

Abstract
The virulence of Agrobacterium tumefaciens depends on both chromosome- and Ti plasmid-encoded gene products. In this study, we characterize a chromosomal locus, chvH, previously identified by TnphoA mutagenesis and shown to be required for tumor formation. Through DNA sequencing and comparison of the sequence with identified sequences in the database, we show that this locus encodes a protein similar in sequence to elongation factor P, a protein thought to be involved in peptide bond synthesis in Escherichia coli. The analysis of vir-lacZ and vir-phoA translational fusions as well as Western immunoblotting revealed that the expression of Vir proteins such as VirE2 was significantly reduced in the chvH mutant compared with the wild-type strain. The E. coli efp gene complemented detergent sensitivity, virulence, and expression of VirE2 in the chvH mutant, suggesting that chvH and efp are functionally homologous. As expected, ChvH exerts its activity at the posttranscriptional level. Southern analysis suggests that the gene encoding this elongation factor is present as a single copy in A. tumefaciens. We constructed a chvH deletion mutant in which a 445-bp fragment within its coding sequence was deleted and replaced with an omega fragment. On complex medium, this mutant grew more slowly than the wild-type strain, indicating that elongation factor P is important but not essential for the growth of Agrobacterium.
Agrobacterium tumefaciens causes crown gall disease in a wide range of dicotyledonous plants. The disease, characterized by neoplastic transformation at the site of infection, results from the transfer and expression of oncogenes from the bacterium to susceptible plant cells (for a review, see reference 27). This transfer process is governed primarily by the products of the vir genes located on the Ti plasmid. These genes are tightly regulated and are expressed to a significant level only in the presence of plant signal molecules synthesized by wounded plant cells. The vir genes are transcriptionally regulated by the Ti plasmid-encoded VirA/VirG two-component regulatory system (26, 52). The VirA protein senses the plant signal molecules and then transduces the signal by phosphate transfer to the response regulator, the VirG protein. The activated VirG protein is a positive transcriptional activator of itself and all other Ti plasmid-encoded vir operons.
Numerous chromosomal virulence genes (chv) have also been shown to play important roles in the ability of Agrobacterium to transform plants (for a review, see reference 39). In general, the functions of chromosomal virulence genes have not been well elucidated, and mutations in these genes are pleiotropic. Consequently, their precise roles in tumor formation have been difficult to assess. An analysis of a limited number of chv mutants suggests that whereas vir genes on the Ti plasmid are dedicated solely to specific steps in the interaction of Agrobacterium with host plants, the chromosomal virulence genes play important roles in the general physiology of Agrobacterium and have been conscripted to play ancillary but significant roles in the interaction of this bacterium with its hosts.
The best-understood chromosomal virulence gene is chvE, which codes for a glucose-galactose periplasmic binding protein. It normally functions in the uptake of a number of monosaccharides into the bacterial cell and is also involved in chemotaxis towards these sugars. In the transformation process, this periplasmic protein interacts with these same monosaccharides, all of which are components of the plant cell wall. It then binds to the periplasmic domain of the VirA sensor molecule (26), a requirement for maximum activation of VirG and the subsequent activation of all Ti plasmid-encoded vir genes. Depending on the strain, chvE mutants are either avirulent or severely attenuated in tumor formation on a wide variety of host plants.
Another chv locus, acvB, is unusual in that some strains of A. tumefaciens have a functional copy (virJ) on the Ti plasmid. Only by studying a strain that lacks a copy on the Ti plasmid was this chv locus identified as one that is required for T-DNA transfer (29, 41, 54). The relationship between the chromosomal locus and its Ti plasmid counterpart is unknown, as is the precise role that this locus plays in tumor formation. However, the identification of functionally redundant loci raises the possibility that a similar situation may hold for other Ti plasmid or chv loci, which complicates their isolation and identification.
Some chv genes have homologues in other bacteria that also display close interactions with host cells, either plant or animal. A. tumefaciens, Brucella abortus, and Sinorhizobium meliloti all belong to the same α-2 subdivision of the proteobacteria according to 16S rRNA sequence analysis. These three genera require similar chromosomal loci to establish a relationship between the bacteria and their hosts. One set of such genes required for the virulence of A. tumefaciens is a chromosomally encoded two-component regulatory system, chvG and chvI (12, 38). Insertion mutations in either chvG (the sensor histidine protein kinase) or chvI (the response regulator) render A. tumefaciens avirulent. Similar two-component regulatory systems critical for endosymbiosis or virulence were found in S. meliloti (15, 40) and B. abortus (48). The similarity of these two-component regulatory systems is accentuated further by the contiguous phosphoenol-pyruvate carboxykinase gene in all three species.
Another set of genes required for tumor formation by A. tumefaciens is chvA/chvB. These two genes are concerned with either the synthesis (chvB) or the transport (chvA) of a cyclic polysaccharide, β-1,2 glucan, into the periplasm. Both S. meliloti and B. abortus synthesize β-1,2 glucan, and both ndvA and ndvB are required for effective nodule invasion by S. meliloti (20). For B. abortus, the cgs gene, which complements an S. meliloti ndvB mutant and an A. tumefaciens chvB mutant, is also required for virulence (28).
In this report we characterize another chromosomal locus, chvH, previously shown to be required for tumor formation. We show that this locus encodes a homologue of the Escherichia coli elongation factor P (2, 3, 4).
