

Abstract
Guanosine triphosphate binding protein 4 (GTPBP4) is a key regulator of cell cycle progression and MAPK activation. However, how its biological properties intersect with cellular metabolism in hepatocellular carcinoma (HCC) development remains poorly unexplained. Here, high GTPBP4 expression is found to be significantly associated with worse clinical outcomes in patients with HCC. Moreover, GTPBP4 upregulation is paralleled by DNA promoter hypomethylation and regulated by DNMT3A, a DNA methyltransferase. Additionally, both gain- and loss-of-function studies demonstrate that GTPBP4 promotes HCC growth and metastasis in vitro and in vivo. Mechanically, GTPBP4 can induce dimeric pyruvate kinase M2 (PKM2) formation through protein sumoylation modification to promote aerobic glycolysis in HCC. Notably, active GTPBP4 facilitates SUMO1 protein activation by UBA2, and acts as a linker bridging activated SUMO1 protein and PKM2 protein to induce PKM2 sumoylation. Furthermore, SUMO-modified PKM2 relocates from the cytoplasm to the nucleus may also could contribute to HCC progression through activating epithelial-mesenchymal transition (EMT) and STAT3 signaling pathway. Shikonin, a PKM2-specific inhibitor, can attenuate PKM2 dependent HCC glycolytic reprogramming, growth and metastasis promoted by GTPBP4, which offers a promising therapeutic candidate for HCC patients. Our findings indicate that GTPBP4-PKM2 regulatory axis plays a vital role in promoting HCC proliferation as well as metastasis by aerobic glycolysis and offer a promising therapeutic target for HCC patients.
Keywords: Hepatocellular carcinoma, GTP binding protein 4, Pyruvate kinase M2, Aerobic glycolysis
Graphical abstract
Highlights
-
•
GTPBP4 promotes HCC progression and metastasis in vitro and in vivo.
-
•
Promoter hypomethylation upregulated GTPBP4 expression.
-
•
GTPBP4 promotes the glycolysis in HCC via inducing dimeric PKM2 formation and sumoylation through UBA2-SUMO1 axis.
-
•
GTPBP4 induces PKM2 nuclear transfer to act as a protein kinase to activate STAT3 and EMT signaling pathway.
-
•
Shikonin induces significant anti-tumor effect in orthotopic mouse models harboring activation of GTPBP4/PKM2 axis.
1. Background
Hepatocellular carcinoma (HCC) is a primary malignancy of the liver and a common and deadly malignancy worldwide [1,2]. Recently, comprehensive treatment based on surgery, and molecular targeted therapy have considerably improved the prognosis of patients with HCC. However, the 2-year recurrence rate for patients with HCC is still close to 50%, and goes up to 75% within 5 years after hepatectomy [3]. Therefore, it is necessary to search for a plausible predictive biomarker for the early recurrence of HCC. Understanding the molecular mechanism of HCC progression and metastasis to identify cancer-specific targets for HCC therapy is the key to improving the effectiveness of HCC treatment.
The guanosine triphosphate binding protein (GTPBP) family proteins act as GTP hydrolases from active (GTP bound) to the inactive (GDP bound) states [4]. GTPBP4 is a member of the human GTPBP family highly conserved across eukaryotes from yeast to humans [5]. Previous studies have shown that GTPBP4 is involved in 60S ribosome biogenesis, cell cycle and DNA mismatch repair system [6]. Moreover, GTPBP4 overexpression is correlated with reduced survival time in breast tumors [7]. In a preliminary study, Liu et al. had found that GTPBP4 is correlated with unfavorable prognosis in patients with HCC [8]. However, the role of GTPBP4 in metabolic regulation and the underlying mechanisms involved in HCC development and metastasis have still not been investigated.
In the 1920s, Otto Warburg found that, even in the presence of adequate oxygen, tumor cells preferentially rely on glycolysis for rapid adenosine triphosphate (ATP) production and large amounts of other biomolecules rather than mitochondrial oxidative phosphorylation (OXPHOS). This metabolic reprogramming phenomenon is a hallmark of cancer cells and known as the Warburg effect, or aerobic glycolysis [9,10]. Despite of the inefficiency of ATP production per mol of glucose, aerobic glycolysis provides various glucose metabolites that are the building blocks for macromolecular biosynthesis and support cell proliferation [11]. Alongside its role in giving carbon for building blocks and energy generation, aerobic glycolysis can facilitate local tumor local invasion and metastasis and inhibit anti-tumor immunity, which is also observed in HCC [12,13]. However, the underlying mechanism remains unclear.
Pyruvate kinase is a rate-limiting enzyme and catalyzes the final step in glycolysis by converting phosphoenolpyruvate (PEP) to pyruvate [10]. Pyruvate kinase (PK) has four mammalian isoforms which are regulated in a specific temporal and spatial manner. These isoforms include pyruvate kinase liver (PKL), pyruvate kinase red blood cells (PKR), pyruvate kinase isozymes M1 (PKM1) and pyruvate kinase isozymes M2 (PKM2) [14,15]. PKM2 is the predominant isoform in the majority of adult human body tissues [16]. Moreover, numerous studies demonstrate that PKM2 is highly expressed in carcinoma of the lung, colon, breast, and prostate, and renal cell carcinoma and can be used as a prognostic marker for signet ring cell gastric cancer [[17], [18], [19], [20], [21]]. Accumulating evidence demonstrates that PKM2 plays a key role in tumorigenesis and metastasis through promoting the Warburg effect [22]. PKM2 exists in three forms: inactive monomers, less active dimers, and active tetramers. Tetrameric PKM2 is involved in oxidative phosphorylation for rapid ATP production. Dimeric PKM2 can not only promotes the Warburg effect by redirecting carbons derived from glucose towards macromolecular biosynthesis, but also display non-glycolytic functions as a protein kinase in the nucleus acting on gene transcription [23].
In the current study, we that high GTPBP4 expression in patients with HCC correlates with poor outcome. We also showed that GTPBP4 upregulation is paralleled by DNA promoter hypomethylation induced by DNA methyltransferase 3a (DNMT3A). Moreover, GTPBP4 promote HCC progression and metastasis by inducing PKM2 dimer conformation through protein sumoylation modification to promote aerobic glycolysis in HCC. Shikonin, a PKM2 inhibitor, efficiently suppressed tumor growth in orthotopic xenograft HCC mouse models and may have a potential therapeutic effect in patients with HCC.
2. Methods
2.1. Patients and specimens
A total of 182 patients with HCC undergoing curative hepatectomy in Zhongshan Hospital Fudan University, Shanghai, China, between January 1, 2009 and January 1, 2010 were enrolled in the study. Matched tumor and adjacent normal tissue samples were collected from these 182 enrolled patients for construction of the tissue microarray (TMA). Of the 182 paired frozen samples, 109 were randomly selected for the detection of GTPBP4 mRNA expression using quantitative reverse-transcription PCR (qRT-PCR).
The inclusion criteria for patients with HCC, clinicopathological characteristics, and follow-up data were described in our previous study [24]. OS was defined as the time interval between the last follow-up or death and surgery. RFS was defined as the time interval between surgery and tumor recurrence or last follow-up.
2.2. Analysis of public clinical datasets
Microarray expression files and clinical information for GSE14520, consisting of 220 HCC specimens, were downloaded from the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). Additionally, RNA-sequencing and clinical data of 427 tissues (377 primary HCCs and 50 paired normal liver specimens) were downloaded from the TCGA database (https://portal.gdc.cancer.gov) in May 2019, and 370 cases of these 377 patients with primary HCC have survival data available. Analyses of GTPBP4 expression in paired tumor and normal tissues samples, and of OS were performed in the R programming language (Version 3.6.2, http://www.r-project.org). Promoter methylation and genomic regulation of GTPBP4 expression in HCC were evaluated by using the UALCAN platform (http://ualcan.path.uab.edu/index.html).
2.3. Cell lines
The normal hepatocyte cell line (L02) and PLC/PRF/5, SMMC7721, and Huh7 HCC cell lines were purchased from the cell bank of the Chinese Academy of Sciences (Shanghai, China). HCCLM3 cells were established at the liver cancer institute, Zhongshan Hospital, Fudan University (Shanghai, China) [25]. Cells were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Gibco) at 37 °C in a humidified environment with 5% CO2.
2.4. Cell transfection
Human GTPBP4 overexpression lentiviruses were created using GV341 vectors and lentiviral-based small hairpin RNA (shRNA) targeting GTPBP4 and TET2 were constructed using GV112 vectors (GeneChem Co., Ltd, Shanghai, China). Small interfering RNA (siRNA) targeting DNMT3A, DNMT3B, and PKM2 were synthesized and purified by OBiO Technology (Shanghai) Corp., Ltd. Human DNMT3A, SENP1, SUMO1, and PKM2 were cloned into pcDNA3.1(+), pcDNA3.1(+) -MCS -6xHis, pcDNA3.1(+) -MCS -Myc, and pcDNA3.1(+) -MCS-GST, and PKM2 point mutations were established as described in our previous study [26].
2.5. Chromatin immunoprecipitation (ChIP) coupled with qRT-PCR (ChIP-qPCR)
ChIP-qPCR was performed as previously described [27]. For more details, please see Supplementary Methods.
2.6. Metabolite extraction and spectrometry analysis
Glycolysis and tricarboxylic acid (TCA) cycle were analyzed using metabolite-specific mass spectrometry methodologies. For more details, please see Supplementary Methods.
2.7. Glucose-induced extracellular acidification rate (ECAR) and fuel flex assay
ECAR was analyzed by Seahorse and the fuel flex assay was performed to assess glucose in glycolysis and the TCA cycle, as described in the supplementary methods.
