

Abstract
Recently, we presented a whole-cell kinetic model of the genetically minimal bacterium JCVI-syn3A that described the coupled metabolic and genetic information processes and predicted behaviors emerging from the interactions among these networks. JCVI-syn3A is a genetically reduced bacterial cell that has the fewest number and smallest fraction of genes of unclear function, with approximately 90 of its 452 protein-coding genes (that is less than 20%) unannotated. Further characterization of unclear JCVI-syn3A genes strengthens the robustness and predictive power of cell modeling efforts and can lead to a deeper understanding of biophysical processes and pathways at the cell scale. Here, we apply computational analyses to elucidate the functions of the products of several essential but previously uncharacterized genes involved in integral cellular processes, particularly those directly affecting cell growth, division, and morphology. We also suggest directed wet-lab experiments informed by our analyses to further understand these “missing puzzle pieces” that are an essential part of the mosaic of biological interactions present in JCVI-syn3A. Our workflow leverages evolutionary sequence analysis, protein structure prediction, interactomics, and genome architecture to determine upgraded annotations. Additionally, we apply the structure prediction analysis component of our work to all 452 protein coding genes in JCVI-syn3A to expedite future functional annotation studies as well as the inverse mapping of the cell state to more physical models requiring all-atom or coarse-grained representations for all JCVI-syn3A proteins.
1. Introduction
JCVI-syn3A is a genetically reduced bacterium containing 493 genes and 543 kbp DNA, that was reduced from its parent organism Mycoplasma mycoides subsp. capri GM12,1−3 giving it one of the most minimal genomes of any living cell. To briefly describe its development, in 2008, the J. Craig Venter Institute (JCVI) demonstrated the chemical synthesis of an entire bacterial genome.4 This was followed by the synthesis of a M. mycoides subsp. capri GM12 genome and its later transplantation into M. capricolum recipient cells to produce JCVI-syn1.0.1,5 Further cycles of the synthetic biology “design, build, test” strategy of the JCVI to reduce the genome by omission of nonessential genes followed by genome synthesis, transplantation, and growth testing gave JCVI-syn3.0.2 Because JCVI-syn3.0 cells were fragile and difficult to manipulate and also because they appeared pleomorphic (with irregular shapes across a population), 19 genes from JCVI-syn1.0 were reintroduced to give JCVI-syn3A.3 Crucially, only approximately 90 of its 452 protein coding genes (or less than 20%) remain without an annotated function3 (compared to approximately 40% for Escherichia coli).These 90 genes can be further subdivided into approximately equivalent thirds which are essential, quasi-essential, and nonessential to JCVI-syn3A development, survival, and proliferation.3 Thus, JCVI-syn3A has one of the most well-characterized genomes of any living cell. The simplicity and small number of genes with unclear function along with genome scale proteomics and essentiality assignments3 makes JCVI-syn3A an attractive system from which to probe the fundamental principles of cellular life (i.e., in essence a “hydrogen atom” for cell biology) as well as a potential platform from which to base future efforts in metabolic engineering of useful industrial or biomedical compounds.6
While experimental efforts to characterize JCVI-syn3A genes of unknown function have been undertaken,7 these remain time-consuming and costly, relative to guidance that might be obtained by computational means. Many bioinformatics approaches exist for characterization of genes of unknown function in relatively understudied organisms such as JCVI-syn3A or the mycoplasmas as a whole. Several previous efforts have sought to further clarify the contents of the JCVI-syn3A genome and that of its precursor JCVI-syn3.0. First, Danchin and Fang8 studied the genome of JCVI-syn3.0 via an engineering perspective, by streamlining their investigative approach to the paleome content, that is the functional units common to all bacteria of this type, thereby suggesting the necessary generic cellular functions of JCVI-syn3.0. In this way they filled in previously unconsidered gaps connecting genes to macro-scale cellular functions and assigned probable functions to 13 of the unknown genes that could be interrogated further. In a second effort, Yang and Tsui9 arrived at functional annotations based on secondary structure element alignments with the program SSEalign. This program connects prediction of structural motifs to a database of sequence motifs, leveraging the fact that amino acid sequences have evolved in a much more expansive fashion relative to protein secondary structures and individual protein folds. The algorithm can also take into account the protein–protein interaction network of JCVI-syn1.010 and generates support for an assignment if the identified E. coli homologue of the JCVI-syn1.0 protein from the SSE procedure shares a similar interaction network.
Uptake of nutrients and metabolic building blocks from the growth media or external environment has been demonstrated to be crucial for JCVI-syn3A by both experiments and theory.3,11 With this in mind, Antczak et al.12 used several approaches, including domain identification,13 prediction of transmembrane helices,14 structural modeling (Phyre2),15 ligand prediction,16 among others,17 that identified 24 genes likely coding for transporters. Along the same way of thinking, we examined cellular response to growth medium changes in JCVI-syn3A via the construction of whole-cell models that can respond to environmental changes.11,18
Most recently, Zhang et al. employed a deep-learning, contact-assisted structure prediction method, followed by structure-based annotation to suggest biological roles and protein–protein interactions for several JCVI-syn3A proteins19 by the C-I-TASSER-COFACTOR method. This method initially uses the I-TASSER protein structure prediction program20 combined with deep-learning based protein residue–residue contact map predictions. Then, protein–ligand binding sites were predicted using COFACTOR21 to help elucidate possible gene product functions.
The previous bioinformatic processes, while often high-performance and high-throughput in their ability to be applied across the entire JCVI-syn3A genome, fail to account for certain factors in casting a wider net. For example, pipelines that depend heavily on mapping to existing structural motifs can suffer when no experimental structures for analogous bacterial proteins are available from the relevant databases. Using the recent and highly successful deep-learning based protein structure prediction method AlphaFold2,22 these missing structures can be generated. While structure can offer hints as to the function of a protein, additional computational and experimental studies are required. Energy landscape theory23 has shown the importance of nearby conformations in considering both the folding pathways as well as a function of a protein. For this reason, we provide predicted structures relaxed via AMBER24 and use a flexible structural alignment approach when comparing predicted structures to experimental structures.25 In taking a holistic and individualized view that accounts for the genome locality (i.e., which genes are neighboring to an unknown gene), interactomics, and where a gene product may fit in as a missing component of a larger cellular network and combining this with recent protein structure prediction tools,22 we are able to provide increased resolution and predictive power for specific and essential JCVI-syn3A genes of unknown function.
The processes of cell growth and division in JCVI-syn3A are of keen interest following recent work, both computational11 and experimental.26 First, Pelletier and co-workers26 demonstrated that, of the 19 genes retained in JCVI-syn3A but not JCVI-syn3.0, 7 (including 5 genes of unclear function) were required together to reduce morphological variation in JCVI-syn3A. Two of the genes, ftsZ/0522 and sepF/0521, contribute to cell division in most bacteria, while five do not have a known biomolecular function. Then, Thornburg et al.11 constructed a whole-cell kinetic model of JCVI-syn3A, describing growth emerging from metabolism and gene expression (including the contribution of integral membrane proteins and lipids etc.). Currently, the model has few instances of direct regulation of growth by specific genes, such as phoU/0428. Regulation of growth could further be influenced by the products of genes of unknown function. With this in mind, we sought to further clarify gene products involved in regulating JCVI-syn3A growth and division, to inform future experimental and modeling efforts. For these processes to be characterized fully, it is important to examine directly the individual molecular actors that contribute to the required increase of biomass and physical force necessary to produce daughter cells.
In addition to those genes implicated in cell growth and division, we examined all of the 30 genes of unclear function that were classified as essential for JCVI-syn3A growth by transposon mutagenesis and computational analysis.3 We demonstrate that several of these genes code for products involved in membrane maintenance, assembly of protein complexes, and regulatory and other crucial developmental processes of JCVI-syn3A cells. Of the approximately 90 protein-coding genes of unclear function reported by Breuer et al.,3 we have upgraded the annotations for 25 genes with high confidence, while providing additional information for tens of the remaining protein-coding JCVI-syn3A genes. Our computational analysis was applied to the sequences of all the 452 proteins giving us complete set of structural models for JCVI-syn3A. We then analyzed our results to generate a selection of gene product annotations of interest provided in Supporting Information section S1, in addition to a larger annotation summary given in Supporting Information Spreadsheet S1 and Supporting Information section S3, and structural database (with an associated analysis notebook) found in Supporting Information section S2.
2. Materials and Methods
Methods
Critical to the computational workflow that we apply is the development and usage of the AlphaFold2 protein structure prediction software.22 This novel, deep-learning based method utilizes existing sequence analysis tools (such as HHsuite327 and HHSearch27) in conjunction with a massive genetic database and structural training data that propelled it to win the CASP14 structure prediction contest as well as being named the Nature “Method of the Year”.28 Here we review the techniques and concepts that we utilized in concert with AlphaFold2 to develop functional characterizations and structure predictions of JCVI-syn3A protein-coding genes.
Sequence Alignment of Conserved Motifs
Sequence Alignment of key sequence motifs can also be a powerful tool as we will demonstrate in the case of secDF/0412. Multiple sequence alignment tools such as CLUSTALW29 and the HMMER Server from the Max Planck Institute (MPI) Bioinformatics Toolkit,30 which allows for sensitive sequence based searching using profile hidden Markov models (HMMs), provide an avenue for such investigations. Another useful tool, the HHPred server from the MPI Bioinformatics toolkit allows for homology detection by multiple sequence alignment followed by HMM-HMM comparison.31,32 Such a protein sequence analysis was generated during prediction runs using the HH-suite327 capabilities present within AlphaFold2, the results of which are provided in the Supplemental Database given in Supporting Information section S2. Finally, amino acid sequence to KEGG functional orthology analysis by BlastKOALA33 and previous bioinformatic analysis of genes in the related Mycoplasma organism Mesoplasma florum L1(34) allowed for a narrowing based on their findings of the general functional categories in which genes of unknown function may lie. The continuing expansion of available sequence data alongside novel coevolutionary analysis methods that examine individual sequence-structural components35 will allow further leveraging of sequence motif conservation for gene functional identification.
Protein Structure Prediction
To improve upon previous work, we used a protein structure prediction based pipeline via the software package AlphaFold222 to further analyze annotation updates for JCVI-syn3A genes leveraging structure-to-function relationships (with comparison to the related method RoseTTAFold36 for gpsB/0353). This software suite, which has revolutionized protein structure prediction via deep learning-based methodologies following their performances in vastly outpacing all other competitors at the Critical Assessment of Structure Prediction (CASP) contest CASP14, allows for a prediction of structure from amino acid sequence with remarkable accuracy when comparing to experimentally solved structures from X-ray crystallography, nuclear magnetic resonance (NMR), or cryo-electron microscopy (cryo-EM).
