

Abstract
Photodynamic therapy (PDT) is an effective anti-cancer modality approved by the U.S. Food and Drug Administration (FDA). Anti-tumor immunity can be augmented during PDT by inducing sterile inflammation in an acute manner, and this process is characterized by interleukin 17 (IL-17)-mediated neutrophil infiltration to tumor-draining lymph nodes (TDLNs). However, the inflammatory factors that influence IL-17 expression in TDLNs are poorly understood. Prior studies have linked the cyclooxygenase 2 (COX2)-driven prostaglandin E2 (PGE2) pathway to IL-17 expression. Here, we report that an immune-activating PDT regimen (imPDT) induces COX2/PGE2 expression in TDLNs, whereby IL-17 expression is facilitated without corresponding effects on the expression of RORγt, the transcriptional driver of the canonical IL-17 pathway. Pharmacologic inhibition with NS398, a COX2 inhibitor, was utilized to demonstrate that imPDT-induced COX2 regulates RORγt-independent expression of IL-17 by B cells and neutrophil entry into TDLNs. Depletion of B cells prior to imPDT significantly reduced neutrophil entry into TDLNs following treatment, and diminishes the efficacy of imPDT, which is dependent upon anti-tumor immunity. These findings are suggestive of a novel role for B cells in the augmentation of anti-tumor immunity by imPDT.
Keywords: Cyclooxygenase 2, Prostaglandin E2, IL-17, B cells, neutrophils, tumor-draining lymph nodes
INTRODUCTION
Neutrophils are the first responders to sites of sterile and pathogen-induced inflammation. During inflammation, neutrophils migrate to draining lymph nodes and contribute to the activation of the adaptive immune system (1–3). Immune-activating photodynamic therapy (imPDT), which involves a low dose of light delivered at slow rate, requires interleukin 17 (IL-17)-dependent neutrophil migration to tumor-draining lymph nodes (TDLNs) for the stimulation of anti-tumor immunity and efficacy (4,5). Presently, the mechanism by which imPDT regulates IL-17 expression in TDLNs is not known.
Prostaglandin E2 (PGE2) is generated from membrane phospholipids via the cyclooxygenase (COX) pathway. There are two forms of COX; COX1 is expressed constitutively and catalyzes the rate-limiting step of PGE2 synthesis under homeostatic conditions, and COX2 is induced rapidly during inflammation (6). imPDT induces COX2 expression in tumor tissue (7). Earlier research also has shown that PGE2 facilitates neutrophil accumulation at inflamed sites in an IL-17-dependent manner in an experimental mice model of rheumatoid arthritis (RA) (8).
IL-17 is expressed by innate lymphoid cells (ILCs), T helper 17 (Th17) cells, and myeloid cells (9–11). In addition, IL-17 expressing B cells have been reported in RA patients and in a mouse model of Trypanosoma cruzi infection (9). Unlike ILC or Th17, B cell expression of IL-17A was found to be independent of the transcription factor retinoic acid-related orphan receptor γt (RORγt) (9). IL-17 expressing B cells have not been reported in instances of cancer to the best of our knowledge. Here, we newly report on imPDT induction of COX2 expression in TDLNs and concordant imPDT-induced COX2-dependent and RORγt-independent expression of IL-17 in TDLNs. Pharmacologic inhibition of COX2 with NS398 reduced neutrophil infiltration into the TDLNs. B cells appeared to be the primary source of IL-17 in TDLNs following imPDT. Subsequent experiments involving antibody-mediated B cell depletion prior to imPDT led to reductions in neutrophil infiltration into TDLNS and impaired efficacy of imPDT. Importantly, these studies are suggestive of a novel role for COX2 and B cells in the induction of neutrophil migration and augmentation of anti-tumor immunity following imPDT.
MATERIALS AND METHODS
Animal models and the tumor system
Pathogen-free female BALB/c mice were obtained from the Jackson laboratory (Bar Harbor, ME). CT26 cells, which were obtained from the American Type Culture Collection (ATCC, Bethesda, MD), were cultured in RPMI media containing 1% penicillin-streptomycin-glutamate (P/S/G) and 10% fetal bovine serum (FBS). COX2−/− CT26 cells generated by the CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9) method were generously donated by Dr. Caetano Reis e Sousa from the Francis Crick Institute, London, UK.
The 8 to 10 week old mice were inoculated subcutaneously on the right shoulder with 1 × 106 tumor cells. Tumor volume (V) was measured as , where l is the longest axis of the tumor and w is the axis perpendicular to l. The Roswell Park Comprehensive Cancer Center (RPCCC) Institutional Animal Care and Use Committee approved all procedures carried out in this study.
Reagents and antibodies
Clinical grade, pyrogen-free 2-[1-hexyloxyethyl]-2-devinyl pyropheophorbide-a (HPPH) was obtained from the Roswell Park Pharmacy and reconstituted in pyrogen-free water containing 5% dextrose (D5W; Baxter, Deerfield, IL) to a concentration of 0.4 mmol/L (12). COX2 inhibitor NS398 (Cayman Chemicals, Ann Arbor, MI) was dissolved in dimethylsulfoxide (DMSO) to obtain a stock solution of 8 mg/mL. Purified anti-mouse monoclonal antibody for CD19 (clone 1D3), CD22 (clone Cy34.1), and B220 (clone RA3.3A1/6.1) depletion, and anti-rat kappa (clone MAR 18.5) antibody and all manufacturer recommended isotypes (BioXCell, Lebanon, NH) were diluted to 0.5 μg/μL in phosphate buffered saline (PBS).
In vivo PDT treatment
The photosensitizer HPPH was injected at a dose of 0.4 μmol/kg into the tail vein of mice when the tumor size reached 5–7 mm in any dimension. Depilatory ointment Nair was used to remove fur around the tumor. Subsequently, 18–24 hours following HPPH administration, the tumor was illuminated with a 1.1 cm2 spot of 665 nm wavelength light generated by a diode laser (Modulight, Tampere, Finland) to a total dose of 88 J/cm2 delivered at a rate of 14 mW/cm2. Control mice were administered the photosensitizer (HPPH) only.
Following imPDT, tumor growth was measured by caliper. Tumor volume (V) was measured as , where l is the longest axis of the tumor and w is the axis perpendicular to l. Mice were euthanized when tumors reached 800mm3.
COX2 inhibition
In vivo COX2 inhibition was carried out as described previously (7). NS398 stock solution was diluted in 1:8 v/v DMSO:PBS to obtain a final concentration of 1 mg/mL. NS398 was administered by intraperitoneal (i.p.) injection at a dose of 10 mg/kg 1 hour before tumor illumination. Control mice for COX2 inhibition received only the vehicle used for drug dissolution. Ex vivo inhibition of COX2 was achieved by diluting the stock solution of NS398 to a final concentration of 50 μM/L in culture media (13).
PGE2 ELISA
Axial and brachial TDLNs were flash frozen in dry ice and stored at −80°C. Whole tissue lysates were obtained by thawing and homogenizing tissues followed by sonication. Homogenization was performed in a buffer containing phosphate and indomethacin (Millipore Sigma, Burlington, MA) following the manufacturer’s instructions for the Prostaglandin E2 ELISA Kit (Cayman Chemical, Ann Arbor, MI). PGE2 concentrations were determined by following the manufacturer’s instructions and collecting the data on a VersaMax Microplate Reader (Molecular Devices, San Jose, CA). Raw data were normalized to the initial protein concentration, which was determined by Bradford’s method.
