

Abstract
Doxorubicin (DOX) is a class of effective chemotherapeutic agents widely used in clinical practice. However, its use has been limited by cardiotoxicity. The mechanism of DOX-induced cardiotoxicity (DIC) is complex, involving oxidative stress, Ca2+ overload, inflammation, pyroptosis, ferroptosis, apoptosis, senescence, etc. Exosomes (EXOs), as extracellular vesicles (EVs), play an important role in the material exchange and signal transmission between cells by carrying components such as proteins and RNAs. More recently, there has been a growing number of publications focusing on the protective effect of EXOs on DIC. Here, this review summarized the main mechanisms of DIC, discussed the mechanism of EXOs in the treatment of DIC, and further explored the value of EXOs as diagnostic biomarkers and therapeutic strategies for DIC.
1. Introduction
The survival duration of cancer patients has increased as medical technology has advanced, yet cardiovascular toxicity has emerged as one of the most significant side effects of cancer treatment [1]. Cancer survivors are eight times more likely than the normal population to suffer cardiovascular disease [2]. DOX is one of the most effective chemotherapeutic agents used in clinical practice, with indications for a wide range of cancers. Nevertheless, its usage has been limited due to the high risk of cardiotoxicity [3], which may be acute, early, or late, including pericarditis, heart failure, and arrhythmia. Studies have shown that the incidence of DOX-induced left ventricular dysfunction ranges from 3% to 48% in a dose-dependent manner [2, 4–6]. Therefore, fully understanding the mechanism of DIC, conducting effective monitoring at an early stage, and taking effective prevention and intervention strategies are the key issues to improve the quality of life of cancer patients.
However, the pathogenesis of DIC is complex, involving oxidative stress, inflammation, Ca2+ dysregulation, senescence, apoptosis, pyroptosis, ferroptosis, etc. [7–9] Besides, there is currently a lack of biomarkers for early diagnosis of DIC that are both specific and sensitive [10]. Although dexrazoxane is the only cardioprotective agent recommended by the FDA to reduce DIC, its application is only for adults with advanced or metastatic breast cancer who have received a cumulative dose of >300 mg/m2 DOX [4, 11], and its protective effect has been questioned in some studies [12].
EXOs are EVs with a diameter of 50 nm to 150 nm formed by cells through a series of regulatory processes (endocytosis, fusion, and excretion), which play an important role in cell communication and tissue microenvironment regulation [13]. Increasing evidence suggests that some components in EXOs, such as miRNAs, lncRNAs, and proteins, are transferred into cardiomyocytes and exert cardioprotective effects through different signaling pathways [14–16].
In this review, we outline the evidence on EXOs' mechanisms for mitigating DIC. As well, we deliver an overview regarding the diagnostic biomarkers and therapeutic effects of EXOs, hoping to provide a theoretical basis for the clinical application of EXOs.
2. Introduction to EXOs and EVs
EVs are membrane-derived vesicles ranging from 50 nm to 2,000 nm in diameter released by cells into the extracellular space [17]. All different types of cells in mammals including neuronal cells, endothelial cells (ECs), mesenchymal stem cells (MSCs), and epithelial cells can release EVs, and EVs are widely distributed in the body and can be detected in urine, blood, saliva, and other body fluids [18]. EXOs are EVs with a size range of ~40 to 160 nm (average ~100 nm) in diameter with an endosomal origin. Depending on the cell of origin, EVs, including EXOs, can contain many constituents of a cell, including DNA, RNA, lipids, metabolites, and cytosolic and cell surface proteins [19].
Generally speaking, EXOs originate from the endosomal pathway. First, the cytoplasmic membrane invaginates to form early-sorting endosomes (ESEs) containing some extracellular components and membrane surface proteins. The contents of the ESEs are obtained by fusing with the previously existing ESEs or exchanging substances with other organelles. ESEs then develop further within the cell into late-sorting endosomes (LSEs). LSEs can eventually form multivesicular bodies (MVBs), which contain intraluminal vesicles (ILVs) that are formed by budding from the inward depression of the multivesicular body membrane. The fusion of MVBs and the cytoplasmic membrane leads to the secretion of ILVs outside the cell, and these ILVs are EXOs [20] (Figure 1).
Figure 1.
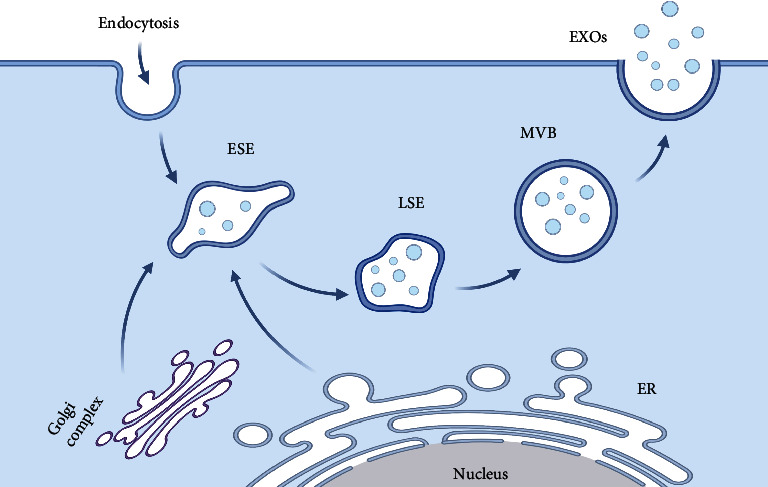
The biosynthesis of EXOs. ESE: early-sorting endosome; LSE: late-sorting endosome; MVB: multivesicular body; EXOs: exosomes; ER: endoplasmic reticulum.
EXOs have different sizes, contents, and origins, leading to complex heterogeneity. EXOs are rich in proteins, lipids, miRNAs, and other substances, and the contents enriched in different EXOs are different [21]. Differences in EXO composition, especially in cell surface proteins, can have different effects on recipient cells. EXOs can mediate intercellular communication under physiological and pathological conditions [22]. Parental cells that released EXOs are absorbed by receptor cells, through the exchange of substances or release of inclusions to achieve the exchange of substances and signals, to play an important role in reproductive development, immune response, disease occurrence, and other biological processes [18, 19, 21, 23]. Studies have shown that EXOs are closely related to cardiovascular diseases, neurodegenerative diseases, tumor growth and metastasis, and drug resistance [19, 24, 25].
3. The Mechanism of DIC
The mechanism of DIC is complex, and there is no clear mechanism to explain it. Multiple pathways may be involved, including oxidative stress, apoptosis, autophagy, pyroptosis, ferroptosis, and senescence, which are thought to be interconnected and act together to cause myocardial damage [26]. Herein, we have summarized molecular mechanisms which are involved in DIC briefly (Figure 2).
Figure 2.
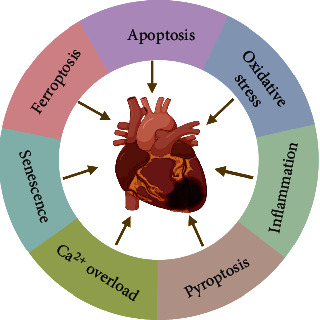
The main mechanisms of DIC.
3.1. Oxidative Stress
The oxidative stress hypothesis involving the intramyocardial production of reactive oxygen species (ROS) has gained the most widespread acceptance [27, 28]. Generally speaking, under the action of nitric oxide synthase (NOS) and NADPH oxidase (NOX), DOX is reduced to semiquinone DOX, which reduces oxygen to superoxide anion; this radical is converted into hydrogen peroxide under the action of superoxide dismutase (SOD). Hydrogen peroxide can be cleaved into hydroxyl radicals [28–32]. The above-mentioned ROS, especially hydroxyl radicals, are active and highly toxic, which can cause lipid peroxidation, thereby destroying the biofilm structure and causing myocardial cell damage and death.
3.2. Apoptosis
The role of apoptosis in DIC is well-established. Many studies have shown that DOX can activate cardiomyocytes to undergo apoptosis. DOX upregulated the expression of heat shock protein 25 (HSP25), which transactivated p53, leading to the expression of Bax, therefore inducing the apoptotic death of cardiomyocytes [33]. DOX also activated caspase-3 [34, 35]. Besides, DOX can induce cardiomyocyte apoptosis by activating the nuclear factor kappa-B (NF-κB) signaling pathway and producing ROS [36–38]. DOX significantly upregulated the expression of death receptors (DRs) (TNFR1, Fas, DR4, and DR5) and subsequently induced apoptosis in iPS-derived cardiomyocytes, and the apoptosis could be accelerated by physiologically relevant death ligands including TNF-related apoptosis inducing ligand (TRAIL) [39]. DOX also downregulated caspase recruitment domain ARC, leading to Bax translocated from the cytosol to mitochondria, resulting in loss of mitochondrial membrane potential, which led to cytochrome C release, thus inducing apoptosis [40].
3.3. Inflammation
Inflammation also plays a role in DOX-triggered cardiac injury. DOX can increase the levels of proinflammatory cytokines such as interleukin- (IL-) 1β, IL-18, IL-6, and tumor necrosis factor-alpha (TNF-α) [41, 42], and their elevations are associated with the activation of the p38/MAPK/NF-κB pathway [41, 43]. Besides, improving nuclear factor erythroid2-related factor 2 (Nrf2) signaling protected the heart from NF-κB-mediated inflammatory injury [43]. One study suggested that DOX could cause upregulation of the proinflammatory toll-like receptor 4 (TLR4) in macrophages and endotoxin leaking from gut flora into the circulation, which combined to cause a systemic inflammatory response [44]. In addition, DOX can cause inflammation by activating the sirtuin 1-NOD-like receptor protein 3 pathway [45].
3.4. Pyroptosis
Pyroptosis in DIC is evidenced by increased cell death, upregulated expression levels of NLR family pyrin domain containing 3 (NLRP3), caspase-3, IL-1β, IL-18, and GMDSD-N, and morphological features [46–50]. Mechanistically, DOX upregulated the lncRNA TINCR, which can recruit IGF2BP1 to upregulate the expression of NLRP3 to induce pyroptosis [46]. In addition, by upregulating BH3-only protein Bcl-2/adenovirus E1B 19-kDa-interacting protein 3 (Bnip3), DOX induced the activation of caspase-3, causing GSDME-dependent pyroptosis [48].
3.5. Senescence
In recent years, many studies have demonstrated that low doses of DOX (≤0.5 μM) preferentially induce cardiovascular cell senescence, rather than apoptosis [51–53]. In general, the mechanism of DOX-induced cardiovascular cell senescence involves oxidative stress, telomere damage, DNA damage, etc. In rat neonatal cardiomyocytes, low levels of DOX can induce senescence through oxidative stress [54]. It can also lead to telomere damage through p38-mediated reduction of telomere binding factors 2 (TRF2) and JNK/p53-mediated reduction of telomere binding factors 1 (TRF1), thereby inducing senescence [55]. 0.25, 0.5, and 1 μM DOX can induce human primary vascular smooth muscle cell senescence [53, 56, 57]. Mechanistically, DOX upregulated uPAR, which led to TRF2 ubiquitination and proteasomal degradation, leading to senescence [56] and DOX-mediated elevation of ROS also aggravated senescence [57]. In addition, 0.25 μM DOX activated MAPK-p38 to induce p16 (INK4A) expression and cytoskeleton remodeling, therefore inducing senescence [58]. In ECs, DOX induced senescence by upregulating p53-dependent XIAP-associating factor 1 expression [59]. In addition to the above in vitro and in vivo models, studies have shown that human cardiac progenitor cells also exhibited aging characteristics in DIC patients [60].
3.6. Ferroptosis
Ferroptosis is a new form of cell death proposed by Stockwell in 2012 [61]. Many scholars have found that DOX can induce ferroptosis in cardiomyocytes [47, 62]. Preadministration of ferroptosis inhibitors largely prevents DIC [62], whereas a high-iron diet can exacerbate DIC [63]. In general, the mechanism of DOX-induced ferroptosis can be divided into two aspects: induction of iron overload in cardiomyocytes and lipid peroxidation. Specifically, DOX can induce iron overload in cardiomyocytes by upregulating the level of transferrin receptor [64, 65] and heme oxygenase 1-mediated heme degradation [62]. DOX can also regulate iron metabolism-related genes by acting on iron-responsive elements/iron-responsive proteins [66–68]. Besides, the quinine moiety of DOX accepted electrons from NOX and NOS to become semiquinone, which was accompanied by the production of ROS [29], and DOX can inhibit the activity of intracellular glutathione peroxidase 4 (GPX4), reducing its antioxidant capacity [69, 70]. Free iron complexes with DOX and through the Fenton reaction create more ROS, thereby inducing ferroptosis. To go a step further, some scholars have focused their attention on mitochondria. They suggested that mitochondria are the main site of DOX-induced ferroptosis [71–73] and that targeting mitochondrial antioxidants can inhibit DIC [62]. DOX can cause mitochondrial iron overload by affecting mitochondrial ferritin [74], ABC protein-B8 [71, 75], frataxin [76], etc. and mitochondrial GPX4 is more important in antiferroptosis than cytoplasmic GPX4 [77].
3.7. Ca2+ Dysregulation
DOX can also induce DIC by affecting Ca2+ homeostasis in cardiomyocytes. DOX can increase the Ca2+ intake by increasing the current of L-type calcium channel, activate the RyR2 receptor to increase the Ca2+ release from sarcoplasmic reticulum (SR), inhibit SERCA2A to reduce the Ca2+ reuptake of SR, and activate CaMKII to cause SR Ca2+ leakage, which lead to the accumulation of intracellular Ca2+, thereby inducing DIC [78–82]. One study suggested that the degradation of titin caused by the accumulation of intracellular Ca2+ through the activation of calpain is an early event in DIC [83]. In addition, DOX-induced Ca2+ disturbance is closely related to ROS and apoptosis. Elevated ROS induced by DOX lead to intracellular Ca2+ overload. Accumulated Ca2+ results in nuclear translocation of NFAT by activating calcineurin, which activated Fas/FasL-mediated apoptosis, while DOX-induced apoptosis can be reversed by Ca2+ chelator and antioxidant [84]. Besides, increased Ca2+ also induced cardiomyocyte apoptosis by activating calcium-dependent CaMKII [85].
4. The Mechanism of EXOs against DIC
In recent years, it has been demonstrated that EXOs harbor a variety of miRNAs, lncRNAs, and proteins which may be transferred to cardiomyocytes through cell endocytosis or membrane fusion and modulate their function [10, 15, 86]. Different components contained in EXOs from different parent cells can inhibit DIC by acting on different signaling pathways. The mechanisms of EXOs against DIC are as follows:
4.1. Antiapoptosis
Several studies have shown that EVs can alleviate DOX-induced cardiomyocyte apoptosis (Figure 3). Current studies mostly focus on EXO-loaded miRNAs. Generally speaking, EXOs acted as miRNA carriers, delivering miRNAs from parental cells to cardiomyocytes, altering the expression of their target genes and thus exerting antiapoptotic effects. EXOs derived from trophoblast stem cells (TSC-EXOs) are abundant in let-7i, which could exert antiapoptosis and antifibrosis effects in DOX-induced dilated cardiomyopathy (DCM) models both in vivo and in vitro by inhibiting the yes associated protein (YAP) signaling pathway. In let-7i mimic-treated cardiomyocytes group, apoptosis-related biomarkers Annexin V and cleaved caspase 3 were decreased, the expression of Bcl2 was increased, and related biomarkers (YAP1, CTGF, and TEAD1) in the YAP signaling pathway were inhibited, and myocardial fibrosis in pathological staining is mitigated [87]. Ni et al. demonstrated that TSC-EXOs protected the heart from DOX-induced apoptosis through the miR-200b/zinc finger E-box-binding protein 1 (Zeb-1) pathway. Specifically speaking, TSC-EXOs downregulated the expression of miR-200b in cardiomyocytes, thereby increasing the transcription of Zeb1. However, the mechanism by which TSC-EXOs downregulated miR-200b was still unclear, and they thought that it might be related to the lncRNAs in EXOs [88]. EVs, acting as miRNA carriers, can also carry miRNAs to target cells [89, 90]. MicroRNAs encapsulated in EVs are the important genetic material that drives cardiac repair. There was a study that demonstrated treatment with EVs derived from MSCs (MSC-EVs) before DOX treatment enhanced H9C2 cell viability. Mechanistically, miR-199a-3p in MSC-EVs upregulated p-Akt levels, thereby inhibiting the activation of transcription factor P53 and promoting the activation of Sp1, thus increasing the expression of antiapoptotic factor survivin to alleviate DIC [91]. One study showed that miR-100-5p in MSC-EVs exerted antiapoptotic effects in AC16 cells by inhibiting the expression of its target gene NOX4. This protective effect of MSC-EVs was reversed when MSC-EVs were transfected with miR-100-5p inhibitors or NOX4 was overexpressed [92].
Figure 3.
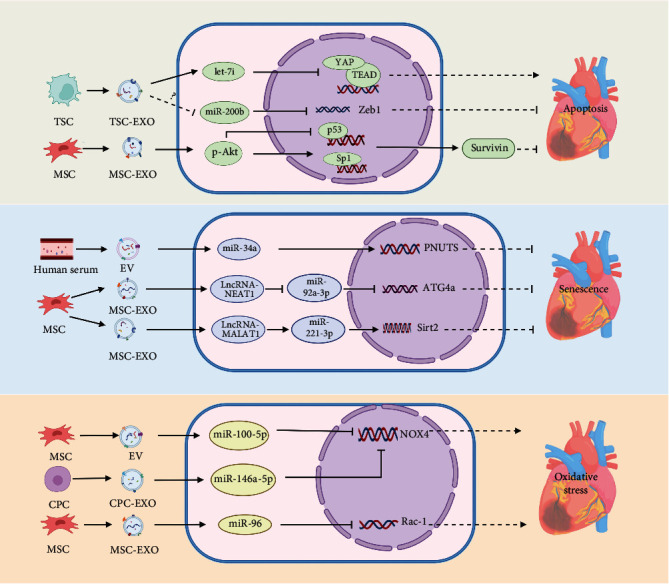
The mechanisms of EXOs against DIC involved in antiapoptosis, antisenescence, and antioxidative stress. TSC: trophoblast stem cell; TSC-EXO: exosome derived from trophoblast stem cell; MSC: mesenchymal stem cell; MSC-EXO: exosome derived from mesenchymal stem cell; YAP: yes associated protein; Zeb1: zinc finger E-box-binding protein 1; Sp1: specificity protein 1; EV: extracellular vesicle; PNUTS: phosphatase 1 nuclear targeting subunit; ATG4a: autophagy-related genes 4a; Sirt2: silent information regulator 2; CPC: cardiac progenitor cell; CPC-EXO: exosome derived from cardiac progenitor cell; NOX4: NADPH oxidase 4; Rac1: ras-related C3 botulinum toxin substrate 1.
4.2. Antisenescence
The mechanism by which EXOs alleviate DIC can be achieved by the antisenescence of cardiomyocytes, which is characterized by more cells escaping from the G0/G1 phase, the decreased expression of the cellular senescence-related genes p27, p53, p21, and p16, a lower percentage of SA-β-gal-positive cells, and the increase in telomere length and activity [93–95]. There was a study showed that it involved the EXOs/lncRNA–NEAT1/miR-221-3p/Sirt2 pathway. Abundant lncRNA–NEAT1 in EXOs derived from MSCs pretreated with macrophage migration inhibitory factor (MIF) (MSC-EXOsMIF) can improve the content of Sirt2 by targeting miR-221-3p, thus playing the antisenescence role. Moreover, siRNA-lncRNA-NEAT1 and miR-221-3p mimic transfection blocked the protective effect of MSC-EXOsMIF [95]. Xia et al. suggested that EXOs secreted by MSCs pretreated with hypoxia (MSC-EXOshypoxia) exerted antisenescence effects through the lncRNA-MALAT1/miR-92a-3p/ATG4a axis. MSC-EXOshypoxia transferred lncRNA-MALAT1 to cardiomyocytes, reducing the expression of targeted miR-92a-3p through ceRNA mechanisms, upregulating the expression of the ATG4a gene, thus exerting a rejuvenation effect. In addition, MSC-EXOshypoxia can also improve the mitochondrial metabolic disorder caused by DOX, which is manifested as the decrease of Fabp3, Fabp4, and Mtfp1 and the increase of Cox4i2, HSPa1a, and Atp1b2. After the lncRNA-MALAT1 knockdown, miR-92a-3p overexpression, or silencing of ATG4a, the above two protective mechanisms of MSC-EXOshypoxia were inhibited. Notably, cardiomyocytes without hypoxic preconditioning were slightly protective, and hypoxic preconditioning enhanced the cardioprotective effect of MSC-EXOs [93]. Besides, Liu et al.'s research suggested that human serum EVs exerted the antiaging effect in DIC H9C2 cells models. It was achieved by suppressing the expression of miR-34a and promoting the expression of its target gene phosphatase 1 nuclear targeting subunit (PNUTS). The miR-34a mimics and silence PNUTS can reverse the antiaging effect of EVs [94]. (Figure 3).
4.3. Antioxidative Stress
EXOs can combat DOX-induced oxidative stress (Figure 3). This was related to the antioxidant proteins and miRNAs contained in EXOs. Proteomics analysis revealed that EXOs from human right cardiac atrial appendage tissue contained more than 70 proteins involved in redox processes, especially SOD2, thrombospondin 1, and collagen 1A1 [96]. miR-96 in MSC-EXOs protected the heart from oxidative stress both in vivo and in vitro, and this effect was achieved through the inhibition of its target gene Rac-1. Compared with the EXOs+miR-96 inhibitor group and NC siRNA group, SOD and GSH-Px were increased, and malondialdehyde was decreased [97]. In addition, it was suggested that MSC-EVs inhibited DOX-induced oxidative stress in AC16 cells through the downregulation of NOX4 induced by miR-100-5p. When MSC-EVs were transfected with miR-100-5p inhibitors or NOX4 was overexpressed, the antioxidant effect was rescued [92].
4.4. Anti-inflammation and Antipyroptosis
There have been many studies showing that EXOs can improve the inflammatory microenvironment of cardiomyocytes and reduce the production of proinflammatory mediators (such as TNF-α, IL-1β, IL-1, and IL-6) [49, 87, 88, 96–99]. CPC-EXOs derived from human cardiac atrial appendage specimens were rich in miR-146a-5p and can enter cardiomyocytes, thereby inhibiting the transcription of their target genes Traf6 and Irak-1 and reducing the infiltration of CD68+ inflammatory cells [96]. In DOX-induced DCM mice models, after being injected with EVs derived from ECs overexpressing Krüppel-like factor 2 (EC-EVsKLF2), the levels of proinflammatory cytokines (IL-1β and TNF-α) were decreased, and the levels of anti-inflammatory cytokine IL-10 were increased, due to its ability to inhibit C-C chemokine receptor 2-mediated the migration of Ly6Chigh Mo/Mø in bone marrow [99]. Besides, in the same mice models, MSC-EXOs could also alleviate the inflammatory environment. Mechanistically, MSC-EXOs decreased proinflammatory Ly6Chigh monocytes and increased anti-inflammatory Ly6Clow macrophages by activating the transcription factor JAK2 and its downstream STAT6 [98]. In addition, a study showed that the anti-inflammatory effect of MSC-EXOs was related to their loaded miR-96, which inhibited DOX-induced activation of the Rac1 gene in cardiomyocytes, thereby reducing TNF-α, IL-6, and IL-1β [97].
As an inflammatory programmed cell death, pyroptosis is also involved in the occurrence and development of DIC. Researches were suggesting that embryonic stem cell-derived EXOs (ESC-EXOs) inhibited the activation of TLR4 and inhibit the formation of NLRP3, thereby improving the inflammatory environment and protecting cardiomyocytes from pyroptosis both in the DIC mice models and H9C2 cells models [49, 100]. These protective effects may be related to ESC-EXOs containing more anti-inflammatory cytokines (IL-4, IL-9, and IL-13) and fewer proinflammatory cytokines (TNF-α, TNFR1, IL-12, and Fas ligand) [49]. Singla et al. believed that the inflammatory microenvironment caused by DOX was related to the activation of the MAPK signaling pathway, and ESC-EXOs could inhibit the activation of these signaling pathway proteins (MyD88, p-P38, and p-JNK). In addition, the anti-inflammatory effect of ESC-EXOs may also be related to promoting the conversion of MI to M2 macrophages [100] (Figure 4).
Figure 4.
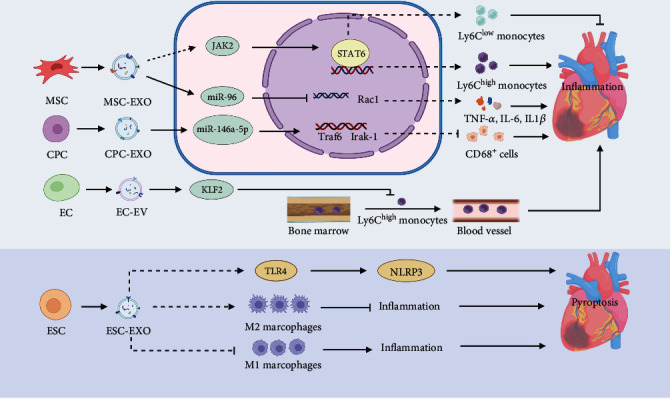
The mechanisms of EXOs against DIC involved in anti-inflammation, and antipyroptosis. MSC: mesenchymal stem cell; MSC-EXO: exosome derived from mesenchymal stem cell; CPC: cardiac progenitor cell; CPC-EXO: exosome derived from cardiac progenitor cell; EC: endothelial cell; EC-EV: extracellular vesicle derived from endothelial cell; JAK2: janus kinase 2; STAT6: signal transducer and activator of transcription 6; Rac1: ras-related C3 botulinum toxin substrate 1; TNF-α: tumor necrosis factor-alpha; IL-6: interleukin-6; IL-1β: interleukin-1β; ESC: embryonic stem cell; ESC-EXO: exosome derived from embryonic stem cell; TLR4: toll-like receptor-4; NLRP3: NLR family pyrin domain containing 3; KLF2: Krüppel-like factor 2.
The mechanisms underlying DIC are complicated; in addition to the above mechanism, autophagy, topoisomerase 2β inhibition, necroptosis, etc. are also involved [47, 101, 102]. However, unfortunately, there are no relevant studies on the protective effects of EXOs or EVs from DIC by regulating these mechanisms.
5. EXOs as Diagnostic Biomarkers for DIC
DIC can be divided into early-onset and late-onset. For early-onset, it is mostly reversible. Early detection and active intervention can prevent irreversible cardiac damage [4]. For delayed cardiotoxicity, if DIC is detected and intervened early, the patient is likely to have good functional recovery [103]. Strategies for screening and detection of cardiotoxicity include echocardiography, nuclear imaging, cardiac magnetic resonance, and biomarkers (troponin and natriuretic peptides) [4, 10]. The Cardio-Oncology Study Group of the Heart Failure Association and the Cardio-Oncology Council of the European Society of Cardiology recommend high-sensitivity troponin (hs-cTN) as a biomarker for early DIC because it can sensitively identify early myocardial damage caused by DOX and predict left ventricular dysfunction [104]. However, the specificity of hs-cTN in the diagnosis of DIC is not satisfactory, as it can also be elevated in other cases, such as hypertensive emergencies and renal failure [105, 106]. And the increase in troponin means that the cardiomyocytes have been damaged. Therefore, biomarkers, which are more sensitive and specific than troponin, need to be explored.
In recent years, EXOs have attracted the attention of many scholars as biomarkers for disease diagnosis. Firstly, EXOs are existed almost in all biological fluids and can be secreted by almost all cells, so it is theoretically possible to isolate EXOs from a patient's body fluids, such as serum or urine, for the diagnosis of disease. Secondly, the molecular characteristics of cargoes in EXOs reflect the phenotype of the cells from which they originated. Thirdly, the biomarkers in EXOs are more stable due to the encapsulation of the plasma membrane [19, 23, 107].
Table 1 shows potential EXOs or EVs cargoes that could be used as biomarkers for DIC. After DOX treatment, damaged myocardial tissue released EVs containing brain/heart glycogen phosphorylase (PYGB) into the blood. It can serve as a potential early biomarker of DIC that is more sensitive than cTnI, which could be detected in serum by proteomics. Yarana et al. and Zhu and Gius demonstrated significant differences in EVs-PYGB between saline and DOX-treated mice as early as 24 hours after treatment, whereas differences in cTnI were not detected until 72 hours after treatment in DIC mice models [107, 108]. Besides, in the DIC dog models, DOX caused changes in the expression profiles of miRNAs in circulating EXOs, as shown by miR107 and miR-146a which were significantly decreased, and the level of miR-502 was increased. Notably, during a total of 5 DOX treatments, the elevation of miR-502 appeared before the 3rd treatment, which was earlier than the changes in cTnI and echocardiographic parameters. Therefore, miR-502 in circulating EXOs can serve as a potential biomarker for DIC [109]. In addition, Li et al. thought that exosomal circ-SKA3 can be a candidate biomarker for DIC. After being exposed to 5 μM DOX, human cardiomyocyte-like AC16 cells secrete EXOs enriched in circ-SKA3 internalized by recipient AC16 cells by docking and fusing to the cytomembrane, which can enhance cardiotoxicity via the miR-1303/TLR4 axis [110].
Table 1.
Summary of studies using EXOs/EVs as biomarkers for DIC diagnosis.
| EVs type | Component | Parts of EXOs/EVs | Study model | DOX administration | Distribution | Ref |
|---|---|---|---|---|---|---|
| EXOs | circ-SKA3 | circRNA | Human cardiomyocyte-like AC16 cells | 5 μM for 24 h | — | [110] |
| EVs | PYGB | Protein | Male C57BL/6J mice | 20 mg/kg, IP; a single dose | Serum | [107] |
| EXOs | miR-502 | miRNA | Dogs diagnosed with sarcoma | 30 mg/m2 for dogs > 15 kg and 1 mg/kg for dogs < 15 kg, IV; 2 to 3 weeks for 5 injections | Serum | [109] |
EXOs: exosomes; EVs: extracellular vesicles; PYGB: brain/heart glycogen phosphorylase; IP: intraperitoneal injection; IV: intravenous injection.
Components such as proteins, lipids, RNA, and miRNAs in EXOs may serve as diagnostic and harbingers of DIC. However, there are few related studies in this field at present, and we have only found a few articles. Moreover, there is a lack of relevant studies of serum EXOs in humans. In addition, it is unclear whether different tumor types and different underlying cardiac function states of patients affect the changes in the content of EXOs derived from cardiomyocytes in serum.
6. Prevention and Treatment of EXOs for DIC
The strategies of EXOs to alleviate DIC can be summarized into two major aspects: first, EXOs as nonimmunogenic nanosized vesicles (EXOs-DOX) improve the delivery efficiency of DOX and the uptake capacity of tumor cells to DOX, enhancing the anticancer effect of DOX, thereby reducing the dosage of DOX, indirectly reducing DIC; second, some contents in EXOs can directly act on the heart, inhibit cardiac damage caused by DOX, and directly treat DIC [10]. There are many studies on EXOs enhancing DOX's anticancer efficacy [24, 111–114], and the therapeutic part of this article focuses on the direct effect of the EXOs described above, as well as the direct evaluation of cardiotoxicity in the article on the role of EXOs as DOX delivery carriers (Table 2).
Table 2.
Summary of studies using EXOs/EVs for the treatment of DIC.
| Parent cell | EVs type | Cargo | