

Abstract
Hundreds of nucleoside-based natural products have been isolated from various microorganisms, several of which have been utilized in agriculture as pesticides and herbicides, in medicine as therapeutics for cancer and infectious disease, and as molecular probes to study biological processes. Natural products consisting of structural modifications of each of the canonical nucleosides have been discovered, ranging from simple modifications such as single-step alkylations or acylations to highly elaborate modifications that dramatically alter the nucleoside scaffold and require multiple enzyme-catalyzed reactions. A vast amount of genomic information has been uncovered the past two decades, which has subsequently allowed the first opportunity to interrogate the chemically intriguing enzymatic transformations for the latter type of modifications. This review highlights (i) the discovery and potential applications of structurally complex pyrimidine nucleoside antibiotics for which genetic information is known, (ii) the established reactions that convert the canonical pyrimidine into a new nucleoside scaffold, and (iii) the important tailoring reactions that impart further structural complexity to these molecules.
1. Introduction
Nucleoside/nucleotide metabolism is an essential and ubiquitous component of life. Not surprisingly, synthetic and natural product-based discovery efforts have yielded a large number of derivatives of the canonical nucleosides with desirable biological activities that have been exploited in the clinic, agriculture, and biotechnology. Nucleoside-based natural products, which have primarily been isolated from microorganisms, encompass derivatives of pyrimidines and purines alike, and range from relatively simple (e.g., methylation) to highly complex structural modifications of the nucleoside scaffold. Several of these nucleoside-based natural products were catalogued in 1988,1 which was subsequently updated in 1991.2 Since the update, dozens more have been discovered, including several uridine-derived nucleoside antibiotics that have been identified by their potent inhibition of the enzyme bacterial translocase I, an essential enzyme involved in peptidoglycan biosynthesis.3, 4
In the past two decades, the biosynthetic genes—which are often clustered in a microorganism—for a number of nucleoside-based natural products have been uncovered, particularly for nucleoside derivatives that fall into the category of chemically complex modifications of the parent nucleoside. In line with the expectations from the analysis of the structures, the assembly of these structurally complex nucleoside derivatives appears to involve dozens of enzyme-catalysed steps, some of which have been shown to be novel or highly unusual. For most of these nucleoside antibiotics, the majority of the biosynthetic steps remain unknown despite the availability of the putative gene products for bioinformatic analysis. This review focuses on the biosynthetic mechanism of pyrimidine derived nucleoside antibiotics, providing a nearly comprehensive list and description of nucleoside derivatives for which the biosynthetic gene clusters have been identified. Importantly, we cover the characterization of the biosynthetic genes and highlight the established, key enzyme-catalysed transformations that divert the canonical nucleosides/nucleotides into unique analogues that serve as the scaffold for further decoration. Furthermore, downstream modifications of the nucleoside-derived core scaffolds—often referred to as tailoring steps for other groups of natural products—are discussed, including the assembly and attachment of unusual sugars, fatty acids and polyketides, nonribosomally derived peptides, and sulfate or phosphate groups.
2. Pyrimidine nucleoside antibiotics
2.1. Uridine-derived nucleosides
2.1.1. Pacidamycin, mureidomycin, napsamycin, and sansanmycin.
The pacidamycins (1-10) were initially isolated from Streptomyces coeruleorubidus in 1989.5, 6 They share a common structural scaffold with the mureidomycins (11-14), which were isolated from Streptomyces flavidovirens SANK 60486 in 1989;7-9 the napsamycins (15-18), which were isolated from Streptomyces sp. HIL Y-82 in 1994;10 and the more recently identified sansanmycins (19-26), which were isolated from Streptomyces sp. Strain SS in 2007.11, 12 This family is classified as peptidyl uridine antibiotics (also called uridyl peptide antibiotics) and are known to inhibit the enzyme translocase I (MraY),13 a transmembrane protein involved in bacterial cell wall biosynthesis and hence essential for bacterial survival. Although narrow in spectrum, they have modest-to-potent antibacterial activity against Pseudomonas aeruginosa (MIC 0.1-3 μg/ml for mureidomycins,14, 15 4-64 μg/ml for pacidamycins,16 12.5-25 μg/ml for napsamycins,10 and 10-12.5 μg/ml for sansanmycins);11 and the mureidomycins have been shown to be effective in a mouse model of infection.16
The structures of the peptidyl uridine family of nucleoside antibiotics all consist of an unusual 3′-deoxyuridine nucleoside core, which is covalently linked via a 4′,5′-enamide bond to a central N-methyl-2,3-diaminobutyric acid (N-methyl-DABA) residue within a tetra- or pentapeptide scaffold (Fig. 1). The direction of the peptide chain of the peptidyl uridine antibiotics is reversed twice, which is a highly unusual feature for nonribosomally derived peptides (Fig. 1). Peptidyl nucleosides 1-10 are structurally differentiated from the rest of the family by the addition of an l-Ala residue at position A4 and contain one of three different aromatic amino acid (l-Trp, l-Phe, and the nonproteinogenic amino acid l-m-Tyr) at the C-terminus (position A5).6 Contrastingly, 11-25 contain an l-Met (or l-Met sulfoxide, l-MetSO) or, in two instances, l-Leu at A4 and have either a l-Trp or l-m-Tyr at A5 of the peptide chain.8, 12 An N-terminal bicyclic amino acid, 6-hydroxy-tetrahydro-isoquinoline carboxylic acid, or the mono-C-methyl variant, is incorporated into the four congeners of napsamycins (15/17 and 16/18, respectively). A related bicyclic amino acid is also found in structures 5, 24, and 25, the last two of which contain a geminal di-C-methyl bicyclic amino acid. A final structural variation of note occurs within the 3′-deoxyuridine nucleoside core, wherein dihydrouracil is sometimes found in place of uracil (exemplified by 12, 14, 17, and 18).
Figure 1.

Structures of representative nucleoside antibiotics of the peptidyl uridine family.
The biosynthetic gene clusters for three of the peptidyl-uridine nucleosides have been identified (Fig. 2A).17-20 In 2010 two independent groups identified the 1-10 biosynthetic gene cluster,17, 18 which spanned approximately 30.3-kb of contiguous DNA and consisted of 22 open reading frames (orfs) named pac1-pac22 or pacA-pacV (for simplicity, only the letter annotation will be used). In both instances the putative cluster was located by genome scanning of the entire sequence that was obtained from 454 sequencing. Inactivation of pacO and pacP encoding proteins with domains with sequence similarity to nonribosomal peptide synthetases (NRPS) abolished pacidamycin production, the former gene of which was complemented in trans to restore production.18 The minimal necessary genes for 10 biosynthesis were also determined by heterologous production within the host Streptomyces lividans TK24.17 Inactivation of a putative DABA synthase gene (pacS) abolished the production of 1-10 in the heterologous host, which was chemically complemented by feeding DABA. In addition to determination of the genomic boundaries, heterologous production resulted in the isolation of a new congener named pacidamycin S that is identical to 10 except for A5 substitution of l-Trp with l-Phe. The 15-18 biosynthetic gene cluster from Streptomyces sp. DSM 5940 was identified by using PCR-probes designed from a putative peptidyl-uridine biosynthetic cluster found in Streptomyces roseosporus NRRL 15998.19 Annotation of the locus revealed 29 hypothetical genes (npsA-npsV) that are likely involved in the biosynthesis of the 15-18. Heterologous expression of the gene cluster in Streptomyces coelicolor M1154 led to the production of 15 and 17 as well as 11 and 12. By thoroughly examining the metabolic profile of the heterologous producer, several novel congeners were identified by MS-MS including mureidomycins containing L-Phe instead of m-Tyr at A5, MetSO-containing variants, and an N-terminal, N-acylated mureidomycin.21 By using the biosynthetic genes for 1-10 as probes for whole genome scanning, the gene cluster for 19-25 biosynthesis was identified and predicted to consist of 25 orfs (ssaA-ssaY) within approximately 33-kb of contiguous DNA (Fig. 2A).20
Figure 2.

Biosynthesis of the peptidyl uridine family of nucleoside antibiotics. (A) Genetic organization of the biosynthetic gene clusters. NCBI accession numbers are napsamycin (nps) from Streptomyces sp. DSM 5940, HQ287563; pacidamycin (pac) from Streptomyces coeruleorubidus strain NRRL 18370, HM855229; and sansanmycin (ssa) from Streptomyces sp. SS, KC188778. (B) Pathway for the biosynthesis of the nucleoside core. (C) Pathways for the biosynthesis of the pseudopeptide and attachment to the nucleoside core. Proteins labelled in bold blue have been functionally assigned in vitro using recombinant enzymes. PLP, pyridoxal-5’-phosphate; DABA, diaminobutyric acid; A, adenylation; C, condensation, T, thiolation.
Although biosynthetic investigations for 15-18 have been reported, insight into the formation of peptidyl-uridine nucleosides has been primarily established using 1-10 biosynthesis as the model, represented with select congeners in Fig. 2B.18, 22-24 Generation of the nucleoside core, 5′-amino-3′,5′-dideoxy-4′,5′-dehydrouridine (27), is orchestrated by three enzymes (PacK, PacM, and PacE) that have been characterized in vitro (Fig. 2B).25 Uridine is the apparent precursor of this three-enzyme pathway, which contrasts most other pyrimidine nucleoside antibiotics that originate from a nucleotide 5’-monophosphate precursor and do not proceed via a canonical nucleoside intermediate. PacK is a flavin-dependent dehydrogenase that initiates the pathway by oxidizing uridine to uridine-5′-aldehyde (28). Subsequently, PacM of the cupin superfamily and a pyridoxal-5’-phosphate (PLP)-dependent aminotransferase PacE catalyse dehydration and transamination, respectively, to produce 27.25, 26 Both PacM and PacE were shown to have flexibility with respect to substrate selection, thus the reaction order could not be established.
Despite DABA being found in several other peptide natural products, the general biosynthetic steps leading to (2S,3S)-DABA are mostly unknown. Prior to the cloning of any peptidyl uridine antibiotic gene clusters, a DABA synthase activity using l-Thr and ammonia was detected using cell free extracts of the 11-14 producing strain, suggesting the biosynthesis proceeds through a β-substitution reaction with ammonia as a nucleophile.27 The reaction was stimulated by the addition of PLP, consistent with PLP as a necessary cofactor for this conversion. Although the specific activity was moderately higher with l-Thr, a variety of β-substituted amino acids were also substrates including O, S, and Cl-substituted l- or d-amino acids. Following the cloning of the biosynthetic gene clusters, a four gene subcluster was uncovered whose gene products were potentially involved in (2S,3S)-DABA biosynthesis. This includes PacQ, with sequence similarity to argininosuccinate lyases; PacR, with similarity to archaeal threonine kinases and bacterial shikimate kinases; PacS, a didomain protein with similarity to 2,3-diaminopropionate synthase and ATP-grasp proteins (which includes argininosuccinate lyases); and PacT, a threonine aldolase. Although pacS was shown to be essential for the biosynthesis of 1-10 by gene inactivation,18 the function of these four gene products has not been determined. Nonetheless, it is likely that the biosynthesis starts from l-Thr and l-Asp and ends with elimination of fumarate via lyase chemistry, thereby generating the nonproteinogenic amino acid (2S,3S)-DABA as a precursor for downstream enzymes (Fig. 2C).
As previously mentioned, the peptide component of the peptidyl uridine antibiotics has several unusual structural features, including a ureido functionality and a β-amide linkage through DABA that effectively reverses the peptide orientation twice. Not surprisingly, the gene clusters encode for several proteins with similarity to nonribosomal peptide synthetases (NRPS), which are enzymes involved in the biosynthesis of numerous bioactive peptide metabolites from relatively simple amino acids or carboxylic acid building blocks. NRPS are often found as large proteins containing multiple modules (which are, in turn, divided into domains), with each module responsible for incorporation of one (amino) acid unit into the peptide. The modular organization has enabled a predictive model for peptide assembly termed the NRPS code, which has been reviewed in detail—along with the general NRPS mechanism that includes the function of individual domains—elsewhere.28-32 Unlike the typical NRPS system, however, the NRPS components encoded within the peptidyl uridine gene clusters are found as several individual proteins with a nonmodular architecture, thus making functional predictions based on sequence challenging. Nonetheless, the pathway has been well studied by using recombinant proteins including eight of the NRPS-related proteins harbouring minimally twelve domains (Fig. 2C).33
The (2S,3S)-DABA component serves as the centerpiece for peptide elongation, wherein amino acid building blocks are added to both α- and β-amino functionalities. To initiate this process, PacP, a tridomain protein consisting of adenylation (A), thiolation (T), and thioesterase (TE) domains, first activates (2S,3S)-DABA and loads it to the T domain in cis to generateDABA-S-PacP (29). PacV catalyses the β-N-methylation of 29 using (S)-adenosyl-l-methionine (AdoMet or SAM) as the cosubstrate.18 Importantly, PacV was unable to methylate free (2S,3S)-DABA, demonstrating the significance of thioesterification to initiate the pathway. Methylated DABA is then transferred to the free-standing T domain PacH via a transthioesterifiation reaction. The formation of N-methyl-DABA-S-PacH (30) appears to be noncatalytic since the TE domain in PacP does not contain the prerequisite catalytic triad for activity nor does the cluster encode for any other obvious transacylation catalyst. The transthioesterifcation to PacH has been shown to be essential, however, since the standalone A enzyme PacU, which has an unusual dual catalytic function by activating l-Ala and forming the amide bond, is specific for a PacH-loaded substrate as an acyl acceptor to generate the dipeptidyl-S-PacH. For the biosynthesis of most congeners, a mechanistically identical reaction occurs wherein the standalone A enzyme PacW functions in place of PacU; but in this case m-Tyr is activated and condensed to form the dipeptide bound to PacH (31). Rather interestingly, PacU and PacW have a very high sequence similarity (87%) despite the differences in substrate selectivity, and structural studies will likely be necessary toward understanding the specificity difference.
Four genes, pacJ, L, N, and O, are required for the biosynthesis of C-terminal ureido-containing dipeptide and attachment to the α-amine of the DABA moiety. PacL, A tridomain NRPS containing a truncated and presumably non-functional condensation (C) domain, A, and T domain, catalyses the activation of aromatic amino acids (m-Tyr, l-Phe, and l-Trp) and loading to the T domain to form the aminoacyl-S-PacL (32). Importantly, the PacL reaction was shown to be dependent upon the inclusion of PacJ, which has sequence similarity to proteins annotated as MbtH-like proteins.34 The discovery of the ‘auxiliary’ function of PacJ was one of the first of the now many reported examples wherein an MbtH homolog is required for the activity of an A domain.35, 36 In parallel with PacL, the standalone A domain PacO activates and loads l-Ala in trans to the T domain of the C-T didomain protein PacN to form l-Ala-S-PacN (33). When incubated with 32 and bicarbonate, 33 is converted to the ureido-containing dipeptidyl-S-PacN (34) using bicarbonate as the source of the ureido group that effectively reverses the chain orientation.
The pseudo-dipeptide 34 can be condensed with the α-amine of DABA-S-PacH 30 to generate 35, a reaction that is catalysed by PacD, a standalone C domain protein. However, the order of addition to the α- and β-amino functionalities of 30 has not been determined, and it is possible that the biosynthesis proceeds through intermediate 31. Nonetheless, it is known that the pseudo-tetrapeptide that is formed following both condensation reactions remains thioesterified to PacH to generate 36 for further downstream chemistry. If the pacidamycin intermediate is processed to a pentapeptide, PacB catalyses the addition of final l-Ala in a reaction that is dependent upon l-Ala-tRNA.37 PacI, which is yet another standalone C domain protein, uses the modified nucleoside 27 as the nucleophile to release the pseudo-pentapeptide-containing pacidamycins from PacH, thus completing the biosynthetic assembly line. Alternatively, PacI can utilize 36 as an acyl acceptor to generate the pseudo-tetrapeptide-containing pacidamycins. Interestingly, PacI is the first identified C-domain enzyme that has been characterized to condense a peptide and nucleoside component. In addition to using an amine nucleophile that is found in the native acceptor substrate, PacI was shown to be able to utilize uridine or 3’-deoxyuridine as alternate substrates.33
The involvement of a tRNA-dependent process to modify an NRPS-derived peptide is an interesting feature of the biosynthesis of 1-10. The function of PacB was predicted using secondary structural homology predictions, suggesting a similarity to FemABX peptidyl transpeptidases that catalyse tRNA-dependent addition of an amino acid to peptidoglycan. Remarkably, the PacB-catalysed reaction was shown to occur prior to PacI-catalysed release of the peptide from the carrier protein PacH, thereby suggesting an importance for both tRNA and protein (PacH) as elements in substrate recognition. The addition of l-Ala occurred with either 36 or 31 in vitro, further supporting the importance of PacH in substrate recognition. It remains unknown whether Gly is introduced by the same enzyme using the same mechanism, but this seems highly likely due to a lack of alternative candidates for this transformation.
Studies aimed at defining the biosynthesis of the peptide component of 1-10 have uncovered several interesting features with respect to NRPS function and mechanism. The overall nonmodularity of the NRPS system and the requirement for an MbtH-like protein for A domain activity are two such examples. A perhaps less appreciated yet significant discovery upon characterizing this unusual assembly line is the role of PacH, the standalone T domain that harbors the (2S,3S)-DABA component and serves as a key recognition element for minimally five independent proteins: two distinct C domain enzymes that add amino acids to the α- and β-amines of DABA, an N-methyltransferase, a tRNA-dependent transacylase PacB, and the off-loading enzyme PacI. Thus, understanding the molecular details behind the enzyme-PacH interactions may open up future efforts toward pathway engineering.
Peptidyl nucleosides 11-26 likely share the general biosynthetic pathway that has been revealed for 1-10. Nonetheless, a couple of differences have been reported. NpsB, a homologue of which is not found encoded within the 1-10 nor 19-26 biosynthetic gene clusters, has been characterized as an acetyltransferase and is responsible for modification of the N-terminal amino acid to generate N-acetyl-12 (in other words, R1 = acetyl).21 This acetylation modification was proposed to be used as a mechanism for self-resistance. A second, distinct structural feature is the dihydrouracil moiety found in 17 and 18. NpsU, which has sequence similarity to FMN-dependent, pyridoxamine 5’-phosphate synthases, was shown to be responsible for uracil reduction: inactivation of npsU resulted in the abolishment of 17 and 18 production with accumulation of 15 and 16.19
The basic understanding regarding the biosynthetic mechanism for nonribosomally-derived peptides and, more specifically, 1-10, has enabled an innovative combinatorial biosynthetic-semisynthetic approach to access novel Trp-derivatized 4 analogues (Fig. 3).23 The prnA gene encoding a Trp-7-halogenase involved in pyrrolnitrin biosynthesis was introduced into the native 4 producer, which resulted in a strain able to produce a chlorinated 4-analogue 37 along with the standard nonhalogenated congeners. The halogen was subsequently used as a handle for further functionalization using Suzuki-Miyaura-type cross-coupling reactions under mild and aqueous conditions, thereby generating Trp-substituted pacidamycins 38-41.
Figure 3.

Generation of unnatural pacidamycins. A combination of combinatorial biosynthesis via heterologous expression of the foreign halogenase-encoding gene prnA and semisynthesis via Suzuki-Miyaura coupling was used to generate Trp-substituted pacidamycins.
Studies have also been undertaken to better define the regulatory network utilized in peptidyl uridine antibiotic biosynthesis. SsaA in sansanmycin biosynthesis pathway has been identified as a novel class of pathway-specific transcriptional activators.20, 38 SsaA has an N-terminal fork head-associated (FHA) domain and a C-terminal LuxR-type helix-turn-helix domain that was proposed to bind to several putative promoter regions within the gene cluster. As predicted, the inactivation of ssaA greatly reduced the expression of the structural genes required for 19-26 biosynthesis. SsaA DNA-binding ability was shown to be reversed by 19 in a concentration-dependent manner, thus suggesting SsaA controls flux through a mechanism involving feedback inhibition. Finally, PacC (corresponding to NpsO for 15-18 biosynthesis) has high sequence similarity to major facilitator transporters and was proposed to be involved in 1-10 export. The function of PacC, however, has not been experimentally assigned.
2.1.2. Caprazamycin, liposidomycin, A-90289, and muraminomicin.
Liposidomycins (42-52) and the more recently identified caprazamycins (53-59), A-90289s (60 and 61) and muraminomicins (62-71) belong to the lipouridine family of nucleoside antibiotics. The liposidomycins, which were initially discovered from the culture broth of Streptomyces griseosporeus in 1985,39 are potent inhibitors of bacterial peptidoglycan biosynthesis and have notable antibacterial activity against pathogenic strains of Mycobacteria (MIC 1.5-2.0 μg/ml). Compound 44 was shown to directly and potently inhibit E. coli MraY with an IC50 of 0.038 μg/ml.40 The caprazamycins were isolated from Streptomyces sp. MK730F-62F2 in 2003 utilizing a screen aimed at discovering novel anti-tuberculosis antibiotics.41, 42 Muraminomicin F (62) and A-90289s, isolated from Streptosporangium amethystogenes SANK 60709 in 2004 and Streptomyces sp. SANK 60405 in 2010, respectively,43-45 were discovered using a specific activity-based screen for identifying MraY inhibitors and have similar antibacterial spectrum as liposidomycins and caprazamycins. More recently, the muraminomicin family has been expanded upon with the discovery of nine knew compounds from the culture broth of Streptosporangium sp. SANK 60501.44
The lipouridine family share four structural components: a ribose-modified uridine, an aminoribose, a diazapenone ring, and fatty acyl moiety (Fig. 4).40, 42, 43, 45, 46 The first two of these components are combined to form a disaccharide core that is also found in the nucleoside antibiotics FR-90049347 and muraymycins, the latter of which is discussed in the next section. The addition of the diazepanone ring to the disaccharide yields a 3-component product that has been named (+)-caprazol (72).48, 49 Attached to the diazepanone ring of 72 are β-hydroxy fatty acids of different chain length and degree of unsaturation, and structure-activity relationship studies have shown that the nature of the fatty acyl group strongly influences antimicrobial activity.50, 51 The β-hydroxyl of the fatty acid is further modified with an unusual 3-methylglutaryl moiety that is also critical for optimal antibacterial activity. The differences between members of the lipouridine family are in the presence or absence of a permethylated l-rhamnose linked to the 3-methylglutaryl moiety and the incorporation of sulfate on one of the two furanoses. Compounds 62-71 hold the distinction in that they consist of 2-deoxy furanoses and have a unique succinylated heptopyranose in place of the permethylated l-rhamnose.
Figure 4.

Structures of representative nucleoside antibiotics of the lipouridine family. *The sulfate was initially assigned to R2; however, recent data is consistent with sulfonation at R1 (ref. 312).
The gene cluster of each member of the lipouridine member has been identified, sequenced, and, in two instances, heterologously expressed to confirm the genetic identity (Fig. 5A). The gene clusters of 53-59 (cpz) and 42-52 (lpm, the latter from Streptomyces sp. SN-1061M) were cloned and characterized in 2009 and 2010, respectively.52, 53 Heterologous expression of the gene cluster for 53-59 biosynthesis in Streptomyces coelicolor M512 resulted in the production of non-glycosylated, desulfo-caprazamycins, and the boundaries of the cpz gene cluster were subsequently identified by a series of gene deletions using the heterologous production system.54 Sequence analysis initially revealed the cpz gene cluster consists of 23 orfs spanning 28.1-kb DNA, which was later extended to 33.7-kb DNA to include 5 orfs (cpz4–8) involved in the biosynthesis of sulfo-53-59.53 Additionally, a 6.6-kb DNA segment consisting of 7 orfs (cpzDI - cpzDVII) located elsewhere in the genome was determined to be essential for L-rhamnose biosynthesis.55 The insertional inactivation of cpzDIII, encoding for a glucose-4,6-dehydratase, demonstrated its involvement in 53-59 biosynthesis. The gene cluster for 42-52 biosynthesis was located by genome scanning targeting a hypothetical N-methyltransferase similar to Cpz11. A 29.7-kb DNA region containing 25 orfs was identified, and heterologous expression confirmed the identity of the lpm gene cluster.53 The gene cluster for 60 and 61 biosynthesis from Streptomyces sp. SANK 60405 was uncovered, in part, by genome scanning using sequence comparisons with the known cpz gene cluster.56 A 35.3-kb DNA region containing minimally 28 orfs (lipA – lipB1) was identified, nearly all of which were homologous to the genes found in the cpz gene cluster. Gene inactivation of lipB (encoding a putative aryl sulfotransferase near the upstream boundary), lipK (encoding a putative serine hydroxymethyltransferase), and lipB1 (encoding a putative glycosyltransferase near the downstream boundary) using the producing strain of 60 and 61 confirmed the identity of the cluster. Based on the isolation of desulfo-variants from the ΔlipB mutant strain, LipB was determined to be essential for sulfation as predicted from bioinformatics. The gene cluster for 62-71 biosynthesis was localized and cloned in 2013 by using degenerate primers for the gene lipK, which was shown to be essential for the biosynthesis of 60 and 61.57 The gene cluster responsible for 62-71 biosynthesis includes 24 orfs spanning 31.8-kb DNA.
Figure 5.

