

Abstract
In a phase 1/2, open-label dose escalation trial, we delivered rAAVrh74.MCK.GALGT2 (also B4GALNT2) bilaterally to the legs of two boys with Duchenne muscular dystrophy using intravascular limb infusion. Subject 1 (age 8.9 years at dosing) received 2.5 × 1013 vector genome (vg)/kg per leg (5 × 1013 vg/kg total) and subject 2 (age 6.9 years at dosing) received 5 × 1013 vg/kg per leg (1 × 1014 vg/kg total). No serious adverse events were observed. Muscle biopsy evaluated 3 or 4 months post treatment versus baseline showed evidence of GALGT2 gene expression and GALGT2-induced muscle cell glycosylation. Functionally, subject 1 showed a decline in 6-min walk test (6MWT) distance; an increase in time to run 100 m, and a decline in North Star Ambulatory Assessment (NSAA) score until ambulation was lost at 24 months. Subject 2, treated at a younger age and at a higher dose, demonstrated an improvement over 24 months in NSAA score (from 20 to 23 points), an increase in 6MWT distance (from 405 to 478 m), and only a minimal increase in 100 m time (45.6–48.4 s). These data suggest preliminary safety at a dose of 1 × 1014 vg/kg and functional stabilization in one patient.
Keywords: Duchenne muscular dystrophy, AAV, gene therapy, GALGT2, B4GALNT
Graphical abstract
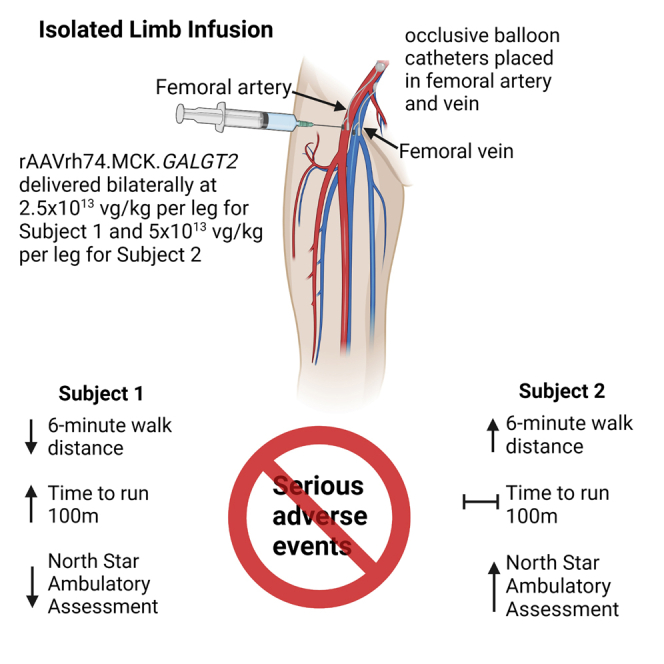
Results of a first-in-human phase 1/2a clinical trial are reported wherein two patients with Duchenne muscular dystrophy were given GALGT2 gene therapy. The study shows possible stabilization of muscle function in one of two subjects, who was treated with a higher dose and at a younger age.
Introduction
Duchenne muscular dystrophy (DMD), a severe X-linked form of muscle degeneration, occurs in approximately 1 in 5,200 live male births.1 Mutations that disrupt the open reading frame of the DMD gene result in absent or severely abnormal expression of dystrophin at the subsarcolemmal membrane in muscle fibers; as a result, muscle fiber membrane integrity is impaired, leading to muscle dysfunction by a variety of structural and cell signaling pathways (as reviewed elsewhere).2 Muscle fiber degeneration and subsequent replacement of muscle by fat and fibrosis lead to progressive motor impairments that are typically apparent to parents around 3–5 years of age but progress to loss of ambulation, typically by the age of 12 years in the pre-steroid treatment era, although current standard treatments (of prednisone or deflazacort) may delay loss of ambulation by up to 2–5 years.3 Following loss of ambulation, symptoms of respiratory muscle weakness and cardiomyopathy become more prominent, typically leading to death in the third decade.4
Although corticosteroids remain the mainstay of treatment, therapies directed at restoring dystrophin expression include recently approved therapies comprising phosphorodiamidate morpholino oligomers (PMOs) intended to induce altered pre-mRNA splicing to restore an open reading frame and thus expression of an internally deleted but partially functional protein. Several such PMOs have gained US Food and Drug Administration (FDA) approval, each based largely on a small but statistically significant increase in dystrophin expression, but each of these agents addresses only a specific exon within the gene; as a result, this therapy is limited only to subjects with mutations amenable to skipping exons 45, 51, or 53.5, 6, 7 Expression of dystrophin by adeno-associated viral (AAV) delivery is complicated by the ∼11.5-kb size of the DMD coding region; as a result, efforts have been concentrated on delivery of a micro-dystrophin gene that fits within the ∼5-kb packaging limit of single-stranded AAV genomes. These microdystrophins generally lack the majority of the dystrophin central rod domain, which consists of 24 modular spectrin-like repeat elements, as well as the C-terminal domains potential important to cell signaling, while maintaining the critical N-terminal actin-binding domain 1 (ABD1) and the C-terminal cysteine-rich domain critical to binding to β-dystroglycan (βDG); several versions of AAV-micro-dystrophin vectors are currently in clinical trials,8 and preliminary results suggest efficacy using measures of motor function such as the North Star Ambulatory Assessment (NSAA).9
Here we report the results of a first-in-human trial using an alternate approach intended as a surrogate gene therapy for muscular dystrophy. The rAAVrh74.MCK.GALGT2 vector contains a transgene encoding human GALGT2—alternatively called B4GALNT2—expressed under control of the skeletal and cardiac muscle-specific MCK promoter. In adult skeletal muscle, the GALGT2 protein acts as a β1,4 N-acetylgalactosaminyltransferase to glycosylate α-dystroglycan,10, 11, 12, 13, 14, 15, 16, 17, 18 and it expression is normally confined to the neuromuscular and myotendinous junctions, analogous to the similar domain-restricted expression of other synaptic dystroglycan-binding proteins, such as utrophin, plectin 1f, laminin α4, laminin α5, and agrin, in adult skeletal muscle.19, 20, 21, 22, 23, 24 Multiple lines of evidence provide a rationale for its use in DMD patients. First, overexpression of GALGT2 not only induces glycosylation of α-dystroglycan but increases the ectopic expression of its normally synaptic binding partners in both mdx and wild-type skeletal muscles, and overexpression of many of these partners has been shown to inhibit the development of muscular dystrophy.12,25, 26, 27 Second, GALGT2 overexpression prevents eccentric contraction-induced muscle injury in both dystrophic mdx and wild-type mouse muscles.11,26 Third, GALGT2 overexpression in mdx mouse heart can prevent the loss of cardiac function as the mice age, a finding of potentially significant clinical relevance in the DMD population.18 Finally, GALGT2 overexpression has been shown to inhibit the development of muscle pathology in mouse models of four different forms of muscular dystrophy, the mdx model of DMD,11,12,14,17 the dyW model of congenital muscular dystrophy 1A (MDC1A),15 the Sgca−/− model of limb-girdle muscular dystrophy 2D (LGMD2D),16 and the FKRPP448L model of limb-girdle muscular dystrophy 2I (LGMD2I),13 raising the possibility of a membrane-stabilizing therapeutic that is broadly applicable to different genetic MD forms. GALGT2 overexpression has also shown improvements in muscles of some golden retriever muscular dystrophy dogs, although these functional results did not reach significance in the four subjects studied.28
For the current study, we delivered the vector into both femoral arteries using an isolated limb infusion (ILI) approach. The choice of this method was determined in part by feasibility, as the amount of clinical vector was limited. The clinical lot had originally been produced with an intent to perform the first-in-human study as a safety study of intramuscular (IM) injection into the extensor digitorum brevis (EDB) muscle of each foot, following the model of a prior gene transfer trial in a limb-girdle muscular dystrophy.29 However, subsequent AAV therapeutic trials demonstrated sufficient safety information regarding systemic AAV delivery to render an IM trial—with its obligate generation of immune responses to the AAVrh74 capsid—unacceptable. Delivery of rAAVrh74.MCK.GALGT2 via ILI to non-human primates demonstrated sufficient transgene expression to anticipate the possibility of clinical benefit,17 and the trial was redesigned prior to enrollment of any subjects. Subsequent dose ranging studies in mice further supported safety and expression following systemic delivery of the vector.30
Results
Enrollment
Sufficient vector manufactured under current Good Manufacturing Practic (cGMP) conditions was available to treat an anticipated two subjects. Four subjects were enrolled via the informed consent process, but only two proceeded to gene transfer, as subject GAL-DMD-02 failed screening due to an unexplained laboratory abnormality and subject GAL-DMD-03 had an AAVrh74 antibody titer of 1:1,600. Both subject GAL-DMD-01 (henceforth subject 1) and GAL-DMD-04 (henceforth subject 2) were dosed with rAAVrh74.MCK.GALGT2 using ILI at the vector genome (vg)/kg doses indicated in Table 1. Balloon catheters were inserted into both the femoral artery and vein and inflated in the upper leg to occlude blood flow, with heparin added to prevent clotting. After a 1-min 2-mL/kg flush of lactated Ringer’s solution, AAV was injected into the femoral artery of the leg, below the balloon, over 1–2 min and allowed to dwell in the leg for 10 min. Following delivery, an additional flush of lactated Ringer’s was given over 1 min, after which the balloons were deflated and catheters removed, restoring leg circulation to the rest of the body. Both legs were treated in each subject. Subject 1 carried an out-of-frame DMD exon duplication and was treated at age 8.9 years old. At the time of enrollment, he was on a daily but non-standard corticosteroid dose of 0.26 mg/kg of prednisone. Corticosteroids are standard of care for DMD patients, as treatment can delay loss of ambulation for several years.31 After gene therapy, he was treated per protocol with prednisone 1 mg/kg/day, an increase geared toward dampening immune response to the vector, and a taper was initiated at day 50 with a decrease to the standard dose of 0.75 mg/kg/day. Due to weight gain (discussed in adverse events), tapering continued until a dose of 0.21 mg/kg/day, roughly equivalent to his pre-enrollment dosing regimen, was reached by day 211. Subject 2 carried a nonsense mutation within exon 19 and was treated at 6.9 years old. At the time of enrollment, he was on a standard daily corticosteroid dose of deflazacort 0.9 mg/kg/day. Following treatment per protocol with prednisolone 1 mg/kg/day after gene transfer, he was tapered on day 63 to deflazacort 0.8 mg/kg day.
