

Abstract
There have been dramatic changes worldwide in the attitudes and consumption of recreational and medical cannabis. Cannabinoid receptors, which mediate the actions of cannabis, are abundantly expressed in brain regions known to mediate neural processes underlying reward, cognition, emotional regulation and stress responsivity relevant to addiction vulnerability. Despite debates regarding potential pathological consequences of cannabis use, cannabis use disorder is a clinical diagnosis with high prevalence in the population and that often has its genesis in adolescence and in vulnerable populations associated with psychiatric comorbidity, genetic and environmental factors. Integrated information from human and animal studies are beginning to expand insights regarding neurobiological systems associated with cannabis use disorder which often share common neural characteristics as other substance use disorders that could inform prevention and treatment strategies.
Editorial summary
The increasing use of cannabis has brought significant attention to cannabis use disorder (CUD) and its neurobiological underpinnings. Here Ferland and Hurd discuss risk factors related to the development of CUD its neurobiological characteristics.
Marijuana (Cannabis) is one of the most popular recreational drugs worldwide, with between 128 and 238 million people reporting use1. Over the past 30 years there has been a dramatic shift in attitudes toward the use of cannabis, steered in large part by rapidly changing sociopolitical perceptions and laws regarding this divisive drug. Over 30 states in the USA and 22 countries have legalized some form of marijuana, either for medical use and/or recreational consumption, and these numbers are growing. The dramatic pendulum shift in the broad societal use of cannabis has now prompted vigorous research efforts focused on cannabis to obtain more in-depth biological insights about its potential risk and/or health benefits. Significant knowledge exists regarding the pharmacological actions of prominent cannabinoids in the cannabis plant, but scientific attention specifically focused on neurobiological underpinnings of cannabis addiction has been somewhat limited. Indeed, there remains contention as to whether it is even possible to develop an addiction to cannabis, though based on clinical diagnostic criteria, it is estimated that past-year diagnosis of cannabis use disorder (CUD) amongst cannabis users is approximately 30%2. This number is similar to drugs such as heroin (25%)3 and cocaine (36.5%)4. However, the relatively high percentage of CUD diagnosis relates to the greater prevalence of cannabis use in the general population2 and not to enhanced addiction liability, which is low for cannabis5. Additionally, cannabis use does not lead to fatal overdoses as those other drugs can, but as with most substance use disorders (SUDs), CUD carries a significant psychiatric burden6.
CUD develops as a consequence of chronic neuroadaptations that occur over time to the repeated use of cannabis. The primary psychoactive component of cannabis contributing to its euphorigenic effects is delta-9-tetrahydrocannabinol (Δ9-THC), which is one of ~140 cannabinoids identified in the cannabis plant, along with over 440 additional compounds including terpenoids that make up the complex effects of the plant7. THC concentrations in common recreational plants have increased 700–2,000% over recent decades8, with current strains containing up to 29% THC9. As with all drugs of abuse, as the concentration of the psychoactive component increases, so does the risk for developing a SUD, thus prompting current concerns of increased CUD risk. The consumption of cannabis directly targets the body’s natural endogenous endocannabinoid (eCB) system, which comprises the receptors that mediate the direct actions of cannabinoids, as well as the eCB lipid ligands (arachidonoyl ethanolamide, also called anandamide; and 2-arachidonoylglycerol, also called 2-AG) and the associated enzymes responsible for their synthesis and degradation10. The stimulation of cannabinoid receptors by THC initiates a cascade of broad biological events due to their abundant expression throughout the brain and body10,11. The Gi/Go-protein-coupled cannabinoid receptor subtype 1 (CB1R) is the predominant form in the brain that mediates the psychoactive effects of cannabis10,11, and it is expressed in multiple brain areas including in the cerebral cortex, basal ganglia, hippocampus, cerebellum, amygdala, brainstem and hypothalamus12,13. CB2 receptors, though very low in abundance in the brain, are implicated in neurobiological actions of THC relevant to addiction14, but are predominantly in immune cells in the periphery. Activation of cannabinoid receptors impacts multiple physiological functions and behaviors that extend beyond the acute intoxication effects of cannabis and that can promote the development of CUD via alterations of neural systems regulating cognition, memory, reward, mood and stress sensitivity.
In this review, we provide an overview of CUD, focusing on different aspects of its risk, and explore neurobiological adaptations (neural activity, morphology, neurochemical and molecular events) relevant to CUD on the basis of human studies and animal models. Altogether, the accruing evidence reflects neurobiological signatures mirroring key components of addiction pathology.
Deconstructing the vulnerability to cannabis use disorder
Similarly to other SUDs, a diagnosis of CUD is characterized by problematic use that includes escalation with loss of control over use, repeated failures to reduce use or quit, and continued use despite negative consequences15. Additionally, the Diagnostic and Statistical Manual of Mental Disorders (DSM)-5 includes the diagnostic criteria of “craving” and “withdrawal” among the symptoms15. Both symptoms are common in CUD, with ~60% of CUD individuals experiencing craving for the drug and 32% undergoing withdrawal6. These and other negative features of CUD appear to contribute to high relapse (~67%)16. While the high prevalence of CUD is partially attributed to a greater number of people consuming the drug due to its wide availability2, emerging research has begun to identify factors that may contribute to vulnerability and thus could provide insights about neurobiological processes relevant to CUD sensitivity. The complex and interrelated etiological factors that appear to contribute to risk range from genetics to psychiatric and psychological susceptibility and environmental factors.
Heredity estimates for a diagnosis of cannabis abus and dependence (DSM-III, DSM-IV) range from 21–78%17,18. This wide range may relate to distinct vulnerabilities relevant to different stages of drug use and/or prevalence of the disorder depending on the incidence of other factors (for example, drug availability, as shown in twin studies that cannabis availability explains most of the shared environmental risk of cannabis initiation and abuse19). Attempts to identify biological contributions underlying the genetic etiology of CUD have historically used candidate gene association strategies based on specific a priori neurobiological hypotheses, but current efforts have prioritized hypothesis-free genome-wide association study (GWAS) approaches. Early GWAS studies of cannabis abuse and dependence failed to achieve genome-wide significance, but more recent large GWAS investigations have identified risk loci associated with cannabis pathology. Unfortunately, none of the risk loci overlap across studies. Agrawal and colleagues identified a cluster of correlated single-nucleotide polymorphisms (SNPs) within chromosome 10 (spanning multiple genes) associated with cannabis dependence20. Demontis and colleagues identified one genome-wide significant intergenic risk locus on chromosome 8 (index variant rs56372821) associated with CUD. rs56372821 is an expression quantitative trait locus (eQTL) for the gene encoding cholinergic receptor nicotinic alpha-2 subunit (CHRNA2)21. CHRNA2 is implicated in cigarette smoking-related phenotypes22, and individuals with CUD are highly comorbid for nicotine use (~70–90% report smoking cigarettes). However, rs56372821 does not meet genome-wide significance for nicotine phenotypes23, indicating a potential specific relationship to CUD. Another large meta-analysis GWAS—the International Cannabis Consortium study—focusing on lifetime cannabis use24, not CUD, revealed eight independent SNP associations in multiple chromosome regions. The majority of these were located in or near genes that encode cell adhesion proteins: CADM2 (cell adhesion molecule 2; rs2875907 top SNP hit), NCAM1 (neural cell adhesion molecule; rs9919557) and SDK1 (sidekick cell adhesion molecule; rs10085617). The lack of replication even with GWAS strategies can be due to factors similar to candidate gene approaches, such as heterogeneity of environment (prenatal and/or postnatal), psychiatric comorbidity and behavioral traits, and the cannabis-related phenotype being studied since, for example, lifetime cannabis use and CUD diagnosis represent different aspects of cannabis use pathology including severity of use. Importantly, CUD is a polygenic disorder with multiple contributing genes. Furthermore, comparison control groups often differ between studies and may or may not include individuals who have ever experimented with the drug. Interestingly, excluding control subjects with no cannabis exposure enhanced the strength of genetic association with cannabis dependence observed by Sherva et al.; nevertheless, the top SNP identified (rs143244591; in RP11–206M11.7, an antisense transcript) still did not match other GWAS findings25.
In addition to genetics, multiple factors contribute to addiction vulnerability, such as the high comorbidity with other psychiatric conditions. An extensive literature documents a strong relationship between cannabis use and the development of psychosis and schizophrenia, particularly with increasing THC content26 and in individuals with certain genetic risks27. There is also a spectrum of other pathologies comorbid with CUD6. Aside from other substances of abuse (mainly nicotine and alcohol), the most common psychiatric comorbidities with CUD are mood disorders (primarily bipolar I), post-traumatic stress disorder, personality disorders (in particular borderline and schizotypal) and generalized anxiety disorder (Fig. 1). The strongest associations are in individuals with severe CUD, where there is a six- to ten-fold increase in the chance of such co-occurrence6. It is still unclear whether comorbid disorders, which also have strong genetic correlates, predate or are a consequence of cannabis use. Monitoring individuals longitudinally suggest that frequent cannabis use and its abuse are associated with increased risk of a subsequent mental health disorder28,29. Conversely, having any mood, anxiety or other substance-use pathology at baseline also predicts future cannabis use and dependence28. Behavioral and neurocognitive traits are also an important feature of CUD. As with other drugs of abuse, sensation-seeking and reward sensitivity have been implicated with the initiation of use and severity of the CUD30. The recent International International Cannabis Consortium GWAS also revealed a genetic correlation between lifetime cannabis use and traits such as risk-taking behavior24, highlighting the contribution of genetic variants to phenotypes associated with drug use.
Fig. 1 |.