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
The bacterial strains and plasmids used in this study are listed in Table 1. A. tumefaciens strains were grown in MG/L (35) or induction medium (IM) (11) at either 22 or 28°C. E. coli strains were grown in Luria-Bertani medium (46) at 37°C. The following antibiotics were used at the indicated concentrations when added to solid medium (in micrograms per millimeter): for A. tumefaciens, kanamycin (100), gentamicin (100), and spectinomycin (250); and for E. coli, carbenicillin (100), kanamycin (30), gentamicin (5), spectinomycin (50), and tetracycline (15). These concentrations were reduced by one-half for liquid medium.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| Agrobacterium strains | ||
| A348 | A136(pTiA6) | 21 |
| A6007 | A348 Pho− Smr | 11 |
| A6880 | A6007 chvH::TnphoA | 12 |
| At13000 | A348 ΔchvH::Ω fragment, contains pWT187kan | This study |
| At13001 | At13000 without pWT187kan | This study |
| E. coli strains | ||
| DH5α | endA1 hsdR17 supE44 thi-1 recA1 gyrA96 relA1 Δ(argF-lacZYA)U169 φ80dlacZΔM15 | Gibco-BRL |
| TG1 | supE hsdΔ5 thi Δ(lac-proAB) F′ (traD36 proAB+ lacIqlacZΔM15) | U.S. Biochemical Corp. |
| Plasmids | ||
| pACL2 | pRK7813 cosmid clone which complements A6880 | 12 |
| pSW172 | IncP broad-host-range vector with lac promoter | 14 |
| pSW213 | IncP broad-host-range vector with lac promoter and lacIq | 14 |
| pTC110 | pUCD2 ΔPvuII-EcoRV | 13 |
| pBBR1MCS-4 | Broad-host-range vector | 32 |
| pUC18 | Multicopy vector | 55 |
| pUC19Ω | SmaI Ω fragment in pUC19 | 10 |
| pPR1068 | pMAL-c2 derivative; NdeI at the start of MalE | New England Biolabs |
| pSP329 | pTJS75 derivative with α-complementation group and multicloning site from pUC18, IncP, Tcr | 11 |
| pSP329Gm | pSP329 derivative, IncP, Gmr | D. M. Raineri |
| pJQ200SK | sacB suicide vector | 44 |
| pUCD2 | IncW broad-host-range vector | 16 |
| pBSIIKS+.NdeI | pBluescriptIIKS+ derivative; NdeI at the start of lacZ′ | 9 |
| pAB2002 | Vector for transcriptional fusion with lacZ | 7 |
| pTC111 | virE2::lacZ translational fusion in pTC110 | T. C. Charles, unpublished |
| pTC112 | virB1::lacZ translational fusion in pTC110 | T. C. Charles, unpublished |
| pUFR047 | IncW broad-host-range vector | 17 |
| pPR1068-E1+E2 | Ptac-virE operon in pPR1068 | 18 |
| pUFR047-E1+E2 | Ptac-virE operon in pUFR047 | 18 |
| pSL59 | virA-phoA translational fusion | 19 |
| pTC234 | 7.4-kb KpnI-SacI fragment from pACL2 in pSP329 | This study |
| pTC234Ω9A::Tn5-B20 | Tn5-B20 insertion into chvH gene of pTC234 | This study |
| pWT131 | 4.4-kb KpnI-BamHI fragment in pBluescriptIISK+ | This study |
| pWT142 | pUC18 carrying a 7.6-kb SalI fragment from A6880, Kmr | This study |
| pWT151 | 2.0-kb EcoRI fragment of pTC234 in pSW172 | This study |
| pWT154 | Ptac-chvH in pPR1068 | This study |
| pWT155 | Ptac-chvH in pUCD2 | This study |
| pWT158 | 2-kb HpaI-ClaI fragment of pPR1068-E1+E2 in pBBR1MCS-4 | This study |
| pWT159 | Ptac-virE1 virE2-lacZ transcriptional fusion in pBBR1MCS-4 | This study |
| pWT160 | virG-lacZ translational fusion in pTC110 | This study |
| pWT179 | Ptac-driven E. coli efp in pPR1068 | This study |
| pWT181 | Ptac-driven E. coli efp in pSP329Gm | This study |
| pWT183 | Plac-chvH in pBSIIKS+.NdeI | This study |
| pWT187 | 1.6-kb PvuII fragment containing Plac-chvH in pSW213 | This study |
| pWT187kan | pWT187 plus a kanamycin resistance cassette | This study |
| pWT188 | 445-bp chvH coding region replaced by Ω in pWT183 | This study |
| pWT191 | pWT131 with the 1.29-kb NdeI-EcoRV fragment replaced by 2.8-kb NdeI-EcoRV fragment from pWT188 | This study |
| pWT193 | 6-kb KpnI-SstI fragment from pWT191 in pJQ200SK | This study |
Construction of plasmids.
Several plasmids were constructed to demonstrate that chvH is the only gene responsible for the defects of the chvH mutant. A 2.0-kb EcoRI fragment from pTC234 containing the chvH gene was cloned into pSW172 to give pWT151. A 1.4-kb NdeI-EcoRI fragment starting from the predicted start site of the chvH gene was ligated to a 5.3-kb NdeI-EcoRI fragment of pPR1068 to create pWT154. In this construct, the chvH gene is under the control of the Ptac promoter. A 2-kb EcoRV fragment of pWT154 containing the chvH gene was cloned into pUCD2 to create pWT155.
To test whether the E. coli efp gene can complement the defects of strain A6880, we placed the E. coli efp gene under the control of the Ptac promoter. Two primers, efp-1, GGCCATATGGCAACGTACTATAGCAAC, and efp-2, ACACTGCAGTTACTTCACGCGAGAGAC, were used to amplify the efp coding region from DH5α genomic DNA with Pfu. NdeI and PstI restriction sites were introduced into primers efp-1 and efp-2, respectively. PCR amplification followed the usual methods using Pfu: denaturation temperature, 95°C for 45 s; annealing temperature, 55°C for 60 min; and polymerization temperature, 72°C for 2 min. The 0.57-kb E. coli efp PCR product (digested with NdeI and PstI) was ligated with a 5.3-kb NdeI-PstI fragment of pPR1068 to create pWT179. A 1.2-kb EcoRV-PstI fragment of pWT179 containing the Ptac-driven efp gene was cloned into pSP329Gm which had been digested with SmaI and PstI to create pWT181.
To determine the transcription of the Ptac-virE operon, pWT159 was constructed as follows. A 2-kb HpaI-ClaI fragment of pPR1068-E1+E2 was cloned into SmaI- and ClaI-digested pBBR1MCS-4, giving pWT158. A 4.5-kb EcoRI fragment containing the lacZ-Gmr cassette from pAB2002 was ligated with EcoRI-digested pWT158 to create pWT159.
DNA sequencing and analysis.
Restriction fragments were subcloned into pBluescript II KS+ or pBluescript II SK+. All double-stranded DNA templates were prepared with a Qiagen kit. Sequencing was completed with universal forward and reverse primers as well as synthetic oligonucleotides deduced from already determined sequences (BRL). DNA sequencing was performed with a BigDye Terminator cycle sequencing ready reaction kit (PE Applied Biosystems, Foster City, Calif.), and the reactions were run on an ABI 377 automated DNA sequencer at the DNA sequence facility of the Biochemistry Department, University of Washington. Both strands were sequenced. DNA sequences were analyzed with GeneJockey. Protein homology searches were performed using the Blastp program at the National Center for Biotechnology Information.
To determine the precise insertion site of TnphoA in the original chvH mutant A6880, we cloned the phoA and flanking chvH region. Total chromosomal DNA of A6880 was digested with SalI, ligated with SalI-digested pUC18, and transformed into strain DH5α, selecting for simultaneous resistance to carbenicillin and kanamycin. The resulting plasmid, pWT142, was used as the template to sequence across the insertion junction by using an oligonucleotide primer, 5′-ACCCGTTAAACGGCGAGCACCGCCGGG-3′ (part of the phoA sequence).
vir gene expression assays.
The reporter plasmid pTC111 is derived from pSM358cd (49, 50), which contains a Tn3HoHo1 insertion in the virE2 gene. This resulted in the production of a VirE2-LacZ fusion protein. The reporter plasmid pTC112 is derived from pSM243cd (49, 50), which contains a Tn3HoHo1 insertion in the virB1 gene, resulting in the production of a VirB1-LacZ fusion protein. pWT160 is derived from pSM321cd (49, 50) and contains a Tn3HoHo1 insertion in the virG gene, resulting in a chimeric VirG-LacZ fusion protein that has approximately 130 amino acids of the VirG protein located at the amino terminus. pSL59 is a virA-phoA translational fusion plasmid (19). The reporter plasmids were introduced into A348 and A6880 by electroporation. vir gene expression assays were performed basically according to published methods (43). Alkaline phosphatase assays were performed as described previously (19).
Protein gels and Western immunoblotting.