Biosynthesis of the lipouridine family of nucleoside antibiotics. (A) Genetic organization of the biosynthetic gene clusters. NCBI accession numbers are caprazamycin (cpz) from Streptomyces sp. MK730F-62F2, FJ490409 and HM051054; liposidomycin (lpm) from Streptomyces griseoporeus, GU219978; A-90289 (lip) from Streptomyces sp. SANK 60405, AB530986; muraminomicin (mra) from Streptosporangium amethystogenes SANK 60709, AB746937. (B) Pathway for the biosynthesis of the disaccharide core. (C) Pathway for the biosynthesis of the diazapenone ring and subsequent acylation steps. Proteins labelled in bold blue have been functionally assigned in vitro using recombinant enzymes. Orthologs of the respective caprazamycin proteins are indicated in parenthesis. X (90-93) indicated the different saturated or unsaturated aliphatic chains shown in Figure 4. αKG, α-ketoglutarate; PPi, inorganic pyrophosphate; Succ., succinate; 2-oxo-MTB, 2-oxo-4-methylthio-butanoate; MTA, methylthioadenosine; AdoMet, S-adenosyl-l-methionine; SAH, S-adenosyl-l-homocysteine; PLP, pyridoxal-5’-phosphate; CoA, coenzyme A.; β-ha-CoA β-hydroxyacyl-CoA. *Respective homologs are LpmH, I, R, X, and Y; LipG, H, Q, W, and X; Mra18, 17, 13, 7, and 6.
The biosynthetic steps to the shared disaccharide core were initially defined using recombinant enzymes from the 60 and 61 biosynthetic pathway (Fig. 5B).58-61 Similar to what was discovered for the biosynthesis of the peptidyl uridine antibiotic family, the first intermediate in the pathway is 28. However, in contrast to the flavin-dependent oxidation of uridine catalyzed by PacK and its orthologs, here the pathway begins with UMP in a reaction catalysed by the non-haem, Fe(II)-dependent αKG:UMP dioxygenase LipL.58 LipL has low sequence similarity to TauD, which has been biochemically characterized in extensive detail and is considered the model enzyme for the non-haem, Fe(II)-dependent dioxygenase superfamily. TauD couples the O2-dependent oxidative decarboxylation of α-ketoglutarate (α-KG) to succinate with regiospecific hydroxylation of taurine, the so-called prime substrate for TauD, which leads to the formation of sulphite and aminoacetaldehyde. Similar to TauD, the activity of LipL was determined to be dependent on Fe(II), α-KG, and O2, stimulated by ascorbic acid, and inhibited by several divalent metals. As a result, the mechanism of LipL was proposed to proceed in the mode of TauD by first catalysing C5′-hydroxylation of UMP, which leads to phosphate elimination and concomitant aldehyde formation. With the aim of trapping this hypothetical hydroxylated intermediate, the phosphonate derivative of UMP (UMcP) was utilized as a surrogated substrate with LipL. However, no reaction was observed, which was consistent with the previous biochemical characterization of LipL that demonstrated a high substrate specificity toward UMP. UMcP was instead shown to be a strong competitive inhibitor with respect to UMP (Ki = 800 nM compared to the KM = 14 μM for UMP).62 Subsequent studies with the homologous enzyme Cpr19 involved in capuramycin biosynthesis, the details of which will be discussed in section 2.1.5, suggest LipL and orthologous proteins indeed catalyse a regio- and stereo-selective hydroxylation of UMP as part of the reaction coordinate.62 This cryptic hydroxylation strategy represents a fundamental different dephosphorylation mechanism than the traditional hydrolytic mechanism of phosphatases.
Bifurcation of the pathway occurs upon the formation of 28. In one branch LipK utilizes 28 and catalyses the formation of the high carbon sugar nucleoside (5′S,6′S)-5′-C-glycyluridine (73)60. Based on sequence similarity to serine hydroxymethyltransferases, LipK was initially predicted to catalyse a PLP-dependent aldol-type condensation using Gly as an aldol donor. Although PLP was demonstrated to be essential, biochemical analysis revealed that LipK was instead an l-Thr:28 transaldolase. The formation of 73 was dependent upon the inclusion of l-Thr, and no activity was observed with other amino acids (including d-Thr or l-allo-Thr) as alternative aldol donors. The transaldolase-like reaction produces acetaldehyde while introducing two adjacent stereocenters into 73, which was assigned as the threo diastereomer by modification with phosgene and comparison to authentic product prepared using chemical synthesis.
The other branch from 28 leads to the formation of the aminoribose component, and this pathway is initiated by the aminotransferase LipO.59 LipO has a 37% sequence identity with PacE that is required for the production of 27 (Fig. 2B), and the activity of the recombinant enzyme was confirmed to be a PLP-dependent 28 aminotransferase that generates 5′-amino-5′-deoxyuridine (74). Biochemical characterization of LipO revealed this enzyme utilizes a variety of amine donors, however L-Met is the preferred amine source. Following the formation of 74, the nucleoside phosphorylase LipP catalyses the formation of 5-amino-5-deoxy-1-phospho-α-d-ribose (75). This enzyme chemistry is well known as part of nucleoside salvage pathways but has not previously been reported as a strategy to make sugar-1-phosphates for generating glycosylated natural products. LipM, which has sequence similarity to nucleotidylyltransferases that use sugar-1-phosphates and nucleotide-5′-triphosphates to generate nucleotide-5′-diphosphate (NDP)-sugars, catalyses the formation of UDP-5-amino-5-deoxy-α-d-ribose (76). The activity of LipM is dependent upon the amine functionality within the ribose, with no activity observed using the hydroxylated counterpart. Although other nucleotidylyltransferases (wild-type and engineered, mutant versions) have been found to utilize aminosugars, the catalytic efficiency or specific activity is comparable to or significantly less than the hydroxylated counterpart. The final step that reunites the two branches is catalysed by the ribosyltransferase LipN, combining 73 with the aminoribose component of 76 to form the shared disaccharide core 77. Although the acceptor is likely 73, the functional assignment and biochemical characterization of LipN was performed with uridine as a surrogate acceptor in the initial study.
The core structure of 62-71 consists of 2-deoxyriboses suggesting some potential, interesting differences in the biosynthesis. Notably, from bioinformatic analysis, the biosynthetic gene cluster lacks a gene homologous to lipL. However, homologous enzymes for the remaining steps are encoded within the gene cluster, suggesting a comparable pathway is followed that includes bifurcation from 2′-deoxy-28 (78) and proceeds through the 2′-deoxy variants 79-83. To explore this possibility, Mra20 (homologous to LipP) and Mra23 (homologous to LipM) have been characterized in vitro (Fig. 5B).57 Similar to the respective orthologues, Mra20 catalyses phosphorolysis to initiate “salvage” of the aminoribose to form 81, and Mra23 activates the resulting sugar-1-phosphate for subsequent ribosyl transfer. Prior studies demonstrated LipP has comparable catalytic efficiency with uridine or 74, and similar results with Mra20 were observed using 2'-deoxyuridine and 80. In contrast to LipP, however, the relative catalytic efficiency for 2′-deoxypyrimidine nucleosides was significantly greater than the hydroxylated counterpart (for example, a 7.6-fold increased efficiency with 2′-deoxyuridine relative to uridine was observed with Mra20 compared to 0.05-fold for the respective substrates with LipP). Additional investigation into the substrate specificity of Mra20 revealed a 5-fold increase in catalytic efficiency with the thymidine-containing variant of 80, suggesting the possibility that thymidine is the pyrimidine precursor leading to 81. This realization contradicts the assumption that 78 serves as a precursor for both branches to generate the sugar acceptor 79, which is not expected to originate from a thymidine nucleoside, and the sugar donor 82. While kinetic analysis of Mra20 established the biochemical imperative for incorporation of 2′-deoxysugars, biochemical studies with the nucleotidylyltransferase Mra23 revealed the amine functionality—similar to LipM—was essential for the generation of the NDP-sugar. The results firmly establish a nucleotidylyltransferase family that has absolute discrimination for the amine over the hydroxyl functionality. Furthermore, ribosides are typically generated using 5-phosphoribosyl-1-pyrophosphate as the ‘activated’ sugar, and the formation of an NDP-ribose precursor by Mra20/LipM established a new donor for ribosyl transfer.
The initial biosynthetic step from 77 and 83 to the dimethylated diazapenone of the hypothetical intermediate 72 and the deoxy variant, respectively, was originally hypothesized to involve the transfer of 3-amino-3-carboxypropyl group from AdoMet. The use of AdoMet in such capacity is known to occur in the biosynthesis of the antibiotic nocardicin63, 64 and the plant metabolite nicotianamine and related metallophores.65-67 Decarboxylated AdoMet is also the direct precursor for 3-aminopropyl group transfer during polyamine biosynthesis in several microorganisms.68 Sequence and structural analysis of these and other known amino(carboxy)propyltransferases demonstrate they have clear similarity to the AdoMet-dependent methyltransferase (MTase) superfamily. The gene cluster for the biosynthesis of 60 and 61 encodes two candidate proteins with similarity to MTases, LipH and LipW, and it was speculated that one of these catalyse 3-amino-3-carboxypropyl group transfer while the other catalyses one or both N-methylations of the diazapenone. In contrast to these expectations, however, neither methyltransferase-like protein is involved in 3-amino-3-carboxypropyl group transfer. Instead, LipJ, which has sequence similarity to plant 1-aminocyclopropane-1-carboxylate synthase, was recently shown to transfer 3-amino-3-carboxypropyl from AdoMet onto 3”-O-phospho-77 (84).69 The phosphoryl group was essential, suggesting the requirement for a phosphotransferase to catalyse the formation of the cryptic intermediate 84. Although the responsible phosphotransferase involved in the biosynthesis of muraymycin (section 2.1.3) was characterized, no such homologous gene is encoded in the gene clusters. Nonetheless, the generation of 84 is consistent with the isolation of 3”-O-phospho-72 from the caprazamycin producer following inactivation of a putative acyltransferase (cpz23, homologous to lipT).70 Presumably, the biosynthesis of 62-71 involves the comparable phosphorylation of 83 to generate the proposed pathway intermediate 85. LipJ function and mechanism is discussed in more detail in section 2.1.3.
Further processing following aminocarboxypropyl incorporation to generate 86 (or 87) requires not only the aforementioned methylations but also hydroxylation, a reaction potentially catalysed by LipG, a putative β-hydroxlyase, and intramolecular amide bond formation, potentially catalysed by LipQ, a putative acyl-CoA synthetase. Acylation of 88 (or 89 for 62-71 biosynthesis) with a β-hydroxy fatty acid is likely catalyzed by the aforementioned Cprz23/LipT, which has modest sequence similarity to lipases and carboxyesterases of the SGNH-hydrolase family. Subsequent acylation of the β-hydroxy functionality of the fatty acid of dephosho-90 (91) with the unusual 3-methylglutaryl component has also been established as the last step toward the synthesis of desulfo-42-52. Gene inactivation of cpz21 (lipR), whose gene product has sequence to enzymes of the α/β-hydrolase fold, led to the isolation of 91 in a heterologous production strain, also suggesting that dephosphorylation occurs at some point following the formation of 84 and prior to the Cpz21-catalyzed reaction. Although Cpz21 likely uses a CoA-acylated substrate as the donor to generate 42-51, the identity of the cosubstrate remains unknown.55 It is envisioned that comparable acylations (to make 92 and 94) and dephosphorylation (to make 93) occurs during 62-71 biosynthesis.
Formation of the permethylated L-rhamnose found in many of the lipouridine antibiotics is initiated by enzymes encoded by four genes (cpzDII, cpzDIII, cpzDVI and cpzDIV) located outside the gene cluster.55 Their participation in caprazamycin biosynthesis was confirmed by insertional inactivation of cpzDIII in the producing strain, yielding a strain that was unable to produce 53-59 but instead the non-glycosylated variants that were also isolated using the heterologous production system. When these four genes were introduced into heterologous producer along with the gene cluster, 53-59 production was restored. Based on bioinformatics analysis, CpzDII is a dTDP-glucose synthase, CpzDIII, a dTDP-glucose 4,6-dehydratase, CpzDVI, a 3,5-epimerase, and CpzDIV, a 4-ketoreducase, that together generate a TDP-l-rhamnose precursor from the primary metabolite, glucose-1-phosphate. Following the formation of the activated sugar, the rhamnosyltransferase Cpz31, which was expressed and verified in vitro, attaches the L-rhamnose to the 53-59 aglycone. Subsequently, three putative methyltransferases, Cpz28 (LipY), Cpz29 (LipZ) and Cpz30 (LipA1), are candidates for sequential O-methylation of the sugar to yield 53-59. These methylations likely occur following rhamnosyltransfer based on precedence from other permethylated glycosides. Two notable variations of this pathway are found in 62-71 biosynthesis, wherein the rhamnose is C-methylated at C6 to generate a heptose and acylated with succinate instead of alkylated at the 3-hydroxyl. As expected, the gene cluster for 62-71 biosynthesis does not encode an orthologue to the proposed 3-O-methyltrasferase Cpz28, but instead the sugar biosynthetic subcluster contains two additional genes, one encoding a putative C-methyltransferase of the radical SAM superfamily (Mra19) and the other a membrane-associated succinyltransferase (Mra20).
In addition to the permethylated rhamnose, a distinctive feature of certain lipouridine antibiotics is the sulfate functionality. Typically, sulfonation occurs by an enzyme family that utilize 3’-phophoadenosine-5’-phosphsulfate (PAPS) as a sulfonate (SO3−) donor. However, no such member of this protein family is encoded within the gene clusters. In contrast a gene encoding a putative arylsulfate sulfotransferase, a small group of PAPS-independent enzymes that are known to utilize an arylsulfate as a SO3− donor and an aryl substrate acceptor, was uncovered near a gene (cpz6) encoding a putative type III polyketide synthase (PKS) just upstream of the proposed boundary. Inactivation of the gene lipB56 or cpz471 encoding this putative arylsufate sulfotransferase from the 60 and 61 and 53-59 gene clusters yielded a mutant strain that produced desulfo-nucleosides, consistent with a role of this gene product in sulfonation. In vitro characterization of LipB revealed it catalysed 2′-O-sulfonation of uridine and desulfo-A-90289s using p-nitrophenylsulfate as an artificial SO3− donor.56 Mechanistic investigations with the LipB-ortholog Cpz4 confirmed this enzyme as the ultimate sulfotransfer catalyst and revealed, for the first time, a non-aryl acceptor substrate for this enzyme type (Fig. 6).71 Like all arylsulfate sulfotransferases, however, the identity of the in vivo SO3− donor remained a mystery. Realizing that type III PKS are known for the production monocyclic aromatic metabolites, the possibility that the PKS product was involved in the SO3− relay was explored. Following gene inactivation and heterologous production, Cpz6 was demonstrated to produce a series of novel aromatic triketides that were termed presulficidins exemplified by presuflicidin A (95).72 Additionally, cpz8, encoding for a hypothetical protein with secondary structure predictions suggesting similarity to mammalian SULTs yet lacking a hallmark PAPS binding motif, was cloned and expressed to reveal the recombinant protein was in fact a PAPS:95 sulfotransferase that generates sulficidin (96). Thus, a novel pathway and role for a polyketide as a genuine SO3− donor was discovered, wherein SO3− is shuttled from PAPS through 96 to form desulfo-nucleosides.
Figure 6.

Sulfonation mechanism using a novel polyketide as a sulfate shuttle. Proteins labelled in bold blue have been functionally assigned in vitro using recombinant enzymes.
The sulfonation of the lipouridine renders the antibiotic inactive against model bacterial strains, suggesting sulfonation may play a role in self-resistance. Furthermore, two proteins LipI and LipX have similarity to TmrB, a tunicamycin resistance protein discussed in 2.1.6. A final, putative resistance mechanism involves transport by LipS, a probable ABC transporter.
2.1.3. Muraymycin and sphaerimicin.
The muraymycins (97-115) were isolated from Streptomyces sp. NRRL 30471 in 2002 73, 74 using a screen to identify inhibitors of peptidoglycan biosynthesis. Sphaerimicin A (116) was isolated from Sphaerisporangium sp. SANK 60911 in 2013,75 and, in contrast to muraymycins, was initially targeted for characterization based solely on the potential that the genomic DNA encoded for proteins involved in nucleoside antibiotic biosynthesis. The crystal structure of Aquifex aeolicus MraY was solved with 111 bound, thus enabling the first structure-based model for nucleoside antibiotic binding to the transmembrane protein.76 Both 97-115 and 116 have modest antibiotic activity against Staphylococci (MIC 2-16 μg/mL), Enterococci (MIC 2-64 μg/mL), and several Gram-positive bacteria. Nucleosides 97-115 also possess activity against several clinically relevant Gram-negative bacteria (MIC 8-64 μg/mL), while sphaerimicin is inactive against representative Gram- negative bacteria.75, 77
The structures of 97-116 consist of the same disaccharide core that is a defining characteristic of the lipouridine family of nucleoside antibiotics (Section 2.1.2) and are often categorized as such (Fig. 7). The primary difference between 97-115 and 116, which also differentiates them from the lipouridine family, are the appendages to the disaccharide core. Nucleosides 97-115 contain a pseudo-tripeptide in place of the diazapenone. Like the peptidyl nucleoside antibiotics, the term “pseudo” is used due to the ureido function that reverses the orientation of the peptide sequence, in this instance yielding a peptide with two carboxy termini. The Leu of the pseudo-tripeptide can be non-hydroxylated (D series of congeners) or hydroxylated (C series), and hydroxylated Leu can be further acylated with guanidinium-containing acyl groups (B series) or saturated fatty acids (A series). Compound 116 contains an unusual, dihydroxy-piperidine ring attached to the aminoribose that is further acylated with a highly reduced, branched polyketide side chain.
Figure 7.

Structures of representative muraymycins and sphaerimicin A.
The biosynthetic gene cluster for 97-115 was identified in 2011 utilizing degenerate primers designed to amplify the sequence of LipK/Cpz14 (l-Thr:28 transaldolase),which produces the proposed biosynthetic intermediate 73.78 This probe was used to identify a single cosmid spanning 43.4-kb DNA, and bioinformatics analysis revealed 33 orfs, 26 of which (mur11-mur36 are likely involved in the biosynthesis (Fig. 8A). The identity of the genetic locus was verified via individual inactivation of two genes, mur16 and mur17, which abolished the production of 105 and 106, the major congeners isolated from the wild-type strain in this study. Similar to the process for identifying the 97-115 gene cluster, the genetic locus for 116 biosynthesis was identified using a distinct set of degenerate primers designed for amplifying an l-Thr:28 transaldolase.75 Contrastingly, however, the process was performed without prior knowledge of the existence and structure of 116. Sequencing of four overlapping cosmids revealed a 44.7-kb DNA region consisting of 33 orfs (sphA-sphW) proposed to be involved in 116 biosynthesis (Fig. 8A).
Figure 8.

Biosynthesis of the muraymycin nucleoside antibiotics. (A) Genetic organization of the biosynthetic gene clusters. NCBI accession numbers are muraymycin (mur) from Streptomyces sp. NRRL 30471, HQ257512; and sphaerimicin (sph) from Sphaerisporangium sp. SANK 60911, AB830104. (B) Pathway for the biosynthesis and convergence of the disaccharide core and pseudo-tripeptide. Proteins labelled in bold blue have been functionally assigned in vitro using recombinant enzymes. A, adenylation; C, condensation; T, thiolation; MTA, methylthioadenosine; AdoMet, S-adenosyl-l-methionine; PLP, pyridoxal-5’-phosphate.
The gene clusters for 97-115 and 116 predictably encodes for protein orthologs used to assemble the disaccharide core of the lipouridine family (Fig. 5B). Most of the biosynthetic steps to produce the disaccharide core of 97-115 have now been defined using recombinant enzymes, thus confirming the shared biosynthetic route.79 To summarize the pathway begins with the conversion of UMP to 28 catalysed by Mur16 (LipL homolog), a non-haem Fe(II), αKG-dependent dioxygenase. The carbon chain of 28 is extended in a reaction catalysed by Mur17 (LipK), a PLP-dependent L-Thr:28 transaldolase that forms the high carbon sugar nucleoside 73. The biosynthesis of the aminoribose also involves 28 as an intermediate with Mur20 (LipO), a PLP-dependent aminotransferase, catalysing transamination to form 74. In contrast to LipO, which prefers L-Met as the amine donor,59 Mur20 preferentially utilizes l-Tyr although has significant activity with a variety of other amine donors. The uracil component of 74 is removed by Mur26 (LipP), a nucleoside phosphorylase, to yield the sugar-1-phosphate 75, followed by Mur18 (LipM)-catalysed nucleotidyl transfer yielding the activated sugar 76. Mur19 (LipN) uses 76 as a sugar donor, transferring the amino sugar to the acceptor 73 to form the ADR-GlyU disaccharide 77. Unlike LipN, which was characterized with uridine as a sugar acceptor, Mur19 was directly shown to use 73.79 Based on bioinformatic analysis suggesting the 116 gene cluster encodes for homologous proteins, the disaccharide core is proposed to be biosynthesized in the same manner. In support of this, the gene product SphJ (Mur17/LipK/Cpr14 homologue) was biochemically confirmed to stereoselectivity catalyse the conversion of 28 to 73 (Fig. 5B).
Mur28, a 3″-O-phosphotransferase, catalyses the phosphorylation of muraymycins, and single-substrate kinetic analysis revealed a clear preference for the biosynthetic intermediate 77 over 111.80 Thus, phosphorylation of the disaccharide is proposed to occur at this stage in the biosynthesis, leading to 84 (Fig. 8B). The alkyltransferase Mur24 (LipJ) catalyses aminobutryl group transfer.69 As previously noted, Mur24 and homologs have sequence similarity to PLP-dependent ACC synthases that utilize AdoMet to form 1-aminocylopropyl-1-carboxylate and MTA. The reaction catalysed by ACC synthases involves formation of an external aldimine between PLP and AdoMet, leading to Cα-deprotonation and intramolecular attack on Cγ to eliminate MTA and form the cyclopropyl ring (Fig. 8B).81 Similar to ACC synthase, preliminary mechanistic studies with Mur24 suggest the reaction is initiated by the formation of an external aldimine followed by Cα-deprotonation.69 However, subsequent chemistry involves a second deprotonation at Cβ that promotes elimination of MTA. Following C-S bond breakage, the C-6’ amine of 84 attacks Cγ in an aza-Michael-type addition, leading to a new C-N bond and—following hydrolysis of PLP—the pathway intermediate 86. The γ-substitution reaction catalysed by Mur24 is followed by Mur23-catalyzed decarboxylation to yield the aminopropyl-containing 117. The decarboxylation, which also requires a PLP cofactor, is the diverging point for the biosynthesis of muraymycins.
The pseudo-tripeptide of 97-115 is highlighted by a ureido functionality and two nonproteinogenic amino acids, β-OH-l-Leu (found in the A-C series) and epicarpreomycidine (118), the latter of which is likely derived from l-Arg. Sequence analysis of the gene cluster uncovered six genes believed to encode proteins containing domains of NRPS and were thus proposed to be involved in the biosynthesis of the pseudo-tripeptide (Fig. 8B). Similar to the peptidyl uridine antibiotics, the NRPS-related proteins consist of a disjointed, nonmodular architecture whose specific functions were not entirely obvious based solely on sequence. Additionally, epicapreomycidine appears to be biosynthesized by a mechanism that is different than established for the diastereomer capreomycidine, which is found in viomycin and capreomycin.82, 83 Capreomycidine biosynthesis involves sequential catalysis by a non-haem Fe(II)- and αKG-dependent l-Arg dioxygenase and a PLP-dependent dehydratase:cyclase,84-86 and the nonproteinogenic amino acid is subsequently incorporated into the peptide by an NRPS.27, 35, 87, 88 However, no such homologous proteins are encoded in the 97-115 gene cluster. A mechanism was proposed wherein l-Arg is converted to 118 catalysed in part by Mur15, a member of the cupin 4 family of proteins.78 Mur12, consisting of a C, A, and T domain, is predicted to activate and load 118. In parallel, the adenylation enzyme Mur27 is predicted to activate and load l-Val to a discrete T domain-containing protein, Mur14. The two thioesterified amino acids, along with a bicarbonate unit, are then condensed by an unclear mechanism that potentially follows the known ureido-incorporation strategy reported for SyrC involved in syringolin A biosynthesis.89 Mur13, a single C domain-containing protein, is potentially involved in this condensation. Mur21, consisting of an A and T domain, activates and loads l-Leu that putatively undergoes hydroxylation following thioesterification, initially proposed to be catalysed by the aforementioned dioxygenase Mur1678 but is likely catalysed by an as-of-yet unidentified hydroxylase. Mur25, a C domain protein, is proposed to catalyse the condensation of thioesterified β-OH-L-Leu with the pseudo-dipeptide to create the Mur21-linked pseudo-tripeptide. Mur30, which has similarity to class C β-lactamases, is proposed to catalyse the coupling of the acyl acceptor 117 with the pseudo-tripeptide. Dephosphorylation occurs prior to or following the condensation of the nucleoside and pseudo-tripeptide components to generate the C series compounds 105, 110, 112, and 115. Finally, the β-OH-L-Leu, if present, is acylated to make the most potent muraymycins of the A and B series. Like the other steps occurring after the biosynthesis of 117, the acylating enzyme has not been functionally assigned.
The dihydroxypiperidine of 116 is a unique functionality in natural products and consequently expected to involve unusual catalytic transformations. Not surprisingly, the gene cluster contains a set of genes (sphM-sphT) that are not found in clusters for other nucleoside antibiotics with the identical nucleoside core. The first modification of the disaccharide core, however, is the same as 97-115: a PLP- and AdoMet-dependent alkyltransferase SphJ (Mur24/LipJ homologs) catalyses aminobutryl group transfer to a phosphorylated disaccharide.69 The transfer is strictly dependent on the phosphate functionality, yet no phosphotransferase homolog is encoded in the 116 gene cluster. The remaining steps include extension of the carbon chain, hydroxylation, and two C-N bond formations. A modular type I PKS within two large proteins (SphU and SphV) was uncovered that consists of a domain architecture and functional predictions that are consistent with the synthesis of the highly reduced, branched polyketide component of 116. The piperidine ring is likely acylated with this polyketide in a reaction catalysed by SphW, which has sequence similarity to C domains of NRPS. SphW possesses 28% sequence similarity to PacI that is responsible for the condensation between the peptide and nucleoside component during 1–10 biosynthesis. Finally, there is a lack of a clear candidate for sulfonation of the 3′-OH of 116, which is not surprising given the different regiochemistry than that found in the lipouridine antibiotics. However, an unshared gene encoding a putative arylsulfatase (SphB) is located at one boundary of the biosynthetic gene cluster and may be involved in this reaction.
Self-resistance toward 97-115 is proposed to occur in part by covalent modification and transport. For the former, Mur29 has been characterized as a muraymycin D series 3"-O-adenylyltransferase that yields products with significantly lower MraY inhibitory activity and antibiotic activity. The latter includes Mur31, which has sequence similarity to proteins of the major facilitator superfamily of transporters. The 116 gene cluster encodes for an ABC transporter-like protein SphD that potentially is involved in self-resistance by efflux transport. Finally, Mur34 was functionally assigned as a negative regulator, wherein deletion of the gene yielded a Δmur34 mutant strain with 10-fold increased production of 97-115.90
2.1.4. Jawsamycin.
Jawsamycin (119), also known as FR-900848, from Streptomyces roseoverticillatus HP-891 was first reported in 1990.91 Nucleoside 119 has excellent activity against several filamentous fungi, initially believed to be a consequence of selective inhibition of cholesteryl ester transfer protein (CETP). Very recent evidence has revealed that 119 targets Spt14, the catalytic subunit of a UDP-glycosyltransferase that catalyses the first step in glycosylphosphatidylinositol biosynthesis.92 The structure of 119 consists of 5′-amino-5′-deoxy-5, 6-dihydrouridine attached via an amide bond to an unusual carbon chain containing multiple cyclopropyl and unsaturated units (Fig. 9). U-106305 (120) from Streptomyces sp. UC 11136 has a similar structure and has been shown to directly inhibit CETP in vitro.93 The relative and absolute stereochemistry has been supported by extensive NMR analyses of the natural products along with total synthesis using stereoselective cyclpropanation.94, 95
Figure 9.