All AlphaFold2 predictions were generated using the default parameters that are most similar to those used in the CASP14 event. This includes number of recycles = 3, number of models = 5, and the “full” 3+ terabyte structural and sequence genetic databases, and with PDB entries deposited on or before 9-15-2021. We provide both “unrelaxed” predictions and structures relaxed via the molecular dynamics platform AMBER,24 as well as the associated lDDT confidence scores22 for each model. Simulations were run on a local heterogeneous CPU-GPU compute cluster containing NVIDIA A40 and NVIDIA Tesla V100 GPUs and Intel Xeon Gold 6154 CPUs, with 1 GPU and 8 CPUs being used for each protein structure predicted.
Protein Sequence-to-Structure Threading
Protein structure prediction methods are useful either containing or in conjunction with other bioinformatic tools such as Phyre2,15 HMMER,30 and LOMETS,37,38 which provide a threading capability that connects amino acid sequence to specific protein fold motifs that are associated with these programs. The predicted number of known amino acid sequences is several orders of magnitude greater than the approximately 1000 predicted similar protein fold families (such as those deposited in Pfam39) that have been observed in experimental structures deposited in the RCSB PDB (Protein Data Bank).40 This sequence-to-structure threading investigation routine is useful in guiding candidates for pairwise structural alignment to extract significant structural similarity in conjunction with AlphaFold2 outputs. Such structural analysis was also generated during prediction runs using the HHSearch27 capabilities present within AlphaFold2, the results of which are provided in the supplemental database given in Supporting Information section S2.
Protein Pairwise Structural Alignment
Various tools then exist that can be used to compare proteins via structural alignment methods such as STAMP,41 and aligned structures can then be visualized in VMD.42 When significant agreement between the top ranking predicted structures from AlphaFold2 to previously solved crystal structures from related bacterial organisms deposited in RCSB PDB40 is obtained by this methodology, such a result motivates an increased gene functional annotation and the development of experiments to more specifically determine the function the gene in question. The quality of alignment can be interrogated and visually represented using Qres scoring and coloring, which calculates the fraction of similar native contacts between aligned residues in two or more protein structures via an energy landscape theory approach described by Eastwood et al.,43 derived from the original theory.44,45 An alternative structural alignment tool that is preferable for quantitative pairwise structural alignment is FATCAT.25 FATCAT provides a flexible protein structural alignment algorithm that uses rotations and translations of one protein structure to minimize the root-mean square deviation (RMSD) between the two structures being aligned, thereby accounting for structural rearrangements due to crystallization conditions or the presence of various biological isoforms within a living cell. The FATCAT algorithm uses a scheme that creates various linked fragments of a protein in its optimization routine, which are chained and twisted to arrive at a final structural alignment with a p-value describing significance of structural similarity. The equations involved in this process are given in further detail in the Formulas section and in eqs 1, 3, and 4. The FATCAT alignment process was attempted for genes given in the main text and in Tables S1 and S2).
Interactomics and Genomic Locality
Interactomics data, such as that presented in the SynWiki resource compilation of JCVI-syn3A-related data10 and deposited in the STRING database,46 is helpful in determining the function of a gene of unknown function when genes from a related cellular process or with known physical interactions are observed in its interactomic network. Important to note is that this resource takes the STRING data for a highly phylogentically related organism, Mycoplasma mycoides subsp. mycoides SC PG1, to populate data for JCVI-syn3A genes. In conjunction with this, biological insight into genome locality (i.e., operonal or transcription unit structure) of related bacterial organisms can assist in clarifying the function of unknown JCVI-syn3A genes since genome architecture/gene locality is often significantly conserved in related organisms.47,48 This is especially useful for proteins where experimentally determined structures are absent or sparse and sequence is not necessarily well conserved (e.g., atpI/0797, which is part of a similar operon structure in Bacillus subtilis(49)) that can be visualized in SubtiWiki.50
Formulas
Structural alignment via the FATCAT pairwise alignment tool25 is a key metric by which we were able to assess confidence in functional assignments of JCVI-syn3A genes of unknown function based on the alignments of their predicted structures from AlphaFold222 and RoseTTAFold36 to experimentally determined structures. For clarity, the process of FATCAT alignment scoring metric calculation is summarized below. FATCAT uses a flexible alignment methodology that takes advantage of aligned fragment pairs (AFPs), which define transformations of local structural elements of a protein. This representation allows for alignment that is not detracted by potential artifacts that may be due to crystallization conditions or regions of high error for a structure generated by protein structure prediction. These constructs are then translated to the mathematical graph formulation used for the structural scoring method detailed in eq 1. For a more detailed explanation, we direct the reader to the work by Li and co-workers.25
FATCAT alignments are initially characterized with the chaining score given by
| 1 |
where S(k) is the best score along the AFP k, a(k) is the score of AFP k itself, c(m → k) is the score of introducing a connection between AFP m and AFP k, and T(k) is the number of twists required to connect the chain of AFPs leading up to S(k).
Such that
| 2 |
And with similarity score:
| 3 |
where cs is the previously mentioned chaining score from eq 1, L is the number of equivalent positions in the alignment, RMSD is the overall RMSD between the two structures when one structure is rearranged at the positions where twists are detected by FATCAT, and N is the number of blocks in the alignment (numberoftwists + 1).
Generating p-value:
| 4 |
where the location and the scale parameter of the extreme value distribution of FATCAT similarity scores of unrelated structures were determined by empirical simulation described by Li et al.25
3. Results
From Mother to Daughter: Characterization of Molecular Actors of Cell Growth, Division, and Morphology
Of keen interest are the additional biomolecular actors regulating cell division in JCVI-syn3A. Recently, it was shown that a set of seven genes that were deleted in the genome reduction from JCVI-syn1.0 to JCVI-syn3.0 and restored in the construction of JCVI-syn3A are necessary to maintain proper cell morphology.26 Of this group, one gene, JCVISYN3A_0520 (that from now on will be referred to as /0520, and likewise for the other JCVI-syn3A genes discussed) is adjacent to genes of the highly conserved division and cell wall (dcw) cluster51−53 and has been previously characterized for Mycoplasma genitalium.54 Another of these genes (/0527) immediately precedes the dcw cluster on the reverse strand of the circular genome. From previous bioinformatic investigations,12,19 /0520 is characterized as a putative member of the α–β hydrolase superfamily while /0527 is annotated as a protein of unknown function containing a domain of the DUF177 family whose members have been posited to participate in membrane protein biosynthesis among other roles.26,55 Other dcw cluster region genes include sepF/0521, ftsZ/0522, and ftsA/0523 that are known to participate in cell division,56mraW/0524 which codes for a regulatory protein that can methylate the 16S rRNA, and mraZ/0525 that acts as a transcriptional repressor of the dcw cluster53,54 but curiously was not detected in JCVI-syn3A proteomics.3 In the following paragraphs, we suggest annotations for genes coding for common products of the Gram-positive divisome and provide possible explanations for the causation of irregular morphologies by deletion of the aforementioned dcw cluster adjacent genes.
gpsB/0353
Gene/0353 was previously annotated as a quasi-essential gene of unknown function. Here, we demonstrate that this gene codes for a gpsB-like protein that interacts with ftsZ/0522 to ensure proper cell division behavior. In many bacterial species, GpsB and its homologue DivIVA contribute to cell division, cell growth, and chromosome segregation.57 Predicted structures for gene gpsB/0353 from AlphaFold222 (and the related protein structure prediction software RoseTTAFold36) align nearly exactly with the crystal structure of the GpsB protein from B. subtilis (RCSB PDB: 4UG3(58)) when aligned only at the N-terminal coiled-coil domain using STAMP41 and visualizing in VMD42 as is shown in Figure 1A. A long 65 amino acid, completely α-helical C-terminal domain of /0353, extending beyond the length of the crystal structure chosen for structural alignment, is not shown. The C-terminal domain of GpsB has yet to be experimentally determined in conjunction with the N-terminal domain to our knowledge. Nonetheless, even without the C-terminal domain of the experimental structure present, the predicted protein structure from AlphaFold2 and the experimental structure from B. subtilis are still judged as significantly similar by FATCAT alignment25 with a p-value of 2.72 × 10–6 and all 60 residues of the crystal structure being evaluated as matching the predicted structure with a RMSD of only 1.10 Å and 1 twist (via the flexible alignment procedure described in the Formulas section). The gene locality of gpsB/0353 near that of recU/0351 within the JCVI-syn1.0 genome has been demonstrated previously in B. subtilis(59) and is thought to be significant due to the fact that recU is required for the segregation of chromosomes into daughter cells60 in conjunction with cell division that is mediated by GpsB. Further support exists to distinguish gene gpsB/0353 from its orthologue divIVA, that is also involved in the Gram-positive cell division landscape and has been included as a potential functional assignment via previous bioinformatic work.12 GpsB proteins are observed to have approximately 120 amino acids or less across a variety of organisms (gpsB/0353 has 125 residues) and contain the conserved “TNFDILK” consensus sequence in the C-terminal region (the portion not shown in structural modeling but analyzed here that can be seen aligned to sequences from B. subtilis and Staphylococcus aureus in Figure 1C), while cell division DivIVA homologs are always greater than 160 amino acids in length.59
Figure 1.

