured neurons exposed to metformin to respond to energetic demands. However, under these culture conditions and time scale it appears that axon regeneration can still be augmented. Thus, metformin was acting as predicted to partially block electron flow within the respiratory chain and activate AMPK [29]. These data support the suggestion that AMPK activators such as metformin or resveratrol could serve as therapies to enhance axon regeneration after peripheral nerve injury.
Figure 3. Metformin augmented neurite outgrowth and activated AMPK in adult sensory neurons.
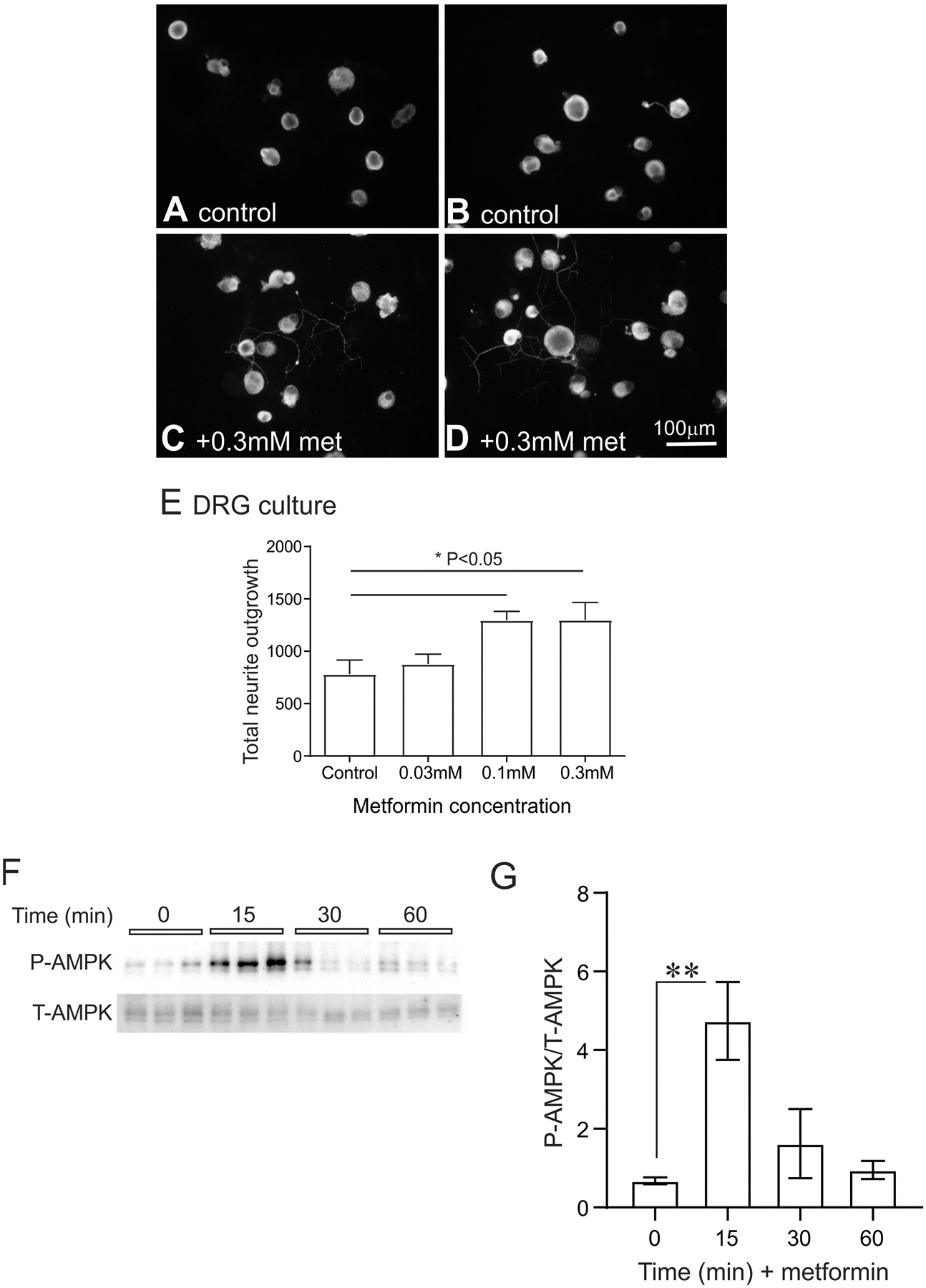
In (A) and (B) are fluorescent images of adult DRG neuron cultures maintained for 24 hours without neurotrophic growth factors. The size marker indicates 100 μm. In both (C) and (D) are fluorescent images of adult rat sensory neurons that were treated with 0.3 mM metformin. In (E) is quantification of total neurite outgrowth in response to a range of metformin doses (0.03 mM, 0.1 mM and 0.3 mM). The pixel density used to quantify neurite outgrowth was adjusted to cell number. See [126] for details on this in vitro culture system. Values are means ± SEM (n = 6 replicate cultures). *P < 0.05 vs control. Statistics were performed by one-way ANOVA with Dunnett’s post hoc test. DRG = dorsal root ganglia; met = metformin. In (F) are presented Western blots derived from total protein samples of DRG cultures exposed to 0.3 mM metformin for 0–60 min and probed for total AMPK (T-AMPK) and phosphorylated AMPK (P-AMPK). (G) reveals the data for P-AMPK normalized to T-AMPK. Values are means ± SEM, n = 3 replicate cultures. *P < 0.01 vs control. Statistics were performed by one-way ANOVA with Dunnett’s post hoc test. Similar effect of metformin was observed when P-AMPK was normalized to total protein on the blot (not shown). We confirm that all ethical standards were followed according to Institutional Animal Care and Use Committee of the University of California, San Diego, and by the University of Manitoba Animal Care Committee following Canadian Council of Animal Care rules.
Figure 4. Metformin inhibits mitochondrial function in cultured adult sensory neurons.
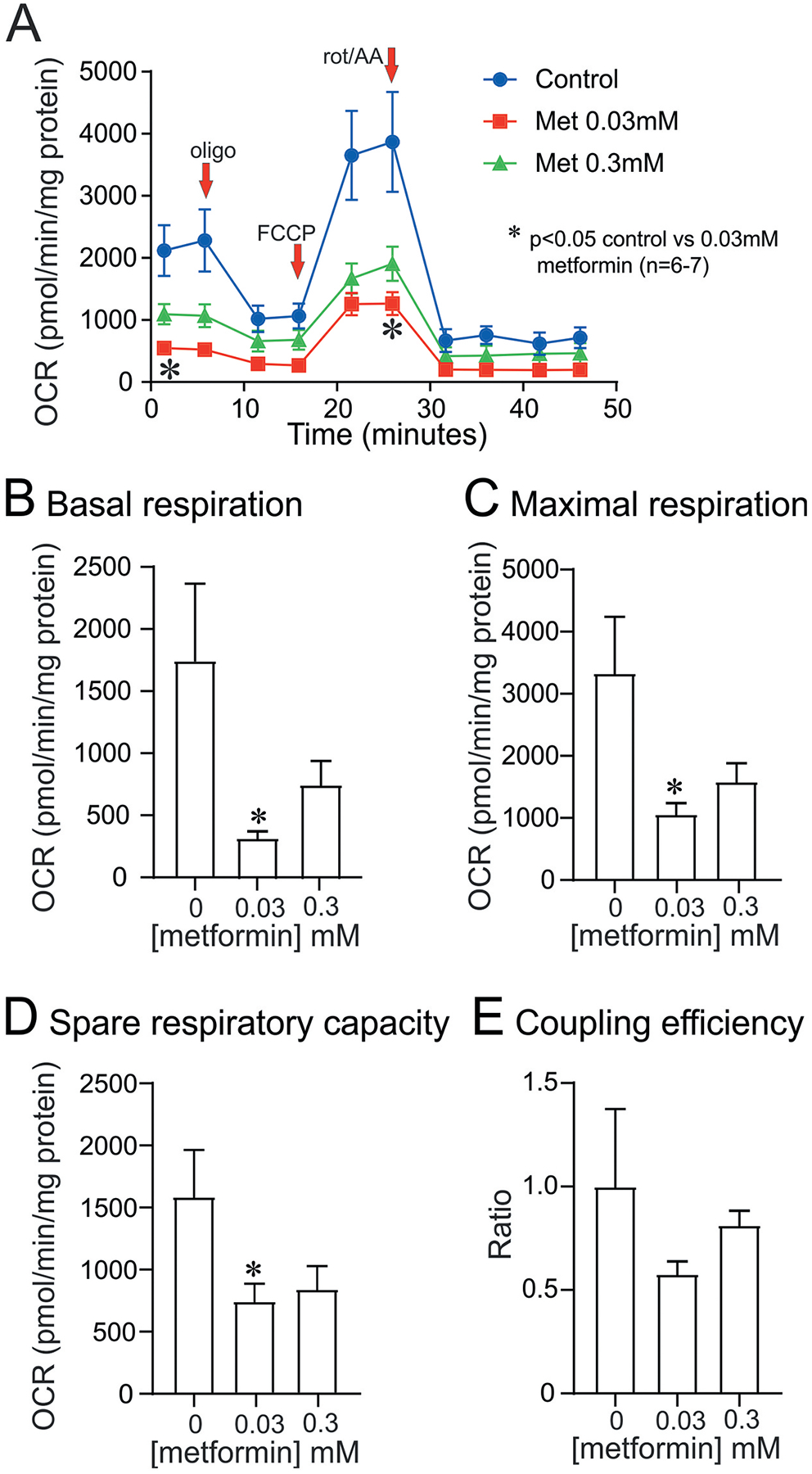
DRG sensory neurons derived from a control rat were cultured for 24 hours in the absence of neurotrophic factors and then treated with 0.03 mM or 0.3 mM metformin for 6 hours. The cultures were then analyzed for cellular bioenergetics in the Seahorse XF24 Analyzer as described previously [57,126]. The various bioenergetic parameters were determined and are explained as previously published [127]. In (A) the oxygen consumption rate (OCR) is displayed and the addition of drugs including oligomycin (oligo), FCCP and rotenone/antimycin A (rot/AA) indicated. All OCR values were normalized to total protein in the culture. Values are means ± SEM; n = 6–7 replicate cultures. *P < 0.05 vs control. Statistics were performed by one-way ANOVA with Dunnett’s post hoc test. (B) to (E) present data for each of the bioenergetic parameters.
4.1.2. Resveratrol and stimulation of axon regeneration
Resveratrol is a more potent activator of AMPK than metformin [58] and activated AMPK in Neuro2a cells (an immortalized sensory neuron clonal cell line), primary DRG neurons and the brain accompanied by AMPK-dependent mitochondrial biogenesis and robust neurite outgrowth that was blocked by genetic and pharmacologic inhibition of AMPK [60]. Resveratrol also increased neurite outgrowth from adult sensory neurons derived from streptozotocin (STZ)-induced diabetic rats, without having effect on neurite outgrowth from normal adult sensory neurons [57]. It is proposed that this differential effect is due to the preexisting decreased expression and activity of AMPK and PGC-1α under diabetic conditions [28,57,111,128,129] so that low AMPK activity prior to resveratrol treatment rendered the cells more responsive to resveratrol-induced AMPK activation [57]. Studies have indicated that nutrient excess under diabetic conditions, mediates AMPK/PGC-1α pathway down-regulation, which is reversed by resveratrol treatment [57,129].
4.2. Neuropathic pain
Neuropathic pain and its management are complex, involving numerous potential mechanisms and pathways [130]. Recent research has investigated the role of AMPK activators as potential therapeutics capable of modulating the development and course of assorted neuropathic pain states [131].
4.2.1. Studies with resveratrol
The polyphenol resveratrol has been of interest to pain researchers due to its anti-inflammatory and neuroprotective effects and its efficacy has been tested in a variety of animal models of neuropathic pain [132]. For example, daily intraperitoneal injections of resveratrol significantly increased the anti-inflammatory markers IL-4Rα, IL-10 R and others in the dorsal spinal cord while improving mechanical allodynia and thermal hyperalgesia in the rat chronic constriction injury (CCI) model [133]. Allodynia induced by CCI of the trigeminal nerve in rats is also amenable to resveratrol treatment [134] with efficacy being prevented by the AMPK inhibitor Compound C, suggesting that AMPK inactivation was involved. Resveratrol has also shown promise in rodent models of diabetic neuropathy. Following daily treatment with resveratrol for a period of 4 weeks in the streptozotocin (STZ)-induced rat model of type 1 diabetes, significant improvements in thermal hyperalgesia were recorded [135].
4.2.2. Treating neuropathic pain with metformin
The capacity of metformin to attenuate neuropathic pain has been studied in a variety of models. In a mouse model of low back pain achieved by lumbar disc puncture, metformin and the direct AMPK activator O304 significantly improved pain thresholds, as assessed by von Frey filament testing of the hind paws [136]. A similar lumbar disc herniation model in rats characterized by increased thermal hyperalgesia and mechanical allodynia was significantly improved following daily metformin treatment that began 1 week prior to disc herniation [137]. Interestingly, the signaling molecule mTOR was upregulated in the DRG of these rats following lumbar disc herniation. Both metformin and the mTOR inhibitor rapamycin reduced these levels to baseline and improved pain indices [137]. However, while mTOR complex 1 inhibitors were developed to treat chronic pain, some can cause debilitating pain syndromes [138]. For example, mTORC1 inhibition with rapamycin resulted in dose-dependent mechanical allodynia in mouse and rat models of spared nerve injury and spinal nerve ligation. Metformin treatment achieved a complete reversal of previously established neurologic pain while AMPK activators metformin and A769662 achieved a remarkable reversal of the mechanical allodynia produced by spared nerve injury or sciatic nerve ligation [139]. Apart from the mTOR pathway, the opioid receptor system may also be involved in metformin’s anti-nociceptive action as the opioid receptor antagonist naltrexone attenuated the reduced mechanical allodynia provided by metformin in the CCI model [140]. Metformin also produces an anti-hyperalgesic effect in a mouse model of post-operative pain with P-AMPK levels being restored to pre-surgery levels [141].
4.3. Chemotherapy-induced peripheral neuropathy
A number of cytotoxic chemotherapeutic drugs cause debilitating side effects including chemotherapy-induced peripheral neuropathy (CIPN). CIPN commonly presents as shooting and stabbing pain, allodynia and numbness in a stocking-and-glove fashion that is similar to diabetic neuropathy [142]. Accumulation of chemotherapeutics in the central and peripheral nervous systems and resultant mitochondrial and neurologic dysfunction are believed to be key mechanisms behind CIPN [109,110,112] and AMPK activators have been tested in the prevention and treatment of CIPN. Paclitaxel-induced mechanical hypersensitivity and hyperalgesic priming in mice were both prevented by the AMPK activator narciclasine while metformin attenuated hyperalgesic priming, but was unable to reverse mechanical allodynia [143]. In a cisplatin model of CIPN, metformin effectively prevented mechanical allodynia when given concurrently with cisplatin whereas delaying metformin treatment until after establishment of CIPN failed to reverse neuropathy [144]. Intraepidermal nerve fiber loss was also attenuated by metformin treatment [144]. In oxaliplatin-induced neuropathy in mice, metformin achieved partial prevention of mechanical hyperalgesia and a significant reduction in cold allodynia [145]. In the DRG of these mice, significantly elevated levels of c-Fos and ATF3, markers of neuronal activation and damage respectively, were prevented by metformin. In a bortezomib model of CIPN, elevated levels of hypoxia inducible factor α (HIF1α) were detected in lumbar DRG of male ICR mice [146]. HIF1α levels appear to be important in the pathogenic cascade, as HIF1α interruption with echinomycin or anti-HIF1α siRNA prevented allodynia. After metformin treatment, HIF1α levels were significantly reduced and neuropathic pain was prevented. There is little current data on the effects of resveratrol on CIPN. One study in cisplatin-induced neuropathy in rats showed that when given together, resveratrol and monosodium glutamate significantly attenuated thermal hyperalgesia produced by cisplatin treatment [147].
4.4. Metformin and diabetic peripheral neuropathy
Uncontrolled chronic hyperglycemia, loss of insulin signaling in peripheral tissues and dyslipidemia are considered the primary catalysts that lead to potentially devastating complications of diabetes such as nephropathy, retinopathy and neuropathy [148]. Intermediate pathogenic mechanisms include multiple aberrations in metabolic function, inflammation, vascular insufficiency, and oxidative stress [149,150]. The most common form of diabetic neuropathy is a distal symmetric polyneuropathy (DSPN) [151]. DSPN is often diagnosed long after onset, once disease has progressed to the point of obvious objective findings. The natural history of DSPN is characterized by progressive loss of vibration and pain sensation, paresthesias and weakness that begins in the feet and hands, extending proximally [130]. Pain also occurs in around 30% of patents [152]. At the conclusion of a ten-year follow up study, patients with DSPN exhibited significantly decreased vibration sense and Achilles tendon reflexes and increased paresthesia and pain compared to non-diabetic controls; poor glycemic control was correlated with disease progression [153]. A recent meta-analysis has shown that duration of diabetes, advancing age, elevated HbA1c and presence of diabetic retinopathy are strong risk factors for the development of DSPN [154]. Elevated BMI and a history of smoking were not found to be risk factors in that study, although other research has suggested the opposite [148]. Early studies suggested disease progression could be halted and even reversed, with conduction velocities in the popliteal and median nerves being improved significantly after only 6-months of medical hyperglycemic management [155]. Subsequent research has found that a basal-bolus insulin management regimen mitigated microvascular complications, including neuropathy, with significant improvements in median nerve conduction velocities after 6.5 years [156].