Structure of jawsamycin and U-106305.
The gene cluster for 119 was reported in 2014.96 The whole genome was sequenced and the biosynthetic locus identified, in part, by searching for genes encoding a modular PKS, predicted to be involved in the biosynthesis of the cyclopropyl-containing chain, as well as orthologues to LipL and LipO, proteins previously demonstrated to convert UMP to 74 during the biosynthesis of the lipouridines 60 and 61.58, 59 The identity of the gene cluster was confirmed by heterologous expression and shown to span approximately 13.3-kb DNA consisting of nine orfs (jaw1-jaw9) including two gene products with sequence similarity to domains of PKS (Jaw4 and Jaw6) and two with sequence similarity to LipL and LipO (Jaw7 and Jaw8, respectively) (Fig. 10A).
Figure 10.

Biosynthesis of jawsamycin. (A) Genetic organization of the biosynthetic gene cluster. NCBI accession number is jawsamycin (jaw) from Streptomyces roseoverticillatus HP-891, AB920328. (B) Pathway for the biosynthesis and convergence of the disaccharide core and pseudo-tripeptide. Proteins labelled in bold blue have been functionally assigned in vitro using recombinant enzymes. aKG, a-ketoglutarate; Succ., succinate; ACP, acyl carrier protein; 2-oxo-MTB, 2-oxo-4-methylthio-butanoate; PLP, pyridoxal-5’-phosphate.
The nucleoside core of 119 was shown to be derived from UMP as expected from the bioinformatics analysis (Fig. 10B). Reactions using recombinant proteins demonstrated Jaw7 is a non-haem Fe(II)-dependent αKG:UMP dioxygenase that produces 28 and Jaw8 is an aminotransferase that converts the LipL product to 74. Similar to LipO, Jaw8 preferentially utilizes l-Met as an amino donor, which is unusual for PLP-dependent aminotransferases.
Jaw2 has sequence similarity to GCN5 N-acyltransferases and was therefore predicted to couple 74 with the polycyclopropanated and unsaturated carbon chain, which is likely thioesterified to the acyl carrier protein (ACP) domain of the PKS Jaw4. Using the recombinant protein, Jaw2 was shown to catalyze a Mg2+-dependent transacylation using stearoyl-CoA as a substitute acyl donor.96 Further biochemical characterization revealed this enzyme utilizes a broad range of aliphatic acyl-CoAs as donors. Similarly to the low specificity for the acyl donor, multiple acyl acceptors were substrates for Jaw2. Nonetheless, 74 was the preferred acceptor in comparison to 5,6-dihydro-74, suggesting that uracil reduction occurs following amide bond formation. This conclusion was supported by biotransformation of dehydro-119 (121) into 119 upon heterologous expression of jaw1 in Streptomyces lividans TK23. Interestingly, Jaw1 has no sequence similarity to the enzyme NpsU catalyzing an identical reduction during the biosynthesis of 17 and 18.
Prior to the identification of the gene cluster, isotopic enrichment studies suggested that 119 and 120 are constructed using a fatty acid or polyketide biosynthetic mechanism with the methylenes of the cyclopropyl groups derived from l-Met.97, 98 As previously noted, the gene cluster harbors two proteins with sequence similarity to domains of PKS, supporting a polyketide origin. Jaw4 is an iterative PKS consisting of sequential ketosynthase, acyltransferase, dehydratase, and ACP domains, and Jaw6 has sequence similarity to 3-ketoacyl-(ACP) reductases that are typically found embedded as a domain of a PKS. Jaw5 has similarity to enzymes of the radical SAM superfamily, and therefore was proposed to catalyse cyclopropylation. The function of these gene products were interrogated through feeding 74 or dihydro-74 to an S. lividans TK23 strain expressing jaw2, jaw4, jaw5, and jaw6 (Fig. 10B).96 Together, these genes were able to produce 119 from either precursor. The PKS system is notably unusual by employing a trans-acting ketoreductase that catalyzes reduction following each extension of the polyketide chain. The probable cyclopropanase, Jaw5, catalyses either cyclopropanation directly after dehydration (Fig. 10B, route a) or after a fully extended polyene is produced (Fig. 10B, route b).94, 97 Evidence for the former, i.e. a stepwise cyclopropanation mechanism, was primarily provided by feeding and incorporation of an N-acetlycystamine thioester of a deuterated, cyclopropyl-containing diketide to the producing strain, leading to the production of labelled 119.97 Additional evidence was provided by an in-frame deletion of jaw5, which abolished production of 119, and no polyunsaturated-containing analogues were detected in the mutant strain despite in vitro data showing that Jaw2 has low substrate specificity.96 Finally, a more recent and thorough examination of the heterologous producing strain revealed the biosynthesis of 23 analogues of 119 with varied polyketide components and, when jaw2 is deleted, 25 analogues of dehydro-119 with similar polyketide variations.99 The structure and yield of these polyketide variants were consistent with a stepwise elongation/cyclopropanation mechanism wherein the first cyclopropanation step can be skipped to yield a terminal conjugated diene. Furthermore, the results confirmed the low acyl donor specificity of the coupling enzyme, Jaw2, and highlighted the potential for using this enzyme as a biocatalyst to make 119 derivatives.
2.1.5. Capuramycin.
Capuramycin (initially named 446-S3; 122) was isolated from Streptomyces griseus 446-S3 in 1986 and shown to have antibiotic activity against Streptococcus pneumoniae (MIC 12.5 μg/ml) and Mycobacterium smegmatis (MIC 3.13 μg/ml).100 While screening for MraY inhibitors, a series of 122 derivatives named A-500359s (123-130) were isolated in 2003 from Streptomyces griseus SANK 60196.101, 102 A-500359 A (123) and A-500359 B (124), the latter of which was subsequently determined to have the same structure as 122, were shown to have IC50 values (18 nM and 17 nM, respectively) comparable to the peptidyl uridine, lipouridine, 97-115, and 116 nucleoside antibiotics.101 The screening campaign also led to the discovery of additional 122 derivatives from distinct strains including A-503083s (131-134) from Streptomyces sp. SANK 62799103 and A-102395 (135) from Amycolatopsis sp. SANK 60206.104
The structures of 122-134 are characterized by three distinct moieties: a uridine-5′-carboxamide (136) nucleoside core, an unsaturated hexuronic acid appended to 136 via a glycosidic bond, and an l-aminocaprolactam (l-ACL) bonded to the hexuronic acid via an amide bond (Fig. 11).105 A few of the discovered capuramycin analogues lack the l-ACL moiety and, as a result, have diminished activity against MraY.103, 106 The two major de-l-ACL analogues terminate with the hexuronic acid as a carboxylic acid (denoted with an F, 128 and 134) or as the methyl ester (denoted with an E, 127 and 133). In contrast to the other members of the capuramycin family, A-102395 (135) has an arylamine-containing polyamide instead of the l-ACL moiety. Although having potent MraY inhibition with IC50 of 11 nM, 135 lacks antimicrobial activity likely due to poor permeability of bacterial membrane.
Figure 11.

Structures of the capuramycin family of nucleoside antibiotics.
Shortly after the discovery of 123-130, the metabolic origin of each component was interrogated using feeding experiments with isotopically labelled precursors.107 The observed incorporation patterns from this study was consistent with the l-ACL, hexuronic acid, and 136 components originating from l-Lys, d-mannose, and a combination of uridine and pyruvate (potentially via phosphoenolpyruvate), respectively. More recent results from the in vitro characterization of recombinant enzymes along with additional feeding experiments are consistent with UMP and l-Thr as the metabolic precursors of 136.61, 108
In 2009 the gene cluster for 123-130 was identified by using an abundantly transcribed NDP-glucose dehydratase (NGDH) gene that was amplified using degenerate primers.109 The gene cluster was predicted to cover approximately 35-kb DNA consisting of a minimum of 24 orfs (orf7-orf30) (Fig. 12A). The orf35, orf36 and orf37 genes were predicted to encode proteins with putative regulatory function, so could not be excluded as essential for 123-130 biosynthesis. Since a genetic system could not be developed, reverse transcriptase PCR (RT-PCR) of the orfs was examined using high-producing, low-producing, and null mutant strains—generated by random chemical mutagenesis—to support the identity of the gene cluster. Genes orf8, encoding a putative truncated carbamoyltransferase, and orf26, encoding a tridomain NRPS, were used as probes to identify the 131-134 gene cluster consisting of 21 homologous orfs annotated as capA-capW.110 The biosynthetic gene cluster for 135 was identified using degenerate primers to amplify a conserved gene encoding an l-Thr:28 transaldolase (at the time, the function of the gene product was unknown and the protein was annotated as a serine hydroxymethyltransferase), which led to the isolation of several overlapping cosmids encompassing 85-kb DNA consisting of 70 orfs (cpr1-cpr70).108 Through bioinformatic analysis, orf17-orf51 were proposed to be essential for 135 biosynthesis.
Figure 12.

Biosynthesis of the capuramycin family of nucleoside antibiotics. (A) Genetic organization of the biosynthetic gene clusters. NCBI accession number are A-500359s (orf) from Streptomyces griseus SANK 60196, AB476988; A-503083s (cap) from Streptomyces sp. SANK 62799, AB538860; and A-102395 (cpr) from Amycolatopsis sp. SANK 60206, KP995196. (B) Pathways for the biosynthesis and convergence of the nucleoside core, the unsaturated hexuronic acid, and aminocaprolactam. The aminocaprolactam is attached via an unusual transacylation reaction catalyzed by ORF24 (CapW or Cpr27), wherein the methyl ester is converted to an amide. (C) Pathway for the biosynthesis of the polyamide component found in A-102395. Proteins labelled in bold blue have been functionally assigned using recombinant enzymes. Orthologs of the respective A-500359 biosynthetic proteins are indicated in parenthesis. PLP, pyridoxal-5’-phosphate; DABA, diaminobutyric acid; A, adenylation; C, condensation, T, thiolation. Abbreviations are αKG, α-ketoglutarate; PPi, inorganic pyrophosphate; Succ., succinate; PLP, pyridoxal-5’-phosphate; AdoMet, S-adenosyl-l-methionine; SAH, S-adenosyl-l-homocysteine; A, adenylation; C, condensation, T, thiolation; ACP, acyl carrier protein.
The first two steps for the biosynthesis of the 136 nucleoside core were unexpectedly found to be identical to that for the lipouridine family of nucleoside antibiotics (Fig. 12B). A gene encoding a protein with high sequence similarity to the dioxygenase LipL was found in all three clusters (Orf7, CapA, and Cpr19, respectively). Cpr19 was functionally assigned as a non-haem, Fe(II)-dependent αKG:UMP dioxygenase catalysing the first step toward 136 synthesis, the formation of 28 from UMP108 The catalytic properties of Cpr19 are similar to LipL except, as previously noted, Cpr19 can regio- and stereospecifically hydroxylate the phosphonate derivative of UMP, which provided evidence for a hydroxylation-phosphorylase mechanism.62 The l-Thr:28 transaldolase (Orf14, CapH, and Cpr25) catalyzes the following step, conversion of 28 to 73. CapH was shown to have virtually indistinguishable properties, including substrate specificity and stereochemical control, when compared to the LipK homolog.60, 108
The conversion of 73 to 136 requires decarboxylation of an α-amino acid, oxygen incorporation, and oxidation. In search of a potential decarboxylase, the protein ORF12’/Cap15, with low sequence similarity to l-seryl-tRNA(Sec) selenium transferase, was identified and targeted for characterization since it was the only remaining protein encoded within the gene cluster that was predicted to be a PLP-dependent enzyme. In contrast to expectations, however, Cap15 was shown to catalyse a PLP- and O2-dependent conversion of (5′S,6′R)-73 (137) directly to 136.111 An atom of O2 is incorporated into 136 that, along with additional biochemical properties, supported the functional assignment of Cap15 as a PLP-dependent 137:O2 monooxygenase-decarboxylase. Preliminary mechanistic studies were consistent with a hydroperoxide mechanism (Fig. 13) wherein the Cα of the external aldimine between PLP and 137 is deprotonated to generate a PLP-stabilized carbanion (137a), an electron is transferred to O2 to form superoxide (137b), which, following protonation, recombines with the carbon-centered radical to form a hydroperoxide (137c). Decarboxylation with elimination of water generates 136 following release of PLP. Cap15 is part of a growing group of enzymes that rely solely on PLP as a redox cofactor for monooxygenation of a substrate.112 The discovery of Cap15 suggests a single catalytic step remains uncharacterized in the CarU-biosynthetic pathway, the isomerization of 73 (the product of ORF15/CapH/Cpr25) to 137, the substrate for ORF12′/Cap15/Cpr23.
Figure 13.

Proposed mechanism of PLP-dependent 136:O2 monooxygenase-decarboxylase Cap15.
The biosynthesis of the hexuronic acid moiety is unclear despite involving seemingly straightforward chemistry. Among the shared proteins encoded within the gene cluster, it was proposed that a putative NDP-hexose 3-ketoreductase (CapC and homologs), a clavaminic acid synthase-like protein (CapD), an NDP-hexose 2,3-dehydratase (CapE), and an NDP-hexose 4-epimerase/dehydratase, are responsible for construction of the unsaturated hexuronic acid (138) beginning with the hypothetical precursor GDP-mannose (Fig. 12B). A glycosyltransferase (CapG and homologs), which has modest sequence similarity to the ribosyltransferases involved in lipouridine biosynthesis (Fig. 5), transfers the activated hexuronic acid to 136 to form 3’-O-desmethyl-128 (130). Although the true identity of the sugar acceptor and donor is currently unknown, it is noteworthy that the glycosyltransferase reaction proceeds with retention of configuration at the anomeric carbon, which is less common than inverting glycosyltransferases in natural product biosynthesis.
Two predicted NRPS proteins (ORF27/CapV and ORF26/CapU) are encoded in the 123-134 gene clusters. Bioinformatic analysis suggests ORF26/CapU is a tri-domain NRPS with C, A, and T domains. However, the C domain contains a point mutation of a predicted, critical His residue, suggesting it is catalytically inactive. Using in vitro assays, the A domain of CapU was found to activate several amino acids but with a preference toward l-Lys as expected.113 Further characterization revealed that CapU alone catalyzed the formation of l-ACL in a reaction that was dependent upon the posttranslational modification of the T domain with the phosphopantetheine group and independent of the N-terminal C domain. Interestingly, the reaction occurred at that same rate regardless of the inclusion ORF27/CapV, which is predicted to consist of a single, active C domain. Thus, it appears that the release of l-ACL occurs via noncatalytic intramolecular aminolysis of a thioesterified l-Lys. Consequently, the function of ORF27/CapV, if any, is unknown.
The attachment mechanism of the l-ACL moiety was serendipitously discovered in 2011.110 ORF28/CapW/Cpr51, which has sequence similarity to class C β-lactamases that are members of the serine protease family that specifically hydrolyze β-lactams, was initially targeted as a possible l-ACL hydrolase due to the bioinformatics prediction. Recombinant CapW indeed converted 132 to a new product, however the identity was revealed to be the glyceryl ester of 134, suggesting CapW catalyzes a transacylation (amide-ester exchange reaction) using the glycerol that was included as a protein stabilizing agent. Given that the methyl ester 133 is isolated from the producing strain, comparable reactions were performed using methanol in place of glycerol, and CapW slowly converted 132 to 133. The transacylation reaction rapidly proceeded using 133 as an acyl donor and l-ACL as an acyl acceptor, and the Ser predicted to be involved in the formation of an acyl-enzyme intermediate—similar to other members of the serine protease family—was shown to be essential for CapW activity. Thus, CapW was assigned as a transacylase catalyzing ATP-independent amide bond formation. The function was confirmed by generating a ΔcapW mutant strain that abolished production of 131 and 132 but not 134, and feeding the ΔcapW mutant strain with l-ACL restored the production of 131 and 132.
The functional assignment of CapW initiated a search for an enzyme capable of generating the methyl ester of 133, CapS was verified in vitro to catalyze carboxyl-methylation thereby converting 134 to 133. It is unclear whether CapS (and by extension, CapW) operates before or after the 2′- or 3’-hydroxyl modifications of 136, and is therefore depicted as a 130:AdoMet carboxymethylatransferase generating 139. CapW, a 139:l-ACL transacylase, subsequently generates 129 that, along with 130, are isolated in minor amounts from the producing strain. Additional studies revealed a low specificity of CapW with respect to the acyl acceptor, enabling a biocatalytic approach to generate several l-ACL-substituted capuramycin analogues starting from 134.114 Alternatively, feeding different unnatural acyl acceptors to the ΔcapW mutant strain established a mutasynthetic approach for generating l-ACL-substituted capuramycin analogues. Finally, in addition to amide-ester exchange, CapW was able to catalyze direct transamidation in vitro starting with 132 and excess, unnatural acyl acceptors.
Three different tailoring steps are possible following the formation of the capuramycin scaffold 129. CapB, a predicted carbamoyl transferase not encoded in the 123-130 and 136 gene clusters, was shown to convert 124 to 132 in vitro, thus, confirming its function as a 2′-O-carbamoyltransferase.110 The predicted O-methyltransferase, CapK and homologs ORF16’ and Cpr29, are proposed to catalyze the methylation of the C3’-hydroxyl, yet the timing of the CapK-catalyzed reaction versus CapW is unknown. Finally, ORF25/CapT, which has sequence similarity to proteins of the radical SAM superfamily, is likely responsible for the stereoselective methylation of l-ACL following the CapW-catalyzed transacylation.
The unique arylamine-containing polyamide found in 135 is likely assembled from gene products of two subclusters of genes. The predicted biosynthesis of the 3-(4-aminophenyl)-2,3-dihydroxypropanoic acid component (140) requires 8 orfs (cpr12, cpr32-cpr38) and utilizes chorismic acid and malonyl-CoA as precursors. In the proposed pathway, Cpr38 and Cpr12 convert chorismic acid to p-aminobenzoic acid (141), both well characterized steps of the folic acid biosynthetic pathway. Interestingly, the gene encoding for Cpr12, which has sequence similarity to p-aminobenzoate synthetase component I that is responsible for l-Gln amidohydrolysis as the amine source, was initially believed to be outside of the gene cluster. Cpr37, which has sequence similarity to A domains that activate aryl amino acids, then activates and loads 141 to the predicted T domain protein, Cpr36. Following thioesterification of 141, Cpr33, which has sequence similarity to ketoreductases, likely catalyzes reduction; and Cpr32, which has similarity to flavin-dependent monooxygenases of the LLM class, catalyzes α-hydroxylation to give 142. The remaining steps in the biosynthesis of 140 require the formation of two amide bonds, the precursors of which are believed to be Gly and l-Asp and catalysed by some combination of Cpr39-Cpr50. Cpr51 has moderate sequence similarity to CapW, suggesting the coupling of 140 likely follows the same transacylation mechanism to convert 139 to 3’-O-desmethyl-135. However, the dramatic structural and chemical differences in the acyl acceptors (aryl amine of 135 and α-amine of l-ACL) warrants further interrogation into the role of Cpr51.In addition to several common biosynthetic steps, the capuramycin-type antibiotics also share an identical mechanism of self-resistance. An ATP-dependent capuramycin phosphotransferase (ORF21, CapP and Cpr17) regio-specifically transfers the γ-phosphate of ATP to the 3”-hydroxyl of the hexuronic acid moiety.108, 115 Kinetic analysis revealed that CapP employs a sequential kinetic mechanism and has a strong preference for l-ACL-containing capuramycins over the deaminocaprolactam variants. Further kinetic analysis with Cpr17 revealed an unexpected preference toward GTP as the phosphate donor. These biochemical characteristics appear to be consistent between homologs. The phosphorylation event yields capuramycins with significantly increased IC50 values against the MraY target and abolished antimycobacterial activity, consistent with phosphorylation as a self-resistance mechanism. Other potential resistance may be conferred by two putative ABC transporters, ORF19/CapN and ORF20/CapO.
2.1.6. Tunicamycin, streptovirudin, and corynetoxin.
Tunicamycins (143-152) were first isolated from Streptomyces lysosuperificus ATCC 31396 in 1971.116, 117 Since this time, 143-152 have been isolated from several other strains of Streptomyces, including Streptomyces chartreusis NRRL 3882, Streptomyces LA-507, and Streptomyces niger NRRL B-3857.118 Tunicamycins are usually produced as a mixture of multiple congeners and are structurally classified as N-acylated disaccharidyl nucleosides (Fig. 14). These congeners differ in their fatty acid-derived acyl side chains or substitution pattern of the uracil base. Other compounds that make up the tunicamycin-type of nucleoside antibiotics include streptovirudins (153-162), 119 corynetoxins (163-176),120 mycospocidins,116 and the antibiotics MM19290 and 24010, 121 all produced by different strains of actinomycetes.
Figure 14.

Structures of the tunicamycin family of nucleoside antibiotics. The unique tunicamine component is highlighted in blue.
The discovery of 143-152 was guided by their antiviral activity against Newcastle disease virus.117 Additional biological testing demonstrated 143-152 to have activity against a broad spectrum of viruses, Gram-positive bacteria, and fungi.122-125 Mechanistic studies revealed 143-152 inhibit the biosynthesis of lipid-linked intermediates found in the construction of several complex carbohydrates, including the bacterial peptidoglycan cell wall.122, 126 Similar to many of the aforementioned nucleoside antibiotics, 143-152 has been demonstrated to inhibit MraY via reversible, competitive inhibition.127 The structure of MraY bound to 144 was recently solved and revealed a direct interaction of tunicamycin with catalytic residues that have been deemed critical for MraY activity.13, 128, 129 In eukaryotes, 143-152 inhibit eukaryotic protein N-glycosylation by binding to UDP-N-acetylglucosamine:dolichol phosphate N-acetylglucosamine-1-phosphate transferase, which results in cytotoxicity against mammalian cells that is not generally observed in other MraY inhibitors.123, 130 Due to its ability to inhibit eukaryotic protein N-glycosylation, 143-152 are heavily used as a laboratory tool to induce endoplasmic reticulum stress.
The primary structural feature that differentiates the tunicamycin family of nucleoside antibiotics is the presence of an N-acetylglucosamine (GlcNAc) moiety linked to a unique 11-carbon aminodialdose sugar called tunicamine (Fig. 14).4, 116, 121, 131 The 4′-amine of tunicamine is acylated with different saturated, mono-unsaturated, or hydroxylated fatty acids of varying lengths and with unique branching patterns, and is the primary difference between the members of the family. Streptovirudins can also vary by having a dihydrouracil (158-162) in place of uracil (153-157). Feeding experiments with [1-13C]-d-glucose and [6,6-2H]-d-glucose implicated uridine and UDP-GlcNAc as the likely precursors of tunicamine and suggested the sugar-derived components were coupled in an unusual tail-to-tail fashion.132 Radiochemical-based feeding experiments using [2-14C]uridine and d-[1-14C]glucosamine provided additional support for the biosynthetic origins of 143-152. The results also implicated a second condensation of UDP-GlcNAc with tunicamine to form the rare α,β-1,1-glycosidic bond of the pseudo-trisaccharide scaffold. Recently, a series of tunicamycin-type nucleoside antibiotics named quinovosamycins were discovered that have an N-acetylquinovosamine substituting for the tunicamine-attached GlcNac (Fig. 14).133 As a consequence of the feeding experiments, 28 was hypothesized to be a potential biosynthetic precursor, similar to the aforementioned nucleoside antibiotics.
The biosynthetic gene cluster for 143-152 from S. chartreusis NRRL 3882 was identified and reported independently in 2010 by two groups using different approaches to locate the genetic locus (Fig. 15A).134, 135 One utilized a bioinformatics search for clustered genes encoding a potential glycosyltransferase and N-deacetylase, among other known nucleoside antibiotic biosynthetic genes. This approach led to a single contig that was confirmed as the biosynthetic gene cluster using heterologous expression in Streptomyces coelicolor. Bioinformatic analysis suggested a single operon with 14 orfs (tunA-tunN) within 12-kb DNA was involved in the construction of 143-152. Contrastingly, the second approach did not rely on bioinformatics, but instead utilized random heterologous expression of a cosmid library in Streptomyces lividans TK24 followed by an antibacterial screen against Bacillus subtilis. This activity-guided approach yielded three cosmids that enabled production of 147 and 149 based on mass spectroscopic analysis of the culture extracts. Several independent groups have identified and characterized the gene cluster following the initial two reports.118, 136, 137 The gene cluster boundary was established by inactivation of tunA and tunL, which generated strains unable to produce 143-152. Inactivation of the upstream orf-1 and downstream orf+1 had no effect on production. Therefore, the minimal set of genes consists of twelve orfs, tunA-tunL, spanning 12-kb DNA, although a possible role for tunN could not be excluded.
Figure 15.

Biosynthesis of tunicamycins. (A) Genetic organization of the biosynthetic gene cluster. NCBI accession number is tunicamycin (tun) from Streptomyces chartreusis NRRL 3882, HQ111437. (B) Pathway for the biosynthesis of the trisaccharide core and subsequence acylation/deacylation steps. Proteins labelled in bold blue have been functionally assigned in vitro using recombinant enzymes; the structure of TunA [Protein Data Bank (PDB) 3VPS] has been solved. PPi, inorganic pyrophosphate; AdoMet, S-adenosyl-l-methionine.
Coinciding with the identification of the gene cluster in 2010, two similar pathways for the early steps of 143-152 biosynthesis were proposed. Following in vitro characterization of TunA and TunF and additional heterologous expression studies, the pathway has now been refined as shown in Fig. 15B. The pathway begins from UTP, and two distinct types of phosphatases, TunG and TunN, catalyze sequential dephosphorylation to generate uridine (Fig. 15B). These two gene products have subsequently been shown to be dispensable, presumably due to cross complementation of enzymes involved in the host primary metabolism.136 In parallel TunA and TunF, a putative UDP-GlcNAc epimerase/dehydratase and a UDP-GlcNAc 4-epimerase, respectively, have been show to sequentially catalyze the conversion of UDP-GlcNAc to the exo-glycal, UDP-6-deoxy-5,6-ene-GlcNAc 177: TunA first catalyzes the NAD+-dependent formation of UDP-6-deoxy-5,6-ene-GlcNAc (178) followed by TunF-catalyzed C-4″ epimerization to yield 177.138, 139 TunB, which has sequence similarity to proteins of the radical SAM superfamily, is proposed to catalyze the coupling of uridine with 177 to generate the characteristic tunicamine-containing nucleoside core 179. TunM, which has sequence similarity to AdoMet-dependent methyltransferases and was initially proposed to be involved in this step, has been re-assigned as a putative resistance-conferring protein that is not essential for the biosynthesis of 143-152.136
The later steps involve four enzyme-catalyzed reactions that have been shown to be essential for biosynthesis in vivo.136 TunH, a putative pyrophosphatase, is proposed to hydrolyse UDP from 179, to generate 180. TunD, which has sequence similarity to group 1 family glycosyltransferases, next catalyzes glycosylation of 180 with GlcNAc thereby generating the pseudo trisaccharide scaffold 181. TunE, a putative GlcNAc-phosphatidylinositol de-N-acetylase, catalyzes deacetylation at the C-10′ hydroxyl of tunicamine to generate 182, which is re-acylated by TunC that has sequence similarity to GCN5-related N-acetyltransferase. A stand-alone acyl carrier protein TunK is encoded within the gene cluster and TunL, a hypothetical phosphatase, have been hypothesized to work with TunC during the N-acetylation process. However, both TunK and TunL have been demonstrated to be nonessential for biosynthesis.
As previously noted, two of the enzymatic steps (TunA and TunF) have been characterized in vitro.138 TunA has closest sequence similarity to proteins annotated as TDP-glucose-4,6-dehydratases, enyzmes that operate by using an NAD+-dependent redox cycle involving minimally three steps: (1) oxidation at C-4″, (2) dehydration at C-6″, and (3) reduction of the resulting 5″-6″ double bond via a 1,4-conjugate addition of a hydride to furnish a 4-keto-6-deoxysugar (Fig. 16). In contrast to the bioinformatics prediction, TunA was demonstrated to convert UDP-GlcNAc to the exo-glycal product, UDP-6″-deoxy-5″,6″-ene-GlcNAc. Preliminary biochemical and mechanistic evaluation of TunA was consistent with an NAD+-dependent mechanism that parallels the first two steps of TDP-glucose-4,6-dehydratases, however the regeneration of NAD+ in the final step is accompanied by reduction of the 4″-keto functionality (Fig. 16). The structure of TunA bound with NAD+ and UDP-GlcNAc revealed that most active site residues are shared with 4,6-dehydratases, yet the relative orientation of the substrate and cofactor is subtly shifted thereby enabling the appropriate hydride delivery to C-4″ as opposed to C-6″ in the final step. A comparison of the structures revealed that TunA has a unique hydrogen bond between the carboxamide of the nicotinamide and the backbone amide of a Val residue (Asn in 4,6-dehydratases), which differentially rotates the nicotinamide ring relative to the co-substrate UDP-GlcNAc and possibly accounts for the different regiochemistry of hydride addition. TunF was demonstrated to have 4-epimerase activity toward several substrates including UDP-GlcNAc, UDP-6″-deoxy-5″,6″-ene-GlcNAc, and their respective galacto-epimers. However, single-substrate kinetic analysis revealed TunF was seven times more efficient with UDP-6″-deoxy-5″,6″-ene-GlcNAc, establishing this enzyme as an ‘exo-glycal epimerase’. The results set the stage for exploring the proposed TunB-catalyzed intermolecular C-C bond formation between C-5’ of uridine and C-6” of the exo-glycal of UDP-6-deoxy-5,6-ene-GalNAc, a mechanism that potentially resembles the recently discovered intramolecular C-C bond forming step in nikkomycin and polyoxin biosynthesis (section 2.1.8).
Figure 16.

Comparison of the mechanisms for 4,6-dehydratases and TunA. Depicted in blue are catalytic residue conserved between the two enzymes. The last step in the mechanism for 4,6-dehydratses is shown with dashed areas and unique residues in red.
A 143-152 resistance protein has been isolated from Bacillus substilis and other microorganisms.140 This transmembrane protein, annotated as TmrB, is proposed to function as a kinase, thereby inactivating 143-152 via phosphorylation.141, 142 In vitro studies have demonstrated TmrB can bind ATP, yet the phosphorylation activity has never been reconstituted for unknown reasons.143 Nonetheless a TmrB homolog is not encoded within the gene cluster, suggesting a distinct mechanism of self-resistance for the producing organism Indeed, two unrelated transporter-like proteins, TunI and TunJ, are encoded within the cluster that have been shown to impart self-resistance along with the aforementioned TunM.136 Further experimentation will be necessary to fully elucidate the function of TunI, TunJ, and TunM and their role in resistance.
2.1.7. A-94964.
A-94964 (183), isolated from the culture broth of Streptomyces sp. SANK 60404, was discovered in 2008 during a screen to identify MraY inhibitors.144 An IC50 value of 1.1 μg/ml was determined against TL1 in vitro, and subsequent screening revealed 183 to have antibacterial activity against of Staphylococcus aureus and Enterococcus faecalis with MIC of 100 and 50 μg/ml, respectively. The structure of 183 consists of a high-carbon sugar nucleoside that is glycosylated and further modified with an N-acylated sugar (Fig. 17).145 Noteworthy structural features that distinguishing A-94964 from other nucleoside antibiotics include 1) an unusual octo-uronic acid in place of a typical ribose within the nucleoside core, 2) a hybrid fatty acid-polyketide-derived acyl chain that is connected to the sugar with a glycine linker, and 3) the presence of a phosphoric acid diester connecting the nucleoside core to the N-acylated sugar. A similar phosphoric acid diester moiety is found in moenomycin, antibiotics that also inhibit peptidoglycan biosynthesis but through a different mechanism of action.146, 147
Figure 17.

Structure of A-94964.
The biosynthetic gene cluster for 183 was identified by searching the whole genome for homologs of tunE, which encodes a 181 N-deacetylase.148 A single contig was identified, and sequencing out from the TunE homolog (initially denoted orf17) revealed 23 open reading frames were identified including two, orf8 and orf9, that are to be homologous to tunI and tunJ, respectively. To confirm the role of this genomic locus in 183 biosynthesis, two cosmids, both containing orf9 and orf17, were separately introduced into the heterologous host Streptomyces albus G153 to generate recombinant strains capable of 183 production. Using gene deletions with heterologous production, the minimum biosynthetic gene cluster was determined to be orf5–19 spanning ~20.8-kb of genomic DNA. The open reading frames deemed essential for 183 biosynthesis were renamed to anb1-15 (Fig. 18A).
Figure 18.

Biosynthesis of A-94964. (A) Genetic organization of the biosynthetic gene cluster. NCBI accession number is A-94964 (anb) from Streptomyces sp. SANK 60404, LC431526. (B) Pathway for the biosynthesis of the nucleoside core and acylation with a polyketide putatively derived from the indicated polyketide synthase. T, thiolation; ER, enoylreductase; KS, ketosynthase; AT, acyltransferase; KR, ketoreductase; DH, dehydratase.
The biosynthetic pathway for 183 has been proposed based on bioinformatic predictions and preliminarily supported by gene inactivation experiments (Fig. 18B).148 The nucleoside core (184) is believed to originate from UMP and pyruvate using three enzymes Anb1 (a predicted dehydrogenase), Anb2 (a flavin-dependent monooxygenase), and Anb10 (oxidoreductase). Inactivation of anb10 abolished production of 183, and no intermediates accumulated, consistent with an early role in the pathway. This data, along with the lack of any candidates with sequence similarity to proteins involved in the extension of the ribose carbon chain for the other uridine-derived nucleoside antibiotics, suggests a fundamentally distinct mechanism for generating a high-carbon sugar nucleoside. A mutant with a deleted anb11 gene was also generated, which led to the isolation of 184, demonstrating the pathway intermediacy of 184. Anb11, which is predicted to have an N-terminal phosphotransferase domain and a C-terminal domain with similarity to proteins annotated as hypothetical, was proposed to transfer phospho-GlcNAc from UDP-GlcNAc to 184, generating the pathway intermediate 185. Deacetylation by Anb13, the aforementioned TunE homolog, and Abn12-catalyzed re-acylation with Gly—presumably in an ATP-dependent reaction—yields the pathway intermediate 186. The existence of 186 was supported by gene inactivations, wherein deletion of anb8, anb9, or anb3 yielded a strain that produced 186. Anb8, predicted to have three domains (ketosynthase, ketosynthase, and acyltransferase, from N-terminus to C-terminus), and Anb9, predicted to have two domains (ketoreductase and dehydratase), were proposed to work with Anb6, a predicted acyl carrier protein, and Anb7, a predicted enoylreductase, to generate the acyl donor. Anb3, which has low sequence similarity to proteins that function as N-acetyltransferases, is proposed to catalyse the acyl transferase reaction to afford the penultimate intermediate 187. Finally, Anb15, which has low sequence similarity to ArnT that catalyses glycosylation of lipid A, is proposed to catalyse the final step to generate the final product 183. Finally, Anb4 and Anb5, the aforementioned TunI and TunJ homologs, are predicted to contribute to self-resistance by facilitating 183 transport.
2.1.8. Nikkomycin and polyoxin.
Nikkomycins (188-203) were isolated from Streptomyces tendae Tü 901 in 1976,149, just over a decade after polyoxins (204-219) were first isolated from the Streptomyces cacaoi var. asoensis in 1965.150 Analogues of both have subsequently been isolated from several other strains including Streptomyces aureochromogenes and Streptomyces ansochromogenes 7100.151-154 This family of nucleoside antibiotics has a distinct biological activity relative to others: they are selective, competitive inhibitors of chitin synthase and, as a result, have fungicidal, insecticidal, and acaricidal activities.155-159 Members of this family are particularly active against various phytopathogenic fungi such as Botrytis cinerea (MIC 5 μg/mL for nikkomycins), Rhizopus circinans (MIC 1 μg/mL for nikkomycins), Alternaria kikuchiana (MIC 0.8 μg/mL for polyoxins) and Piricularia oryzae (MIC 6.25 μg/mL for polyoxins).158, 160-162 Nikkomycins are also highly effective at killing Coccidioides, the causative agent of Valley fever, and there remains some effort at developing nikkomycins to treat this rare disease.163-166 Members of this family exhibit low toxicity towards mammals and bees and are easily degraded in nature; as such, both are useful in agriculture as antifungal agents.
The shared structural feature of 188-219 is an aminohexuronic acid in place of a typical ribose within the nucleoside core (Fig. 19).167-171 Nucleosides 188-203 consist primarily of two series of congeners termed the Z and X series, consisting of a uracil base (for example, nikkomycin Z, J, and LZ; 188-195) or a 4-formyl-4-imidazolin-2-one base (for example, nikkomycin X, I, and LX; 196-203), respectively. While a uracil base is also found in some 204-219 congeners, 5-(hydroxymethyl)uracil, uracil-5-carboxylic acid, and thymine bases are also commonly observed. The 204-219-producing strain S. cacaoi var. asoensis was also identified as a producer of nikkomycin-type compounds with an imidazolidinone base, and these metabolites were named neopolyoxins (for example, polyoxin N, 219).170
Figure 19.

Structures of nikkomycin and polyoxin family of nucleoside antibiotics.
Nucleosides 188-203 and 204-219 also differ in other components bound to the aminohexuronic acid. Nucleosides 188-203 contain a nonproteinogenic amino acid, typically 4-(4′-hydroxy-2′-pyridinyl)-homothreonine, appended to the nucleoside core via an amide bond at the C-5′ amine, while 204-214 and 219, contain the unusual amino acid named carbamoylpolyoxamic acid. Furthermore, some congeners for both have been isolated with an amino acid bonded at the C-6′ carboxylate through a second amide bond. For the nikkomycins this amino acid moiety is l-Glu or l-homoserine represented by 189 and 193, respectively. For polyoxins this moiety is another unusual amino acid that has been named polyoximic acid, found in 204207 and 218.
The metabolic origin of 188-219 was initially examined by utilizing feeding experiments with isotopically labelled substrates. The isotope enrichment was consistent with the shared aminohexuronic acid of the nucleoside core originating through the condensation of a ribonucleoside with phosphoenolpyruvate (PEP) or related metabolite that is derived from pyruvate.172-176 For 188-203 the data suggested that the nucleoside base originates from l-His or uracil, and the precursor of the pyridyl component of the 4-(4′-hydroxy-2′-pyridinyl)-homothreonine moiety is derived from l-Lys.177, 178 l-Ile and l-Glu are the proposed precursors for the two modified amino acids polyoximic acid and carbamoylpolyoxamic acid, respectively, in 204-219.175, 176 In 1996 the gene cluster for the biosynthesis of 188-203 from Streptomyces tendae Tü901 was cloned and expressed in the heterologous host, Streptomyces lividans TK23 (Fig. 20A).179 The minimal gene cluster was determined by gene inactivation and found to include 23 orfs and span 29-kb genomic DNA. Genes found to be essential to for the biosynthesis from S. tendae Tü901 were annotated ‘nik’. Nucleosides 188-203 were subsequently isolated from S. ansochromogenes 7100, and the genes from this producer were annotated ‘san’ (for clarity, when discussing the biosynthesis, the genes will be described with respect to the nik annotation). The gene cluster required for the biosynthesis of 204-219 from Streptomyces cacaoi was identified by searching for a gene homologous to nikO, the gene product of which was demonstrated to be essential for the shared aminohexuronic acid moiety.180 The putative gene cluster was confirmed by heterologous expression in Streptomyces lividans TK24, and genetic analysis revealed the biosynthesis involves 20 putative pol genes encompassing 24-kb DNA.
Figure 20.

Biosynthesis of the nikkomycin and polyoxin family of nucleoside antibiotics. (A) Genetic organization of the biosynthetic gene clusters. NCBI accession number are nikkomycin (nik) from Streptomyces tendae Tü 901, AJ250581, AJ250878, AJ251438, Y18574; nikkomycin (san) from Streptomyces aureochromogenes, AF322179, AF469956, MF055656; polyoxin (pol) from Streptomyces cacaoi var. asoensis, EU158805.(B) Pathway for the biosynthesis of the imidazolinone base found in the nikkomycin X series. (C) Pathway for the biosynthesis of the nucleoside core. During the conversion of 223 to 225, C3 of phosphoenolpyruvate (indicated with l) is retained. (D) Pathway for the biosynthesis of the nonproteinogenic amino acid component of nikkomycins and attachment to nucleoside core. (E) Pathway for the biosynthesis of the nonproteinogenic amino acid component of polyoxins and attachment to nucleoside core. Proteins labelled in bold have been functionally assigned in vitro using recombinant enzymes; the structures of NikD (PDB 3VPS) and NikO (PDB 4FQD) have been solved. Orthologs of the respective nikkomycin biosynthetic proteins are indicated in parenthesis. #, MbtH-like protein; A, adenylation; T, thiolation; TE, thioesterase; AMT, aminotransferase; αKG, α-ketoglutarate; Succ., succinate; αKB, α-ketobutyrate; PRPP, phosphoribosylpyrophosphate; PEP, phosphoenolpyruvate; dA, 5’-deoxyadenosine; PPi, inorganic pyrophosphate; CoA, coenzyme A.
A combination of in vivo and in vitro approaches have been used to determine the role of many of the nik genes.181, 182 The biosynthesis of the imidazole base of the nikkomycin X series is initiated by three enzymes, NikQ, NikP1 and NikP2 (Fig. 20B).183 Gene disruption of nikQ, a putative cytochrome P450 monooxygenase, had no effect on Z series 188-195 production yet abolished the production of X series 196-203. The production 196 was restored by feeding the imidazolinone base, suggesting nikQ is essential for its biosynthesis. Functional characterization of NikQ was further carried out in vitro with two additional enzymes: NikP1, a tridomain NRPS that includes A and T domains and an unusual N-terminal domain with similarity to MbtH proteins, and NikP2, a putative type II (free-standing) TE.184 The A domain of NikP1 was shown to activate and load l-His to the T domain to generate His-S-NikP1. NikQ utilizes the thioesterified-His intermediate to catalyze an O2-dependent β-hydroxylation to form β-OH-His-S-NikP1. The reaction catalyzed by NikQ is both regio- and stereospecificity and does not occur with free L-His. NikP2 subsequently catalyzes thioester hydrolysis to release (2S,3R)-β-OH-His (220) from NikP1. The identity of the enzymes catalyzing the remaining steps, which includes oxidation of the imidazole ring and a retro-aldol-like reaction to simultaneously generate Gly and the imidazolinone base (221), are still unknown.
NikR, which has sequence similarity to uracil phosphoribosyltransferases involved in pyrimidine salvage pathways, is responsible for the transfer of 221 to 5-phosphoribosyl-1-pyrophosphate (PRPP), yielding the nucleoside monophosphate 222 (Fig. 20C). The activity of NikR was confirmed using uracil, and it remains to be seen whether 222 is an efficient substrate.183 The homologous proteins are not encoded in the polyoxin gene cluster, suggesting that 219 is assembled by proteins encoded elsewhere in the genome or the biosynthesis is accomplished by a different mechanism.
Feeding experiments were consistent with the extension of the carbon chain of ribose following the formation of the nucleoside. NikO (PolA), which has sequence similarity to enolpyruvyltransferases, was shown to catalyze the transfer of an enolpyruvyl moiety from phosphoenolpyruvate to UMP.185, 186 The transfer of the pyruvoyl group occurred at the 3′-OH group of the nucleoside core to generate 3′-O-enoylpyruvyl-UMP (223), the regiochemistry of which is similar the reaction catalyzed by MurA as a step in peptidoglycan cell wall biosynthesis. The X-ray crystal structure and modeling studies of NikO, as well as site-directed mutagenesis, indicate that NikO and MurA share important conserved active site residues to direct C-O bond formation at the 3′-OH of a sugar.186 PolA, the homolog of NikO involved in 204-219 biosynthesis, has been verified to have the same function.180 Furthermore, the nikO gene was revealed to be able to cross-complement the polA mutant strain, confirming identical functions.
Gene inactivation experiments have indicated that nikI (polG), nikJ (polH), nikK (polI), nikL (polJ), nikM (polK), and nikN (polQ1) are involved in the processing of 223 to form 217 (also known as nikkomycin CZ) and the respective 221-containing nucleoside called nikkomycin CX.181 NikJ (PolH), which has sequence similarity to proteins of the radical SAM family, was functionally assigned as the enzyme responsible for C-C bond formation during carbon chain extension of 223 to generate 5′-O-phospho-octosyl acid nucleoside 224.187-189 In contrast to predictions that C5’-extension proceeded through a C-5′-aldehyde intermediate similar to the biosynthesis of 41-70, 96-115, and 121-134, the characterization of NikJ, which included analysis by electron paramagnetic resonance spectroscopy, was consistent with a radical-based mechanism wherein the reaction starts by formation of a 5′-deoxyadenosine radical from AdoMet that is common among the radical SAM family, followed by the abstraction of a C-5′ hydrogen atom from the primary substrate 223 (Fig. 21). The carbon-centered radical then attacks C-3″ to generate the new C-C bond, and the radical is quenched by hydrogen abstraction of a side-chain Cys residue to generate 224. Site-directed mutagenesis of C209 of PolH to either C209A and C209S gave mutant proteins that produced a relatively small amount of 224 and epi-224, consistent with C209 serving as a reducing agent in the last step. How the thiyl radical is ultimately reduced, and the identity of the exogenous reductant, is unknown.
Figure 21.

Mechanism of NikJ and PolH. Abbreviations are AdoMet, S-adenosyl-l-methionine; 5-dA, 5’-deoxyadenosinie.
The remaining steps for converting 224 to 217 (or nikkomycin Cx) require dephosphorylation, C-C and C-O bond breaking steps, decarboxylation, oxidations, and transamination. Of the unassigned enzymes predicted to be involved in generating 217 or nikkomycin Cx, only NikK, a putative PLP-dependent aminotransferase, has been partially characterized in vitro.190 NikK was able to catalyze PLP-dependent transamination with l-Ala as an amine donor and αKG as an amine acceptor, and several amino acids functioned as amino donors when half-transamination reactions were examined. However, the true amine acceptor was not determined, and it is believed that NikK converts the ketohexuronic acid 225 to 217/nikkomycin Cx using l-Glu as the amino donor, although this specific activity has not been confirmed.
The biosynthesis of 4-(4′-hydroxy-2′-pyridinyl)-homothreonine of 188-203 is initiated by NikC, which catalyzes PLP-dependent transamination of the α-amino group of l-Lys to generate piperideine-2-carboxylic acid (226) (Fig. 20D).191 NikD, which has sequence similarity to monomeric sarcosine oxidases, subsequently catalyzes a four-electron oxidation to convert 226 to picolinic acid (227).192-194 The reaction requires an FAD cofactor, which is covalently bound to NikD, and concomitantly reduces two moles of molecular oxygen to two moles of hydrogen peroxide. Biochemical studies have shown that NikD requires a cyclic C-N bond but not the double bond (imine) functionality. For example, NikD catalyzes the oxidation of l-pipecolate and l-Pro as alternative substrates, although the products of the reaction were not rigorously characterized. Following the formation of 227, NikE uses ATP to activate the carboxylic acid to form picolinoyl-CoA (228).195, 196 NikA, which has sequence similarity to acetaldehyde dehydrogenases, was shown to catalyze an NADH-dependent reduction of benzoyl-CoA to form benzaldehyde in vitro.197 By extension, NikA is assumed to reduce 228 to generate the pathway intermediate piconaldehyde (229). NikB catalyzes the C-C bond forming step using 229 as an aldol acceptor and 2-oxobutyrate as an aldol donor, resulting in the generation of 4-(2'-pyridinyl)-2-oxo-4-hydroxyisovalerate (230).198 Based on the isolation of 191, 192, 199, and 200, NikB also catalyze the aldol reaction using pyruvate as an aldol donor. NikE potentially has a second function in the biosynthetic pathway by activating/loading 230 to the T domain of NikT, a didomain protein with an N-terminal T domain and a C-terminal aminotransferase (AMT) domain.195, 199 Once loaded, NikT presumably catalyzes transamination to generate a thioesterified 4-(2’-pyridinyl)-homothreonine-S-NikT (231). However, NikT was shown to have low aminotransferase activity with 230 in the absence of NikE, thus forming the free acid of 231.199 Therefore it cannot be concluded whether thioesterification of 230 is essential. The enzyme responsible for coupling 231 with 217/nikkomycin Cx remains unknown, although NikS, which has similarity to proteins of the ATP grasp superfamily, has been proposed to fulfill this function to generate.200 Finally, gene inactivation of nikF, encoding a putative cytochrome P450 protein, resulted in the production of 195 and 203 that contain 4-(2′-pyridinyl)-homothreonine instead of the hydroxylated counterpart, 4-(4′-hydroxy-2′-pyridinyl)-homothreonine.201, 202 Thus NikF, likely in combination with NikG, putative ferredoxin protein, is proposed to be responsible for the final hydroxylation step to form 188 and 196, the major nikkomycin congeners isolated from the producing strain.
The biosynthesis of the polyoximic acid and carbamoylpolyoxamic acid components of 204-207 and 218 has been delineated using the aforementioned feeding experiments, in vivo gene inactivations,203 and/or most recently, in vitro characterization of the recombinant enzymes (Fig. 20E). The biosynthesis of carbamoylpolyoxamic acid originates from l-Glu and is proposed to involve five proteins, PolL-PolP.204 PolN, which has similarity to N-acetyltransferases of the GNAT family, catalyzes transfer of an N-acetyl group for acetyl-CoA to l-Glu to form N-acetyl-l-glutamate (232), a reaction that proceeds through a seryl-enzyme intermediate. PolP, which has sequence similarity to acetylglutamate kinases (ArgB), then catalyzes phosphorylation of 232 to form N-acetyl-l-glutamoyl phosphate (233). PolM, which has sequence similarity to N-acetyl-γ-glutamyl-phosphate reductase (ArgC), catalyzes an NADPH-dependent reduction of 233. Unlike ArgC that catalyzes a two-electron reduction to generate N-acetyl-l-glutamate 5-semialdehyde (234), PolM catalyzes 2 sequential two-electron reductions to generate 2-acetamido-5-hydroxyvaleric acid (235) via 234 as an intermediate. PolN was determined to have a second function in the biosynthetic pathway by catalyzing transacetylation with 235 as the donor and l-Glu as the acceptor to form 2-amino-5-hydroxyvaleric acid (236) and 232. PolO, which has similarity to carbamoyltransferases, converts 236 to 2-amino-5-O-carbamoylhydroxyvaleric acid (237). PolL, which has sequence similarity to non-haem Fe2+- and αKG-dependent dioxygenases, catalyzes two sequential hydroxylations, first 4-hydroxylation to generate 2-amino-4-hydroxy-5-O-carbamoylhydroxyvaleric acid followed by 3-hydroxylation to form the nonproteinogenic amino acid carbamoylpolyoxamic acid (238) that is characteristic of polyoxins. Finally, PolG, which has sequence similarity to NikS and other members of the ATP grasp superfamily, catalyzes ATP-dependent amide bond formation between 238 and 215, 216, 217, or 221 to give 208, 212, 213, or 219, respectively.205
As previously noted, isotopic enrichment experiments are consistent with l-Ile as the precursor of polyoximic acid. The pathway putatively involves three proteins: PolF, a putative molybdopterin oxidoreductase, PolC, a putative non-haem Fe2+- and αKG-dependent dioxygenase, and PolE, a hypothetical protein. There is no clear candidate for the amide bond-forming enzyme that catalyzes the ligation of polyoximic acid and 208, 209, 212, or 213 to make the final product, 204-207, respectively.
The regulatory network controlling the biosynthesis of 188-219 has been partially characterized. One study demonstrated that production of the respective metabolites can be significantly increased with the introduction of multiple copies of sanG (orfR for the nik cluster)/polR into the producing strain.206, 207 The gene product of sanG was subsequently revealed to function as an ATPase/GTPase that positively regulates nikkomycin biosynthesis by binding to the sanN–sanO intergenic region. Another study of 204-219 biosynthetic regulation showed that polY, encoding a protein that consists of an OmpR-like DNA binding domain at the N-terminus and a central ATPase domain, is essential for biosynthesis.208 Additional experiments indicated that PolY bound to the promoter region of polR, thereby upregulating polyoxin production by activating the transcription of other operons in the gene cluster. Gel retardation assays indicated that PolR has specific DNA-binding activity for the promoter regions of polC and polB, consistent with the regulatory prediction.207 Finally, the genes polQ1 and polQ2 are two co-transcribed transport genes encoding membrane proteins that are predicted to be involved in polyoxin transport.180 Neither gene product has been characterized in vitro.
2.1.9. Pseudouridimycin.
Pseudouridimycin (239) was discovered upon screening 3,000 actinobacteria and fungal extracts for compounds that inhibited bacterial RNA polymerase but not the structurally unrelated bacteriophage RNA polymerase.209 Examination of the activity revealed 239 is a relatively broad spectrum antibiotic with activity against several pathogenic strains. Furthermore, 239 was determined to be highly selective for bacterial RNA polymerase with an IC50 of 0.1 μM and did not exhibit any cross resistance with other clinically used bacterial RNA polymerase inhibitors, suggesting a novel mechanism of action. Consistent with this, 239 was shown to bind to the NTP addition site of the polymerase,210, 211 which differs from other RNA polymerase inhibitors that bind to sites related to NTP ligation and chain extension.
Structurally, 239 is categorized as a peptidyl nucleoside antibiotic consisting of a 5′-aminopseudouridine core connected to a dipeptide by an amide bond (Fig. 22).209 The dipeptide consists of guanidinoacetic acid and l-Gln that are linked by an N-hydroxylated amide bond. The gene cluster for 239 was identified by sequencing and bioinformatics analysis of the genome from one of the two producers, Streptomyces sp. ID38640.212 A genetic locus encoding a protein with similarity to pseudouridine synthases, predicted to be required for the generation of a C-nucleoside, and a protein with similarity to glycine amidinotransferase, predicted to catalyse the formation of guanidinoacetic acid, was uncovered and predicted to be involved in 239 biosynthesis. Analysis of the locus suggested the biosynthetic gene cluster spans 20-kb DNA and consists of 15 orfs annotated as pumA-pumO (Fig. 23A).
Figure 22.

Structure of pseudouridimycin.
Figure 23.

Biosynthesis of pseudouridimycin. (A) Genetic organization of the biosynthetic gene cluster. NCBI accession number is pseudouridimycin (pum) from Streptomyces sp. ID38640, MG266907. (B) Pathway for the biosynthesis of the nucleoside core and subsequent acylation steps. Oxa, oxaloacetate
The biosynthesis of 239 has been interrogating using a combination of bioinformatic predictions and gene inactivation experiments. The pathway likely begins from either UMP or potentially UDP (Fig. 23B). PumH, with sequence similarity to adenylate kinases, is proposed to catalyse 3′-O-phosphorylation to generate a 3’,5’-diphospho-uridine nucleoside (240) in the phosphorylation state resembling RNA, the substrate for known pseudouridine synthases.213 PumJ, which resembles known pseudouridine synthases, then likely catalyses the isomerization of the nucleoside base to form phosphorylated pseudouridine (241). A ΔpumJ mutant strain did not produce 239 but instead accumulated guanidinoacetic acid (242); feeding pseudouridine (243) to the ΔpumJ mutant strain restored the production of 239, consistent with the proposed functional assignment of PumJ and suggesting that 243 is a biosynthetic intermediate. PumD, which has sequence similarity to proteins of the HAD family of hydrolases, was proposed to function as the phosphatase that converts 241 to 243.
Following the formation of 243, PumI, which has sequence similarity to FAD-dependent glucose-methanol-choline oxidoreductases, is proposed to catalyse the oxidation of C-5’ of the ribose moiety to generate the aldehyde 244. PumG, a putative PLP-dependent aspartate aminotransferase, is predicted to catalyse transamination thereby converting 244 to 5’-amino-5’-deoxypseudouridine (245). Following the generation of the nucleoside core, PumK and PumM, both of which show sequence similarity to ATP-dependent carboxylate-amine ligases of the ATP grasp superfamily, are predicted to attach l-Gln and 242. Inactivation of either pumK or pumM yielded mutant strains unable to produce 239. However, the ΔpumK mutant strain produced low titres of 243 and 245, while the ΔpumM mutant strain produced relatively high titres of a new metabolite identified as l-Gln-245 (246). This result is consistent with PumK first catalysing the attachment of l-Gln to 245 followed by PumM-catalysed attachment of 242 to generate deoxypseudouridimycin (247). In turn PumN, which has sequence similarity to amidinotransferases, is proposed to generate 242 from Gly and l-Arg. Inactivation of pumN yielded a mutant strain that produced 246, and 239 production was restored by feeding 242 to the ΔpumN mutant strain. The final step in the biosynthesis is N-hydroxylation proposed to be catalysed by PumE, a flavin-dependent oxidoreductase. Consistent with the proposed function, the ΔpumE mutant strain produced a metabolite that was 16 amu less than 239, although the identity of the metabolite was not confirmed by NMR spectroscopy. Enzyme-catalysed N-hydroxylation of an amide functionality in natural product biosynthesis is rare, and the possibility remains that PumE functions prior to PumM-catalysed amide bond formation.
Based on bioinformatic analysis, PumB and PumL, both with sequence similarity to transporters of the major facilitator superfamily, are likely responsible for the transport of 239 out of the cell. PumC, which has similarity to DeoR-transcriptional regulators, is potentially involved in gene expression and regulation. The remaining genes—pumA, pumF, and pumO—encode hypothetical proteins with no obvious connection to 239 biosynthesis, although PumF does show some homology to a protein of unknown function in the 96-114 gene cluster, suggesting it could be involved in the biosynthesis. The mechanism of 239 self-resistance remains unknown.
2.2. Cytidine-derived nucleosides
2.2.1. Albomycin.
Albomycins (248-250) are broad-spectrum antibiotics isolated from Streptomyces sp. ATCC 700974.214 and consist of two structural components: a nucleoside core and a peptide-based siderophore moiety.215, 216 Both of these components play an important role in the antibiotic activity of 248-250.217 The siderophore component binds ferric iron, and the complex is actively transported into the cell by many species of bacteria as a mechanism to acquire this relatively scarce metal. Upon internalization, 248-250 is enzymatically hydrolyzed, releasing the siderophore from the nucleoside, which inhibits seryl-tRNA synthetase activity leading to cell death.218-222 The active transport mechanism renders 248-250 with excellent antibiotic activity against several pathogenic bacteria including Streptococcus and Yersinia ssp., among others 223-225
The nucleoside core (named SB-217452) was independently isolated from the same Streptomyces strain guided by an activity-based screen designed to identify inhibitors of seryl-tRNA synthetase.226, 227 Albomycin 249, which contains the SB-217452 component covalently attached to the siderophore, has no inhibitory effect against seryl-tRNA synthetase; likewise, SB-217452 alone has no antibiotic properties in whole cell assays, suggesting the nucleoside cannot pass through the cell membrane and highlighting the importance of both components for activity.
The structure of nucleoside core of 248-250 consists of several features not found in other nucleoside antibiotics (Fig. 24). The nucleoside core is derived from cytosine, and each congener contains a different modification at C-4 of the 3-N-methylated nucleoside base: an imine (albomycin ε, 248), a carbamylated imine (albomycin δ2, 249), or carbonyl (albomycin δ1, 250). The typical ribose of the nucleoside is modified to a thiofuranose containing a C-5′-glycyl unit; this sugar possesses different relative stereochemistry at C-3’ and C-6’ compared to the previously discussed lipouridine family of nucleoside antibiotics (Section 2.1.2). The peptide component of 248-250 consists of a three N-hydroxylated, N-acetylated ornithines that are connected to the nucleoside core via a Ser linker. An identical peptide moiety is found in the fungal-derived siderophore, ferrichrome.228
Figure 24.

Structure of albomycins.
The gene cluster for 248-250 was identified by scanning the sequenced genome of Streptomyces sp. ATCC 700974.229 A potential genomic locus was identified that included proteins AbmA, a putative l-Orn N-acetyltransferase, AbmB, a putative l-Orn N-hydroxylase, and AbmQ, a putative multidomain NRPS, all of which were expected to assemble the siderophore ferrichrome. Additionally, AbmK, which has sequence similarity to seryl-tRNA synthetases, was identified and is likely involved in self-resistance.230 Based on this bioinformatic analysis, the gene cluster was proposed to span a minimum of 23.7-kb DNA and consist of 18 orfs annotated as abmA-R (Fig. 25A).
Figure 25.

Biosynthesis of albomycins. (A) Genetic organization of the biosynthetic gene cluster. NCBI accession number is albomycin (abm) from Streptomyces sp. ATCC 700974, JN252488. (B) Pathway for the biosynthesis of the nucleoside core. (C) Pathway for the biosynthesis of the tripeptide siderophore component and subsequent acylation to the nucleoside core. Proteins labelled in bold blue have been functionally assigned in vitro using recombinant enzymes. PLP, pyridoxal-5’-phosphate; AdoMet, S-adenosyl-l-methionine; SAH, S-adenosyl-l-homocysteine; PPi, inorganic pyrophosphate; dA, 5’-deoxyadenosine; CoA, coenzyme A; C, condensation; A, adenylation; T, thiolation.
The function of the majority of the orfs has now been defined by gene inactivation and/or characterization of the recombinant enzymes. Initial insight into how the nucleoside core is assembled was provided by the identification and characterization of AbmH, which has moderate sequence similarity to 27:L-Thr transaldolases involved in 41-71 and 121-134 biosynthesis. AbmH was shown to catalyze the PLP-dependent conversion of 27 to 72, but in contrast to the other transaldolases that specifically generate (5’S,6’S)-72, a 1:1 mixture of the (5’S,6’S) and (5’R,6’S) epimers was observed.231 Additional studies revealed AbmH utilized the 4’-thionucleoside variant of 27 (251), generating the 4’-thiohepturonic acid-containing nucleoside (252) (Fig. 25B). Kinetic analysis revealed AbmH preferred 251 over 27, suggesting that S atom incorporation occurs at an earlier stage in the biosynthesis. Additionally, the (5’S,6’S)-isomer of 252 was exclusively produced, revealing the need for an epimerase to generate the 5’S,6’R configuration that is found in 248-250. The PLP-dependent protein AbmD was subsequently shown to catalyze this epimerization, converting the AbmH-product to (5’S,6’R)-252 (253).231 AbmD was unable to isomerize (5’S,6’S)- or (5’R,6’S)-72, providing further evidence that S atom incorporation occurs prior to the AbmH-catalyzed reaction. A likely candidate for S incorporation is AbmM, a protein with sequence similarity to enzymes of the radical SAM superfamily including lipoyl synthase and biotin synthase, both of which mobilize and incorporate sulfur into the respective metabolites.
The nucleoside base of 253 is proposed to be modified in sequential steps catalyzed by the methyltransferase, AbmI, and carbamoyltransferase, AbmE, to generate 254 and 255, respectively. Although the function of these two gene products has been supported by gene inactivation,229 the timing of the AbmI- and AbmE-catalyzed reactions remains unknown and might occur at a later stage in the pathway. AbmF, which has low sequence similarity to aminoacyl-tRNA synthetases, was demonstrated to catalyze amide bond formation between l-Ser and 255 to generate the dipeptide-containing nucleoside core (256).232 AbmF utilizes a seryl-adenylate as the acyl donor, which is supplied mostly by the seryl-tRNA synthetase AbmK that was initially characterized as a self-resistance protein.230 Importantly, AbmF is highly specific for the acyl donor (other aminoacyl adenylates were not substrates), and evidence was provided to show that the AbmF-catalyzed reaction is independent of tRNA. With respect to the acyl acceptor, AbmF was specific for the (5’S,6’R) configuration but had relaxed specificity with the base and could catalyze the conversion with the uracil-containing derivative of 255. The final step is C3’-epimerization to generate SB-217452 (257). Inactivation of abmJ yielded a mutant strain that produced 256 and C3’-epi-249, consistent with AbmJ functioning as the epimerase.232 In vitro characterization revealed AbmJ indeed catalyzed C3’-epimerization and was specific for 256: neither C3’-epi-249 nor 255 were substrates. However, the specificity of AbmF toward the nucleoside base was not examined.
The tripeptide component of 248-250 is assembled from l-Orn and involves AbmA, a putative acetyltransferase, AbmB, a putative monooxygenase, and AbmQ, a multidomain NRPS. Based on precedent during the biosynthesis of related siderophores,233 AbmB is expected to first catalyze l-Orn hydroxylation followed by AbmA-catalyzed acetylation. AbmQ has a domain organization consisting of one A domain, two C domains, and three T domains. The lone, N-terminal A domain is predicted to activate and load N-acetyl-N-hydroxy-l-Orn to each T domain that are separated by the C domains. NRPS usually have a C-terminal C or TE domain for releasing the peptide via hydrolysis, lactamization, or lactonization. However, AbmQ terminates with a T domain, suggesting that the tripeptide-S-AbmQ serves as the substrate for downstream N-acylation. AbmC, which has sequence similarity to cysteine proteases and several proteins annotated as hypothetical, was proposed to catalyze the N-acylation of 257. Consistent with this proposal, a ΔabmC mutant strain lost its ability to produce 248-250, yet retained the ability to produce the likely acyl acceptor 257.232 Finally, 250 can be generated from 249 by mild alkaline hydrolysis, suggesting its formation is likely an artifact of workup.
Self-resistance to 248-250 is mediated by transport and target modification. With respect to the former, abmN, abmO, abmP, and abmQ encode proteins with sequence similarity to ABC transporters. With respect to the latter, AbmK, the previously noted seryl-tRNA synthetase, is less susceptible to 249 inhibition than the housekeeping seryl-tRNA synthetase.230
2.2.2. Amicetin, bamicetin, oxyplicacetin, SF2457, oxamicetin, 40551, streptcytosine, and cytosaminomycin.
Amicetin (258) is a disaccharide pyrimidine nucleoside antibiotic initially isolated from Streptomyces vinaceusdrappus and Streptomyces fasciculatis in 1953.234, 235 Since this time, 258 has been isolated from several other actinomycetes. Amicetin has been shown to be a peptidyl transferase inhibitor in protein biosynthesis, and has near universal antibiotic activity against microorganisms found in domains archaea, bacteria, protozoa and fungi.236-242 Amicetin binds to a conserved structural motif of 23S rRNAs, thereby inhibiting peptide bond formation during the translation process.243
The 258 family continues to expand and now includes several different groups isolated from several strains including 258 from multiple strains; bamicetin (259), plicacetin (260), and norplicacetin (261) from Streptomyces plicatus;244 oxyplicacetin (262) from Streptomyces ramuloses;245 SF2457 (263) from Nocardia brasiliensis SF2457;246 oxamicetin (264) from Arthrobacter oxamicetus sp.;247, 248 40551s (265-271) from Streptomyces sp. OPMA40551 and Streptomyces sp. SSA28;249 cytosaminomycins (272-276) from Streptomyces amakusaensis KO-8119 and Streptomyces sp. SSA28;250, 251 and streptcytosine A (277; also called rocheicoside A when isolated from Streptomyces rochei 06CM016) and other strep cytosines (278-291) from Streptomyces sp. TPU1236A and Streptomyces sp. SSA28.252, 253 The general structure of the 258 family consists of a deoxy sugar called amicetose glycosydically linked to an acylated—or in one instance alkylated—cytosine base (Fig. 26).
Figure 26.

Structures of the amicetin family of nucleoside antibiotics.
The parent 258 is acylated with p-aminobenzoic acid (PABA) that is further modified with an α-methyl-Ser.234, 254 A second notable variation found in the family is the modification of the amicetose, wherein most are modified with a sugar called amosamine that is linked via an α-(1→4)-glycosidic bond.
In 2012 the 258 biosynthetic gene cluster from Streptomyces vinaceusdrappus NRRL 2363 was reported and partially characterized by gene inactivation (Fig. 27A).255 The gene cluster was located using degenerate primers designed to amplify genes encoding an NDP-glucose 4,6-dehydratase and a PABA synthase. A single cosmid was positive for both fragments, and heterologous expression in Streptomyces lividans TK64 resulted in the production of 258, 260, and 261. Bioinformatic analysis of the sequence along with inactivation of the predicted downstream and upstream orfs revealed 21 essential genes (amiA to amiU) spanning 27-kb DNA.
Figure 27.

Biosynthesis of amicetin. (A) Genetic organization of the biosynthetic gene cluster. NCBI accession number is amicetin (ami) from Streptomyces vinaceusdrappus strain NRRL 2363, HM748814. (B) Pathway for the biosynthesis of the nucleoside core and subsequent acylation of the base. (C) Pathway for the biosynthesis of TDP-amicetose and TDP-amosamine. Proteins labelled in bold blue have been functionally assigned in vitro using recombinant enzymes. PLP, pyridoxal-5’-phosphate; AdoMet, S-adenosyl-l-methionine; SAH, S-adenosyl-l-homocysteine; PPi, inorganic pyrophosphate; THF, N5,N10-methylene tetrahydrofolic acid.
Based in part on comparisons with the blasticidin gene cluster that was identified in 2003 (Section 2.2.4) the nucleoside core was proposed to originate from CMP (Fig. 27B). The protein AmiI, which has sequence similarity to cytidine deoxyribosyltransferases including BlsM,256 was proposed to hydrolyse the glycosidic bond to provide cystosine that is eventually attached to amicetose to form the nucleoside core (292). Inactivation of amiI led to a mutant strain unable to produce 258 or derivatives, consistent with this function early in the biosynthetic pathway. Seven genes, amiC-amiE, amiJ, amiK, amiN, and amiU, were proposed to be involved in amicetose biosynthesis and attachment (Fig. 27C). The thymidylyltransferase AmiE initiates the pathway by converting D-glucose-1-phosphate to TDP-D-glucose. A TDP-glucose 4,6-dehydratase AmiU then catalyzes the formation of TDP-4-keto-6-deoxy-D-glucose, the intermediate in essentially all bacterial deoxy sugar biosynthetic pathways. This intermediate serves as a diverging point for the biosynthesis of both the TDP-D-amicetose (293) and TDP-D-amosamine (294). The former is proposed to involve four enzymes: AmiC, a putative NDP-hexose 2, 3-dehydratase; AmiD, a putative NDP-hexose 3-ketoreductase; AmiN, a putative TDP-4-keto-6-deoxyglucose-3-dehydratase; and AmiK, a putative NDP-hexose 4-ketoreductase. AmiC and AmiD work in concert to convert TDP-4-keto-6-deoxy-D-glucose to TDP-4-keto-2,6-dideoxy-D-glucose. Subsequently, AmiN is predicted to catalyze C-3 deoxygenation, which is followed by AmiK catalyzed keto reduction to afford the activated NDP-sugar 293. The putative glycosyltransferase AmiJ, which has sequence similarity to the cytosylglucuronic acid synthase BlsD,257 likely catalyzes the coupling of amicetose (via 293) and cytosine to form 292 (Fig. 27B).
Amosamine biosynthesis also begins from TDP-4-keto-6-deoxy-D-glucose (Fig. 27C). AmiB, a putative PLP-dependent aminotransferase, first generates TDP-D-viosamine (295). Subsequently, AmiH, which has sequence similarity to methyltransferases, catalyzes iterative methylations to first form the monomethylated TDP-D-viosamine (296) and then 294. Gene inactivation of amiH yielded a mutant strain that produced desmethyl-258, thus consistent with the functional assignment.255 However, when N-desmethyl-258 was fed to the ΔamiI mutant strain, 258 production was not restored,255, 258 suggesting methylation occurs prior to transfer of the sugar to the acceptor. Inactivation of amiG, encoding a putative glycotransferase, yielded a ΔamiG mutant strain that produced the desamosamine precursor 297, consistent with acylation of cytosine occurring prior to glycosyltransfer (Fig. 27C).258 In vitro characterization revealed AmiG is a retaining glycosyltransferase able to preferentially glycosylate the acceptor 297 using 294 as the donor, thereby forming an α-(1→4)-glycosidic bond. Further characterization revealed AmiG is able to use compound 298 as an acceptor and several activated sugars as donors. Interestingly, AmiG can also catalyze the reverse reaction, enabling the enzymatic exchange of the acceptor and the sugar to generate several derivatives of 258. This process, termed glycorandomization, has been successfully employed with inverting glycosyltransferase, and the results with AmiG have now expanded this technology to include retaining glycosyltransferases.258
Based on the kinetic characterization of AmiG, acylation of cytosine is expected to come before addition of the amosamine (Fig. 27C). For the biosynthesis of PABA, found in 258-266, 277, and 291, the process begins from chorismic acid and requires four enzymes. For the biosynthesis of 258, AmiM, which has similarity to PABA synthases, is predicted to catalyze the formation of 4-amino-4-deoxychlorismate (ADC) using l-Gln as an amino donor. AmiA, which has sequence similarity to ADC lyases, catalyzes the elimination of pyruvate from 4-amino-4-deoxychlorismate to produce PABA. AmiL, which has sequence similarity to benzoyl coenzyme A ligases, subsequently generates PABA-S-CoA. Finally, AmiF, which has sequence similarity to GCN5-related N-acetyltransferases, is predicted to catalyze the acylation step. Gene inactivation of amiL or amiF yielded a mutant strain unable to produce 258 but instead accumulated the shunt product amosamine-modified 299, consistent with the proposed roles and further confirming the relaxed substrate specificity of the glycosyltransferase AmiG.255 Three gene candidates, amiS, amiT, and amiR, were identified for the biosynthesis and incorporation of (2S)-α-methylserine. AmiS, which has sequence similarity to serine hydroxymethyltransferases, is proposed to convert L-Ala to (2S)-α-methylserine using N5,N10-methylene tetrahydrofolic acid. AmiT, which has sequence similarity to NRPS and is predicted to contain an unknown domain, an A domain, and a T domain, activates and loads (2S)-α-methylserine to the phosphopantetheinyl group of the T domain. AmiR, which has sequence similarity to malonyl CoA-acyl carrier protein transacylases, is predicted to catalyze the condensation of α-D-methylserine to 298 to generate the AmiG substrate, 297. The variability of the cytosine modifications suggests alternative mechanisms might be employed for other members in the family.
Lastly, the genes amiO, amiP, and amiQ are proposed to be involved in transport and regulation of 258 biosynthesis. The amiO and amiQ genes encode a putative ABC transporter and a transporter belonging to major facilitator superfamily, respectively. AmiP is predicted to belong to the putative TetR transcriptional regulator family.
2.2.2. Blasticidin, mildiomycin, cytomycin, and arginomycin.
Blasticidin S (300) is a peptidyl nucleoside antibiotic that was initially isolated from Streptomyces griseochromogenes in 1958.259 Several variants have subsequently been isolated from other bacterial sources, including arginomycin (301) from Streptomyces arginensis,260 cytomycin (302) from Streptomyces sp. HKI-0052,261 mildiomycin (303) from Streptoverticillum remofaciens ZJU5119,262, 263 and blasticidin P10 (304) from extracts of the marine sponge Theonella swinhoei.264 Members of the family inhibit the growth of a wide array of prokaryotes and eukaryotes.242, 265, 266 Both 300 and 303 have been used as agricultural fungicides, finding specific utility in the prevention and treatment of rice blast. The general mechanism of action is through binding to a highly conserved peptidyl transferase site of the 70S ribosome, thereby inhibiting the release of the peptide during termination of translation.267, 268 Structural studies have demonstrated that 300 causes a restructuring of the peptidyl transferase site, which prevents the proper positioning of release factor 1, a protein that is involved in hydrolysis of the peptidyl-tRNA bound at the peptidyl site and therefore peptide release. This process is shared across the domains of life, hence explaining the broad spectrum of activity.
The common structural feature of the family is a cytosine base bonded to an unsaturated aminohexuronic acid by a glycosidic bond, thus generating the nucleoside core (Fig. 28A).269-273 The aminohexuronic acid of 300 is N-acylated with an N-methyl-β-Arg. Several potential intermediates or shunt products of 300 biosynthesis have also been isolated from S. griseochromogenes, including demethylblasticidin S (305),274 blasticidin H (306),275 pentopyranic acid (307) (also called cytosylgluruconic acid,276-278 pentapyranine A (308),279 and pentapyranine C (309) (Fig. 28B).277, 279 Blasticidin P10 (304) differs by containing a carboxamide instead of a carboxylic acid in the aminosugar.264 Both 301 and 302 have uniquely modified Arg derivatives in place of the β-arginine found in 300.260, 261 Mildiomycin (303) is distinct from the rest of the family in that the carboxylic acid component of the aminoheruxonic acid is substituted with a 5-guanidino-2,4-dihydroxyvalerate side chain.262, 280 Furthermore, 303 contains l-Ser in place of the Arg derivative of 300-302, and the cytosine base is modified with a C5-hydroxymethyl group. Hydroxymethylated congeners of 300 and 302 have also been reported.261, 281, 282
Figure 28.
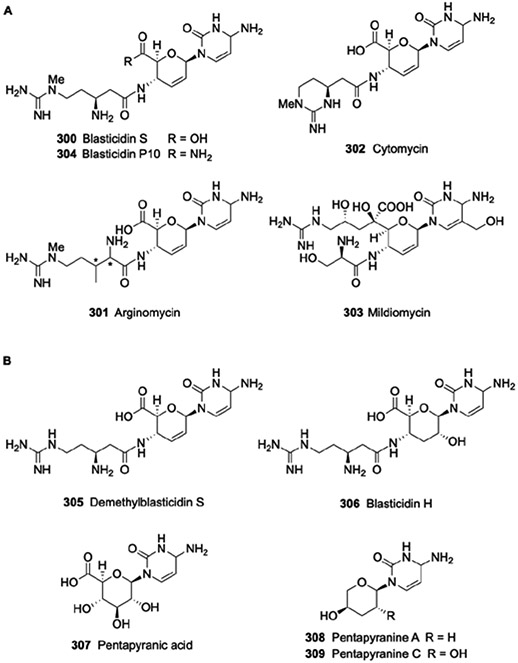
Structures of the blasticidin family of nucleoside antibiotics. (A) Structures of related compounds isolated from different strains. (B) Structures of biosynthetic precursors or shunt products isolated from the blasticidin producing strain.
In 1998 a portion of the gene cluster for 300 was identified.283 Two cloned DNA fragments (2.6-kb and 4.8-kb DNA) from the genome of S. griseochromogenes were found to confer resistance to 300 when heterologously expressed in Streptomyces lividans. Further heterologous expression of larger, surrounding DNA fragments yielded heterologous strains that produced 307 and 305. A restriction digest analysis of cosmids was used in conjunction with metabolite profiling to reveal a contiguous DNA fragment containing 28 open reading frames, 19 of which within 22-kb DNA were likely involved in 300 biosynthesis (Fig. 29A). In 2008, the gene cluster for 303 was identified by searching the sequenced genome for milB, a homolog of blsM encoding a functionally confirmed CMP hydrolase.284 The cosmid containing the proposed gene cluster was heterologous expressed in Streptoverticillum remofaciens ZJU5119 leading to the production of both 303 and deshydroxymethyl-303. A later study found two cosmids containing milB, and both were independently expressed in Streptomyces lividans 1326 to yield a strain capable of 303 production.285 The boundaries and several essential genes of the biosynthetic gene cluster were identified by gene deletion, resulting in 16 ORFs (milA to milQ) encompassing ~20-kb DNA required for 303 biosynthesis. The only gene deemed nonessential was milI: inactivation of mill, encoding a hypothetical protein with similarity to BlmF, did not have any effect on 303 production.285 The biosynthetic gene cluster for 301 was reported in 2014 and encompasses ~16-kb DNA encoding 14 ORFs (argA to argN).286
Figure 29.