(A) Predicted structures of gpsB/0353 from AlphaFold2 (red) and RoseTTAFold (blue) aligned with the experimental structure of gpsB from B. subtilis (RCSB PDB: 4UG3)58 at the N-terminal coiled-coil domain using STAMP.41 (B) Predicted structure of lemA/0604 (see section 3) from RoseTTAFold (blue) aligned by FATCAT25 with the lemA two-component cell growth regulator from T. maritima (yellow, RCSD PDB: 2ETD(61)). 142 of the 210 residues (68%) are well-aligned with a RMSD of only 1.67 Å. (C) Multiple sequence alignment of /0353 from JCVI-syn3A with sequences of gpsB from B. subtilis (strain 168) and S. aureus (strain NCTC 8325/PS 47) shows the conserved C-terminal domain consensus sequence,59 lending further evidence to the assignment of gpsB/0353 instead of its paralogue divIVA. All images visualized in VMD.42
ezrA/0239
When considering candidate genes for those unaccounted for in the Gram-positive bacterial divisome, which are excellently reviewed in a study regarding ftsZ,56 we considered the fact that some of these genes may interact with the well-known proteins ftsA/0523 and ftsZ/0522. Of this pool of candidates, one showed similarities to /0239 in terms of both sequence and structural motifs, namely the ftsZ protofilament bundle regulator ezrA. This protein has been demonstrated to prevent aberrant FtsZ Z-ring formation in low GC Gram-positive bacteria that are closely related to JCVI-syn3A.62 First, ezrA is well-known to interact with ftsZ via the sequence motif known as the “QNR” patch,63,64 that is conserved across several bacterial organisms including B. subtilis, Staphylococcus aureus, Streptococcus pnuemonia, Lactobacillus acidophilus, and Enterococcus faecilis. When the amino acid sequence corresponding to /0239 is aligned against the sequences of ezrA in B. subtilis and S. aureus via CLUSTALW,29 the “QNR” region from residue 504–510 is relatively conserved at the same residues as it is for the other bacteria. Most importantly, the central asparagine residue that is conserved across nearly all bacterial species (see Figure 2C) is present. When a structural alignment of the closely phylogenetically related Gram-positive bacterium B. subtilis ezrA to the predicted protein structure of /0239 from JCVI-syn3A is generated by AlphaFold2, a high degree of similarity is observed (Figure 2A). By FATCAT alignment, the structures are characterized as significantly similar to a p-value of approximately 3.01 × 10–10 (with values < 5.00 × 10–2 indicating significant structural similarity) and with 498 equivalent positions with a RMSD of 4.64 Å with 4 twists (see Figure 2A). Gene /0239 contains 595 residues, meaning that approximately 84% of these residues are well-aligned to the EzrA protein from B. subtilis. Furthermore, the predicted structure for /0239 is heavily covered with hydrophobic residues at both the N and C terminus regions and the same sites that are predicted to bind to the membrane and also have high hydrophobicity in the experimental structure from B. subtilis (see Figure 2B). Finally, further support is provided by the genome architecture of ezrA in the closely related Gram-positive bacterial organism B. subtilis where the ezrA gene is located just upstream of thiL on the reverse strand (separated by only a few genes) and immediately precedes rpsD on the forward strand. This exact genome arrangement of ezrA between thiL and rpsB is seen for ezrA/0239 in JCVI-syn3A (see Figure 2D).
Figure 2.

(A) Structural alignment by FATCAT25 of the predicted AlphaFold2 structure for gene /0239 (blue) to the structure of the cell division regulatory protein EzrA from B. subtilis (yellow, RCSB PDB: 4UXV(65)) with 498 or 84% of the residues of /0239 being well-aligned to the experimental structure with a p-value of 3.01 × 10–10 for significant structural similarity. (B) Hydrophobicity coloring (red, hydrophobic resiudes; blue, hydrophilic residues) for the AlphaFold2 predicted structure for /0239. Hydrophobic regions at the N and C termini match the previous findings that EzrA binds the membrane at each terminal domain.65 (C) Sequence Alignment of gene /0239 with the sequences for ezrA from S. aureus and B. subtilis with BLOSUM50 similarity score coloring. While the entire characteristic “QNR” patch63,64 is not present in JCVI-syn3A, significant sequence similarity remains and the nearly universally conserved asparagine residue located centrally in the patch is present. (D) The genome architecture of JCVI-syn3A also lends support to the assignment of ezrA/0239. Just as in the related Gram-positive organism B. subtilis, the gene coding for EzrA lies between the genes for thiL coding for thiamine monophosphate kinase and rpsD coding for ribosomal protein S2 on opposing strands of the circular genome.
Briefly, EzrA acts a regulator of the bundling and formation of the FtsZ protofilament. In B. subtilis, EzrA increases the rate at which FtsZ hydrolyzes GTP and decreases the binding affinity of FtsZ for GTP.64 In EzrA deletion strains, an aberrant long cell phenotype, likely due to improper cell division caused by the absence of the aforementioned regulation, is observed.64
yqkD/0520
If we consider gene /0520 in particular, while it is expressed well below the value of approximately 200 protein copies observed to be expressed on average for JCVI-syn3A genes,3 it still retains a proteomic value on par with the dcw gene adjacent to it, sepF/0521, and the nearby ftsA/0523 having 50 and 51 copies respectively, with /0520 having 30 copies measured in a proteomic analysis of JCVI-syn3A.3 Previously, Pelletier et al. and others19,26 reported this gene as being a member of the α–β hydrolase family. Interestingly, this superfamily, containing proteins whose functions are wide-ranging and are reviewed in ref (66), has members with functions related to cell growth, such as lipases and serine aminopeptidases that may significantly modify lipids and proteins that make up the JCVI-syn3A membrane and bacterial membranes in general. A review of the similarity in sequence and structural motifs between α–β fold lipases and peptidases in this type has been conducted,67 that is especially of interest in terms of the prolyl-oligopeptidase family proteins. Of the aforementioned candidates, the predicted structure of gene /0520 aligns especially well to the AlphaFold2 predicted structure of the esterase/α–β hydrolase yqkD from B. subtilis(68) with over 96% of residues well-aligned, a RMSD of 3.00 Å, and a p-value for similarity greater than 1.00 × 10–15 (see Figure 3A and Table 2). Providing further support, structural alignment of the AlphaFold2 predicted protein structure for /0520 to a Pfam PF2001 α–β hydrolase from Pyrococcus furiosis (RCSB PDB: 5G59(69)), that also has esterase-like properties, shows that the hydrolase helical bundle is highly structurally conserved, which can been seen by Qres coloring of the STAMP structural alignment in VMD (see Figure 3B, C). This catalytic site conservation is additionally corroborated by previous bioinformatic work by Zhang et al.19 via the COFACTOR analysis pipeline where peptidase ester or lipase cosubstrate binding sites are predicted at residues 96 and 166–168 especially. Amino acid sequence analysis27 also detects the previously observed lipase sequence motifs HG and GXSXG (where X is a wildcard) from Mycoplasma mycoides subsp. mycoides LC.70 Experimentally determined interactomics data reported in SynWiki10 shows interactions of /0520 orthologues with the membrane protease lon/0394, that degrades FtsZ among other related substrates in Mycoplasma pneumoniae,71 so /0520 and/or Lon could be involved in degrading controllers of cell division. If /0520 does indeed code for a yqkD-like serine peptidase and is of the chymotrypsin like substrate-specificity family, it would then degrade membrane proteins (thus explaining the presence of Lon in interactomic data) in which medium sized hydrophobic residues such as Tyr, Phe, and Trp are exposed.72 Interestingly, this gene has also been observed to be positively correlated with the expression of other lipases and esterases in B. subtilis.73 We have previously demonstrated that the JCVI-syn3A membrane is quite protein-rich11 with over 10% of the proteome and approximately 50% of cell membrane surface area composed of membrane proteins. Thus, the absence of such a yqkD-like aminopeptidase/protease could substantially modify the membrane composition of JCVI-syn3A cells that give it regular daughter cell morphologies. Additionally, if /0520 was to have lipase activity, one could also anticipate that its deletion could have substantial effects on daughter cell morphologies, due to the elimination of membrane remodeling abilities that may allow for scavenging or transfer of acyl chains between lipids that may have played a key role in the pathogenic behaviors of the JCVI-syn3A Mycoplasma parent organism as it does in related organisms.74
Figure 3.

(A) Structural alignment of the AlphaFold2 predicted structure of the YqkD esterase/lipase/serine aminopeptidase from B. subtilis,68 (yellow) to the predicted protein structure of /0520 from AlphaFold2 (blue) using FATCAT 2.0.25 The alignment characterizes the structures as being significantly similar to more than 95% of the residues of /0520 being well-aligned to the B. subtilis protein predicted structure. (B) Structural alignment via STAMP of P. furiosis PF2001 hydrolase (yellow, RCSB PDB: 5G59(69)) to the predicted structures for /0520 from both AlphaFold2 (red) and RoseTTAFold (blue). (C) The alignment with Qres coloring43 shows that the hydrolase helical bundle is highly conserved (blue-white-red color scheme of decreasing conservation), supporting the assignment of /0520 as an aminopeptidase/esterase/lipase, of the α–β hydrolase superfamily (see ref (66) and Pfam), and suggesting that PF2001 might play a similar role beyond its confirmed general esterase activity.
Table 2. FATCAT25 Structural Alignment Statistics for Proteins of Formerly Unknown Function in JCVI-syn3A Cell Growth, Maintenance, and Divisiona.
| gene | exp. struct. PDB ID | p-value | RMSD, Å (% aligned) |
|---|---|---|---|
| ezrA/0239 | 4UXV65 | 3.01 × 10–10 | 4.64 (84%) |
| gpsB/0353 | 4UG358 | 2.72 × 10–6 | 1.10 (100%) |
| secD/0412 | 5XAM102 | 9.20 × 10–4 | 6.10 (41%) |
| secF/0412 | 5MG3103 | 6.49 × 10–5 | 3.05 (53%) |
| yloU/0421 | B. subtilis (AF2)104 | 1.11 × 10–11 | 2.29 (99%) |
| ylxM/0430 | 1S7O82 | 3.21 × 10–10 | 1.99 (99%) |
| yqgP-gplG/0516 | B. subtilis (AF2)87 and 2NR989 | 7.19 × 10–9 | 3.07 (82%) |
| yqkD/0520 | B. subtilis (AF2)68 | 0.00 × 10–0 | 3.00 (96%) |
| ylbN/0527 | B. subtilis (AF2)76 | 3.03 × 10–7 | 2.45 (84%) |
| lemA-gacS/0604 | 2ETD61 | 6.78 × 10–5 | 1.67 (68%) |
| atpI/0797 | B. subtilis (AF2)98 and 6FKI101 | 1.81 × 10–11 | 1.22 (92%) |
| gabP/0878 | B. subtilis (AF2105) | 4.47 × 10–11 | 3.23 (87%) |
| yidC/0908 | M. pneumoniae (AF2106) | 0.00 × 10–0 | 2.33 (92%) |
Alignment values generated from experimentally solved crystal structures or with AlphaFold2 (AF2) structures where this abbreviation is indicated, aligned with predicted AlphaFold2 structures for JCVI-syn3A genes. A p-value of less than 5.0 × 10–2 demonstrates significant structural similarity. A value of 0.00 indicates a p-value of less than 1.0 × 10–20. The percent aligned relates to the percentage of residues in the protein of shorter length that are well-aligned to the larger structure by FATCAT, with the associated RMSD. Equations generating these statistical values are given in the Formulas section.