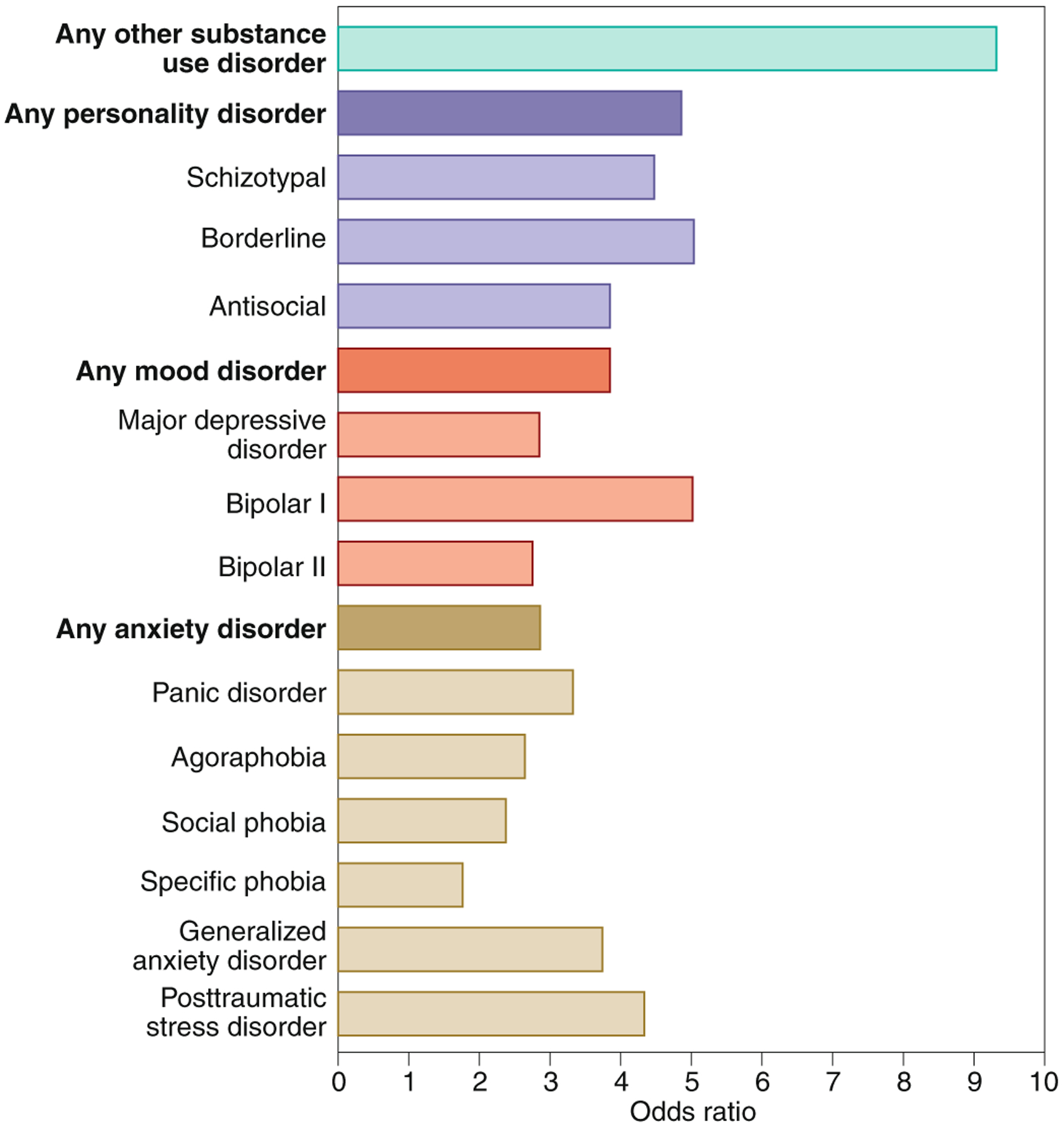
Odds ratios of psychiatric conditions associated with CUD. Data based on the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC) study6. Figures: Debbie Maizels/Springer Nature.
Consistent with the characteristics of a complex psychiatric disorder, environmental factors also modulate predisposition and the course of cannabis use and CUD. For example, increased incidence of traumatic or challenging experiences (including childhood trauma or family or neighborhood adverse events17,31) strongly influences CUD risk. Studies incorporating genetic, environmental and behavioral characteristics have also demonstrated complex interactions between genes and environment32,33, emphasizing multiple intrinsic and extrinsic factors that collectively mediate CUD susceptibility. Irrespective of other factors, age of onset is a strong predictive variable, with significant correlations evident between younger age of cannabis use initiation and CUD liability. Importantly, the risk for CUD onset normally peaks in late adolescence and the early twenties34, when the brain has not achieved full adult maturity; this is a sensitive developmental window for psychiatric vulnerability. Sex also has important implications for CUD. Men have higher rates of CUD than women, a pattern that is present from childhood, when more boys than girls develop CUD2,35. Despite the high prevalence of cannabis-dependent males, the escalation of use and severity of CUD is greater in females, as with many substances of abuse36.
Deconstructing the neural underpinnings of cannabis use disorder
The neurobiology underlying CUD can be explored at multiple in vivo and postmortem levels of assessments, based on diverse experimental strategies that have been employed investigating cannabis use. While results are not always equivocal, due to various confounds—including issues of the frequency or severity of cannabis use, duration of use and abstinence, comorbid disorders, co-use of other psychoactive drugs, sex and genetics—several consistent patterns have begun to emerge that begins to establish a neurobiological framework for CUD.
Neurochemical signature of CUD
The eCB system, being the prime site for THC’s action, has been the focus for positron emission tomography (PET) neuroimaging studies, which consistently demonstrate that CB1Rs37–39 are dynamically altered depending on the phase of chronic cannabis use (Fig. 2). CB1Rs are generally downregulated in cannabis users, with strongest alteration evident shortly after cannabis use. These changes are predominantly localized to the neocortex and limbic cortices37,38, which regulate cognition and emotional processing, as well as the ventral striatum38, which is critically involved in reward and goal-directed behavior40. Longer duration of use associates with lower CB1R densities37. Postmortem brain analysis substantiates downregulation of CB1R binding in cannabis abusers as well as reduced mRNA expression of the gene (CNR1) encoding CB1R41. However, reduction of CB1R in cannabis dependent subjects is not permanent, and it normalizes ~2 to 28 days after drug use ceases39. The return to apparent normal levels after protracted abstinence might, however, not be paralleled by normalization of receptor function, based on animal studies (see below), suggesting that CB1R may remain functionally altered during abstinence. Nevertheless, reduced CB1R during early stages of the disorder is expected to impact the cascade of downstream neurobiological systems that can maintain persistent changes underling the behaviors characteristic of CUD. Aside from that for CB1R, little in vivo evidence exists for other components of the eCB system, but preliminary data indicate reduced binding of fatty acid amide hydrolase (FAAH), the enzyme that metabolizes anandamide, suggesting altered anandamide levels within corticolimbic brain regions in CUD individuals42.
Fig. 2 |.
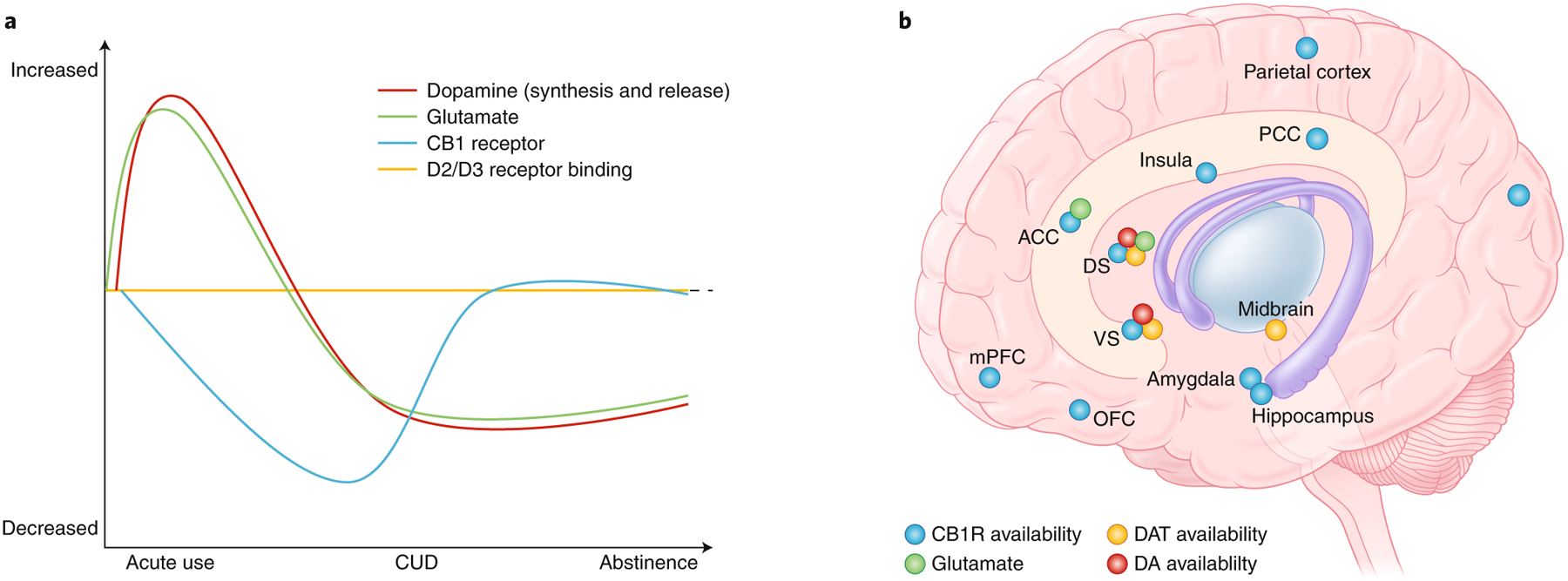
Overview of the dynamic patterns of the in vivo neurochemical-related alterations (based on PET, functional MRI, and H-MRS studies) associated with CUD. a, Line graphs indicate specific alterations during acute use as compared to neurobiological state in individuals with CUD, including during periods of abstinence. b, Dots indicate brain regions in which neurochemical marks reflective of CB1R, dopamine transporter (DAT) and dopamine (DA) synthesis have been detected in individuals with CUD. ACC, anterior cingulate cortex; DS, dorsal striatum; PCC, posterior cingulate cortex; VS, ventral striatum. Figures: Debbie Maizels/Springer Nature.
Of the neurotransmitter systems linked with addiction, dopamine has received most attention, given its strong role in reward, motivation and goal-directed behavior. Similarly to other substances of abuse, acute THC increases dopamine release in the striatum of healthy subjects43 (Fig. 2). Following chronic use, there is a reduction of stimulated dopamine levels in CUD44, as in other SUDs (e.g., psychostimulants, nicotine, alcohol and opioids; Fig. 2). Early age of onset or longer duration of cannabis use correlates with reduced stimulated striatal dopamine release (evoked by psychostimulant administration)45. The lower striatal dopamine release apparent in heavy cannabis users relates to inattention and greater negative symptoms46, and it inversely correlates with negative emotionality and addiction severity47. Reduced release also corresponds with decreased dopamine synthesis in cannabis-dependent individuals48, an effect associated with greater apathy49. PET measures of dopamine transporters (necessary for presynaptic dopamine reuptake) also reveal reduced availability in multiple brain areas, including the striatum, in cannabis-dependent people50. Altogether, the presynaptic hypodopaminergic state evident with CUD would be consistent with the classic ‘amotivational syndrome’ and negative affect characteristic of this disorder. However, a feature of many SUDs that often accompanies blunted dopamine release is reduced striatal dopamine D2/D3 receptor availability51, but this is relatively normal in CUD individuals44, suggesting potentially unique aspects of dopaminergic transmission with CUD.