Biosynthesis of the blasticidin family of nucleoside antibiotics. (A) Genetic organization of the biosynthetic gene clusters. NCBI accession number are blasticidin (bls) from Streptomyces griseochromogenes, AY196214; mildiomycin (mil) from Streptoverticillum remofaciens ZJU5119, JN999998; and arginomycin (arg) from Streptomyces arginensis, KC181124.(B) Pathway for the biosynthesis of the nucleoside core. (C) Pathway for the biosynthesis of the nonproteinogenic amino acid component of blasticidin and attachment to the nucleoside core. (D) Pathway for the biosynthesis of the nonproteinogenic amino acid component of arginomycin and mildiomycin and attachment to the nucleoside core. Proteins labelled in bold blue have been functionally assigned in vitro using recombinant enzymes; the structures of BlsM (PDB 5VTO), MilA (PDB 5JNH), and MilB (PDB 4OHR) have been solved. Orthologs of the respective biosynthetic proteins are indicated in parenthesis. 5hm-CMP, 5-hydroxymethyl-CMP; AdoMet, S-adenosyl-l-methionine; SAH, S-adenosyl-l-homocysteine; PPi, inorganic pyrophosphate.
Prior to identifying the gene cluster, the potential precursors of 300 were probed by isotopic enrichment experiments. The results indicated that cytosinine, the name of nucleoside core, originates from glucose and cytosine.287 Furthermore, feeding with d-[6-14C]glucose followed by degradative analysis of the resultant 300 provided strong evidence that the carboxylic acid of cytosinine originates from C-6 of glucose. The cytosine base of cytosinine was also enriched when feeding with cytidine, suggesting hydrolysis of the nucleoside is a feasible source of the base. As expected, N-methyl-β-arginine was enriched when feeding l-[U-14C]Arg. However, whether the β-amine migrates from Cα or is derived from a distinct metabolic precursor was not examined. Finally, the methyl group was clearly enriched following feeding l-[methyl-14C]Met, consistent with a traditional N-methylation event using AdoMet as the methyl donor.
The biosynthesis of the nucleoside core of 300 begins with the hydrolysis of cytidine-5’-phosphate (CMP) to form cytosine, a reaction shown to be catalysed by BlsM (Fig. 29B).256 The BlsM-homolog MilB catalyses the same reaction during the biosynthesis of 303 except 5-hydroxymethyl-CMP serves as the substrate, generating 5-hydroxymethyl-cytosine (310).284 Inactivation of the gene encoding MilA, which has sequence similarity to thymidylate synthases and is unique to the 303 gene cluster, abolished the production of 303 and yielded a strain that produced deshydroxymethyl-303, consistent with MilA functioning as a hydroxymethyltransferase to generate the MilB substrate.284 Characterization of MilA in vitro confirmed an N5,N10-methylene-tetrahydrofolate-dependent hydroxymethyltransferase activity and revealed the enzyme only uses CMP as the methyl acceptor while dCMP, CDP, and other nucleotides were not recognized. MilB subsequently hydrolyses the glycosidic bond to form 310. Once the free base is generated, BlsD or the homolog MilC catalyse glycosylation using UDP-glucuronic acid as the sugar donor and the respective base as a sugar acceptor.278, 285 The product of BlsD, 307, was demonstrated to be a pathway intermediate by generating radiolabelled material that was subsequently incorporated into 287 using the producing strain.278 Furthermore pathway intermediate 307 is a known minor metabolite isolated from the producing strain 276 Recombinant MilC was shown to catalyse the comparable reaction to give hydroxymethyl-cytosylglucuronic acid (311),285 further supporting the glucuronidated base as a biosynthetic intermediate.
Little information is known about the steps following the formation of 307/311 that yield the characteristic unsaturated nucleoside core of the family. Cytosinine (312) has been routinely stated to be a pathway intermediate,257, 285 yet the citations that are given as proof for its intermediacy do not provide such evidence. Instead, 312 is an established hydrolytic product of 300 when treated with acid (Fig. 29C),273 and attempts at biotransformation of the chemically prepared material revealed radioactive 312 is not incorporated into 300.288 In lieu of this information, the biosynthesis is proposed here to continue with 4’-oxidation/3’-dehydration of 307/311 in a reaction catalysed by BlsE/MilG, which have sequence similarity to proteins of the radical SAM superfamily. This chemical transformation would be comparable to the reaction catalysed by a recently reported radical SAM diol dehydratase, AprD4.289 However, this proposal is at odds with the report characterizing recombinant BlsE, which was shown to catalyse C5’ decarboxylation of 307 in vitro to generate a pentapyranose 3.290, 291 The in vitro activity, if relevant for biosynthesis, would necessitate an additional carboxylation step and is inconsistent with the feeding experiments that demonstrated clear C-6’ enrichment from d-[6-14C]glucose. Hence, we hypothesize that decarboxylation is a side reaction that is a consequence of the in vitro conditions. Nonetheless, whether decarboxylation is part of the biosynthetic pathway, and what is the significance (if any) of the decarboxylated metabolites 308 and 309, remains one of several biosynthetic unknowns. Following the proposed BlsE-catalysed formation of the β-keto acid-containing nucleoside (314/315), BlsH/MilD, which have sequence similarity to PLP-dependent aminotransferases, are hypothesized to catalyse transamination to generate 3’,4’-dideoxy-4’-aminoglucuronic acid-containing nucleoside (316/317). The same series of steps is predicted to occur during the biosynthesis of the nucleoside core of 301 (Fig. 29B).
The remaining steps in 300 biosynthesis involve formation of the Arg-derived component and acylation to the nucleoside core. The timing of C2’ dehydration of the nucleoside core, and whether this occurs with 316 or after acylation, is unknown. Notably, the aforementioned BlsH/MilD was speculated to catalyse both β-elimination of water along with transamination,277, 288 not unreasonable for enzymes employing PLP cofactors. Consequently, 312 was suggested to be a biosynthetic intermediate. Here, we propose that dehydration occurs following acylation. Regardless of the identity of the acyl acceptor, BlsG, which has sequence similarity to PLP-dependent aminomutase, initiates the biosynthesis of the acyl group by converting L-Arg to β-Arg 318 (Fig. 29C). By feeding a variety of stereoselectively labelled l-Arg to the producing strain of 300, the transformation of l-Arg to 300 was shown to occur with retention of the α-amine and migration of the pro-3R hydrogen to Cα (with concomitant loss of the Cα hydrogen),292 suggesting the BlsG-catalysed reaction proceeds like the well characterized l-Lys 2,3-aminomutase from Clostridium subterminale.293 Leucyldemethyl-300 (319) is a known metabolite of the producing strain,294 suggesting that 318 undergoes N-acylation with l-Leu in addition to amide bond formation with the nucleoside core. BlsI, which has sequence similarity to proteins of the ATP grasp superfamily, likely catalyses one of these steps, and we propose the condensation of l-Leu and 318 to first form a dipeptide (320). Subsequently, BlsK, a hypothetical protein, is proposed to catalyse ligation of the dipeptide to the nucleoside core 316 to generate the hypothetical precursor 321. The order of the two condensations and the identity of the corresponding catalyst, however, remains unknown, and it is possible that ligation of l-Arg to 316 precedes l-Leu attachment. BlsF, which has low sequence similarity to glycosyltransferases and is one of the only remaining, unassigned protein that is shared in the 300, 301, and 303 gene clusters, is proposed to catalyse C2’ dehydration to form 319. The BlsF-catalysed reaction could potentially proceed through a cryptic glycosylation followed by lyase chemistry to give the unsaturated sugar that is characteristic of the family.
The protein BlsL, which has sequence similarity to guanidinoacetate methyltransferases, catalyses the penultimate step to generate Leucylblasticidin S (322). Inactivation of blsL in a heterologous production strain resulted in the accumulation of 319, consistent with the proposed function and timing of the BlsL-catalysed reaction.295 Recombinant BlsL efficiently methylated 319 to form 322, confirming the functional assignment as an AdoMet-dependent 319 methyltransferase.295 An unidentified amidohydrolase, the activity of which has been demonstrated in cell free extracts of the producing strain, hydrolyses l-Leu from 322 to generate the final product, 300.287, 294 Similar to the substrate permissiveness observed for many of the enzymes in the pathway, the amidohydrolase also utilizes 319 and blasticidin H, the latter of which is the 2’-hydrated form of 300.275 Contrastingly, in vitro characterization of BlmL revealed N-methylation almost exclusively requires 319, suggesting that hydrolysis of l-Leu is the last step in the pathway.295
The other members of the blasticidin family have distinct substituents in place of the β-Arg. Compound 301 contains a β-C-methyl-Arg that putatively requires two unique enzyme-catalysed transformations (Fig. 29D). ArgM, a PLP-dependent aminotransferase that has been assigned in vitro, catalyses transamination of l-Arg to generate the corresponding α-keto acid, 5-guanidino-2-oxovaleric acid (323).286 ArgN, an AdoMet-dependent methyltransferase that has similarly been characterized in vitro, converts 323 to 3-methyl-5-guanidino-2-oxovaleric acid (324). Finally, ArgM reintroduces the amine to generate 3-methyl-Arg (325); analysis of NMR data supported the ArgM-product as the (2S,3R)-325 isomer. Bioinformatic analysis of the 301 gene cluster revealed homologs to BlsI (ArgJ), BlsK (ArgK), and BlsL (ArgL), suggesting the remaining attachment and processing of 325 is comparable to the biosynthesis of 300.
Like the other members of the family, 303 has an l-Arg derived unit; however, this unit is uniquely bonded to C5’ of the nucleoside core. Isotopic enrichment studies revealed l-Arg is incorporated intact, and the attachment is likely coupled with decarboxylation of the 3’,4’-dideoxy-4’-aminogluronic acid-containing nucleoside (317) (Fig. 29D).285 MilM, which has 29% sequence identity to ArgM, likely catalyses transamination of l-Arg to generate 5-guanidino-2-oxovaleric acid. MilH, which has 26% sequence identity to BlsK, is potentially responsible for a decarboxylative, intermolecular aldol reaction to attach 5-guanidino-2-oxovaleric acid to generate the hypothetical intermediate 326. MilJ, which has low sequence similarity to aldose-ketose isomerases, and MilN, which has sequence similarity to 4-hydroxy-tetrahydrodipicolinate synthase, are both unassigned and could be involved in this series of steps. The enzyme responsible for hydroxylation to generate 326, and whether this occurs before or after attachment, is unknown. However, it is worth noting that MppP and homologs, which were initially identified as PLP-dependent aminotransferases by bioinformatics, have been shown to directly convert L-Arg into 5-guanidino-4-hydroxy-2-oxovalerate during the biosynthesis of unrelated natural products.296, 297 MppP is part of a growing family of PLP-dependent enzymes that utilize O2 as a cosubstrate to oxidize an organic substrate,298 and it is intriguing to speculate that MilM has the same activity as MppP, thereby bypassing the need for a separate hydroxylation catalyst. MilI, which has an N-terminus with sequence similarity to T domains of NRPS and a C-terminus with sequence similarity to seryl-tRNA synthetases, likely catalyses the penultimate step of 303 biosynthesis, the activation and attachment of l-Ser to form 327. Whether this event is dependent on the T domain or requires another enzyme for condensation to 326—one potential candidate is the aforementioned MilH—remains to be determined. Finally, the BlsF-homolog MilL catalyses 2’-dehydration of 327 to form 303.
The remaining genes in the biosynthetic gene clusters either have unknown function or appear to be involved in the regulation, resistance, and transportation of the respective nucleosides. In the gene cluster for 300, blsJ is proposed to encode a transmembrane protein involved in metabolite efflux. Sequence analysis of blsO and blsP indicates that they encode membrane proteins that may also be involved in self-resistance and/or efflux. The genes blsQ and blsC are predicted to encode transcriptional regulatory proteins. In the gene cluster for 303, milO and milK, the latter homologous to blsJ, have been demonstrated to encode regulatory proteins. Additional, potential regulatory, resistance, and/or transport genes include milE, encoding a putative aminoglycoside phosphotransferase; milQ, encoding a distinct aminoglycoside phosphotransferase, and milP, encoding a putative ABC transporter.
2.2.4. Gougerotin, ningnanmycin, and bagougeramine.
Gougerotin (328), also known as aspiculamycin and asteromycin, is a peptidyl nucleoside antibiotic first isolated from Streptomyces gougerotii in 1962 and later identified from the culture broth of Streptomyces toyocaensis var. aspiculamyceticus, Streptomyces S-514, and Streptomyces graminearus.299 Ningnanmycin (329), isolated from Streptomyces noursei var. xichangensis,300 and bagougeramine A (330) and bagougeramine B (331), isolated from Bacillus circulans TB-2125,301, 302 are structural variants of 328 reported in 1995 and 1986, respectively. The family possesses nonselective biological activity including antibacterial, antiviral, and anticancer activity.238, 242, 299, 303 Similar to 258, 328 is a specific inhibitor of protein synthesis by binding a highly conserved region of the 23S rRNA thereby interfering with the proper orientation of the 3’ end of the P site-bound tRNA.304 Nucleoside 329 has found commercial utility and is available in China as an antiviral for agricultural purposes.305
The structure of 328 consists of a nucleoside core consisting of cytosine base and 4-amino-4-deoxyglucuronamide and a 4’-N-appended sarcosyl-d-serine dipeptide (Fig. 30).306-308 Ningnamycin 329 is a stereoisomer of 328 wherein l-Ser is found in the dipeptide moiety. For bagougeramines the Ser is substituted with an N-guanidino-d-Ala. While 330 contains the C-6’-carboxamide like 328 and 329,301 331 contains a spermidine attached to the nucleoside core at this position.
Figure 30.

Structures of the gougerotin family of nucleoside antibiotics.
The biosynthetic gene cluster for 328 from Streptomyces graminearus strain AS4.506 was reported in 2013.309 Based on the structural similarity with the 300-304, 307 was hypothesized to be an upstream pathway intermediate. Consequently, the gene cluster was located by searching the sequenced genome for a blsD homolog. One cosmid containing the blsD homolog was introduced into Streptomyces coelicolor M1146, yielding a recombinant strain that produced 328 as the major metabolite along with moderate amounts of 307. The identity and boundaries were further speculated by sequence analysis and confirmed by gene inactivation, leading to a proposed gene cluster that consists of 15 ORFs annotated as gouA-gouN spanning ~17-kb DNA (Fig. 31A).
Figure 31.