Total RNA extraction from TDLNs and tumors
TDLNs and tumors were harvested at indicated time points and flash frozen in TRIzol (Invitrogen, Carlsbad, CA). Total mRNA was extracted by using the manufacturer’s protocol. Briefly, tissue was homogenized in TRIzol and extracted with chloroform. The RNA was precipitated in isopropanol and washed with ethanol, and the RNA pellet was reconstituted in molecular grade water (Millipore Sigma, Burlington, MA). The RNA preparations were quantified by A260 measurements on a spectrophotometer (SmartSpec Plus, Bio-Rad, Hercules, CA), and samples were stored at −80°C. The OD260/OD280 ratios of the samples were between 1.8 and 2.0.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed by using the comparative CT (i.e., 2-ΔCT, where CT refers to the cycle threshold) method to analyze target gene expression. An iSCRIPT cDNA Synthesis Kit from Bio-Rad (Hercules, CA) was used for reverse transcription by following the manufacturer’s instructions and employing a Bio-Rad T100 Thermal Cycler. For each reaction, a total of 100 ng RNA was used. Following reverse transcription, qPCR for murine COX2 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was performed by using the SsoAdvanced Universal SYBR Green Supermix reagent (Bio-Rad, Hercules, CA) and following the manufacturer’s protocol; this step was carried out in a Bio-Rad CFX Connect Real Time System with a 20 μL reaction volume. The following primer sets were used: COX2 5’-CAG ACA ACA TAA ACT GCG CCT T-3’ (forward) and 5’-GAT ACA CCT CTC CAC CAA TGA CC-3’ (reverse); GAPDH 5’-ACG GCA AAT TCA ACG GCA CAG TCA-3’ (forward ) and 5’-TGG GGG CAT CGG CAG AAG G-3’ (reverse). ΔCt was determined by subtracting the GAPDH Ct value from the COX2 Ct value. The relative amount of COX2 mRNA is reported here, and these data were calculated by using the equation 2−ΔCt.
In vivo B cell depletion
B cells were depleted as described previously (14). Briefly, 150 μG of CD19, CD22, and B220 depleting antibodies (BioXcell) were administered intraperitoneally; then, 48 hours later, 100 μG anti-rat kappa antibody was administered. Tumor illumination for imPDT was performed 24 hours after administration of the anti-rat kappa antibody. In survival studies, B cell depletion was repeated 5 and 9 days after imPDT.
Ex vivo primary B cell stimulation
Single cell suspensions of splenocytes were generated from spleen of naïve female BALB/c mice. B cells were negatively sorted by magnetic separation with an EasySep™ Mouse B Cell Isolation Kit from Stemcell Technologies (Vancouver, Canada) following the manufacturer’s instructions. Cells were plated at a density of 2 × 106 cells per 1 mL of B cell media (RPMI containing 1% P/S/G, 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate, and 1X β-mercaptoethanol prepared from a 1000X stock solution). B cells were stimulated with 10 μg lipopolysaccharide (LPS), Sigma-Aldrich, St. Louis, MO) for 72 hours.
Flow cytometry
Tumor tissue and axial and brachial TDLNs were harvested at indicated time points, and then, single cell suspensions were generated. B cell cultures were harvested at 72 hours following plating and washed twice with PBS. Cells were stained with the following fluorochrome conjugated antibodies for surface markers: CD11b-BV421 (BD Biosciences, San Jose, CA, clone M1/70), Gr1-FITC (BD Biosciences, San Jose, CA, clone RB6-8C5), CD19-APC (Biolegend, San Diego, CA, clone 6D5), and B220-Pacific Blue (BD Biosciences, San Jose, CA, clone RA3-6B2). Additionally, the following antibodies were used to stain for intracellular markers: IL-17A-PE (BD Biosciences, San Jose, CA, clone TC11-18H10), RORγt-APC (BD Biosciences, San Jose, CA, clone Q31-378), and COX2-Ax488 (Santa Cruz Biotechnology, Dallas, TX, clone H-3). We used a FOXP3 Fixation/Permeabilization Kit from Invitrogen (Carlsbad, CA) for the above staining procedure. A BD LSRII Flow Cytometer System was used for the flow cytometric analyses. Data were acquired from 200,000 TDLN cells by using the BD-LSRII instrument, and these data were analyzed by using FlowJo software. Positive gates (gates containing antibody binding cells) were established using fluorescence minus one (FMO) controls (15). FMO controls are the experimental cells stained with all the fluorophores minus one.
Statistical evaluations
All measured values are represented with the standard error of the mean (SEM). The unpaired Student’s t-test with Welch’s correction was used to compare values between two groups in all experiments. In all cases, significance was defined as P <0.05. Statistical significance in tumor regrowth experiments was determined by log-rank (Mantel-Cox) tests of Kaplan-Meir plots.
RESULTS
COX2 regulates IL-17 expression in TDLNs following imPDT
imPDT induces IL-17 expression in TDLNs (12) via an unknown mechanism; COX2 has been previously shown to regulate IL-17 (8). To determine if COX2 regulates IL-17 expression in TDLNs following imPDT, the expression of COX2 in TDLNs was assessed.
COX2 mRNA expression increased in TDLNs following imPDT (Fig. 1A). Functional activity of imPDT-induced COX2 was evaluated by assaying for the expression of PGE2, which was increased in TDLNs (Fig. 1B). Administration of the COX2 inhibitor NS398 one hour prior to imPDT significantly reduced the number of IL-17 expressing cells in TDLNs (Fig. 1C, 1D). However, COX2 inhibition did not impact the expression of RORγt, the key transcription factor of the conventional IL-17 pathway (Supplementary Fig. 1; see Supporting Materials). Thus, imPDT-induced COX2 appears to regulate IL-17 expression in TDLNs independent of RORγt expression.
Figure 1.