Table 1.
Enrollment status
| Subject ID | Mutation | At gene transfer |
||
|---|---|---|---|---|
| Age (years) | Weight (kg) | Dose | ||
| Subject 1 | duplication exon 22–41 | 8.9 | 37.8 | 2.5 × 1013 vg/kg per leg (5.0 × 1013 vg/kg bilateral total) |
| Subject 2 | nonsense mutation, exon 19 (c.2332C>T/p.Gln778Ter) | 6.9 | 20.9 | 5.0 × 1013 vg/kg per leg (1.0 × 1014 vg/kg bilateral total) |
Enrollment and dosage information for subjects 1 and 2. All AAV vector genome (vg) doses were relative to kilograms of total body weight.
Adverse events
The ILI procedure was generally well tolerated. During infusion, neither subject showed signs of acute toxicity, and no subject reached the predefined procedural stopping rules of significant deterioration in vital signs or medical judgment of intolerability. Both subjects had adverse events (AEs) related to ILI. Subject 1 had bruising at the groin site that was still present at the day 14 examination but resolved by day 30. Subject 2 had bleeding at the femoral catheterization site on the day of gene transfer, and more extensive bruising that resolved by the day 60 examination point. He also had a single episode of vomiting on day 1.
A full list of AEs is found in Table S1. Both subjects had transient decreases in lymphocyte count following gene transfer, although the timing of these abnormalities (as noted in Table S1) cannot clearly be associated with gene transfer. Subject 2 had a mildly low hemoglobin level for days 1–14 post gene transfer, which was attributed to the bleeding at the femoral site on top of the intensive laboratory blood sample regimen; it resolved without treatment. Subject 1 was noted to have a minimally Cushingoid face (first noted at day 60), and weight gain (noted at day 90), leading to a continued decrease in his steroid dose to pre-enrollment levels (as discussed above). During the course of the study, he was noted to have a decreased cardiac ejection fraction of 54% at 18 months post gene transfer (at age 10.5 years), which was attributed to progression of disease; subject 2’s cardiac status showed no significant change.
Serum biochemistry, serum hematology, and urinalysis
Serum chemistry studies are shown in Tables S2A and S2B, and Figure 1. There were no significant unexpected abnormalities noted. Consistent with typical findings in patients with DMD,32 serum transaminase levels (aspartate transaminase [AST], alanine transaminase [ALT]) in both subjects were elevated at baseline, as was serum creatine kinase (CK). Serum CK diminished compared with baseline in both subjects, more strikingly in subject 1, decreasing from 21,583 U/L to 5,930 U/L. However, in this subject, this may reflect diminished activity due to loss of ambulation (discussed below). Subject 2 had serum CK values at month 24 (27,776 U/L) that remained decreased compared with the screening value (32,819 U/L). No elevation in serum gamma-glutamyl transferase (GGT), or other evidence of hepatic dysfunction, was noted. Hematologic and coagulation study results are shown in Tables S3 and S4. No major abnormalities were noted for either subject; specifically, no negative impact on platelet count was seen. Urinalysis (Tables S5 and S6) showed no significant clinical findings, although red blood cells were found in subject 1 at baseline and at days 1 and 30 and a urobilinogen level of 2mg/dL was present in subject 2 at days 1 and 2 post treatment.
Figure 1.

Serum chemistry values
Serum alanine transaminase (ALT), aspartate transaminase (AST), and gamma-glutamyl transferase (GGT) are shown in the upper panel for both subjects. Normal values are as follows: <40 U/L ALT, 15–50 U/L AST, and 8–78 U/L GGT. Creatine kinase (CK) values are plotted in the lower panel for both subjects. Dotted horizontal lines mark the upper boundary of the normal CK range (37–430 U/L). The dashed vertical steroid taper line marks the time point when gradual steroid dose tapering from the post-gene-transfer dose (of 1 mg/kg) began for each subject.
Immune responses
Both subjects demonstrated the expected anti-AAVrh74 antibody responses after gene transfer, as shown in Figure 2. rAAVrh74 antibody titers were not detectable for either subject prior to treatment (<1:12.5). The AAVrh74 antibody titer for subject 1 rose to a maximum of 1:819,200 at the month 6 visit. For subject 2, the maximum was 1:204,800 at multiple visits (day 90, month 6, and month 18), with an intervening value of 1:102,400 at the month 12 and 24 visits. T cell responses as measured by enzyme-linked immune absorbent spot (ELISpot) (Figure 2) showed minimally positive T cell responses to AAVrh74 capsid and GALGT2 peptide pools at varying time points. Overlapping peptides encompassing the entire protein coding region were divided into three pools for rAAV capsid and two pools for GALGT2. An expected peak was observed to all three capsid pools at day 14 for both subjects that subsided by day 30. For subject 2, and additional peak above 100 SFC/106 peripheral blood mononuclear cells (PBMCs) was observed at 18 months in two of three capsid pools and one of two GALGT2 pools that then resolved by 24 months. At no point were these considered clinically significant or correlated to clinical or biochemical findings.
Figure 2.

Immune response markers
(A) AAVrh74 antibody titer by ELISA immunoassay for both subjects. ELISA titers >1:50 (horizontal line) are considered positive. (B) ELISpot immunoassay values for both subjects for three AAVrh74 peptide pools and two GALGT2 peptide pools. Values ≥50 SPF/1 × 106 PBMCs (horizontal line) are considered positive. The dashed vertical steroid taper line marks the time point when gradual steroid dose tapering from the post-gene-transfer dose (of 1 mg/kg) began for each subject.
Histopathology
Open muscle biopsies were taken at baseline and at 3 months (subject 2) or 4 months (subject 1) post treatment, and Vacora needle biopsies were taken at 6 and 12 months post treatment (both subjects). Hematoxylin and eosin staining of biopsy muscle sections showed more dystrophic changes, in particular fat infiltration, in subject 1 than in subject 2, both at baseline and following treatment (Figure S1), consistent with subject 1’s more advanced age at the time of treatment.
AAV muscle biodistribution
As shown in Table S7, pre-infusion baseline biopsies showed no detectable vg present (or less than 50 vg/μg of genomic DNA). All post-treatment biopsies showed vg signals, suggesting AAV transduction of -muscle tissue. Subject 1, who received a total ILI dose of 5 × 1013 vg/kg, showed lower transduction at the 4-month time point (2.5 ± 0.6 × 104 vg/kg or 0.16 vg/nucleus, n = 2) than did subject 2, who received a total ILI dose of 1 × 1014 vg/kg, at the 3-month time point (2.3 ± 0.7 × 105 vg/μg or 1.5 vg/nucleus). AAV transduction was also evident in Vacora needle biopsies for both subjects at 6 and 12 months; however, transduction levels were reduced at these time points relative to measures in the 3- or 4-month open-biopsy samples. Such differences may reflect the high biopsy-to-biopsy variability in transduction found with the ILI method in non-human primates.17
GALGT2 gene expression
Baseline muscle specimens from both subject 1 and subject 2 showed no difference in GALGT2 gene expression compared with three healthy control samples (Figure S2). Post treatment, however, muscle from both subjects showed elevated GALGT2 gene expression with two primer-probe sets at all time points. GALGT2 gene expression was maximal in the 4- or 3-month open biopsy. For subject 1, gene expression was elevated on average 6- to 8-fold, while in subject 2 (treated with the higher dose) expression was elevated 25- to 30-fold above healthy control and pre-treatment baseline levels.