Involvement of AMPK signaling pathways in the pathogenesis of diabetic neuropathy is suggested by studies that investigated AMPK expression in the spinal cord and DRG of rats fed a high fat diet (HFD) to model early T2D [157]. HFD-fed rats displayed increased pain sensitivity and elevated levels of calcitonin gene-related peptide, the primary neurotransmitter of nociceptive C fibers. Elevating AMPK activity with the AMPK activator, AICAR, led to decreased pain sensitivity and mechanical allodynia in HFD fed rats compared to rats fed a low-fat diet.
Use of metformin to treat indices of diabetic neuropathy in rodents has yielded mixed findings to date. Positive metformin effects on neuropathy and neuropathic pain have been demonstrated by multiple research groups. Metformin reduced the deposition of destructive glycotoxins in the sciatic nerves of STZ-diabetic rats, resulting in improved conduction velocity compared to healthy controls [158]. Metformin also dose-dependently improved heat and mechanical hyperalgesia in STZ-diabetic rats without having any effect in healthy controls [159]. Activation of AMPK and key target proteins, SIRT3, nNOS, and PGC-1α was also observed in sciatic nerve and reduced oxidative stress, as determined by reduction in glycotoxin formation and increased production of superoxide dismutase, suggested a protective mechanism [159]. In the dorsal horn of diabetic rats, an increase in synaptic number was considered suggestive of increased sensitivity and neuropathic pain [160]. Metformin reduced hyperalgesia and mitigated the increase in synapse number in the L5 segment of the spinal cord of diabetic rats [161]. Attenuation of the inflammatory response to hyperglycemia in peripheral nerves was another consequence of metformin treatment [162]. Thus, upregulation of the pro-inflammatory cytokine IL-1β, in the peripheral nerves of diabetic mice, was dose-dependently attenuated by metformin treatment, concurrent with an increase in neurotrophic and angiogenic factors, whereas the upregulation of TNF-α was not affected by metformin [162]. Most recently, metformin treatment prevented development of tactile allodynia and paw pressure hyperalgesia but not heat hyperalgesia in rats with combined STZ and HFD induced diabetes [163]. This was accompanied by protection of large diameter fiber in the sciatic nerve and loss of small sensory fibers in the skin.
Negative impacts of metformin on the course and progression of diabetic neuropathy have also been suggested in both humans and animals, with a report of more severe indices of neuropathy in patients receiving metformin treatment [164] and a claim of exacerbated myelin damage in HFD mice [165]. However, while prolonged metformin use has been associated with worsening DSPN, so is prolonged diabetes, making it difficult to delineate whether metformin is contributory or not [166]. As discussed below, metformin is also regularly associated with vitamin B12 deficiency, another potential cause of neuropathy [167].
4.5. Resveratrol and diabetic peripheral neuropathy
The therapeutic effects of resveratrol have been studied in a variety of conditions including cardiovascular disease, longevity, and metabolic disturbances [168]. The free radical scavenging capacity and anti-inflammatory properties of resveratrol also make it of interest as a potential treatment for complications of diabetes such as hypertension, retinopathy, and cardiovascular disease. Resveratrol weakly improves insulin sensitivity and decreases markers of oxidative stress in patients with type 2 diabetes [169]. Activation of Akt, an intracellular signaling pathway activated by insulin, also occurs. In another study, BMI was found to be significantly reduced after resveratrol treatment [170]. Production of superoxide anion was also significantly reduced with associated upregulation of superoxide dismutase and Nrf2, a regulator of antioxidant gene expression [170]. In contrast, no change in peripheral and hepatic insulin sensitization was detected in patients with appropriately managed type 2 diabetes following 30 days of resveratrol supplementation [171].
Given the effects of metformin described above, the therapeutic profile of resveratrol on nerve function and metabolic parameters in diabetes has also been investigated. Conduction velocity in the sciatic nerves of diabetic rats significantly improved after only 2 weeks of resveratrol treatment [172]. This was associated with increased sciatic blood flow, decreased oxidative stress and DNA fragmentation and improvements in hyperalgesia and mechanical allodynia. Further studies replicated the capacity of resveratrol to correct nerve conduction slowing and also demonstrated decreased pro-inflammatory cytokine and NF-κB production in a dose-dependent manner [173]. Plasma glucose levels were not affected by resveratrol. Increased neurite outgrowth and AMPK activation following resveratrol treatment has been reported in DRG cultures derived from STZ-induced type 1 diabetic rats. Over-expression of dominant negative AMPK mutants blocked any effect of resveratrol treatment suggesting that AMPK activation was an essential mechanistic step [57]. Respiratory complex impairment and mitochondrial phenotype in STZ-diabetic rats were also significantly improved by systemic resveratrol treatment alongside reductions in thermal hypoalgesia.
4.6. Metformin, B12 deficiency, and neuropathy
The potential of metformin as a therapy for diabetic neuropathy has been complicated by reports of an association between long-term use of metformin and the development of vitamin B12 (cobalamin) deficiency [174]. Deficiency in B12 can lead to a form of neuropathy that is clinically indistinguishable from DSPN, characterized by symmetrical paresthesia, loss of sensation, and gait disturbances [175]. Characteristics such as age, race, time since diabetes diagnosis, and metformin dose and duration of use all likely affect the development of B12 deficiency, as well as metformin-related neuropathy [176]. However the association has been considered strong enough to prompt the American Diabetes Association to recommend periodic testing of B12 levels in those with a history of neuropathy or anemia and long-term metformin use [5]. Early research suggested that malabsorption of B12 may be the primary mechanism of deficiency, occurring in a manner independent of intrinsic factor (IF) levels [177]. Supplementation with calcium, an essential mineral for ileal B12-IF receptor function, produced partial reversal of B12 deficiency in diabetic subjects as determined by measured holotranscobalamin levels, an earlier and more sensitive measure of deficiency than serum B12, offering more support for the putative malabsorption mechanism [178]. Other mechanisms for B12 malabsorption have also been investigated [179].
Various RCTs and observational studies have also investigated the clinical effects of metformin on diabetic neuropathy and B12 deficiency. A cross-sectional study demonstrated a significant inverse relationship between metformin dose and serum B12 levels [180]. Length of metformin use did not appear to be correlated with B12 levels. Furthermore, 64% of study patients with diabetic neuropathy had low or borderline levels of B12. In another observational study, those subjects with definite B12 deficiency experienced a greater incidence of diabetic neuropathy that was also correlated with increasing length of metformin use [166]. A follow-up to the HOME trial was the first long-term study to show permanent changes to B12 levels following metformin use [181]. Metformin dose and treatment duration were found to be significant determinants of B12 levels, which were demonstrably lower at study conclusion. Homocysteine, a more sensitive measure of B12 deficiency was also elevated, further supporting overall deficiency in B12. Elevated methylmalonic acid, another marker of B12 deficiency, was also positively correlated with cumulative metformin use while clinical presentation of neuropathy as determined by the validated Toronto Clinical Severity Score, was worsened along with lower conduction velocities in sural and superficial peroneal nerves [164]. More assessment of the HOME trial data showed that rising methylmalonic acid levels were associated with increasing cumulative dose of metformin [7]. Surprisingly, there was no apparent effect of metformin on the presence of neuropathy and it was hypothesized that the glycemic effect of metformin mitigated the deleterious effects of rising methylmalonic acid levels. Further definitive research is required to fully delineate the effects of metformin on the development of DSPN, separate from potential associated B12 deficiency.
5. Expert opinion
Metformin has proven protective in a wide variety of animal models of neurological disease that exhibit an array of differing etiologies. The primary target of metformin action is activation of AMPK and the subsequent downstream suppression of mTOR pathway activity, accompanied by upregulation of autophagy. In PD and MS, the likely therapeutic pathway is metformin-dependent augmentation of mitochondrial function, with no clear evidence of mTOR pathway involvement. The literature provides compelling support for metformin acting via suppression of mTOR pathway activity to reduce toxic protein translation, in HD, and aberrant excitability, in epilepsy. Of note, in a cohort of patents with HD exhibiting T2D, the presence of metformin therapy was protective [86]. Mechanisms of metformin-dependent protection in stroke are less clear.