The compulsive perpetuation of the addiction cycle is also characterized by alterations of glutamate, which plays a major role in mediating inhibitory control and drug-seeking behaviors52. Glutamatergic transmission is highly regulated by eCB signaling53 (through presynaptic terminal CB1Rs that reduce glutamate release) and thus sensitive to THC54. Proton magnetic resonance spectroscopy reveals increased glutamate levels with acute THC use55, but following chronic use, steady-state glutamate levels are reduced in various brain regions in both adults and teens56,57 (Fig. 1). Hypoglutamatergic transmission is also evident in other SUDs58,59, particularly during drug abstinence, and is considered to underlie the impaired decision-making that in turn contributes to continued drug-seeking52,60.
Morphological characteristics of CUD
Human structural MRI (sMRI) studies consistently document architectural alterations particularly within corticolimbic structures such as the prefrontal cortex (PFC), hippocampus and amygdala with CUD (Fig. 3). The integrity of the orbitofrontal cortex (OFC; localized within the PFC), which contributes to cognitive flexibility, valuation and decision-making61, is commonly impaired in SUD62 and strongly relates to problematic cannabis use. Overall, greater disease severity, regularity of use and duration of cannabis use are associated with reduced volume of the medial OFC63,64. Reduced OFC volume in young teens predicts initiation of cannabis use in later adolescence, suggestive of a preexisting neurobiological risk factor65. A similar sensitivity is evident in the hippocampus, a region central to learning and memory63,66, in which hippocampal gray matter is inversely associated with the amount of THC consumed, emphasizing the direct contribution of CB1R stimulation. Interestingly, a functional variant of the CNR1 gene (rs2023239 G allele) linked with higher cortical CB1R is associated with smaller hippocampal volume in chronic cannabis users, but not healthy controls, indicating a potential gene × drug interaction67. Likewise, reduced cortical thickness evident in some teens appears to relate to genetics—for example, the negative impact of cannabis use in early adolescence on cortical maturation is only in individuals with a high genetic risk of schizophrenia68. Cannabis use and CUD severity also associate, in a graded manner, with deficits of amygdala gray matter volume, with gray matter volume inversely related to CUD severity69,70. Given the involvement of the amygdala in emotional regulation, craving and drug-seeking behavior40, these morphological alterations may relate to the prominent mood and anxiety comorbid characteristics of CUD (Fig. 3). However, the significance of these morphological changes is debated, since twins of cannabis users who themselves do not use cannabis can have lower amygdala volume, suggesting that decreased amygdala volume may be predispositional71. Nevertheless, animal models have established a direct causal relationship between THC and changes in cortical structure72,73 (see below). Overall, preexisting morphological differences in brain regions regulating cognition and emotion may enhance the risk for cannabis use, and they are also impacted by the severity and duration of subsequent cannabis exposure.
Fig. 3 |.
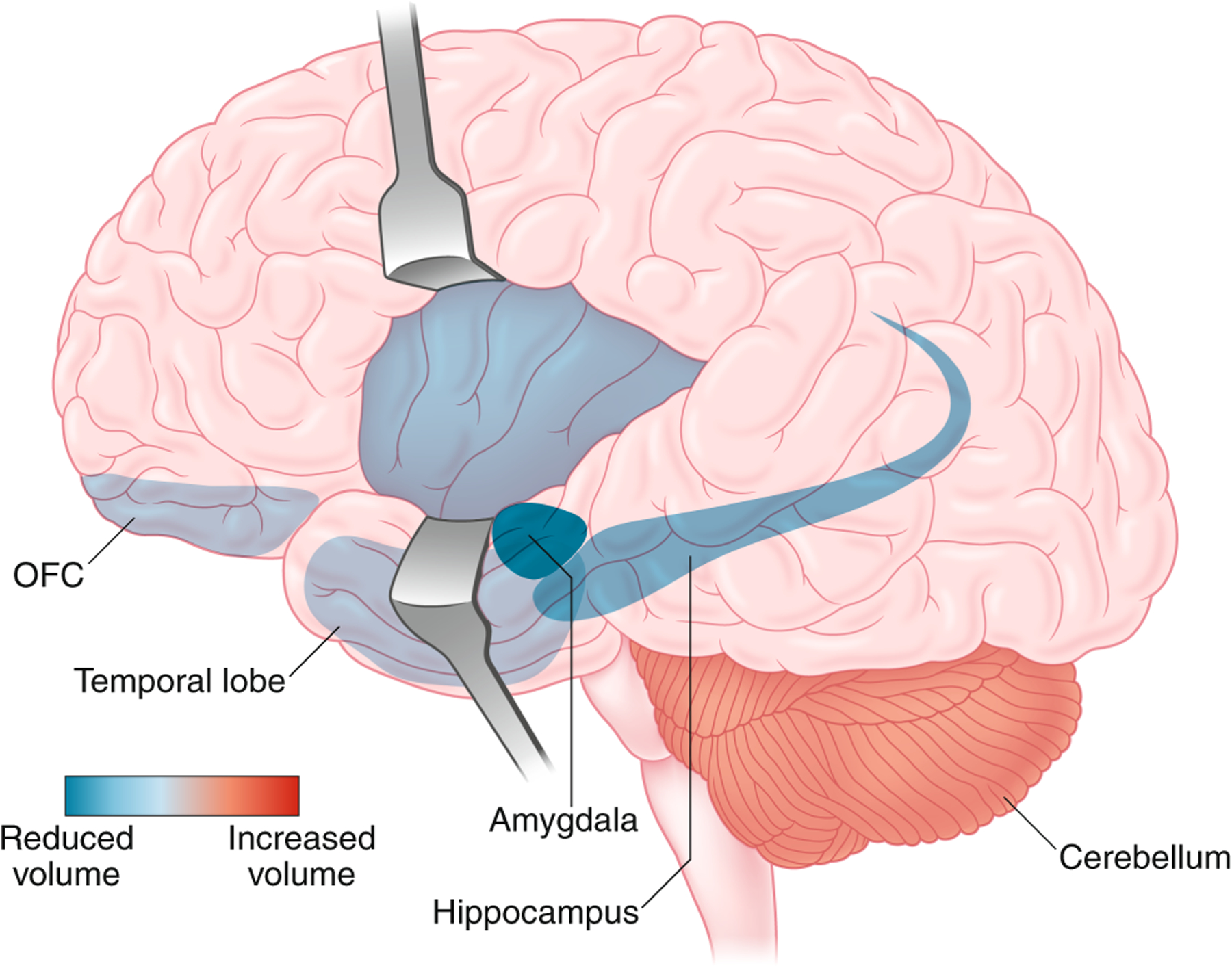
Alterations of gray matter volume (based on MRI studies) detected in individuals with CUD. Colors denote decreased (red) and increased (blue) gray matter volume. Figures: Debbie Maizels/Springer Nature.
The deficits in gray matter volume observed in CUD are mirrored in many SUDs74–76. One major difference is the cerebellum, which has increased gray matter volume in CUD subjects63,69, an effect maintained even a month of abstinence in adolescent cannabis users and which is associated with poor executive functioning63,69,77. Although the cerebellum is not typically linked with SUD pathophysiology, a significant body of evidence emphasizes cerebellar involvement in cognition, impulsivity and emotional processing in line with its strong anatomical projections to frontal cortical regions78. In contrast to consistent cerebellar findings, the striatum, though critical to reward and habit formation40, shows no clear pattern of gray matter structural alterations related to CUD79. Overall, the CUD neuronal architecture, at least that visible with MRI spatial resolution, primarily indicates sensitivity of structures linked to cognition and emotional processing (Fig. 3 and Table 1).
Table 1 |.
Alterations detected in specific brain regions of in vivo measures of gray matter volume and functional activity, associated with certain tasks and stimuli, in abstinent individuals with CUD.
| Region / circuit | Morphology | Neural activity | Association with function- or task-based activation |
|---|---|---|---|
| Frontal cortex/prefrontal cortex | Decrease in volume (OFC)63,64,142 | Decrease60,86,98,143 or increase90–92,97 in activity based on task and absinence85 | Decreased in decision-making60; decreased activity with uncertain reward143; decreased during cognitive appraisal of emotional stimuli98; decreased activity on attention task85,86, but increased activity after 1 year of abstinence is associated with better treatment outcome85; increased activity on working memory tasks90–92 and responsivity to negative emotional stimuli97 |
| Ventral striatum | * 79 | Decrease85,# or increase in activity144, based on task and abstinence | Decreased activity on an attentional task85; increased activity during reward anticipation144 |
| Dorsal striatum | * 79 | Decrease in activity85,# | Decreased activity during attentional processing85 |
| Hippocampus/temporal lobe | Decrease in volume63,66 | Decrease in activity (hipp)85,# | Attentional processing85 |
| Amygdala | Decrease in volume69,70,145 | Decrease85,# or increase97 in activity based on task | Decreased functionality associated with attentional performance85, increased activation in response to negative emotional stimuli97 |
| Cerebellum | Increase in volume63,69 | Decrease86,# or increase60,# in activity based on task | Increased activation during decision-making60, decreased activity associated with attentional processing86 |
| Mesocorticolimbic pathway | Increase in activity93,94,146,147 | Cannabis cue reactivity93,94,146,147 |
The majority of individuals with CUD were studied during periods of short abstinence from ~12 h to 4–5 days.
longer periods of abstinence, from ~1 to 36 months;
inconsistent observations.
Functional brain activity of CUD
In addition to morphological changes, various lines of functional MRI evidence reveal specific brain activity signatures with cannabis use relevant to CUD, both during resting conditions and when individuals are engaged in specific tasks (Fig. 4). One of the most common patterns of resting state brain activity identified in chronic cannabis users, as with other SUDs80, is increased functional connectivity associated with the default mode network (which primarily includes the posterior cingulate cortex, adjacent medial PFC (mPFC) and portions of the parietal cortex, the precuneus and angular gyri) and insula networks81,82. The default mode and insula networks are highly interconnected and are primarily activated during self-referential and introspective thought83. Importantly, resting-state alterations in frontal and sensory systems persist even following a month of abstinence, but with blunting of the changes81,84, suggesting the potential of the brain to reverse some of the effects of prior chronic cannabis use.
Fig. 4 |.
