

Abstract
Introduction
Few studies have investigated how neuroinflammation early in the disease course may affect Alzheimer's disease (AD) progression over time despite evidence that neuroinflammation is associated with AD.
Methods
Research participants with cerebrospinal fluid (CSF) biomarkers from the Alzheimer's Disease Neuroimaging Initiative (ADNI) were included in this study. Cox models were used to investigate whether baseline CSF neuroinflammation was associated with incident mild cognitive impairment (MCI) or AD. Moderating effects of sex and apolipoprotein E (APOE) ε4 were also examined.
Results
Elevated levels of tumor necrosis factor α (TNF‐α), interleukin (IL)‐9, and IL‐12p40 at baseline were associated with higher rates of conversion to MCI/AD. Interactions with sex and APOE ε4 were observed, such that women with elevated TNF‐α and all APOE ε4 carriers with elevated IL‐9 levels had shorter times to conversion. In addition, TNF‐α mediated the relationship between elevated IL‐12p40 and IL‐9.
Discussion
Elevated neuroinflammation markers are associated with incident MCI/AD, and the factors of sex and APOE ε4 status modify the time to conversion.
Keywords: ADNI, Alzheimer's disease, conversion, CSF inflammation, sex differences
1. BACKGROUND
Alzheimer's disease (AD) is increasingly recognized as a complex disease with heterogenous pathologies that affects populations and individuals differently. Numerous studies have demonstrated the importance of cerebrospinal fluid (CSF) biomarkers amyloid beta (Aβ) 42, phosphorylated‐tau (p‐tau), and total‐tau (t‐tau) as predictors of conversion to mild cognitive impairment (MCI) or AD. However, there is less certainty about the role of neuroinflammation in conversion, especially with regard to the interactions between neuroinflammation and sex or apolipoprotein E (APOE) ε4 status, which is a major predictor of AD.
A growing body of evidence indicates that neuroinflammatory responses exacerbate AD progression 1 , 2 , 3 and that inflammatory markers co‐localize with AD pathology postmortem in AD clinical cohorts. 4 , 5 , 6 , 7 Nuclear factor kappa B (NF‐κB) is a protein complex that regulates a large array of genes involved in different immune processes and inflammatory responses. Postmortem studies of the AD brain indicate overexpression and/or activation of NF‐κB, particularly in regions with high levels of Aβ plaques and neurofibrillary tangles (NFTs). 8 NF‐κB is a key transcriptional regulator of inflammation and, more recently, has also been found to have roles in learning and memory via neuronal signaling. 9 , 10 NF‐κB has binding sites in the promoter region of the genes involved in amyloidogenesis and inflammation 8 and, as such, inflammatory markers associated with the NF‐κB pathway are of particular interest and relevance to AD.
The cytokine tumor necrosis factor α (TNF‐α), implicated in many AD studies, is among the most well‐studied and well‐known activators of NF‐κB in both neuronal and non‐neuronal cells 11 , 12 , 13 and plays an important role in regulating long‐term potentiation (LTP). 9 In addition to TNF‐α, interleukin 9 (IL‐9) is also a part of the NF‐κB inflammatory pathway and has been associated with AD risk. IL‐9 is an understudied pro‐inflammatory cytokine known for promoting cell proliferation and prevention of apoptosis and whose expression is regulated by NF‐κB. 13 Collectively, this pathway may play a central role in inflammation associated with AD risk. Furthermore, we hypothesize that there could be factors that influence the role of inflammation on AD. Sex and APOE ε4 status present differential risks to both AD and inflammation. 14 , 15 , 16 Ultimately, they may sit at the interface between neurodegeneration, inflammation, and the spread of pathologies through the brain, which is why their consideration can help with the prediction of disease onset.
In this longitudinal observational study we examined whether the baseline level of each CSF neuroinflammatory marker was associated with incident MCI or AD, and whether these markers interact to participate in the same inflammatory pathway. We hypothesized that neuroinflammatory markers (TNF‐α, IL‐9), along with the classic CSF biomarkers of AD (Aβ42, t‐tau, and p‐tau), would predict clinical conversion to MCI/AD. Finally, accompanying the current literature on sex and APOE ε4 differences in AD, 17 we investigated the role of these two risk factors on neuroinflammation and conversion in an exploratory analysis.
2. METHODS
2.1. Subjects
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 as a public‐private partnership, led by Principal Investigator Michael W. Weiner, MD, with the primary goal of testing whether magnetic resonance imaging (MRI), positron emission tomography (PET), biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. ADNI is a convenience sample comprising predominantly highly educated, non‐Hispanic whites.
Participants were selected by the availability of CSF data at baseline, and the availability of at least one additional time point for diagnosis as of January 2021. Additional participant demographics included APOE status, age, sex, and diagnosis at time of CSF collection. The combined sample excluded participants with AD diagnosis at baseline visit. Participants who converted to MCI or AD during follow‐up were categorized as converters. Participants without diagnostic change throughout the observation period were categorized as non‐converters. The frequency of follow‐up visits consisted of a baseline visit and 3‐month, 6‐month, and annual visits. Information regarding those at risk and those who converted during a certain time frame can be found in Table S1. Local institutional review boards approved the ADNI study across the United States and Canada, and each subject provided written informed consent.
RESEARCH IN CONTEXT
Systematic Review: Authors reviewed the literature using traditional sources (e.g., PubMed, Google Scholar) as well as meeting abstracts and presentations. Cerebrospinal fluid (CSF) and blood‐based inflammatory markers of the nuclear factor kappa B (NF‐κB) pathway are elevated in older adults and those with early mild cognitive impairment (MCI). However, whether elevated inflammation in older adults predicts clinical conversion is ill‐understood.
Interpretation: Higher levels of inflammatory markers tumor necrosis factor α (TNF‐α) and interleukin (IL)‐9 are associated with increased risk of conversion to MCI and AD in older adults. This relationship is exacerbated in apolipoprotein E (APOE) ε4 carriers.
Future Directions: These inflammatory biomarkers could point to new biological pathways that may play an early role in dementia progression.