Drivers of Cell Growth: Ribosomal Assembly and Protein Translocation Machinery
Previously, we have developed with our dynamic whole-cell model of JCVI-syn3A a cellular growth module that predicts doubling times across a population cells. The growth of the JCVI-syn3A cells is dependent on lipid uptake and synthesis, and membrane protein translocation, since both of these components contribute surface area to the membrane as it doubles.11 For this reason, clarifying those genes coding for proteins involved the processes of translocation and degradation of membrane proteins and remodeling of the membrane is of paramount importance in improving our modeling efforts and deepening our understanding of JCVI-syn3A cell growth.
ylbN/0527
With gene yqkD/0520 identified, this leaves only one gene unannotated from the unannotated genes surrounding the JCVI-syn3A dcw cluster. This gene, /0527, had previously been bioinformatically characterized to contain a DUF177 domain.26 Not far from the dcw operon in the genome of B. subtilis is the gene ylbN that is involved in the regulation and assembly of the 23S rRNA Large Subunit (LSU) component. Like gene /0527, YlbN contains a reported DUF177 domain, from residues 56–165 in B. subtilis, with a conserved 158Arg residue75 that is also present in JCVI-syn3A when the sequence of /0527 is aligned to the B. subtilis sequence (see Figure 4C). The locality of gene /0527 adjacent to LSU component rpmF/0526 coding for ribosomal protein L32 (and that is also adjacent to ylbN in B. subtilis; see Figure 4A) lends additional support to this hypothesis. Structural alignment of the Alphafold2 prediction for /0527 to the predicted structure from AlphaFold2 for ylbN from B. subtilis,76 that is the best available structure since none have been experimentally solved, results in an excellent agreement demonstrated in Figure 4B. Also, the interactome of /0527 reported in SynWiki10 includes several LSU ribosomal proteins (L1, L13, L21, L32) including rplA/0809 (protein L1) that binds directly to the 23S rRNA.
Figure 4.

(A) Genome locality of ylbN and ylbN/0527 in B. subtilis and JCVI-syn3A, respectively. The ylbN genes are colored in red, while adjacent genes located prior to the dcw cluster in both organisms include rpmF, mraZ, and rsmH (also known as mraW) (blue), with additional genes only present in B. subtilis shown (green). Both gene clusters show similar orientation and arrangement on the reverse strand. (B) Structural alignment of AlphaFold2 prediction of ylbN/0527 (blue) and the predicted AlphaFold2 structure for ylbN in B. subtilis 168 (yellow76). (C) CLUSTALW sequence alignment of ylbN/0527 and ylbN from B. subtilis with BLOSUM50 similarity coloring. The conserved 158R reported in ref (75) is aligned in the “DPR” region of both sequences.
Due to its reduced genome and pathogenic origins, JCVI-syn3A has been considered to be a cell that “lives on the edge” in terms of maintaining its energetic requirements and cellular homeostasis.11 Further investigation into these biological roles may shed some light onto why ylbN/0527 is a pivotal component in regulating JCVI-syn3A daughter cell morphology and proper development behavior.26 A study in which a DUF177 domain ylbN-orthologue knockout strain was generated in Zea mays led to the observation of a significant decrease in 23S rRNA production.55 Previously, in B. subtilis,77ylbN has been observed to be downregulated by the stringent response (for example, by the alarmone metabolite ppGpp that is synthesized by a gene present in JCVI-syn3A relA/041478), whereby a cell reduces its growth activity to conserve energy often by decreasing the generation of ribosomes.79 These two studies implicate YlbN in 23S rRNA accumulation and in ribosome biogenesis. However, at this point, this remains a speculative suggestion as to the possible linkage between gene ylbN/0527 removal and the generation of JCVI-syn3A daughter cells with irregular morphologies and further experimental study is needed (see section 4).
secDF/0412
Related to cell surface growth and more specifically to protein translocation across the membrane, we have further support to annotate /0412 as a putative secDF protein export enhancing membrane component. SecDF has been demonstrated to interact with the SecYEG translocon, which comprises secY/0652, secE/0839, and secG/0774, and enhance its translocation efficacy by proton-motive force that is transduced to SecYEG.80 We have determined that SecF likely comprises the amino acids from 948 to 1384 while SecD comprises the residues from 1 to 947 given in the JCVI-syn3A NCBI entry (NCBI GenBank: CP002027.1). Each of these two regions contains conserved sequence motifs that have been observed for both SecD and SecF across a variety of organisms,81 that previously supported the putative assignment of the analogous gene in M. florum L1 as secDF-like.34 Structural alignments of the AlphaFold2 predicted protein structures for both SecD and SecF (blue) were aligned with FATCAT25 to the analogous structures from Deinococcus radiodurans and E. coli, respectively. The secF portion has 52% of its residues well-aligned to the experimental structure with a RMSD of 3.05 Å, while the SecD portion has 43% alignment and a p-value of 9.20 × 10–4 denoting structural significance (see Table 2). The interactome of this gene including other translocation related genes such as secY/0652, secA/0095, and the translocation regulator ftsY/0429 as well as the gene we will discuss next also support secDF assignment. There is additional support present for this functional assignment in conserved genome architecture of genes: relA/0414 and apt/0413 are adjacent to secDF/0412, that is also seen in B. subtilis (see Figure 5A).
Figure 5.
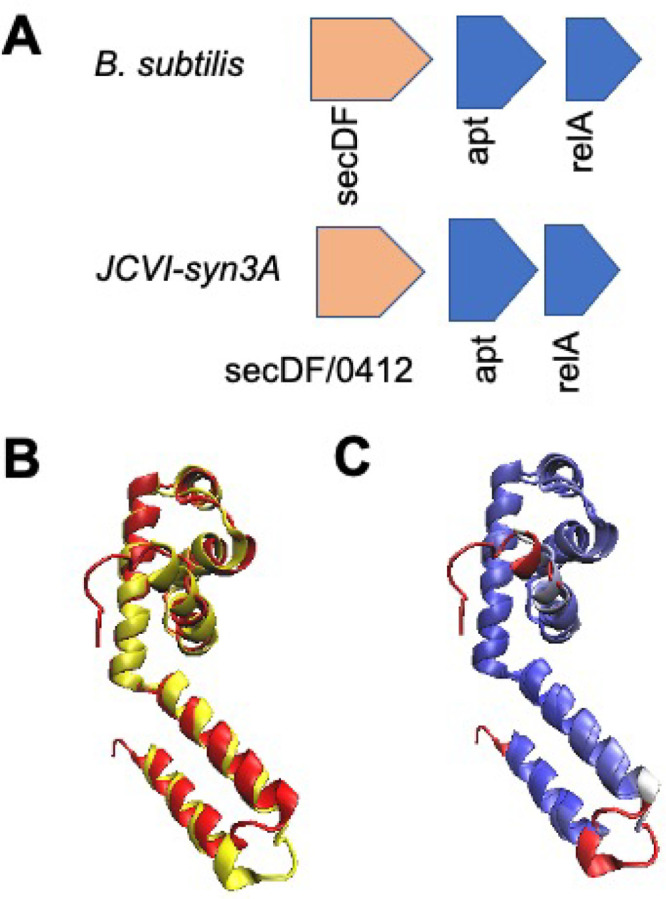
(A) Genome architecture of the related Gram-positive bacterium B. subtilis near the gene coding for SecDF translocon components and that adjacent to gene/0412 show conservation. (B) STAMP structural alignment with visualization in VMD of the AlphaFold2 predicted structure of gene 0430/ylxM (red) and the experimentally determined structure for YlxM-like effector of the signal recognition particle from S. aureus (yellow, RCSD PDB: 1S7O(82)). (C) Qres coloring43 (with a blue-white-red color palette of decreasing structural similarity) shows a high degree of structural conservation between the predicted and experimental structures.
ylxM/0430
Additionally, a previously unknown essential JCVI-syn3A gene that likely has a critical role in cell growth, gene ylxM/0430, has been identified as a putative effector of the signal recognition particle (SRP) ribonucleoprotein (whose RNA component is coded for by gene ffs/0049). When the predicted structure from AlphaFold222 is aligned to the crystal structure of the YlxM-like effector of the SRP from Streptococcus pygenes (RCSB PDB: 1S7O(82)), significant structural conservation is observed (see Figure 5B and C, regions where structural motifs are shaded blue via the Qres43 coloring scheme). The effector of SRP, YlxM, is thought to modulate GTPase activity and thereby influence recycling of SRP components in the membrane protein translocation process that has been observed in Streptococcus mutans.83 Improved kinetic modeling attained by including this protein in the JCVI-syn3A cell growth module implemented in Thornburg et al.11 may have substantial effects via interactions with the 4.5S RNA signal recognition particle ffs/0049. The SRP system consists of the secYEG translocon consisting of the genes secY/0652, secE/0839, and secG/0774 that is responsible for receiving newly translated membrane proteins and translocating them through the lipid bilayer via the SecY polypetide conducting channel, while also modulating the activity of the SRP receptor GTPase ftsY/0429 that negatively regulates the translocation of membrane proteins,84 as well as yidC/0908 discussed in the following paragraphs, all of which are observed in its JCVI-syn3A interactome.10
yidC/0908
Besides YlxM and the SecYEG,DF proteins, other biomolecules play a key role in membrane protein translocation. One such protein seems to be coded for by gene /0908 that was previously annotated as a probable translocase component.2,3 Here, we further characterize this gene based on structural comparison, genome locality and interactomics. In the JCVI-syn3A genome, gene /0908 lies adjacent to rnpA/0907, the tRNA processing RNase P. In both of the related organisms B. subtilis and M. pneumoniae, RNase P exists immediately prior on the reverse strand to the gene coding for the translocase associated protein YidC. When an AlphaFold2 prediction is generated for /0907, an excellent agreement in terms of structural similarity is found to the analogous AlphaFold2 prediction for YidC from M. pneumoniae (see Table 2). Lending further support is the observed interactomic data for this gene with SecA, SecY, FtsY, and ffh, that are all members of the translocon and signal recognition particle machinery.10 YidC specifically acts to facilitate the translocation of membrane proteins across the membrane via the SRP-mediated network, by acting in conjunction with SecYEG to reduce the hydrophobicity experienced by newly translated membrane proteins as they are inserted,85,86 making it a valuable component of the JCVI-syn3A translocation system to model moving forward.