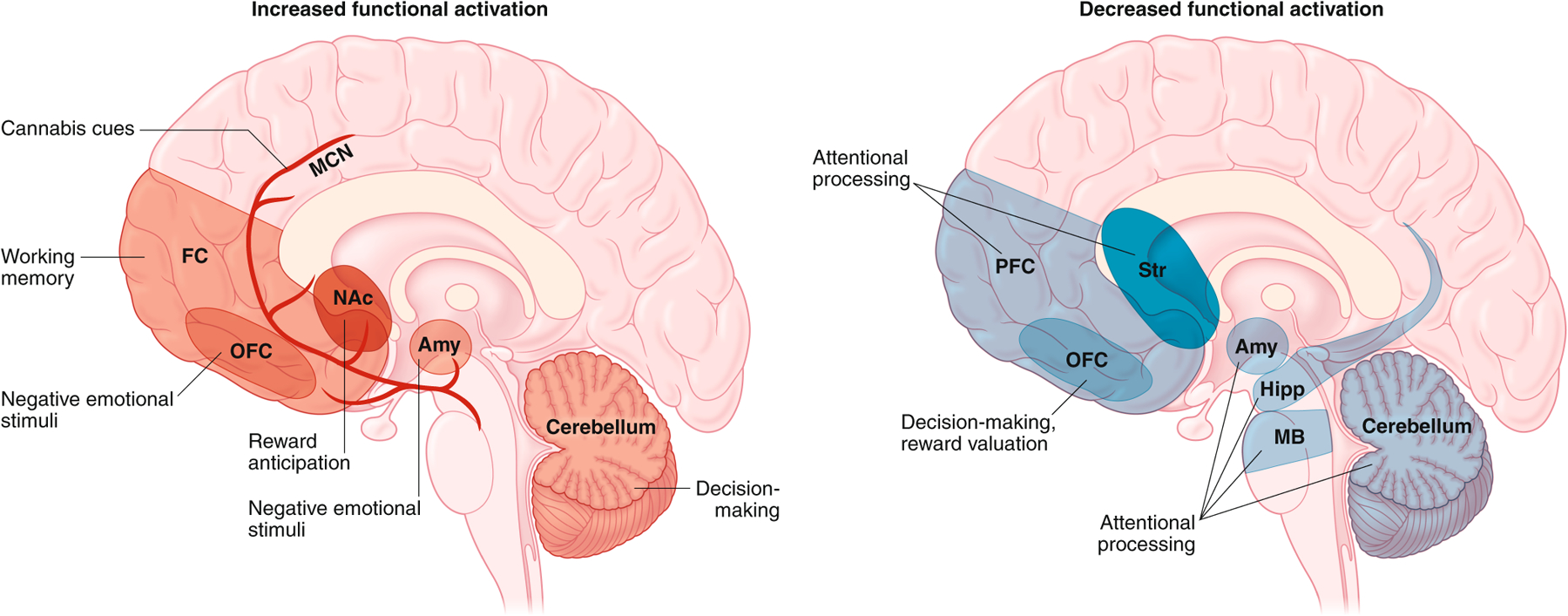
Differences in functional activity (based on functional MRI and electroencephalogram studies) detected in the brain of abstinent individuals with CUD during exposure to specific tasks and stimuli. Increased activation in specific regions is indicated in the left panel in red, and reduced activation is depicted in the right panel in blue. The specific task- or stimulus-driven alteration in activity is indicated in each region and circuit. Amy, amygdala; hipp, hippocampus; MCN, mesocorticolimbic network; FC, frontal cortex; NAc, nucleus accumbens; Str, striatum; MB, midbrain. Figures: Debbie Maizels/Springer Nature.
Functional MRI studies also document differential brain activity during neurocognitive tasks in CUD individuals (Table 1). For example, cost–benefit decision-making conditions in heavy cannabis users associate with reduced activity of the OFC and dorsolateral PFC, but also with increased cerebellar activity (Fig. 4), an effect that tracks tightly with impaired choice outcome60. Compromised attentional control is also a feature of SUDs, and multiple brain areas are underactive during cognitive interference conditions (for example, the Stroop task) in cannabis-dependent individuals, including the PFC, striatum, amygdala–parahippocampal gyrus, cerebellum and midbrain85–87 (Fig. 4). However, the under-recruitment of these circuits during attentional processes often occurs even with no outward deficits in attention85,86. Interestingly, the hypoactivity in these neural circuits appear to be mutable, as the pattern reverses after a year of abstinence, with increased activation within many of these areas85; this could also suggest a link between enhanced activation during attentional processes with the ability to abstain from drug. For working memory, despite the well-documented negative cognitive impact of acute THC in drug-naïve individuals and infrequent cannabis users, cannabis-experienced users often have normal working memory performance88,89. However, neural networks associated with such cognitive function are not normal: chronic and heavy cannabis use is associated with hyperactivation of frontal regions and networks underlying working memory90. Similar alterations are also evident in abstinent adolescent cannabis users91,92. Collectively, these modifications suggest an overcompensation of neural networks in individuals with CUD to achieve apparent normal executive function when cognitive demand is required.
Heightened sensitivity to drug cues is a hallmark of addiction that often contributes to craving and relapse, and, consistent with other SUDs, greater activation of the mesocorticolimbic reward pathway (ventral tegmental area, striatum, OFC, anterior cingulate gyrus) to drug cues is observed with CUD93,94. Higher response to cannabis cues is associated with greater cannabis-related problems94,95. The activity within these reward-related structures in response to cannabis cues has been associated with genetic variants of CNR1 (rs2023239) and FAAH (rs324420), where the heightened response to cues increases as the number of eCB risk alleles increases96, again suggesting genetic contributions to neural processes relevant to CUD.
Alterations in neurobiological systems associated with emotional processing are also common with cannabis use pathology, influencing the responsivity to anxiety, threat, depression, and maladaptive coping with stress that are associated with relapse risk. Both the PFC and amygdala are particularly reactive to negative stimuli in CUD; stronger medial OFC activity is associated with negative, but not positive, emotional stimuli after nearly a month of abstinence97. In contrast, the cognitive appraisal of affective stimuli in heavy cannabis abusers is associated with decreased activity in the mPFC (anterior cingulate and ventromedial PFC)98. The greater OFC and amygdala activation in CUD for negative stimuli, concomitant with decreased PFC activation for cognitive evaluation, would suggest an overactive affective neural loop with attenuation of appraisal networks. Combined with heightened sensitivity to drug cues and reward anticipation, these changes would classically reflect an addiction neurobiological signature of relapse vulnerability.
Translational insights from animal studies
While significant neurobiological insights have been garnered about the human brain from in vivo imaging strategies related to CUD, debates continue as to the direct role of cannabis to these apparent perturbations, considering complex issues such as genetic and environmental factors that might account for neurobiological differences predating cannabis use. THC animal models corroborate CB1R alterations observed in humans, establishing a causal impact of THC on CB1R fluctuations across time with proof of receptor desensitization (reflecting attenuated receptor coupling)99,100. Such studies also demonstrate that female rats exhibit greater attenuation of CB1R function in most brain regions than males, especially when chronic THC is administered during adolescence rather than in adulthood100,101, potentially relevant to the greater escalation and severity of CUD in human females than males. As in humans, there is (over time) a recovery to normal levels of CB1R following cessation of THC administration102. However, even at time points when apparent receptor binding or the number of CB1R returns to normal, there remain persistent perturbations of intracellular signaling and downstream molecular processes. For example, downstream effectors such as cAMP response element binding (CREB) protein are reduced in the hippocampus103 following chronic THC administration, an effect that persists weeks after drug exposure. An interesting downstream target of cAMP and CREB are cell adhesion molecules known to play key roles in long-term potentiation (LTP) and synaptic plasticity104,105. Concomitant with disturbances of genes related to synaptic plasticity, rats with chronic THC administration exhibiting impaired learning and memory have reduced hippocampal neuronal cell adhesion molecule, NCAM106. Importantly, reduced NCAM is well documented in different THC treatment regimens107,108. Moreover, chronic THC exposure in animals with genetic deficiency of polysialylated NCAM exacerbates the genetically induced memory deficits into adulthood109, supporting a gene × environmental interaction in line with the recent GWAS identifying genetic variants of genes encoding cell adhesion molecules, including NCAM, with lifetime cannabis use110. Overall, THC-induced alterations of CB1R, CREB and NCAM (Fig. 5) strongly suggest significant modification of synaptic plasticity consistent with the critical role of the eCB system in regulating synaptic transmission.
Fig. 5 |.
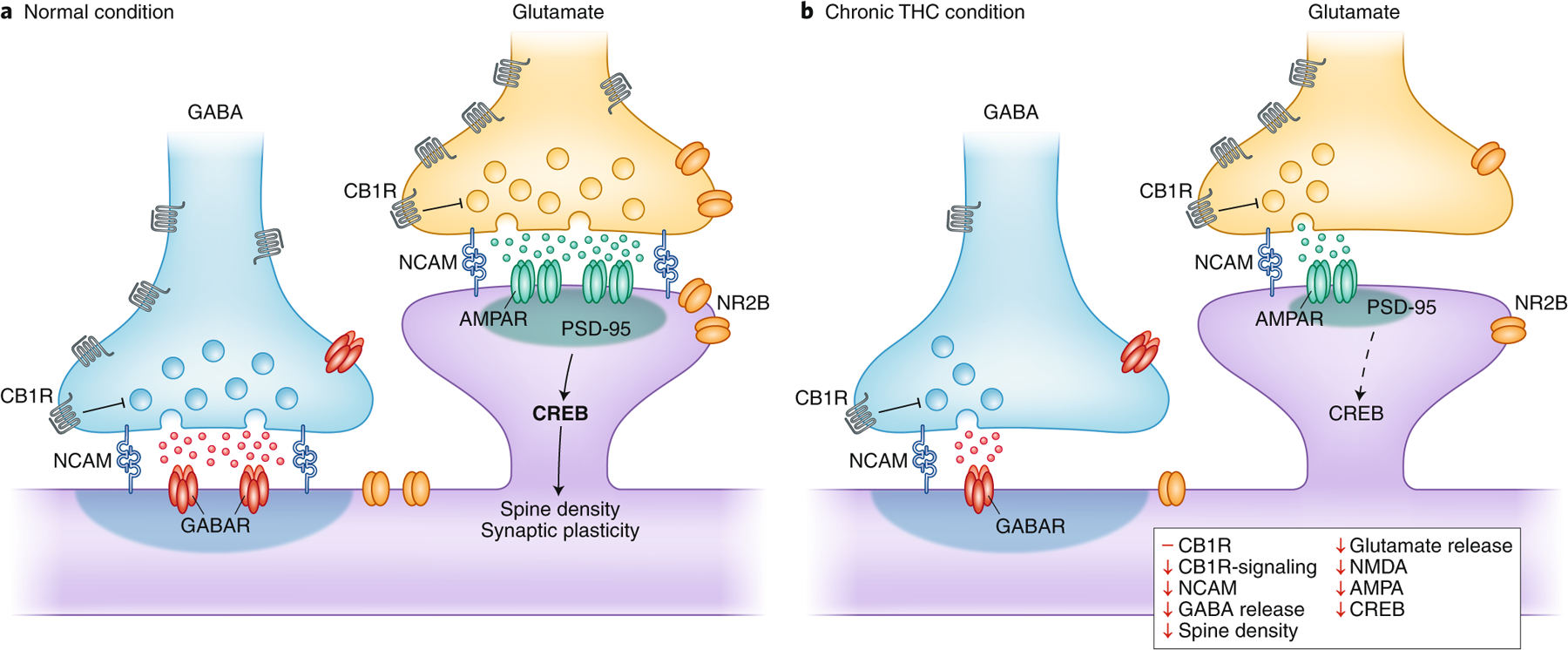
Synaptic perturbations based on animal models associated with chronic THC exposure (right) as compared to control condition (left) in glutamate and GABA synapses in the cortex. THC is known to have a greater effect on the interneuronal GABA microcircuit, most likely due to the greater (~20-fold) number of CB1R on cortical GABAergic interneuron axon terminals compared to glutamatergic terminals141. AMDAR, AMDA receptor; GABAR, GABA receptor; NCAM, neural cell adhesion molecule; NMDAR, NMDA receptor. Figures: Debbie Maizels/Springer Nature.