2.2. APOE ε4 carrier status
The details of APOE genotyping have been published previously. 18 Participants with at least one copy of the ε4 allele were considered APOE ε4 carriers, which consisted of primarily heterozygous APOE ε3/ε4 genotypes (78% APOE ε3/ε4; 19% APOE ε4/ε4; and 0.03% APOE ε2/ε4).
2.3. CSF markers
According to ADNI protocol, blinded CSF samples were shipped from Biomarker Core to Emory University to be examined by William Hu and J. Christina Howell, Department of Neurology. All markers were measured in duplicate using commercially available multiplex immunoassays (Millipore Sigma, Burlington, MA, USA). 19 Values are given in pg/mL. CSF neuroinflammatory protein levels were normalized across plates using the six CSF standard values. ADNI CSF samples were first randomized across twelve 96‐well plates, and each batch was analyzed for levels of all 15 proteins during the same 2‐day block to avoid freeze‐thawing. Primary analyses examined CSF markers of interest TNF‐α and IL‐9 to test the hypothesis that higher levels of neuroinflammatory markers associated with transcription factor NF‐κB would be associated with conversion. Exploratory analyses examined the CSF biomarker IL‐12p40 as well as IL‐6, tumer necrosis factor receptor (TNFR)‐1, TNFR‐2, intercellular adhesion molecule (ICAM)‐1, and vascular cell adhesion molecule (VCAM)‐1 (null results are reported in Table S2). Detailed information can be found in Hu et al. 19
CSF AD biomarkers Aβ42 and t‐tau and p‐tau proteins were analyzed with the multiplex xMAP Luminex platform and Innogenetics/Fujirebio AlzBio3 immunoassay reagents, details of which have been described previously. 20
2.4. Statistical analyses
Analyses were conducted using SPSS Statistics (v27), R (v4.1), and PASS (v14) software. Baseline differences in CSF neuroinflammatory marker levels between converters and non‐converters were assessed by Wilcoxon rank‐sum test. An association was considered statistically significant if the two‐sided P‐value corresponding to the test statistic was less than 0.05. Cox proportional hazards models were used to investigate whether the baseline levels for each CSF neuroinflammatory marker and pathology markers (Aβ42, p‐tau, and t‐tau) were associated with hazard rate of conversion, illustrated via forest plots as well as survival curve graphs. Survival in this context refers to the lack of disease progression during the follow‐up period and is therefore defined as not converting to AD or MCI. Lower CSF Aβ42 values is an indicator of a greater presence of Aβ in the brain, which is contrary to the other CSF markers in which higher levels indicate greater AD risk. Therefore, for Aβ42 values only, the inverse hazard ratio (HR) is reported (1/HR) to align lower Aβ42 values with increased risk of AD, placing all CSF markers on the same scale of HR > 1.0. CSF neuroinflammatory markers were scaled to have a median of 0 and interquartile range (IQR) of 1 for interpretation purposes; effect estimates, expressed as HRs are therefore interpreted per IQR. Baseline age, sex, and APOE ε4 status were included in the analyses as covariates. Schoenfeld residuals were visually inspected to test for violation of the proportional hazards’ assumption. Plots of Martingale residuals against continuous covariates were used to detect non‐linearity. Participants with outlier marker values (determined from the residual plots) were excluded from the analyses. The potential moderating effects of sex and APOE ε4 were tested by assessing CSF marker*sex and CSF marker*APOE ε4 interactions using females and APOE ε4 non‐carriers as the corresponding reference groups. Further stratification and repeated analyses were conducted if the P‐value corresponding to an interaction term was below 0.15. 21
The assumption for validity of the proportional hazards model was tested before carrying out power analyses. Standard deviations of the variables of interest were calculated for power analyses. The sample size was adjusted based on the variability in the continuous variables of interest explained by the additional covariates in the Cox regression. The sample size was adjusted for the anticipated event rate. We assumed 80% power and a two‐sided alpha of 0.05 and calculated the minimum detectable regression coefficient (in ln(HR)) for the analyses at ± 0.2410. This corresponds to an HR of 1.27 (0.79 for −0.2410).
A mediation analysis was carried out using the PROCESS macro 22 version 3.5.2 for SPSS to test the significance of associations between CSF neuroinflammatory markers in the NF‐κB pathway. TNF‐α was treated as a potential mediator in IL‐12p40 and IL‐9's relationship (x = IL‐12p40, y = IL‐9) based on a priori knowledge that IL‐12p40 stimulates production of TNF‐α 23 , 24 using age and sex as covariates and APOE ε4.
3. RESULTS
3.1. Demographic characteristics of the cohort
A total of 292 participants (152 converters, 140 non‐converters) were included in this study excluding 3 participants due to outlying CSF values. Of the total sample, 18 were non‐White (5 participants of Asian descent and 13 African American participants). A total of 118 participants (40%) were female, 123 (42%) were APOE ε4 carriers, and 108 (37%) were cognitively unimpaired. The CSF markers Aβ42, t‐tau, and p‐tau differed between groups, with increased levels of pathology in converters. Between converters and non‐converters, there were no significant differences in age or CSF levels of TNF‐α, IL‐9 or IL‐12‐p40 at baseline (Table 1). Variables of interest that yielded significant results, namely, TNF‐α, IL‐9, or IL‐12‐p40, are summarized in Table 1. Exploratory variables that yielded a null result are reported in Table S2.
TABLE 1.