Stewards of Cellular Maintenance: Protease, Assembly, Uptake and Regulatory Components
In a reduced genome such as that of JCVI-syn3A, regulatory components that are typically present in other bacterial organisms may not be present. For this reason, cellular maintenance molecules play a key role in preserving a stable biophysical environment as we have discussed in our previous whole-cell modeling work.11 Here, we further characterize the players involved in these cellular housekeeping processes.
yqgP-gplG/0516
Gene /0516, located not far downstream on the reverse strand from yqkD/0520 and ylbN/0527, is also of interest as a quasi-essential gene for JCVI-syn3A viability with multiple predicted transmembrane helices (Figure 6).3 When the gene is analyzed via HHPred31 and a predicted structure is generated via AlphaFold2, it aligns very well with the structure for Haemophilus influenzae gplG membrane protease, especially at the α-helical portion of the protein. When the /0516 structure is aligned to the predicted AlphaFold2 structure of the paralogous yqgP membrane protease87 from the more closely phylogenetically related organism B. subtilis, an excellent FATCAT alignment is achieved across the entire structure with high coverage and statistically significant structural similarity (see Table 2). yqgP-glpG is a membrane protease that partners with the membrane protease ftsH/0039 and is responsible for degradation of metal transporters such as mgtA/0787 to prevent ion toxicity.88,89 This assignment is supported by the observation of ftsH/0039 in the interactomics for JCVI-syn3A10 as well as the observation of the JCVI-syn1.0 ion transporter mgtE/0157 (deleted in genome reduction from JCVI-syn1.0 to JCVI-syn3A).
Figure 6.

(A) FATCAT25 structural alignment of AlphaFold2 predicted structure of /0516 (blue) and gplG protease from H. influenzae (RCSB PDB: 2NR9(89)), where only the lower helical portion of the protease is available in the experimental structure. (B) FATCAT structural alignment of the predicted structure from (A) with the Alphafold2 predicted structure for the gplG paralogue protease yqgP from B. subtilis shows excellent agreement to both the upper β- sheet and the lower -a-helical regions of the JCVI-syn3A structure, with a p-value of 7.19 × 10–9 showing significant structural similarity.
gabP/0878
Gene /0878 was previously described as encoding a quasi-essential transmembrane protein of unknown function.2 More recent work has determined that this protein could have likely amino acid binding and transport functionality.3,19 Here, we strengthen the certainty of this functional assignment by characterizing the gene as coding for a GabP-like amino acid permease. When the AlphaFold2 predicted structure for /0878 is aligned to the AlphaFold2 predicted structure for GabP from B. subtilis, a strong agreement is achieved, with 87% of residues being well-aligned with a p-value of 4.47 × 10–11 for significant similarity from FATCAT alignment.25 The specific functionality of GabP lies in the uptake of proline, that is present in the JCVI-syn3A growth media11 and is necessary to take up for generation of proteins, since no JCVI-syn3A amino acid synthesis capability exists. The speculated ability of this protein to bind amino acids nonspecifically (albeit with a preference for proline)90 may shed some light onto the quasi-essential nature of this gene along with the other identified amino acid permeases and transporters gltP/0886 (with glutamine preference) and the Opp system coded for by genes oppB/0165–oppA/0169 that bind amino acids at an ATP cost3 unlike the previously mentioned proton-symport permease systems.
yloU/0421
Gene/0421 presents an interesting case, in that it is an essential gene of unknown function adjacent to the fatty acid kinase fakA/0420 and lipid metabolism acyltransferase gene plsX/0419. Upon further examination, due to both genome locality and structural alignment, the suspicion that this gene may be involved in cellular processing of fatty acids is confirmed. When the AlphaFold2 prediction for /0421 is structurally aligned to the predicted structure for the fatty acid metabolism regulatory element YloU from B. subtilis, a convincing agreement is obtained (see Figure 7B) with a significant similarity p-value exceeding 10–11 and 99% of the residues being well-aligned by FATCAT25 (see Table 2). In addition, in B. subtilis, the gene yloU immediately precedes the fatty acid kinase gene fakA on the forward strand, just as it does in JCVI-syn3A in yet another case of conserved genome architecture. Previous bioinformatic work12 predicted /0421 to be a member of the Asp23 protein family, that includes YloU and whose members are often involved in stress response processes. YloU is thought to regulate the processing of fatty acids and phospholipid generation as a whole, however its exact function remains uncertain.91
Figure 7.

(A) Structural alignment of AlphaFold2 prediction for gabP/0878 (blue) with the predicted structure of the B. subtilis GabP permease (yellow). (B) FATCAT structural alignment of the predicted structure of yloU/0421 from JCVI-syn3A (blue) with the AlphaFold2 predicted structure for B. subtilis YloU fatty acid regulatory protein (yellow). (C) The genomic context in which yloU is found, adjacent to the fatty acid processing genes fakA and plsX, is conserved for gene yloU/0421 in JCVI-syn3A and has been observed to be conserved previously in the Firmicutes.91 (D) FATCAT structural alignment of predicted structure for gene yidC/0908 and predicted structure for M. pneumoniae show significant structural similarity.
lemA-gacS/0604
Gene /0604 was among the seven genes necessary to restore normal cell division in JCVI-syn3.0, but its biomolecular function in JCVI-syn3A remains unknown.26 When the predicted AlphaFold2 structure of /0604 is aligned via FATCAT to the structure of lemA from Thermotoga maritima a convincing agreement is obtained, that can be seen in Figure 1B. Typically, lemA is part of a two-component regulatory system, which have been well-studied across bacterial systems92−94 (but that have not yet been observed in Mycoplasma to our knowledge), and are involved in regulation of various cellular processes varying from phosphate uptake, to respiration, to sporulation. While the exact function of this gene remains unclear, several two-component systems present in B. subtilis contribute to regulation of cell growth and division processes including the pairs comA and comP and the only essential two-component system in the bacterium walK and walR.95
atpI/0797
Gene /0797 provides an important case-study in the computational analysis of JCVI-syn3A genes, in taking a holistic view in characterizing genes, rather than using a more high-throughput sequence or structurally based approach that can be applied with relative speed to the entire genome. When techniques such as the TASSER-I based structural pipeline19 or HHPred31 are applied to /0797, a wide-ranging set of possible functionalities are generated, ranging from secretory proteins to sugar transporter components. However, when a closer look was taken at its position within the genome, we observed that it lies just prior to the genes coding for the ATP synthase complex, a critical molecular machine for maintaining an appropriate membrane pH gradient in Mycoplasmas96,97 as well as in some instances generating ATP for cellular energy expenses,11 on the reverse strand. When the ATP synthase containing operon is examined in B. subtilis,49 we observe that the gene atpI is located immediately prior the remainder of the ATP synthase coding genes, in the identical order as they appear in JCVI-syn3A. With this in mind, we sought to compare the predicted protein structure of /0797 to existing structures of AtpI; however, existing structural information is sparse to nonexistent, especially for related organisms like B. subtilis. Faced with this dilemma, we utilized a predicted structure for the analogous gene in B. subtilis,98 and when it is structurally aligned with the AlphaFold2 structure of /0797, the structures resemble each other strongly (see Figure 8A, B), with a significantly similar FATCAT p-value of 1.81 × 10–11 and 122 equivalent positions (approximately 92% of the 132 total residues in /0797) with a RMSD of 2.99 Å without twists. In terms of specific function, atpI is thought to possibly be involved in guiding the assembly of the ATP synthase complex, with mutants of this gene causing a 20% decrease in overall synthase efficiency99 and a modulation of atpB/0796 expression, likely via a post-translational regulatory process.100
Figure 8.
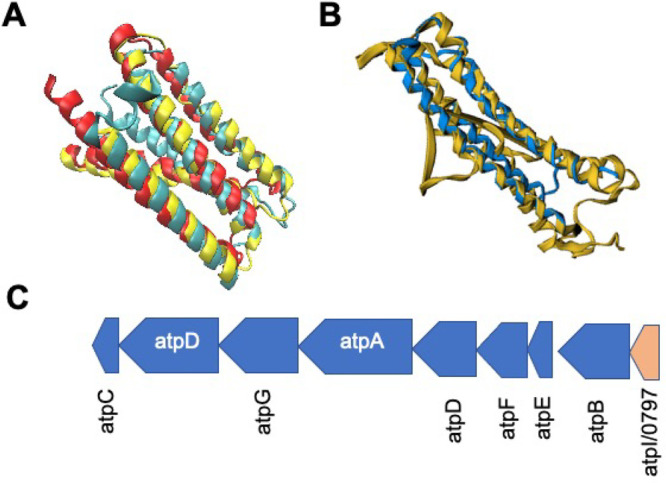
(A) STAMP structural alignment of predicted structure for B. subtilis atpI (yellow) with predicted protein structures for atpI/0797 from AlphaFold2 (red) and RoseTTAFold (blue). Images generated using VMD.42 Alignment of these two structures obtained a FATCAT25p-value of greater than 1.0 × 10–11, showing significant similarity with 92% of the JCVI-syn3A structure being well-aligned to that predicted for B. subtilis. (B) FATCAT alignment of the Spinicia oleracea atpI structure (RSCB PDB: 6FKI(101)) with the AlphaFold2 predicted structure of atpI/0797 with conservation of the central two α-helical regions especially. Alignment of these two structures obtained a FATCAT25 gave 130 residues or 99% of the JCVI-syn3A structure being well-aligned to the larger 247 residue cryo-EM structure for Spinicia oleracea. (C) Genomic organization from JCVI-syn3A and B. subtilis of the ATP synthase operon3,49 is identical lending further support to the assignment of atpI/0797 (red, bottom-right).
Summary of Computationally Annotated JCVI-syn3A Genes of Unknown Function
Table 1 contains functional annotations and generalized gene process descriptions for each of the previously unannotated JCVI-syn3A genes (of essential and quasi-essential nature3) involved in the crucial processes of cell growth, cell division, and membrane maintenance that we elucidated in this work. This knowledge can be used to extend the experimental and computational work in characterizing JCVI-syn3A given in previous works.2,3,11
Table 1. Table of Gene Products Characterized Computationallya.
| gene | cellular process | function |
|---|---|---|
| ezrA/0239 | cell division | FtsZ filament regulator |
| gpsB/0353 | cell division | divisome localization regulator |
| secDF/0412 | cell membrane | protein translocase subunit |
| yloU/0421 | cell growth | lipid synthesis regulation |
| ylxM/0430 | cell membrane | effector of translocation SRP |
| yqgP-gplG/0516 | cell membrane maintenance | membrane protease |
| yqkD/0520 | cell growth and morphology | membrane lipase/aminopeptidase |
| ylbN/0527 | cell growth and morphology | 23S rRNA regulator |
| lemA-gacS/0604 | cell growth and morphology | two component sensor/membrane |
| atpI/0797 | membrane pH gradient | ATP synthase component |
| gabP/0878 | nutrient uptake | amino acid transporter |
| yidC/0908 | cell membrane | protein translocon component |
JCVI-syn3A genes involved in cell growth, maintenance, and division in ascending order of gene locus tag (NCBI Entry - NCBI GenBank: CP002027.1), with associated bacterial orthologue gene names given. The general cellular process of each gene is given as well as its more specific cellular function.