In addition to providing neurochemical and molecular insights, animal models confirm a direct impact of repeated THC exposure to alter neuronal architecture. For instance, long-term (90 days) exposure to high dose THC strongly reduces neuronal number, as well as the number of synapses and the dendritic length of hippocampal neurons, even months after the last THC dose72. Moreover, adolescent THC administration of a dose comparable to low-to-moderate cannabis use reduces the spine number and complexity of branching of PFC pyramidal neurons in adulthood73. These morphological changes are accompanied by marked disturbances of genes linked to dendritic development and cytoskeleton organization, as well as reprogramming of the epigenetic transcriptome73. Thus, the animal literature largely lends support to the idea of structural alterations being prominent within the cortex and hippocampus following repeated cannabis or THC exposure, emphasizing the cortical sensitivity of chronic use.
Animal models also reveal, at cellular-level resolution, responses relevant to alteration of neural activity detected in individuals with CUD. Notably, chronic THC exposure in most electrophysiological animal studies confirms indices of persistently reduced cortical neuronal activity similar to deficits in neural oscillations reported in humans with CUD111. For example, adult rats with adolescent THC exposure exhibit long-term reduction of cortical oscillations in the rostral mPFC specifically mediated by CB1R mechanisms112. CB1R-mediated disruption of neural oscillations are linked to reduced transmission of GABAergic interneurons in cortical circuits113. Chronic THC exposure also depresses glutamatergic synaptic responsiveness, as evident in the hippocampus where parallel downregulation of NMDA and AMPA glutamate receptor expression could contribute to reduced LTP103. Fig. 5 provides an overview of the neurochemical cortical changes generally observed in chronic THC animal models. These alterations are not, however, mirrored in all brain areas, and cell-type- and synapse-specific differences are actively being explored regarding the long-term subregional effects of THC. Such complexity is exemplified by recent evidence demonstrating that long-term THC exposure weakens mPFC inputs to the ventral striatum but strengthens those from the hippocampus and amygdala114, in line with reduced prefrontal cognitive control but enhanced emotional lability in cannabis users.
The use of animal models to inform neurobiological underpinnings associated with phenotypes relevant to CUD-comorbid disorders, such as addiction to other substances, have firmly established that adolescent THC exposure increases the sensitivity in adulthood to opioid reward115–118. There remains, however, a surprising paucity of animal studies evaluating the interaction of THC with other drugs, despite the high comorbidity of CUD with alcohol and nicotine addiction119. Of the few existing studies found that short-term THC exposure increased the likelihood of rats to acquire nicotine self-administration120. Unfortunately, study of chronic THC animal models focused on psychiatric-related phenotypes such as anxiety-like behavior is also limited, despite the tight link between the eCB and stress systems121. In the published behavioral data, the effects of THC exposure are variable, but might reflect important aspects of dose, neurodevelopmental sensitivity and sex that are also evident in humans. For instance, high THC doses increase anxiety-like behaviors, whereas low doses lead to a decrease122. Additionally, females appear more sensitive to the anxiogenic properties of THC than males, even during THC-abstinent periods122. Moreover, chronic cannabinoid administration during adolescence, but not adulthood, increases anxiety behavior weeks after exposure123.
Although THC animal models substantiate a number of alterations detected in humans, there remains a significant gap of in-depth knowledge critical to advance fundamental mechanistic underpinnings of CUD. A major challenge for translational efforts is that the prevalent short-term and often excessive THC doses used in many animal models do not reflect the human condition, thus lacking face validity for CUD. Moreover, the gold standard self-administration animal models normally used in the addiction field are challenging for cannabis because rodents experience THC as aversive and thus do not readily acquire or maintain stable self-administration behavior124. To circumvent such challenges, most animal research has mainly used passive parenteral THC administration routes. New animal inhalation self-administration models for THC125 and cannabis cigarette126 will help bridge the translational gap to advance molecular and cellular knowledge relevant to CUD.
Treatment: reconstructing neurobiological systems impacted by cannabis
Despite the millions of people diagnosed with CUD there are presently no approved FDA pharmacotherapies. Psychosocial interventions, which are normally the first line of treatment, have the potential to modify gray matter volume as well as to increase the functional and structural connectivity between frontal and limbic cortices127,128 as well as cerebellar cognitive-related circuits129. However, no studies to date have examined whether cognitive–behavioral strategies can improve the neural alterations and behavioral outcomes in individuals with CUD.
The majority of pharmacological interventions being explored for CUD are substitution therapies with cannabinoid agonists as well as agents targeting neurotransmitter systems known to be aberrant in addiction. Table 2 provides a summary of these agents. In addition, recent attention has focused on cannabidiol (CBD), a non-intoxicating cannabinoid which has anxiolytic properties130–132 and appears to reverse behaviors and neural systems relevant to the effects of cannabis. For instance, cognitive and anxiety-like behaviors induced by chronic THC administration during adolescence in rodent models are prevented by the co-administration of CBD133. Additionally in humans, CBD blocks THC-induced anxiety134 and reduces wanting and liking of cannabis-related stimuli in cannabis users130. On a neurobiological level, animal studies demonstrate that CBD normalizes glutamatergic systems135, known to be dysregulated in SUDs including CUD. Moreover, CBD activates corticolimbic structures and circuits that are reduced during attentional processing in individuals with CUD136. Concomitant to reducing anxiety, CBD also reduces stress-induced activation of the hippocampus and other cortical areas132 and restores hippocampal volume in current cannabis users, particularly those with greater lifetime cannabis exposure137. However, acute administration of CBD does not impact the reinforcing or physiological effects of smoked cannabis in cannabis users138. Thus, CBD’s potential therapeutic value might relate more to alleviating craving and stress-related responses that contribute to relapse in CUD6 rather than blocking the acute rewarding effects of THC. Overall, the current medications are still in relatively early phases of exploration as potential CUD pharmacotherapeutics, but the list is expected to grow as greater insights are gleaned about the underlying pathophysiology of CUD.
Table 2 |.
Putative pharmacotherapeutics that have been investigated for CUD.
| Pharmacotherapy (market name(s)) | Formulation | Mechanism of action | Mitigates intoxication | Mitigates withdrawal symptoms | Mitigates relapse | Citation |
|---|---|---|---|---|---|---|
| Dronabinol (Marinol, Syndros) | Extracted Δ9-THC in capsule | CB1 agonist | Yes, at high doses | Yes | No | 148 |
| Nabilone (Cesamet) | Synthetic Δ9-THC mimetic in capsule | CB1 agonist | - | Yes | Yes | 148 |
| Nabiximols (Sativex) | Δ9-THC + CBD in nasal spray | CB1 agonist | - | Yes | No | 148 |
| CBD | Cannabis extract in capsule | No | Yes, but limited | - | 138,149 | |
| PF-04457845 | Capsule | FAAH inhibitor | - | Yes | Yes | 150 |
| Gabapentin (Neurontin) | Capsule | GABA analog, voltage-gated Ca2+ antagonist | - | Yes | Yes | 148 |
| N-acetylcysteine | Capsule | Glutamate agonist | - | - | Yes, in adolescents | 148 |
| Topiramate (Topamax) | Capsule | GABA agonist, glutamate antagonist | Decreased amount smoked, but not overall relapse, and poorly tolerated | 148 | ||
| Guanfacine (Tenex) | Capsule | Alpha-2a adrenergic agonist | - | Yes | - | 148 |
Summary and future directions
CUD is a multifactorial complex disorder in which there is interaction among genetic and environmental factors that, in combination with psychiatric comorbidity and the continued use of cannabis, contribute to neurobiological alterations that underlie the addiction phenotype (Fig. 6). There is still much to learn about the neurobiology of CUD, but contrary to the growing nonchalance regarding cannabis use by many in society, the neurobiological evidence demonstrates multilevel divergent patterns in neurochemistry, morphology and neural activity in brain regions that are highly implicated in other SUDs. The shared neurobiological signature may underlie common addiction behavioral phenotypes, such as compromised decision-making and attentional processing, marked drug craving and sensitivity to drug cues, as well as anxiety and negative affect particularly associated with drug withdrawal.
Fig. 6 |.
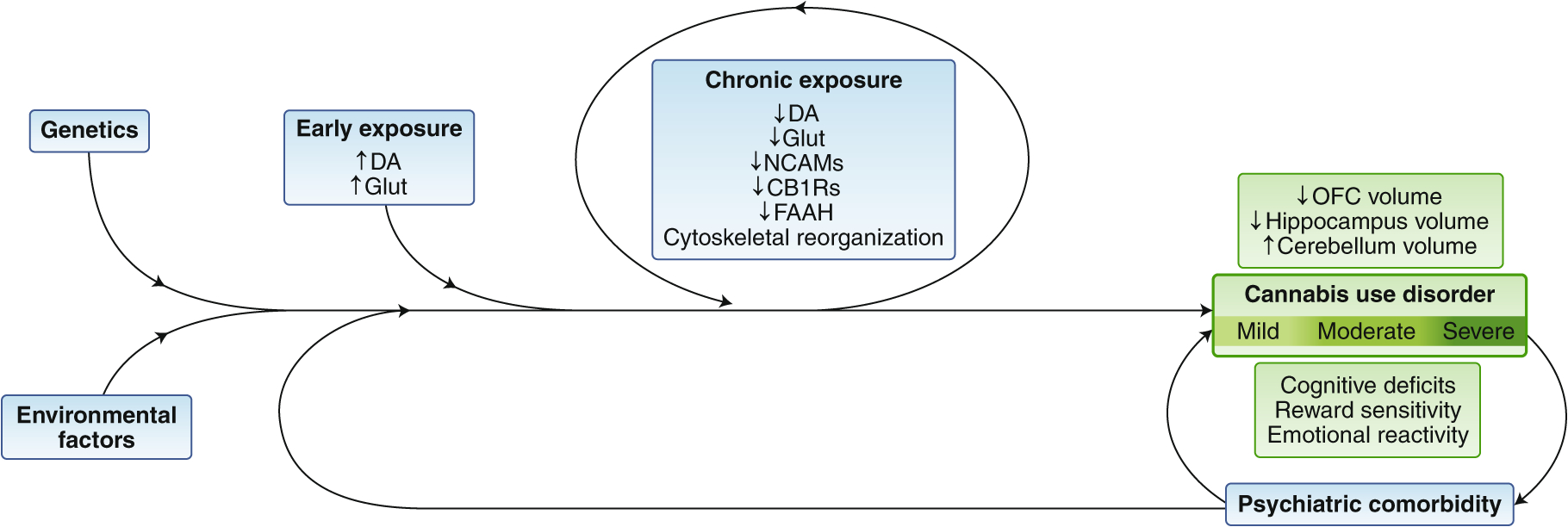
Factors contributing to CUD. Schematic summary of multiple factors that contribute to the neurobiological patterns documented in relation to cannabis use and eventual CUD, where the more pronounced neurobiological alterations are associated with greater severity of the disorder and behavioral consequences. FAAH, fatty acid amide hydrolase; Glut, glutamate; Vol, volume. Figures: Debbie Maizels/Springer Nature.