Baseline demographic and biomarker values of research participants
| Baseline variable | All (n = 292) | Converter (n = 152) | Non‐converter (n = 140) | Effect size | P‐value |
|---|---|---|---|---|---|
| Sex (M/F) | 174/118 | 97/55 | 77/63 | 0.08 | .16 |
| APOE ε4 (carrier/non‐carrier) | 123/169 | 76/76 | 47/93 | 0.16 | <.01 |
| Race (% non‐Hispanic, White) | 91.78% | 92.11% | 91.43% | 0.04 | .39 |
| Age, years | 74.85 (6.78) | 74.50 (7.07) | 75.23 (6.46) | 0.1 | .05 |
| TNF‐α, pg/mL | 1.74 (0.52) | 1.80 (0.53) | 1.68 (0.51) | 0.23 | .19 |
| IL‐9, pg/mL | 3.44 (1.55) | 3.58 (1.69) | 3.30 (1.38) | 0.18 | .07 |
| IL‐12p40, pg/mL | 1.31 (1.76) | 1.52 (2.21) | 1.08 (1.06) | 0.25 | <.01 |
| Aβ‐42, pg/mL | 1018.82 (581.56) | 870.28 (545.27) | 1180.09 (578.58) | 0.55 | <.01 |
| p‐tau, pg/mL | 27.70 (13.10) | 31.42 (13.72) | 23.76 (11.11) | 0.61 | <.01 |
| t‐tau, pg/mL | 287.48 (114.95) | 318.49 (119.16) | 252.82 (99.58) | 0.6 | <.01 |
Note: Effect size and P‐value correspond to group differences between participants who go on to convert (converters) and those that remain at the same impairment stage (non‐converters). Medians and interquartile ranges were reported for continuous variables and counts, and corresponding percentages were reported for categorical variables. Continuous variable medians were compared using Wilcoxon rank‐sum test and effect size was calculated according to Cohen's D. Categorical variable counts were compared using χ 2 test and the respective size was calculated according to Cramer's V. CSF neuroinflammatory marker sample count for TNF‐α, IL‐9, and IL‐12 was 292, 212, and 212, respectively. CSF pathology markers sample count for Aβ42, p‐tau and t‐tau was 288. Note: the P‐value for age not TNF‐α has been rounded down to 0.05. There were no significant differences in median TNF‐α levels between converters and non‐converters.
The median follow‐up period was 2.13 years (IQR = 3.21) after the baseline visit for converters and 3.22 years (IQR = 4.44) for non‐converters. For converters, median time to conversion to AD was 1.99 years (IQR = 2.97 years), whereas conversion to MCI was 5.32 years (IQR = 3.67 years). Fourteen CN participants (4.79%) converted to AD, 22 CN participants (7.53%) converted to MCI, and 114 MCI participants (39.04%) converted to AD.
3.2. CSF biomarkers at baseline predict conversion to MCI/AD
3.2.1. Amyloid beta‐42/phosphorylated‐tau/total‐tau
As expected, baseline Aβ42, p‐tau, and t‐tau were significantly associated with increased rate of conversion to MCI/AD after adjusting for baseline age, sex, and APOE ε4 carrier status. For further details on APOE ε4 and sex interactions see Tables S3–S5.
3.3. Elevated CSF neuroinflammatory markers at baseline predict conversion to MCI/AD
3.3.1. Tumor necrosis factor‐α
Baseline TNF‐α level was significantly associated with the rate of combined conversion to MCI/AD (HR = 1.24 per 1 IQR, 95% confidence interval [CI] = 1.01, 1.52), P = .04, Figure 1). A significant TNF‐α*sex interaction on combined conversion was observed (HR = 0.81 per 1 IQR, 95% CI = 0.62, 1.06, P = .13, Figure 2A) as described in the Methods section (indicating significance if the P‐value corresponding to an interaction term was below .15). This would suggest that the association between TNF‐α and conversion was higher among women (HR = 1.38 per 1 IQR, 95% CI = 1.05, 1.83, P = .02), but not among men (HR = 1.06 per 1 IQR, 95% CI = 0.99, 1.127, P = .06) in sex‐stratified models.
FIGURE 1.
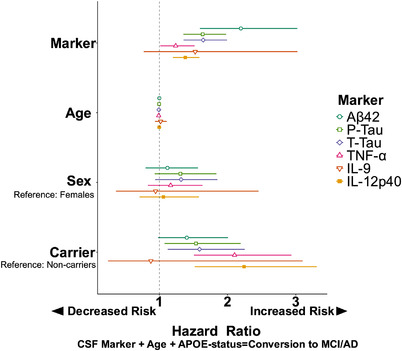
Forest plot highlighting predictive power of CSF neuroinflammatory markers as predictors of conversion. Each model tested is represented by a line on the plot where color and shape differentiate our variables of interest. Line of no effect (HR = 1.0) is represented by dotted line. Variable hazard ratios (HRs; per IQR) visualize how the ratio magnitude of neuroinflammatory markers TNF‐α, IL‐9, and IL‐12p40 compared to the magnitudes of the ratios of AD biomarkers. A relationship was significant if the corresponding 95% confidence interval did not cross HR = 1.0 vertical line
FIGURE 2.
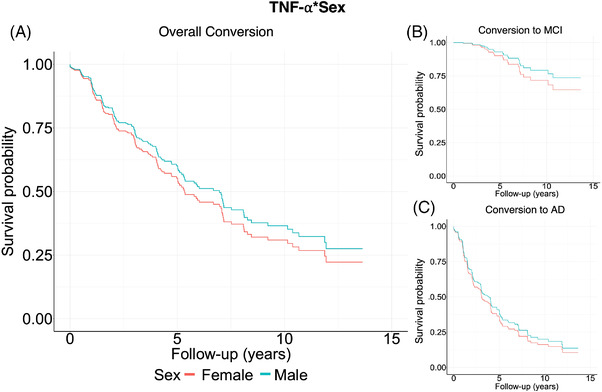
Survival curves for CSF neuroinflammatory marker, TNF‐α. Panel A shows survival probabilities split between sex where survival = no conversion to MCI/AD (this is also referred as event‐free survival [EFS] in literature). Red represents females and blue, males, to better show the interaction effect between sexes of overall conversion to either MCI or AD. Females with high levels of TNF‐α tended to convert at higher rates. Results were split by conversion to either MCI (panel B) or AD (panel C) with conversion to MCI not significant
There was a significant main effect of TNF‐α on the rate of conversion to AD (HR = 1.33 per 1 IQR, 95% CI = 1.04, 1.71, P = .02). A TNF‐α*sex interaction on conversion to AD (HR = 0.69 per 1 IQR, 95% CI = 0.42, 0.86, P = .15, Figure 2C) indicated that the association between TNF‐α and conversion was higher among women (HR = 1.66 per 1 IQR, 95% CI: = 1.1, 2.48) P = .01) than men (HR = 1.08 per 1 IQR, 95% CI= 0.78, 1.5, P = .63).