Immunostaining for the cytotoxic T cell glycan antigen expression and by western blotting for the GALGT2 protein
The percentage of fibers staining for cytotoxic T cell (CT) glycan made by GALGT2, marked by GalNAc-positive Wisteria floribunda agglutinin (WFA) staining, was increased from baseline in both subjects at the first biopsy time point (4 months in subject 1, and 3 months in subject 2) compared with healthy control (Figure 3). The proportion of positive fibers increased from 1.59% at baseline to 11.68% at 4 months (a 7.4-fold change) in subject 1, and from 0.49% at baseline to 5.76% at 3 months (an 11.69-fold change) in subject 2 (Figure 3A). The mean intensity of fibers (Figure 3B) was minimally increased at baseline compared with healthy control tissue (1.50 in subject 1 and 1.33 in subject 2), with minimal further increases of approximately 0.4 at the first time point (1.95 in subject 1 and 1.72 in subject 2).
Figure 3.

CT glycan antigen expression by WFA-FITC labeling of GalNAc in biopsy sections
(A) The percentage of GalNAc-positive fibers for both subjects across time points is shown on the left, and the same data are represented as a fold change versus baseline (horizontal dashed line) for each subject on the right. Data for fibers from all blocks representing a given time point was pooled to produce a single value for each time point. (B) GalNAc intensity in all fibers normalized to the healthy control median intensity (horizontal dashed line) shown for each time point on the left, and for only GalNAc-positive muscle fibers on the right. Each point represents a single muscle fiber, and the solid horizontal line at each time point represents the mean of all muscle fibers. (C) WFA-FITC fluorescence (green) labeling GalNAc in representative muscle regions counter-stained for spectrin (magenta) at baseline and the first post-treatment time point for each subject, compared with a representative healthy control sample. Fiber GalNAc positivity and normalized fiber GalNAc intensity analysis results are shown in the third and fourth columns using color-coded heatmaps. Color keys indicating the conversion scale for percentage GalNAc-positive perimeter and fold GalNAc intensity (normalized to healthy control median) are shown below each column. Gray color indicates regions of high connective tissue accumulation that were masked to avoid interference with the automated analysis. All regions are 2 × 3 mm.
Considerable inhomogeneity in the distribution of GalNAc-positive signal was observed in biopsy samples from both subjects at the first time point (Figure 4). Regional compartmentalization of whole-section images from the first post-treatment time point by apparent fascicular organization resulted in identification of highly expressing regions (up to 24.1% positive fibers) and lower expressing regions (as low as 1.83% positive fibers), which may reflect differences in microvascular supply. In addition, staining was reduced at later 6- and 12-month time points (Figure S3), much as was seen for AAV biodistribution (Table S7) and GALGT2 gene expression (Figure S2).
Figure 4.

Regional variability of GalNAc expression at the first post-treatment time point
(A) GalNAc signal quantification for high- and low-expressing regions separated along the fascia in whole-muscle sections from the first treated time point. On the right, each point represents a single muscle fiber and the solid horizontal line at each time point represents the mean of all muscle fibers. The horizontal dashed line represents the healthy control median GalNAc intensity. (B) Images of tissue sections from subjects 1 and 2 showing variable regional GalNAc expression (green), with high-expressing fiber regions outlined in red and low-expressing regions outlined in cyan. Fiber GalNAc positivity analysis results are shown in the lower row of images using color-coded heatmaps. The color key indicating the conversion scale for percentage GalNAc-positive perimeter is shown below the images. Scale bar, 1mm.
Because GALGT2 is normally only expressed at the neuromuscular junction (in synaptic Golgi), it is often not possible to reproducibly observe protein via western blots in whole-cell muscle lysates. We therefore concentrated GALGT2 protein by precipitation with fetuin agarose, a GALGT2 substrate, to assess protein expression (Figure S4). Only sufficient biopsy material from subject 1 was available for completion of this analysis. GALGT2 protein was elevated in both post-treatment samples of the biopsy taken from subject 1 relative to two baseline samples and relative to two different biopsy samples from healthy controls. GALGT2 protein can be expressed in a long form (62 kDa), which was the form used for the protein standard, or a shorter form (56 kDa), which is the form expressed by the GALGT2 transgene. Unexpectedly, rAAVrh74.MCK.GALGT2 treatment elevated expression of both forms. If both bands were quantified together, this resulted in a 3.2 ± 0.9-fold elevation in protein expression after treatment (4 months) for subject 1 relative to pre-treatment baseline (p = 0.057 by Student’s unpaired two-tailed t test).
Expression of dystrophin surrogate genes and proteins
We compared relative gene expression for surrogate genes known to be induced by GALGT2 gene overexpression in mouse or macaque skeletal muscles15, 16, 17 as well as dystrophin. These included probes for utrophin, dystroglycan, plectin 1, laminin α2, laminin α4 and laminin α5, integrin α7, and integrin β1 (Figure S5). None of these genes showed significant induction of gene expression as the result of treatment in either patient relative to baseline pre-treatment levels. Baseline levels of gene expression, however, were decreased for dystrophin and plectin 1 in subject 1 and subject 2 relative to healthy controls, while they were increased for laminin α4 and laminin α5.
We analyzed expression of surrogate proteins by western blot (Figure 5). 427-kDa native protein bands for dystrophin were identified in all normal controls, but no dystrophin protein was identified in any samples from subject 1 or subject 2, as expected for DMD. Western blots for utrophin (utrophin and DRP2) showed bands that were elevated in subject 1 and subject 2 in baseline biopsies relative to healthy controls, again as expected for DMD, but there was no further induction of utrophin protein expression by treatment in either subject. Α-Dystroglycan (αDG, IIH6), β-dystroglycan (βDG), and α-sarcoglycan (αSG) signals were present in all control muscles. βDG and αSG blots were highly reduced in both subject 1 and subject 2 samples, while IIH6 blots were variable and sometimes increased. As with utrophin, however, there was no induction of these proteins by treatment.
Figure 5.

Protein expression of dystrophin, dystrophin-associated glycoproteins, and utrophin
(A) Western blot images of dystrophin (DYS1 and DYS2), utrophin (DRP2 and utrophin), α-dystroglycan (IIH6), β-dystroglycan (βDG), and α-sarcoglycan (αSG) bands are shown compared with their respective GAPDH bands for both subjects and for three healthy controls. (B) Quantification of the blots shown in (A), with mean ± SEM shown for each set of replicates. Individual points for subject 1 and 2 represent different blocks from the same muscle biopsy.
Because DMD subjects often have spontaneous exon skipping that may lead to low levels of dystrophin expression, expression that may affect their disease progression, we also performed a quantitative analysis of dystrophin immunofluorescence in subjects 1 and 2, both before and 4 or 3 months post treatment, respectively, and compared these data with dystrophin staining from biopsies of three healthy controls (Figure S6). Subject 1 showed between 6% and 8% of muscle fibers with positive dystrophin staining before and after treatment, while subject 2 showed 1% or fewer dystrophin-positive fibers. By contrast, healthy controls averaged 99.3% dystrophin-positive fibers. Measures of mean dystrophin signal intensity in subjects 1 and 2, normalized to staining intensity in healthy controls, correlated almost exactly with the percentage of positive fibers found in each instance (Figure S6).
Functional clinical findings
The results of clinical assessments for both subjects are summarized in Figure 6, Figure 7, and Figure S7. At time of gene transfer, subject 1—treated at the low dose—was 8.9 years old, with significant motor impairment (Figure 6). His 6-min walk test (6MWT) distance was 357 m. The time for him to walk 100 m was 103.6 s, or 28.0% of the predicted value for age-matched control cohorts. Consistent with these results, his NSAA was 18 out of a possible 34 points. During the 24 months post gene transfer, his loss of independent motor function progressed; he lost the ability to ambulate independently, with functional loss of ambulation occurring between time points representing age 10.4 and 10.9 years. His NSAA similarly diminished to a score of 2 at his 24-month visit. Nevertheless, his forced vital capacity percentage (FVC%) predicted value did not decline, and in fact increased slightly from 85% at enrollment to 93% (Figure S7). It is important to note here that, while the ILI treatment focuses transduction in the leg for 10 min, remaining vector is released into the general circulation after that time, and can stimulate expression in non-targeted muscles, including the diaphragm and heart.17
Figure 6.