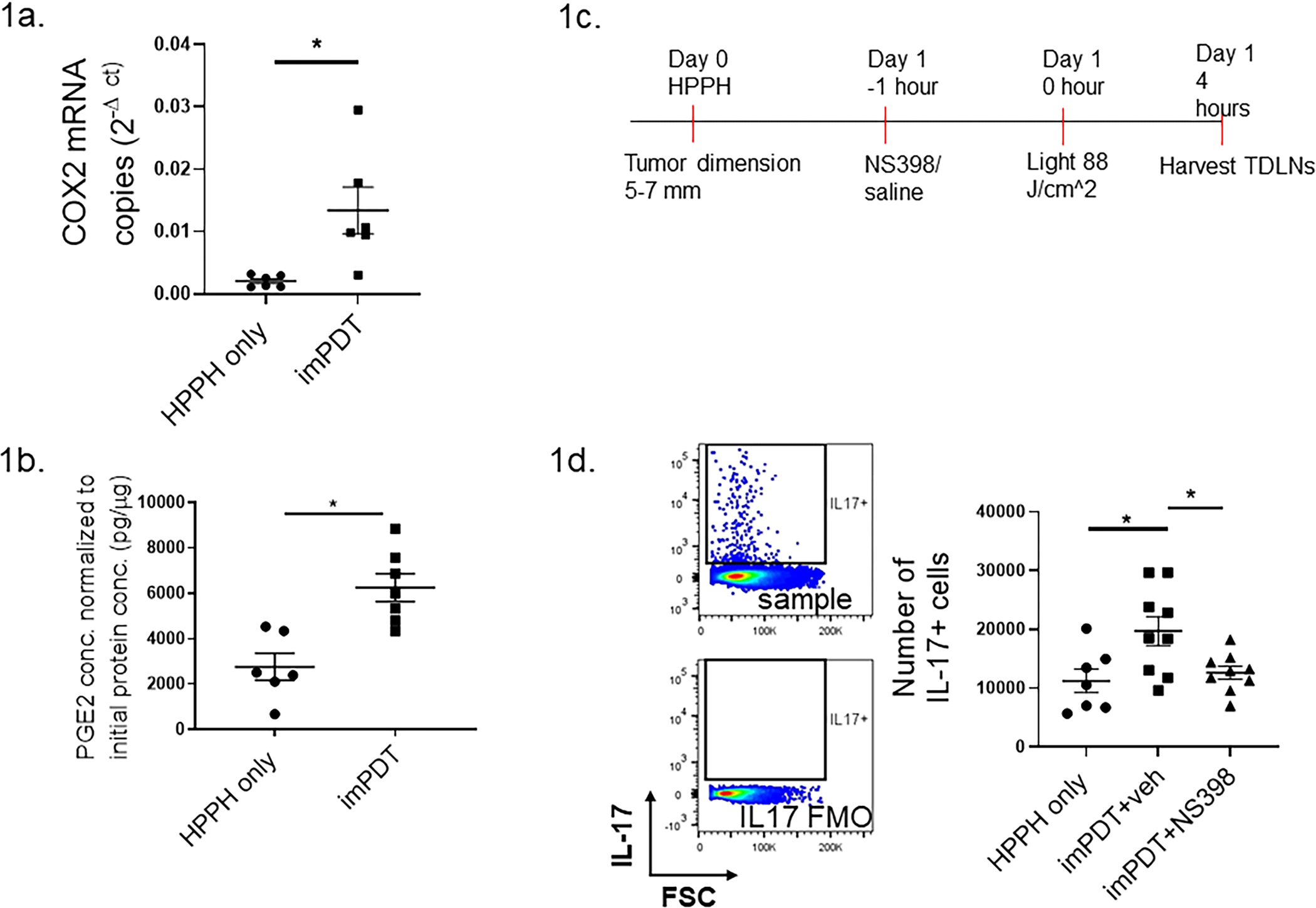
imPDT-induced COX2 regulates IL-17A expression in TDLNs. (a,b) CT26 tumor-bearing mice underwent imPDT; TDLNs were harvested 4 hours later. Control mice were administered the photosensitizer (HPPH) only. (a) COX2 mRNA expression in TDLNs normalized to total protein concentrations (n = 6, two experiments). (b) Concentrations of PGE2 measured by ELISA (n = 6–7, two experiments). (c) Schematic for administration of the COX2 inhibitor NS398 prior to imPDT. (d) CT26 tumor-bearing mice received either NS398 or the vehicle followed by imPDT; TDLNs were harvested 4 hours post treatment. Numbers of IL-17 expressing cells in TDLNs were evaluated by flow cytometry (n = 7–9, three experiments). Gating strategy is shown via example histograms: Upper histogram shows cells stained with anti-IL-17; lower histogram shows cells stained without the anti-IL-17 antibody. Error bars represent the mean ± SEM. *P <0.05
imPDT-induced COX2 regulates IL-17 expressing B cells in TDLNs
B cells express IL-17 independent of RORγt (9). In this study, imPDT increased the number of COX2 expressing B (CD19+B220+) cells in TDLNs after treatment (Fig. 2A). B cells made up ~30% of IL-17-A expressing cells in TDLNs following imPDT; most of the IL-17 expressing B cells also expressed COX2 (Fig. 2B). Pharmacologic blockade of COX2 with NS398 prior to imPDT reduced the number of IL-17 expressing B cells in TDLNs (Fig. 2C). Depletion of B cells (Supplementary Fig. 2) reduced the overall number of IL-17 expressing cells in TDLNs following imPDT (Fig. 2D). Thus, imPDT increased IL-17 expressing B cells in TDLNs in a COX2-dependent manner.
Figure 2.
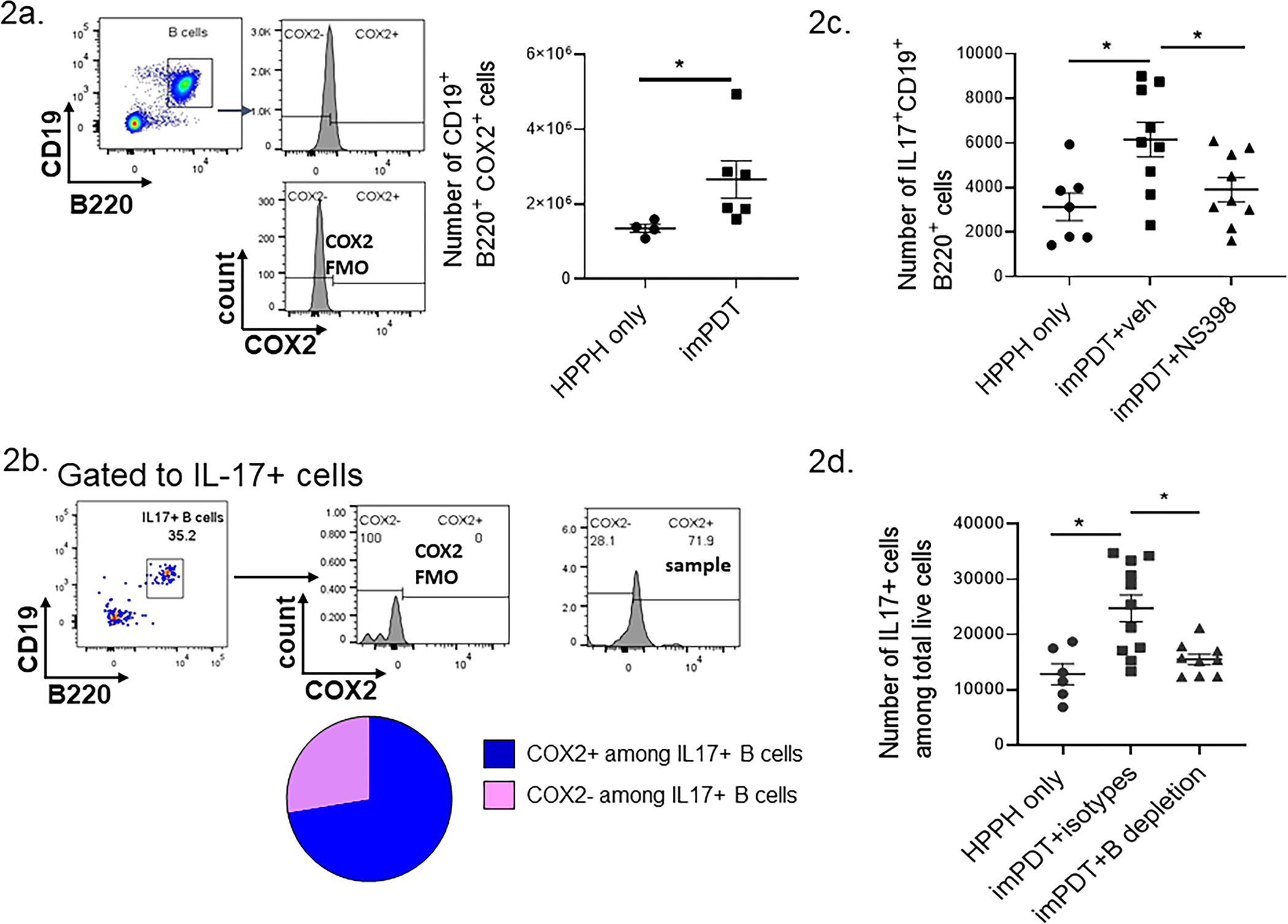
COX2 regulates B cell IL-17 expression in TDLNs following imPDT. (a,b) CT26 tumor-bearing mice underwent imPDT; TDLNs were harvested 4 hours later. (a) Numbers of COX2 expressing B cells were evaluated by flow cytometry (n = 4–6, two experiments). The gating strategy is shown on the left; B cells (CD19+, B220+) were selected and analyzed for COX2 expression by comparing staining with anti-COX2 (upper) with cells stained in the absence of anti-COX2 (lower). (b) Frequency of COX2 expressing cells among IL-17 expressing B cells in imPDT-treated mice as evaluated by flow cytometry (mean of n = 3, one experiment). The gating strategy is shown on the in the upper panels; B cells (CD19+, B220+) were selected among IL-17 expressing cells and analyzed for COX2 expression by comparing staining with anti-COX2 (far right panel) with cells stained in the absence of anti-COX2 (center panel). (c) CT26 tumor-bearing mice received either NS398 or the vehicle followed by imPDT; TDLNs were harvested 4 hours post treatment. Numbers of IL-17 expressing B cells were evaluated by flow cytometry (n = 7–9, three experiments). (d) CT26 tumor-bearing mice underwent B cell depletion followed by imPDT. Numbers of IL-17 expressing cells were evaluated by flow cytometry (n = 6–11, three experiments). Error bars represent the mean ± SEM. *P <0.05