TNF‐α was not associated significantly with rate of conversion to MCI (HR = 1.36 per 1 IQR, 95% CI = 0.87, 2.11, P = .18, Figure 2B).
There were no significant interactions with APOE ε4 carrier status in the models.
3.3.2. Interleukin‐9
Baseline IL‐9 levels were not significantly associated with the rate of combined conversion to MCI/AD (HR = 1.07 per 1 IQR, 95% CI = 0.84, 1.37) P = .56, Figures 1, 3A). No significant interactions with sex or APOE ε4 and IL‐9 on combined conversion to MCI/AD and on conversion to AD were observed.
FIGURE 3.
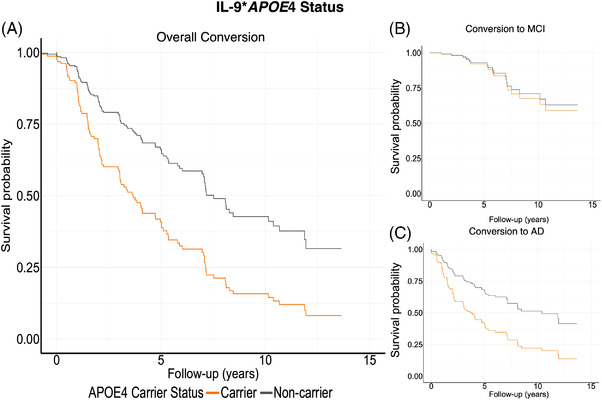
Survival curves for CSF neuroinflammatory marker, IL‐9. Panel A displays overall conversion to either MCI or AD, split between APOE ε4 carriers and noncarriers. Orange represents APOE ε4 carriers, and grey, non‐carriers, illustrate that APOE ε4 carriers convert at higher rate. When split between conversion status, these results were driven by conversion to MCI (panel B). Conversion to AD was not significant (panel C)
There was no significant main effect of IL‐9 on the rate of conversion to AD (HR = 1.07 per 1 IQR, 95% CI = 0.81, 1.39, P = .65) (Figure 3C).
Baseline IL‐9 levels were significantly associated with a higher rate of conversion to MCI (HR = 1.53 per 1 IQR, 95% CI = 3.97, 5935.36, P = .01) (Figures 1, 3B). There was a significant IL‐9* APOE ε4 interaction (HR = 0.001 per 1 IQR, 95% CI = < 0.001, 0.17), P = .01) on CN to MCI conversion, whereby IL‐9 significantly related to conversion in APOE ε4 carriers (HR = 1.39 per 1 IQR, 95% CI = 0.37, 52713, P = .10), but not in non‐carriers (HR = 1.04 per 1 IQR, 95% CI = 0.5, 2.23, P = .91) in APOE ε4‐stratified models.
There was no significant interaction with IL‐9*sex on conversion to MCI.
3.3.3. Interleukin‐12p40
Baseline levels of IL‐12p40 were significantly associated with a higher rate of combined conversion to MCI/AD (HR = 1.38 per 1 IQR, 95% CI = 1.20, 1.59, P < .01, Figures 1, 4A).
FIGURE 4.
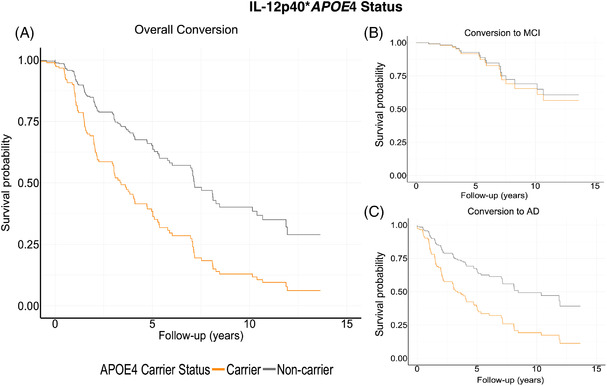
Survival curves for CSF neuroinflammatory marker, IL‐12p40. Panel A displays conversion to either MCI or AD, split between APOE ε4 carriers and noncarriers. Orange represents APOE ε4 carriers, and gray represents non‐carriers. When split between conversion status, these results were driven by conversion to AD (panel C). Conversion to MCI was not significant (panel B)
IL‐12p40 was significantly associated with conversion to AD (HR = 1.36 per 1 IQR, 95% CI = 1.18, 1.57, P < .01, Figure 4C).
IL‐12p40 was not significantly associated with conversion to MCI (HR = 1.42 per 1 IQR, 95% CI = 0.72, 2.815, P = .31, Figure 4B).
No sex or APOE ε4 interactions with IL‐12p40 on any conversion were observed.
3.4. TNF‐α mediates the association between IL‐12p40 and IL‐9
Based on a priori knowledge of the NF‐κB relationship between the three inflammatory markers (IL‐12p40, IL‐9, and TNF‐α), a bootstrapped mediation analysis was conducted to investigate whether TNF‐α (M) was a significant mediator of IL‐12p40 (X) and IL‐9 (Y). With adjustments for age, APOE ε4, and sex, IL‐12p40 was significantly associated with TNF‐α (β = 0.07 per 1 IQR, 95% CI = 0.04, 0.11, P < .001) and with IL‐9 (β = .12 per 1 IQR, 95% CI = 0.00, 0.24, P = .04). TNF‐α was a significant mediator in IL‐12p40 – IL‐9 relationship. The indirect effect (average mediated effect) was 0.11 (95% CI = 0.05, 0.18, P < .001). Average direct effect was 0.01 (95% CI = −0.10, 0.12, P = .81). Total effect was 0.13 (95% CI = 0.01, 0.24, P = .04). TNF‐α mediated 88.64% of the relationship (95% CI = 0.33, 5.22, P = .04, Figure 5).