Functional tests
The 6-min walk (6MW) distance, 100-m time, time to rise, and North Star Ambulatory Assessment (NSAA) scores for both subjects are shown at baseline screening and at times after treatment. The “X” symbol represents time points at which the test could not be performed due to loss of function.
Figure 7.

Maximal voluntary isometric contraction testing
The maximum voluntary isometric contraction (MVIC) force is shown for right (R) and left (L) knee flexion and extension for both subjects. The plotted lines trace the average value of the R and L sides for each measure.
In contrast, subject 2—treated at the higher dose at the age of 6.9 years—showed an improvement in his 6MWT distance from 405 to 478 m, even though the percentage predicted value for the 100-m walk test (100MWT) diminished from 67.5% to 57.8% (Figure 6). Notably, the NSAA increased from 20 to 23 over the 24 months following gene therapy, representing improvements in scores for transition from lying to sitting (increasing from 1 to 2) and in the scores for the ability to hop for each leg (increasing from 0 to 1 on both the left and right legs). His FVC% at 24 months (81%) was only slightly diminished compared with enrollment (85%) (Figure S7). Notably, his parents report increased activity and diminished fatigability in play and at school over the same period.
Maximal voluntary isometric contraction testing (MVICT) results are shown in Figure 7. Subject 1 showed no improvement compared with his baseline values, with only one of four functions (left knee extension) remaining stable. Subject 2 showed improvement in force generation in knee extensor and flexor strength (averaging both limbs) through 18 months (+24% and +31%, respectively), with persistent improvement in the right leg at the 24 month examination.
Cardiac function as measured by ejection fraction (EF) was 64.7% at screening in subject 1; it decreased to 54% by month 18, but was 63% at 24 months. In subject 2, the EF at baseline was measured at 74%, which likely represented an overestimate; the lowest subsequent EF was 64% at days 90 and 180, with a value of 67% at month 24.
Discussion
This limited study of only two subjects sought to assess the safety of rAAVrh74.MCK.GALGT2 delivered by the ILI method to both legs. In these two subjects, our data show that such delivery is safe and well tolerated in this small cohort. Such a conclusion is necessarily preliminary, given the small number of subjects, but neither one demonstrated evidence for significant liver or other organ toxicity. Unfortunately, resources were limited at the time of production of the clinical lot, originally generated in anticipation of an IM study. Repurposing of the limited clinical lot for ILI allows an initial assessment of tolerability, with the most prominent AEs limited to the cannulization during the procedure itself.
Evidence for tolerability is provided by an absence of significant elevations in serum transaminase values beyond the values associated with DMD itself. Despite the large total dose each received, neither developed hematologic abnormalities such as thrombocytopenia, or developed sustained detectable T cell responses to transgene-encoded or viral antigens. In the case of the GALGT2 protein itself, this is as expected. Unlike the case with micro-dystrophin gene therapies, where expression of a non-self-dystrophin isoform occurs, GALGT2 is endogenously expressed, with very high expression, for example, in the colon.33 Its therapeutic effect occurs instead by overexpression specifically in heart and skeletal muscle, where its endogenous expression is typically quite low, in the case of skeletal muscle being typically confined to regions at the neuromuscular and myotendinous junction in adults.34,35
This study was not sufficiently powered to assess measures of clinical efficacy, except as exploratory outcomes. Nevertheless, functional outcomes measures in subject 2 are promising. Treated at a younger age than subject 1, and with a higher dose, subject 2 showed an increase in his NSAA score since treatment, a result inconsistent with the natural progression of NSAA scores over this age range in untreated DMD cohorts, where an average decline of eight points per year is expected.36 Parental reports of his activity—particularly decreased fatigability—are consistent with the significant increase in the 6MWT distance over the same period. Such results are consistent with micro-dystrophin gene therapy studies that suggest a greater impact for treatment for patients younger than 7 years, the age at which DMD subjects begin to show a pronounced decline in ambulation based on natural history studies. The relative lack of spontaneous dystrophin expression in muscle biopsies from subject 2 further suggests that residual dystrophin expression could not account for their minimal disease progression after treatment.
The evidence for significant CT glycan antigen expression—the marker and product of GALGT2 glycosyltransferase activity—in post-treatment biopsies was underwhelming, but clearly present. Increased expression was present in each biopsy at the first post-treatment biopsy, which was the predefined point for assessing expression. The absence of a clear increase in expression at 6 and 12 months must be interpreted in the light of differences in how the biopsy was obtained. Baseline and month 3 or 4 biopsies were open surgical biopsies, allowing a greater cross-sectional area to be examined, whereas 6- and 12-month biopsies were performed using the Vacora needle biopsy device. The smaller sampling area with the Vacora may result in random sampling of areas that express less GALGT2, a hypothesis suggested by regional analysis of the larger open-biopsy samples and by a previous study of this method using rAAVrh74.MCK.GALGT2 in rhesus macaques, where biopsy-to-biopsy variability in a heatmap taken throughout individual muscles showed significant variability in AAV transduction after ILI delivery.17 Similarly, sampling of elevations in expression of dystrophin surrogate proteins such as utrophin, which was not obviously increased in treated muscles, may result from sampling of biopsies with relatively low and variable GALGT2 expression (and from the high elevation in utrophin expression at baseline compared with healthy controls). Here, it is important to note that GALGT2 can still have therapeutic benefits in dystrophin-deficient mice where utrophin is not present.14
We suspect that these results may simply reflect insufficient dose escalation, a hypothesis that can be addressed by future systemic intravenous (i.v.) delivery trials. Notably, regional variability in expression across muscle sections was not seen in recent pre-clinical studies of systemic delivery designed to assess dose response, toxicity, and biodistribution in mice.30 There, i.v. delivery of 4.3 × 1014 vg/kg resulted in GALGT2-induced glycosylation in the majority of skeletal myofibers throughout the body and in almost all cardiomyocytes, importantly, without evidence of toxicity. Such pre-clinical i.v. studies suggest a target for a future clinical trial in DMD subjects.30
Materials and methods
Subjects
Subjects were identified among patients seen in the muscular dystrophy clinic at Nationwide Children’s Hospital. Due to the need for an invasive procedure (arterial and venous cannulation, as discussed below), the Data Safety Monitoring Board required the first subject to be no younger than prior research patients who had received gene transfer at our institution using a similar approach in a different trial37; following review of safety findings from the first subject, the age for enrollment was lowered to 4 years or older. Subjects were required to be ambulant and have completely defined DMD gene mutation using clinically accepted methods that interrogated all exons. In addition, they were required to have measurably impaired muscle function (defined as less than 80% of the predicted value for age in the 100MWT), but with sufficient muscle preservation to ensure assessment of muscle transfection based on clinical evaluation by the principal investigator (PI) and expert colleagues. This degree of preservation included ability to extend the knee fully against gravity, preserved ambulation with ability to walk ≥350 m during the 6MWT, a magnetic resonance image of the quadriceps showing preservation of sufficient muscle mass to permit transfection, ability to cooperate with muscle testing, and a stable regimen of corticosteroid therapy (including either prednisone, prednisolone, deflazacort or their generic forms) for at least 12 weeks prior to gene transfer. The major exclusionary criterion was the presence of rAAVrh74 binding antibody titers ≥1:50 as determined by ELISA immunoassay. Subjects were also screened to ensure that they did not have serum antibodies to the Sd(a) blood group antigen, an antigen whose synthesis requires GALGT2 expression.
Following informed consent obtained under a Nationwide Children’s Hospital (NCH) Institutional Review Board-approved protocol (IRB#17-00395), subjects underwent screening in two visits. The first visit included all relevant serologic, biochemical, and clinical testing. After these data were reviewed to confirm eligibility, muscle MRI and baseline muscle biopsy were performed.
Regardless of pre-gene transfer regimen, each subject was placed on a standardized regimen of 1.0 mg/kg of oral prednisone, beginning on day −1 and continuing thereafter. Tapering of prednisone to the pre-treatment regimen began after day 30, with a time course that varied depending on the biochemistry, hematologic, and immunologic laboratory results.
Vector delivery via ILI
Two subjects received vector in an ascending dose fashion. The first subject received the minimally efficacious dose as established by non-human primate studies, consisting of 2.5 × 1013 vg/kg per leg, delivered bilaterally (to total 5.0 × 1013 vg/kg per subject). A second subject received 5 × 1013 vg/kg per leg, delivered bilaterally (to total 1.0 × 1014 vg/kg per subject). The total vg dose for each subject was adjusted by rounding up the subject body weight to the closest kilogram. Biochemical and clinical safety data from five time points (days 1, 2, 7, 14, and 30) were reviewed and discussed with the DSMB prior to dose escalation to the higher dose.
Subjects participating in this trial received an isolated limb infusion of rAAVrh74.MCK.GALGT2 delivered via the femoral artery to both legs using methodology previously studied in non-human primates17 and used in another clinical trial.37 Under standard anesthetic care and using Doppler ultrasound guidance, the femoral artery and vein were accessed percutaneously via the Seldinger approach in a retrograde direction with side port containing vascular sheaths. Each subject was anticoagulated with a dose of 200 IU heparin/kg via a peripheral arm i.v. or vascular sheaths placed in the femoral vessels of the groin. The heparin was allowed to circulate for 3–5 min, at which time a blood sample was drawn and an activated clotting time (ACT) measured with the goal of >200 s from an anticipated base line of 90–120 s drawn systemically (peripheral i.v., peripheral artery line, or vascular sheaths placed in the femoral vessels of the groin). If the post heparin ACT was <200 s, a second 50-IU heparin/kg dose was administered. An immediate follow-up ACT was obtained with the targeted ACT goal remaining >200 s. This was repeated until ACT values were within the designated range.
Once the vascular sheaths were positioned and the subject anticoagulated with heparin, occlusive balloon catheters were placed and positioned in the femoral artery and vein such that, on balloon inflation, stasis of the lower limb vasculature occurred. This was assessed by fluoroscopy. With balloons inflated and stasis confirmed, a pre-flush volume of lactated Ringer’s solution (2 mL/kg/limb) was delivered over 1 min. The vector (8 mL/kg/limb) was then infused into the side port of the arterial vascular sheath over approximately 1–2 min and the leg remained vascularly isolated (balloons inflated) for the 10-min dwell following infusion. After completion of the vector infusion and dwell, a post-flush volume was delivered over 1 min (2 mL/kg/limb lactated Ringer’s) with the balloons still inflated. At the conclusion of the post-flush, the balloon catheters were deflated and removed along with the vascular sheaths. At this point, remaining vector was released systemically. Pressure was maintained on the entry sites and accessed vessels for 10–15 min or as long as necessary to maintain hemostasis.
Throughout the infusion, subjects were closely monitored for side effects, including continuous heart rate, respiratory rate, pulse oximetry, and blood pressure monitoring. Heart rate, respiratory rate, pulse oximetry, temperature, and blood pressure were measured before and immediately after the infusion, and at least every 5 min during the infusion, and repeated at 15 min post infusion. Subjects were monitored for bleeding, infusion to the extremity, and compartment syndrome. Evidence for compartment syndrome was assessed by direct limb visualization and palpation. Subjects were transferred from the catheterization suite to the cardiac stepdown unit when determined to be hemodynamically stable by the consulting anesthesiologist, and remained admitted to the hospital for up to 48 h after gene transfer. Vital signs (heart rate, respiratory rate, temperature, and blood pressure) were obtained hourly for 4 h following the injection and then every 4 h until discharge.
Muscle biopsy
Muscle biopsy was performed using standard clinical protocols as either open biopsy (at baseline and at 3–4 months post transfer) or needle biopsy using the Vacora probe device (at 6 and 12 months). Notably, the protocol defined a 3-month post-gene-transfer time point for biopsy, but, due to intervening familial circumstances, one subject could not return for biopsy until 4 months post gene transfer. This was discussed with the Data Safety Monitoring Board (DSMB).
In each case, muscle MRI was reviewed to ensure adequate muscle tissue to target for muscle biopsy. Baseline and 3-month biopsies were performed in the vastus medialis muscles of either leg for the first biopsy and the opposite leg for the second. Needle biopsies were performed in the vastus lateralis muscles in the same alternating fashion, with the exception of subject 1 at 6 months, where the medial gastrocnemius muscle was biopsied following review of the MRI images that suggested inadequate contractile tissue for assessment in the intended vastus lateralis region. Muscle biopsy tissue was divided in half for mounting, and snap frozen in isopentane cooled in liquid nitrogen using standard techniques. For all evaluations listed below, the investigators were blinded to baseline versus post-treatment sample identification until each appropriate analysis was complete.
Gene expression
Samples were blinded before RNA extraction. RNA was extracted from 10–25 mg of muscle with Trizol and the RNA was purified using the Direct-zol RNA Miniprep kit (Zymo Research, Irvine, CA). Then 300 ng of RNA was used with a high-capacity cDNA archive kit to reverse transcribe cDNA according to manufacturer’s guidelines. Samples were subjected to real-time PCR in duplicate on an ABI 7500 Sequence Detection system (Applied Biosystems) using TaqMan gene expression master mix (Applied Biosystems, Foster City, CA). We used two primer-probe sets to analyze expression of the human GALGT2 gene. One primer-probe set spanned exons 2 and 3, in the stem domain, and one primer/probe set spanned exons 9 and 10, in the catalytic domain. qRT-PCR was run for 40 cycles. Undetected values were given a crossing threshold (Ct) assignment of 40, which was the value found in almost all healthy (otherwise normal) controls and in both patients at baseline. As such, the elevation in GALGT2 gene expression in treated biopsies is a minimal estimate of fold induction, based on the 40-cycle protocol. These healthy control values were set to 1 to show the fold induction in all other conditions. TaqMan gene expression assays were used to analyze the following genes: dystrophin (assay ID Hs00240712_m1), dystroglycan (assay ID Hs00189308_m1), utrophin (assay ID Hs01125994_m1), Plectin1 (assay ID Hs00356986_g1), laminin α2 (assay ID Hs00166308_m1), laminin α4 (assay ID Hs00935293_m1), laminin α5 (assay ID Hs00966585_m1), integrin α7 (assay ID Hs01056475_m1), integrin β1 (assay ID Hs01127536_m1), and GALGT2 (assay ID Hs00396440_m1) (Applied Biosystems; Foster City, CA). A second GALGT2 assay was designed and the primers and probe were synthesized (Integrated DNA Technologies, Coralville, IA) as follows: forward 5′-CTACGATGGAATCTGGCTGTT-3′, probe 5′-/56-FAM/AGCCAACAA/ZEN/AGAGCAGGGAGGTTA/3IABkFQ/-3′, reverse 5′-GCCATAGGCATCCTGAAAGT-3′. Relative transcription levels were assessed using the 2−ΔΔC(T) method with 18S rRNA as an internal reference. Samples were unblinded after delta C(T) calculations.
Biodistribution of the rAAVrh74.MCK.GALGT2 vector
qPCR was performed on genomic DNA extracted from sections of muscle biopsy from subject 1 and subject 2 biopsies performed at baseline, 4 (subject 1) or 3 (subject 2) months, 6 months, and 12 months post transfer. Then 100 ng of DNA was run with TaqMan gene expression master mix (Applied Biosystems, Foster City, CA), 500 nM forward primer (5′-CCTCAGTGGATGTTGCCTTTA-3′), 500 nM reverse primer (5′-ATCTTGAGGAGCCACAGAAATC-3′), 250 nM probe (5′-/56-FAM/AAAGCTGCG/ZEN/GAATTGTACCCGC/3IABkFQ/-3′). qPCR was performed on an ABI 7500 Sequence Detection system (Applied Biosystems). qPCR results were compared with a standard curve of linear plasmid ranging from 50 vg to 5 × 106 vg in log increments. This standard curve was highly linear, with R2 = 0.99. All measures were standardized to 1 μg of total genomic DNA, and vg/diploid genome was calculated assuming there are 1.67 × 105 nuclei in 1 μg of human genomic DNA. Each sample was run in triplicate and the values were averaged.