Definitive conclusions about specific neurobiological features of CUD still remain elusive due to complex heterogeneous factors, such as genetics, environment and sex, evaluated across studies. While the ‘chicken or egg’ pre-existing or direct cannabis cause of such factors can continue to be debated, it is clear that individuals with and without pre-existing neurobiological risk are being exposed more than ever to high THC doses. This is of particular concern for the vulnerable neurodevelopmental periods—prenatal, childhood and adolescence. Longitudinal projects, such as the Adolescent Brain Cognitive Development (ABCD) Study and new trans-NIH baby Brain Cognitive Development (bBCD) project to monitor normative and drug-exposed neurodevelopmental trajectories starting from adolescent and prenatal stages, respectively, are important steps to identify specific early risk factors and expand knowledge about the long-term impact of cannabis (and other drug) exposure. Data from the Dutch Generation R Study, a population-based prospective cohort study from fetal life until young adulthood, is already being leveraged to understand the influence of the prenatal environment on brain maturation and childhood behavior139. Moreover, accrued data has established that the adolescent brain is not impervious to cannabis effects, with early onset of use often associated with greater changes in brain structure and activity.
What is nevertheless also apparent is the potential for some neurobiological alterations to be reversed during extended abstinence. This emphasizes the adaptive and continued dynamic nature of the brain when drug-free. However, animal models overwhelmingly demonstrate protracted molecular alterations subsequent to THC use, suggesting that underlying molecular and cellular processes may remain sensitive to subsequent drug use later in life even if general neurobiological features appear relatively normal. The neurobiological mechanisms that maintain these protracted effects are likely epigenetic processes140, which means that they are malleable. As such, the drug-abstinence window may be a critical period for targeting these molecular processes to improve long-term outcomes.
Despite significant insights gleaned about the in vivo neurobiological patterns of CUD, there still remains a surprising large gap of knowledge from animal studies. In-depth preclinical molecular interrogations based on the human brain findings are limited. For instance, the cerebellum is not well studied in animals, though individuals with CUD have clear differences in this region. Moreover, despite the growing animal literature on THC, dosing regimen and treatment strategies more reflective of the human condition are lacking in the field. Enhanced integration of human and preclinical animal research would significantly expand neurobiological knowledge important to guiding prevention and treatment strategies.
Finally, the ability to expedite scientific understandings of CUD is unfortunately hindered today in a sociopolitical quagmire in the US, where the public perceives little risk of cannabis use, while governmental regulations still consider it a Schedule I drug, which impedes critical research necessary to provide guidance about cannabis risk–benefit and potential treatments. Reducing such impediments will greatly advance scientific and clinical discoveries toward treatment development and to promote evidence-based policies.
Acknowledgments
This work was supported by a grant from the US National Institutes of Health (NIH) DA030359.
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Competing interests
The authors declare no competing interests.
References
- 1.UN Office on Drugs and Crime World Drug Report 3: Market Analysis of Plant-Based Drugs: Opiates, Cocaine, Cannabis. (UNODC Research, 2017). [Google Scholar]
- 2.Hasin DS et al. Prevalence of marijuana use disorders in the United States between 2001–2002 and 2012–2013. JAMA Psychiatry 72, 1235–1242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martins SS et al. Changes in US lifetime heroin use and heroin use disorder: prevalence from the 2001–2002 to 2012–2013 National Epidemiologic Survey on Alcohol and Related Conditions. JAMA Psychiatry 74, 445–455 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kerridge BT et al. Changes in the prevalence and correlates of cocaine use and cocaine use disorder in the United States, 2001–2002 and 2012–2013. Addict. Behav 90, 250–257 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Nutt DJ, King LA & Phillips LD; Independent Scientific Committee on Drugs. Drug harms in the UK: a multicriteria decision analysis. Lancet 376, 1558–1565 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Hasin DS et al. Prevalence and correlates of DSM-5 cannabis use disorder, 2012–2013: findings from the National Epidemiologic Survey on Alcohol and Related Conditions-III. Am. J. Psychiatry 173, 588–599 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanuš LO, Meyer SM, Muñoz E, Taglialatela-Scafati O & Appendino G Phytocannabinoids: a unified critical inventory. Nat. Prod. Rep 33, 1357–1392 (2016). [DOI] [PubMed] [Google Scholar]
- 8.ElSohly MA et al. Potency trends of delta9-THC and other cannabinoids in confiscated marijuana from 1980–1997. J. Forensic Sci 45, 24–30 (2000). [PubMed] [Google Scholar]
- 9.Chandra S et al. New trends in cannabis potency in USA and Europe during the last decade (2008–2017). Eur. Arch. Psychiatry Clin. Neurosci 269, 5–15 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Mechoulam R, Hanuš LO, Pertwee R & Howlett AC Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat. Rev. Neurosci 15, 757–764 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Mechoulam R & Parker LA The endocannabinoid system and the brain. Annu. Rev. Psychol 64, 21–47 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Herkenham M et al. Cannabinoid receptor localization in brain. Proc. Natl. Acad. Sci. USA 87, 1932–1936 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Busquets-Garcia A, Bains J & Marsicano G CB1 receptor signaling in the brain: extracting specificity from ubiquity. Neuropsychopharmacology 43, 4–20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xi ZX et al. Brain cannabinoid CB2 receptors modulate cocaine’s actions in mice. Nat. Neurosci 14, 1160–1166 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. (American Psychiatric Association, 2013). [Google Scholar]
- 16.Feingold D, Fox J, Rehm J & Lev-Ran S Natural outcome of cannabis use disorder: a 3-year longitudinal follow-up. Addiction 110, 1963–1974 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Verweij KJ et al. Genetic and environmental influences on cannabis use initiation and problematic use: a meta-analysis of twin studies. Addiction 105, 417–430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lynskey MT et al. An Australian twin study of cannabis and other illicit drug use and misuse, and other psychopathology. Twin Res. Hum. Genet 15, 631–641 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gillespie NA, Neale MC & Kendler KS Pathways to cannabis abuse: a multi-stage model from cannabis availability, cannabis initiation and progression to abuse. Addiction 104, 430–438 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agrawal A et al. Genome-wide association study identifies a novel locus for cannabis dependence. Mol. Psychiatry 23, 1293–1302 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Demontis D et al. Genome-wide association study implicates CHRNA2 in cannabis use disorder. Nat. Neurosci 22, 1066–1074 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J et al. The contribution of rare and common variants in 30 genes to risk nicotine dependence. Mol. Psychiatry 20, 1467–1478 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKay JD et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat. Genet 49, 1126–1132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasman JA et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat. Neurosci 21, 1161–1170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherva R et al. Genome-wide association study of cannabis dependence severity, novel risk variants, and shared genetic risks. JAMA Psychiatry 73, 472–480 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Forti M et al. The contribution of cannabis use to variation in the incidence of psychotic disorder across Europe (EU-GEI): a multicentre case-control study. Lancet Psychiatry 6, 427–436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Radhakrishnan R, Wilkinson ST & D’Souza DC Gone to pot - a review of the association between cannabis and psychosis. Front. Psychiatry 5, 54 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wittchen HU et al. Cannabis use and cannabis use disorders and their relationship to mental disorders: a 10-year prospective-longitudinal community study in adolescents. Drug Alcohol Depend. 88 Suppl 1, S60–S70 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Blanco C et al. Cannabis use and risk of psychiatric disorders: prospective evidence from a US national longitudinal study. JAMA Psychiatry 73, 388–395 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Emery NN & Simons JS A reinforcement sensitivity model of affective and behavioral dysregulation in marijuana use and associated problems. Exp. Clin. Psychopharmacol 25, 281–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ridenour TA et al. Neurobehavior disinhibition, parental substance use disorder, neighborhood quality and development of cannabis use disorder in boys. Drug Alcohol Depend. 102, 71–77 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hines LA et al. Overlap of heritable influences between cannabis use disorder, frequency of use and opportunity to use cannabis: trivariate twin modelling and implications for genetic design. Psychol. Med 48, 2786–2793 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Rogosch FA, Oshri A & Cicchetti D From child maltreatment to adolescent cannabis abuse and dependence: a developmental cascade model. Dev. Psychopathol 22, 883–897 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stinson FS, Ruan WJ, Pickering R & Grant BF Cannabis use disorders in the USA: prevalence, correlates and co-morbidity. Psychol. Med 36, 1447–1460 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Haberstick BC et al. Prevalence and correlates of alcohol and cannabis use disorders in the United States: results from the national longitudinal study of adolescent health. Drug Alcohol Depend. 136, 158–161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khan SS et al. Gender differences in cannabis use disorders: results from the National Epidemiologic Survey of Alcohol and Related Conditions. Drug Alcohol Depend. 130, 101–108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirvonen J et al. Reversible and regionally selective downregulation of brain cannabinoid CB1 receptors in chronic daily cannabis smokers. Mol. Psychiatry 17, 642–649 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ceccarini J et al. [18F]MK-9470 PET measurement of cannabinoid CB1 receptor availability in chronic cannabis users. Addict. Biol 20, 357–367 (2015). [DOI] [PubMed] [Google Scholar]