FIGURE 5.
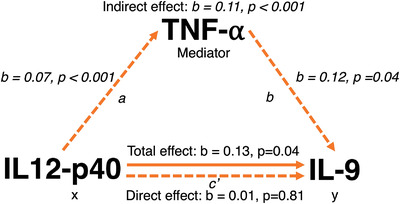
TNF‐α mediates relationship between IL12‐p40 and IL‐9. The figure depicts the relationship between the independent, mediator, and dependent variables. The mediation analysis partitions the total effect of the predictor into a part that is independent of the mediating variable (direct effect) and a part that is accounted for via the mediating variable (indirect effect). a represents the “a” path and b represents the “b” path. A bootstrapped mediation analysis revealed that TNF‐α significantly mediated the relationship between neuroinflammatory cytokines IL12‐p40 (independent) and IL‐9 (dependent) variables
4. DISCUSSION
Chronic low‐grade inflammation is a hallmark feature of AD, as evidenced by the increased expression of proinflammatory cytokines in postmortem AD brains. 25 Yet outside of examining known CSF biomarkers, few studies have examined the contribution of elevated neuroinflammatory levels on future incidence of clinical conversion. In the present study, the CSF biomarkers Aβ42, p‐tau, and t‐tau predicted conversion to MCI or AD, especially in APOE ε4 carriers, which has been shown previously. 26 , 27 Unique to this study, neuroinflammatory CSF markers TNF‐α, IL‐9, and IL‐12p40 were associated with conversion to MCI/AD. These factors interacted with sex and APOE ε4, independently, such that APOE ε4 carriers and females with higher CSF analyte levels were at the highest risk of progression to MCI/AD.
Although neural degeneration and the chronic presence of pathology can stimulate inflammation, evidence suggests that inflammation itself can both generate and increase pathology. Modulatory proteins associated with innate immunity are connected directly to Aβ production, 28 indicating the importance of neuroimmune actions on AD. Therefore, it is possible that inflammatory markers may be important indicators of MCI and AD risk, possibly accelerating decline. Several interactions involving sex and APOE ε4 status among these neuroinflammatory markers were observed, suggesting that these genetic factors may influence the relationship between early inflammation and future AD risk, providing key insight for precision‐medicine approaches. The association between these markers suggests a common tie to the NF‐kB inflammatory pathway, which may be activated in AD.
CSF TNF‐α, one of the inflammatory markers most consistently implicated in AD, was associated with conversion in individuals who were cognitively normal at baseline. Previous work has shown increased levels of TNF‐α in both the brains and plasma of patients with clinical AD. 29 This may be partially related to Aβ, which can directly stimulate microglial production of TNF‐α through activation of the transcription factor NF‐κB. 30 In addition, early increases in TNF‐α can increase Aβ burden through the upregulation of β‐secretase production and increase γ‐secretase activity, 31 demonstrating how TNF‐α and Aβ can perpetuate a vicious cycle of increasing pathology and chronic inflammation. In corroboration for TNF‐α's role in AD, a strong association between elevated TNF‐α levels and decreased functional connectivity in an AD susceptible cohort has been observed at a whole‐brain level. 32 Aside from TNF‐α as a consistently elevated proinflammatory marker in AD, its regulation of the NF‐κB inflammatory cascade is well characterized. 33 , 34
When decomposing the association between TNF‐α levels in cognitively normal individuals and clinical converters, the effect was driven by conversion to AD, but not conversion to MCI, indicating that elevated levels of TNF‐α resulted in accelerated onset of Alzheimer's dementia. Given that TNF‐α both promotes and is regulated by NF‐κB, it could be that TNF‐α accumulates at a faster rate than other inflammatory factors. Previous research has shown that in AD patients who experienced systemic inflammatory events (SIEs), rapid cognitive decline was observed over 6 months, creating a 2‐fold decrease in cognitive function. In addition, SIE AD patients also exhibited an association between elevated plasma TNF‐α levels and a 10‐fold greater rate of cognitive decline over a 6‐month observation period. 35
A sex*TNF‐α interaction was observed in which women with higher TNF‐α levels had a shorter time to conversion to MCI or AD, confirming recent evidence of sex differences in immune parameters. This includes brain responses to inflammatory mediators and markers of microglial disruption, which may contribute to a higher risk of AD in women. 36 For instance, previous research has shown that given an acute endotoxin challenge, women responded with higher levels of pro‐inflammatory plasma cytokines like TNF‐α, compared to men. 37 This may be due to a putative link between levels of TNF‐α and sex hormones. A significant positive correlation between luteinizing hormone and TNF‐α levels has been observed (albeit only in males) providing some evidence that sex hormones exert their influence on AD by modulating systemic TNF‐α levels. 38 There are also sex differences among function, morphology, and volume of microglia, with evidence suggesting that women later in life have higher numbers of microglia than men, creating an exaggerated immune response.39 Of interest, our finding that women with higher TNF‐α levels have a shorter time to conversion to MCI/AD suggests that elevated neuroinflammation is associated with AD risk. It has also been suggested that elevated inflammatory factors, including TNF‐α, are associated with lower brain levels of Aβ and tau in cognitively normal individuals, 40 meaning that neuroinflammation may be protective for a time before eventually exhausting this compensatory effect and converting to AD at an accelerated rate. Studies have started showing prevention of cognitive loss with anti‐TNF‐α therapy in a mouse model of compromised cognition, 41 lending additional support to an important role of TNF‐α in the initiation and amplification of the inflammatory cascade.