In the PNS, metformin elevated AMPK activity and corrected various clinically relevant endpoints in models of nerve regeneration, neuropathic pain, CIPN and diabetic neuropathy. The involvement of optimized mitochondrial function remains unclear, although, studies with resveratrol demonstrate a strong link between up-regulation of AMPK and improved mitochondrial bioenergetics. However, caution must be used when comparing metformin’s effects directly with those generated by resveratrol; both activate AMPK in the CNS and PNS. However, in the case of metformin there is a direct negative impact on mitochondrial electron transport which then triggers compensatory mechanisms, involving AMPK, which then act to enhance mitochondrial performance, biogenesis and dynamics. Resveratrol can achieve all of these effects while not initially causing a negative impact on mitochondrial function. Study of the differential effects of metformin vs resveratrol on mitochondrial bioenergetics in various disease settings and durations will be critical. In nerve regeneration, neuropathic pain and CIPN metformin therapy is protective, in part, via suppression of mTOR pathway activity. The reduction in translation of specific proteins linked to pain pathway activation and/or the triggering of autophagy combine to optimize nerve regeneration and function. It remains uncertain how autophagy is being regulated by metformin in these various neurological settings. It is feasible that AMPK activation is directly driving elevated autophagy independently of any involvement of the mTOR pathway [182].
There is a significant need for additional investigations of the effects of short-term vs long term therapy with metformin in various models of neurological disease. Over the short-term it is apparent that metformin interference with mitochondrial function can trigger a neuroprotective AMPK-dependent response. However, over the longer term it is plausible that the ‘assault’ on the mitochondrial respiratory chain eventually proves too damaging, leading to neurodegeneration. Coupled to this approach is the need to carefully assess metformin dose. In HD, low doses of metformin impacted the mTOR pathway without concurrent impact on AMPK [89]. Only at higher doses of metformin was AMPK activation observed. In this scenario, htt translation was suppressed via an mTOR pathway independent of upstream AMPK activation. The same complexities are likely operating in other neurodegenerative diseases.
Any potential therapeutic approach involving metformin will have to be carefully adjusted for dose and duration of treatment given that metformin’s primary action is to block electron flow within the mitochondrion. The therapeutic aim should be to minimize the impact on electron flow while maximizing activation of downstream protective pathways, such as AMPK. The array of preclinical studies cited in this review that demonstrate efficacy of metformin against a range of clinically relevant endpoints strongly support the feasibility of such an approach. With an appropriate dose, route and duration of treatment it appears that the downstream activation of protective pathways can overcome any deficit arising from disturbance of the electron transport pathway. This is presumably the consequence of increased mitochondrial refurbishment, optimized dynamics/trafficking and enhanced quality control via mitophagy. If the mitochondrial pool in the neuron or support cell is viewed as one homogenous entity, then partial inhibition of electron transport in each mitochondrial unit, which contains multiple Complex I units, may be overcome by the benefits of AMPK activation and its positive downstream effects on mitochondrial fidelity and function. Alternatively, there may be pools of mitochondria within a cell that are differentially targeted or impacted by the drug. For example, mitochondria of unmyelinated neurons in the epidermis exist in an environment that lacks direct vascular supply so that exposure to metformin following systemic delivery may be expected to be minimal. In contrast, mitochondria in the perikarya of the same neurons may be inhibited by systemic metformin due to the plentiful blood supply to the DRG. A metformin-triggered signal in the perikarya could spread anterogradely to the nerve ending via refurbished and rapidly transported mitochondria to enhance bioenergetic performance in the distal axonal compartment despite the absence of local metformin-induced injury. Mitochondria in the nerve ending or synapse drive plasticity and, if impaired in disease, could trigger distal dying-back of the axon or synaptic pruning; metformin therapy could overcome this mitochondrial deficit through either route described above.
The HD field has performed a number of mechanistic studies using transgenic knockout of AMPK subunits or delivery of siRNA to components of the putative metformin pathway followed by assessment of the effects of metformin on clinically relevant endpoints – for example, the elegant work by Vazquez-Manrique and colleagues [88]. These in vivo mechanistic approaches would be equally valuable if applied to models of MS, stroke and peripheral nerve disease to dissect metformin signaling pathways.
There is also a need for more detailed descriptions of mitochondrial dysfunction in neurological diseases of the CNS and PNS, with analysis of mitochondrial bioenergetics to determine impact of metformin on electron transport. Figure 4 shows in adult DRG cultures that metformin has a profound inhibitory effect on mitochondrial electron flow but whether metformin operates in this manner in vivo is unknown. The downstream consequences of metformin diverting cellular metabolism from oxidative phosphorylation via use of glucose as a substrate and toward fatty acid oxidation and/or glycolysis also need to be established. For example, fatty acid oxidation could presumably be limited due to the metformin block at Complex I, but due to its higher yield of ATP could still provide the energy needed for neuroprotection.
The importance of autophagy and mitophagy in these neurological diseases also requires further investigation. There are hints from numerous studies that metformin is mobilizing autophagy to trigger nerve regeneration and protection from degeneration. However, detailed and mechanistic experiments are required to dissect the activation pathways and their downstream targets and consequences. A key question is how AMPK activation is driving autophagy – is perhaps mobilization of AMPK by metformin directly activating ULK1 to enhance autophagy independent of the mTOR pathway? In addition, any modulatory role for AMPK signaling in the etiology of neurodegenerative disease remains to be determined. No genetic links with altered AMPK function have been identified. Given the intricate linkage between cellular energy status and AMPK activation status there is a need for experiments to dissect out any primary role of AMPK in the neurodegenerative process. What is clear, irrespective of any putative role for AMPK modulation in the disease process, is that activation of AMPK is protective and/or reparative in many disease settings. In HD the issue is complex with different roles for modulation of AMPK in early vs late disease progression. In other diseases, for example diabetic neuropathy, there is clear evidence that deactivation of AMPK is associated with development of pain and neuronal injury. However, there remains no definitive proof that this deactivation caused the pain and neuronal injury, although drug-induced activation of AMPK was neuroprotective.
Finally, more studies in patients with T2D under metformin therapy are required to assess impact on cognitive function or other neurological impairments in the CNS and PNS. Clinical trials in carefully selected neurologically impaired patient cohorts containing a sub-population suffering from T2D, similar to the study in HD reported by Hervas et al [86]. Obvious areas for further study are PD and Alzheimer’s disease (for example see [183]), where there is an aged population in whom T2D will be a common co-morbidity.
Article highlights.
In animal models of PD, metformin improved neurological symptoms, including lowering dyskinesias. The therapeutic pathways involved elevation of mitochondrial function and ROS scavenging, with evidence of AMPK-independent signaling
In early HD and epilepsy, metformin-dependent suppression of the mTOR pathway was neuroprotective
Metformin signaling via AMPK and associated elevation of PGC-1α activity can prevent disease in cuprizone and PTX-induced mouse models of MS
In animal models of stroke and epilepsy, treatment with metformin raised AMPK activity and afforded protection
Peripheral nerve regeneration is augmented by metformin acting via Schwann cells and also with direct neuronal effects that include optimization of autophagy
In a variety of animal models of neuropathic pain, including CIPN, metformin prevented tactile and cold allodynia with suppression of mTOR signaling a major target
The diabetic state depresses AMPK signaling in the DRG and thus treatment with metformin, or resveratrol, is protective against fiber loss and pain in multiple rodent models
Metformin through subtle adjustment of the balance between AMPK activation and mTOR pathway suppression provides neuroprotection in a variety of neurological diseases
Enhancing mitochondrial function, to surmount metabolic failure, combined with stimulation of autophagy are two major neuroprotective pathways mobilized by metformin to augment axonal/dendritic plasticity and suppress toxic protein aggregation
Funding
This work was supported by grant # MOP-130282 from the Canadian Institutes of Health Research to P.F. and grant NS081082 from the National Institutes of Health to NA Calcutt. The authors are grateful to St Boniface Hospital Research for support. S Demaré was supported by a Natural Sciences and Engineering Research Council (NSERC) undergraduate research award. A Kothari was supported by an award from the Rady Faculty of Health Sciences BSc Med program.