Protein analysis using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed according to standard protocols (1). Gels were either stained with Coomassie blue or processed for Western blot analysis. Proteins were transferred to polyvinylidene difluoride membranes (Millipore) using the Trans-Blot SD semidry electrophoretic transfer cell (Bio-Rad Laboratories) and detected with the ECL Western blotting analysis system (Amersham Life Science). To detect VirA and VirB proteins, proteins were transferred to nitrocellulose in a Tris-glycine-methanol transfer buffer using a Transblot apparatus (Hofer, San Francisco, Calif.). vir genes in A. tumefaciens were induced as described previously (11). A348 and A6880 cells were harvested from overnight cultures grown in MG/L medium and washed once with induction medium containing 200 μM AS. The cells were then diluted into fresh induction medium containing 200 μM acetosyringone (AS) to an optical density at 600 nm (OD600) of 0.1 and then induced for 20 h at 28°C (or 16 h at 22°C for VirA and VirB proteins). Total crude extracts were prepared and subjected to electrophoresis (the polyacrylamide concentrations were 7% for VirA; 10% for VirB, VirE2, and VirD2; and 12% for VirG, VirJ, ChvE, and Ros) and immunoblotting analysis as described previously (6, 43). Polyclonal antibodies against proteins of VirA (a gift from S. Winans) or VirB (6), VirE2 and VirD2 (18), and VirJ (41) were used to detect Vir proteins. A monoclonal VirG antibody (a gift from S. Jin) was used to detect VirG. Antiserum specific to ChvE (43) or Ros (34) was used to detect ChvE and Ros, respectively. Protein concentrations were determined by using the Bio-Rad protein assay kit (Bio-Rad Laboratories, Richmond, Calif.) with bovine serum albumin as the standard.
Southern blot analysis.
A. tumefaciens genomic DNA for hybridization analysis was prepared by a published method (12). Genomic DNA was digested with restriction enzymes, subjected to gel electrophoresis, and transferred to a Hybond-N+ membrane under alkaline conditions. The membranes were hybridized in Church buffer (0.5 M NaHPO4, 7% SDS, 1 mM EDTA) at 65°C. Two 30-min washes were performed in 2× SSC–0.1% SDS (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) at 65°C for low-stringency conditions. For high-stringency washes, two 30-min washes in 0.5× SSC–0.1% SDS and one 30-min wash in 0.1× SSC–0.1% SDS were performed. Radiolabeled probes were prepared by a random oligonucleotide labeling procedure using the Ready To Go DNA labeling beads (without dCTP) from Amersham Pharmacia Biotech Inc.
Construction of strain At13000.
To determine whether chvH is essential for viability of Agrobacterium, strains were constructed that contained a single functional copy of chvH under the control of the inducible lac promoter. The 1.4-kb NdeI-EcoRI fragment starting from the predicted start site of chvH was ligated with pBSIIKS+.NdeI digested with NdeI and EcoRI to create pWT183. In this construct the expression of chvH was under the control of the lac promoter. A 1.6-kb PvuII fragment containing the Plac-driven chvH was cloned into blunt-ended EcoRI-digested pSW213, which can replicate in Agrobacterium and contains lacIq, resulting in pWT187. A 1.6-kb BamHI fragment containing a kanamycin resistance cassette was inserted at the BamHI site of pWT187 to give pWT187kan.
To construct a deleted version of the chvH mutation, pWT183 was digested with HindIII and ClaI, deleting a 445-bp fragment within the chvH coding region. A 2-kb SmaI fragment containing the spectinomycin-resistant Ω cassette was inserted between these sites to give pWT188. The 2.8-kb NdeI-EcoRV fragment containing Ω was subcloned into the large NdeI-EcoRV fragment of pWT131 to create pWT191. The 6-kb KpnI-SstI fragment of pWT191 was treated with T4 DNA polymerase and cloned into SmaI-digested pJQ200SK, which resulted in construct pWT193. pJQ200SK contains two selection markers for the subsequent homologous recombination step: aacC1, conferring gentamicin (Gm) resistance, and sacB, conferring sucrose sensitivity. In construct pWT193, a 445-bp HindIII-ClaI fragment within the coding sequence of chvH was replaced by the Ω fragment. Both pWT187kan and pWT193 were transformed into A348, and Kanr Spr colonies were selected (single crossover). The resulting strains had a total of three chvH copies: a functional chvH copy controlled by the lac promoter, a chvH wild-type gene, and a deleted version of the chvH gene replaced by the Ω fragment. To isolate strains that carried a single functional chvH gene controlled by the lac promoter, sucrose selection was carried out on these Kanr Spr colonies. The Kanr Spr colonies were grown overnight in MG/L medium containing 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) and then spread onto AB plates (11) containing 5% sucrose, 1 mM IPTG, spectinomycin, and kanamycin. The Sucr Kanr Spr colonies were then tested for the loss of the Gmr marker on plates containing IPTG. The Sucr Kanr Spr Gms colonies were further characterized by Southern blotting to identify the strain (At13000) which had a single functional chvH gene controlled by the lac promoter in a plasmid and a deleted version of chvH in the chromosome.
Assays for virulence.
Virulence assays were performed on Kalanchoe daigremontiana leaves. A. tumefaciens cells were grown in liquid MG/L to mid-log phase, then pelleted by centrifugation, concentrated, and deposited onto leaves wounded with a toothpick. Inoculated plants were grown for 2 to 5 weeks before tumor formation was scored.
Nucleotide sequence accession number.
The nucleotide sequence of the 4.4-kb KpnI-BamHI fragment carried on pTC234 has been submitted to the GenBank database and assigned accession number AF177860.
RESULTS
General features of chvH mutant A6880.
The chvH mutant A6880, obtained by TnphoA mutagenesis of A6007 (11, 12), is an avirulent, pleiotropic mutant. It is far more sensitive to detergents such as SDS, sodium deoxycholate, and Sarkosyl than the parent strain (12) and also to carbenicillin compared to the parental strain (data not shown). This suggests that the integrity of the outer membrane is impaired. The growth rate of the mutant on an enriched medium is somewhat slower than that of the wild-type strain (Fig. 1).
FIG. 1.

Growth curves. At time zero, overnight cultures were diluted in MG/L at a starting OD600 of 0.06 and incubated at 28°C with shaking. At the indicated times, the OD600 was measured and plotted against time. The growth curves shown represent a typical experiment.
chvH locus encodes a protein similar to elongation factor P.
To gain insight into the possible function of chvH, we first isolated a cosmid clone, pACL2, which complemented the detergent sensitivity and restored virulence to chvH mutant A6880 (12). We subcloned a 7.4-kb KpnI-SacI fragment from pACL2 into pSP329 to make plasmid pTC234. This plasmid also complemented the chvH mutant. A partial restriction map of pTC234 was then constructed (Fig. 2). TnphoA (36, 37) and Tn5-B20 (47) were used to mutagenize the 7.4-kb fragment in order to localize the region required for complementation of strain A6880. Several insertions in a 2.0-kb EcoRI fragment (the insert in pWT151) (Fig. 2) abolished complementation (data not shown).
FIG. 2.

Restriction map of A. tumefaciens chromosomal DNA inserts in pTC234, pWT151, and pWT155. Restriction sites are labeled as follows: B, BamHI; E, EcoRI; EV, EcoRV; H, HindIII; K, KpnI; N, NdeI; S, SacI; X, XhoI. The arrowhead under ORF2 indicates the site of the TnphoA insertion, while the arrow underneath indicates its orientation.
The 4.4-kb KpnI-BamHI fragment of pTC234 was sequenced (GenBank accession number AF177860), and three open reading frames (ORFs) were identified (ORF1, ORF2, and ORF3) (Fig. 2). The first one, extending from nucleotides 771 to 2225, is preceded by a ribosome-binding site (GGGAAA) and encodes a putative protein of 484 amino acids with a predicted molecular mass of 51,000 Da. The second ORF (nucleotides 2346 to 2915, complementary strand) with its potential ribosome-binding site (AGGAAG) encodes a putative protein of 189 amino acids with a predicted molecular mass of 21,000 Da. A third ORF was identified between nucleotides 3167 and 4231, which codes for a predicted protein of 354 amino acids with a molecular mass of 39,000 Da.