Baseline elevated IL‐9 levels were associated with conversion, but in contrast to TNF‐α, the results were driven by conversion to MCI specifically and not AD. In addition, no interaction with sex was observed. IL‐9 is an understudied pro‐inflammatory cytokine that promotes cell proliferation and whose expression is driven by NF‐κB. 42 Its pleiotropic effects have been implicated in both neurological (i.e., multiple sclerosis 43 ) and systemic disorders (i.e., psoriasis 44 ). Furthermore, neurons are thought to possess the ability to induce T cells to release IL‐9. 45 Thus, IL‐9 may represent a critical link in the crosstalk between neurons and perivascular T cells in the brain. Changes related to IL‐9 have not been reported consistently in AD, 46 , 47 but an APOE ε4 knock‐in mouse model has shown greater IL‐9 production than the wild‐type ε3 allele. 45 We add to this evidence by reporting an interaction between APOE ε4 and IL‐9 levels, such that APOE ε4 carriers with higher IL‐9 levels at baseline had higher rates of conversion.
Higher baseline levels of the proinflammatory cytokine IL‐12p40 were also significantly associated with conversion to AD. IL‐12, which has different subunits, including p40, is involved in the differentiation of naive T cells into Th1 cells 48 and plays an important role in the activities of natural killer (NK) cells and T lymphocytes, enhancing their activity. IL‐12 also stimulates production of TNF‐α from T and NK cells, 49 and recent evidence indicates a role for IL‐12 in amyloid‐induced neurodegeneration. 50 The p40 subunit of IL‐12 is produced by microglia and was found to be increased in transgenic mouse models, demonstrating a significant correlation between cognitive performance and CSF IL‐12p40 levels. 50 More support of its role in AD was shown by elevated CSF IL‐12p40 levels in patients with AD. 51 Of interest, the potential association of IL‐12p40 levels with the diagnosis of MCI or AD was also highlighted by a recent plasma multianalyte profiling study. 52 Together, these findings imply that IL‐12p40 is involved in AD progression.
To investigate the interrelationships between cytokines showing significant associations with conversion to MCI/AD, a mediation analyses using a priori knowledge of the markers and their role in the NF‐κB pathway was conducted. As mentioned previously, IL‐12p40 has been shown to stimulate the production of TNF‐α, 24 a canonical promotor of the NF‐κB inflammatory pathway (34), and evidence shows that IL‐9 production is mediated by NF‐κB. 23 TNF‐α was tested as a significant mediator between IL12‐p40 and IL‐9, and our mediation analysis results found that TNF‐α mediated 89% of the association between IL12‐p40 and IL‐9. We interpret this effect to mean that elevated IL12‐p40 leads to increasing IL‐9 levels via a mediating effect of TNF‐α. Based on the available literature, this study is the first showing that these cytokines may be involved in the same NF‐κB inflammatory pathway, thereby further implicating the inflammatory cascade's importance in AD detection.
Current limitations of this study include an inability to test specific demographic variables, including race, which we know play a significant role in inflammation and AD and yet have not been prioritized in ADNI recruitment. The investigation of longitudinal cognitive data from this sample and other data sets could help determine if differences exist between converters and non‐converters, and shed light on specific cognitive domains that may be more closely tied to neuroinflammation. We also note that because this was interval censored data, we are not able to know the actual time of conversion and are restricted to examining data at discrete time points. Competing risks were not discussed (i.e., death occurring prior to follow‐up). In addition, because we had only baseline CSF neuroinflammatory markers, we were restricted to a cross‐sectional mediation analysis that limits our confidence in the temporality of events. However, as more data become available, longitudinal analyses of change in neuroinflammatory markers can be evaluated. Finally, a liberal significance cutoff of 0.15 was used to explore interactions as referenced in Buckley et al., 21 so the results should be considered carefully given that the P‐value cut point was different for interaction analyses. Because of the nature of ADNI cohort, we simplified our analyses so as to not have to correct for mulitple comparisons. Future studies aimed at replication of these findings in other independent cohorts, including those that are more nationally representative of the AD population, would help validate these results.
This work demonstrates the need to investigate neuroinflammatory markers and their role in conversion to AD. Investigations focused on neuroinflammation differences between key groupings, based on clinical diagnosis, sex, and APOE ε4 status may provide insight into key underlying processes relevant to future clinical conversion. As a testament to the importance of longitudinal analysis, no baseline differences in CSF neuroinflammatory marker levels were observed between CN and MCI groups, or between APOE ε4 carriers and non‐carriers in the current study. The strength of this study comes from the rich longitudinal data set because changes occurring within individual participants may be more useful for characterizing disease progression than cross‐sectional comparisons, which are confounded/rendered less sensitive by large amounts of interindividual variation. These neuroinflammatory biomarkers could prove to be easily accessible targets to gauge the efficacy of therapeutic interventions and be crucial to point to new biological pathways that may play a role in dementia.
CONSENT FOR PUBLICATION
This manuscript meets the guidelines put forth by the Alzheimer's Disease Neuroimaging Initiative Data Sharing and Publication Policy.
CONFLICT OF INTEREST
The authors declare no conflict of interest. Author disclosures are available in the supporting information.
AUTHOR CONTRIBUTIONS
Conceptualization: Daniel S. Albrecht; Implementation and computational methods: Joey A. Contreras and Vahan Aslanyan; Paper writing: Joey A. Contreras; Paper review and supervision: Judy Pa. All authors read and approved the final manuscript.