Footnotes
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. [DOI] [PubMed] [Google Scholar]
- 2.Montvida O, Shaw J, Atherton JJ, et al. Long-term trends in anti-diabetes drug usage in the U.S.: real-world evidence in patients newly diagnosed with type 2 diabetes. Diabetes Care. 2018;41:69–78. [DOI] [PubMed] [Google Scholar]
- 3.Evans M, Morgan AR, Yousef Z. What next after metformin? Thinking beyond glycaemia: are SGLT2 inhibitors the answer? Diabetes Ther. 2019;10:1719–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipscombe L, Booth G, Butalia S, et al. Pharmacologic glycemic management of type 2 diabetes in adults. Can J Diabetes. 2018;42 Suppl 1:S88–S103. [DOI] [PubMed] [Google Scholar]
- 5.American Diabetes Association. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes-2019. Diabetes Care. 2019;42:S90–S102. [DOI] [PubMed] [Google Scholar]
- 6.Buse JB, Wexler DJ, Tsapas A, et al. 2019 update to: management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2020;63:221–228. [DOI] [PubMed] [Google Scholar]
- 7.Out M, Kooy A, Lehert P, et al. Long-term treatment with metformin in type 2 diabetes and methylmalonic acid: post hoc analysis of a randomized controlled 4.3year trial. J Diabetes Complications. 2018;32:171–178. [DOI] [PubMed] [Google Scholar]
- 8.DePalo VA, Mailer K, Yoburn D, et al. Lactic acidosis associated with metformin use in treatment of type 2 diabetes mellitus. Geriatrics. 2005;60:36–41. [PubMed] [Google Scholar]
- 9.Donnan K, Segar L. SGLT2 inhibitors and metformin: dual antihyperglycemic therapy and the risk of metabolic acidosis in type 2 diabetes. Eur J Pharmacol. 2019;846:23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Frid A, Sterner GN, Londahl M, et al. Novel assay of metformin levels in patients with type 2 diabetes and varying levels of renal function: clinical recommendations. Diabetes Care. 2010;33:1291–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw JS, Wilmot RL, Kilpatrick ES. Establishing pragmatic estimated GFR thresholds to guide metformin prescribing. Diabet Med. 2007;24:1160–1163. [DOI] [PubMed] [Google Scholar]
- 12.Moin T, Schmittdiel JA, Flory JH, et al. Review of metformin use for type 2 diabetes prevention. Am J Prev Med. 2018;55:565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diabetes Prevention Program Research Group. The diabetes prevention program (DPP): description of lifestyle intervention. Diabetes Care. 2002;25:2165–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.UKPDS. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). UK prospective diabetes study (UKPDS) Group. Lancet. 1998;352:854–865. [PubMed] [Google Scholar]
- 15.Holman RR, Paul SK, Bethel MA, et al. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med. 2008;359:1577–1589. [DOI] [PubMed] [Google Scholar]
- 16.Panagiotopoulos C, Hadjiyannakis S, Henderson M. Type 2 diabetes in children and adolescents. Can J Diabetes. 2018;42 Suppl 1:S247–S54. [DOI] [PubMed] [Google Scholar]
- 17.Jones KL, Arslanian S, Peterokova VA, et al. Effect of metformin in pediatric patients with type 2 diabetes: a randomized controlled trial. Diabetes Care. 2002;25:89–94. [DOI] [PubMed] [Google Scholar]
- 18.Gerstein HC, Miller ME, Byington RP, et al. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meneilly GS, Knip A, Miller DB, et al. Diabetes in Older People. Can J Diabetes. 2018;42 Suppl 1:S283–S95. [DOI] [PubMed] [Google Scholar]
- 20.Cusi K, Consoli A, DeFronzo RA. Metabolic effects of metformin on glucose and lactate metabolism in noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1996;81:4059–4067. [DOI] [PubMed] [Google Scholar]
- 21.Hundal RS, Krssak M, Dufour S, et al. Mechanism by which metformin reduces glucose production in type 2 diabetes. Diabetes. 2000;49:2063–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stumvoll M, Nurjhan N, Perriello G, et al. Metabolic effects of metformin in non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333:550–554. [DOI] [PubMed] [Google Scholar]
- 23.Luo T, Nocon A, Fry J, et al. AMPK Activation by metformin suppresses abnormal extracellular matrix remodeling in adipose tissue and ameliorates insulin resistance in obesity. Diabetes. 2016;65:2295–2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang X, Giacomini KM. Transporters involved in metformin pharmacokinetics and treatment response. J Pharm Sci. 2017;106:2245–2250. [DOI] [PubMed] [Google Scholar]
- 25.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Canto C, Auwerx J. AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci. 2010;67:3407–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feige JN, Auwerx J. Transcriptional coregulators in the control of energy homeostasis. Trends Cell Biol. 2007;17:292–301. [DOI] [PubMed] [Google Scholar]
- 29.Foretz M, Guigas B, Bertrand L, et al. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953–966. [DOI] [PubMed] [Google Scholar]; •• Excellent review of pharmacology of metformin. Covers mechanism of AMPK activation but also reviews alternate sites of metformin action. Discusses cardioprotective and anti-neoplastic properties of metformin.
- 30.Vial G, Detaille D, Guigas B. Role of mitochondria in the mechanism(s) of action of metformin. Front Endocrinol (Lausanne). 2019;10:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez-Marcos PJ, Auwerx J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr. 2011;93:884S–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu Y, Chen H, Zhang L, et al. The AMPK-MFN2 axis regulates MAM dynamics and autophagy induced by energy stresses. Autophagy. 2020;1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Investig. 2001;108:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foretz M, Hebrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120:2355–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Owen MR, Doran E, Halestrao AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- 37.El-Mir MY, Nogueira V, Fontaine E, et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. [DOI] [PubMed] [Google Scholar]
- 38.Bridges HR, Jones AJ, Pollak MN, et al. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. 2014;462:475–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alshawi A, Agius L. Low metformin causes a more oxidized mitochondrial NADH/NAD redox state in hepatocytes and inhibits gluconeogenesis by a redox-independent mechanism. J Biol Chem. 2019;294:2839–2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailey CJ, Mynett KJ, Page T. Importance of the intestine as a site of metformin-stimulated glucose utilization. Br J Pharmacol. 1994;112:671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCreight LJ, Bailey CJ, Pearson ER. Metformin and the gastrointestinal tract. Diabetologia. 2016;59:426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker J, Jijon HB, Diaz H, et al. 5-aminoimidazole-4-carboxamide riboside (AICAR) enhances GLUT2-dependent jejunal glucose transport: a possible role for AMPK. Biochem J. 2005;385:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem. 2011;286:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boyle JG, Logan PJ, Jones GC, et al. AMP-activated protein kinase is activated in adipose tissue of individuals with type 2 diabetes treated with metformin: a randomised glycaemia-controlled crossover study. Diabetologia. 2011;54:1799–1809. [DOI] [PubMed] [Google Scholar]
- 45.Moreno-Navarrete JM, Ortega FJ, Rodriguez-Hermosa JI, et al. OCT1 Expression in adipocytes could contribute to increased metformin action in obese subjects. Diabetes. 2011;60:168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karise I, Bargut TC, Del Sol M, et al. Metformin enhances mitochondrial biogenesis and thermogenesis in brown adipocytes of mice. Biomed Pharmacother. 2019;111:1156–1165. [DOI] [PubMed] [Google Scholar]
- 47.Breining P, Jensen JB, Sundelin EI, et al. Metformin targets brown adipose tissue in vivo and reduces oxygen consumption in vitro. Diabetes Obes Metab. 2018;20:2264–2273. [DOI] [PubMed] [Google Scholar]
- 48.Chiang MC, Cheng YC, Chen SJ, et al. Metformin activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against Amyloid-beta-induced mitochondrial dysfunction. Exp Cell Res. 2016;347:322–331. [DOI] [PubMed] [Google Scholar]
- 49.Inyang KE, Szabo-Pardi T, Wentworth E, et al. The antidiabetic drug metformin prevents and reverses neuropathic pain and spinal cord microglial activation in male but not female mice. Pharmacol Res. 2019;139:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung MM, Nicol CJ, Cheng YC, et al. Metformin activation of AMPK suppresses AGE-induced inflammatory response in hNSCs. Exp Cell Res. 2017;352:75–83. [DOI] [PubMed] [Google Scholar]
- 51.Wang S, Kobayashi K, Kogure Y, et al. Negative regulation of TRPA1 by AMPK in primary sensory neurons as a potential mechanism of painful diabetic neuropathy. Diabetes. 2018;67:98–109. [DOI] [PubMed] [Google Scholar]
- 52.Chau-Van C, Gamba M, Salvi R, et al. Metformin inhibits adenosine 5ʹ-monophosphate-activated kinase activation and prevents increases in neuropeptide Y expression in cultured hypothalamic neurons. Endocrinology. 2007;148:507–511. [DOI] [PubMed] [Google Scholar]
- 53.El-Mir MY, Detaille D, RV G, et al. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J Mol Neurosci. 2008;34:77–87. [DOI] [PubMed] [Google Scholar]
- 54.Kulkarni SS, Canto C. The molecular targets of resveratrol. Biochim Biophys Acta. 2015;1852:1114–1123. [DOI] [PubMed] [Google Scholar]
- 55.Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bonnefont-Rousselot D Resveratrol and cardiovascular diseases. Nutrients. 2016;8:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roy Chowdhury SK, Smith DR, Saleh A, et al. Impaired adenosine monophosphate-activated protein kinase signalling in dorsal root ganglia neurons is linked to mitochondrial dysfunction and peripheral neuropathy in diabetes. Brain. 2012;135:1751–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]; • First to show suppression of AMPK signaling in sensory neurons in diabetes. Neurite outgrowth in adult neurons mediated by AMPK pathway. In rats treatment with resveratrol activated AMPK in DRG, augmented mitochondrial function and protected from diabetic neuropathy.
- 58.Zang M, Xu S, Maitland-Toolan KA, et al. Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice. Diabetes. 2006;55:2180–2191. [DOI] [PubMed] [Google Scholar]
- 59.Suchankova G, Nelson LE, Gerhart-Hines Z, et al. Concurrent regulation of AMP-activated protein kinase and SIRT1 in mammalian cells. Biochem Biophys Res Commun. 2009;378:836–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.*Dasgupta B, Milbrandt J Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104:7217–7222. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Initial demonstration that resveratrol and AICAR via AMPK activation elevated neurite outgrowth in neuronal cell line and embryonic primary neurons. AMPK activation mediated by LKB1 in neurons.
- 61.Breen DM, Sanli T, Giacca A, et al. Stimulation of muscle cell glucose uptake by resveratrol through sirtuins and AMPK. Biochem Biophys Res Commun. 2008;374:117–122. [DOI] [PubMed] [Google Scholar]
- 62.Price NL, Gomes AP, Ling AJ, et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012;15:675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gantois I, Popic J, Khoutorsky A, et al. Metformin for treatment of fragile X syndrome and other neurological disorders. Annu Rev Med. 2019;70:167–181. [DOI] [PubMed] [Google Scholar]
- 64.Lipton JO, Sahin M. The neurology of mTOR. Neuron. 2014;84:275–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Christie KJ, Webber CA, Martinez JA, et al. PTEN inhibition to facilitate intrinsic regenerative outgrowth of adult peripheral axons. J Neurosci. 2010;30:9306–9315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Park KK, Liu K, Hu Y, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Aghanoori MR, Smith DR, Shariati-Ievari S, et al. Insulin-like growth factor-1 activates AMPK to augment mitochondrial function and correct neuronal metabolism in sensory neurons in type 1 diabetes. Mol Metab. 2019;20:149–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Koley S, Rozenbaum M, Fainzilber M, et al. Translating regeneration: local protein synthesis in the neuronal injury response. Neurosci Res. 2019;139:26–36. [DOI] [PubMed] [Google Scholar]; • Informative and concise review highlighting role of mTOR pathway in regulating protein synthesis in the axon. Good discussion of complexity of mTOR and associated pathways in directing neuronal growth.
- 69.Rotermund C, Machetanz G, Fitzgerald JC. The therapeutic potential of metformin in neurodegenerative diseases. Front Endocrinol (Lausanne). 2018;9:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Riching AS, Major JL, Londono P, et al. The brain-heart axis: alzheimer’s, diabetes, and hypertension. ACS Pharmacol Transl Sci. 2020;3:21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wahl D, Solon-Biet SM, Cogger VC, et al. Aging, lifestyle and dementia. Neurobiol Dis. 2019;130:104481. [DOI] [PubMed] [Google Scholar]
- 72.Markowicz-Piasecka M, Sikora J, Szydlowska A, et al. Metformin - a future therapy for neurodegenerative diseases: theme: drug discovery, development and delivery in alzheimer’s disease guest editor: davide brambilla. Pharm Res. 2017;34:2614–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Torres-Odio S, Key J, Hoepken HH, et al. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation. 2017;14:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Curry DW, Stutz B, Andrews ZB, et al. Targeting AMPK signaling as a neuroprotective strategy in Parkinson’s disease. J Parkinsons Dis. 2018;8:161–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zheng B, Liao Z, Locascio JJ, et al. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med. 2010;2:52ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu M, Su C, Qiao C, et al. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol. 2016;19:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Katila N, Bhurtel S, Shadfar S, et al. Metformin lowers alpha-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology. 2017;125:396–407. [DOI] [PubMed] [Google Scholar]
- 78.Patil SP, Jain PD, Ghumatkar PJ, et al. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience. 2014;277:747–754. [DOI] [PubMed] [Google Scholar]
- 79.Bayliss JA, Lemus MB, Santos VV, et al. Metformin prevents nigrostriatal dopamine degeneration independent of AMPK activation in dopamine neurons. PLoS One. 2016;11:e0159381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dias V, Junn E, Mouradian MM. The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis. 2013;3:461–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fitzgerald JC, Zimprich A, Carvajal Berrio DA, et al. Metformin reverses TRAP1 mutation-associated alterations in mitochondrial function in Parkinson’s disease. Brain. 2017;140:2444–2459. [DOI] [PubMed] [Google Scholar]; • First to identify the mitochondrial protein TRAP1, a regulator of energy metabolism, is mutated in late onset PD. Patient fibroblasts exhibited mitochondrial unfolded response and mitochondrial depolarization that was prevented by metformin.
- 82.Ryu YK, Park HY, Go J, et al. Metformin inhibits the development of L-DOPA-induced dyskinesia in a murine model of Parkinson’s disease. Mol Neurobiol. 2018;55:5715–5726. [DOI] [PubMed] [Google Scholar]
- 83.Schulte J, Littleton JT. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr Trends Neurol. 2011;5:65–78. [PMC free article] [PubMed] [Google Scholar]
- 84.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–1555. [DOI] [PubMed] [Google Scholar]
- 85.Steffan JS, Kazantsev A, Spasic-Boskovic O, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hervas D, Fornes-Ferrer V, Gomez-Escribano AP, et al. Metformin intake associates with better cognitive function in patients with Huntington’s disease. PLoS One. 2017;12:e0179283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Parker JA, Connolly JB, Wellington C, et al. Expanded polygluta-mines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc Natl Acad Sci U S A. 2001;98:13318–13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vazquez-Manrique RP, Farina F, Cambon K, et al. AMPK activation protects from neuronal dysfunction and vulnerability across nematode, cellular and mouse models of Huntington’s disease. Hum Mol Genet. 2016;25:1043–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]; • Elegant work using genetically modified nematode and mouse models of early HD to reveal that AMPK activation and treatment with metformin are neuroprotective.
- 89.Arnoux I, Willam M, Griesche N, et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. Elife. 2018;7:e38744. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• Compelling study of Hdh150 knockin mouse model of early HD using 2-photon microscopy revealing aberrant visual cortex circuit activity was prevented by metformin. Htt load was reduced by metformin, independently of AMPK, via suppression of mTOR-dependent phosphorylation of S6.
- 90.Ju TC, Chen HM, Lin JT, et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J Cell Biol. 2011;194:209–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Antel JP, Lin YH, Cui QL, et al. Immunology of oligodendrocyte precursor cells in vivo and in vitro. J Neuroimmunol. 2019;331:28–35. [DOI] [PubMed] [Google Scholar]
- 92.Ludwin SK, Rao V, Moore CS, et al. Astrocytes in multiple sclerosis. Mult Scler. 2016;22:1114–1124. [DOI] [PubMed] [Google Scholar]
- 93.Largani SHH, Borhani-Haghighi M, Pasbakhsh P, et al. Oligoprotective effect of metformin through the AMPK-dependent on restoration of mitochondrial hemostasis in the cuprizone-induced multiple sclerosis model. J Mol Histol. 2019;50:263–271. [DOI] [PubMed] [Google Scholar]
- 94.Smirnova LP, Krotenko NV, Grishko EV, et al. State of antioxidant system in patients with multiple sclerosis during therapy. Biomed Khim. 2011;57:661–670. [DOI] [PubMed] [Google Scholar]
- 95.Paintlia AS, Paintlia MK, Mohan S, et al. AMP-activated protein kinase signaling protects oligodendrocytes that restore central nervous system functions in an experimental autoimmune encephalomyelitis model. Am J Pathol. 2013;183:526–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Collaborators GMaCoD. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1459–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang S, Zhai H, Wei L, et al. Socioeconomic status predicts the risk of stroke death: a systematic review and meta-analysis. Prev Med Rep. 2020;19:101124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leech T, Chattipakorn N, Chattipakorn SC. The beneficial roles of metformin on the brain with cerebral ischaemia/reperfusion injury. Pharmacol Res. 2019;146:104261. [DOI] [PubMed] [Google Scholar]
- 99.Xia CY, Zhang S, Gao Y, et al. Selective modulation of microglia polarization to M2 phenotype for stroke treatment. Int Immunopharmacol. 2015;25:377–382. [DOI] [PubMed] [Google Scholar]
- 100.Jin Q, Cheng J, Liu Y, et al. Improvement of functional recovery by chronic metformin treatment is associated with enhanced alternative activation of microglia/macrophages and increased angiogenesis and neurogenesis following experimental stroke. Brain Behav Immun. 2014;40:131–142. [DOI] [PubMed] [Google Scholar]
- 101.Zhu J, Liu K, Huang K, et al. Metformin improves neurologic outcome via AMP-activated protein kinase-mediated autophagy activation in a rat model of cardiac arrest and resuscitation. J Am Heart Assoc. 2018;7:e008389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nandini HS, Paudel YN, Krishna KL. Envisioning the neuroprotective effect of Metformin in experimental epilepsy: a portrait of molecular crosstalk. Life Sci. 2019;233:116686. [DOI] [PubMed] [Google Scholar]
- 103.Citraro R, Leo A, Constanti A, et al. mTOR pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol Res. 2016;107:333–343. [DOI] [PubMed] [Google Scholar]
- 104.Yang Y, Zhu B, Zheng F, et al. Chronic metformin treatment facilitates seizure termination. Biochem Biophys Res Commun. 2017;484:450–455. [DOI] [PubMed] [Google Scholar]
- 105.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf). 2009;196:65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mehrabi S, Sanadgol N, Barati M, et al. Evaluation of metformin effects in the chronic phase of spontaneous seizures in pilocarpine model of temporal lobe epilepsy. Metab Brain Dis. 2018;33:107–114. [DOI] [PubMed] [Google Scholar]
- 107.Zeyghami MA, Hesam E, Khadivar P, et al. Effects of atorvastatin and metformin on development of pentylenetetrazole-induced seizure in mice. Heliyon. 2020;6:e03761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Heinrich C, Lahteinen S, Suzuki F, et al. Increase in BDNF-mediated TrkB signaling promotes epileptogenesis in a mouse model of mesial temporal lobe epilepsy. Neurobiol Dis. 2011;42:35–47. [DOI] [PubMed] [Google Scholar]
- 109.Bennett GJ, Doyle T, Salvemini D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat Rev Neurol. 2014;10:326–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cashman CR, Hoke A. Mechanisms of distal axonal degeneration in peripheral neuropathies. Neurosci Lett. 2015;596:33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chowdhury SK, Smith DR, Fernyhough P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiol Dis. 2013;51:56–65. [DOI] [PubMed] [Google Scholar]
- 112.Trecarichi A, Flatters SJL. Mitochondrial dysfunction in the pathogenesis of chemotherapy-induced peripheral neuropathy. Int Rev Neurobiol. 2019;145:83–126. [DOI] [PubMed] [Google Scholar]
- 113.Bernstein BW, Bamburg JR. Actin-ATP hydrolysis is a major energy drain for neurons. J Neurosci. 2003;23:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75:762–777. [DOI] [PubMed] [Google Scholar]
- 115.Li Z, Okamoto K, Hayashi Y, et al. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. [DOI] [PubMed] [Google Scholar]
- 116.Wang SS, Shultz JR, Burish MJ, et al. Functional trade-offs in white matter axonal scaling. J Neurosci. 2008;28:4047–4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Chen H, Chan DC. Critical dependence of neurons on mitochondrial dynamics. Curr Opin Cell Biol. 2006;18:453–459. [DOI] [PubMed] [Google Scholar]
- 118.Breathnach AS. Electron microscopy of cutaneous nerves and receptors. J Invest Dermatol. 1977;69:8–26. [DOI] [PubMed] [Google Scholar]
- 119.Liu L, Tian D, Liu C, et al. Metformin enhances functional recovery of peripheral nerve in rats with sciatic nerve crush injury. Med Sci Monit. 2019;25:10067–10076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Piermarini E, Cartelli D, Pastore A, et al. Frataxin silencing alters microtubule stability in motor neurons: implications for Friedreich’s ataxia. Hum Mol Genet. 2016;25:4288–4301. [DOI] [PubMed] [Google Scholar]
- 121.Huber K, Patel P, Zhang L, et al. 2-[(1-methylpropyl)dithio]-1H-imidazole inhibits tubulin polymerization through cysteine oxidation. Mol Cancer Ther. 2008;7:143–151. [DOI] [PubMed] [Google Scholar]
- 122.Wang H, Zheng Z, Han W, et al. Metformin promotes axon regeneration after spinal cord injury through inhibiting oxidative stress and stabilizing microtubule. Oxid Med Cell Longev. 2020;2020:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pereira JA, Lebrun-Julien F, Suter U. Molecular mechanisms regulating myelination in the peripheral nervous system. Trends Neurosci. 2012;35:123–134. [DOI] [PubMed] [Google Scholar]
- 124.Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol. 2014;36:121–129. [DOI] [PubMed] [Google Scholar]
- 125.Ma J, Liu J, Yu H, et al. Effect of metformin on Schwann cells under hypoxia condition. Int J Clin Exp Pathol. 2015;8:6748–6755. [PMC free article] [PubMed] [Google Scholar]
- 126.Calcutt NA, Smith DR, Frizzi K, et al. Selective antagonism of muscarinic receptors is neuroprotective in peripheral neuropathy. J Clin Invest. 2017;127:608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Puigserver P, Wu Z, Park CW, et al. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. [DOI] [PubMed] [Google Scholar]
- 129.Fernyhough P Mitochondrial dysfunction in diabetic neuropathy: a series of unfortunate metabolic events. Curr Diab Rep. 2015;15:89. [DOI] [PubMed] [Google Scholar]
- 130.Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, patho-physiological mechanisms, and treatment. Lancet Neurol. 2010;9:807–819. [DOI] [PubMed] [Google Scholar]
- 131.Price TJ, Das V, Dussor G. Adenosine monophosphate-activated protein kinase (AMPK) activators for the prevention, treatment and potential reversal of pathological pain. Curr Drug Targets. 2016;17:908–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bastianetto S, Menard C, Quirion R. Neuroprotective action of resveratrol. Biochim Biophys Acta. 2015;1852:1195–1201. [DOI] [PubMed] [Google Scholar]
- 133.Xu M, Cheng Z, Ding Z, et al. Resveratrol enhances IL-4 receptor-mediated anti-inflammatory effects in spinal cord and attenuates neuropathic pain following sciatic nerve injury. Mol Pain. 2018;14:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yang YJ, Hu L, Xia YP, et al. Resveratrol suppresses glial activation and alleviates trigeminal neuralgia via activation of AMPK. J Neuroinflammation. 2016;13:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Sharma S, Kulkarni SK, Chopra K. Effect of resveratrol, a polyphenolic phytoalexin, on thermal hyperalgesia in a mouse model of diabetic neuropathic pain. Fundam Clin Pharmacol. 2007;21:89–94. [DOI] [PubMed] [Google Scholar]
- 136.Das V, Kroin JS, Moric M, et al. AMP-activated protein kinase (AMPK) activator drugs reduce mechanical allodynia in a mouse model of low back pain. Reg Anesth Pain Med. 2019;44:1010–1014. [DOI] [PubMed] [Google Scholar]
- 137.Liu Y, Li J, Li H, et al. AMP-activated protein kinase activation in dorsal root ganglion suppresses mTOR/p70S6K signaling and alleviates painful radiculopathies in lumbar disc herniation rat model. Spine (Phila Pa 1976). 2019;44:E865–E72. [DOI] [PubMed] [Google Scholar]
- 138.Melemedjian OK, Khoutorsky A, Sorge RE, et al. mTORC1 inhibition induces pain via IRS-1-dependent feedback activation of ERK. Pain. 2013;154:1080–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Melemedjian OK, Asiedu MN, Tillu DV, et al. Targeting adenosine monophosphate-activated protein kinase (AMPK) in preclinical models reveals a potential mechanism for the treatment of neuropathic pain. Mol Pain. 2011;7:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Augusto PSA, Braga AV, Rodrigues FF, et al. Metformin antinociceptive effect in models of nociceptive and neuropathic pain is partially mediated by activation of opioidergic mechanisms. Eur J Pharmacol. 2019;858:172497. [DOI] [PubMed] [Google Scholar]
- 141.Das V, Kroin JS, Moric M, et al. Antihyperalgesia effect of AMP-activated protein kinase (AMPK) activators in a mouse model of postoperative pain. Reg Anesth Pain Med. 2019;44:781–786. [DOI] [PubMed] [Google Scholar]
- 142.Ma J, Kavelaars A, Dougherty PM, et al. Beyond symptomatic relief for chemotherapy-induced peripheral neuropathy: targeting the source. Cancer. 2018;124:2289–2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Inyang KE, McDougal TA, Ramirez ED, et al. Alleviation of paclitaxel-induced mechanical hypersensitivity and hyperalgesic priming with AMPK activators in male and female mice. Neurobiol Pain. 2019;6:100037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mao-Ying QL, Kavelaars A, Krukowski K, et al. The anti-diabetic drug metformin protects against chemotherapy-induced peripheral neuropathy in a mouse model. PLoS One. 2014;9:e100701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pereira AF, Pereira LMS, Silva CMP, et al. Metformin reduces c-Fos and ATF3 expression in the dorsal root ganglia and protects against oxaliplatin-induced peripheral sensory neuropathy in mice. Neurosci Lett. 2019;709:134378. [DOI] [PubMed] [Google Scholar]
- 146.Ludman T, Melemedjian OK. Bortezomib and metformin opposingly regulate the expression of hypoxia-inducible factor alpha and the consequent development of chemotherapy-induced painful peripheral neuropathy. Mol Pain. 2019;15:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]; • In mouse CIPN model the mTOR target protein HIF1α triggered onset and maintenance of tactile allodynia. Metformin prevented onset of pain via down-regulation of HIF1α (associated with elevated AMPK activity and reduced phosphorylated S6). However, metformin could not reverse tactile allodynia in CIPN.