Biosynthesis of gougerotin. (A) Genetic organization of the biosynthetic gene cluster. NCBI accession number is gougerotin (gou) from Streptomyces graminearus strain AS4.506, JQ307220. (B) Pathway for the biosynthesis of the nucleoside core. (C) Pathway for the biosynthesis of the amino acid component and subsequent acylation to the nucleoside core.
The biosynthetic pathway for 298 was proposed using bioinformatics analysis along with the identification of accumulated intermediates from mutant strains (Fig. 31B).309, 310 The origin of the cytosine base is unknown since, unlike the CMP hydrolases BlsM and AmiI encoded in the 300 and 258 gene clusters, respectively, a homologous protein is not encoded in the 328 gene cluster. Instead, GouF, which has sequence similarity to BlsD and AmiJ, catalyzes the first step: the coupling of cytosine with UDP-glucuronic acid to form 307. GouA, which has sequence similarity to NAD(P)-dependent alcohol dehydrogenases, catalyzes C-4’ oxidization of glucuronic acid. The resulting oxidized product 332 subsequently undergoes transamination to form 4’-amino-4’-deoxy-307 (333), and two aminotransferases GouH and GouI, with 28% sequence identity to each other, are encoded within the gene cluster. It is believed that GouH is this catalyst due to the higher sequence identity (32% vs. 27%, respectively) to the 300 biosynthetic counterpart BlsH. Inactivation of gouH abolished the production of 328 and yielded a mutant strain that produced solely 307,309 thus supporting the proposed function.
The biosynthesis and attachment of the sarcosyl-D-serine moiety of 328 is highly speculative at this point (Fig. 31C). There is evidence to suggest, however, that d-Ser is first attached to 333: inactivation of gouN encoding a putative methyltransferase yielded a mutant strain that accumulated the advanced intermediate 334.309 This data also suggests that GouN is the methyltransferase that converts Gly to sarcosine. How sarcosine is attached to 334 is unknown, however one of two proteins (GouJ and GouK) with sequence similarity to acyl-CoA N-acyltransferases could function in this capacity. Based on bioinformatic analysis, the origin of d-Ser does not appear to be a product of racemization. One potential pathway parallels the biosynthesis of l-Ser from 3-phosphoglycerate. GouL, which has sequence similarity to β-hydroxyacid dehydrogenases, first catalyzes the conversion of 3-phosphoglycerate to 3-phosphohydroxypyruvate (335). Inactivation of gouL yielded a mutant strain that produced a mixture of 307 and 333, consistent with a role in d-Ser biosynthesis. The aforementioned aminotransferase-like protein GouI catalyzes transamination to form 3-phospho-d-Ser (336). Subsequently, GouG, which has sequence similarity to phosphoglycerate mutase enzymes that are in the same superfamily as alkaline phosphatases, catalyzes dephosphorylation to generate d-Ser. How the carboxylate is activated is unclear, but ligation to form 334 likely involves one of the aforementioned acyl-CoA N-acyltransferases.
The isolation of intermediate 334 suggests conversion of the C-6’ carboxylate to the carboxamide occurs after peptide attachment. GouB, which has sequence similarity to asparagine synthetases, likely catalyzes this reaction. The proteins GouC, GouD, and GouE, which have sequence similarity to proteins annotated as conserved hypothetical proteins, glycosyltransferases, and hypothetical Rossmann fold proteins, respectively, do not have an obvious role in 328 biosynthesis and remain unassigned.
GouR shows homology to the a TetR transcriptional regulator family, and transcriptional analysis via real-time PCR indicated that GouR represses the transcription of 11 genes (gouB-gouL) and promotes the transcription of gouM, which encodes a major facilitator superfamily transporter.311 Electrophoresis mobility shift assays (EMSAs) and DNase I footprinting experiments revealed GouR specifically binds the promoter regions of gouL, gouM, and gouR. Interestingly, gouR and gouM are located at opposite ends of the gene cluster, which is in contrast to typical repressor-efflux pump genes pairs that are typically located in close proximity.
3. Conclusions
A common structural theme of most uridine-derived nucleoside antibiotics is the presence of a high-carbon sugar nucleoside. Studies described here have confirmed that the biosynthesis is achieved by extension of the carbon chain of ribose by C-5’ oxidation of uridine/UMP precursors. Interestingly, however, is the realization that C-5’ oxidation is not limited to one type of mechanism but occurs by several distinct enzymatic strategies, including non-haem Fe2+- and αKG-dependent dioxygenases, radical SAM enzymes, and flavin-dependent enzymes. Similar to the several ways nature achieves C-5’ oxidation as entry points into the biosynthesis, the downstream steps showcase the many ways that catalytic machinery exploits the power of the PLP cofactor to effect metabolic transformations. In addition to the well-established PLP-dependent transaminations, the biosynthesis of uridine-derived nucleoside antibiotics involves less-common, PLP-dependent enzyme activities, including transaldolase, monooxygenase:decarboxylase, and alkyltransferase chemistries. Comparable catalysts, particularly the PLP-dependent oxidases, are emerging as important contributors for the biosynthesis of several unrelated natural products.
Similar to the uridine-derived nucleoside antibiotics, the cytidine-derived nucleoside antibiotics are structurally characterized by a high-carbon sugar nucleoside. In one instance (albomycins), the high carbon sugar is biosynthesized in an identical manner as the uridine-derived nucleoside antibiotics. However, the other pathways feature a distinct mechanism for installing a high carbon sugar, namely by hydrolytic elimination of the ribose followed by glycosylation of the nucleoside base with a “pre-formed” high carbon sugar. It will be interesting to see if this remains a common theme for the many other cytidine-derived nucleoside antibiotics whose biosynthetic gene clusters have yet to be identified.
Nearly 70 of the 100-plus proteins involved in the biosynthesis of nucleoside antibiotics discussed here have been functionally assigned and partially characterized in vitro. Although many biosynthetic questions have been answered the past decade, several enzymatic steps remain unclear. For example, the origin and catalytic conversions for making the high-carbon sugar nucleoside 184 of A-94964 are unknown and, based on bioinformatics, appears to be unique compared to the other uridine-derived nucleoside antibiotics. The formation of other high-carbon sugar nucleosides, notably tunicamycins, nikkomycins, and polyoxins, also have outstanding steps that need to be assigned. Even more steps remain uncharacterized with respect to the tailoring of the nucleoside cores. Notable examples include the NRPS involved in muraymycin biosynthesis and the PKS involved in A-94964 biosynthesis. Both of these systems consist of functional domains found as discrete proteins instead of as multiple domains in a single, large protein. Thus, studying these systems should cast light on the molecular details for how the protein ensembles orchestrate the specific tailoring of the respective nucleoside cores, which is a critical first step toward engineering the biosynthetic machinery to make novel analogues. The biosynthesis of sphaerimicin and albomycin also require PKS/NRPS and NRPS, respecitively, and both have unusual features that differentiate them from canonical systems. Finally, the formation of the diazepanone of X, which includes an amide bond formatting event, remains unknown. Bioinformatic analysis of the gene clusters would imply an atypical mechanism is used for amide bond formation.
Although over 70 proteins have been functionally assigned, the structures for only 6 proteins have been solved and reported. Future structural studies will be critical for elucidating the mechanism of key enzymes and establishing platforms for structure-guided protein engineering. The latter is expected to open up avenues for using enzymes as biocatatlytic tools for structural modifications with the hope that the already privileged, nucleoside-derived scaffolds can be optimized for desired applications.
4. Acknowledgements
We would like to acknowledge Bertukan Van Lanen for help with the figures. Research in the S.V.L. laboratory is funded in part by the National Institute of Allergy and Infectious Disease grants AI087849 and AI128862, National Cancer Institute grant CA217255, and National Institute of General Medical Science grant GM130456.
5 Notes and References
- 1.Isono K, J. Antibiot. (Tokyo), 1988, 41, 1711–1739. [DOI] [PubMed] [Google Scholar]
- 2.Isono K, Pharmacol. Ther, 1991, 52, 269. [DOI] [PubMed] [Google Scholar]
- 3.Winn M, Goss RJ, Kimura K and Bugg TD, Nat. Prod. Rep, 2010, 27, 279. [DOI] [PubMed] [Google Scholar]
- 4.Kimura K and Bugg TD, Nat. Prod. Rep, 2003, 20, 252. [DOI] [PubMed] [Google Scholar]
- 5.Fernandes PB, Swanson RN, Hardy DJ, Hanson CW, Coen L, Rasmussen RR and Chen RH, J. Antibiot. (Tokyo), 1989, 42, 521. [DOI] [PubMed] [Google Scholar]
- 6.Chen RH, Buko AM, Whittern DN and McAlpine JB, J. Antibiot. (Tokyo), 1989, 42, 512. [DOI] [PubMed] [Google Scholar]
- 7.Isono F, Katayama T, Inukai M and Haneishi T, J. Antibiot. (Tokyo), 1989, 42, 674. [DOI] [PubMed] [Google Scholar]
- 8.Isono F, Inukai M, Takahashi S, Haneishi T, Kinoshita T and Kuwano H, J. Antibiot. (Tokyo), 1989, 42, 667. [DOI] [PubMed] [Google Scholar]
- 9.Inukai M, Isono F, Takahashi S, Enokita R, Sakaida Y and Haneishi T, J. Antibiot. (Tokyo), 1989, 42, 662. [DOI] [PubMed] [Google Scholar]
- 10.Chatterjee S, Nadkarni SR, Vijayakumar EK, Patel MV, Ganguli BN, Fehlhaber HW and Vertesy L, J. Antibiot. (Tokyo), 1994, 47, 595. [DOI] [PubMed] [Google Scholar]
- 11.Xie Y, Chen R, Si S, Sun C and Xu H, J. Antibiot. (Tokyo), 2007, 60, 158. [DOI] [PubMed] [Google Scholar]
- 12.Xie Y, Xu H, Si S, Sun C and Chen R, J. Antibiot. (Tokyo), 2008, 61, 237. [DOI] [PubMed] [Google Scholar]
- 13.Mashalidis EH, Kaeser B, Terasawa Y, Katsuyama A, Kwon DY, Lee K, Hong J, Ichikawa S and Lee SY, Nat. Commun, 2019, 10, 2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isono F, Kodama K and Inukai M, Antimicrob. Agents Chemother, 1992, 36, 1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inukai M, Isono F and Takatsuki A, Antimicrob. Agents Chemother, 1993, 37, 980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karwowski JP, Jackson M, Theriault RJ, Chen RH, Barlow GJ and Maus ML, J. Antibiot. (Tokyo), 1989, 42, 506. [DOI] [PubMed] [Google Scholar]
- 17.Rackham EJ, Grüschow S, Ragab AE, Dickens S and Goss RJ, Chembiochem, 2010, 11, 1700. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W, Ostash B and Walsh CT, Proc. Natl. Acad. Sci. U. S. A, 2010, 107, 16828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaysser L, Tang X, Wemakor E, Sedding K, Hennig S, Siebenberg S and Gust B, Chembiochem, 2011, 12, 477. [DOI] [PubMed] [Google Scholar]
- 20.Li Q, Wang L, Xie Y, Wang S, Chen R and Hong B, J. Bacteriol, 2013, 195, 2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang X, Gross M, Xie Y, Kulik A and Gust B, Chembiochem, 2013, 14, 2248. [DOI] [PubMed] [Google Scholar]
- 22.Gruschow S, Rackham EJ, Elkins B, Newilll PLA, Hill LM and Goss RJM, Chembiochem, 2009, 10, 355. [DOI] [PubMed] [Google Scholar]
- 23.Deb Roy A, Gruschow S, Cairns N and Goss RJ, J. Am. Chem. Soc. 2010, 132, 12243. [DOI] [PubMed] [Google Scholar]
- 24.Walsh CT and Zhang W, ACS Chem. Biol, 2011, 6, 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ragab AE, Grüschow S, Tromans DR and Goss RJ, J. Am. Chem. Soc, 2011, 133, 15288. [DOI] [PubMed] [Google Scholar]
- 26.Michailidou F, Chung CW, Brown MJB, Bent AF, Naismith JH, Leavens WJ, Lynn SM, Sharma SV and Goss RJM, Angew. Chem. Int. Ed, 2017, 56, 12492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam WH, Rychli K and Bugg TD, Org. Biomol. Chem, 2008, 6, 1912. [DOI] [PubMed] [Google Scholar]
- 28.Stachelhaus T, Mootz HD and Marahiel MA, Chem. Biol, 1999, 6, 493. [DOI] [PubMed] [Google Scholar]
- 29.Challis GL, Ravel J and Townsend CA, Chem. Biol, 2000, 7, 211. [DOI] [PubMed] [Google Scholar]
- 30.Süssmuth RD and Mainz A, Angew. Chem. Int. Ed, 2017, 56, 3770. [DOI] [PubMed] [Google Scholar]
- 31.Reimer JM, Haque AS, Tarry MJ and Schmeing TM, Curr. Opin. Struct. Biol, 2018, 49, 104. [DOI] [PubMed] [Google Scholar]
- 32.McErlean M, Overbay J and Van Lanen S, J. Ind. Microbiol. Biotechnol, 2019, 46, 493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang W, Ntai I, Bolla ML, Malcolmson SJ, Kahne D, Kelleher NL and Walsh CT, J. Am. Chem. Soc, 2011, 133, 5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W, Heemstra JR, Walsh CT and Imker HJ, Biochemistry, 2010, 49, 9946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Felnagle EA, Barkei JJ, Park H, Podevels AM, McMahon MD, Drott DW and Thomas MG, Biochemistry, 2010, 49, 8815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schomer RA and Thomas MG, Biochemistry, 2017, 56, 5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang W, Ntai I, Kelleher NL and Walsh CT, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang L, Wang L, Zhang J, Liu H, Hong B, Tan H and Niu G, Sci. Rep, 2015, 5, 14111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Isono K, Uramoto M, Kusakabe H, Kimura K, Isaki K, Nelson CC and McCloskey JA, J. Antibiot. (Tokyo), 1985, 38, 1617. [DOI] [PubMed] [Google Scholar]
- 40.Kimura K, Ikeda Y, Kagami S, Yoshihama M, Suzuki K, Osada H and Isono K, J. Antibiot. (Tokyo), 1998, 51, 1099. [DOI] [PubMed] [Google Scholar]
- 41.Igarashi M, Nakagawa N, Doi N, Hattori S, Naganawa H and Hamada M, J. Antibiot. (Tokyo), 2003, 56, 580. [DOI] [PubMed] [Google Scholar]
- 42.Igarashi M, Takahashi Y, Shitara T, Nakamura H, Naganawa H, Miyake T and Akamatsu Y, J. Antibiot. (Tokyo), 2005, 58, 327. [DOI] [PubMed] [Google Scholar]
- 43.Fujita Y, Kizuka M, Funabashi M, Ogawa Y, Ishikawa T, Nonaka K and Takatsu T, J. Antibiot. (Tokyo), 2011, 64, 495. [DOI] [PubMed] [Google Scholar]
- 44.Kagoshima Y, Tokumitsu A, Masuda T, Namba E, Inoue H, Sugihara C, Yokoyama M, Yamamoto Y, Suzuki K, Iida K, Tamura A, Fujita Y, Takatsu T, Konosu T and Koga T, J. Antibiot. (Tokyo), 2019, 72, 956. [DOI] [PubMed] [Google Scholar]
- 45.Fujita Y, Kagoshima Y, Masuda T, Kizuka M, Ogawa Y, Endo S, Nishigoori H, Saito K, Takasugi K, Miura M, Murakami R, Muramatsu Y, Tokumitsu A, Koga T, Iizuka M, Aoyagi A, Suzuki T, Suzuki Y, Ishida O, Nakahira T, Miyakoshi S, Konosu T and Takatsu T, J. Antibiot. (Tokyo), 2019, 72, 943. [DOI] [PubMed] [Google Scholar]
- 46.Ubukata K, Kimura K, Isono K, Nelson CC, Gregson JM and Mcclosky JA, J. Org. Chem, 1992, 6392. [Google Scholar]
- 47.Hirano S, Ichikawa S and Matsuda A, Tetrahedron, 2007, 63, 2798. [Google Scholar]
- 48.Hirano S, Ichikawa S and Matsuda A, Angew. Chem. Int. Ed, 2005, 44, 1854. [DOI] [PubMed] [Google Scholar]
- 49.Ichikawa S and Matsuda A, Nucleosides Nucleotides Nucleic Acids, 2005, 24, 319. [DOI] [PubMed] [Google Scholar]
- 50.Hirano S, Ichikawa S and Matsuda A, J. Org. Chem, 2008, 73, 569. [DOI] [PubMed] [Google Scholar]
- 51.Wiker F, Hauck N, Grond S and Gust B, Int. J. Med. Microbiol, 2019, 309, 319. [DOI] [PubMed] [Google Scholar]
- 52.Kaysser L, Lutsch L, Siebenberg S, Wemakor E, Kammerer B and Gust B, J. Biol. Chem, 2009, 284, 14987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kaysser L, Siebenberg S, Kammerer B and Gust B, Chembiochem, 2010, 11, 191. [DOI] [PubMed] [Google Scholar]
- 54.Flinspach K, Westrich L, Kaysser L, Siebenberg S, Gomez-Escribano JP, Bibb M, Gust B and Heide L, Biopolymers, 2010, 93, 823. [DOI] [PubMed] [Google Scholar]
- 55.Kaysser L, Wemakor E, Siebenberg S, Salas JA, Sohng JK, Kammerer B and Gust B, Appl. Environ. Microbiol,. 2010, 76, 4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Funabashi M, Baba S, Nonaka K, Hosobuchi M, Fujita Y, Shibata T and Van Lanen SG, Chembiochem, 2010, 11, 184. [DOI] [PubMed] [Google Scholar]
- 57.Chi X, Baba S, Tibrewal N, Funabashi M, Nonaka K and Van Lanen SG, Medchemcomm, 2013, 4, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Z, Chi X, Funabashi M, Baba S, Nonaka K, Pahari P, Unrine J, Jacobsen JM, Elliott GI, Rohr J and Van Lanen SG, J. Biol. Chem, 2011, 286, 7885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chi X, Pahari P, Nonaka K and Van Lanen SG, J. Am. Chem. Soc, 2011, 133, 14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barnard-Britson S, Chi X, Nonaka K, Spork AP, Tibrewal N, Goswami A, Pahari P, Ducho C, Rohr J and Van Lanen SG, J. Am. Chem. Soc, 2012, 134, 18514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang Z, Unrine J, Nonaka K and Van Lanen SG, Methods Enzymol., 2012, 516, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goswami A, Liu X, Cai W, Wyche TP, Bugni TS, Meurillon M, Peyrottes S, Perigaud C, Nonaka K, Rohr J and Van Lanen SG, FEBS Lett., 2017, 591, 468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reeve AM, Breazeale SD and Townsend CA, J. Biol. Chem, 1998, 273, 30695. [DOI] [PubMed] [Google Scholar]
- 64.Gunsior M, Breazeale SD, Lind AJ, Ravel J, Janc JW and Townsend CA, Chem. Biol, 2004, 11, 927. [DOI] [PubMed] [Google Scholar]
- 65.Herbik A, Koch G, Mock HP, Dushkov D, Czihal A, Thielmann J, Stephan UW and Bäumlein H, Eur. J. Biochem, 1999, 265, 231. [DOI] [PubMed] [Google Scholar]
- 66.Dreyfus C, Lemaire D, Mari S, Pignol D and Arnoux P, Proc. Natl. Acad. Sci. U. S. A, 2009, 106, 16180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ghssein G, Brutesco C, Ouerdane L, Fojcik C, Izaute A, Wang S, Hajjar C, Lobinski R, Lemaire D, Richaud P, Voulhoux R, Espaillat A, Cava F, Pignol D, Borezée-Durant E and Arnoux P, Science, 2016, 352, 1105. [DOI] [PubMed] [Google Scholar]
- 68.Michael AJ, Biochem. J, 2016, 473, 2315. [DOI] [PubMed] [Google Scholar]
- 69.Cui Z, Overbay J, Wang X, Liu X, Zhang Y, Bhardwaj M, Lemke A, Wiegmann D, Niro G, Thorson JS, Ducho C and Van Lanen SG, Nat. Chem. Biol, 2020, 16, 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shiraishi T, Hiro N, Igarashi M, Nishiyama M and Kuzuyama T, J. Gen. Appl. Microbiol, 2016, 62, 164. [DOI] [PubMed] [Google Scholar]
- 71.Kaysser L, Eitel K, Tanino T, Siebenberg S, Matsuda A, Ichikawa S and Gust B, J. Biol. Chem, 2010, 285, 12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang X, Eitel K, Kaysser L, Kulik A, Grond S and Gust B, Nat. Chem. Biol, 2013, 9, 610. [DOI] [PubMed] [Google Scholar]
- 73.Lin YI, Li Z, Francisco GD, McDonald LA, Davis RA, Singh G, Yang Y and Mansour TS, Bioorg. Med. Chem. Lett, 2002, 12, 2341. [DOI] [PubMed] [Google Scholar]
- 74.McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G and Williamson RT, J. Am. Chem. Soc, 2002, 124, 10260. [DOI] [PubMed] [Google Scholar]
- 75.Funabashi M, Baba S, Takatsu T, Kizuka M, Ohata Y, Tanaka M, Nonaka K, Spork AP, Ducho C, Chen WC and Van Lanen SG, Angew. Chem. Int. Ed, 2013, 52, 11607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chung BC, Mashalidis EH, Tanino T, Kim M, Matsuda A, Hong J, Ichikawa S and Lee SY, Nature, 2016, 533, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koppermann S and Ducho C, Angew. Chem. Int. Ed, 2016, 55, 11722. [DOI] [PubMed] [Google Scholar]
- 78.Cheng L, Chen W, Zhai L, Xu D, Huang T, Lin S, Zhou X and Deng Z, Mol. Biosyst, 2011, 7, 920. [DOI] [PubMed] [Google Scholar]
- 79.Cui Z, Liu X, Overbay J, Cai W, Wang X, Lemke A, Wiegmann D, Niro G, Thorson JS, Ducho C and Van Lanen SG, J. Org. Chem, 2018, 83, 7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cui Z, Wang XC, Liu X, Lemke A, Koppermann S, Ducho C, Rohr J, Thorson JS and Van Lanen SG, Antimicrob. Agents Chemother, 2018, 62, e00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.White MF, Vasquez J, Yang SF and Kirsch JF, Proc. Natl. Acad. Sci. U. S. A, 1994, 91, 12428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bycroft BW, Cameron D, Croft LR, Hassanali-Walji A, Johnson AW and Webb T, Nature, 1971, 231, 301. [DOI] [PubMed] [Google Scholar]
- 83.Bycroft BW, Cameron D, Croft LR, Johnson AW, Webb T and Hassanali-Walji A, Experientia, 1971, 27, 501. [DOI] [PubMed] [Google Scholar]
- 84.Ju J, Ozanick SG, Shen B and Thomas MG, Chembiochem, 2004, 5, 1281. [DOI] [PubMed] [Google Scholar]
- 85.Yin X, McPhail KL, Kim KJ and Zabriskie TM, Chembiochem, 2004, 5, 1278. [DOI] [PubMed] [Google Scholar]
- 86.Yin X and Zabriskie TM, Chembiochem, 2004, 5, 1274. [DOI] [PubMed] [Google Scholar]
- 87.Felnagle EA, Podevels AM, Barkei JJ and Thomas MG, Chembiochem, 2011, 12, 1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fei X, Yin X, Zhang L and Zabriskie TM, J. Nat. Prod, 2007, 70, 618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Imker HJ, Walsh CT and Wuest WM, J. Am. Chem. Soc, 2009, 131, 18263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu D, Liu G, Cheng L, Lu X, Chen W and Deng Z, PLoS One, 2013, 8, e76068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yoshida M, Ezaki M, Hashimoto M, Yamashita M, Shigematsu N, Okuhara M, Kohsaka M and Horikoshi K, J. Antibiot. (Tokyo), 1990, 43, 748. [DOI] [PubMed] [Google Scholar]
- 92.Fu Y, Estoppey D, Roggo S, Pistorius D, Fuchs F, Studer C, Ibrahim AS, Aust T, Grandjean F, Mihalic M, Memmert K, Prindle V, Richard E, Riedl R, Schuierer S, Weber E, Hunziker J, Petersen F, Tao J and Hoepfner D, Nat. Commun, 2020, 11, 3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pietruszka J, Chem. Rev, 2003, 103, 1051. [DOI] [PubMed] [Google Scholar]
- 94.Falck J, Mekonnen B, Yu J and Lai J-Y, J. Am. Chem. Soc, 1996, 118, 6096. [Google Scholar]
- 95.Charette AB and Lebel H, J. Am. Chem. Soc, 1996, 118, 10327. [Google Scholar]
- 96.Hiratsuka T, Suzuki H, Kariya R, Seo T, Minami A and Oikawa H, Angew. Chem. Int. Ed, 2014, 53, 5423. [DOI] [PubMed] [Google Scholar]
- 97.Tokiwano T, Watanabe H, Seo T and Oikawa H, Chem. Commun. (Camb), 2008, 6016. [DOI] [PubMed] [Google Scholar]
- 98.Watanabe H, Tokiwano T and Oikawa H, J. Antibiot. (Tokyo), 2006, 59, 607. [DOI] [PubMed] [Google Scholar]
- 99.Hiratsuka T, Suzuki H, Minami A and Oikawa H, Org. Biomol. Chem, 2017, 15, 1076. [DOI] [PubMed] [Google Scholar]
- 100.Yamaguchi H, Sato S, Yoshida S, Takada K, Itoh M, Seto H and Otake N, J. Antibiot. (Tokyo), 1986, 39, 1047. [DOI] [PubMed] [Google Scholar]
- 101.Muramatsu Y, Ishii MM and Inukai M, J. Antibiot. (Tokyo), 2003, 56, 253. [DOI] [PubMed] [Google Scholar]
- 102.Muramatsu Y, Muramatsu A, Ohnuki T, Ishii MM, Kizuka M, Enokita R, Tsutsumi S, Arai M, Ogawa Y, Suzuki T, Takatsu T and Inukai M, J. Antibiot. (Tokyo), 2003, 56, 243. [DOI] [PubMed] [Google Scholar]
- 103.Muramatsu Y, Ohnuki T, Ishii MM, Kizuka M, Enokita R, Miyakoshi S, Takatsu T and Inukai M, J. Antibiot. (Tokyo), 2004, 57, 639. [DOI] [PubMed] [Google Scholar]
- 104.Murakami R, Fujita Y, Kizuka M, Kagawa T, Muramatsu Y, Miyakoshi S, Takatsu T and Inukai M, J. Antibiot. (Tokyo), 2007, 60, 690. [DOI] [PubMed] [Google Scholar]
- 105.Seto H, Otake N, Sato S, Yamaguchi H, Takada K, Itoh M, Lu HSM and Clardy J, Tetrahedron Lett., 1988, 29, 2343. [Google Scholar]
- 106.Muramatsu Y, Miyakoshi S, Ogawa Y, Ohnuki T, Ishii MM, Arai M, Takatsu T and Inukai M, J. Antibiot. (Tokyo), 2003, 56, 259. [DOI] [PubMed] [Google Scholar]
- 107.Ohnuki T, Muramatsu Y, Miyakoshi S, Takatsu T and Inukai M, J. Antibiot. (Tokyo), 2003, 56, 268. [DOI] [PubMed] [Google Scholar]
- 108.Cai W, Goswami A, Yang Z, Liu X, Green KD, Barnard-Britson S, Baba S, Funabashi M, Nonaka K, Sunkara M, Morris AJ, Spork AP, Ducho C, Garneau-Tsodikova S, Thorson JS and Van Lanen SG, J. Biol. Chem, 2015, 290, 13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Funabashi M, Nonaka K, Yada C, Hosobuchi M, Masuda N, Shibata T and Van Lanen SG, J. Antibiot. (Tokyo), 2009, 62, 325. [DOI] [PubMed] [Google Scholar]
- 110.Funabashi M, Yang Z, Nonaka K, Hosobuchi M, Fujita Y, Shibata T, Chi X and Van Lanen SG, Nat. Chem. Biol, 2010, 6, 581. [DOI] [PubMed] [Google Scholar]