CT glycan antigen expression by immunofluorescence
Muscle biopsies from all subject time points and from four healthy controls were cryosectioned at 10 μm and placed on slides for immunofluorescence staining. Slides were blinded prior to staining and imaging. Staining was performed using the conjugated lectin W. floribunda agglutinin linked to fluorescein isothiocyanate (WFA-FITC), to label GalNAc moieties, which are part of the CT glycan antigen made by GALGT2 [GalNAcβ1-4[Neu5Acα2-3]Galβ1-4GlcNAcβ-], and a mouse monoclonal antibody targeting spectrin to label muscle fibers. Tissue sections on slides were circled with PAP pen and incubated in blocking buffer (1× PBS with 10% donkey serum) for 1 h at room temperature. After blocking, tissue sections were incubated for 2 h in the primary antibody solution of blocking buffer with 1:100 mouse anti-spectrin primary antibody (Leica Biosystems NCL-SPEC1). One secondary-only control section per slide was incubated in blocking buffer without antibody during this time. Following primary antibody staining, slides were submerged in fresh 1× PBS on a shaker and washed three times for 10 min. Tissue sections were then incubated in the secondary antibody solution of blocking buffer with 1:2,000 WFA-FITC (Vector Laboratories FL-1351) and 1:200 goat anti-mouse Alexa Fluor 647 secondary antibody (Invitrogen A-21235) for 1 h in the dark at room temperature. The secondary-only control sections were incubated in blocking buffer with only the goat anti-mouse secondary antibody and without WFA-FITC during this time. Slides were then washed again three times in fresh 1× PBS for 10 min each, and coverslips were mounted using ProLong Gold antifade mountant (Invitrogen P36934). Slides were kept protected from light until imaging, which was done within 36 h of staining.
Multichannel whole-section images were collected in a blinded fashion on a fully motorized Nikon Ti2-E widefield fluorescence microscope with a Lumencor SOLA LED light engine and Hamamatsu ORCA Fusion camera at 10× magnification with a pixel resolution of 0.64 μm/pixel. Identical exposures and other imaging settings were used for capturing all images. WFA image quantification was performed using a custom automated analysis work flow developed in NIS-Elements software with the General Analysis 3 (GA3) software module (Nikon). Individual muscle fibers were detected using sarcolemmal spectrin signal, and GalNAc signal intensity and positivity were measured around the perimeter of each muscle fiber. GalNAc-positive pixels were considered to be those with markedly higher GalNAc signal than untreated healthy control muscle. These pixels were identified automatically using a threshold equal to the 99.5th percentile intensity in the GalNAc channel of healthy control images. Subsequently, the total length of GalNAc-positive sarcolemmal segments was divided by the total length of the spectrin-positive fiber perimeter to produce a measurement of the percentage GalNAc-positive perimeter for each muscle fiber. Muscle fibers having GalNAc-positive signal around ≥30% of their perimeter were considered overall positive for GalNAc. The mean intensity of the GalNAc signal was also measured in the sarcolemmal region of each muscle fiber independently of GalNAc positivity, and the measurement was normalized to the mean of the median sarcolemmal GalNAc intensity in each of the healthy controls. Since multiple biopsy blocks of unequal sizes were available for each subject at most time points, the measurements from all fibers in blocks representing the same subject and time point were pooled and treated as a single large dataset.
Fetuin-agarose precipitation of GALGT2 and GALGT2 western blots
Muscle biopsies were shaved on a cryostat and lysed in buffer (50 mM Tris pH 7.5, 20 mM MnCl2, 1% NP40, cOmplete protease inhibitors; Roche, Basel, Switzerland) using 2.3-mm stainless steel beads (BioSpec Products; Bartlesville, OK), shaking five times at 30 Hz for 30 s each time using the Tissue Lyser II (Qiagen, Germantown, MD). Samples were rocked gently overnight at 4°C, then centrifuged at 15,000 × g at 4°C for 10 min. Supernatant was collected and protein concentration was measured using the Pierce Coomassie Plus Assay kit (Thermo Scientific, Waltham, MA). Fetuin-agarose beads (EY Laboratories, San Mateo, CA) were washed using GlcNAc buffer (50 mM Tris pH 7.5, 20 mM MnCl2, 85 mM N-acetyl-D-glucosamine, 1% NP-40, cOmplete protease inhibitors). GlcNAc buffer was added to 300 μg of protein lysate and 75 μL of fetuin-agarose beads, and rocked overnight at 4°C. Beads were washed three times with TBST, then resuspended with NuPage LDS sample buffer (Invitrogen, Carlsbad, CA) with β-mercaptoethanol. Precipitated protein was boiled for 10 min and then analyzed by western blot for GALGT2 protein expression.
Precipitated samples or 1.0 ng of recombinant human GALGT2 (also B4GALNT2) purified protein (Novus Biologicals, Littleton, CO) were loaded on a NuPAGE 4%–12% Bis-Tris Plus Gel (Invitrogen, Carlsbad, CA) and run at 120 V in NuPAGE MOPS SDS Running Buffer until the dye front ran off the bottom of the gel. The proteins were transferred to a nitrocellulose membrane. The membrane was blocked for 1 h in 5% nonfat dry milk in 1× TBST and then probed overnight with anti-B4GALNT2 antibody (Abnova, Taipei, Taiwan). After washing, blots were probed with a peroxidase-conjugated anti-rabbit secondary antibody (Jackson Immunoresearch, West Grove, PA) for 1 h, washed again, and imaged using chemiluminescent reagent (Lumigen, Southfield, MI) with various exposure times on radiography film. Bands were quantitated using ImageJ software, version 1.53e. The amount of B4GALNT2 protein in each band was calculated as a ratio of peak area of unknown to peak area of 1 ng of B4GALNT2 standard. It is important to note here that the long form of GALGT2, the only purified protein available commercially, is predicted to be 62kDa, while the cDNA used for the transgene expresses the short form of GALGT2, which is predicted to be 56 kDa,33 6 kDa smaller in overall size.
Western blot analysis of expression of dystrophin, dystrophin-associated glycoproteins, and dystrophin surrogate proteins
Portions of muscle biopsies were solubilized in 1% NP-40 extraction buffer (1% NP-40, 50 mM Tris, 1 mM EDTA, cOmplete EDTA-free protease inhibitor cocktail; Roche, Basel, Switzerland) overnight. Solubilized protein was then measured via BCA assay and 25 μg of sample was denatured in NuPAGE SDS sample buffer and run on a 4%–12% Bis-Tris polyacrylamide gel. Separated proteins were transferred to nitrocellulose and probed by western blot using the antibodies shown. Blots were probed with one of two monoclonal antibodies to dystrophin (Dys1 and Dys2), a monoclonal (Drp2) or a polyclonal (utrophin) antibody to utrophin, or with monoclonal antibodies to α-dystroglycan (IIH6), βDG, or αSG. An antibody to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was also used on all blots as an internal control, which was visualized in a separate fluorescence channel from the other probed proteins. Bands were then quantified using ImageJ software and reported as signals relative to GAPDH. Three samples from healthy control muscle biopsies (HC1, HC2, and HC3) were compared with samples from each subject.
Endogenous dystrophin expression by immunofluorescence
Muscle biopsies from subject 1 and subject 2 baseline (n = 2) and from subject 1 at 4 months post transfer (n = 2) and subject 2 at 3 months post transfer (n = 1) were cryosectioned at 10 μm and placed on slides for immunofluorescence staining. Three healthy controls were also included. Staining was performed using a rabbit monoclonal antibody to dystrophin and rat antibodies to laminin α-2 and laminin γ-1 to label muscle fibers. Tissue sections on slides were circled with PAP pen and incubated for 1 h in PBS with 1:200 rabbit anti-dystrophin antibody (Abcam ab154168), 1:500 rat anti-laminin α-2 (Sigma L0663), and 1:500 rat anti-laminin γ-1 (Abcam ab80580). One secondary-only control section per slide was incubated in PBS without antibody during this time. Following primary antibody staining, slides were submerged in fresh 1× PBS on a shaker and washed two times for 5 min. Tissue sections were then incubated in the secondary antibody solution of PBS with 1:500 Alexa Fluor 555 donkey anti-rabbit (Invitrogen A32794) and 1:500 donkey anti-rat Alexa Fluor 647 secondary antibody (Invitrogen A21247) for 30 min in the dark at room temperature. Slides were then washed again two times in fresh 1× PBS for 5 min each, and coverslips were mounted using ProLong Gold antifade mountant with DAPI (Invitrogen P36935). Slides were kept protected from light until imaging, which was completed within 24 h of staining.
Multichannel whole-section images were collected on a fully motorized Nikon Ti2-E widefield fluorescence microscope with a Lumencor SOLA LED light engine and Hamamatsu ORCA Fusion camera at 10× magnification with a pixel resolution of 0.64 μm/pixel. Identical exposures and other imaging settings were used for capturing all images. Dystrophin image quantification was performed using laminin as the sarcolemmal marker as previously described.38 Briefly, laminin-positive pixels and dystrophin-positive pixels were identified automatically using thresholds calculated from sarcoplasmic signal within the same image, and muscle fiber perimeters were defined based on laminin-positive signal. The total length of dystrophin-positive sarcolemmal segments was divided by the total length of the laminin-positive fiber perimeter to calculate the percentage dystrophin-positive perimeter of each muscle fiber. Muscle fibers with dystrophin-positive signal around ≥30% of their perimeter were considered overall positive in calculating the percentage dystrophin-positive fibers (PDPFs). Mean dystrophin channel signal intensity was also measured within the sarcolemmal region of each muscle fiber, and was normalized to the average of the median fiber dystrophin intensities in the three healthy controls. Since multiple biopsy blocks of unequal sizes were available for each subject at most time points, the measurements from all fibers in blocks representing the same subject and time point were pooled and represented as a single dataset.