COX2–B cell axis facilitates neutrophil infiltration to TDLNs following imPDT
Neutrophil infiltration to TDLNs following imPDT is dependent on IL-17 expression in TDLNs (12). In this study, blockade of COX2 prior to imPDT reduced neutrophil (CD11b+, Gr1Hi) infiltration to TDLNs (Fig. 3A). A similar reduction in neutrophil infiltration was observed in TDLNs when B cell depleted mice underwent imPDT (Fig. 3B). imPDT increased COX2 mRNA in tumor tissue (Supplementary Fig. 3A). To eliminate the possibility that the effect of NS398 on IL-17 expression by B cells and neutrophil infiltration to TDLNs is an artifact of inhibiting COX2 expression by tumor cells, mice were inoculated with COX2 deficient (COX2−/−) CT26 cells. imPDT of COX2−/− CT26 tumors increased the number of neutrophils and IL-17 expressing B cells in TDLNs (Supplementary Figs. 3B and 3C). Thus, it appears that the COX2–B cell axis in TDLNs that is induced upon imPDT regulates neutrophil infiltration to TDLNs.
Figure 3.
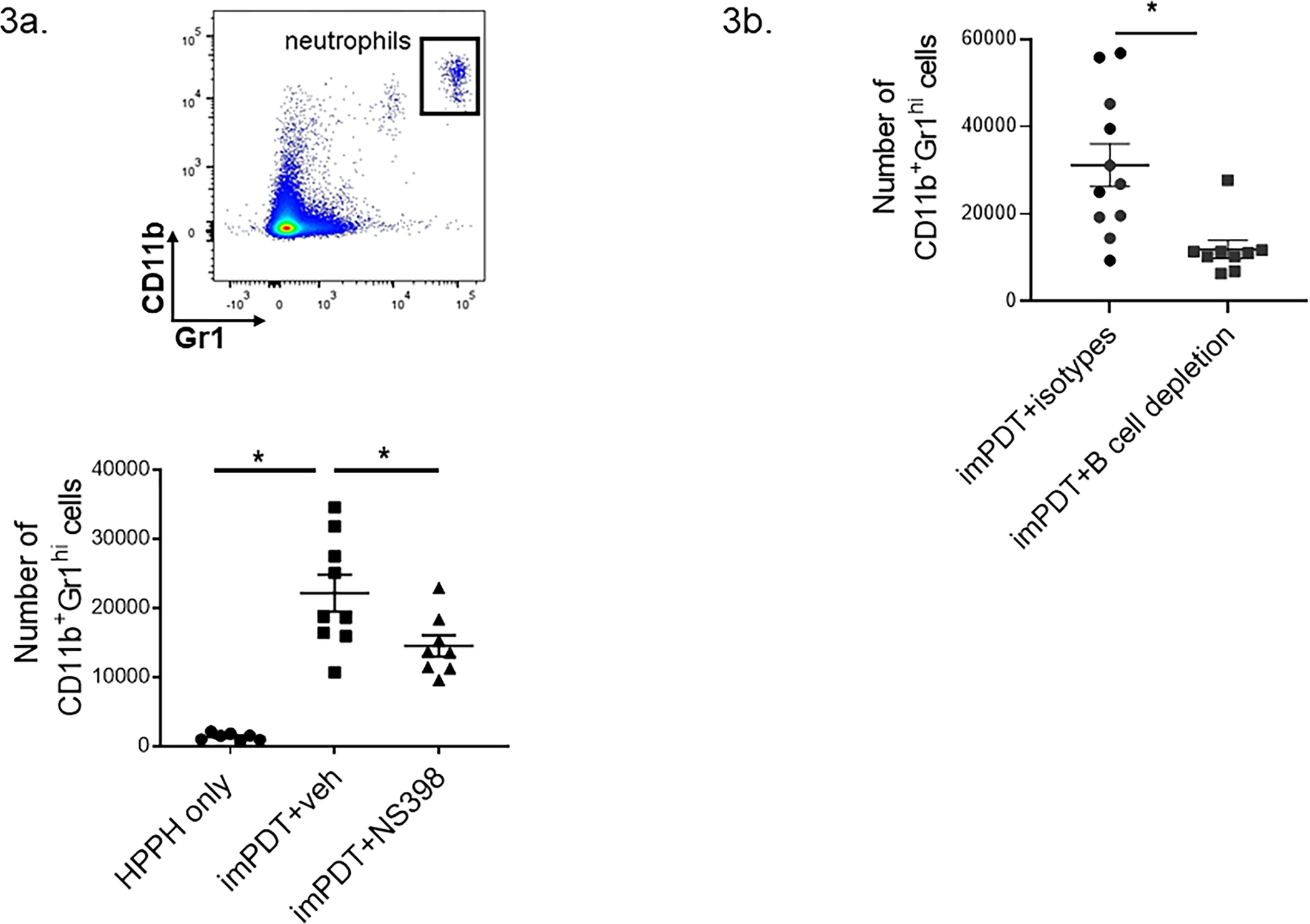
imPDT-induced COX2–B cell axis regulates neutrophil infiltration to TDLNs. (a) CT26 tumor-bearing mice received either NS398 or the vehicle followed by imPDT; TDLNs were harvested 4 hours post treatment. Numbers of neutrophils (CD11b+ Gr1-hi) were evaluated by flow cytometry (n = 7–9, three experiments). (b) CT26 tumor-bearing mice underwent B cell depletion followed by imPDT. Numbers of neutrophils were evaluated by flow cytometry (n = 10–11, three experiments). Error bars represent the mean ± SEM. *P <0.05
LPS stimulation induces COX2-regulated IL-17 expression by B cells ex vivo
Sterile inflammation is initiated by the release of damage-associated molecular patterns (DAMPs) from host cells through a process known as immunogenic cell death (ICD). Pathogenic inflammation is initiated by pathogen-associated molecular patterns (PAMPs) derived from microorganisms. To evaluate the possibility of COX2 regulation over B cell IL-17 expression in pathogen-induced inflammation, B cells were isolated from spleens of naïve mice (Supplementary Fig. 4) and stimulated with Escherichia coli derived PAMP LPS for 72 hours. The LPS stimulation induced COX2 expression and IL-17 expression by B cells (Fig. 4A, 4B), and the majority of IL-17 expressing B cells (~90%) expressed COX2 (Fig. 4C). The COX2 blockade significantly reduced the percentage of IL-17 expressing B cells upon LPS stimulation (Fig. 4D). Thus, COX2 regulated B cell IL-17 expression in an in vitro model of pathogen-induced inflammation.
Figure 4.
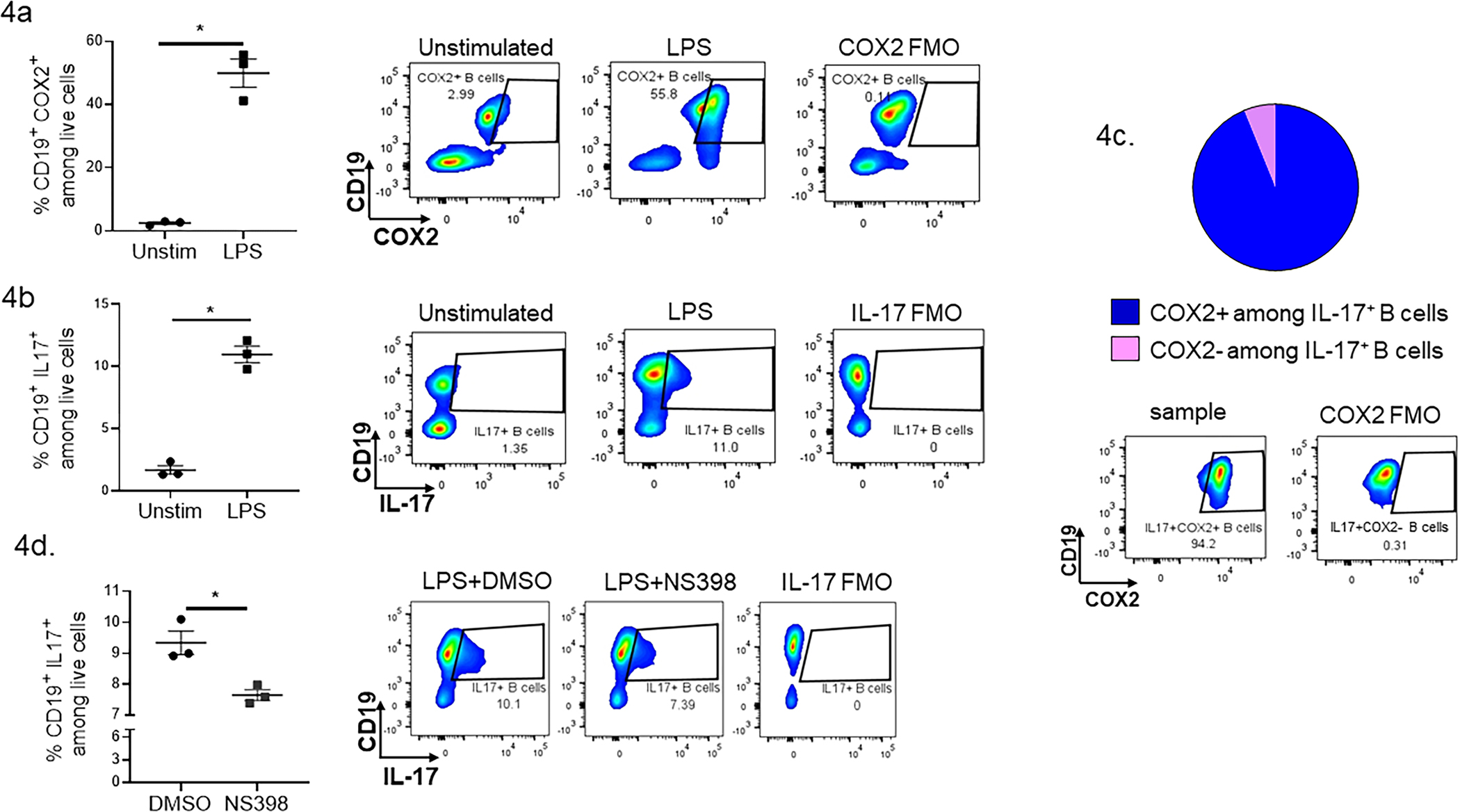
COX2 regulates IL-17 expression by B cells ex vivo. (a–d) B cells isolated from naïve BALB/c spleen were stimulated with LPS for 72 hours. (a) Percentages of COX2 expressing B cells and (b) IL-17 expressing B cells were evaluated by flow cytometry (data from six spleens pooled together, assayed in triplicate). (c) Pie chart showing the distribution of COX2+ and COX2- populations among IL-17+ B cells after LPS stimulation. (d) IL-17 expressing B cell populations in LPS stimulated B cells in the presence of the vehicle (DMSO) or NS398. Histograms accompany each plot and depict examples of sample plots (unstimulated, LPS, LPS + NS398) as well as an example of a sample plot stained with all but the COX2 or IL-17 specific antibody (COX2 or IL-17 FMO). Error bars represent the mean ± SEM. *P <0.05
B cell depletion reduces the efficacy of imPDT
The efficacy of imPDT is dependent upon neutrophil infiltration into TDLNs and the subsequent enhancement of anti-tumor immunity (5,16). To evaluate whether B cells contribute to the efficacy of imPDT, CT26 tumor bearing mice were subjected to B cell depletion 4 days before and 5 and 9 days after imPDT (Figure 5). Control animals received isotype control antibodies 4 days before and 5 and 9 days after imPDT. Animal survival was significantly improved by treatment with PDT (Isotype control + HPPH vs. Isotype control + PDT and B cell depletion + HPPH vs. B cell depletion + PDT; P≤0.0001). Depletion of B cells led to a significant reduction in imPDT efficacy as measured by animal survival (Isotype control + PDT vs. B cell depletion + PDT; P≤0.011).
Figure 5.