- 39.D’Souza DC et al. Rapid changes in CB1 receptor availability in cannabis dependent males after abstinence from cannabis. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 1, 60–67 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koob GF & Volkow ND Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3, 760–773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Villares J Chronic use of marijuana decreases cannabinoid receptor binding and mRNA expression in the human brain. Neuroscience 145, 323–334 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Boileau I et al. Fatty acid amide hydrolase binding in brain of cannabis users: imaging with the novel radiotracer [11C]CURB. Biol. Psychiatry 80, 691–701 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bossong MG et al. Delta 9-tetrahydrocannabinol induces dopamine release in the human striatum. Neuropsychopharmacology 34, 759–766 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Thiruchselvam T, Malik S & Le Foll B A review of positron emission tomography studies exploring the dopaminergic system in substance use with a focus on tobacco as a co-variate. Am. J. Drug Alcohol Abuse 43, 197–214 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Urban NB et al. Dopamine release in chronic cannabis users: a [11c]raclopride positron emission tomography study. Biol. Psychiatry 71, 677–683 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van de Giessen E et al. Deficits in striatal dopamine release in cannabis dependence. Mol. Psychiatry 22, 68–75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Volkow ND et al. Decreased dopamine brain reactivity in marijuana abusers is associated with negative emotionality and addiction severity. Proc. Natl. Acad. Sci. USA 111, E3149–E3156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bloomfield MA et al. Dopaminergic function in cannabis users and its relationship to cannabis-induced psychotic symptoms. Biol. Psychiatry 75, 470–478 (2014). [DOI] [PubMed] [Google Scholar]
- 49.Bloomfield MA, Morgan CJ, Kapur S, Curran HV & Howes OD The link between dopamine function and apathy in cannabis users: an [18F]-DOPA PET imaging study. Psychopharmacology (Berl.) 231, 2251–2259 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Leroy C et al. Striatal and extrastriatal dopamine transporter in cannabis and tobacco addiction: a high-resolution PET study. Addict. Biol 17, 981–990 (2012). [DOI] [PubMed] [Google Scholar]
- 51.Volkow ND, Fowler JS, Wang GJ, Baler R & Telang F Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology 56 Suppl 1, 3–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kalivas PW The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci 10, 561–572 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Katona I Cannabis and endocannabinoid signaling in epilepsy. Handb. Exp. Pharmacol 231, 285–316 (2015). [DOI] [PubMed] [Google Scholar]
- 54.Colizzi M, McGuire P, Pertwee RG & Bhattacharyya S Effect of cannabis on glutamate signalling in the brain: A systematic review of human and animal evidence. Neurosci. Biobehav. Rev 64, 359–381 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Colizzi M et al. Delta-9-tetrahydrocannabinol increases striatal glutamate levels in healthy individuals: implications for psychosis. Mol. Psychiatry 10.1038/s41380-019-0374-8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Muetzel RL et al. In vivo 1H magnetic resonance spectroscopy in young-adult daily marijuana users. Neuroimage Clin. 2, 581–589 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prescot AP, Locatelli AE, Renshaw PF & Yurgelun-Todd DA Neurochemical alterations in adolescent chronic marijuana smokers: a proton MRS study. Neuroimage 57, 69–75 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mon A, Durazzo TC & Meyerhoff DJ Glutamate, GABA, and other cortical metabolite concentrations during early abstinence from alcohol and their associations with neurocognitive changes. Drug Alcohol Depend. 125, 27–36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang S et al. Lower glutamate levels in rostral anterior cingulate of chronic cocaine users - A (1)H-MRS study using TE-averaged PRESS at 3 T with an optimized quantification strategy. Psychiatry Res. 174, 171–176 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bolla KI, Eldreth DA, Matochik JA & Cadet JL Neural substrates of faulty decision-making in abstinent marijuana users. Neuroimage 26, 480–492 (2005). [DOI] [PubMed] [Google Scholar]
- 61.Izquierdo A Functional heterogeneity within rat orbitofrontal cortex in reward learning and decision making. J. Neurosci 37, 10529–10540 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guttman Z, Moeller SJ & London ED Neural underpinnings of maladaptive decision-making in addictions. Pharmacol. Biochem. Behav 164, 84–98 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Battistella G et al. Long-term effects of cannabis on brain structure. Neuropsychopharmacology 39, 2041–2048 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chye Y et al. Orbitofrontal and caudate volumes in cannabis users: a multi-site mega-analysis comparing dependent versus non-dependent users. Psychopharmacology (Berl.) 234, 1985–1995 (2017). [DOI] [PubMed] [Google Scholar]
- 65.Cheetham A et al. Orbitofrontal volumes in early adolescence predict initiation of cannabis use: a 4-year longitudinal and prospective study. Biol. Psychiatry 71, 684–692 (2012). [DOI] [PubMed] [Google Scholar]
- 66.Chye Y et al. Cannabis-related hippocampal volumetric abnormalities specific to subregions in dependent users. Psychopharmacology (Berl.) 234, 2149–2157 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Schacht JP, Hutchison KE & Filbey FM Associations between cannabinoid receptor-1 (CNR1) variation and hippocampus and amygdala volumes in heavy cannabis users. Neuropsychopharmacology 37, 2368–2376 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.French L et al. Early cannabis use, polygenic risk score for schizophrenia and brain maturation in adolescence. JAMA Psychiatry 72, 1002–1011 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cousijn J et al. Grey matter alterations associated with cannabis use: results of a VBM study in heavy cannabis users and healthy controls. Neuroimage 59, 3845–3851 (2012). [DOI] [PubMed] [Google Scholar]
- 70.Koenders L et al. Grey matter changes associated with heavy cannabis use: a longitudinal sMRI study. PLoS One 11, e0152482 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pagliaccio D et al. Shared predisposition in the association between cannabis use and subcortical brain structure. JAMA Psychiatry 72, 994–1001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scallet AC et al. Morphometric studies of the rat hippocampus following chronic delta-9-tetrahydrocannabinol (THC). Brain Res. 436, 193–198 (1987). [DOI] [PubMed] [Google Scholar]
- 73.Miller ML et al. Adolescent exposure to Δ9-tetrahydrocannabinol alters the transcriptional trajectory and dendritic architecture of prefrontal pyramidal neurons. Mol. Psychiatry 24, 588–600 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zahr NM & Pfefferbaum A Alcohol’s effects on the brain: neuroimaging results in humans and animal models. Alcohol Res. 38, 183–206 (2017). [PMC free article] [PubMed] [Google Scholar]
- 75.Wollman SC et al. Gray matter abnormalities in opioid-dependent patients: A neuroimaging meta-analysis. Am. J. Drug Alcohol Abuse 43, 505–517 (2017). [DOI] [PubMed] [Google Scholar]
- 76.Mackey S et al. Mega-analysis of gray matter volume in substance dependence: general and substance-specific regional effects. Am. J. Psychiatry 176, 119–128 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Medina KL, Nagel BJ & Tapert SF Abnormal cerebellar morphometry in abstinent adolescent marijuana users. Psychiatry Res. 182, 152–159 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schmahmann JD The cerebellum and cognition. Neurosci. Lett 688, 62–75 (2019). [DOI] [PubMed] [Google Scholar]
- 79.Nader DA & Sanchez ZM Effects of regular cannabis use on neurocognition, brain structure, and function: a systematic review of findings in adults. Am. J. Drug Alcohol Abuse 44, 4–18 (2018). [DOI] [PubMed] [Google Scholar]
- 80.DeWitt SJ, Ketcherside A, McQueeny TM, Dunlop JP & Filbey FM The hyper-sentient addict: an exteroception model of addiction. Am. J. Drug Alcohol Abuse 41, 374–381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pujol J et al. Functional connectivity alterations in brain networks relevant to self-awareness in chronic cannabis users. J. Psychiatr. Res 51, 68–78 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Wetherill RR et al. Cannabis, cigarettes, and their co-occurring use: Disentangling differences in default mode network functional connectivity. Drug Alcohol Depend. 153, 116–123 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mak LE et al. The default mode network in healthy individuals: a systematic review and meta-analysis. Brain Connect. 7, 25–33 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Blanco-Hinojo L et al. Attenuated frontal and sensory inputs to the basal ganglia in cannabis users. Addict. Biol 22, 1036–1047 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Kober H, DeVito EE, DeLeone CM, Carroll KM & Potenza MN Cannabis abstinence during treatment and one-year follow-up: relationship to neural activity in men. Neuropsychopharmacology 39, 2288–2298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chang L, Yakupov R, Cloak C & Ernst T Marijuana use is associated with a reorganized visual-attention network and cerebellar hypoactivation. Brain 129, 1096–1112 (2006). [DOI] [PubMed] [Google Scholar]
- 87.Broyd SJ, van Hell HH, Beale C, Yücel M & Solowij N Acute and chronic effects of cannabinoids on human cognition-a systematic review. Biol. Psychiatry 79, 557–567 (2016). [DOI] [PubMed] [Google Scholar]
- 88.Kanayama G, Rogowska J, Pope HG, Gruber SA & Yurgelun-Todd DA Spatial working memory in heavy cannabis users: a functional magnetic resonance imaging study. Psychopharmacology (Berl.) 176, 239–247 (2004). [DOI] [PubMed] [Google Scholar]
- 89.Smith AM, Longo CA, Fried PA, Hogan MJ & Cameron I Effects of marijuana on visuospatial working memory: an fMRI study in young adults. Psychopharmacology (Berl.) 210, 429–438 (2010). [DOI] [PubMed] [Google Scholar]
- 90.Sagar KA & Gruber SA Interactions between recreational cannabis use and cognitive function: lessons from functional magnetic resonance imaging. Ann. NY Acad. Sci 1451, 42–70 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schweinsburg AD et al. The influence of recency of use on fMRI response during spatial working memory in adolescent marijuana users. J. Psychoactive Drugs 42, 401–412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Padula CB, Schweinsburg AD & Tapert SF Spatial working memory performance and fMRI activation interaction in abstinent adolescent marijuana users. Psychol. Addict. Behav 21, 478–487 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cousijn J et al. Neural responses associated with cue-reactivity in frequent cannabis users. Addict. Biol 18, 570–580 (2013). [DOI] [PubMed] [Google Scholar]
- 94.Filbey FM, Schacht JP, Myers US, Chavez RS & Hutchison KE Marijuana craving in the brain. Proc. Natl. Acad. Sci. USA 106, 13016–13021 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Filbey FM et al. fMRI study of neural sensitization to hedonic stimuli in long-term, daily cannabis users. Hum. Brain Mapp 37, 3431–3443 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Filbey FM, Schacht JP, Myers US, Chavez RS & Hutchison KE Individual and additive effects of the CNR1 and FAAH genes on brain response to marijuana cues. Neuropsychopharmacology 35, 967–975 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zimmermann K et al. Altered orbitofrontal activity and dorsal striatal connectivity during emotion processing in dependent marijuana users after 28 days of abstinence. Psychopharmacology (Berl.) 235, 849–859 (2018). [DOI] [PubMed] [Google Scholar]