- 111.Huang Y, Liu X, Cui Z, Wiegmann D, Niro G, Ducho C, Song Y, Yang Z and Van Lanen SG, Proc. Natl. Acad. Sci. U. S. A, 2018, 115, 974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hoffarth ER, Rothchild KW and Ryan KS, FEBS J., 2020, 287, 1403. [DOI] [PubMed] [Google Scholar]
- 113.Liu X, Jin Y, Cui Z, Nonaka K, Baba S, Funabashi M, Yang Z and Van Lanen SG, Chembiochem, 2016, 17, 804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu X, Jin Y, Cai W, Green KD, Goswami A, Garneau-Tsodikova S, Nonaka K, Baba S, Funabashi M, Yang Z and Van Lanen SG, Org. Biomol. Chem, 2016, 14, 3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang Z, Funabashi M, Nonaka K, Hosobuchi M, Shibata T, Pahari P and Van Lanen SG, J. Biol. Chem, 2010, 285, 12899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Takatsuki A, Arima K and Tamura G, J. Antibiot. (Tokyo), 1971, 24, 215. [DOI] [PubMed] [Google Scholar]
- 117.Takatsuki A and Tamura G, J. Antibiot. (Tokyo), 1971, 24, 224–231. [PubMed] [Google Scholar]
- 118.Doroghazi JR, Ju KS, Brown DW, Labeda DP, Deng Z, Metcalf WW, Chen W and Price NP, J. Bacteriol, 2011, 193, 7021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Thrum H, Eckardt K, Bradler G, Fügner R, Tonew E and Tonew M, J. Antibiot. (Tokyo), 1975, 28, 514. [DOI] [PubMed] [Google Scholar]
- 120.Vogel P, Petterson DS, Berry PH, Frahn JL, Anderton N, Cockrum PA, Edgar JA, Jago MV, Lanigan GW, Payne AL and Culvenor CC, Aust. J. Exp. Biol. Med. Sci, 1981, 59, 455. [DOI] [PubMed] [Google Scholar]
- 121.Kenig M and Reading C, J. Antibiot. (Tokyo), 1979, 32, 549. [DOI] [PubMed] [Google Scholar]
- 122.Bettinger GE and Young FE, Biochem. Biophys. Res. Commun, 1975, 67, 16. [DOI] [PubMed] [Google Scholar]
- 123.Tkacz JS and Lampen O, Biochem. Biophys. Res. Commun, 1975, 65, 248. [DOI] [PubMed] [Google Scholar]
- 124.Kuo SC and Lampen JO, Biochem. Biophys. Res. Commun, 1974, 58, 287. [DOI] [PubMed] [Google Scholar]
- 125.Takatsuki A, Shimizu KI and Tamura G, J. Antibiot. (Tokyo), 1972, 25, 75. [DOI] [PubMed] [Google Scholar]
- 126.Ward JB, FEBS Lett., 1977, 78, 151. [DOI] [PubMed] [Google Scholar]
- 127.Brandish PE, Kimura KI, Inukai M, Southgate R, Lonsdale JT and Bugg TD, Antimicrob. Agents Chemother, 1996, 40, 1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mashalidis EH and Lee SY, J. Mol. Biol, 2020, 432, 4946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yamamoto K, Katsuyama A and Ichikawa S, Bioorg. Med. Chem, 2019, 27, 1714. [DOI] [PubMed] [Google Scholar]
- 130.Lehle L and Tanner W, FEBS Lett., 1976, 72, 167. [DOI] [PubMed] [Google Scholar]
- 131.Eckardt K, Wetzstein H, Thrum H and Ihn W, J. Antibiot. (Tokyo), 1980, 33, 908. [DOI] [PubMed] [Google Scholar]
- 132.Tsvetanova BC, Kiemle DJ and Price NP, J. Biol. Chem, 2002, 277, 35289. [DOI] [PubMed] [Google Scholar]
- 133.Price NP, Labeda DP, Naumann TA, Vermillion KE, Bowman MJ, Berhow MA, Metcalf WW and Bischoff KM, J. Antibiot. (Tokyo), 2016, 69, 637. [DOI] [PubMed] [Google Scholar]
- 134.Chen W, Qu D, Zhai L, Tao M, Wang Y, Lin S, Price NP and Deng Z, Protein Cell, 2010, 1, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wyszynski FJ, Hesketh AR, Bibb MJ and Davis BG, Chem. Sci, 2010, 1, 581. [Google Scholar]
- 136.Widdick D, Royer SF, Wang H, Vior NM, Gomez-Escribano JP, Davis BG and Bibb MJ, Antimicrob. Agents Chemother, 2018, 62, e00130–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Karki S, Kwon S-Y and Kwon H-J, J. Kor. Soc. Appl. Biol. Chem, 2011, 54, 136. [Google Scholar]
- 138.Wyszynski FJ, Lee SS, Yabe T, Wang H, Gomez-Escribano JP, Bibb MJ, Lee SJ, Davies GJ and Davis BG, Nat. Chem, 2012, 4, 539. [DOI] [PubMed] [Google Scholar]
- 139.Goddard-Borger ED and Withers SG, Nat. Chem, 2012, 4, 520. [DOI] [PubMed] [Google Scholar]
- 140.Noda Y, Yoda K, Takatsuki A and Yamasaki M, J. Bacteriol, 1992, 174, 4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Noda Y, Takatsuki A, Yoda K and Yamasaki M, Biosci. Biotechnol. Biochem, 1995, 59, 321. [DOI] [PubMed] [Google Scholar]
- 142.Noda Y, Yoda K and Yamasaki M, Biosci. Biotechnol. Biochem, 1994, 58, 196. [DOI] [PubMed] [Google Scholar]
- 143.Zutz A, Hoffmann J, Hellmich UA, Glaubitz C, Ludwig B, Brutschy B and Tampé R, J. Biol. Chem, 2011, 286, 7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Murakami R, Fujita Y, Kizuka M, Kagawa T, Muramatsu Y, Miyakoshi S, Takatsu T and Inukai M, J. Antibiot. (Tokyo), 2008, 61, 537. [DOI] [PubMed] [Google Scholar]
- 145.Fujita Y, Murakami R, Muramatsu Y, Miyakoshi S and Takatsu T, J. Antibiot. (Tokyo), 2008, 61, 545. [DOI] [PubMed] [Google Scholar]
- 146.Yuan Y, Fuse S, Ostash B, Sliz P, Kahne D and Walker S, ACS Chem. Biol, 2008, 3, 429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Huber G and Nesemann G, Biochem. Biophys. Res. Commun, 1968, 30, 7. [DOI] [PubMed] [Google Scholar]
- 148.Shiraishi T, Nishiyama M and Kuzuyama T, Org. Biomol. Chem, 2019, 17, 461. [DOI] [PubMed] [Google Scholar]
- 149.Dähn U, Hagenmaier H, Höhne H, König WA, Wolf G and Zähner H, Arch. Microbiol, 1976, 107, 143. [DOI] [PubMed] [Google Scholar]
- 150.Suzuki S, Isono K, Nagatsu J, Mizutani T, Kawashima Y and Mizuno T, J. Antibiot. (Tokyo), 1965, 18, 131. [PubMed] [Google Scholar]
- 151.Zhai L, Lin S, Qu D, Hong X, Bai L, Chen W and Deng Z, Metab. Eng, 2012, 14, 388. [DOI] [PubMed] [Google Scholar]
- 152.Qi J, Liu J, Wan D, Cai YS, Wang Y, Li S, Wu P, Feng X, Qiu G, Yang SP, Chen W and Deng Z, Biotechnol. Bioeng, 2015, 112, 1865. [DOI] [PubMed] [Google Scholar]
- 153.Li J, Li L, Tian Y, Niu G and Tan H, Metab. Eng, 2011, 13, 336. [DOI] [PubMed] [Google Scholar]
- 154.Wang G and Tan H, Biotechnol Lett, 2004, 26, 229–233. [DOI] [PubMed] [Google Scholar]
- 155.Kim MK, Park HS, Kim CH, Park HM and Choi W, Yeast, 2002, 19, 341. [DOI] [PubMed] [Google Scholar]
- 156.Gaughran JP, Lai MH, Kirsch DR and Silverman SJ, J. Bacteriol, 1994, 176, 5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Schlüter U, Wilehm Roux Arch. Dev. Biol, 1982, 191, 205. [DOI] [PubMed] [Google Scholar]
- 158.Endo A, Kakiki K and Misato T, J. Bacteriol, 1970, 104, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Endo A and Misato T, Biochem. Biophys. Res. Commun, 1969, 37, 718. [DOI] [PubMed] [Google Scholar]
- 160.Tariq VN and Devlin PL, Fungal Genet. Biol, 1996, 20, 4. [DOI] [PubMed] [Google Scholar]
- 161.Hector RF, Clin. Microbiol. Rev, 1993, 6, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chapman T, Kinsman O and Houston J, Antimicrob. Agents Chemother, 1992, 36, 1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Goughenour KD and Rappleye CA, Virulence, 2017, 8, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Shubitz LF, Trinh HT, Perrill RH, Thompson CM, Hanan NJ, Galgiani JN and Nix DE, J. Infect. Dis, 2014, 209, 1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Shubitz LF, Roy ME, Nix DE and Galgiani JN, Med. Mycol, 2013, 51, 747. [DOI] [PubMed] [Google Scholar]
- 166.Hector RF, Zimmer BL and Pappagianis D, Antimicrob. Agents Chemother, 1990, 34, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Isono K, Asahi K and Suzuki S, J. Am. Chem. Soc, 1969, 91, 7490. [DOI] [PubMed] [Google Scholar]
- 168.Isono K and Suzuki S, Tetrahedron Lett., 1968, 2, 203. [DOI] [PubMed] [Google Scholar]
- 169.Heitsch H, König WA, Decker H, Bormann C, Fiedler HP and Zähner H, J. Antibiot. (Tokyo), 1989, 42, 711. [DOI] [PubMed] [Google Scholar]
- 170.Uramoto M, Kobinata K, Isono K, Jenkins EE, McCloskey JA, Higashijima T and Miyazawa T, Nucleic Acids Symp. Ser, 1980, s69. [PubMed] [Google Scholar]
- 171.Bormann C, Mattern S, Schrempf H, Fiedler HP and Zähner H, J. Antibiot. (Tokyo), 1989, 42, 913. [DOI] [PubMed] [Google Scholar]
- 172.Isono K, Crain PF and McCloskey JA, Biomed. Mass Spectrom, 1978, 5, 89. [DOI] [PubMed] [Google Scholar]
- 173.Isono K, Sato T, Hirasawa K, Funayama S and Suzuki S, J. Am. Chem. Soc, 1978, 100, 3937. [Google Scholar]
- 174.Funayama S and Isono K, Biochemistry, 1977, 16, 3121. [DOI] [PubMed] [Google Scholar]
- 175.Funayama S and Isono K, Biochemistry, 1975, 14, 5568. [DOI] [PubMed] [Google Scholar]
- 176.Isono K, Funayama S and Suhadolnik RJ, Biochemistry, 1975, 14, 2992. [DOI] [PubMed] [Google Scholar]
- 177.Evans DR, Herbert RB, Baumberg S, Cove JH, Southey EA, Buss AD, Dawson MJ, Noble D and Rudd BAM, Tetrahedron Lett., 1995, 36, 2351. [Google Scholar]
- 178.Fiedler HP, Kurth R, Langhärig J, Delzer J and Zähner H, J. Chem. Technol. Biotechnol, 1982, 32, 271. [Google Scholar]
- 179.Bormann C, Möhrle V and Bruntner C, J. Bacteriol. 1996, 178, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chen W, Huang T, He X, Meng Q, You D, Bai L, Li J, Wu M, Li R, Xie Z, Zhou H, Zhou X, Tan H and Deng Z, J. Biol. Chem, 2009, 284, 10627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Bormann C, Microbial Fundamentals of Biotechnology, 2001, 102. [Google Scholar]
- 182.Liao G, Li J, Li L, Yang H, Tian Y and Tan H, Microb. Cell Fact, 2010, 9, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lauer B, Russwurm R and Bormann C, Eur. J. Biochem, 2000, 267, 1698. [DOI] [PubMed] [Google Scholar]
- 184.Chen H, Hubbard BK, O'Connor SE and Walsh CT, Chem. Biol, 2002, 9, 103. [DOI] [PubMed] [Google Scholar]
- 185.Ginj C, Rüegger H, Amrhein N and Macheroux P, Chembiochem, 2005, 6, 1974. [DOI] [PubMed] [Google Scholar]
- 186.Oberdorfer G, Binter A, Ginj C, Macheroux P and Gruber K, J. Biol. Chem, 2012, 287, 31427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Yokoyama K and Lilla EA, Nat. Prod. Rep, 2018, 35, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lilla EA and Yokoyama K, Nat. Chem. Biol, 2016, 12, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.He N, Wu P, Lei Y, Xu B, Zhu X, Xu G, Gao Y, Qi J, Deng Z, Tang G, Chen W and Xiao Y, Chem. Sci, 2017, 8, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Binter A, Oberdorfer G, Hofzumahaus S, Nerstheimer S, Altenbacher G, Gruber K and Macheroux P, FEBS J., 2011, 278, 4122. [DOI] [PubMed] [Google Scholar]
- 191.Bruntner C and Bormann C, Eur. J. Biochem, 1998, 254, 347. [DOI] [PubMed] [Google Scholar]
- 192.Carrell CJ, Bruckner RC, Venci D, Zhao G, Jorns MS and Mathews FS, Structure, 2007, 15, 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bruckner RC, Zhao G, Venci D and Jorns MS, Biochemistry, 2004, 43, 9160. [DOI] [PubMed] [Google Scholar]
- 194.Venci D, Zhao G and Jorns MS, Biochemistry, 2002, 41, 15795. [DOI] [PubMed] [Google Scholar]
- 195.Moon M and Van Lanen SG, Biopolymers, 2010, 93, 791. [DOI] [PubMed] [Google Scholar]
- 196.Niu G, Liu G, Tian Y and Tan H, Metab. Eng, 2006, 8, 183. [DOI] [PubMed] [Google Scholar]
- 197.Ling HB, Wang GJ, Li JE and Tan HR, J. Microbiol. Biotechnol,. 2008, 18, 397. [PubMed] [Google Scholar]
- 198.Ling H, Wang G, Tian Y, Liu G and Tan H, Biochem. Biophys. Res. Commun, 2007, 361, 196. [DOI] [PubMed] [Google Scholar]
- 199.Jia L, Tian Y and Tan H, Biochem. Biophys. Res. Commun, 2007, 362, 1031. [DOI] [PubMed] [Google Scholar]
- 200.Li Y, Zeng H and Tan H, Curr. Microbiol, 2004, 49, 128. [DOI] [PubMed] [Google Scholar]
- 201.Bruntner C, Lauer B, Schwarz W, Möhrle V and Bormann C, Mol. Gen. Genet, 1999, 262, 102. [DOI] [PubMed] [Google Scholar]
- 202.Xie Z, Niu G, Li R, Liu G and Tan H, Curr. Microbiol, 2007, 55, 537. [DOI] [PubMed] [Google Scholar]
- 203.Chen W, Dai D, Wang C, Huang T, Zhai L and Deng Z, Microb. Cell Fact, 2013, 12, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Qi J, Wan D, Ma H, Liu Y, Gong R, Qu X, Sun Y, Deng Z and Chen W, Cell Chem. Biol, 2016, 23, 935. [DOI] [PubMed] [Google Scholar]
- 205.Gong R, Qi J, Wu P, Cai YS, Ma H, Liu Y, Duan H, Wang M, Deng Z, Price NPJ and Chen W, Appl. Environ. Microbiol, 2018, 84, e00501–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.He X, Li R, Pan Y, Liu G and Tan H, Microbiology, 2010, 156, 828. [DOI] [PubMed] [Google Scholar]
- 207.Li R, Xie Z, Tian Y, Yang H, Chen W, You D, Liu G, Deng Z and Tan H, Microbiology, 2009, 155, 1819. [DOI] [PubMed] [Google Scholar]
- 208.Li R, Liu G, Xie Z, He X, Chen W, Deng Z and Tan H, Mol. Microbiol, 2010, 75, 349. [DOI] [PubMed] [Google Scholar]
- 209.Maffioli SI, Zhang Y, Degen D, Carzaniga T, Del Gatto G, Serina S, Monciardini P, Mazzetti C, Guglierame P, Candiani G, Chiriac AI, Facchetti G, Kaltofen P, Sahl HG, Dehò G, Donadio S and Ebright RH, Cell, 2017, 169, 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.O'Malley PA, Clin. Nurse Spec, 2018, 32, 114. [DOI] [PubMed] [Google Scholar]
- 211.Chellat MF and Riedl R, Angew. Chem. Int. Ed, 2017, 56, 13184. [DOI] [PubMed] [Google Scholar]
- 212.Sosio M, Gaspari E, Iorio M, Pessina S, Medema MH, Bernasconi A, Simone M, Maffioli SI, Ebright RH and Donadio S, Cell Chem. Biol, 2018, 25, 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Chen J and Patton JR, RNA, 1999, 5, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Gause GF, Br. Med. J, 1955, 2, 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Turkova J, Mikes O, Sraml I, Knessl O and Sorum F, Antibiotiki, 1964, 9, 506. [PubMed] [Google Scholar]
- 216.Turkova J, Mikes O and Sorum F, Experientia, 1963, 19, 633. [DOI] [PubMed] [Google Scholar]
- 217.Braun V, Pramanik A, Gwinner T, Köberle M and Bohn E, Biometals, 2009, 22, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hartmann A, Fiedler HP and Braun V, Eur. J. Biochem, 1979, 99, 517. [DOI] [PubMed] [Google Scholar]
- 219.Stapley EO and Ormond RE, Science, 1957, 125, 587. [DOI] [PubMed] [Google Scholar]
- 220.Maehr H and Berger J, Biotechnol. Bioengineering, 1969, 11, 1111. [DOI] [PubMed] [Google Scholar]
- 221.Sensi P and Timbal MT, Antibiot. Chemother. 1959, 9, 160. [PubMed] [Google Scholar]
- 222.Paulsen H, Brieden M and Benz G, Liebigs Annalen der Chemie, 1987, 1987, 565. [Google Scholar]
- 223.Pramanik A, Stroeher UH, Krejci J, Standish AJ, Bohn E, Paton JC, Autenrieth IB and Braun V, Int. J. Med. Microbiol, 2007, 297, 459. [DOI] [PubMed] [Google Scholar]
- 224.Krechmer BB, Val'ter EM and Baiandina SA, Sov. Med, 1951, 10, 10. [PubMed] [Google Scholar]
- 225.Lin Z, Xu X, Zhao S, Yang X, Guo J, Zhang Q, Jing C, Chen S and He Y, Nat. Commun, 2018, 9, 3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Saha A, Dutta S and Nandi N, J. Biomol. Struct. Dyn, 2020, 38, 2440. [DOI] [PubMed] [Google Scholar]
- 227.Stefanska AL, Fulston M, Houge-Frydrych CS, Jones JJ and Warr SR, J. Antibiot. (Tokyo), 2000, 53, 1346. [DOI] [PubMed] [Google Scholar]
- 228.Zalkin A, Forrester JD and Templeton DH, J. Am. Chem. Soc, 1966, 88, 1810. [DOI] [PubMed] [Google Scholar]
- 229.Zeng Y, Kulkarni A, Yang Z, Patil PB, Zhou W, Chi X, Van Lanen S and Chen S, ACS Chem. Biol, 2012, 7, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Zeng Y, Roy H, Patil PB, Ibba M and Chen S, Antimicrob. Agents Chemother, 2009, 53, 4619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ushimaru R and Liu HW, J. Am. Chem. Soc, 2019, 141, 2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Ushimaru R, Chen Z, Zhao H, Fan PH and Liu HW, Angew. Chem. Int. Ed, 2020, 59, 3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Emery TF, Biochemistry, 1966, 5, 3694. [DOI] [PubMed] [Google Scholar]
- 234.Flynn EH, Hinman JW, Caron EL and Woolf DO, J. Am. Chem. Soc, 1953, 75, 5867. [Google Scholar]
- 235.DeBoer C, Caron EL and Hinman JW, J. Am. Chem. Soc, 1953, 75, 499. [Google Scholar]
- 236.Leviev IG, Rodriguez-Fonseca C, Phan H, Garrett RA, Heilek G, Noller HF and Mankin AS, EMBO J., 1994, 13, 1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.González A, Vázquez D and Jiménez A, Biochim. Biophys. Acta, 1979, 561, 403. [DOI] [PubMed] [Google Scholar]
- 238.Contreras A, Vazquez D and Carrasco L, J. Antibiot. (Tokyo), 1978, 31, 598. [DOI] [PubMed] [Google Scholar]
- 239.Lichtenthaler FW, Cerná J and Rychlik I, FEBS Lett., 1975, 53, 184. [DOI] [PubMed] [Google Scholar]
- 240.Bloch A and Coutsogeorgopoulos C, Biochemistry, 1966, 5, 3345. [DOI] [PubMed] [Google Scholar]
- 241.Carrasco L and Vázquez D, Med. Res. Rev, 1984, 4, 471. [DOI] [PubMed] [Google Scholar]
- 242.Contreras A and Carrasco L, J. Virol, 1979, 29, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Leviev IG, Rodriguez-Fonseca C, Phan H, Garrett RA, Heilek G, Noller HF and Mankin AS, EMBO J., 1994, 13, 1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Haskell TH, Ryder A, Frohardt RP, Fusari SA, Jakubowski ZL and Bartz QR, J. Am. Chem. Soc, 1958, 80, 743. [Google Scholar]
- 245.Chen Y, Zeeck A, Chen Z and Zahner H, Kangshengsu, 1985, 10, 285. [Google Scholar]
- 246.Itoh J and Miyadoh S, J. Antibiot. (Tokyo), 1992, 45, 846. [DOI] [PubMed] [Google Scholar]
- 247.Konishi M, Naruishi M, Tsuno T, Tsukiura H and Kawaguchi H, J. Antibiot. (Tokyo), 1973, 26, 757. [DOI] [PubMed] [Google Scholar]
- 248.Konishi M, Kimeda M, Tsukiura H, Yamamoto H and Hoshiya T, J. Antibiot. (Tokyo), 1973, 26, 752. [DOI] [PubMed] [Google Scholar]
- 249.Xu C-D, Zhang H-J and Ma Z-J, J. Nat. Prod, 2019, 82, 2509. [DOI] [PubMed] [Google Scholar]
- 250.Haneda K, Shinose M, Seino A, Tabata N, Tomoda H, Iwai Y and Omura S, J. Antibiot. (Tokyo), 1994, 47, 774. [DOI] [PubMed] [Google Scholar]
- 251.Shiomi K, Haneda K, Tomoda H, Iwai Y and Omura S, J. Antibiot. (Tokyo), 1994, 47, 782. [DOI] [PubMed] [Google Scholar]
- 252.Aksoy S, Uzel A and Bedir E, J. Antibiot. (Tokyo), 2016, 69, 51. [DOI] [PubMed] [Google Scholar]
- 253.Bu YY, Yamazaki H, Ukai K and Namikoshi M, Mar. Drugs, 2014, 12, 6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Stevens CL, Nagarajan K and Haskell TH, J. Org. Chem, 1962, 27, 2991. [Google Scholar]
- 255.Zhang G, Zhang H, Li S, Xiao J, Zhu Y, Niu S, Ju J and Zhang C, Appl. Environ. Microbiol, 2012, 78, 2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Grochowski LL and Zabriskie TM, Chembiochem, 2006, 7, 957. [DOI] [PubMed] [Google Scholar]
- 257.Cone MC, Yin X, Grochowski LL, Parker MR and Zabriskie TM, Chembiochem, 2003, 4, 821. [DOI] [PubMed] [Google Scholar]
- 258.Chen R, Zhang H, Zhang G, Li S, Zhu Y, Liu J and Zhang C, J. Am. Chem. Soc, 2013, 135, 12152. [DOI] [PubMed] [Google Scholar]
- 259.Takeuchi S, Hirayama K, Ueda K, Sakai H and Yonehara H, J. Antibiot. (Tokyo), 1958, 11, 1. [PubMed] [Google Scholar]
- 260.Argoudelis AD, Baczynskyj L, Kuo MT, Laborde AL, Sebek OK, Truesdell SE and Shilliday FB, J. Antibiot. (Tokyo), 1987, 40, 750. [DOI] [PubMed] [Google Scholar]
- 261.Neumann T, Ihn W, Ritzau M, Vettermann R, Fleck WF and Grafe U, Nat. Prod. Lett, 1996, 8, 137. [Google Scholar]
- 262.Harada S and Kishi T, J. Antibiot. (Tokyo), 1978, 31, 519. [DOI] [PubMed] [Google Scholar]
- 263.Iwasa T, Suetomi K and Kusaka T, J. Antibiot. (Tokyo), 1978, 31, 511. [DOI] [PubMed] [Google Scholar]
- 264.Davison JR, Lohith KM, Wang X, Bobyk K, Mandadapu SR, Lee SL, Cencic R, Nelson J, Simpkins S, Frank KM, Pelletier J, Myers CL, Piotrowski J, Smith HE and Bewley CA, Antimicrob. Agents Chemother, 2017, 61, e02635–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Yamaguchi H, Yamamoto C and Tanaka N, J. Biochem, 1965, 57, 667. [PubMed] [Google Scholar]
- 266.Huang KT, Misato T and Asuyama H, J. Antibiot. (Tokyo), 1964, 17, 65. [PubMed] [Google Scholar]
- 267.Svidritskiy E and Korostelev AA, 2018, 430, 591–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Feduchi E, Cosín M and Carrasco L, J. Antibiot. (Tokyo), 1985, 38, 415. [DOI] [PubMed] [Google Scholar]
- 269.Fox JJ and Watanabe KA, Tetrahedron Lett., 1966, 9, 897. [DOI] [PubMed] [Google Scholar]
- 270.Otake N, Takeuchi S, Endo T and Yonehara H, Tetrahedron Lett., 1965, 1405. [DOI] [PubMed] [Google Scholar]
- 271.Otake N, Takeuchi S, Endo T and Yonehara H, Tetrahedron Lett., 1965, 1411. [DOI] [PubMed] [Google Scholar]
- 272.Endo T, Otake N, Takeuchi S and Yonehara H, J. Antibiot. (Tokyo), 1964, 17, 172. [PubMed] [Google Scholar]
- 273.Yonehara H, Takeuchi S, Otake N, Endo T, Sakagamiand Y Sumiki Y, J. Antibiot. (Tokyo), 1963, 16, 195. [PubMed] [Google Scholar]
- 274.Seto H and Yonehara H, J. Antibiot. (Tokyo), 1977, 30, 1022. [DOI] [PubMed] [Google Scholar]
- 275.Seto H and Yonehara H, J. Antibiot. (Tokyo), 1977, 30, 1019. [DOI] [PubMed] [Google Scholar]
- 276.Seto H, Furihata K and Yonehara H, J. Antibiot. (Tokyo), 1976, 29, 595. [DOI] [PubMed] [Google Scholar]
- 277.Gould SJ and Guo J, J. Am. Chem. Soc, 1992, 114, 10176. [Google Scholar]
- 278.Guo J and Gould SJ, J. Am. Chem. Soc, 1991, 113, 5898. [Google Scholar]
- 279.Seto H, Otake N and Yonehara H, Agr. Biol. Chem, 1973, 37, 2421–2426. [Google Scholar]
- 280.Harada S, Mizuta E and Kishi T, J. Am. Chem. Soc, 1978, 100, 4895. [Google Scholar]
- 281.Larsen SH, Berry DM, Paschal JW and Gilliam JM, J. Antibiot. (Tokyo), 1989, 42, 470. [DOI] [PubMed] [Google Scholar]
- 282.Cooper R, Conover M and Patel M., J. Antibiot. (Tokyo), 1988, 41, 123. [DOI] [PubMed] [Google Scholar]
- 283.Cone MC, Petrich AK, Gould SJ and Zabriskie TM, J. Antibiot. (Tokyo), 1998, 51, 570. [DOI] [PubMed] [Google Scholar]
- 284.Li L, Xu Z, Xu X, Wu J, Zhang Y, He X, Zabriskie TM and Deng Z, Chembiochem, 2008, 9, 1286. [DOI] [PubMed] [Google Scholar]
- 285.Wu J, Li L, Deng Z, Zabriskie TM and He X, Chembiochem, 2012, 13, 1613. [DOI] [PubMed] [Google Scholar]
- 286.Feng J, Wu J, Gao J, Xia Z, Deng Z and He X, Appl. Environ. Microbiol, 2014, 80, 5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Seto H, Yamaguchi I, Ōtake N and Yonehara H, Agr. Biol. Chem, 1968, 32, 1292. [Google Scholar]
- 288.Gould SJ, Guo J, Geitmann A and Dejesus K, Can. J. Chem. 1994, 72, 6. [Google Scholar]
- 289.Kim HJ, LeVieux J, Yeh YC and Liu HW, Angew. Chem. Int. Ed, 2016, 55, 3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Liu L, Ji X, Li Y, Ji W, Mo T, Ding W and Zhang Q, Chem. Commun. (Camb), 2017, 53, 8952. [DOI] [PubMed] [Google Scholar]
- 291.Feng J, Wu J, Dai N, Lin S, Xu HH, Deng Z and He X, PLoS One, 2013, 8, e68545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Aberhart DJ, Gould SJ, Lin HJ, Thiruvengadam TK and Weiller BH, J. Am. Chem. Soc, 1983, 105, 5461. [Google Scholar]
- 293.Frey PA, Ballinger MD and Reed GH, Biochem. Soc. Trans, 1998, 26, 304. [DOI] [PubMed] [Google Scholar]
- 294.Zhang Q, Cone MC, Gould SJ and Mark Zabriskie T, Tetrahedron, 2000, 56, 693. [Google Scholar]
- 295.Wang X, Du A, Yu G, Deng Z and He X, Front. Microbiol, 2017, 8, 1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Han L, Vuksanovic N, Oehm SA, Fenske TG, Schwabacher AW and Silvaggi NR, Biochemistry, 2018, 57, 3252. [DOI] [PubMed] [Google Scholar]
- 297.Han L, Schwabacher AW, Moran GR and Silvaggi NR, Biochemistry, 2015, 54, 7029. [DOI] [PubMed] [Google Scholar]
- 298.Du YL and Ryan KS, Nat. Prod. Rep, 2019, 36, 430. [DOI] [PubMed] [Google Scholar]
- 299.Clark JM and Gunther JK, Biochim. Biophys. Acta, 1963, 76, 636. [PubMed] [Google Scholar]
- 300.Niu G and Tan H, Trends Microbiol., 2015, 23, 110. [DOI] [PubMed] [Google Scholar]
- 301.Takahashi A, Ikeda D, Naganawa H, Okami Y and Umezawa H, J. Antibiot. (Tokyo), 1986, 39, 1041. [DOI] [PubMed] [Google Scholar]
- 302.Takahashi A, Saito N, Hotta K, Okami Y and Umezawa H, J. Antibiot. (Tokyo), 1986, 39, 1033. [DOI] [PubMed] [Google Scholar]
- 303.Casjens SR and Morris AJ, Biochim. Biophys. Acta, 1965, 108, 677. [DOI] [PubMed] [Google Scholar]
- 304.Kirillov SV, Porse BT and Garrett RA, RNA, 1999, 5, 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Wang T, Yin S and Hu J, Bull. Environ. Contam. Toxicol, 2013, 90, 256. [DOI] [PubMed] [Google Scholar]
- 306.Dolak L, J. Antibiot. (Tokyo), 1979, 32, 1346. [DOI] [PubMed] [Google Scholar]
- 307.Fox JJ, Kuwasa Y and Watanabe KA, Tetrahedron Lett., 1968, 6029. [DOI] [PubMed] [Google Scholar]
- 308.Fox JJ, Kuwada Y, Watanabe KA, Ueda T and Whipple EB, Antimicrob. Agents Chemother. (Bethesda), 1964, 10, 518. [PubMed] [Google Scholar]
- 309.Niu G, Li L, Wei J and Tan H, Chem. Biol, 2013, 20, 34. [DOI] [PubMed] [Google Scholar]
- 310.Jiang L, Wei J, Li L, Niu G and Tan H, Appl. Microbiol. Biotechnol, 2013, 97, 10469. [DOI] [PubMed] [Google Scholar]
- 311.Wei J, Tian Y, Niu G and Tan H, Appl. Environ. Microbiol, 2014, 80, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Wilker F, Konnerth M, Helmle I, Kulik A, Kaysser L, Gross H and Gust B, ACS Chem. Biol 2019, 14, 1972. [DOI] [PubMed] [Google Scholar]