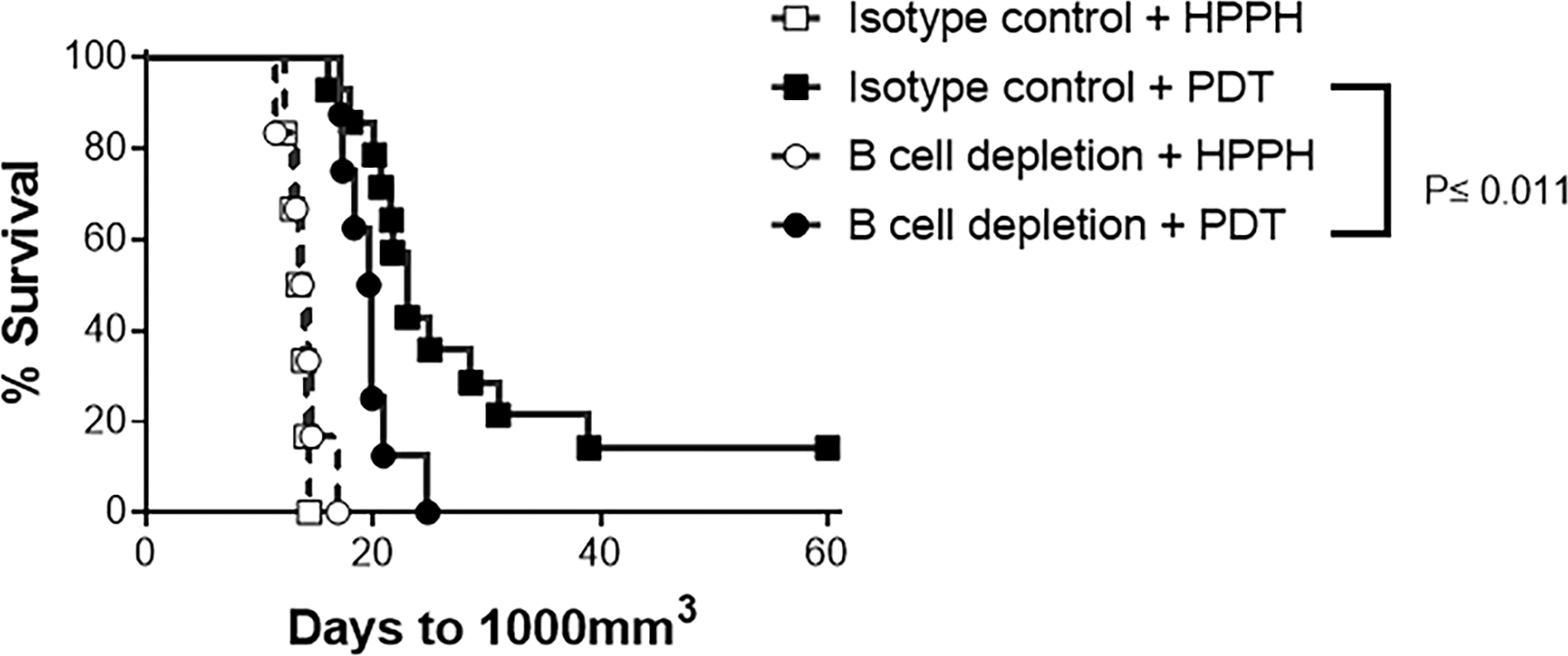
B cells augment imPDT efficacy. CT26 tumor bearing mice underwent B cell depletion on 4 days before and 5 and 9 days after imPDT. Tumor growth was monitored; data are presented as a survival curve, wherein mice were terminated by humane euthanasia once their tumor had reached an upper size endpoint of 1000 cm3. B cell depletion significantly reduced PDT efficacy; P≤ 0.011.
DISCUSSION
The inflammatory cytokine IL-17 offers protective immunity against many infectious agents (17–19) but contributes to the disease pathogenesis of sterile inflammatory conditions such as autoimmune arthritis, colitis, multiple sclerosis, and some cancers (20–22). imPDT-induced IL-17 drives neutrophil infiltration to TDLNs, which is a critical factor for augmentation of imPDT-enhanced anti-tumor immunity (5). In this study, we have evaluated the effects of imPDT-induced COX2 on IL-17 expression and neutrophil infiltration. The results show that i) imPDT induces activation of the COX2 pathway within a few hours of treatment resulting in IL-17 expression in B cells, ii) B cells facilitate neutrophil infiltration to TDLNs following imPDT, and iii) B cells contribute to the efficacy of imPDT. To the best of our knowledge, this is the first report on COX2-driven augmentation of IL-17 expressing B cells and the role of B cells in neutrophil infiltration to TDLNs.
The role of the COX2/PGE2 axis in regulating IL-17 expression is context dependent. Studies have reported increased IL-17 expression by the COX2/PGE2 axis in ex vivo models of T cell differentiation and activation (23,24), in lymph node cultures of murine experimental RA (8), and in ex vivo cultures of γδT cells from RA patients (25). Conversely, negative regulation of IL-17 by COX2/PGE2 has been demonstrated in murine models of leishmaniasis (26), infectious colitis (27), and cryptococcosis (28). Our observations of increases in IL-17 transcription and the number of IL-17 expressing cells by cancer therapy-induced COX2 adds to the functional complexity of the COX2/PGE2 axis during IL-17 expression.
IL-17 is expressed by multiple cells including Th17 cells, γδT cells, innate lymphoid cells (ILC), invariant natural killer T cells (iNKT), and natural killer cells (9–11). The transcription factor RORγ plays a central role in inducing IL-17 in these cells (9,10,29). In experimental models where COX2/PGE2 increased IL-17 expression, RORγt expression was also augmented (23,24), while COX2/PGE2-regulated reductions in IL-17 coincided with decreases in RORγt expression (28). Our observation that imPDT-induced COX2 had no effect on RORγt expression is indicative of a mechanism in which IL-17 expression is independent of RORγt.
The role of IL-17 in neutrophil recruitment during pathogen-induced inflammation and sterile inflammation is well known (30,31). We have previously reported on the critical role of IL-17 neutrophil infiltration to TDLNs following imPDT (12). In a murine model of RA, Lemos et al. showed that COX2/PGE2 regulation over IL-17 expression drives neutrophil recruitment to inflamed sites (8). Thus, our current observation that imPDT-induced COX2, which enhances IL-17 expression in TDLNs, facilitates neutrophil recruitment to TDLNs is in line with these previous studies. In contrast, an in vitro study by Armstrong showed that interaction of PGE2 with its receptors on neutrophils impeded neutrophil chemotaxis toward the chemoattractant N-formyl-methionyl-leucine-phenylalanine (FMLP) (32). In our model, i.p. administration of the COX2 inhibitor NS398 limits the amount of PGE2 available for signaling through receptors on neutrophils. However, the key driver of neutrophil infiltration to TDLNs in our model is IL-17, which is upregulated downstream of COX2/PGE2. Thus, it seems that the role of the COX2/PGE2 axis in IL-17 expression overrides any direct negative role of the axis in neutrophil chemotaxis. In fact, Lemos et al. have shown that once IL-17 is expressed in the system, COX2 inhibition has no effect on neutrophil migration (8). Their observation further bolsters our argument that role of COX2/PGE2 in neutrophil migration to inflamed lymph nodes is exerted via IL-17 expression.
Our observation that imPDT-induced COX2 augments IL-17 expressing B cells in TDLNs is significant because i) it confirms IL-17 induction independent of RORγt in B cells as was reported previously by Bermejo et al. in an experimental model of Trypanozoma cruzi infection, and ii) it brings forward downstream targets of the COX2/PGE2 pathway as a potential mechanism for B cell expression of IL-17 (9). Schlegel et al. have reported IL-17 expressing B cells in RA patients. Our communication is the first to report IL-17 expressing B cells in the context of cancer (33). The most well studied roles of B cells in immunity are as antigen presenting cells and antibody producers (34–37); presently, IL-17 expression by B cells is poorly understood. Thus, B cell expression of IL-17 requires further research, especially in regard to diseases where IL-17 is a prognostic marker.
Previous studies have reported a role for neutrophils in regulating B cell survival, maturation, differentiation, and function (38–42). However, the effects of B cells on neutrophil biology are poorly understood. In one study by Kim et al., physical interaction of B cells and neutrophils in the lungs, via β-integrin, induced neutrophil apoptosis and macrophage-mediated clearance (43). Genetic deletion of B cells in this model had a long-term effect on neutrophil accumulation in the lung. Conversely, short-term antibody-mediated B cell depletion in our study reduced the immediate effect of neutrophil infiltration to TDLNs, potentially in response to the reduced IL-17. Thus, our study shows the immediate impact of soluble inflammatory factors from B cells on neutrophil migration, while the work of Kim et al. [41] demonstrates the homeostatic effect of the physical interaction between B cells and neutrophils. The difference in the outcome of neutrophil accumulation in these studies is indicative of the complex relationship between B cells and neutrophils, which depends on the context and nature of the interaction.
DAMPs are components of host cells that are sequestered inside the cells and are exposed upon cell death. Recognition of DAMPs by pattern recognition receptors (PRRs) initiates sterile inflammatory pathways (44,45). imPDT-induced sterile inflammation is set into motion by the release of DAMPs from cells dying in the tumor tissue in response to phototherapy (16,46). Pathogen-induced inflammation is initiated by recognition of PAMPS from the infectious agent by PRRs. Therefore, it is conceivable that additional parallels might exist between initiation of sterile and pathogenic inflammation. Our evaluation of an in vitro model of pathogenic inflammation involving stimulation of B cells with LPS resulted in a COX2-dependent increase in B cell IL-17 expression. Considering the critical role of IL-17 in immunity against infectious agents, further studies into the role of COX2-dependent IL-17 expression by B cells in infectious diseases are necessary.
Activation of anti-tumor immunity along with the advantage of tumor specificity makes imPDT a potential candidate for adjuvant immunotherapy during the treatment of cancer. Rapid inflammation characterized by neutrophil infiltration to TDLNs within 4 hours of treatment is critical for enhancing anti-tumor immunity by imPDT (4,5). This study advances our knowledge of the early events of inflammation upstream of neutrophil infiltration to TDLNs and lays a foundation for studies of parallel events during the initiation of pathogenic inflammation.
Supplementary Material
Acknowledgements:
The authors thank Kimberley Ramsey for technical assistance with animal experiments. Research reported in this publication was supported in part by the National Cancer Institute of the National Institute of Health under Awards 5P01CA98156 (SG), 5T32CA085183 (RF-M) and the Roswell Park Alliance Foundation. The study used shared resources supported by the Roswell Park Cancer Comprehensive Cancer Center Support Grant (P30CA016056).
Footnotes
Conflicts of interest: The authors declare that they have no conflict of interest.
Ethics approval: The RPCCC Institutional Animal Care and Use Committee approved both animal care and experiments.
Data availability:
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Hampton HR, Chtanova T. The lymph node neutrophil. Seminars in Immunology 2016;28(2):129–36. [DOI] [PubMed] [Google Scholar]
- 2.Hampton HR, Bailey J, Tomura M, Brink R, Chtanova T. Microbe-dependent lymphatic migration of neutrophils modulates lymphocyte proliferation in lymph nodes. Nat Commun 2015;6:7139 doi 10.1038/ncomms8139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogoslowski A, Butcher EC, Kubes P. Neutrophils recruited through high endothelial venules of the lymph nodes via PNAd intercept disseminating Staphylococcus aureus</em>. Proceedings of the National Academy of Sciences 2018;115(10):2449 doi 10.1073/pnas.1715756115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Henderson BW, Gollnick SO, Snyder JW, Busch TM, Kousis PC, Cheney RT, et al. Choice of Oxygen-Conserving Treatment Regimen Determines the Inflammatory Response and Outcome of Photodynamic Therapy of Tumors. Cancer Res 2004;64(6):2120–6. [DOI] [PubMed] [Google Scholar]
- 5.Kousis PC, Henderson BW, Maier PG, Gollnick SO. Photodynamic therapy enhancement of antitumor immunity is regulated by neutrophils. Cancer Res 2007;67(21):10501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalinski P Regulation of immune responses by prostaglandin E2. Journal of immunology (Baltimore, Md : 1950) 2012;188(1):21–8 doi 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferrario A, Von Tiehl K, Wong S, Luna M, Gomer CJ. Cyclooxygenase-2 inhibitor treatment enhances photodynamic therapy-mediated tumor response. Cancer research 2002;62(14):3956–61. [PubMed] [Google Scholar]
- 8.Lemos HP, Grespan R, Vieira SM, Cunha TM, Verri WA Jr., Fernandes KS, et al. Prostaglandin mediates IL-23/IL-17-induced neutrophil migration in inflammation by inhibiting IL-12 and IFNgamma production. Proceedings of the National Academy of Sciences of the United States of America 2009;106(14):5954–9 doi 10.1073/pnas.0812782106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bermejo DA, Jackson SW, Gorosito-Serran M, Acosta-Rodriguez EV, Amezcua-Vesely MC, Sather BD, et al. Trypanosoma cruzi trans-sialidase initiates a program independent of the transcription factors RORγt and Ahr that leads to IL-17 production by activated B cells. Nature Immunology 2013;14(5):514–22 doi 10.1038/ni.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cua DJ, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nature Reviews Immunology 2010;10(7):479–89 doi 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- 11.Gaffen SL. Recent advances in the IL-17 cytokine family. Curr Opin Immunol 2011;23(5):613–9 doi 10.1016/j.coi.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brackett CM, Muhitch JB, Evans SS, Gollnick SO. IL-17 promotes neutrophil entry into tumor-draining lymph nodes following induction of sterile inflammation. Journal of Immunology (Baltimore, Md : 1950) 2013;191(8):4348–57 doi 10.4049/jimmunol.1103621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrario A, Fisher AM, Rucker N, Gomer CJ. Celecoxib and NS-398 Enhance Photodynamic Therapy by Increasing In vitro Apoptosis and Decreasing In vivo Inflammatory and Angiogenic Factors. Cancer research 2005;65(20):9473 doi 10.1158/0008-5472.CAN-05-1659. [DOI] [PubMed] [Google Scholar]
- 14.Keren Z, Naor S, Nussbaum S, Golan K, Itkin T, Sasaki Y, et al. B-cell depletion reactivates B lymphopoiesis in the BM and rejuvenates the B lineage in aging. Blood 2011;117(11):3104–12 doi 10.1182/blood-2010-09-307983. [DOI] [PubMed] [Google Scholar]
- 15.Perfetto SP, Chattopadhyay PK, Roederer M. Seventeen-colour flow cytometry: unravelling the immune system. Nature reviews Immunology 2004;4(8):648–55 doi 10.1038/nri1416. [DOI] [PubMed] [Google Scholar]
- 16.Falk-Mahapatra R, Gollnick SO. Photodynamic Therapy and Immunity: An Update. Photochemistry and photobiology 2020;96(3):550–9 doi 10.1111/php.13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kelly MN, Kolls JK, Happel K, Schwartzman JD, Schwarzenberger P, Combe C, et al. Interleukin-17/interleukin-17 receptor-mediated signaling is important for generation of an optimal polymorphonuclear response against Toxoplasma gondii infection. Infection and immunity 2005;73(1):617–21 doi 10.1128/iai.73.1.617-621.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye P, Garvey PB, Zhang P, Nelson S, Bagby G, Summer WR, et al. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. American journal of respiratory cell and molecular biology 2001;25(3):335–40 doi 10.1165/ajrcmb.25.3.4424. [DOI] [PubMed] [Google Scholar]
- 19.Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. The Journal of infectious diseases 2004;190(3):624–31 doi 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- 20.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, et al. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 2005;201(2):233–40 doi 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. The Journal of clinical investigation 1999;103(9):1345–52 doi 10.1172/jci5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuen D-S, Kim B-S, Chung Y IL-17-Producing Cells in Tumor Immunity: Friends or Foes? Immune Netw 2020;20(1):e6–e doi 10.4110/in.2020.20.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Napolitani G, Acosta-Rodriguez EV, Lanzavecchia A, Sallusto F. Prostaglandin E2 enhances Th17 responses via modulation of IL-17 and IFN-gamma production by memory CD4+ T cells. European journal of immunology 2009;39(5):1301–12 doi 10.1002/eji.200838969. [DOI] [PubMed] [Google Scholar]
- 24.Boniface K, Bak-Jensen KS, Li Y, Blumenschein WM, McGeachy MJ, McClanahan TK, et al. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J Exp Med 2009;206(3):535–48 doi 10.1084/jem.20082293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Du B, Zhu M, Li Y, Li G, Xi X. The prostaglandin E2 increases the production of IL-17 and the expression of costimulatory molecules on γδ T cells in rheumatoid arthritis. Scandinavian Journal of Immunology 2020;91(5):e12872 doi 10.1111/sji.12872. [DOI] [PubMed] [Google Scholar]
- 26.Saha A, Biswas A, Srivastav S, Mukherjee M, Das PK, Ukil A. Prostaglandin E2 negatively regulates the production of inflammatory cytokines/chemokines and IL-17 in visceral leishmaniasis. Journal of immunology (Baltimore, Md : 1950) 2014;193(5):2330–9 doi 10.4049/jimmunol.1400399. [DOI] [PubMed] [Google Scholar]
- 27.Dejani NN, Orlando AB, Niño VE, Penteado LdA, Verdan FF, Bazzano JMR, et al. Intestinal host defense outcome is dictated by PGE2 production during efferocytosis of infected cells. Proceedings of the National Academy of Sciences 2018;115(36):E8469 doi 10.1073/pnas.1722016115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valdez PA, Vithayathil PJ, Janelsins BM, Shaffer AL, Williamson PR, Datta SK. Prostaglandin E2 suppresses antifungal immunity by inhibiting interferon regulatory factor 4 function and interleukin-17 expression in T cells. Immunity 2012;36(4):668–79 doi 10.1016/j.immuni.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Venken K, Jacques P, Mortier C, Labadia ME, Decruy T, Coudenys J, et al. RORγt inhibition selectively targets IL-17 producing iNKT and γδ-T cells enriched in Spondyloarthritis patients. Nat Commun 2019;10(1):9 doi 10.1038/s41467-018-07911-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 2008;28(4):454–67 doi 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu JJ, Ruddy MJ, Wong GC, Sfintescu C, Baker PJ, Smith JB, et al. An essential role for IL-17 in preventing pathogen-initiated bone destruction: recruitment of neutrophils to inflamed bone requires IL-17 receptor–dependent signals. Blood 2007;109(9):3794–802 doi 10.1182/blood-2005-09-010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Armstrong RA. Investigation of the inhibitory effects of PGE2 and selective EP agonists on chemotaxis of human neutrophils. British Journal of Pharmacology 1995;116(7):2903–8 doi 10.1111/j.1476-5381.1995.tb15943.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlegel PM, Steiert I, Kötter I, Müller CA. B cells contribute to heterogeneity of IL-17 producing cells in rheumatoid arthritis and healthy controls. PloS one 2013;8(12):e82580–e doi 10.1371/journal.pone.0082580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen X, Jensen PE. The role of B lymphocytes as antigen-presenting cells. Arch Immunol Ther Exp (Warsz) 2008;56(2):77–83 doi 10.1007/s00005-008-0014-5. [DOI] [PubMed] [Google Scholar]
- 35.Adler LN, Jiang W, Bhamidipati K, Millican M, Macaubas C, Hung S-c, et al. The Other Function: Class II-Restricted Antigen Presentation by B Cells. 2017;8(319) doi 10.3389/fimmu.2017.00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong S, Zhang Z, Liu H, Tian M, Zhu X, Zhang Z, et al. B Cells Are the Dominant Antigen-Presenting Cells that Activate Naive CD4(+) T Cells upon Immunization with a Virus-Derived Nanoparticle Antigen. Immunity 2018;49(4):695–708.e4 doi 10.1016/j.immuni.2018.08.012. [DOI] [PubMed] [Google Scholar]
- 37.Häusser-Kinzel S, Weber MS. The Role of B Cells and Antibodies in Multiple Sclerosis, Neuromyelitis Optica, and Related Disorders. Front Immunol 2019;10:201 doi 10.3389/fimmu.2019.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Costa S, Bevilacqua D, Cassatella MA, Scapini P. Recent advances on the crosstalk between neutrophils and B or T lymphocytes. Immunology 2019;156(1):23–32 doi 10.1111/imm.13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Puga I, Cols M, Barra CM, He B, Cassis L, Gentile M, et al. B cell-helper neutrophils stimulate the diversification and production of immunoglobulin in the marginal zone of the spleen. Nat Immunol 2011;13(2):170–80 doi 10.1038/ni.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scapini P, Nardelli B, Nadali G, Calzetti F, Pizzolo G, Montecucco C, et al. G-CSF-stimulated neutrophils are a prominent source of functional BLyS. J Exp Med 2003;197(3):297–302 doi 10.1084/jem.20021343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scapini P, Bazzoni F, Cassatella MA. Regulation of B-cell-activating factor (BAFF)/B lymphocyte stimulator (BLyS) expression in human neutrophils. Immunology letters 2008;116(1):1–6 doi 10.1016/j.imlet.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 42.Huard B, McKee T, Bosshard C, Durual S, Matthes T, Myit S, et al. APRIL secreted by neutrophils binds to heparan sulfate proteoglycans to create plasma cell niches in human mucosa. The Journal of clinical investigation 2008;118(8):2887–95 doi 10.1172/jci33760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JH, Podstawka J, Lou Y, Li L, Lee EKS, Divangahi M, et al. Aged polymorphonuclear leukocytes cause fibrotic interstitial lung disease in the absence of regulation by B cells. Nat Immunol 2018;19(2):192–201 doi 10.1038/s41590-017-0030-x. [DOI] [PubMed] [Google Scholar]
- 44.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. Journal of leukocyte biology 2007;81(1):1–5 doi 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 45.Garg AD, Nowis D, Golab J, Vandenabeele P, Krysko DV, Agostinis P. Immunogenic cell death, DAMPs and anticancer therapeutics: an emerging amalgamation. Biochimica et biophysica acta 2010;1805(1):53–71 doi 10.1016/j.bbcan.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 46.Garg AD, Nowis D, Golab J, Agostinis P. Photodynamic therapy: illuminating the road from cell death towards anti-tumour immunity. Apoptosis : an international journal on programmed cell death 2010;15(9):1050–71 doi 10.1007/s10495-010-0479-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.