Protein Structure Prediction Alignment Data
Table 2 contains structural alignment data for each of the previously unannotated genes implicated in cell division, growth, and membrane maintenance that we have clarified here in this work. Data comparing alignment and structural similarity of experimental structures of these genes from related bacterial organisms such as B. subtilis compared to AlphaFold2 prediction protein structures for JCVI-syn3A genes is presented with analysis conducted via FATCAT.25
4. Discussion
Benefits of Computational Analysis and Suggested Directed Functional Experiments
We have hereby demonstrated the effectiveness of a comprehensive approach integrating protein structure prediction, interactomics, and genome architecture to determine the function of previously unannotated genes in JCVI-syn3A. Of the approximately 90 protein-coding genes of unclear function reported by Breuer et al. in 2019,3 we have upgraded the annotations for 25 genes, while providing additional information for tens of the remaining protein-coding JCVI-syn3A genes, that will be useful in future studies. This workflow can be further used in elucidating the functional roles of the remaining JCVI-syn3A genes of unclear and unknown function by the design and execution of “directed wet lab assays” developed hand-in-hand with bioinformatics and simulation. Through this work, we believe the community can discover the roles of the relevant enzymes that are “missing puzzle pieces” in the biological reaction networks of JCVI-syn3A,3 which serves as a platform from which to understand the fundamental behaviors of living cells.
Nearly all of the genes annotated in this work were denoted as either essential or quasi-essential by analysis of transposon mutagenesis of JCVI-syn3A,3 underscoring their importance in JCVI-syn3A cellular processes. We note that phenotypes of such gene products are difficult to study due to the inability to obtain deletion strains of the genes in question due to their essentiality. Several of these newly annotated components may play an important role in a more detailed model for cell growth and morphological development for JCVI-syn3A. First, incorporation of gpsB/0353 and ezrA/0239 regulatory effects into a cell division and filamentation model based on the activities of ftsZ/0522 and ftsA/0523 may be crucial in modeling the proper dynamics of this process. Previously, we have demonstrated the coupling of metabolism and gene expression via whole cell modeling of JCVI-syn3A,11 and recent work in B. subtilis(107) suggests that similar couplings exist between metabolism and cell division, with pyruvate possibly allowing for sensitivity to metabolic activity by cell division components. In analyzing the possible functionality of ezrA/0239 as coding for an EzrA-like cell division protein, it is interesting to to consider its connection to the metabolic enzyme pdhA/0225 which was deleted in the reduction from JCVI-syn1.0 to JCVI-syn3.0. A previous study conducted in B. subtilis(107) demonstrated that the presence of either ezrA or pdhA, which mediates pyruvate production in the central metabolism as a signaling molecule, maintains the morphology of daughter cells, while a double knockout strain of these genes causes oblong cells.
Cellular actors involved in cell growth and specifically the translocation of membrane proteins have also been clarified, including secDF/0412 which is part of the Sec translocase system, ylxM/0430 which is a regulator of the SRP which assists in recognition of proteins to be translocated across the membrane, and yidC/0908 which also mediates the SRP based translocation process. yqgP-gplG/0516 additionally plays a role in cell growth by its activity in conjunction with ftsH/0039 in the degradation of membrane proteins, thereby negatively regulating the addition of membrane proteins that is carried out by the Sec system. Inclusion of these specific proteins and their associated gene expression profiles from experimental proteomics3 and theory11 allows for enhanced molecular detail of the growth and membrane translocation model presented by Thornburg et al.,11 making the model even more responsive to changes in metabolism by these biophysical avenues.
By explicitly modeling the interactions of proteins involved in cell division and cell growth, we can go beyond the sizer model108,109 which was assumed in previous work11 that modeled cell doubling. In this way, we can consider more carefully the specific cell-to-cell doubling time variation in the populations of JCVI-syn3A cells that may be due to the aforementioned cell division and growth processes. In parallel to construction of such models, experiments to more confidently assign annotations to these genes can be undertaken.
Of special interest are genes yqkD/0520 and ylbN/0527 which were demonstrated to cause significant changes in JCVI-syn3A daughter cell morphologies when these genes were singly deleted.26 Our predictions suggest that the gene coding for yqkD/0520 may be involved in maintaining cell morphology due to its role in processing membrane proteins, while the gene product ylbN/0527 is involved in this process through its role in regulating accumulation of the 23S (large subunit associated) rRNA and thus generation of ribosomes overall.
A possible experiment by which to further understand the role of ylbN/0527 would be a proteomic or transcriptomic experiment that gauges the number of ribosomal proteins or rRNA that is present in a ylbN/0527 deletion strain, such as the one reported by Pelletier et al. in that work.26 With such an investigation, researchers might further understand the role of this gene in regulating accumulation of the 23S rRNA and how this might affect other properties tied to cell growth behavior, such as number of membrane proteins and number of cellular ribosomes present. Additionally, introduction of B. subtilis ylbN/0527 into the JCVI-syn3A Lox-insertion site genetic “landing pad” in a gene ylbN/0527 deletion strain provides another experimental avenue. Ideally, this gene complementation experiment would result in JCVI-syn3A cells with normal morphology and validate our hypothesis that gene /0527 encodes a YlbN orthologue.
Gene yqkD/0520 remains a more difficult case to examine experimentally. However, the membrane protease activity of this gene could be observed in E. coli or a cell-free system using a recoded version of the gene that uses the standard genetic code (such a recoded set of JCVI-syn3A genes is available through the “Free Genes” Initiative110) and then is subjected to a membrane protease activity assay such as that developed by Yoshitani et al.,111 or by a more specialized aminopeptidase assay using solid-phase fluorophore chemistry.112 The secondary assignment of prolyl-oligopeptidase superfamily lipase activity could then be tested with the free genes purified protein in a similar manner using established lipase activity assays.113
Reconstitution of /0239 in E. coli via the codon-optimized platform “FreeGenes”110 might provide an avenue by which to test the GTPase activity of gene /0239 if it indeed codes for an EzrA-like protein that modulates the GTPase activity of FtsZ and formation of FtsZ filaments. This could be accomplished with an experiment such as a Malachite Green Phosphate Assay such as the one described in ref (7) to observe the changes in FtsZ GTPase activity that were seen in B. subtilis.64
Confirming the annotation of gene atpI/0797 could be ascertained by fluorescence microscopy or immunoprecipitation experiments, whereby atpI/0797 could be colocalized alongside labeled copies of one of the ATP synthase genes that provides the base component of the molecular assembly, such as atpB/0796, consistent with the role of atpI in modulating assembly of the ATP synthase complex.99
Likewise, gene ylxM/0430 could be confirmed as an effector of the regulation of the SRP by such colocalization to the SRP itself (ffs/0049) as well as the other members of the Sec translocase system such as secA/0095 and secY/0652.
The predicted membrane protease yqgP-gplG/0516 might be subjected to “Free Genes”110 reconstitution in E. coli or a similar technique and then be tested for activity on purified membrane proteins from E. coli that are also present in JCVI-syn1.0 and JCVI-syn3A such as mgtA/0787 and mgtE/0157. The assay could use the previously mentioned method developed by Yoshitani et al.,111 which might be transferable to proteases such as this.
The presence in the JCVI-syn3A genome of moonlighting proteins, i.e., multifunctional proteins that are not the result of gene fusions, could confound efforts to predict protein functions. Because of evolutionary pressure to delete nonessential genes, mycoplasmas are known for having such multifunctional proteins.114−117 Some predict that mycoplasmas will have more moonlighting proteins than bacteria not under evolutionary pressure to shed genes.118,119 This may result in a protein evolving to have part of its structure or amino acid sequence be similar to that of functionally characterized proteins with one function and another part similar to a different protein with a different function. Thus, protein function prediction software may offer more than one prediction with significant confidence. It is vital that both computational and experimental biologists keep this in mind when evaluating predictions of protein function. Furthermore, dissimilar protein function predictions with varying confidence levels may both be correct for moonlighting proteins. Additionally, an analysis of some transporter proteins such as that coded for by ptsG/0779 predicted via AlphaFold2 serves as a cautionary note for computational methodologies and motivation for further experimentation. Low prediction confidence (see Supporting Information section S2 analysis notebook) can be observed especially in and around membrane embedded domains where it is less likely that experimental structures were available at the time of CASP14 and during AlphaFold2 development.
Note that we also provide in Supporting Information section S2 a database containing AlphaFold2 protein structure predictions that we have generated for all of the approximately 450 protein-coding genes present in the JCVI-syn3A genome, along with an associated Jupyter analysis and visualization notebook. We hope that these predictions in conjunction with the computational workflow we have demonstrated here and with additional directed experiments will help clarify and resolve our understanding of the remaining genes of unknown function in the JCVI-syn3A genome, moving us as a scientific community toward the complete characterization of the genetically minimal cell JCVI-syn3A. Additionally, we provide a supplemental table summarizing gene functionality for all of the genes of previously unclear function3 in Supporting Information section S3 and Supporting Information Spreadsheet S1. Assignments of gene functional annotation were made when agreement of a predicted structure to an experimentally solved structure was observed via FATCAT.25 These assignments were further supported by sequence analysis and genome architecture examination, in a similar manner to that demonstrated in this work (such high confidence predictions from the main text and Supporting Information Table S1 are highlighted).
5. Conclusions
Individualized computational analysis of JCVI-syn3A genes allowed us to further characterize the JCVI-syn3A genome. We uncovered the functions of several genes whose products include important players in fundamental cellular processes, such as cell growth, membrane pH maintenance, divisome formation, and membrane protein translocation, to name a few. These findings combined with ongoing work will result in better modeling and experimental characterization of these processes and their dependencies in the context of this minimized genome. Further results from this treatment and associated directed experiments can inform the “design, build, test” synthetic biology development of JCVI-syn3A moving forward. Bioinformatic, computational, and experimental efforts related to the remaining JCVI-syn3A genes of unknown function can precipitate a self-improving cycle, where in silico analysis and predictions can inform more robust computational models for cell growth and division, that in turn generate predictions that can be examined by experiments. We hope that the techniques used in this work and the suggested annotation updates and directed characterization experiments will drive increased understanding and motivation to uncover the secrets of the genetically minimal cell, JCVI-syn3A. In this way, we can elucidate the outstanding unknowns of its biological milieu and work toward the complete characterization of the genome of a living cell.