- 98.Wesley MJ, Lile JA, Hanlon CA & Porrino LJ Abnormal medial prefrontal cortex activity in heavy cannabis users during conscious emotional evaluation. Psychopharmacology (Berl.) 233, 1035–1044 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Breivogel CS et al. Chronic delta9-tetrahydrocannabinol treatment produces a time-dependent loss of cannabinoid receptors and cannabinoid receptor-activated G proteins in rat brain. J. Neurochem 73, 2447–2459 (1999). [DOI] [PubMed] [Google Scholar]
- 100.Burston JJ, Wiley JL, Craig AA, Selley DE & Sim-Selley LJ Regional enhancement of cannabinoid CB₁ receptor desensitization in female adolescent rats following repeated Delta-tetrahydrocannabinol exposure. Br. J. Pharmacol 161, 103–112 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rubino T et al. Chronic delta 9-tetrahydrocannabinol during adolescence provokes sex-dependent changes in the emotional profile in adult rats: behavioral and biochemical correlates. Neuropsychopharmacology 33, 2760–2771 (2008). [DOI] [PubMed] [Google Scholar]
- 102.Sim-Selley LJ et al. Prolonged recovery rate of CB1 receptor adaptation after cessation of long-term cannabinoid administration. Mol. Pharmacol 70, 986–996 (2006). [DOI] [PubMed] [Google Scholar]
- 103.Fan N, Yang H, Zhang J & Chen C Reduced expression of glutamate receptors and phosphorylation of CREB are responsible for in vivo Delta9-THC exposure-impaired hippocampal synaptic plasticity. J. Neurochem 112, 691–702 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang H & Zhang M The role of Ca2+-stimulated adenylyl cyclases in bidirectional synaptic plasticity and brain function. Rev. Neurosci 23, 67–78 (2012). [DOI] [PubMed] [Google Scholar]
- 105.Barco A & Marie H Genetic approaches to investigate the role of CREB in neuronal plasticity and memory. Mol. Neurobiol 44, 330–349 (2011). [DOI] [PubMed] [Google Scholar]
- 106.Steel RW, Miller JH, Sim DA & Day DJ Delta-9-tetrahydrocannabinol disrupts hippocampal neuroplasticity and neurogenesis in trained, but not untrained adolescent Sprague-Dawley rats. Brain Res. 1548, 12–19 (2014). [DOI] [PubMed] [Google Scholar]
- 107.Kittler JT et al. Large-scale analysis of gene expression changes during acute and chronic exposure to [Delta]9-THC in rats. Physiol. Genomics 3, 175–185 (2000). [DOI] [PubMed] [Google Scholar]
- 108.Grigorenko E et al. Assessment of cannabinoid induced gene changes: tolerance and neuroprotection. Chem. Phys. Lipids 121, 257–266 (2002). [DOI] [PubMed] [Google Scholar]
- 109.Tantra M et al. St8sia2 deficiency plus juvenile cannabis exposure in mice synergistically affect higher cognition in adulthood. Behav. Brain Res 275, 166–175 (2014). [DOI] [PubMed] [Google Scholar]
- 110.Stringer S et al. Genome-wide association study of lifetime cannabis use based on a large meta-analytic sample of 32 330 subjects from the International Cannabis Consortium. Transl. Psychiatry 6, e769 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Skosnik PD, Cortes-Briones JA & Hajós M It’s all in the rhythm: the role of cannabinoids in neural oscillations and psychosis. Biol. Psychiatry 79, 568–577 (2016). [DOI] [PubMed] [Google Scholar]
- 112.Raver SM & Keller A Permanent suppression of cortical oscillations in mice after adolescent exposure to cannabinoids: receptor mechanisms. Neuropharmacology 86, 161–173 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hajós M, Hoffmann WE & Kocsis B Activation of cannabinoid-1 receptors disrupts sensory gating and neuronal oscillation: relevance to schizophrenia. Biol. Psychiatry 63, 1075–1083 (2008). [DOI] [PubMed] [Google Scholar]
- 114.Hwang EK & Lupica CR Altered corticolimbic control of the nucleus accumbens by long-term Δ9-tetrahydrocannabinol exposure. Biol. Psychiatry S0006–3223(19)31559–8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Morel LJ, Giros B & Daugé V Adolescent exposure to chronic delta-9-tetrahydrocannabinol blocks opiate dependence in maternally deprived rats. Neuropsychopharmacology 34, 2469–2476 (2009). [DOI] [PubMed] [Google Scholar]
- 116.Stopponi S et al. Chronic THC during adolescence increases the vulnerability to stress-induced relapse to heroin seeking in adult rats. Eur. Neuropsychopharmacol 24, 1037–1045 (2014). [DOI] [PubMed] [Google Scholar]
- 117.Solinas M, Panlilio LV & Goldberg SR Exposure to delta-9-tetrahydrocannabinol (THC) increases subsequent heroin taking but not heroin’s reinforcing efficacy: a self-administration study in rats. Neuropsychopharmacology 29, 1301–1311 (2004). [DOI] [PubMed] [Google Scholar]
- 118.Hurd YL, Michaelides M, Miller ML & Jutras-Aswad D Trajectory of adolescent cannabis use on addiction vulnerability. Neuropharmacology 76 Pt B, 416–424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Blanco C, Flórez-Salamanca L, Secades-Villa R, Wang S & Hasin DS Predictors of initiation of nicotine, alcohol, cannabis, and cocaine use: results of the National Epidemiologic Survey on Alcohol and Related Conditions (NESARC). Am. J. Addict 27, 477–484 (2018). [DOI] [PubMed] [Google Scholar]
- 120.Panlilio LV, Zanettini C, Barnes C, Solinas M & Goldberg SR Prior exposure to THC increases the addictive effects of nicotine in rats. Neuropsychopharmacology 38, 1198–1208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hillard CJ, Beatka M & Sarvaideo J Endocannabinoid signaling and the hypothalamic-pituitary-adrenal axis. Compr. Physiol 7, 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harte-Hargrove LC & Dow-Edwards DL Withdrawal from THC during adolescence: sex differences in locomotor activity and anxiety. Behav. Brain Res 231, 48–59 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.O’Shea M, Singh ME, McGregor IS & Mallet PE Chronic cannabinoid exposure produces lasting memory impairment and increased anxiety in adolescent but not adult rats. J. Psychopharmacol 18, 502–508 (2004). [DOI] [PubMed] [Google Scholar]
- 124.Barrus DG, Lefever TW & Wiley JL Evaluation of reinforcing and aversive effects of voluntary Δ9-tetrahydrocannabinol ingestion in rats. Neuropharmacology 137, 133–140 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nguyen JD et al. Inhaled delivery of Δ(9)-tetrahydrocannabinol (THC) to rats by e-cigarette vapor technology. Neuropharmacology 109, 112–120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bruijnzeel AW et al. Behavioral characterization of the effects of cannabis smoke and anandamide in rats. PLoS One 11, e0153327 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Månsson KN et al. Neuroplasticity in response to cognitive behavior therapy for social anxiety disorder. Transl. Psychiatry 6, e727 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yang Z et al. Cognitive behavioral therapy is associated with enhanced cognitive control network activity in major depression and posttraumatic stress disorder. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 3, 311–319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yuan Minlan, et al. Cerebellar neural circuits involving executive control network predict response to group cognitive behavior therapy in social anxiety disorder. Cerebellum 16, 673–682 (2017). [DOI] [PubMed] [Google Scholar]
- 130.Morgan CJ, Schafer G, Freeman TP & Curran HV Impact of cannabidiol on the acute memory and psychotomimetic effects of smoked cannabis: naturalistic study: naturalistic study [corrected]. Br. J. Psychiatry 197, 285–290 (2010). [DOI] [PubMed] [Google Scholar]
- 131.Bergamaschi MM et al. Cannabidiol reduces the anxiety induced by simulated public speaking in treatment-naïve social phobia patients. Neuropsychopharmacology 36, 1219–1226 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Crippa JA et al. Neural basis of anxiolytic effects of cannabidiol (CBD) in generalized social anxiety disorder: a preliminary report. J. Psychopharmacol 25, 121–130 (2011). [DOI] [PubMed] [Google Scholar]
- 133.Murphy M et al. Chronic adolescent Δ9-tetrahydrocannabinol treatment of male mice leads to long-term cognitive and behavioral dysfunction, which are prevented by concurrent cannabidiol treatment. Cannabis Cannabinoid Res. 2, 235–246 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zuardi AW, Shirakawa I, Finkelfarb E & Karniol IG Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology (Berl.) 76, 245–250 (1982). [DOI] [PubMed] [Google Scholar]
- 135.Ren Y, Whittard J, Higuera-Matas A, Morris CV & Hurd YL Cannabidiol, a nonpsychotropic component of cannabis, inhibits cue-induced heroin seeking and normalizes discrete mesolimbic neuronal disturbances. J. Neurosci 29, 14764–14769 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Grimm O et al. Probing the endocannabinoid system in healthy volunteers: Cannabidiol alters fronto-striatal resting-state connectivity. Eur. Neuropsychopharmacol 28, 841–849 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Beale C et al. Prolonged cannabidiol treatment effects on hippocampal subfield volumes in current cannabis users. Cannabis Cannabinoid Res. 3, 94–107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Haney M et al. Oral cannabidiol does not alter the subjective, reinforcing or cardiovascular effects of smoked cannabis. Neuropsychopharmacology 41, 1974–1982 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.White T et al. Paediatric population neuroimaging and the Generation R Study: the second wave. Eur. J. Epidemiol 33, 99–125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Szutorisz H & Hurd YL Epigenetic effects of cannabis exposure. Biol. Psychiatry 79, 586–594 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kawamura Y et al. The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J. Neurosci 26, 2991–3001 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Filbey FM et al. Long-term effects of marijuana use on the brain. Proc. Natl. Acad. Sci. USA 111, 16913–16918 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.De Bellis MD et al. Neural mechanisms of risky decision-making and reward response in adolescent onset cannabis use disorder. Drug Alcohol Depend. 133, 134–145 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nestor L, Hester R & Garavan H Increased ventral striatal BOLD activity during non-drug reward anticipation in cannabis users. Neuroimage 49, 1133–1143 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yücel M et al. Regional brain abnormalities associated with long-term heavy cannabis use. Arch. Gen. Psychiatry 65, 694–701 (2008). [DOI] [PubMed] [Google Scholar]
- 146.Charboneau EJ et al. Cannabis cue-induced brain activation correlates with drug craving in limbic and visual salience regions: preliminary results. Psychiatry Res. 214, 122–131 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Goldman M et al. Reward-related brain response and craving correlates of marijuana cue exposure: a preliminary study in treatment-seeking marijuana-dependent subjects. J. Addict. Med 7, 8–16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Brezing CA & Levin FR The current state of pharmacological treatments for cannabis use disorder and withdrawal. Neuropsychopharmacology 43, 173–194 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Morgan CJ, Freeman TP, Schafer GL & Curran HV Cannabidiol attenuates the appetitive effects of Delta 9-tetrahydrocannabinol in humans smoking their chosen cannabis. Neuropsychopharmacology 35, 1879–1885 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.D’Souza DC et al. Efficacy and safety of a fatty acid amide hydrolase inhibitor (PF-04457845) in the treatment of cannabis withdrawal and dependence in men: a double-blind, placebo-controlled, parallel group, phase 2a single-site randomised controlled trial. Lancet Psychiatry 6, 35–45 (2019). [DOI] [PubMed] [Google Scholar]