Interferon gamma ELISpot analysis
ELISpot assays were performed on fresh PBMCs, which were added at a concentration of 2 × 105/well in duplicate wells of a 96-well flat-bottom membrane plate (Millipore, Billerica, MA). Three peptide pools encompassed the rh74 capsid protein, and two peptide pools encompassed the B4GALNT2 protein (Proimmune, Littlemore, Oxford, UK), where pool 1 encompassed peptide 1 (MTSGGSRFLWLLKILVII, aa1–18) through peptide 36 (VIPKLYDPGPERKLRNLV, aa246–263), and pool 2 was peptide 37 (PGPERKLRNLVTIATKTF, aa253–270) to peptide 71 (VQFKLALHYFKNHLQCAA, aa489–506). Concanavalin A (Sigma, St. Louis, MO) served as a positive control and a hepatitis B virus peptide pool as a negative control. Peptides were added directly to the wells at a final concentration of 1 μg/mL in 200 μL of AIM-HS (Aim-V lymphocyte media [Invitrogen, Carlsbad, CA] supplemented with 2% human AB serum [Gemini-BioScience, Basel, Switzerland]). ELISpot kits were purchased from U-CyTech (Utrecht, the Netherlands). After the addition of PBMCs and peptides, the plates were incubated at 37°C for 48 h and then developed according to the manufacturer’s protocol. Interferon gamma (IFN-γ) spot formation was counted using a Cellular Technologies systems analyzer (Cellular Technologies, Cleveland, OH).
Anti-AAVrh74 ELISAs
Plates were coated overnight at 4°C in coating solution (0.2 M bicarbonate buffer, pH 9.4) with or without rAAVrh74 particles at 2 × 109 viral particles/well. Plates were blocked for 1–3 h at 37°C in blocking buffer (5% milk, 1% rabbit serum in PBS). Initial screening of samples was done by diluting the serum 1:12.5 to 1:100 in blocking buffer, and other patient samples were diluted from 1:50 to 1:819,200. Each sample was done in duplicate in wells coated with viral particles and with bicarbonate buffer alone to adjust for background. Samples were incubated at 37°C for 1 h. After washing, horseradish peroxidase (HRP)-conjugated anti-human immunoglobulin G (IgG) (whole molecule) was added at 1:10,000 dilution (Sigma, St. Louis, MO) and incubated at room temperature for 30 min, washed, and developed using 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution (Sigma, St. Louis, MO). The reaction was stopped with 1 N hydrochloric acid and the optical density of each well was read on a Synergy2 Plate Reader (BioTek, Winooski, VT) at 450 nm. For each sample, the mean optical density was calculated by subtracting the background value (minus viral particle wells) from the sample value (plus viral particle wells) and determining the signal/background ratio. The serum sample was called AAV positive if the signal/background ratio was greater than 2.
Acknowledgments
The authors thank the subjects and families for participating in the studies. They are grateful for the assistance of Brad Bolon and Chris Walker for the performance of ELISpots, and Kathrin Meyer and Shibi Likhite for the performance of ELISAs. This study was supported by NIAMS (R21AR068040 and P50 AR070604), with support for vector production provided by the Little Hercules Foundation and the Team Joseph Foundation.
Author contributions
P.T.M. and K.M.F. conceived of and designed the clinical trial, obtained funding, and wrote the manuscript. P.T.M. supervised all molecular studies and K.M.F. supervised all clinical studies. T.A.V. designed and performed quantitative fluorescence studies, assembled figures, and assisted with the manuscript. T.R.R., F.R., and J.H. supervised clinical trial and regulatory matters. M.I. performed clinical muscle function studies. E.C.F. supervised blinding and performed histology studies. L.G.C., B.B., J.P.C., and S.L.C. performed the ILI procedure. M.A.W. performed clinical care. D.A.Z. and D.P. performed molecular studies.
Declaration of interests
P.T.M. and Nationwide Children’s Hospital report a financial conflict of interest related to this study, as they both receive licensing fees from Sarepta Therapeutics for rAAVrh74.MCK.GALGT2. P.T.M. is also co-founder and President of Genosera Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2022.08.009.
Contributor Information
Kevin M. Flanigan, Email: kevin.flanigan@nationwidechildrens.org.
Paul T. Martin, Email: paul.martin@nationwidechildrens.org.
Supplemental information
Data availability
Data supporting the studies presented in this manuscript can be made available by request to the corresponding authors.
References
- 1.Mendell J.R., Shilling C., Leslie N.D., Flanigan K.M., al-Dahhak R., Gastier-Foster J., Kneile K., Dunn D.M., Duval B., Aoyagi A., et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012;71:304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- 2.Duan D., Goemans N., Takeda S., Mercuri E., Aartsma-Rus A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers. 2021;7:13. doi: 10.1038/s41572-021-00248-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moxley R.T., 3rd, Pandya S., Ciafaloni E., Fox D.J., Campbell K. Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. J. Child Neurol. 2010;25:1116–1129. doi: 10.1177/0883073810371004. [DOI] [PubMed] [Google Scholar]
- 4.Eagle M. Report on the muscular dystrophy campaign workshop: exercise in neuromuscular diseases Newcastle, January 2002. Neuromuscul. Disord. 2002;12:975–983. doi: 10.1016/s0960-8966(02)00136-0. [DOI] [PubMed] [Google Scholar]
- 5.Mendell J.R., Goemans N., Lowes L.P., Alfano L.N., Berry K., Shao J., Kaye E.M., Mercuri E., Eteplirsen Study G., Telethon Foundation, DMDIN Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016;79:257–271. doi: 10.1002/ana.24555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Frank D.E., Schnell F.J., Akana C., El-Husayni S.H., Desjardins C.A., Morgan J., Charleston J.S., Sardone V., Domingos J., Dickson G., et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. 2020;94:e2270–e2282. doi: 10.1212/WNL.0000000000009233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clemens P.R., Rao V.K., Connolly A.M., Harper A.D., Mah J.K., Smith E.C., McDonald C.M., Zaidman C.M., Morgenroth L.P., Osaki H., et al. Safety, tolerability, and efficacy of Viltolarsen in boys with Duchenne muscular dystrophy amenable to exon 53 skipping: a phase 2 Randomized clinical trial. JAMA Neurol. 2020;77:982–991. doi: 10.1001/jamaneurol.2020.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duan D. Systemic AAV micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol. Ther. 2018;26:2337–2356. doi: 10.1016/j.ymthe.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mendell J.R., Sahenk Z., Lehman K., Nease C., Lowes L.P., Miller N.F., Iammarino M.A., Alfano L.N., Nicholl A., Al-Zaidy S., et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in Children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 2020 doi: 10.1001/jamaneurol.2020.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chicoine L.G., Montgomery C.L., Bremer W.G., Shontz K.M., Griffin D.A., Heller K.N., Lewis S., Malik V., Grose W.E., Shilling C.J., et al. Plasmapheresis eliminates the negative impact of AAV antibodies on microdystrophin gene expression following vascular delivery. Mol. Ther. 2014;22:338–347. doi: 10.1038/mt.2013.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin P.T., Xu R., Rodino-Klapac L.R., Oglesbay E., Camboni M., Montgomery C.L., Shontz K., Chicoine L.G., Clark K.R., Sahenk Z., et al. Overexpression of Galgt2 in skeletal muscle prevents injury resulting from eccentric contractions in both mdx and wild-type mice. Am. J. Physiol. Cell Physiol. 2009;296:C476–C488. doi: 10.1152/ajpcell.00456.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen H.H., Jayasinha V., Xia B., Hoyte K., Martin P.T. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc. Natl. Acad. Sci. USA. 2002;99:5616–5621. doi: 10.1073/pnas.082613599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas P.J., Xu R., Martin P.T. B4GALNT2 (GALGT2) gene therapy reduces skeletal muscle pathology in the FKRP P448L mouse model of limb girdle muscular dystrophy 2I. Am. J. Pathol. 2016;186:2429–2448. doi: 10.1016/j.ajpath.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu R., Camboni M., Martin P.T. Postnatal overexpression of the CT GalNAc transferase inhibits muscular dystrophy in mdx mice without altering muscle growth or neuromuscular development: evidence for a utrophin-independent mechanism. Neuromuscul. Disord. 2007;17:209–220. doi: 10.1016/j.nmd.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu R., Chandrasekharan K., Yoon J.H., Camboni M., Martin P.T. Overexpression of the cytotoxic T cell (CT) carbohydrate inhibits muscular dystrophy in the dyW mouse model of congenital muscular dystrophy 1A. Am. J. Pathol. 2007;171:181–199. doi: 10.2353/ajpath.2007.060927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu R., DeVries S., Camboni M., Martin P.T. Overexpression of Galgt2 reduces dystrophic pathology in the skeletal muscles of alpha sarcoglycan-deficient mice. Am. J. Pathol. 2009;175:235–247. doi: 10.2353/ajpath.2009.080967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu R., Jia Y., Zygmunt D.A., Cramer M.L., Crowe K.E., Shao G., Maki A.E., Guggenheim H.N., Hood B.C., Griffin D.A., et al. An isolated limb infusion method allows for Broad distribution of rAAVrh74.MCK.GALGT2 to leg skeletal muscles in the rhesus macaque. Mol. Ther. Methods Clin. Dev. 2018;10:89–104. doi: 10.1016/j.omtm.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu R., Jia Y., Zygmunt D.A., Martin P.T. rAAVrh74.MCK.GALGT2 Protects against loss of hemodynamic function in the aging mdx mouse heart. Mol. Ther. 2019;27:636–649. doi: 10.1016/j.ymthe.2019.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohlendieck K., Ervasti J.M., Matsumura K., Kahl S.D., Leveille C.J., Campbell K.P. Dystrophin-related protein is localized to neuromuscular junctions of adult skeletal muscle. Neuron. 1991;7:499–508. doi: 10.1016/0896-6273(91)90301-f. [DOI] [PubMed] [Google Scholar]
- 20.Rupp F., Payan D.G., Magill-Solc C., Cowan D.M., Scheller R.H. Structure and expression of a rat agrin. Neuron. 1991;6:811–823. doi: 10.1016/0896-6273(91)90177-2. [DOI] [PubMed] [Google Scholar]
- 21.Rezniczek G.A., Konieczny P., Nikolic B., Reipert S., Schneller D., Abrahamsberg C., Davies K.E., Winder S.J., Wiche G. Plectin 1f scaffolding at the sarcolemma of dystrophic (mdx) muscle fibers through multiple interactions with beta-dystroglycan. J. Cell Biol. 2007;176:965–977. doi: 10.1083/jcb.200604179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu A.Y., Sanes J.R. Development of basal lamina in synaptic and extrasynaptic portions of embryonic rat muscle. Dev. Biol. 1984;103:456–467. doi: 10.1016/0012-1606(84)90333-6. [DOI] [PubMed] [Google Scholar]
- 23.Patton B.L., Miner J.H., Chiu A.Y., Sanes J.R. Distribution and function of laminins in the neuromuscular system of developing, adult, and mutant mice. J. Cell Biol. 1997;139:1507–1521. doi: 10.1083/jcb.139.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoch W., Ferns M., Campanelli J.T., Hall Z.W., Scheller R.H. Developmental regulation of highly active alternatively spliced forms of agrin. Neuron. 1993;11:479–490. doi: 10.1016/0896-6273(93)90152-h. [DOI] [PubMed] [Google Scholar]
- 25.Xu R., Singhal N., Serinagaoglu Y., Chandrasekharan K., Joshi M., Bauer J.A., Janssen P.M., Martin P.T. Deletion of Galgt2 (B4Galnt2) reduces muscle growth in response to acute injury and increases muscle inflammation and pathology in dystrophin-deficient mice. Am. J. Pathol. 2015;185:2668–2684. doi: 10.1016/j.ajpath.2015.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chicoine L.G., Rodino-Klapac L.R., Shao G., Xu R., Bremer W.G., Camboni M., Golden B., Montgomery C.L., Shontz K., Heller K.N., et al. Vascular delivery of rAAVrh74.MCK.GALGT2 to the gastrocnemius muscle of the rhesus macaque stimulates the expression of dystrophin and laminin alpha2 surrogates. Mol. Ther. 2014;22:713–724. doi: 10.1038/mt.2013.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xia B., Hoyte K., Kammesheidt A., Deerinck T., Ellisman M., Martin P.T. Overexpression of the CT GalNAc transferase in skeletal muscle alters myofiber growth, neuromuscular structure, and laminin expression. Dev. Biol. 2002;242:58–73. doi: 10.1006/dbio.2001.0530. [DOI] [PubMed] [Google Scholar]
- 28.Martin P.T., Zygmunt D.A., Ashbrook A., Hamilton S., Packer D., Birch S.M., Bettis A.K., Balog-Alvarez C.J., Guo L.J., Nghiem P.P., et al. Short-term treatment of golden retriever muscular dystrophy (GRMD) dogs with rAAVrh74.MHCK7.GALGT2 induces muscle glycosylation and utrophin expression but has no significant effect on muscle strength. PLoS One. 2021;16:e0248721. doi: 10.1371/journal.pone.0248721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mendell J.R., Rodino-Klapac L.R., Rosales X.Q., Coley B.D., Galloway G., Lewis S., Malik V., Shilling C., Byrne B.J., Conlon T., et al. Sustained alpha-sarcoglycan gene expression after gene transfer in limb-girdle muscular dystrophy, type 2D. Ann. Neurol. 2010;68:629–638. doi: 10.1002/ana.22251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zygmunt D.A., Xu R., Jia Y., Ashbrook A., Menke C., Shao G., Yoon J.H., Hamilton S., Pisharath H., Bolon B., et al. rAAVrh74.MCK.GALGT2 Demonstrates safety and Widespread muscle glycosylation after intravenous delivery in C57BL/6J mice. Mol. Ther. Methods Clin. Dev. 2019;15:305–319. doi: 10.1016/j.omtm.2019.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matthews E., Brassington R., Kuntzer T., Jichi F., Manzur A.Y. Cochrane Database Syst Rev; 2016. Corticosteroids for the Treatment of Duchenne Muscular Dystrophy; p. CD003725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McMillan H.J., Gregas M., Darras B.T., Kang P.B. Serum transaminase levels in boys with Duchenne and Becker muscular dystrophy. Pediatrics. 2011;127 doi: 10.1542/peds.2010-0929. e132–136. [DOI] [PubMed] [Google Scholar]
- 33.Montiel M.D., Krzewinski-Recchi M.A., Delannoy P., Harduin-Lepers A. Molecular cloning, gene organization and expression of the human UDP-GalNAc:Neu5Acalpha2-3Galbeta-R beta1,4-N-acetylgalactosaminyltransferase responsible for the biosynthesis of the blood group Sda/Cad antigen: evidence for an unusual extended cytoplasmic domain. Biochem. J. 2003;373:369–379. doi: 10.1042/BJ20021892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoyte K., Kang C., Martin P.T. Definition of pre- and postsynaptic forms of the CT carbohydrate antigen at the neuromuscular junction: ubiquitous expression of the CT antigens and the CT GalNAc transferase in mouse tissues. Brain Res. Mol. Brain Res. 2002;109:146–160. doi: 10.1016/s0169-328x(02)00551-x. [DOI] [PubMed] [Google Scholar]
- 35.Martin P.T., Scott L.J., Porter B.E., Sanes J.R. Distinct structures and functions of related pre- and postsynaptic carbohydrates at the mammalian neuromuscular junction. Mol. Cell. Neurosci. 1999;13:105–118. doi: 10.1006/mcne.1999.0737. [DOI] [PubMed] [Google Scholar]
- 36.Ricotti V., Ridout D.A., Pane M., Main M., Mayhew A., Mercuri E., Manzur A.Y., Muntoni F., Network U.K.N.C. The NorthStar Ambulatory Assessment in Duchenne muscular dystrophy: considerations for the design of clinical trials. J. Neurol. Neurosurg. Psychiatry. 2016;87:149–155. doi: 10.1136/jnnp-2014-309405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mendell J.R., Chicoine L.G., Al-Zaidy S.A., Sahenk Z., Lehman K., Lowes L., Miller N., Alfano L., Galliers B., Lewis S., et al. Gene delivery for limb-girdle muscular dystrophy type 2D by isolated limb infusion. Hum. Gene Ther. 2019;30:794–801. doi: 10.1089/hum.2019.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vetter T.A., Nicolau S., Bradley A.J., Frair E.C., Flanigan K.M. Automated immunofluorescence analysis for sensitive and precise dystrophin quantification in muscle biopsies. Neuropathol. Appl. Neurobiol. 2022;48:e12785. doi: 10.1111/nan.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting the studies presented in this manuscript can be made available by request to the corresponding authors.