Supporting information
SUPPORTING INFORMATION
SUPPORTING INFORMATION
ACKNOWLEDGMENTS
Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. J.A., V.A., and J.P. are supported by the National Institute on Aging (R01‐AG054617). Data collection and sharing for this project were funded by the ADNI (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Contreras JA, Aslanyan V, Albrecht DS, Mack WJ, Alzheimer's Disease Neuroimaging Initiative (ADNI), Pa J. Higher baseline levels of CSF inflammation increase risk of incident mild cognitive impairment and Alzheimer's disease dementia. Alzheimer's Dement. 2022;14:e12346. 10.1002/dad2.12346
REFERENCES
- 1. Holtzman DM, Mandelkow E, Selkoe DJ. Alzheimer Disease in 2020. Cold Spring Harb Perspect Med. 2012;2:a011585‐a011585. doi: 10.1101/cshperspect.a011585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol. 2007;7:161‐167. doi: 10.1038/nri2015 [DOI] [PubMed] [Google Scholar]
- 3. Bettcher BM, Kramer JH. Longitudinal inflammation, cognitive decline, and Alzheimer's disease: a mini‐review. Clin Pharmacol Ther. 2014;96:464‐469. doi: 10.1038/clpt.2014.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Janelidze S, Mattsson N, Stomrud E, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91:e867‐e877. doi: 10.1212/WNL.0000000000006082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brosseron F, Traschütz A, Widmann CN, et al. Characterization and clinical use of inflammatory cerebrospinal fluid protein markers in Alzheimer's disease. Alz Res Therapy. 2018;10:25. doi: 10.1186/s13195-018-0353-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Whelan CD, Mattsson N, Nagle MW, et al. Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer's disease. Acta Neuropathol Commun. 2019;7:169. doi: 10.1186/s40478-019-0795-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puzzo D, Privitera L, Leznik E, et al. Picomolar amyloid‐ positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537‐14545. doi: 10.1523/JNEUROSCI.2692-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ju Hwang C, Choi D‐Y, Park MH, Hong JT. NF‐κB as a key mediator of brain inflammation in Alzheimer's disease. CNSNDDT. 2019;18:3‐10. doi: 10.2174/1871527316666170807130011 [DOI] [PubMed] [Google Scholar]
- 9. Jones SV, Kounatidis I. Nuclear factor‐kappa B and Alzheimer disease, unifying genetic and environmental risk factors from cell to humans. Front Immunol. 2017;8:1805. doi: 10.3389/fimmu.2017.01805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Snow WM, Albensi BC. Neuronal gene targets of NF‐κB and their dysregulation in Alzheimer's disease. Front Mol Neurosci. 2016;9:118. doi: 10.3389/fnmol.2016.00118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hayden MS, Ghosh S. Regulation of NF‐κB by TNF family cytokines. Semin Immunol. 2014;26:253‐266. doi: 10.1016/j.smim.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kounatidis I, Ligoxygakis P. Drosophila as a model system to unravel the layers of innate immunity to infection. Open Biol. 2012;2:120075. doi: 10.1098/rsob.120075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Early SB, Huyett P, Brown‐Steinke K, Borish L, Steinke JW. Functional analysis of −351 interleukin‐9 promoter polymorphism reveals an activator controlled by NF‐κB. Genes Immun. 2009;10:341‐349. doi: 10.1038/gene.2009.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hobel Z, Isenberg AL, Raghupathy D, Mack W, Pa J. APOE ɛ4 gene dose and sex effects on Alzheimer's disease MRI biomarkers in older adults with mild cognitive impairment. JAD. 2019;71:647‐658. doi: 10.3233/JAD-180859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16:626‐638. doi: 10.1038/nri.2016.90 [DOI] [PubMed] [Google Scholar]
- 16. Duarte‐Guterman P, Albert AY, Inkster AM, Barha CK, Galea LAM, on behalf of the Alzheimer's Disease Neuroimaging Initiative. Inflammation in Alzheimer's disease: do sex and APOE matter?. JAD 2020;78:627‐641. doi: 10.3233/JAD-200982 [DOI] [PubMed] [Google Scholar]
- 17. Cartier A, Côté M, Lemieux I, et al. Sex differences in inflammatory markers: what is the contribution of visceral adiposity? Am J Clin Nutr. 2009;89:1307‐1314. doi: 10.3945/ajcn.2008.27030 [DOI] [PubMed] [Google Scholar]
- 18. Nation DA, Sweeney MD, Montagne A, et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25:270‐276. doi: 10.1038/s41591-018-0297-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hu WT, Ozturk T, Kollhoff A, et al. Higher CSF sTNFR1‐related proteins associate with better prognosis in very early Alzheimer's disease. Nat Commun. 2021;12:4001. doi: 10.1038/s41467-021-24220-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol. 2009;65:403‐413. doi: 10.1002/ana.21610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Buckley JP, Doherty BT, Keil AP, Engel SM. Statistical approaches for estimating sex‐specific effects in endocrine disruptors research. Environ Health Perspect. 2017;125:067013. doi: 10.1289/EHP334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hayes AF. Introduction to Mediation, Moderation, and Conditional Process Analysis: A Regression‐Based Approach. 3rd ed. The Guilford Press; 2022. [Google Scholar]
- 23. Goswami R, Kaplan MH. A brief history of IL‐9. J Immunol. 2011;186:3283‐3288. doi: 10.4049/jimmunol.1003049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ma X, editor. Regulation of Cytokine Gene Expression in Immunity and Diseases. vol. 941. Dordrecht: Springer Netherlands; 2016. 10.1007/978-94-024-0921-5 [DOI] [Google Scholar]
- 25. Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16:358‐372. doi: 10.1038/nrn3880 [DOI] [PubMed] [Google Scholar]
- 26. Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/2‐Amyloid42 ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:7. [DOI] [PubMed] [Google Scholar]
- 27. Blennow K, Shaw LM, Stomrud E, et al. Predicting clinical decline and conversion to Alzheimer's disease or dementia using novel Elecsys Aβ(1‐42), pTau and tTau CSF immunoassays. Sci Rep. 2019;9:19024. doi: 10.1038/s41598-019-54204-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hur J‐Y, Frost GR, Wu X, et al. The innate immunity protein IFITM3 modulates γ‐secretase in Alzheimer's disease. Nature. 2020;586:735‐740. doi: 10.1038/s41586-020-2681-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chang R, Yee K‐L, Sumbria RK. Tumor necrosis factor α inhibition for Alzheimer's disease. J Cent Nerv Syst Dis. 2017;9:117957351770927. doi: 10.1177/1179573517709278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Combs CK, Karlo JC, Kao S‐C, Landreth GE. β‐Amyloid Stimulation of microglia and monocytes results in TNFα‐Dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J Neurosci. 2001;21:1179‐1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liao Y‐F, Wang B‐J, Cheng H‐T, Kuo L‐H, Wolfe MS. Tumor necrosis factor‐α, interleukin‐1β, and interferon‐γ stimulate γ‐Secretase‐mediated cleavage of amyloid precursor protein through a JNK‐dependent MAPK pathway. J Biol Chem. 2004;279:49523‐49532. doi: 10.1074/jbc.M402034200 [DOI] [PubMed] [Google Scholar]
- 32. Contreras JA, Aslanyan V, Sweeney MD, et al. Functional connectivity among brain regions affected in Alzheimer's disease is associated with CSF TNF‐α in APOE4 carriers. Neurobiol Aging. 2020;86:112‐122. doi: 10.1016/j.neurobiolaging.2019.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383‐421. doi: 10.1016/s0197-4580(00)00124-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fillit H, Ding W, Buee L, et al. Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett. 1991;129:318‐320. doi: 10.1016/0304-3940(91)90490-K [DOI] [PubMed] [Google Scholar]
- 35. Holmes C, Cunningham C, Zotova E, Culliford D, Perry VH. Proinflammatory cytokines, sickness behavior, and Alzheimer disease. Neurology. 2011;77:212‐218. doi: 10.1212/WNL.0b013e318225ae07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Podcasy JL, Epperson CN. Considering sex and gender in Alzheimer disease and other dementias. Dialogues Clin Neurosci. 2016;18:437‐446. 10.31887/DCNS.2016.18.4/cepperson [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Engler H, Benson S, Wegner A, Spreitzer I, Schedlowski M, Elsenbruch S. Men and women differ in inflammatory and neuroendocrine responses to endotoxin but not in the severity of sickness symptoms. Brain Behav Immun. 2016;52:18‐26. doi: 10.1016/j.bbi.2015.08.013 [DOI] [PubMed] [Google Scholar]
- 38. Butchart J, Birch B, Bassily R, Wolfe L, Holmes C. Male sex hormones and systemic inflammation in Alzheimer disease. Alzheimer Dis Assoc Disord. 2013;27:153‐156. doi: 10.1097/WAD.0b013e318258cd63 [DOI] [PubMed] [Google Scholar]
- 39. Schwarz JM, Sholar PW, Bilbo SD. Sex differences in microglial colonization of the developing rat brain: sex differences in microglial colonization. J Neurochem. 2012;120:948‐963. doi: 10.1111/j.1471-4159.2011.07630.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Albrecht DS, Sagare A, Pachicano M, et al. Early neuroinflammation is associated with lower amyloid and tau levels in cognitively normal older adults. Brain Behav Immun. 2021;94:299‐307. doi: 10.1016/j.bbi.2021.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Terrando N, Monaco C, Ma D, Foxwell BMJ, Feldmann M, Maze M. Tumor necrosis factor‐α triggers a cytokine cascade yielding postoperative cognitive decline. Proc Natl Acad Sci USA. 2010;107:20518‐20522. doi: 10.1073/pnas.1014557107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nowak EC, Weaver CT, Turner H, et al. IL‐9 as a mediator of Th17‐driven inflammatory disease. J Exp Med. 2009;206:1653‐1660. doi: 10.1084/jem.20090246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li H, Nourbakhsh B, Ciric B, Zhang G‐X, Rostami A. Neutralization of IL‐9 ameliorates experimental autoimmune encephalomyelitis by decreasing the effector T cell population. J Immunol. 2010;185:4095‐4100. doi: 10.4049/jimmunol.1000986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schlapbach C, Gehad A, Yang C, et al. Human T H 9 cells are skin‐tropic and have autocrine and paracrine proinflammatory capacity. Sci Transl Med. 2014;6:219ra8. doi: 10.1126/scitranslmed.3007828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu Y, Teige I, Birnir B, Issazadeh‐Navikas S. Neuron‐mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med. 2006;12:518‐525. doi: 10.1038/nm1402 [DOI] [PubMed] [Google Scholar]
- 46. Saresella M, Calabrese E, Marventano I, et al. Increased activity of Th‐17 and Th‐9 lymphocytes and a skewing of the post‐thymic differentiation pathway are seen in Alzheimer's disease. Brain Behav Immun. 2011;25:539‐547. doi: 10.1016/j.bbi.2010.12.004 [DOI] [PubMed] [Google Scholar]
- 47. Stertz L, Contreras‐Shannon V, Monroy‐Jaramillo N, Sun J, Walss‐Bass C. BACE1‐Deficient mice exhibit alterations in immune system pathways. Mol Neurobiol. 2018;55:709‐717. doi: 10.1007/s12035-016-0341-1 [DOI] [PubMed] [Google Scholar]
- 48. Hsieh C‐S, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of T H 1 CD4 + T cells through IL‐12 produced by Listeria ‐Induced macrophages. Science. 1993;260:547‐549. doi: 10.1126/science.8097338 [DOI] [PubMed] [Google Scholar]
- 49. Tripp CS, WOLFt SF, Unanue ER. Interleukin 12 and tumor necrosis factor a are costimulators of interferon y production by natural killer cells in severe combined immunodeficiency mice with listeriosis, and interleukin 10 is a physiologic antagonist. Proc Natl Acad Sci USA. 1993;90:3725‐3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tan M‐S, Yu J‐T, Jiang T, Zhu X‐C, Guan H‐S, Tan L. IL12/23 p40 inhibition ameliorates Alzheimer's disease‐associated neuropathology and spatial memory in SAMP8 Mice. JAD. 2013;38:633‐646. doi: 10.3233/JAD-131148 [DOI] [PubMed] [Google Scholar]
- 51.vom Berg J, Prokop S, Miller KR, et al. Inhibition of IL‐12/IL‐23 signaling reduces Alzheimer's disease – like pathology and cognitive decline. Nat Med. 2012;18:1812‐1819. doi: 10.1038/nm.2965 [DOI] [PubMed] [Google Scholar]
- 52. Hu WT, Holtzman DM, Fagan AM, et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 2012;9:897‐905. doi: 10.1212/WNL.0b013e318266fa70 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
SUPPORTING INFORMATION