A database search of the predicted amino acid sequence of the three polypeptides revealed that ORF1 has a high level of identity with NodT from Rhizobium leguminosarum (52% identity and 68% similarity) (51). NodT is a member of a growing family of outer membrane proteins found in a wide variety of gram-negative bacteria, including several pathogens (42). ORF3 shows a high degree of similarity to lysyl-tRNA synthetase from a large number of bacteria.
ORF2 contains the site of the TnphoA insertion of the avirulent mutant A6880, which has been designated chvH. Comparing the predicted amino acid sequence of ChvH with the protein database revealed that ChvH is similar at the amino acid sequence level to a range of elongation factor P proteins. Elongation factor P has been implicated in peptide bond synthesis (2). The predicted amino acid sequences for the A. tumefaciens ChvH and the elongation factor P proteins for Rickettsia prowazekii and E. coli are aligned in Fig. 3 using the Malign program. The identity and similarity at the amino acid level are 39 and 62% for R. prowazekii and 36 and 57% for E. coli, respectively.
FIG. 3.

Amino acid alignment of the A. tumefaciens (Atu) elongation factor P protein with the elongation factor P proteins from R. prowazekii (Rpr) (AJ235271) and E. coli (Eco) (X61676). Amino acids are represented by the single-letter code. The Malign program was used for the comparison. Amino acid positions are indicated on the right. Amino acid residues that are identical in two of the sequences are shaded.
TnphoA is a transposon that can fuse alkaline phosphatase lacking a signal peptide to the amino-terminal sequences of the proteins into whose genes it inserts. Active fusions expressing alkaline phosphatase can arise only when this transposon inserts in genes encoding secreted or membrane-spanning proteins. Sequence analysis suggests that ChvH is a soluble protein. The TnphoA insertion in A6880 is located between bp 123 and 124 of the chvH ORF, and chvH is in the opposite orientation relative to the phoA gene of TnphoA (Fig. 2), although the insertion was initially identified after screening a collection of mutants that expressed alkaline phosphatase activity. However, when we checked colonies of A6880 growing on MG/L plates containing XP (5-bromo-4-chloro-3-indolylphosphate), they were white, as would be predicted. Two potential rho-independent transcriptional terminators (nucleotides 2244 to 2279 and 2299 to 2326) were identified, one downstream of the chvH gene and the other downstream of the nodT homologue. The isoelectric point of ChvH, calculated from the sequence, is 5.13.
Complementation of the chvH mutation.
To prove that the TnphoA insertion is actually responsible for the several defects observed in strain A6880, we complemented the mutation with chvH-containing subclones of the complementing cosmid. Two plasmids, pWT151 and pWT155, were constructed (see Fig. 2 and Materials and Methods). pWT151 contains the chvH gene under the control of its native promoter and flanking DNA sequences, while pWT155 contains the chvH gene under the control of the tac promoter. Both pWT151 and pWT155 complemented the detergent sensitivity (Fig. 4A) and restored virulence of A6880 on K. daigremontiana (for complementation by pWT155, see Fig. 5). These data strongly suggest that disruption of chvH is responsible for both the avirulence and detergent sensitivity of A6880.
FIG. 4.

(A) Functional complementation of chvH mutant A6880. Cells were grown on MG/L plates containing 0.2 g of SDS per liter at 28°C and observed after 3 days. (B) Measurement of VirE2 expression. A. tumefaciens strains were grown in IM containing 200 μM AS at 28°C for 20 h. Cells were pelleted, and total crude extracts were subjected to immunoblotting analysis as described in Materials and Methods. Strains: 1, A6007; 2, A6880(pWT155); 3, A6880(pWT181); 4, A6880(pSP329Gm).
FIG. 5.

Tumorigenesis assay on Kalanchoe leaves. The assay was performed as described in Materials and Methods.
We also tested whether the E. coli efp gene could complement the chvH mutation. The efp gene from E. coli was cloned under the control of the tac promoter to form plasmid pWT181. This gene (pWT181) complemented the detergent sensitivity of the chvH mutant strain (Fig. 4A) and increased the growth rate (data not shown). Most importantly, the E. coli efp gene (pWT181) restored virulence to A6880 (Fig. 5), although it took longer for tumors to appear and the tumors were smaller than those formed by the construct with the tac promoter-driven chvH itself (pWT155). Thus, the tac promoter-driven E. coli efp can phenotypically complement the chvH mutant, although the E. coli gene does not function optimally in A. tumefaciens. chvH and efp are functionally homologous.
Regulation of the chvH gene.
Plasmid pTC234Ω9A::Tn5-B20 is a derivative of pTC234 in which Tn5-B20 has inserted into the 2.0-kb EcoRI fragment containing the chvH gene. The Tn5-B20 insertion also abolished the complementation ability of pTC234. Tn5-B20 forms an operon fusion with the gene into which it inserts. We transformed A348 with pTC234Ω9A::Tn5-B20. The cells were grown in (i) AB (pH 7.5) or AB plus 50 mM MES (morpholineethanesulfonic acid, pH 5.5) or IM with and without AS. The β-galactosidase activities were determined. The results showed that chvH gene is constitutively expressed, independent of acid conditions and phenolic compounds (data not shown).
Reduced expression of virulence genes in chvH mutant.
Since the chvH mutation rendered cells avirulent, we investigated how well the Ti plasmid-encoded vir genes were expressed in the mutant. An appropriate level of expression of the vir genes is critical for tumor formation. Plasmid pTC112 containing the virB1::lacZ translational fusion was introduced into the chvH mutant and the wild-type strain. As shown in Table 2, expression of the virB-lacZ fusion was reduced 80% in the chvH mutant compared with the wild-type strain under the same conditions. We also introduced a virE2-lacZ translational fusion on a plasmid (pTC111) into the same strains. The VirE2-LacZ fusion protein in the chvH mutant was assayed under inducing and noninducing conditions. Under noninducing conditions, we observed that the basal level of VirE2-LacZ expression was reduced approximately 70% in the chvH mutant compared with the wild-type strain. Under inducing conditions, the expression of VirE2-LacZ was reduced approximately 85%.
TABLE 2.
Effect of chvH mutation on expression of lacZ and phoA translational fusions to vir genesa
| Fusion (plasmid) | β-Galactosidase expression (Miller units)
|
|||
|---|---|---|---|---|
|
chvH strain (A348)
|
chvH::TnphoA strain (A6880)
|
|||
| IM | IM + 100 μM AS | IM | IM + 100 μM AS | |
| virB-lacZ (pTC112) | ND | 1,357 ± 5 | ND | 285 ± 6 (80%) |
| virE-lacZ (pTC111) | 46.6 ± 1.0 | 3,208 ± 46 | 15.1 ± 1.0 (70%) | 519 ± 15 (85%) |
| virG-lacZ (pWT160) | 77.5 ± 1.5 | 1,536 ± 39 | 48.0 ± 2.4 (40%) | 360 ± 10 (75%) |
| virA-phoA (pSL59) | 6.9 ± 0.7 | 59 ± 1.5 (8.6) | 6.9 ± 0.2 | 42 ± 1.0 (6) |
Data are means ± standard errors of the mean for three samples. The percentages in parentheses represent the reduction in expression in the chvH mutant relative to the wild-type strain. The numbers in parentheses are the fold increases in induction upon adding AS. The units are Miller units. ND, not determined.
The above results clearly show that the expression of VirB and VirE2 is reduced in the chvH mutant. Because the vir operons are under the control of the VirA/VirG two-component system, chvH might reduce the level of VirB and VirE2 proteins indirectly by affecting the expression of the two-component system. Therefore, the level of VirA was determined by assaying the phoA activity of a virA-phoA translational fusion (pSL59) in the two backgrounds. As shown in Table 2, the induction of VirA expression by AS in the wild-type strain agrees with previous observations (45, 53), while the expression of VirA was only induced slightly less in the chvH mutant. To measure VirG expression, we used virG-lacZ translational fusion plasmid pWT160. Under noninducing conditions, VirG-LacZ expression was reduced 40% in the chvH mutant (Table 2). In the presence of AS, the reduction was substantially greater (approximately 75%), probably in part because of the positive autoregulation of VirG (52) (Table 2).
We also analyzed the expression of vir genes by measuring the accumulation of several Vir proteins by Western immunoblotting. As shown in Fig. 6, the levels of VirB8, -9, -10, and -11 were reduced dramatically in the mutant cells compared with those of the wild-type strain. VirE2 and VirJ were undetectable in the chvH mutant. The level of VirD2 was markedly reduced but still detectable. The levels of both VirA and VirG were significantly reduced. The expression of VirE2 was also measured in the chvH mutant transformed with the Ptac-driven chvH gene (pWT155) and the Ptac-driven E. coli efp gene (pWT181). As shown in Fig. 4B, both the A. tumefaciens chvH gene (pWT155) and the E. coli efp gene (pWT181) restored the expression of VirE2.
FIG. 6.

Effect of the chvH mutation on the production of virulence-related proteins. A. tumefaciens strains A348 and A6880 were grown in IM containing 200 μM AS at 28°C for 20 h (or 22°C for 16 h for VirA and VirB proteins). Cells were pelleted, and total crude extracts were subjected to immunoblotting analysis as described in Materials and Methods. To detect VirB proteins, loading was standardized so that the samples in each lane represent equivalent numbers of cells. To detect other proteins, equal amounts of total protein from A348 and A6880 were loaded. The wild-type strain A348 is on the left, while the chvH mutant A6880 is on the right.
Both kinds of analysis indicate that the expression of the two-component system, especially VirG, the transcriptional activator for all of the vir genes including itself, was reduced. Therefore, the reduced levels of Vir proteins could in part be explained by the reduced levels of the VirA and VirG proteins. We conclude that the wild-type chvH locus is essential for full expression of vir genes encoded by the Ti plasmid.
The levels of two chromosomally encoded proteins were also analyzed in the chvH mutant by Western blotting. As shown in Fig. 6, the level of the ChvE protein was about the same in the chvH mutant as in the wild-type strain, whereas the level of the Ros protein was significantly reduced. It appears that the level of different proteins is affected differently by the chvH mutation.
ChvH functions at the posttranscriptional level.
It has been reported that elongation factor P is involved in peptide bond synthesis in E. coli (2, 3, 4). To confirm the involvement of ChvH at the posttranscriptional level, we measured the expression of the VirE2 protein in A348 and A6880. To bypass the VirA/VirG two-component system, which adds another level of complexity to the expression level, we used the Ptac-virE operon construct pUFR047-E1+E2. Figure 7A shows that the synthesis of VirE2 was significantly reduced in the chvH mutant compared with the wild-type strain. When the transcription of the Ptac-virE operon was determined by using lacZ transcriptional fusion plasmid pWT159, the transcription of the Ptac-virE operon was not affected by the chvH mutation (Fig. 7B). Therefore, we conclude that ChvH functions at the posttranscriptional level. According to the data shown in Fig. 7B, translation of native β-galactosidase was not affected by the chvH mutation.
FIG. 7.

(A) Measurement of VirE2 protein levels with a Ptac-driven virE operon in A348 and A6880. Cells were grown to mid-log phase in MG/L at 28°C, and total crude extracts were prepared as described in Materials and Methods. Equal amounts of total crude extracts (20 μg) from each strain were loaded. Lanes: 1, A348(pUFR047-E1+E2); 2, A6880(pUFR047-E1+E2). (B) Determination of transcription level of the tac promoter in A348 and A6880. A348 and A6880 were transformed with the reporter plasmid pWT159. The cells were grown in AB medium at 28°C and harvested at an OD600 of about 1.0. β-Galactosidase activity (Miller units) was monitored as described in Materials and Methods.
A. tumefaciens gene encoding elongation factor P is present as a single copy in the genome.
Aoki et al. (4) reported that in E. coli, the efp locus is essential for viability. The TnphoA insertion in the chvH mutant is within the coding region of the gene encoding elongation factor P. These results raised the possibility that Agrobacterium might have two copies of the gene encoding elongation factor P.
A genomic Southern blot analysis was performed to determine if the gene encoding elongation factor P is a single copy. The 0.5-kb NdeI-ClaI internal fragment of the chvH gene was used to probe a Southern blot of A348 genomic DNA individually digested with a panel of restriction enzymes. Employing low-stringency wash conditions (see Materials and Methods), only those fragments expected from the restriction map of the chvH locus itself were observed (data not shown). This strongly suggests that only one copy of the gene encoding elongation factor P is present in the A. tumefaciens genome.
Is chvH essential for viability of Agrobacterium?
The above data suggest that only one copy of the gene encoding elongation factor P is present in Agrobacterium. Therefore, two possibilities exist: (i) the gene encoding elongation factor P is essential for the viability of Agrobacterium but the TnphoA insertion is leaky, and (ii) the gene is not essential for the viability of Agrobacterium. To distinguish between these two possibilities, we constructed a deletion mutant of chvH and characterized the resulting strain.
We first placed the chvH gene under the control of the IPTG-inducible lac promoter (in plasmid pWT187kan; see Materials and Methods). This construct was then transferred into A. tumefaciens A348 by electroporation. We then exchanged the wild-type copy of chvH with the deleted version of the chvH gene (a 445-bp HindIII-ClaI fragment was deleted) in the presence of IPTG, giving strain At13000 (see Materials and Methods).
To determine whether chvH is an essential gene, we first streaked out the At13000 cells on MG/L plates lacking IPTG to see whether the depletion of the ChvH protein stopped cell growth. The depletion did not stop cell growth. The cells formed colonies on solid medium and also increased in optical density in liquid medium, suggesting that chvH is not an essential gene. If this is indeed the case, it should be possible to cure strain At13000 of plasmid pWT187kan without affecting cell viability. To this end, we grew strain At13000 under nonselective conditions overnight in liquid medium and plated out the culture for single colonies on MG/L plates containing spectinomycin. Among 300 colonies, 20 colonies were kanamycin sensitive. These were candidates for cells that had lost the plasmid. Southern blot analysis of 10 of these colonies confirmed that they indeed did not contain pWT187kan (data not shown). The cured strain was named At13001. Since a strain which lacked the chvH gene could be isolated, the gene product is not essential for cell viability.
Elongation factor P is necessary for optimum growth of A. tumefaciens.
We investigated the effect of elimination of the chvH locus on cell growth in rich medium, MG/L. As shown in Fig. 1, the deletion mutant At13001 grew significantly more poorly than the TnphoA insertion mutant A6880, suggesting that the original mutant was leaky for elongation factor P activity. The deletion mutant also exhibited a longer lag time than the TnphoA insertion mutant. The effect of an introduced wild-type chvH gene and the E. coli efp gene on the growth of strain At13001 was also studied. As expected, the chvH gene expressed from the Ptac promoter (pWT155) increased the growth rate to nearly the level of the wild-type strain A348 (data not shown). Interestingly, the E. coli efp gene expressed from the Ptac promoter (pWT181) increased the growth rate to about the same extent as the introduced chvH gene (data not shown). As expected, strain At13001 is avirulent. Both pWT155 and pWT181 restored virulence to strain At13001 (data not shown).
A 32-kDa protein accumulates in the chvH mutant.
The effect of the chvH mutation in A6880 on the protein profile was examined on an SDS-PAGE gel. Figure 8 shows representative data for the patterns of soluble proteins in the chvH mutant A6880 and its parental strain A6007 when equal amounts of protein were loaded on the gel. The most striking difference is that a 32-kDa protein accumulated in the chvH mutant. Several other less striking differences were observed between the mutant and parental strains. The 32-kDa protein also accumulated in the chvH deletion mutant At13001 (data not shown). We are in the process of identifying this protein.
FIG. 8.
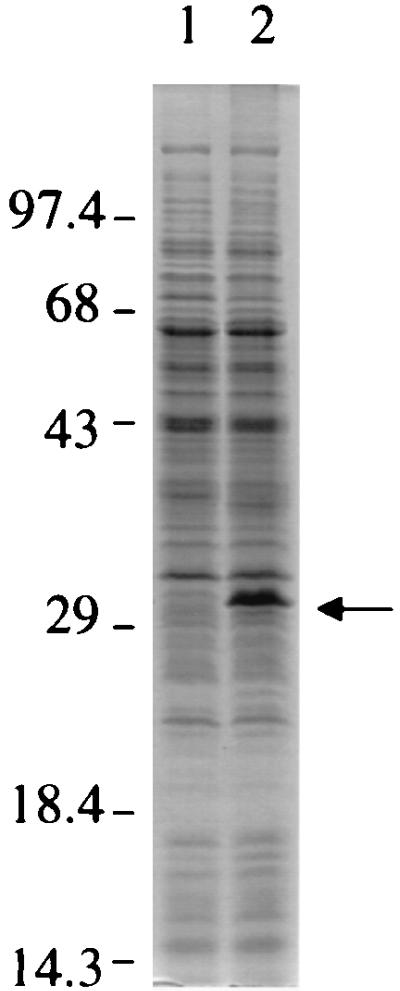
Protein profiles of chvH mutant A6880 and its parental strain A6007. A. tumefaciens A6007 and A6880 were grown in IM containing 200 μM AS at 28°C for 20 h. Cells were pelleted, and total crude extracts were subjected to SDS-PAGE analysis. Equal amounts of total crude extracts (20 μg) from each strain were loaded. Lanes: 1, A6007; 2, A6880. The arrow indicates the 32-kDa protein.
DISCUSSION
In this study, we have characterized the chvH chromosomal virulence locus, which was originally identified by TnphoA mutagenesis as being required for virulence. Sequence analysis indicates that chvH encodes a protein homologous to the elongation factor P protein of E. coli (2, 3, 4). This sequence homology is strongly supported by the observation that expression of the efp gene of E. coli can complement the chvH mutant and restore all of the phenotypic alterations resulting from the mutation in Agrobacterium. This is the first demonstration that an E. coli gene can complement the avirulent phenotype of an A. tumefaciens mutant and speaks to the highly conserved nature of this protein.
The exact role of elongation factor P in protein synthesis is not clear. It was originally isolated from a complex of 70S ribosome-AUG-formyl[35S]Met-tRNA and added puromycin (22, 23). This factor stimulated the synthesis of N-formyl-methionyl-puromycin and was thought to be involved in peptide bond synthesis. Further studies indicated that elongation factor P was more effective in increasing the efficiency of peptide bond formation between formyl[35S]Met-tRNA and amino acids with small R groups, such as glycine (24). A recent study indicates that bacterial elongation factor P is homologous to the eukaryal/archaeal eIF-5A (33), with the highest homology at the N-terminal 90 residues. Factor eIF-5A was originally isolated from a high-salt wash of rabbit reticulocyte ribosomes and was thought to be involved in translation initiation (31). eIF-5A enhanced the synthesis of methionyl-puromycin in vitro, suggesting that eIF-5A is involved in the formation of the first peptide bond in translation (8). In Saccharomyces cerevisiae, two genes (TIF51A and TIF51B) code for eIF-5A. Although deletion of both eIF-5A genes was lethal, the complete depletion of eIF-5A in the cell led to only a 30% drop in the first round of protein synthesis (30). These authors suggested that eIF-5A is not absolutely necessary for general protein synthesis in eukaryotic cells but is essential for the translation of a subset of specific mRNAs.
A recent study concluded that efp was essential for viability and was required for protein synthesis in E. coli (4). We have not found this to be the case in A. tumefaciens. We have clearly shown that A. tumefaciens has a single copy of chvH, which can be disrupted without loss of viability, although the cells grow more slowly and are no longer virulent. Our observations are consistent with the supposition that elongation factor P increases the efficiency of formation of peptide bonds involving aminoacyl acceptors that bind poorly to the ribosome in its absence (25) and argue against the notion that elongation factor P is required for general protein synthesis.
The virulence genes of A. tumefaciens for which chvH-dependent expression was examined in the present study appear to belong to a class of genes whose optimal translation depends on elongation factor P. Expression of other genes that were tested was not affected to the same extent. The elongation factor P-dependent genes might code for particular sequences of amino acids, perhaps near the start of translation, that are exceptionally dependent on elongation factor P for translation. An example of this type of specificity is the observation that when the Bacteroides fragilis efp gene was introduced into E. coli, the B. fragilis glutamine synthetase activity increased in E. coli but the activity of B. fragilis sucrase was unaffected (1). The significance of the control of A. tumefaciens virulence gene expression at the posttranscriptional level is not immediately apparent. Further experimental work is necessary to characterize the role of specific amino acid sequences on the elongation factor P dependence of translation.
We have demonstrated that at least some of the vir genes are regulated posttranscriptionally in a chvH-dependent manner. When virE2 transcription was driven by the tac promoter and thus uncoupled from VirG control, the level of VirE2 protein was drastically reduced in the chvH mutant strain, although the levels of virE2 transcription in the chvH mutant and the wild-type strain were comparable (Fig. 7). The reduction in the levels of Vir proteins in the chvH mutant thus appears to be determined at two stages. In addition to the direct effects on translation of vir mRNA, the reduction in the levels of all Vir proteins could be partly due to the reduced levels of VirA and especially VirG, which is rate limiting for vir gene induction (14). The effect of the chvH mutation on the expression of VirG is likely due to a direct effect on the translation of VirG. However, it is possible that this reduction results from overproduction of the 32-kDa protein acting on the translation of the vir genes. In this case, the role of chvH would be indirect. The reduction in Vir protein levels at the posttranscriptional level raises the possibility that elongation factor P serves as a posttranscriptional regulator of vir gene expression, perhaps in concert with a second regulatory factor. Identification of the 32-kDa protein that accumulated in the chvH mutant might provide an insight into this possibility.
The simplest explanation for the avirulence of the chvH mutant is that the levels of key proteins required for T-DNA transfer are reduced profoundly. These include VirB8, VirB9, VirB10, and VirB11 as well as the single-stranded-DNA-binding protein VirE2. We would predict that the levels of other Vir proteins would also be depressed. However, the possibility that the chvH gene product contributes in other ways to tumorigenesis cannot be ruled out. In this regard, it may be significant that a number of chromosomal avirulent mutants have in common a lack of integrity in their outer membrane (12).
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant GM32618 from the National Institutes of Health to E. W. Nester and by grant MCB9513662 from the National Science Foundation to L. M. Banta.
We thank S. Jin, Y. Machida, and C. I. Kado for antibodies to VirG, CheE, and Ros, respectively. A. Becker, P. Christie, M. Hynes, and K. M. Peterson kindly provided plasmids. Lishan Chen, Derek Wood, Rad Roberts, Mario Pantoja, and Paul de Figueiredo were helpful in giving suggestions and engaging in critical discussions of the data. The technical assistance of T. Jackson and M. Stremlau is gratefully acknowledged. B. R. Glick is thanked for enlightening discussions on early elongation factor P research.
REFERENCES
- 1.Abratt V R, Mbewe M, Woods D R. Cloning of an EF-P homologue from Bacteroides fragilis that increases B. fragilis glutamine synthetase activity in Escherichia coli. Mol Gen Genet. 1998;258:363–372. doi: 10.1007/s004380050742. [DOI] [PubMed] [Google Scholar]
- 2.Aoki H, Adams S L, Chung D G, Yaguchi M, Chuang S E, Ganoza M C. Cloning, sequencing and overexpression of the gene for prokaryotic factor EF-P involved in peptide bond synthesis. Nucleic Acids Res. 1991;19:6215–6220. doi: 10.1093/nar/19.22.6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aoki H, Adams S L, Turner M A, Ganoza M C. Molecular characterization of the prokaryotic efp gene product involved in a peptidyltransferase reaction. Biochimie. 1997;79:7–11. doi: 10.1016/s0300-9084(97)87619-5. [DOI] [PubMed] [Google Scholar]
- 4.Aoki H, Dekany K, Adams S L, Ganoza M C. The gene encoding the elongation factor P protein is essential for viability and is required for protein synthesis. J Biol Chem. 1997;272:32254–32259. doi: 10.1074/jbc.272.51.32254. [DOI] [PubMed] [Google Scholar]
- 5.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. New York, N.Y: John Wiley & Sons; 1996. [Google Scholar]
- 6.Banta L, Bohne M J, Lovejoy S D, Dostal K. Stability of the Agrobacterium tumefaciens VirB10 protein is modulated by growth temperature and periplasmic osmoadaption. J Bacteriol. 1998;180:6597–6606. doi: 10.1128/jb.180.24.6597-6606.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker A, Schmidt M, Jager M, Puhler A. New gentamicin-resistance and lacZ promoter-probe cassettes suitable for insertion mutagenesis and generation of transcriptional fusions. Gene. 1995;162:37–39. doi: 10.1016/0378-1119(95)00313-u. [DOI] [PubMed] [Google Scholar]
- 8.Benne R, Broen-Luedi M L, Hershey J W. Purification and characterization of protein synthesis initiation factors eIF-1, eIF-4C, eIF-4D, and eIF-5 from rabbit reticulocytes. J Biol Chem. 1978;253:3070–3077. [PubMed] [Google Scholar]
- 9.Berger B R, Christie P J. Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J Bacteriol. 1994;176:3646–3660. doi: 10.1128/jb.176.12.3646-3660.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bilge S S, Apostol J M, Jr, Fullner K J, Moseley S L. Transcriptional organization of the F1845 fimbrial adhesin determinant of Escherichia coli. Mol Microbiol. 1993;7:993–1006. doi: 10.1111/j.1365-2958.1993.tb01191.x. [DOI] [PubMed] [Google Scholar]
- 11.Cangelosi G A, Best E A, Martinetti G, Nester E W. Genetic analysis of Agrobacterium. Methods Enzymol. 1991;204:384–397. doi: 10.1016/0076-6879(91)04020-o. [DOI] [PubMed] [Google Scholar]
- 12.Charles T C, Nester E W. A chromosomally encoded two-component sensory transduction system is required for virulence of Agrobacterium tumefaciens. J Bacteriol. 1993;175:6614–6625. doi: 10.1128/jb.175.20.6614-6625.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charles T C, Doty S L, Nester E W. Construction of Agrobacterium strains by electroporation of genomic DNA and its utility in analysis of chromosomal virulence mutations. Appl Environ Microbiol. 1994;60:4192–4194. doi: 10.1128/aem.60.11.4192-4194.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C-Y, Winans S C. Controlled expression of the transcriptional activator gene virG in Agrobacterium tumefaciens by using the Escherichia coli lac promoter. J Bacteriol. 1991;173:1139–1144. doi: 10.1128/jb.173.3.1139-1144.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng H P, Walker G C. Succinoglycan production by Rhizobium meliloti is regulated through the ExoS-ChvI two-component regulatory system. J Bacteriol. 1998;180:20–26. doi: 10.1128/jb.180.1.20-26.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Close T J, Zaitlin D, Kado C I. Design and development of amplifiable broad-host-range cloning vectors: analysis of the vir region of Agrobacterium tumefaciens plasmid pTiC58. Plasmid. 1984;12:111–118. doi: 10.1016/0147-619x(84)90057-x. [DOI] [PubMed] [Google Scholar]
- 17.DeFeyter R D, Yang Y, Gabriel D W. Gene-for-genes interactions between cotton R genes and Xanthomonas campestris pv. malvacearum avr genes. Mol Plant-Microbe Interact. 1993;6:225–237. doi: 10.1094/mpmi-6-225. [DOI] [PubMed] [Google Scholar]
- 18.Deng W, Chen L, Peng W T, Liang X, Sekiguchi S, Gordon M P, Comai L, Nester E W. VirE1 is a specific molecular chaperone for the exported single-stranded-DNA-binding protein VirE2 in Agrobacterium. Mol Microbiol. 1999;31:1795–1807. doi: 10.1046/j.1365-2958.1999.01316.x. [DOI] [PubMed] [Google Scholar]
- 19.Doty S L, Yu M C, Lundin J I, Heath J D, Nester E W. Mutational analysis of the input domain of the VirA protein of Agrobacterium tumefaciens. J Bacteriol. 1996;178:961–970. doi: 10.1128/jb.178.4.961-970.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dylan T, Ielpi L, Stanfield S, Kashyap L, Douglas C, Yanofsky M, et al. Rhizobium meliloti genes required for nodule development are related to chromosomal virulence genes of Agrobacterium tumefaciens. Proc Natl Acad Sci USA. 1986;83:4403–4407. doi: 10.1073/pnas.83.12.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garfinkel D J, Nester E W. Agrobacterium tumefaciens mutants affected in crown gall tumorigenesis and octopine catabolism. J Bacteriol. 1980;144:732–743. doi: 10.1128/jb.144.2.732-743.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glick B R, Ganoza M C. Identification of a soluble protein that stimulates peptide bond synthesis. Proc Nat Acad Sci USA. 1975;72:4257–4260. doi: 10.1073/pnas.72.11.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glick B R, Ganoza M C. Characterization and site of action of a soluble protein that stimulates peptide-bond synthesis. Eur J Biochem. 1976;71:483–491. doi: 10.1111/j.1432-1033.1976.tb11137.x. [DOI] [PubMed] [Google Scholar]
- 24.Glick B R, Chladek S, Ganoza M C. Peptide bond formation stimulated by protein synthesis factor EF-P depends on the aminoacyl moiety of the acceptor. Eur J Biochem. 1979;97:23–28. doi: 10.1111/j.1432-1033.1979.tb13081.x. [DOI] [PubMed] [Google Scholar]
- 25.Glick B R. A molecular model of peptide chain propogation. J Theor Biol. 1980;82:149–155. doi: 10.1016/0022-5193(80)90094-6. [DOI] [PubMed] [Google Scholar]
- 26.Heath J D, Charles T C, Nester E W. Ti plasmid and chromosomally encoded two-component systems important in plant cell transformation by Agrobacterium species. In: Hoch J A, Silhavy T J, editors. Two-component signal transduction. Washington, D.C.: American Society for Microbiology; 1995. pp. 367–385. [Google Scholar]
- 27.Hooykaas P J J, Beijersbergen A G M. The virulence system of Agrobacterium tumefaciens. Annu Rev Phytopathol. 1994;32:157–179. [Google Scholar]
- 28.Inon de Iannino N, Briones G, Tolmasky M, Ugalde R A. Molecular cloning and characterization of cgs, the Brucella abortus cyclic β(1-2) glucan synthetase gene: genetic complementation of Rhizobium meliloti ndvB and Agrobacterium tumefaciens chvB mutants. J Bacteriol. 1998;180:4392–4400. doi: 10.1128/jb.180.17.4392-4400.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kalogeraki V S, Winans S C. The octopine Ti plasmid pTiA6 of Agrobacterium tumefaciens contains a gene homologous to the chromosomal virulence gene acvB. J Bacteriol. 1995;177:892–897. doi: 10.1128/jb.177.4.892-897.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kang H A, Hershey J W. Effect of initiation factor eIF-5A depletion on protein synthesis and proliferation of Saccharomyces cerevisiae. J Biol Chem. 1994;269:3934–3940. [PubMed] [Google Scholar]
- 31.Kemper W M, Berry K W, Merrick W C. Purification and properties of rabbit reticulocyte protein synthesis initiation factors M2Bα and M2Bβ. J Biol Chem. 1976;251:5551–5557. [PubMed] [Google Scholar]
- 32.Kovach M E, Elzer P H, Hill D S, Robertson G T, Farris M A, Roop R M., II Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene. 1995;166:175–176. doi: 10.1016/0378-1119(95)00584-1. [DOI] [PubMed] [Google Scholar]
- 33.Kyrpides N C, Woese C R. Universally conserved translation initiation factors. Proc Natl Acad Sci USA. 1998;95:224–228. doi: 10.1073/pnas.95.1.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lai E M, Kado C I. Processed VirB2 is the major subunit of the promiscuous pilus of Agrobacterium tumefaciens. J Bacteriol. 1998;180:2711–2717. doi: 10.1128/jb.180.10.2711-2717.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lichtenstein C P, Draper J. Genetic engineering of plants. In: Glover D M, editor. DNA cloning: a practical approach. II. Washington, D.C.: IRL Press; 1985. pp. 67–119. [Google Scholar]
- 36.Long S, McCune S, Walker G C. Symbiotic loci of Rhizobium meliloti identified by random TnphoA mutagenesis. J Bacteriol. 1988;170:4257–4265. doi: 10.1128/jb.170.9.4257-4265.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manoil C, Beckwith J. TnphoA: a transposon probe for protein export signals. Proc Natl Acad Sci USA. 1985;82:8129–8133. doi: 10.1073/pnas.82.23.8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mantis N J, Winans S C. The chromosomal response regulatory gene chvI of Agrobacterium tumefaciens complements an Escherichia coli phoB mutation and is required for virulence. J Bacteriol. 1993;175:6625–6636. doi: 10.1128/jb.175.20.6626-6636.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nester E W, Kemner J, Deng W, Lee Y-W, Fullner K, Liang X. Agrobacterium: a natural genetic engineer exploited for plant biotechnology. In: Stacey G, Mullin B, Gresshoff P M, editors. Biology of plant-microbe interactions. Minneapolis, Minn: International Society for Molecular Plant-Microbe Interactions; 1996. pp. 111–120. [Google Scholar]
- 40.Østerås M, Stanley J, Finan T M. Identification of rhizobium-specific intergenic mosaic elements within an essential two-component regulatory system of Rhizobium species. J Bacteriol. 1995;177:5485–5494. doi: 10.1128/jb.177.19.5485-5494.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan S Q, Jin S, Boulton M I, Hawes M, Gordon M P, Nester E W. An Agrobacterium virulence factor encoded by a Ti plasmid gene or a chromosomal gene is required for T-DNA transfer into plants. Mol Microbiol. 1995;17:259–69. doi: 10.1111/j.1365-2958.1995.mmi_17020259.x. [DOI] [PubMed] [Google Scholar]
- 42.Paulsen I T, Park J H, Choi P S, Saier M H., Jr A family of gram-negative bacterial outer membrane factors that function in the export of proteins, carbohydrates, drugs and heavy metals from gram-negative bacteria. FEMS Microbiol Lett. 1997;156:1–8. doi: 10.1111/j.1574-6968.1997.tb12697.x. [DOI] [PubMed] [Google Scholar]
- 43.Peng W-T, Lee Y-W, Nester E W. The phenolic recognition profiles of the Agrobacterium tumefaciens VirA protein are broadened by a high level of the sugar binding protein ChvE. J Bacteriol. 1998;180:5632–5638. doi: 10.1128/jb.180.21.5632-5638.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quandt J, Hynes M F. Versatile suicide vectors which allow direct selection for gene replacement in gram-negative bacteria. Gene. 1993;127:15–21. doi: 10.1016/0378-1119(93)90611-6. [DOI] [PubMed] [Google Scholar]
- 45.Rogowsky P M, Close T J, Chimera J A, Shaw J J, Kado C I. Regulation of the vir genes of Agrobacterium tumefaciens plasmid pTiC58. J Bacteriol. 1987;169:5101–5112. doi: 10.1128/jb.169.11.5101-5112.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 47.Simon R, Quandt J, Klipp W. New derivatives of transposon Tn5 suitable for mobilization of replicons, generation of operon fusions and induction of genes in gram-negative bacteria. Gene. 1989;80:161–169. doi: 10.1016/0378-1119(89)90262-x. [DOI] [PubMed] [Google Scholar]
- 48.Sola-Landa A, Pizarro-Cerda J, Grillo M J, Moreno E, Moriyon I, Blasco J M, Gorvel J P, Lopez-Goni I. A two-component regulatory system playing a critical role in plant pathogens and endosymbionts is present in Brucella abortus and controls cell invasion and virulence. Mol Microbiol. 1998;29:125–38. doi: 10.1046/j.1365-2958.1998.00913.x. [DOI] [PubMed] [Google Scholar]
- 49.Stachel S E, An G, Flores C, Nester E W. A Tn3-lacZ transposon for the random generation of β-galactosidase gene fusions: applications to the analysis of gene expression in Agrobacterium. EMBO J. 1985;4:891–898. doi: 10.1002/j.1460-2075.1985.tb03715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stachel S E, Zambryski P C. virA and virG control the plant-induced activation of the T-DNA transfer process of A. tumefaciens. Cell. 1986;46:325–333. doi: 10.1016/0092-8674(86)90653-7. [DOI] [PubMed] [Google Scholar]
- 51.Surin B P, Watson J M, Hamilton W D O, Economou A, Downie J A. Molecular characterization of the nodulation gene nodT from two biovars of Rhizobium leguminosarum. Mol Microbiol. 1990;4:245–252. doi: 10.1111/j.1365-2958.1990.tb00591.x. [DOI] [PubMed] [Google Scholar]
- 52.Winans S C. Two-way chemical signaling in Agrobacterium-plant interactions. Microbiol Rev. 1992;56:12–31. doi: 10.1128/mr.56.1.12-31.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Winans S C, Kerstetter R A, Nester E W. Transcriptional regulation of the virA and virG genes of Agrobacterium tumefaciens. J Bacteriol. 1988;170:4047–4054. doi: 10.1128/jb.170.9.4047-4054.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wirawan I G, Kang H W, Kojima M. Isolation and characterization of a new chromosomal virulence gene of Agrobacterium tumefaciens. J Bacteriol. 1993;175:3208–3212. doi: 10.1128/jb.175.10.3208-3212.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]