Acknowledgments
This work would not have been possible without support from NSF MCB-1818344 and NSF MCB-1840320. We also acknowledge support from NSF Physics Frontier Center PHY-1430124 “The Center for the Physics of Living Cells” (CPLC) and the NSF Physics of Living Systems (PoLS) Initiative NSF PHY-2014027. J.F.P. was supported by an International Human Frontier Science Program Organization (HFSPO) Cross-Disciplinary Fellowship LT000901/2021-C.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.2c04188.
The authors declare no competing financial interest.
Special Issue
Published as part of The Journal of Physical Chemistry virtual special issue “Jose Onuchic Festschrift”.
Supplementary Material
References
- Gibson D. G.; et al. Science 2010, 329, 52–56. 10.1126/science.1190719. [DOI] [PubMed] [Google Scholar]
- Hutchison C. A.; et al. Science 2016, 351, aad6253. 10.1126/science.aad6253. [DOI] [PubMed] [Google Scholar]
- Breuer M. eLife 2019, 8, e36842. 10.7554/eLife.36842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson D. G.; et al. Science 2008, 319, 1215–1220. 10.1126/science.1151721. [DOI] [PubMed] [Google Scholar]
- Lartigue C.; Glass J. I.; Alperovich N.; Pieper R.; Parmar P. P.; Hutchison C. A.; Smith H. O.; Venter J. C. Science 2007, 317, 632–638. 10.1126/science.1144622. [DOI] [PubMed] [Google Scholar]
- Lachance J.-C.; Rodrigue S.; Palsson B. O. eLife 2019, 8, e45379. 10.7554/eLife.45379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas D.; et al. mBio 2022, 22, e01630 10.1128/mbio.01630-22. [DOI] [Google Scholar]
- Danchin A.; Fang G. Microbial Biotechnology 2016, 9, 530–540. 10.1111/1751-7915.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Tsui S. K.-W. J. Proteome Res. 2018, 17, 2511–2520. 10.1021/acs.jproteome.8b00262. [DOI] [PubMed] [Google Scholar]
- Pedreira T.; Elfmann C.; Singh N.; Stülke J. Protein Sci. 2022, 31, 54–62. 10.1002/pro.4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornburg Z. R.; et al. Cell 2022, 185, 345–360. 10.1016/j.cell.2021.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antczak M.; Michaelis M.; Wass M. N. Nat. Commun. 2019, 10, 3100. 10.1038/s41467-019-10837-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantalapiedra C. P.; Hernández-Plaza A.; Letunic I.; Bork P.; Huerta-Cepas J. Mol. Biol. Evol. 2021, 38, 5825–5829. 10.1093/molbev/msab293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A.; Larsson B.; von Heijne G.; Sonnhammer E. L. J. Mol. Biol. 2001, 305, 567–580. 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Kelley L. A.; Mezulis S.; Yates C. M.; Wass M. N.; Sternberg M. J. E. Nat. Protoc. 2015, 10, 845–858. 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wass M. N.; Kelley L. A.; Sternberg M. J. E. Nucleic Acids Res. 2010, 38, W469–W473. 10.1093/nar/gkq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigrist C. J. A.; de Castro E.; Cerutti L.; Cuche B. A.; Hulo N.; Bridge A.; Bougueleret L.; Xenarios I. Nucleic Acids Res. 2012, 41, D344–D347. 10.1093/nar/gks1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi D. M.; Peterson J. R.; Earnest T. M.; Hallock M. J.; Luthey-Schulten Z. IET Systems Biology 2018, 12, 170–176. 10.1049/iet-syb.2017.0070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Zheng W.; Cheng M.; Omenn G. S.; Freddolino P. L.; Zhang Y. J. Proteome Res. 2021, 20, 1178–1189. 10.1021/acs.jproteome.0c00359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Zhang Y. Curr. Protoc. Bioinf. 2015, 52, 5.8.1–5.8.15. 10.1002/0471250953.bi0508s52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A.; Yang J.; Zhang Y. Nucleic Acids Res. 2012, 40, W471–W477. 10.1093/nar/gks372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J.; et al. Nature 2021, 596, 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuchic J. N.; Luthey-Schulten Z.; Wolynes P. G. Annu. Rev. Phys. Chem. 1997, 48, 545–600. 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Case D. A.; Walker R. C. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 3, 198–210. 10.1002/wcms.1121. [DOI] [Google Scholar]
- Li Z.; Jaroszewski L.; Iyer M.; Sedova M.; Godzik A. Nucleic Acids Res. 2020, 48, W60–W64. 10.1093/nar/gkaa443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier J. F.; Sun L.; Wise K. S.; Assad-Garcia N.; Karas B. J.; Deerinck T. J.; Ellisman M. H.; Mershin A.; Gershenfeld N.; Chuang R.-Y.; et al. Cell 2021, 184, 2430–2440. 10.1016/j.cell.2021.03.008. [DOI] [PubMed] [Google Scholar]
- Steinegger M.; Meier M.; Mirdita M.; Vöhringer H.; Haunsberger S. J.; Söding J. BMC Bioinf. 2019, 20, 473. 10.1186/s12859-019-3019-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nat. Methods 2022, 19, 1. 10.1038/s41592-021-01380-4 [DOI] [PubMed] [Google Scholar]
- Thompson J. D.; Higgins D. G.; Gibson T. J. Nucleic Acids Res. 1994, 22, 4673–4680. 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D.; Clements J.; Eddy S. R. Nucleic Acids Res. 2011, 39, W29–W37. 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann L.; Stephens A.; Nam S.-Z.; Rau D.; Kübler J.; Lozajic M.; Gabler F.; Söding J.; Lupas A. N.; Alva V. J. Mol. Biol. 2018, 430, 2237–2243. 10.1016/j.jmb.2017.12.007. [DOI] [PubMed] [Google Scholar]
- Soding J.; Biegert A.; Lupas A. N. Nucleic Acids Res. 2005, 33, W244–W248. 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M.; Sato Y.; Morishima K. J. Mol. Biol. 2016, 428, 726–731. 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Matteau D.; Lachance J.-C.; Grenier F.; Gauthier S.; Daubenspeck J. M.; Dybvig K.; Garneau D.; Knight T. F.; Jacques P.-É.; Rodrigue S. Mol. Syst. Biol. 2020, 16, e9844 10.15252/msb.20209844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrabiani K. M.; Cheng R. R.; Onuchic J. N. J. Phys. Chem. B 2021, 125, 11408–11417. 10.1021/acs.jpcb.1c07145. [DOI] [PubMed] [Google Scholar]
- Baek M.; et al. Science 2021, 373, 871–876. 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S.; Zhang Y. Nucleic Acids Res. 2007, 35, 3375–3382. 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W.; Zhang C.; Wuyun Q.; Pearce R.; Li Y.; Zhang Y. Nucleic Acids Res. 2019, 47, W429–W436. 10.1093/nar/gkz384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J.; et al. Nucleic Acids Res. 2021, 49, D412–D419. 10.1093/nar/gkaa913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell R. B.; Barton G. J. Proteins: Struct., Funct., Genet. 1992, 14, 309–323. 10.1002/prot.340140216. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Eastwood M.; Hardin C.; Luthey-Schulten Z.; Wolynes P. IBM J. Res. Dev. 2001, 45, 475–497. 10.1147/rd.453.0475. [DOI] [Google Scholar]
- Onuchic J. N.; Wolynes P. G.; Luthey-Schulten Z.; Socci N. D. Proc. Natl. Acad. Sci. U. S. A. 1995, 92, 3626–3630. 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryngelson J. D.; Onuchic J. N.; Socci N. D.; Wolynes P. G. Proteins: Struct., Funct., Genet. 1995, 21, 167–195. 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D.; et al. Nucleic Acids Res. 2021, 49, D605–D612. 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamames J. Genome Biol. 2001, 2, research0020.1. 10.1186/gb-2001-2-6-research0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G.; Rocha E. P.; Danchin A. BMC Genomics 2008, 9, 4. 10.1186/1471-2164-9-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart J. C.; Lalanne J.-B.; Li G.-W. Annu. Rev. Microbiol. 2021, 75, 243–267. 10.1146/annurev-micro-041921-012646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedreira T.; Elfmann C.; Stülke J. Nucleic Acids Res. 2022, 50, D875–D882. 10.1093/nar/gkab943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benders G. A.; Powell B. C.; Hutchison C. A. J. Bacteriol. 2005, 187, 4542–4551. 10.1128/JB.187.13.4542-4551.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcón F.; de Vasconcelos A. T. R.; Yim L.; Zaha A. Genet. Mol. Biol. 2007, 30, 174–181. 10.1590/S1415-47572007000200003. [DOI] [Google Scholar]
- Eraso J. M.; Markillie L. M.; Mitchell H. D.; Taylor R. C.; Orr G.; Margolin W. J. Bacteriol. 2014, 196, 2053–2066. 10.1128/JB.01370-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Torró C.; Torres-Puig S.; Marcos-Silva M.; Huguet-Ramón M.; Muñoz-Navarro C.; Lluch-Senar M.; Serrano L.; Querol E.; Piñol J.; Pich O. Q. Frontiers in Microbiology 2021, 12, 695572. 10.3389/fmicb.2021.695572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J.; Suzuki M.; McCarty D. R. Journal of Experimental Botany 2016, 67, 5447–5460. 10.1093/jxb/erw311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber N.; de Opitz C. L. M.; Mayer C.; Sass P. Future Microbiol. 2020, 15, 801–831. 10.2217/fmb-2019-0348. [DOI] [PubMed] [Google Scholar]
- Eswara P. J.; Brzozowski R. S.; Viola M. G.; Graham G.; Spanoudis C.; Trebino C.; Jha J.; Aubee J. I.; Thompson K. M.; Camberg J. L.; Ramamurthi K. S. eLife 2018, 7, 38856. 10.7554/eLife.38856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismondo J.; Cleverley R. M.; Lane H. V.; Großhennig S.; Steglich A.; Möller L.; Mannala G. K.; Hain T.; Lewis R. J.; Halbedel S. Mol. Microbiol. 2016, 99, 978–998. 10.1111/mmi.13279. [DOI] [PubMed] [Google Scholar]
- Tavares J. R.; de Souza R. F.; Meira G. L. S.; Gueiros-Filho F. J. J. Bacteriol. 2008, 190, 7096–7107. 10.1128/JB.00064-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira A. R.; Reed P.; Veiga H.; Pinho M. G. BMC Microbiol. 2013, 13, 18. 10.1186/1471-2180-13-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joint Center for Structural Genomics (JCSG) .Crystal structure of a lema protein (tm0961) from thermotoga maritima msb8 at 2.28 A resolution; Worldwide PDB, 2005. 10.2210/pdb2etd/pdb (date accessed 2022-03-16). [DOI]
- Singh J. K.; Makde R. D.; Kumar V.; Panda D. Biochemistry 2007, 46, 11013–11022. 10.1021/bi700710j. [DOI] [PubMed] [Google Scholar]
- Haeusser D. P.; Schwartz R. L.; Smith A. M.; Oates M. E.; Levin P. A. Mol. Microbiol. 2004, 52, 801–814. 10.1111/j.1365-2958.2004.04016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung K.-M.; Hsu H.-H.; Yeh H.-Y.; Chang B.-Y. J. Biol. Chem. 2007, 282, 14891–14897. 10.1074/jbc.M605177200. [DOI] [PubMed] [Google Scholar]
- Cleverley R. M.; et al. Nat. Commun. 2014, 5, 5421. 10.1038/ncomms6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ollis D. L.; et al. Protein Eng. Des. Sel. 1992, 5, 197–211. 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- Polgár L. FEBS Lett. 1992, 311, 281–284. 10.1016/0014-5793(92)81120-B. [DOI] [PubMed] [Google Scholar]
- Uncharacterized protein YqkD - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/P54567 (date accessed 2022-04-03).