2.8. Glucose uptake and lactate production
Glucose uptake and lactate production were measured by glucose assay kit (BioVision) and lactate assay kit (BioVision), respectively, as described in the supplementary methods.
2.9. Co-immunoprecipitation and mass spectrometry (Co-IP/MS)
Co-IP assay and MS were conducted as described in the supplementary methods [28].
2.10. Other materials and methods
Details of qRT-PCR, Western blot, luciferase assay, cross-linking assay, cell transfection, cell proliferation, migration and invasion assays, immunofluorescence staining, and immunoprecipitation assays are described in the supplementary materials and methods.
2.11. Mouse xenograft study
In vivo experiments were performed in BALB/c nude mice (male; age range, 4-6 weeks) purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. Mice were bred and maintained in a standard pathogen-free environment. Animal experiments in this study were in accordance with the institutional animal care and use guidelines and were approved by the Animal Care and Use Committee of Zhongshan Hospital, Fudan University. For subcutaneous tumor models, BALB/c nude mice were randomly divided into different groups (6 mice per group) and administered subcutaneous injections with 5 x 106 HCC cells (PLC/PRF/5-Vector, PLC/PRF/5-GTPBP4, HCCLM3- Control, HCCLM3-shGTPBP4) suspended in 200 μl of phosphate-buffered saline. The mice were monitored regularly and the subcutaneous tumors were measured with digital Vernier calipers and tumor volume was calculated: Tumor volume (mm3) = 0.5 × length × width2. Six weeks after injection, mice were euthanized and tumors were collected. For the orthotopic mouse models, subcutaneous tumors from HCC cells (PLC/PRF/5-Vector, PLC/PRF/5-GTPBP4, HCCLM3- Control, HCCLM3-shGTPBP4) were dissected and cut into several 1 mm3. Mice (male; age range, 4-6 weeks) were randomly divided into seven groups (PLC/PRF/5-Vector, PLC/PRF/5-GTPBP4, PLC/PRF/5-GTPBP4+ Shikonin, PLC/PRF/5-Vector + Shikonin, HCCLM3-Control, HCCLM3-shGTPBP4, HCCLM3-Control + Shikonin) and a single cube of the corresponding tumor was implanted into the right lobe of liver parenchyma under anesthesia. For chemosensitivity tests, one week after implantation, mice bearing HCCLM3-Control, PLC/PRF/5-GTPBP4, and PLC/PRF/5-Vector mice were treated with Shikonin. Shikonin (MedChemExpress, USA) was dissolved in 10% DMSO, 40% PEG300, 5% Tween-80, and 45% saline. Shikonin (1 mg/kg) was administrated via intraperitoneal injection two days a week. For the control group, mice were treated with vehicle (0.9% NaCl). At the end of therapy (4 weeks), mice underwent PET-CT scans.
Then, mice were euthanized and tumors were collected. Tumors were fixed with 4% paraformaldehyde for further hematoxylin and eosin (HE) staining and immunohistochemistry (IHC) analysis.
2.12. Statistical analysis
SPSS software (23.0, IBM, Armonk, NY, USA) and Prism software (GraphPad 8 Software) were used for statistical analyses. Continuous variables were analyzed by student's t-test and recorded as means ± standard deviation (SD). Pearson's correlation coefficient was used for measuring the degree of linearity. Categorical variables were analyzed using Pearson χ2 test or Fisher's exact test. OS and RFS were calculated using the Kaplan-Meier method and compared between patients in different groups by log-rank test. Univariate and multivariate Cox proportional hazards regression analyses were used to analyze and screen significant variables related to OS and RFS. P value < 0.05 was considered significant.
3. Results
3.1. GTPBP4 is upregulated in human HCC and correlates with poor prognosis
To identify the potential clinical significance of GTPBP4 in human HCC, we first analyzed GTPBP4 mRNA expression in matched tumor and adjacent normal tissue samples from 109 patients with HCC. The 109 patients were divided into higher and lower GTPBP4 expression group from the median value of relative expression levels. We found that GTPBP4 is upregulated in 77.06% of tumor tissues (84/109) (Supplementary Fig. S1A). The same results were obtained in public database, including data set GSE14520 from the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database and The Cancer Gene Atlas (TCGA) (P < 0.001 for both) (Supplementary Fig. S1B). In addition, high GTPBP4 expression is confirmed to correlate with advanced Tumor-Node-Metastasis (TNM) stages in the GSE14520 dataset (stage I vs. stage III, P = 0.02) and in the TCGA dataset (stage I vs. stage II, P = 0.041; stage I vs. stage III, P < 0.001) (Supplementary Fig. S1C). To investigate GTPBP4 protein expression in HCC tissues, IHC was performed on the TMA (containing 182 specimens from patients with HCC in the Zhongshan cohort). Consistent with our GTPBP4 mRNA results, GTPBP4 protein expression is significantly higher in HCC tissues than that in adjacent tissues (P < 0.001) (Supplementary Fig. S1D). The clinicopathological analysis also revealed that high GTPBP4 expression may correlate with HCC malignant progression (Additional file: Table S1).
In the Zhongshan HCC cohort, IHC staining scores (negative (−), weak (+), moderate (++), and strong (+++)), were used to classify patients into high (++/+++) or low GTPBP4 expression groups (Supplementary Fig. S1D). Univariate and multivariate Cox regression analyses showed that high GTPBP4 expression was an independent prognostic factor for overall survival (OS) (hazard ratio [HR] = 2.407, 95% confidence interval [CI]: 1.486–3.898, P < 0.001) and recurrence free survival (RFS) (HR = 3.690, 95% CI: 2.355–5.784, P < 0.001) (Additional file: Table S2). Survival analysis revealed that patients with HCC and high GTPBP4 expression have noticeably worse OS and shorter RFS than do those with the low GTPBP4 expression (Supplementary Fig. S2A). Similarly, in both GSE14520 datasets and in the TCGA dataset, we further validated the above conclusions (Supplementary Fig. S2B). Together, these results demonstrated that high GTPBP4 expression was significantly associated with poor prognosis in patients with HCC. In this section, we get the same result with a previous study [8]. However, the data in our study are more advanced and adequate.
3.2. Promoter hypomethylation upregulated GTPBP4 expression
HCC is characterized by global DNA hypomethylation and regional hypermethylation in promoter regions. DNA promoter hypermethylation could silence multiple tumor suppressor genes expression, while hypomethylation may involve oncogene upregulation [29,30]. Analysis of TCGA database revealed that GTPBP4 promoter was hypomethylated in tumors when compared to non-tumorous samples (Fig. 1A), and high GTPBP4 expression was related to GTPBP4 promoter hypomethylation (Pearson correlation R = - 0.524, P < 0.001; Fig. 1B). To further investigate how GTPBP4 expression is regulated by promoter methylation, we detected GTPBP4 mRNA and protein expression in five different HCC cell lines (HCCLM3, Hep3B, SMMC-7721, Huh7, and PLC/PRF/5), and the normal human liver cell line, L02 (Supplementary Fig. S3A). GTPBP4 expression in HCC cell lines was upregulated compared to normal liver L02 cells at both mRNA and protein levels. Then, a total of 79 CpG sites in GTPBP4 promoter region were identified by DNA sequence analysis, among which 22 CpG sites can be detected using bisulfite pyrosequencing in five different HCC cell lines. At these 22 CpG sites, the median methylation levels were 20% and 50% in HCC cells and normal human liver cells, L02, respectively. The 22 CpG sites analyzed for methylation surrounding the GTPBP4 promoter region is showed in Fig. S3B. The results showed that compared with L02 cells, the HCC cell lines exhibit low methylation levels (Supplementary Fig. S3C). DNA methyltransferases (DNMTs) and demethylases (TETs) maintain DNA methylation and its dynamic balance. We then inhibited DNMTs and TETs by transfecting PLC/PRF/5 cells with siRNAs specific to DNMT3A, DNMT3B, DNMT1, and TET2. Obvious upregulation of GTPBP4 expression was only observed after knockdown of DNMT3A, which reveals that DNMT3A could negatively regulate GTPBP4 expression via DNA methylation (Fig. 1C–D). We then performed a ChIP-qPCR assay and found that DNMT3A could directly bind to the GTPBP4 gene locus (Fig. 1E–F). Next, based on the DNMT3A-favored binding motif according to the previous study, the potential binding site was identified on the GTPBP4 promoter (GATTGG, Fig. 1G) [31]. The dual-luciferase reporter assay was performed using PLC/PRF/5 cells transfected with the pGL3 mammalian cell expression vector, which includes the luciferase structural gene and binding site containing GTPBP4 promoter (PGL3-GTPBP4 vector) or its mutation (PGL3-GTPBP4 mutation vector). PLC/PRF/5 cells were transfected with pCMV500 plasmid as an empty control vector. Compared with the PGL3 control vector or the PGL3-GTPBP4 vector, luciferase activity was significantly decreased after co-transfection with the DNMT3A vector. However, co-transfection of the PGL3-GTPBP4 mutation vector and the DNMT3A vector did not affect luciferase activity (Fig. 1H). These results show that DNMT3A negatively regulates GTPBP4 expression.
Fig. 1.