- 147.Bhadri N, Sanji T, Madakasira Guggilla H, et al. Amelioration of behavioural, biochemical, and neurophysiological deficits by combination of monosodium glutamate with resveratrol/alpha-lipoic acid/coenzyme Q10 in rat model of cisplatin-induced peripheral neuropathy. ScientificWorldJournal. 2013;2013:565813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Selvarajah D, Kar D, Khunti K, et al. Diabetic peripheral neuropathy: advances in diagnosis and strategies for screening and early intervention. Lancet Diabetes Endocrinol. 2019;7:938–948. [DOI] [PubMed] [Google Scholar]
- 149.Tesfaye S, Selvarajah D. Advances in the epidemiology, pathogenesis and management of diabetic peripheral neuropathy. Diabetes Metab Res Rev. 2012;28 Suppl 1:8–14. [DOI] [PubMed] [Google Scholar]
- 150.Vinik AI, Nevoret ML, Casellini C, et al. Diabetic neuropathy. Endocrinol Metab Clin North Am. 2013;42:747–787. [DOI] [PubMed] [Google Scholar]
- 151.Pop-Busui R, Boulton AJ, Feldman EL, et al. Diabetic neuropathy: a position statement by the american diabetes association. Diabetes Care. 2017;40:136–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Abbott CA, Malik RA, van Ross ER, et al. Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K.. Diabetes Care. 2011;34:2220–2224.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Partanen J, Niskanen L, Lehtinen J, et al. Natural history of peripheral neuropathy in patients with non-insulin-dependent diabetes mellitus. N Engl J Med. 1995;333:89–94. [DOI] [PubMed] [Google Scholar]
- 154.Liu X, Xu Y, An M, et al. The risk factors for diabetic peripheral neuropathy: a meta-analysis. PLoS One. 2019;14:e0212574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ward JD, Fisher DJ, Barnes CG, et al. Improvement in nerve conduction following treatment in newly diagnosed diabetics. Lancet. 1971;297:428–431. [DOI] [PubMed] [Google Scholar]
- 156.Pop-Busui R, Martin C. Neuropathy in the DCCT/EDIC-what was done then and what we would do better now. Int Rev Neurobiol. 2016;127:9–25. [DOI] [PubMed] [Google Scholar]
- 157.Guo X, Tao X, Tong Q, et al. Impaired AMPK-CGRP signaling in the central nervous system contributes to enhanced neuropathic pain in highfat dietinduced obese rats, with or without nerve injury. Mol Med Rep. 2019;20:1279–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tanaka Y, Uchino H, Shimizu T, et al. Effect of metformin on advanced glycation endproduct formation and peripheral nerve function in streptozotocin-induced diabetic rats. Eur J Pharmacol. 1999;376:17–22. [DOI] [PubMed] [Google Scholar]
- 159.Ma J, Yu H, Liu J, et al. Metformin attenuates hyperalgesia and allodynia in rats with painful diabetic neuropathy induced by streptozotocin. Eur J Pharmacol. 2015;764:599–606. [DOI] [PubMed] [Google Scholar]
- 160.Lin JY, Huang XL, Chen J, et al. Stereological study on the number of synapses in the rat spinal dorsal horn with painful diabetic neuropathy induced by streptozotocin. Neuroreport. 2017;28:319–324. [DOI] [PubMed] [Google Scholar]
- 161.Lin JY, He YN, Zhu N, et al. Metformin attenuates increase of synaptic number in the rat spinal dorsal horn with painful diabetic neuropathy induced by type 2 diabetes: a stereological study. Neurochem Res. 2018;43:2232–2239. [DOI] [PubMed] [Google Scholar]
- 162.Los DB, Oliveira WH, Duarte-Silva E, et al. Preventive role of metformin on peripheral neuropathy induced by diabetes. Int Immunopharmacol. 2019;74:105672. [DOI] [PubMed] [Google Scholar]
- 163.Kim SH, Park TS, Jin HY. Metformin preserves peripheral nerve damage with comparable effects to alpha lipoic acid in streptozotocin/high-fat diet induced diabetic rats. Diabetes Metab J. 2020. DOI: 10.4093/dmj.2019.0190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wile DJ, Toth C. Association of metformin, elevated homocysteine, and methylmalonic acid levels and clinically worsened diabetic peripheral neuropathy. Diabetes Care. 2010;33:156–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ciric D, Martinovic T, Petricevic S, et al. Metformin exacerbates and simvastatin attenuates myelin damage in high fat diet-fed C57BL/6 J mice. Neuropathology. 2018;38:468–474. [DOI] [PubMed] [Google Scholar]
- 166.Gupta K, Jain A, Rohatgi A. An observational study of vitamin b12 levels and peripheral neuropathy profile in patients of diabetes mellitus on metformin therapy. Diabetes Metab Syndr. 2018;12:51–58. [DOI] [PubMed] [Google Scholar]
- 167.Miller JW. Proton pump inhibitors, H2-receptor antagonists, metformin, and vitamin B-12 deficiency: clinical implications. Adv Nutr. 2018;9:511S–8S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ozturk E, Arslan AKK, Yerer MB, et al. Resveratrol and diabetes: a critical review of clinical studies. Biomed Pharmacother. 2017;95:230–234. [DOI] [PubMed] [Google Scholar]
- 169.Brasnyo P, Molnar GA, Mohas M, et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br J Nutr. 2011;106:383–389. [DOI] [PubMed] [Google Scholar]
- 170.Seyyedebrahimi S, Khodabandehloo H, Nasli Esfahani E, et al. The effects of resveratrol on markers of oxidative stress in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled clinical trial. Acta Diabetol. 2018;55:341–353. [DOI] [PubMed] [Google Scholar]
- 171.Timmers S, de Ligt M, Phielix E, et al. Resveratrol as add-on therapy in subjects with well-controlled type 2 diabetes: a randomized controlled trial. Diabetes Care. 2016;39:2211–2217. [DOI] [PubMed] [Google Scholar]
- 172.Kumar A, Kaundal RK, Iyer S, et al. Effects of resveratrol on nerve functions, oxidative stress and DNA fragmentation in experimental diabetic neuropathy. Life Sci. 2007;80:1236–1244. [DOI] [PubMed] [Google Scholar]
- 173.Kumar A, Sharma SS. NF-kappaB inhibitory action of resveratrol: a probable mechanism of neuroprotection in experimental diabetic neuropathy. Biochem Biophys Res Commun. 2010;394:360–365. [DOI] [PubMed] [Google Scholar]
- 174.Yang W, Cai X, Wu H, et al. Associations between metformin use and vitamin B12 levels, anemia, and neuropathy in patients with diabetes: a meta-analysis. J Diabetes. 2019;11:729–743. [DOI] [PubMed] [Google Scholar]
- 175.Stabler SP. Vitamin B12Deficiency. N Engl J Med. 2013;368:149–160. [DOI] [PubMed] [Google Scholar]
- 176.Ahmed MA, Muntingh GL, Rheeder P. Perspectives on peripheral neuropathy as a consequence of metformin-induced vitamin B12 deficiency in T2DM. Int J Endocrinol. 2017;2017:2452853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Adams JF, Clark JS, Ireland JT, et al. Malabsorption of vitamin B12 and intrinsic factor secretion during biguanide therapy. Diabetologia. 1983;24:16–18. [DOI] [PubMed] [Google Scholar]
- 178.Bauman WA, Shaw S, Jayatilleke E, et al. Increased intake of calcium reverses vitamin B12 malabsorption induced by metformin. Diabetes Care. 2000;23:1227–1231. [DOI] [PubMed] [Google Scholar]
- 179.Scarpello JHB, Hodgson E, Howlett HCS. Effect of metformin on bile salt circulation and intestinal motility in Type 2 diabetes mellitus. Diabetic Med. 1998;15:651–656. [DOI] [PubMed] [Google Scholar]
- 180.Alvarez M, Sierra OR, Saavedra G, et al. Vitamin B12 deficiency and diabetic neuropathy in patients taking metformin: a cross-sectional study. Endocr Connect. 2019;8:1324–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.de Jager J, Kooy A, Lehert P, et al. Long term treatment with metformin in patients with type 2 diabetes and risk of vitamin B-12 deficiency: randomised placebo controlled trial. BMJ. 2010;340:c2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Campbell JM, Stephenson MD, de Courten B, et al. Metformin use associated with reduced risk of dementia in patients with diabetes: a systematic review and meta-analysis. J Alzheimers Dis. 2018;65:1225–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]