- Varejão N.; De-Andrade R. A.; Almeida R. V.; Anobom C. D.; Foguel D.; Reverter D. Structure 2018, 26, 199–208.e3. 10.1016/j.str.2017.12.004. [DOI] [PubMed] [Google Scholar]
- Rawadi G.; Lalanne J.-L.; Roulland-Dussoix D. Gene 1995, 158, 107–111. 10.1016/0378-1119(95)00160-8. [DOI] [PubMed] [Google Scholar]
- Burgos R.; Weber M.; Martinez S.; Lluch-Senar M.; Serrano L. Mol. Syst. Biol. 2020, 16, e9530 10.15252/msb.20209530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovaere P.; Lippens S.; Vandenabeele P.; Declercq W. Trends Biochem. Sci. 2009, 34, 453–463. 10.1016/j.tibs.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Nicolas P.; et al. Science 2012, 335, 1103–1106. 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- O’Neal A. J.; Butler L. R.; Rolandelli A.; Gilk S. D.; Pedra J. H. eLife 2020, 9, 61675. 10.7554/eLife.61675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsholz A. K. W.; et al. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 7451–7456. 10.1073/pnas.1117483109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uncharacterized protein YlbN - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/O34445 (date accessed 2022-04-05).
- de Jong I. G.; Veening J.-W.; Kuipers O. P. Environmental Microbiology 2012, 14, 3110–3121. 10.1111/j.1462-2920.2012.02892.x. [DOI] [PubMed] [Google Scholar]
- Eymann C.; Homuth G.; Scharf C.; Hecker M. J. Bacteriol. 2002, 184, 2500–2520. 10.1128/JB.184.9.2500-2520.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott M.; Klumpp S.; Mateescu E. M.; Hwa T. Molecular Systems Biology 2014, 10, 747. 10.15252/msb.20145379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukazaki T.; Mori H.; Echizen Y.; Ishitani R.; Fukai S.; Tanaka T.; Perederina A.; Vassylyev D. G.; Kohno T.; Maturana A. D.; Ito K.; Nureki O. Nature 2011, 474, 235–238. 10.1038/nature09980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukazaki T. FEMS Microbiolol. Lett. 2018, 365, fny112. 10.1093/femsle/fny112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oganesyan V.; Pufan R.; DeGiovanni A.; Yokota H.; Kim R.; Kim S.-H. Acta Crystallographica Section D Biological Crystallography 2004, 60, 1266–1271. 10.1107/S0907444904009394. [DOI] [PubMed] [Google Scholar]
- Williams M. L.; Crowley P. J.; Hasona A.; Brady L. J. J. Bacteriol. 2014, 196, 2043–2052. 10.1128/JB.01465-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki R.; Yura T.; Suzuki T.; Dohmae N.; Mori H.; Akiyama Y. Sci. Rep. 2016, 6, 24147. 10.1038/srep24147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petriman N.-A.; Jauß B.; Hufnagel A.; Franz L.; Sachelaru I.; Drepper F.; Warscheid B.; Koch H.-G. Sci. Rep. 2018, 8, 578. 10.1038/s41598-017-19019-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald J.; Njenga R.; Natriashvili A.; Sarmah P.; Koch H.-G. Frontiers in Molecular Biosciences 2021, 8, 664241. 10.3389/fmolb.2021.664241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhomboid protease GluP - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/P54493 (date accessed 2022-04-06).
- Began J.; et al. EMBO J. 2020, 39, 10. 10.15252/embj.2019102935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux M. J.; Fischer S. J.; Cherney M. M.; Bateman K. S.; James M. N. G. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 750–754. 10.1073/pnas.0609981104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaprasis A.; Hoffmann T.; Stannek L.; Gunka K.; Commichau F. M.; Bremer E. J. Bacteriol. 2014, 196, 515–526. 10.1128/JB.01128-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tödter D.; Gunka K.; Stülke J. Front. Microbiol. 2017, 8, 883. 10.3389/fmicb.2017.00883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschiedrich C. P.; Keidel V.; Szurmant H. J. Mol. Biol. 2016, 428, 3752–3775. 10.1016/j.jmb.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dintner S.; Staroń A.; Berchtold E.; Petri T.; Mascher T.; Gebhard S. J. Bacteriol. 2011, 193, 3851–3862. 10.1128/JB.05175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabak E. M.; Willis D. K. J. Bacteriol. 1992, 174, 3011–3020. 10.1128/jb.174.9.3011-3020.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch J. A. Annu. Rev. Microbiol. 2017, 71, 1–19. 10.1146/annurev-micro-030117-020355. [DOI] [PubMed] [Google Scholar]
- Benyoucef M.; Rigaud J.-L.; Leblanc G. Eur. J. Biochem. 1981, 113, 491–498. 10.1111/j.1432-1033.1981.tb05090.x. [DOI] [PubMed] [Google Scholar]
- Benyoucef M.; Rigaud J.-L.; Leblanc G. Eur. J. Biochem. 1981, 113, 499–506. 10.1111/j.1432-1033.1981.tb05091.x. [DOI] [PubMed] [Google Scholar]
- https://www.uniprot.org/uniprot/P37816#structure.
- Gay N. J. J. Bacteriol. 1984, 158, 820–825. 10.1128/jb.158.3.820-825.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu D. K.; Brusilow W. S. FEBS Lett. 1995, 371, 127–131. 10.1016/0014-5793(95)00867-9. [DOI] [PubMed] [Google Scholar]
- Hahn A.; Vonck J.; Mills D. J.; Meier T.; Kühlbrandt W. Science 2018, 360, eaat4318. 10.1126/science.aat4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa A.; Yoshikaie K.; Mori T.; Mori H.; Morimoto Y. V.; Sugano Y.; Iwaki S.; Minamino T.; Sugita Y.; Tanaka Y.; Tsukazaki T. Cell Reports 2017, 19, 895–901. 10.1016/j.celrep.2017.04.030. [DOI] [PubMed] [Google Scholar]
- Botte M. Sci. Rep. 2016, 6, 38399. 10.1038/srep38399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uncharacterized protein YloU - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/O34318 (date accessed 2022-04-09).
- GABA permease GabP- AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/P46349 (date accessed 2022-04-09).
- Membrane protein insertase YidC - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/P75112 (date accessed 2022-04-03).
- Monahan L. G.; Hajduk I. V.; Blaber S. P.; Charles I. G.; Harry E. J. mBio 2014, 5, e00935-14. 10.1128/mBio.00935-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallden M.; Fange D.; Lundius E. G.; Baltekin Ö.; Elf J. Cell 2016, 166, 729–739. 10.1016/j.cell.2016.06.052. [DOI] [PubMed] [Google Scholar]
- Facchetti G.; Chang F.; Howard M. Current Opinion in Systems Biology 2017, 5, 86–92. 10.1016/j.coisb.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JCVI-syn3.0 genes (E. coli codon optimized), 2021; https://stanford.freegenes.org/collections/all/products/jcvi_syn3-0#bionet.
- Yoshitani K.; Hizukuri Y.; Akiyama Y. FEBS Lett. 2019, 593, 842–851. 10.1002/1873-3468.13368. [DOI] [PubMed] [Google Scholar]
- Drag M.; Bogyo M.; Ellman J. A.; Salvesen G. S. J. Biol. Chem. 2010, 285, 3310–3318. 10.1074/jbc.M109.060418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R.; Rathi P.; Gupta N.; Bradoo S. Biotechnology and Applied Biochemistry 2003, 37, 63. 10.1042/BA20020059. [DOI] [PubMed] [Google Scholar]
- Robinson M. W.; et al. Open Biology 2013, 3, 130017. 10.1098/rsob.130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarocki V. M.; Santos J.; Tacchi J. L.; Raymond B. B. A.; Deutscher A. T.; Jenkins C.; Padula M. P.; Djordjevic S. P. Open Biology 2015, 5, 140175. 10.1098/rsob.140175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacchi J. L.; et al. Open Biology 2016, 6, 150210. 10.1098/rsob.150210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widjaja M.; et al. Sci. Rep. 2017, 7, 11227. 10.1038/s41598-017-10644-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daubenspeck J. M.; Liu R.; Dybvig K. PLoS One 2016, 11, e0162505 10.1371/journal.pone.0162505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwille P.; Frohn B. P. Trends in Cell Biology 2022, 32, 102–109. 10.1016/j.tcb.2021.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Joint Center for Structural Genomics (JCSG) .Crystal structure of a lema protein (tm0961) from thermotoga maritima msb8 at 2.28 A resolution; Worldwide PDB, 2005. 10.2210/pdb2etd/pdb (date accessed 2022-03-16). [DOI]
- Uncharacterized protein YqkD - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/P54567 (date accessed 2022-04-03).
- Uncharacterized protein YlbN - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/O34445 (date accessed 2022-04-05).
- Rhomboid protease GluP - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/P54493 (date accessed 2022-04-06).
- Uncharacterized protein YloU - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/O34318 (date accessed 2022-04-09).
- GABA permease GabP- AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/P46349 (date accessed 2022-04-09).
- Membrane protein insertase YidC - AlphaFold structure prediction. https://alphafold.ebi.ac.uk/entry/P75112 (date accessed 2022-04-03).