Promoter hypomethylation upregulated GTPBP4 expression. (A) GTPBP4 promoter methylation status in normal liver and tumor specimens from TCGA. unpaired 2-tailed t-test. (B) The GTPBP4 promoter methylation status inversely correlates with GTPBP4 mRNA expression in tumor tissues in TCGA. Pearson's correlation test. (C–D) PLC/PRF/5 cells were transfected with DNMT3A, DNMT3B, DNMT1, and TET2 specific siRNAs, respectively. Western blotting (C) and qRT-PCR (D) were used to analyze GTPBP4 expression. n = 3, unpaired 2-tailed t-test. (E–F) ChIP-qPCR analysis of DNMT3A binding to the GTPBP4 promoter region in PLC/PRF/5 cells. The chromatin of PLC/PRF/5 cells was immunoprecipitated with anti-DNMT3A antibody (E), and the GTPBP4 promoter region was amplified by qRT-PCR (F). n = 3, unpaired 2-tailed t-test. (G) The vector containing the predicted binding site in the GTPBP4 promoter control or mutation constructed for luciferase reporter assays. (H) PLC/PRF/5 cells were co-transfected with the pGL3-GTPBP4-WT or mutation reporter and DNMT3A overexpression vector, or its mutation as the control. Dual luciferase activity was determined at 48 h after transfection. Error bars represent the mean ± SD from six independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001. n.s, not significant. n = 5, unpaired 2-tailed t-test. Abbreviations: siRNA: small interfering RNA; ChIP-qPCR: Chromatin immunoprecipitation-qPCR.
3.3. GTPBP4 promotes HCC growth and metastasis
To investigate whether GTPBP4 upregulation contributes to HCC progression and metastasis, we generated stable GTPBP4-overexpression (GTPBP4 OE) cells in relatively low GTPBP4 expressing PLC/PRF/5 and SMMC-7721 cell lines. shRNA-GTPBP4 knockdown (GTPBP4 KD) cells were generated in high GTPBP4-expressing HCCLM3 and Huh7 cell lines. The overexpression and knockdown efficiency of GTPBP4 in these cell lines was assessed by western blotting and qRT‐PCR (Fig. 2A; Supplementary Fig. S4A). shRNA-2 showed the best GTPBP4 inhibitory in HCCLM3 and Huh7 cells and was selected for further experiments.
Fig. 2.

GTPBP4 promotes HCC growth and metastasis. (A) GTPBP4-OE PLC/PRF/5 and GTPBP4‐KD HCCLM3 cells were constructed by transfecting overexpressing lentivirus and shRNA and were validated by western blotting and qRT-PCR. n = 3, unpaired 2-tailed t-test. (B) Evaluation of the influence of GTPBP4 overexpression and knockdown on the proliferation of PLC/PRF/5 and HCCLM3 cells by CCK-8. n = 6, one-way ANOVA. (C) The influence of GTPBP4 overexpression and knockdown on migratory and invasive capacities of PLC/PRF/5 and HCCLM3 cells was evaluated by transwell assays. n = 3, one-way ANOVA or unpaired 2-tailed t-test. (D) Evaluation of the influence of GTPBP4 overexpression and knockdown on apoptosis in PLC/PRF/5 and HCCLM3, respectively, was determined by flow cytometry. n = 3, unpaired 2-tailed t-test. (E) Subcutaneous xenograft mouse models were constructed using PLC/PRF/5 Vector or PLC/PRF/5-GTPBP4 cells. Tumor growth curves (middle) and weight (right) are shown. n = 6, unpaired 2-tailed t-test. (F) Subcutaneous xenograft mouse models were constructed by using HCCLM3-Control or HCCLM3-shGTPBP4. Tumor growth curves (middle) and weight (right) are shown. n = 6, Error bars represent the mean ± SD from three or more independent experiments. **P < 0.01, ***P < 0.001; unpaired 2- tailed t-test.
GTPBP4 overexpression significantly promoted PLC/PRF/5 and SMMC-7721 cells proliferation measured by cell counting kit (CCK)-8 assay, while GTPBP4 knockdown markedly decreased proliferation capacity of HCCLM3 and Huh7 cells compared with control cells in vitro (Fig. 2B; Supplementary Fig. S4B). Scratch assays and transwell assays also showed that the migratory and invasive capacities of PLC/PRF/5 and SMMC-7721 cells were greatly promoted by GTPBP4 overexpression, and that GTPBP4 knockdown impeded cell migratory and invasive capacities compared with HCCLM3 and Huh7 cells (Fig. 2C; Supplementary Figs. S4C–E).
Subsequently, the effects of GTPBP4 on apoptosis and cell cycle distribution were measured using flow cytometer. We observed that GTPBP4 inhibits apoptosis (Fig. 2D; Supplementary Fig. S5A). These results indicate that GTPBP4 promotes the proliferation, invasion, and metastasis of HCC cells in vitro. We hypothesized that GTPBP4 may contribute to the epithelial-mesenchymal transition (EMT). We found that GTPBP4 overexpression increased the expression of EMT associated transcription factors, including Snail, N-cadherin and vimentin, and decreased the expression of E-cadherin and ZO-1. The EMT phenotype was suppressed in HCCLM3 cells with knockdown of GTPBP4 (Supplementary Fig. S5B). Together, these data reveal that GTPBP4 contributes to HCC progression and metastasis. To verify these findings in vivo, we established cell-derived xenograft (CDX) models. Then, GTPBP4 overexpressing PLC/PRF/5 cells and knockdown HCCLM3 cells, or their controls, were subcutaneously injected into nude mice, and tumor volumes were measured weekly using a caliper. The growth curves showed that stable overexpression of GTPBP4 markedly promotes the xenograft tumor proliferation (Fig. 2E). In contrast, GTPBP4 knockdown effectively inhibited the tumor growth progression (Fig. 2F). Taken together, these results indicate that GTPBP4 contributes to HCC cell proliferation, tumor progression, and metastasis.
3.4. GTPBP4 promotes aerobic glycolysis in HCC
To explore the molecular mechanism by which GTPBP4 contributes to HCC progression, immunoprecipitation (IP) and liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) analyses were used. GTPBP4‐interacting proteins complexes were isolated by anti-Flag mAbs and paramagnetic beads in GTPBP4 OE PLC/PRF/5 cells, PLC/PRF/5 cells, GTPBP4 OE SMMC-7721 cells and SMMC-7721 cells. Affinity purification and MS analyses further showed that after removing proteins in controls, a total of 101 proteins through a proteomic analysis were identified as GTPBP4 specific binding proteins in GTPBP4 OE PLC/PRF/5 cells, and 136 proteins in GTPBP4 OE SMMC-7721 cells. A total of 61 common interacting proteins were identified (Fig. 3A). Pathway enrichment analysis showed that GTPBP4‐interacting proteins were mostly involved in metabolic pathways, carbon metabolism, glycolysis and the citrate cycle (Fig. 3B). We also searched the TCGA database to perform GTPBP4-related gene set enrichment analysis (GSEA) analysis. Glycolysis and glycolysis-related pathway were significantly elevated in the GTPBP4 high expression group (Supplementary Fig. S5C).
Fig. 3.

GTPBP4 promotes aerobic glycolysis in HCC. (A) Venn diagram illustrating the proteins identified as GTPBP4 binding proteins from GTPBP4 OE PLC/PRF/5 and GTPBP4 OE SMMC-7721 cell groups. (B) Enrichment analysis and functional classification of GTPBP4 specific binding proteins using IPA software (left). The 25 most abundant proteins are listed (right). (C) Flux map and 13C labeled glucose tracing of glycolysis and the TCA cycle. (D) Quantification of intermediate metabolites in glycolysis and the TCA cycle by gas chromatography and mass spectrometry, Q-Exactive-Plus mass-spectrometer, and liquid chromatography coupled with tandem mass spectrometry for different metabolites. n = 6, unpaired 2-tailed t-test. (E) Quantification of isotopes derived from 13C6-labeled glucose intermediates in glycolysis and the TCA cycle was performed using mass spectrometry analysis. M+3 indicates two carbon-labeled PEP, pyruvate, and lactate; M+2 indicates citrate, cis-aconitate, succinate, fumarate, and malate. n = 6, unpaired 2-tailed t-test. (F) The extracellular acidification rate was determined in control and GTPBP4 knockdown HCCLM3 cells. n = 6, one-way ANOVA. (G–H) Lactate production (G) and glucose consumption (H) were measured in HCC cells. Error bars represent the mean ± SD from six independent experiments. n = 6, unpaired 2-tailed t-test. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: IPA: ingenuity pathway analysis; TCA cycle: tricarboxylic acid cycle; ECAR: Extracellular acidification rate.
To comprehensively elucidate whether GTPBP4 plays a vital role in regulating glucose metabolism, we performed metabolomic profiling using different types of mass spectrometry for different metabolites in GTPBP4 OE PLC/PRF/5 cells and relevant controls. Specifically, some key glycolytic intermediate metabolites, including glucose-6-phosphate (G6P), fructose 6-bisphosphate (F1,6P), 3-phosphoglyceric acid (3 PG), 2-phosphoglyceric acid (2 PG), phosphonyl pyruvic acid (PEP), pyruvic acid, acetyl coenzyme A (CoA), and lactate were increased in GTPBP4 OE PLC/PRF/5 cells compared with control group. TCA intermediate metabolites, including citrate, cis-aconitate, ɑ-ketoglutarate, succinate, fumarate, and malate were also increased in GTPBP4 OE PLC/PRF/5 cells compared with control group (Fig. 3C–D). To confirm the glycolysis and TCA cycle metabolic flux changes upon GTPBP4 knockdown, analysis of [U–13C6]-glucose carbon tracing was performed to detect labeled metabolites. [U–13C6]-PEP/Pyruvate/Lactate (M+3) and [U–13C6]-Citrate/Cis-aconitate/Succinate/Fumarate/Malate (M+2) were increased in GTPBP4 OE PLC/PRF/5 cells compared with the levels observed in the control group (Fig. 3E). This shows that glycolytic and TCA cycle glycolytic flux were decelerated in GTPBP4 knockdown cells. Together, these results showed that GTPBP4 is involved in glycolysis and the TCA cycle.
Glycolysis has been confirmed to play important roles in the tumor process. Therefore, we focused on the effect of GTPBP4 on glycolysis. We first measured glucose-induced extracellular acidification rate (ECAR), a proxy for the rate of glycolysis and glycolytic capacity, using the extracellular flux assay (Seahorse), and found that the knockdown of GTPBP4 significantly suppressed the increase of ECAR (Fig. 3F). Then lactate production and glucose consumption assay data showed that GTPBP4 overexpression could promote glucose consumption and lactate production in GTPBP4 OE PLC/PRF/5 and GTPBP4 OE SMMC-7721 cells, and that GTPBP4 knockdown could reduce glucose consumption and lactate production in GTPBP4 KD HCCLM3 and GTPBP4 KD Huh 7 cells (Fig. 3G–H; Supplementary Figs. S5D–E). Taken together, these results show that GTPBP4 can promote glycolysis.
3.5. GTPBP4 promotes HCC growth through dimeric PKM2 dependent Warburg effect
PKM2 is the most abundant of GTPBP4‐interacting proteins and an essential Warburg effect enzyme (Fig. 3B; Supplementary Fig. S6A). We first found that endogenous GTPBP4 and PKM2 proteins can bind to each other in PLC/PRF/5 cell using Co-IP (Fig. 4A). Immunofluorescence staining showed that GTPBP4 and PKM2 colocalized in PLC/PRF/5 cells (Fig. 4B). We next expressed Flag-tagged GTPBP4 and GST-tagged PKM2 in HEK-293T cells and found that Flag-tagged GTPBP4 bounds in GST-tagged PKM2 in vivo (Fig. 4C).
Fig. 4.

GTPBP4 promotes the Warburg effect by regulating PKM2 in HCC. (A) Co-IP assays were conducted in PLC/PRF/5 cells transfected with or without Flag-tagged GTPBP4. n = 3, IgG was used as control. (B) GTPBP4 and PKM2 co-localization were determined by confocal microscopy scanning of immunofluorescence staining in PLC/PRF/5 cells. DAPI was used for nuclear staining. Scale bars = 10 μm. (C) HEK-293T cells were transfected with Flag-tagged GTPBP4 and GST-tagged PKM2 and subjected to co-immunoprecipitation assays. n = 3. (D) Representative IHC figures of GTPBP4 and PKM2 expression in HCC tissue microarray. Scale bars = 200 μm or 20 μm, respectively. (E) Correlation between GTPBP4 and PKM2 expression levels in human clinical HCC samples. Data are compared between different groups using Pearson Chi-squared test. (F) Protein samples from HCC cells were crosslinked with DSS (100 μM) and collected. Monomeric and Dimeric PKM2 expression was analyzed using Western blot. n = 3. (G) The glucose-induce ECAR was determined in control and GTPBP4 OE PLC/PRF/5 cells with or without PKM2 inhibition. n = 6. (H) Lactate production and glucose consumption activity were measured in GTPBP4 OE PLC/PRF/5 cells treated with or without PKM2 inhibition and respective controls. Error bars represent the mean ± SD from six independent experiments. The data presented in (G)–(H) are compared among groups using one-way ANOVA. *P < 0.05, **, P < 0.01, ***, P < 0.001.
Then, to determine if there is a correlation between GTPBP4 and PKM2 expression in clinical HCC specimens, GTPBP4 and PKM2 expression were analyzed by IHC staining in the Zhongshan HCC cohort (Fig. 4D, representative images shown). GTPBP4 expression significantly and positively correlates with that of PKM2 (P < 0.001). Specifically, about 78% (75 of 96) of the tumor tissues with high GTPBP4 expression showed moderate or strong PKM2 staining and 67% (58 of 86) of those with low GTPBP4 expression displayed negative or weak GTPBP4 staining (Fig. 4E).
It has been demonstrated that dimeric PKM2 promotes cancer progression by regulating the Warburg effect. Therefore, we speculated that GTPBP4 promotes HCC growth through the dimeric PKM2 dependent Warburg effect. Cross-linked PKM2 experiments demonstrated that dimeric PKM2 increased following GTPBP4 overexpression in PLC/PRF/5 and SMMC-7721 cells (Fig. 4F, left), and was reduced in GTPBP4 KD HCCLM3 and GTPBP4 KD Huh7 cells (Fig. 4F, right). TEPP-46, a small molecule inhibitor, could limit PKM2 dimer formation [32,33]. We found that TEPP-46 was sufficient to reverse the GTPBP4-overexpression-mediated ECAR increase in GTPBP4 OE PLC/PRF/5 cells (Fig. 4G). Consistently, we found that increased lactate production and glucose consumption activity assay by GTPBP4 overexpression in PLC/PRF/5 and SMMC-7721 cells could be fully antagonized by TEPP-46 (Fig. 4H, Supplementary Fig. S6B). Together, these data indicate that GTPBP4 regulated the Warburg effect through inducing dimeric PKM2 formation. Furthermore, Functional experiments showed that TEPP-46 could abrogate the increased proliferation capacity, invasion and migration ability of PLC/PRF/5 and SMMC-7721 cells caused by GTPBP4-overexpression (Supplementary Figs. S6C–F and Figs. S7A–B). Moreover, TEPP-46 also could elicit restoration of the apoptotic state of GTPBP4 OE PLC/PRF/5 and GTPBP4 OE SMMC-7721 cells (Supplementary Figs. S7C–D).
We also examined the expression of EMT markers and found that EMT-associated markers upregulated by GTPBP4 overexpression are also restored by PKM2 inhibition (Supplementary Fig. S7E). E Cadherin level also returned to control when PKM2 was inhibited. These results suggest that GTPBP4-mediated regulation of HCC growth and metastasis may also be achieved through inducing dimeric PKM2 dependent Warburg effect.
3.6. GTPBP4 induces PKM2 protein sumoylation and dimer formation through UBA2-SUMO1 axis
Previous studies have demonstrated that SUMO modifications contribute to subcellular localization of PKM2 proteins [34,35]. However, the effect of SUMO modifications on dimeric PKM2 formation was still not confirmed. GSEA analysis in the TCGA database shows that sumoylation and sumoylation-related pathways were significantly elevated in GTPBP4 high expression group (Supplementary Fig. S8A). We hypothesized that GTPBP4 regulates dimeric PKM2 formation through sumoylation. We co-expressed GST-tagged PKM2 and Myc-tagged SUMO1 in HEK-293T cells and performed Co-IP. The results showed that PKM2 could interact with SUMO1 (Fig. 5A). Furthermore, IP experiment verifies that GTPBP4 could promote PKM2 protein sumoylation (Fig. 5B). Ginkgolic acid (GA) and 2-D08 are small-molecule inhibitors targeting SUMOylation. GA inhibits SUMO-activating enzyme E1 [36,37], and 2-D08 inhibits SUMO E2 conjugating enzyme UBC9 [38,39]. IP experiment show that GA could inhibit PKM2 protein sumoylation induced by GTPBP4 overexpression (Fig. 5C). Western blot revealed that 2-D08 (50 μM) and GA (50 μM) could directly inhibit dimeric PKM2 formation in PLC/PRF/5 and SMMC-7721 cells, whether GTPBP4 overexpressed or not (Fig. 5D and Supplementary Fig. S8B). These data showed that GTPBP4 facilitates the binding of PKM2 with SUMO1 protein and its sumoylation. Consistently, ECAR was also decreased in PLC/PRF/5 cells regardless of GTPBP4 overexpression, when treated with 2-D08 and GA (Fig. 5E–F and Supplementary Figs. S8C–D). Lactate production and glucose consumption activity were also inhibited by 2-D08 and GA in PLC/PRF/5 and SMMC-7721 cells regardless of GTPBP4 overexpression (Supplementary Figs. S8E–F and Figs. S9A–B). Furthermore, functional experiments showed that 2-D08 and GA could abrogate the increased proliferation, invasion and migration ability of PLC/PRF/5 and SMMC-7721 cells caused by GTPBP4-overexpression (Supplementary Figs. S9C–F, Figs. S10A–D and Figs. S11A–B). Moreover, 2-D08 and GA could directly promote apoptosis of PLC/PRF/5 cells and restore the apoptotic state of GTPBP4 OE PLC/PRF/5 cells (Supplementary Figs. S11C–D).
Fig. 5.

GTPBP4 induces PKM2 protein sumoylation and dimer formation through UBA2/SUMO1 axis. (A) IP and Western blot were performed to assess PKM2 sumoylation in HEK-293T cells. n = 3. (B) IP and Western blot analyses were performed to assess PKM2 sumoylation in GTPBP4 OE PLC/PRF/5 and GTPBP4 KD HCCLM3 cells compared with the respective controls. n = 3. (C) sumoylation inhibitors, GA (50 μM) could inhibit GTPBP4-induced PKM2 protein Sumoylation. n = 3. (D) Monomeric and dimeric PKM2 expression were analyzed using Western blot in PLC/PRF/5 cells with or without GTPBP4 overexpression in the presence of Sumoylation inhibitors, GA (50 μM) and 2-D08 (50 μM). n = 3. (E–F) ECAR were detected in GTPBP4 OE PLC/PRF/5 and control cells in the presence of sumoylation inhibitors, GA and 2-D08. n = 6. (G) GTPBP4 and UBA2 co-localization were determined by Co-IP assays. (H) GTPBP4 has no effect on UBA2 protein expression. n = 3. (I) UBA2 knockdown could inhibit GTPBP4-induced PKM2 Sumoylation. n = 3. (J) GDI could significantly inhibit the activation of SUMO1 protein by GTPBP4 overexpression through UBA2. n = 3. (K) GDI could significantly inhibit the GTPBP4-induced PKM2 protein Sumoylation. n = 3. The data presented in (E)–(F) are compared among groups using one-way ANOVA. Error bars represent the mean ± SD from six independent experiments. *, P < 0.05, **, P < 0.01, ***, P < 0.001.
Ubiquitin-like modifier-activating enzyme 2 (UBA2) is an essential element of SUMO modification system, as it participates specifically at the activation step of SUMO proteins. UBA2 catalyzes SUMO1 activation in the presence of ATP by linking C-terminus of SUMO1 to a cysteine residue in UBA2 through a thioester. The energy required for this activation process derived from ATP hydrolysis, which forms high-energy thioester bond [40]. The guanosine triphosphate binding protein (GTPBP) family proteins act as GTP hydrolases from active (GTP bound) to the inactive (GDP bound) states [4]. GTPBP4 proteins is also a GTPases that, when activated in their GTP-bound form, activate downstream signal pathway. The mass spectrometry results show that UBA2 protein are potential direct GTPBP4 -binding protein (data not shown). Thus, we speculated that the process of GTP hydrolysis by GTPBP4 also could facilitate SUMO1 protein activation by UBA2. We found that endogenous GTPBP4 and UBA2 proteins can bind to each other in PLC/PRF/5 cell using Co-IP (Fig. 5G). However, Western blot indicates that GTPBP4 had no effect on UBA2 protein expression (Fig. 5H). Thus, GTPBP4 might accelerate SUMO1 protein activation through UBA2.
To test this, we first inhibit UBA2 expression by small interfering RNA and found that UBA2 knockdown could inhibit PKM2 protein sumoylation induced by GTPBP4 overexpression (Fig. 5I). GDP dissociation inhibitor (GDI) is a GTPase inhibitor. Western blot revealed that GDI could significantly inhibit the activation of SUMO1 protein by GTPBP4 overexpression through UBA2 (Fig. 5J). Moreover, GDI also could inhibit the sumoylation of PKM2 protein by GTPBP4 overexpression (Fig. 5K).
These results suggest that GTPBP4 can promote the activation of SUMO1 protein by UBA2 protein, and acts as a linker bridging activated SUMO1 protein and PKM2 protein to induce PKM2 sumoylation.
The SUMO moiety was recognized by the SUMO-interacting motif (SIM) for sumoylation of the targeted protein. Previous studies have shown that the PKM2 SIM site is IKII265-268. Point mutants at this SIM site, PKM2I267&268A, abolished SUMO1-induced PKM2 sumoylation [35]. We also validated that IKII265-268 is PKM2 SUMO-modification site. (Fig. 6A). Western blot revealed that transfection of PKM2I267&268A failed to induce dimeric PKM2 formation in both PLC/PRF/5 and SMMC-7721 cells even though GTPBP4 is overexpressed (Fig. 6B). Functional experiments showed that transfection of PKM2I267&268A did not increase proliferation capacity, invasion and migration ability of PLC/PRF/5 cells even though GTPBP4 is overexpressed (Supplementary Figs. S12A and C and Fig. S13A). The same results were obtained in GTPBP4 OE SMMC-7721 and SMMC-7721 cells (Supplementary Figs. S12B and D and Supplementary Fig. S13B). Furthermore, transfection of PKM2I267&268A did not inhibit GTPBP4 OE PLC/PRF/5 and PLC/PRF/5 cells apoptosis (Supplementary Fig. S13C). Taken together, these results demonstrate that GTPBP4 promotes SUMO1 protein activation by UBA2 to induce PKM2 protein sumoylation and dimeric PKM2 formation.
Fig. 6.

GTPBP4 regulates translocation of PKM2 into the nucleus by sumoylation. (A) Co-IP was performed to verify that PKM2 SIM mutants (PKM2I267&268A) could abolish SUMO1-induced PKM2 sumoylation. n = 3. (B) Monomeric and dimeric PKM2 expression was analyzed using Western blot in PLC/PRF/5 (left) and SMMC-7721 cells (right) grouped and treated as showed. n = 3. (C) Western blot was used to detect PKM2 expression in the nucleus in GTPBP4 OE PLC/PRF/5 cells and GTPBP4 KD HCCLM3 cells and corresponding control cells. n = 3. (D) PLC/PRF/5 cells were transfected with Flag-tagged GTPBP4, GST-tagged PKM2, Myc-tagged SUMO1, and HA-tagged SENP1 and subjected to Western blot assay. (E) PLC/PRF/5 cells were transfected with Flag-tagged GTPBP4, GST-tagged PKM2, His-tagged PKM2I267&268A, and Myc-tagged SUMO1 and subjected to Western blot assay. n = 3. (F) ECAR was detected in GTPBP4 OE PLC/PRF/5 and control cells transfected with or without PKM2I267&268A. Error bars represent the mean ± SD from six independent experiments. n = 6. (G–H) Lactate production (G) and glucose consumption activity (H) were measured were detected in GTPBP4 OE PLC/PRF/5 cells and GTPBP4 OE SMMC-7721 cells and their respective control cells transfected both with or without PKM2I267&268A. n = 6. The data presented in (F)–(H) are compared among groups using one-way ANOVA. *P < 0.05, **P < 0.01, ***P < 0.001.
3.7. GTPBP4 regulates translocation of PKM2 into the nucleus by sumoylation
Dimeric PKM2 also promote aerobic glycolysis as a protein kinase and a co-transcription factor in the nucleus acting on gene transcription during tumor malignancy transformation [23,41]. Here, we first found that GTPBP4 could induce PKM2 nuclear localization using Western blot (Fig. 6C), which was further verified by immunofluorescence assays (Supplementary Fig. S14A). Previous studies showed that nuclear PKM2 could act as a protein kinase and phosphorylates STAT3 at Y705.
We also found that GTPBP4 could promote STAT3 phosphorylation (Supplementary Fig. S14B). These results show that GTPBP4 regulates PKM2 expression and its nuclear translocation.
Previous studies have demonstrated that the nuclear translocation of PKM2 is regulated by sumoylation [34]. Combined with these results above, we hypothesized that GTPBP4 regulating dimeric PKM2 formation through sumoylation and nuclear translocation is a dynamic and continuous process. SUMO specific protease 1 (SENP1) is a desumoylation enzyme. We examined PKM2 localization in PLC/PRF/5 cells when transfected with plasmid expressing GTPBP4, PKM2, SUMO1, or SENP1, Western blot and immunofluorescence analysis revealed that GTPBP4-induced sumoylation triggers PKM2 protein nuclear localization and SENP1 could inhibit GTPBP4-mediated PKM2 nuclear localization via sumoylation (Fig. 6D; Supplementary Fig. S14C). Furthermore, we transfected PLC/PRF/5 cells with plasmid expressing GTPBP4, PKM2, SUMO1, or PKM2I267&268A and examined PKM2 localization. Western blot and immunofluorescence analysis revealed that GTPBP4 and SUMO1 co-transfection induced the PKM2 nuclear translocation in PLC/PRF/5 cells, and that this effect was abolished in the presence of PKM2I267&268A (Fig. 6E; Supplementary Fig. S14D). Combining the above results, we concluded that GTPBP4-induced sumoylation triggers dimeric PKM2 protein nuclear localization. Furthermore, we detected no corresponding increase in ECAR in both GTPBP4 OE PLC/PRF/5 and PLC/PRF/5 cells when transfected of PKM2I267&268A (Fig. 6F). Lactate production and glucose consumption activity were not significantly altered in both PLC/PRF/5, SMMC-7721 cells after transfection of PKM2I267&268A even though GTPBP4 is overexpressed (Fig. 6G–H). Taken together, GTPBP4 regulates translocation of PKM2 into the nucleus by sumoylation to promote aerobic glycolysis and activate STAT3 signal way.
3.8. PKM2 inhibitor suppresses HCC progression and metastasis
Shikonin, the major chemical component of Lithospermum erythrorhizon (Purple Cromwell) roots, is a specific PKM2 inhibitor [42,43]. It has been demonstrated that Shikonin exerted a remarkable antitumor effect in many tumors. However, the mechanism underlying this effect remains incompletely understood. We found that, first Shikonin could inhibit the binding of GTPBP4 and PKM2 proteins (Fig. 7A). As a result, Shikonin could inhibit GTPBP4-overexpression-mediated dimeric PKM2 formation and ECAR increase in GTPBP4 OE PLC/PRF/5 cells (Fig. 7B–C). Consistently, we found that increased lactate production and glucose consumption activity by GTPBP4 overexpression in PLC/PRF/5 and SMMC-7721 cells cells could be fully antagonized by Shikonin (Fig. 7D–E).
Fig. 7.

PKM2 inhibitor suppresses HCC cells progression in vitro. (A) IP and Western blot analyses were performed to verify that Shikonin, could reducing the interaction between GTPBP4 and PKM2 proteins in GTPBP4 OE PLC/PRF/5 cells. n = 3. (B) Monomeric and dimeric PKM2 expression was analyzed using Western blot in GTPBP4 OE PLC/PRF/5 and control cells treated with or without Shikonin. n = 3. (C) The glucose-induce ECAR was determined in GTPBP4 OE PLC/PRF/5 and control cells treated with or without Shikonin. n = 6. one-way ANOVA. (D–E) Lactate production (D) and glucose consumption activity (E) were measured in GTPBP4 OE PLC/PRF/5 cells and GTPBP4 OE SMMC-7721 cells treated with or without Shikonin and their respective controls. n = 6. one-way ANOVA. (F) The migratory and invasive capacities of GTPBP4 OE PLC/PRF/5 cells treated with or without Shikonin and PLC/PRF/5 cells were evaluated by transwell assays. n = 6. one-way ANOVA (G) Cell proliferation of GTPBP4 OE PLC/PRF/5 cells treated with or without Shikonin and PLC/PRF/5 cells were assessed by CCK-8. n = 6. one-way ANOVA. (H) Apoptosis was assessed by flow cytometry in GTPBP4 OE PLC/PRF/5 cells treated with or without Shikonin and PLC/PRF/5 cells. n = 3, one-way ANOVA. Error bars represent the mean ± SD from three or more independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001.
Furthermore, functional experiments showed that Shikonin could inhibit the increased proliferation, invasion and migration ability of PLC/PRF/5 and SMMC-7721 cells caused by GTPBP4-overexpression (Fig. 7F–G; Supplementary Figs. S15A–D). Moreover, Shikonin also could elicit restoration of the apoptotic state of GTPBP4 OE PLC/PRF/5 and GTPBP4 OE SMMC-7721 cells (Fig. 7H; Supplementary Fig. S15E). These experiments in vitro demonstrated that Shikonin could inhibit HCC growth and metastasis through repressing the dimeric PKM2 dependent Warburg effect.
To evaluate the inhibition effects of shGTPBP4 and Shikonin on HCC growth and metastasis in vivo, we established orthotopic xenograft mouse models of HCC (6 mice per group). In addition, no significant changes in toxicity in the liver and kidney or average mice body were observed (Supplementary Figs. S16A–D). We found that GTPBP4 promotes tumor growth and metastasis through PKM2 in vivo. The proliferation and lung metastases of GTPBP4 overexpression PLC/PRF/5 xenograft tumors (vs. control) was antagonized by Shikonin treatment and administrations of Shikonin only on PLC/PRF/5 xenograft tumors also significantly suppressed the HCC growth and lung metastases (Fig. 8A, E). GTPBP4 knockdown and Shikonin-treated HCCLM3 xenograft tumors and lung metastases were significantly smaller and less than those of the vector control (Fig. 8C, F). We also detected the effect of GTPBP4 on glycolysis of HCC in vivo through measuring [18F]-FDG uptake by PET-CT. As reflected in Fig. 8B and D, GTPBP4 overexpression could promote HCC growth, glycolysis, while knockdown GTPBP4 expression could suppress HCC glycolysis, growth. Moreover, Shikonin could suppress HCC growth and glycolysis through inhibiting PKM2 dependent glucose metabolism (Fig. 8B and D).
Fig. 8.

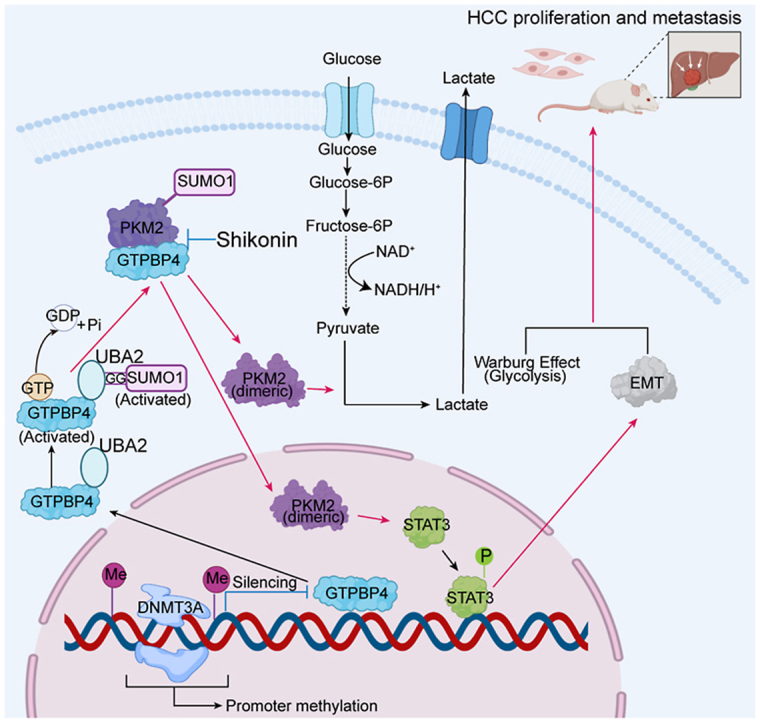
PKM2 inhibitor suppresses HCC cells progression in vivo (A) Orthotopic xenograft models were derived from PLC/PRF/5 Vector and PLC/PRF/5-GTPBP4 cells treated as indicated (left). Tumor volumes are shown (right). n = 6, one-way ANOVA. (B) Glucose metabolism of HCC in each group in (A) was confirmed by representative [18F]-PET-CT images at the end of therapy (4 weeks). (C) Orthotopic xenograft models were derived from HCCLM3 Control and HCCLM3-shGTPBP4 cells treated as indicated (left). Tumor volumes are shown (right). n = 6, one-way ANOVA. (D) Glucose metabolism of HCC in each group was confirmed by representative [18F]-PET-CT images at the end of therapy (4 weeks). n = 6, one-way ANOVA. (E) Representative images of H&E staining of lung tissues (left) and the number of lung metastatic foci (right) from each group shown in (A). n = 6, one-way ANOVA. (F) Representative images of H&E staining of lung tissues (upper) and the number of lung metastatic foci (lower) from each group shown in (B). n = 6, one-way ANOVA. (G) Schematic diagram illustrating role of GTPBP4 in promoting hepatocellular carcinoma progression. DNA methyltransferase, DNMT3A, negatively regulates GTPBP4 expression through GTPBP4 DNA promoter methylation modification. Active GTPBP4 (GTP bound) facilitates SUMO1 protein activation by UBA2, and acts as a linker bridging activated SUMO1 protein and PKM2 protein to induce PKM2 sumoylation to promote aerobic glycolysis in HCC. Furthermore, SUMO-modified PKM2 relocates from the cytoplasm to the nucleus and activates STAT3 signaling pathway and epithelial-mesenchymal transition (EMT). Together, the two ways contribute to the development of HCC. Abbreviations: IHC: immunohistochemical. *P < 0.05, **P < 0.01, ***P < 0.001.
IHC staining was used to validate these observations. Tumors with GTPBP4 knockdown or Shikonin treatment had markedly reduced Ki-67, GTPBP4, PKM2, N-cadherin, and p-STAT3 levels, and increased E-cadherin expression. The increased Ki-67, GTPBP4, PKM2 and p-STAT3 expression was attenuated in GTPBP4 overexpression PLC/PRF/51 xenografts treated with Shikonin. Downregulation of E-cadherin in GTPBP4 overexpression PLC/PRF/51 xenografts was also rescued by Shikonin treatment (Supplementary Fig. S16E and Fig. S17A). Together, these results suggest that GTPBP4 promotes HCC proliferation and metastasis.
4. Discussion
GTPBP members, consisting of G-proteins and the Ras superfamily, are an important class of molecular switch proteins, that flip between active (GTP-bound) and inactive, (GDP-bound) states [4]. When in the activated state, GTPBP acts as a signal to trigger other physiological processes and cellular events. Although a few of these studies revealed that GTPBP4 is associated with an unfavorable prognosis in patients with breast tumors or colorectal carcinoma, the roles of GTPBP4 in tumor progression remain unknown. In this study, we found that GTPBP4 expression may correlate with HCC malignant progression and contributes to the poor OS and short RFS, which is the same with a previous study [8]. However, our study conducted deeper research on GTPBP4 and its clinical significance.
DNA methylation is an important epigenetic regulation mechanism and regulate gene expression in development and disease. Aberrant DNA promoter CpG islands methylation has been extensively studied and is involved in multiple genetic abnormalities in the course of tumorigenesis [44,45]. However, the mechanisms underlying promoter methylation and gene regulation remain unclear. HCC is characterized by global DNA hypomethylation, transcriptional enhancer hypomethylation, and promoter hypermethylation [29,46]. However, DNA promoter hypomethylation involved in oncogene upregulation also has recently been reported [30]. In the present study, we found that GTPBP4 upregulation is paralleled by DNA promoter hypomethylation. Our CHIP-qPCR and luciferase assay results show that this hypomethylation is regulated by DNMT3A. The sequence of binding sites is GATTGG.
Therefore, this result highlights the mechanism of GTPBP4 transcription regulation, and verifies that tumor-promoting gene promoter hypomethylation is involved in HCC progression. Of course, it is worth noting that there are likely to be other transcriptional binding site we have not yet detected. More studies are necessary to validate the way DNMT3A regulates GTPBP4 expression. Furthermore, our study does not examine the factor in the tumor microenvironment directly or indirectly affect epigenetic reprogramming of GTPBP4 promoter. More studies are needed to explore the effect of tumor microenvironment on tumoral cells epigenetic reprogramming for cancer progression and metastasis.
This study identified PKM2 mediated glycolysis pathways regulated by GTPBP4 in promoting HCC proliferation and metastasis. We first found that GTPBP4 is involved in glycolysis and the TCA cycle. It is accepted that cancer cells preferentially utilize glycolysis to bypass the TCA cycle or mitochondrial dysfunction [47,48]. However, accumulating evidence demonstrates that multiple cancer cells, especially metastatic cancer cells, show active OXPHOS and heavily depend on the TCA cycle for energy production and macromolecule synthesis [[49], [50], [51]]. Therefore, that GTPBP4 was found to promote both glycolysis and TCA cycle is not necessarily contradictory. PKM2 is abundant in GTPBP4‐interacting proteins through IP and mass spectrometric analyses. Therefore, we examined the effect of GTPBP4 on PKM2 regulation at the protein level. Dimeric PKM2 promotes the Warburg effect. The Warburg effect is an important hallmark of cancer cells. We first demonstrate that GTPBP4 could induce PKM2 protein sumoylation and dimeric PKM2 formation to promote HCC glycolysis. We also found that active GTPBP4 (GTP bound) facilitates SUMO1 protein activation by UBA2 to induce PKM2 protein sumoylation. We speculate that GTP hydrolysis by GTPBP4 provides conditions for SUMO1 protein and UBA2 protein binding, like energy, and acts as a linker bridging activated SUMO1 protein and PKM2 protein to induce PKM2 sumoylation. Then, we found that GTPBP4 promotes the Warburg effect by inducing PKM2 dimer formation in HCC by using extracellular flux, glucose consumption, and lactate production detection assays. Apart from promoting glycolysis, PKM2 also has non-glycolytic functions. Nuclear PKM2 exits in its dimer conformation and functions as a protein kinase, which could activate some important signaling pathways in tumorigenesis and progression. We also found that high GTPBP4 expression induces the translocation of PKM2 from the cytosol to the nucleus and activates STAT3 and EMT signal pathway, which are important molecular signatures involved in promoting tumor progression and metastasis. Previous studies have demonstrated that PKM2 sumoylation regulates its nuclear translocation [34]. We investigated whether GTPBP4 regulates PKM2 nuclear translocation by sumoylation. Finally, we used CO-IP experiments and Immunofluorescence analysis to demonstrate that GTPBP4 regulates PKM2 expression and translocation of dimer PKM2 into the nucleus through sumolylation.
In conclusion, this study verified that the GTPBP4-PKM2 axis plays a significant role in promoting HCC progression and metastasis and could become a therapeutic target for HCC treatment (Fig. 8G). Shikonin, the major chemical component of Lithospermum erythrorhizon (Purple Cromwell) roots, is a specific PKM2 inhibitor [42]. There is still no research on the effect of Shikonin on the treatment of HCC. We found that Shikonin administration efficiently suppresses tumor growth in orthotopic xenograft mouse models of HCC and could become a promising drug candidate for HCC therapy.
5. Conclusions
Metabolic reprogramming, especial aerobic glycolysis, is viewed as a hallmark of cancer. However, underlying regulatory mechanisms are still elusive. In this study, we found that GTPBP4 promotes HCC progression and metastasis through inducing dimeric PKM2 dependent metabolic reprogramming via protein sumoylation modification. We also found that Shikonin can attenuate HCC progression and metastasis and will make promising drug candidates for HCC therapy.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82073208, 82103521), the Shanghai Shen Kang Hospital Development Center new frontier technology joint project (SHDC12021109), the Shanghai Sailing Program (21YF1407500), China Postdoctoral Science Foundation (2021M690674), the Science and Technology Plan Project of Xiamen City (3502Z20194036), and the Program of Shanghai Academic Research Leader (No. 20XD1400900).
Author contributions
Q. Z. was responsible for drafting of the manuscript, functional experiments, analysis and interpretation of data; Y. Y. was responsible for functional experiments, analysis; M. Y. was responsible interpretation of data; J. S. and Z. Y. were responsible for critical revision of the manuscript for important intellectual content and animal model establishment; M. A. and W. C. were responsible for patient tissue sample collection; J. W. and D. G.was responsible for clinical data analysis; Y. Shen provided technical support for Seahorse studies; W. C. and Q. D. provided technical support for Seahorse studies; C. Z. was responsible for supervision of technical, material support, and manuscript review. N. R. was responsible for supervision of the entire project, technical, or material support, experimental design, and manuscript review.
Ethic approval and consent to participate
The study was approved by the Ethics Committee of Zhongshan Hospital, Fudan University. Informed consent for the use of clinical data in this study was obtained from all patients without financial compensation. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the institution's human research committee. Animal experiments in this study were in accordance with the institutional animal care and use guidelines and were approved by the Animal Care and Use Committee of Zhongshan Hospital, Fudan University.
Consent for publication
All the authors have signed the form of consent to publication.
Data availability statement
This study includes no data deposited in external repositories.
Declaration of competing interest
The authors have declared that no competing interest exists.
Footnotes
All co-authors have contributed to, read and approved the final manuscript for submission and there is no financial interest to report. We certify that the manuscript is original research that has neither been published nor submitted for publication elsewhere.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102458.
Contributor Information
Qiongzhu Dong, Email: qzhdong@fudan.edu.cn.
Chenhao Zhou, Email: zhouchenhao@fudan.edu.cn.
Ning Ren, Email: ren.ning@zs-hospital.sh.cn.
Abbreviations
- HCC
hepatocellular carcinoma
- GTPBP4
Guanosine triphosphate binding protein 4
- OS
overall survival
- RFS
recurrence-free survival
- PKM2
pyruvate kinase M2
- DNMT3A
DNA methyltransferase 3a
- TMA
tissue microarray
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
- shRNA
small hairpin RNA
- ECAR
Glucose-induced extracellular acidification rate
- GEO
- Gene Expression Omnibus
- TCGA
The Cancer Geneome Atlas
- TNM
tumor-nodes-metastasis
- IHC
immunohistochemical
- CCK-8
Cell Counting Kit-8
- OE
overexpressing
- KD
knockdown
- HR
hazard ratio
- CI
confidence interval
- IP
immunoprecipitation
- LC-MS/MS
liquid chromatography coupled with tandem mass spectrometry
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Siegel R.L., Miller K.D., Jemal A. Cancer statistics. CA Cancer J. Clin. 2020;70(1) doi: 10.3322/caac.21590. 2020. [DOI] [PubMed] [Google Scholar]
- 2.Chen W., Zheng R., Baade P.D., Zhang S., Zeng H., Bray F., et al. Cancer statistics in China, 2015. Ca - Cancer J. Clin. 2016;66(2):115–132. doi: 10.3322/caac.21338. 671 2020s. [DOI] [PubMed] [Google Scholar]
- 3.Gluer A.M., Cocco N., Laurence J.M., Johnston E.S., Hollands M.J., Pleass H.C.C., et al. Systematic review of actual 10-year survival following resection for hepatocellular carcinoma. HPB : Off. J. Int. Hepato Pancreato Biliary Assoc. 2012;14(5):285–290. doi: 10.1111/j.1477-2574.2012.00446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chung K.Y., Rasmussen S.G.F., Liu T., Li S., DeVree B.T., Chae P.S., et al. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature. 2011;477(7366):611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim Y.-I., Bandyopadhyay J., Cho I., Lee J., Park D.H., Cho J.H. Nucleolar GTPase NOG-1 regulates development, fat storage, and longevity through insulin/IGF signaling in C. elegans. Mol. Cell. 2014;37(1):51–57. doi: 10.14348/molcells.2014.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chouchana L., Fernández-Ramos A.A., Dumont F., Marchetti C., Ceballos-Picot I., Beaune P., et al. DNAMolecular insight into thiopurine resistance: transcriptomic signature in lymphoblastoid cell lines. Genome Med. 2015;7(1):37. doi: 10.1186/s13073-015-0150-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lunardi A., Di Minin G., Provero P., Dal Ferro M., Carotti M., Del Sal G., et al. A genome-scale protein interaction profile of Drosophila p53 uncovers additional nodes of the human p53 network. Proc. Natl. Acad. Sci. U. S. A. 2010;107(14):6322–6327. doi: 10.1073/pnas.1002447107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu W.-B., Jia W.-D., Ma J.-L., Xu G.-L., Zhou H.-C., Peng Y., et al. Knockdown of GTPBP4 inhibits cell growth and survival in human hepatocellular carcinoma and its prognostic significance. Oncotarget. 2017;8(55):93984–93997. doi: 10.18632/oncotarget.21500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liberti M.V., Locasale J.W. The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci. 2016;41(3):211–218. [Google Scholar]
- 10.Yang W., Xia Y., Hawke D., Li X., Liang J., Xing D., et al. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell. 2014;158(5):1210. doi: 10.1016/j.cell.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 11.Hosios A.M., Hecht V.C., Danai L.V., Johnson M.O., Rathmell J.C., Steinhauser M.L., et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev. Cell. 2016;36(5):540–549. doi: 10.1016/j.devcel.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kitamura K., Hatano E., Higashi T., Narita M., Seo S., Nakamoto Y., et al. Proliferative activity in hepatocellular carcinoma is closely correlated with glucose metabolism but not angiogenesis. J. Hepatol. 2011;55(4):846–857. doi: 10.1016/j.jhep.2011.01.038. [DOI] [PubMed] [Google Scholar]
- 13.Tamada M., Suematsu M., Saya H. Pyruvate kinase M2: multiple faces for conferring benefits on cancer cells. Clin. Cancer Res. 2012;18(20):5554–5561. doi: 10.1158/1078-0432.CCR-12-0859. [DOI] [PubMed] [Google Scholar]
- 14.Noguchi T., Yamada K., Inoue H., Matsuda T., Tanaka T. The L- and R-type isozymes of rat pyruvate kinase are produced from a single gene by use of different promoters. J. Biol. Chem. 1987;262(29):14366–14371. [PubMed] [Google Scholar]
- 15.Noguchi T., Inoue H., Tanaka T. The M1- and M2-type isozymes of rat pyruvate kinase are produced from the same gene by alternative RNA splicing. J. Biol. Chem. 1986;261(29):13807–13812. [PubMed] [Google Scholar]
- 16.Imamura K., Tanaka T. Multimolecular forms of pyruvate kinase from rat and other mammalian tissues. I. Electrophoretic studies. J. Biochem. 1972;71(6):1043–1051. doi: 10.1093/oxfordjournals.jbchem.a129852. [DOI] [PubMed] [Google Scholar]
- 17.De Rosa V., Iommelli F., Monti M., Fonti R., Votta G., Stoppelli M.P., et al. Reversal of Warburg effect and reactivation of oxidative phosphorylation by differential inhibition of EGFR signaling pathways in non-small cell lung cancer. Clin. Cancer Res. 2015;21(22):5110–5120. doi: 10.1158/1078-0432.CCR-15-0375. [DOI] [PubMed] [Google Scholar]
- 18.Huang J.-Z., Chen M., Chen D., Gao X.-C., Zhu S., Huang H., et al. A peptide encoded by a putative lncRNA HOXB-AS3 suppresses colon cancer growth. Mol. cell. 2017;68(1) doi: 10.1016/j.molcel.2017.09.015. [DOI] [PubMed] [Google Scholar]
- 19.Lv L., Xu Y.-P., Zhao D., Li F.-L., Wang W., Sasaki N., et al. Mitogenic and oncogenic stimulation of K433 acetylation promotes PKM2 protein kinase activity and nuclear localization. Mol. cell. 2013;52(3):340–352. doi: 10.1016/j.molcel.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai J., Escara-Wilke J., Keller J.M., Jung Y., Taichman R.S., Pienta K.J., et al. Primary prostate cancer educates bone stroma through exosomal pyruvate kinase M2 to promote bone metastasis. J. Exp. Med. 2019;216(12):2883–2899. doi: 10.1084/jem.20190158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim J.Y., Yoon S.O., Seol S.Y., Hong S.W., Kim J.W., Choi S.H., et al. Overexpression of the M2 isoform of pyruvate kinase is an adverse prognostic factor for signet ring cell gastric cancer. World J. Gastroenterol. 2012;18(30):4037–4043. doi: 10.3748/wjg.v18.i30.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu F., Ma F., Wang Y., Hao L., Zeng H., Jia C., et al. PKM2 methylation by CARM1 activates aerobic glycolysis to promote tumorigenesis. Nat. Cell Biol. 2017;19(11):1358–1370. doi: 10.1038/ncb3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao X., Wang H., Yang J.J., Liu X., Liu Z.-R. Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell. 2012;45(5):598–609. doi: 10.1016/j.molcel.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou C., Liu C., Liu W., Chen W., Yin Y., Li C.W., et al. SLFN11 inhibits hepatocellular carcinoma tumorigenesis and metastasis by targeting RPS4X via mTOR pathway. Theranostics. 2020;10(10):4627–4643. doi: 10.7150/thno.42869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y., Tian B., Yang J., Zhao L., Wu X., Ye S.L., et al. Stepwise metastatic human hepatocellular carcinoma cell model system with multiple metastatic potentials established through consecutive in vivo selection and studies on metastatic characteristics. J. Cancer Res. Clin. Oncol. 2004;130(8):460–468. doi: 10.1007/s00432-004-0564-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou P.P., Luo L.J., Chen H.Z., Chen Q.T., Bian X.L., Wu S.F., et al. Ectosomal PKM2 promotes HCC by inducing macrophage differentiation and remodeling the tumor microenvironment. Mol. Cell. 2020;78(6):1192–1206. doi: 10.1016/j.molcel.2020.05.004. e10. [DOI] [PubMed] [Google Scholar]
- 27.Chen X., Müller G.A., Quaas M., Fischer M., Han N., Stutchbury B., et al. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol. Cell Biol. 2013;33(2):227–236. doi: 10.1128/MCB.00881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H., Li X., Liu S., Guo L., Zhang B., Zhang J., et al. Programmed cell death-1 (PD-1) checkpoint blockade in combination with a mammalian target of rapamycin inhibitor restrains hepatocellular carcinoma growth induced by hepatoma cell-intrinsic PD-1. Hepatology. 2017;66(6):1920–1933. doi: 10.1002/hep.29360. [DOI] [PubMed] [Google Scholar]
- 29.Calvisi D.F., Ladu S., Gorden A., Farina M., Lee J.S., Conner E.A., et al. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Invest. 2007;117(9):2713–2722. doi: 10.1172/JCI31457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stefanska B., Cheishvili D., Suderman M., Arakelian A., Huang J., Hallett M., et al. Genome-wide study of hypomethylated and induced genes in patients with liver cancer unravels novel anticancer targets. Clin. Cancer Res. 2014;20(12):3118–3132. doi: 10.1158/1078-0432.CCR-13-0283. [DOI] [PubMed] [Google Scholar]
- 31.Gao L., Emperle M., Guo Y., Grimm S.A., Ren W., Adam S., et al. Comprehensive structure-function characterization of DNMT3B and DNMT3A reveals distinctive de novo DNA methylation mechanisms. Nat. Commun. 2020;11(1):3355. doi: 10.1038/s41467-020-17109-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angiari S., Runtsch M.C., Sutton C.E., Palsson-McDermott E.M., Kelly B., Rana N., et al. Pharmacological activation of pyruvate kinase M2 inhibits CD4 T cell pathogenicity and suppresses autoimmunity. Cell Metabol. 2020;31(2) doi: 10.1016/j.cmet.2019.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nayak M.K., Ghatge M., Flora G.D., Dhanesha N., Jain M., Markan K.R., et al. The metabolic enzyme pyruvate kinase M2 regulates platelet function and arterial thrombosis. Blood. 2021;137(12):1658–1668. doi: 10.1182/blood.2020007140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spoden G.A., Morandell D., Ehehalt D., Fiedler M., Jansen-Dürr P., Hermann M., et al. The SUMO-E3 ligase PIAS3 targets pyruvate kinase M2. J. Cell. Biochem. 2009;107(2):293–302. doi: 10.1002/jcb.22125. [DOI] [PubMed] [Google Scholar]
- 35.Hou P.-P., Luo L.-J., Chen H.-Z., Chen Q.-T., Bian X.-L., Wu S.-F., et al. Ectosomal PKM2 promotes HCC by inducing macrophage differentiation and remodeling the tumor microenvironment. Mol. Cell. 2020;78(6) doi: 10.1016/j.molcel.2020.05.004. [DOI] [PubMed] [Google Scholar]
- 36.Fukuda I., Ito A., Hirai G., Nishimura S., Kawasaki H., Saitoh H., et al. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009;16(2):133–140. doi: 10.1016/j.chembiol.2009.01.009. [DOI] [PubMed] [Google Scholar]
- 37.Liu K., Wang X., Li D., Xu D., Li D., Lv Z., et al. Ginkgolic acid, a SUMO-1 inhibitor, inhibits the progression of oral squamous cell carcinoma by alleviating SUMOylation of SMAD4. Mol Ther. Oncolytics. 2020;16:86–99. doi: 10.1016/j.omto.2019.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou P., Chen X., Li M., Tan J., Zhang Y., Yuan W., et al. 2-D08 as a SUMOylation inhibitor induced ROS accumulation mediates apoptosis of acute myeloid leukemia cells possibly through the deSUMOylation of NOX2. Biochem. Biophys. Res. Commun. 2019;513(4):1063–1069. doi: 10.1016/j.bbrc.2019.04.079. [DOI] [PubMed] [Google Scholar]
- 39.Kim Y.S., Keyser S.G.L., Schneekloth J.S. Synthesis of 2',3',4'-trihydroxyflavone (2-D08), an inhibitor of protein sumoylation. Bioorg. Med. Chem. Lett. 2014;24(4):1094–1097. doi: 10.1016/j.bmcl.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aichem A., Sailer C., Ryu S., Catone N., Stankovic-Valentin N., Schmidtke G., et al. The ubiquitin-like modifier FAT10 interferes with SUMO activation. Nat. Commun. 2019;10(1):4452. doi: 10.1038/s41467-019-12430-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang W., Xia Y., Ji H., Zheng Y., Liang J., Huang W., et al. Nuclear PKM2 regulates β-catenin transactivation upon EGFR activation. Nature. 2011;480(7375):118–122. doi: 10.1038/nature10598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andújar I., Recio M.C., Giner R.M., Ríos J.L. Traditional Chinese medicine remedy to jury: the pharmacological basis for the use of shikonin as an anticancer therapy. Curr. Med. Chem. 2013;20(23):2892–2898. doi: 10.2174/09298673113209990008. [DOI] [PubMed] [Google Scholar]
- 43.Boulos J.C., Rahama M., Hegazy M.F., Efferth T. Shikonin derivatives for cancer prevention and therapy. Cancer Lett. 2019;459:248–267. doi: 10.1016/j.canlet.2019.04.033. [DOI] [PubMed] [Google Scholar]
- 44.Chapuy B., McKeown M.R., Lin C.Y., Monti S., Roemer M.G., Qi J., et al. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013;24(6):777–790. doi: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mack S.C., Pajtler K.W., Chavez L., Okonechnikov K., Bertrand K.C., Wang X., et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature. 2018;553(7686):101–105. doi: 10.1038/nature25169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiong L., Wu F., Wu Q., Xu L., Cheung O.K., Kang W., et al. Aberrant enhancer hypomethylation contributes to hepatic carcinogenesis through global transcriptional reprogramming. Nat. Commun. 2019;10(1):335. doi: 10.1038/s41467-018-08245-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liberti M.V., Locasale J.W. The Warburg effect: how does it benefit cancer cells? Trends Biochem. Sci. 2016;41(3):211–218. [Google Scholar]
- 48.Warburg O. On the origin of cancer cells. Science. 1956;123(3191):309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 49.Anderson N.M., Mucka P., Kern J.G., Feng H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. 2018;9(2):216–237. doi: 10.1007/s13238-017-0451-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pascual G., Domínguez D., Benitah S.A. The contributions of cancer cell metabolism to metastasis. Dis. Model. Mech. 2018;11(8) doi: 10.1242/dmm.032920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jing Y.Y., Cai F.F., Zhang L., Han J., Yang L., Tang F., et al. Epigenetic regulation of the Warburg effect by H2B monoubiquitination. Cell Death Differ. 2020;27(5):1660–1676. doi: 10.1038/s41418-019-0450-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study includes no data deposited in external repositories.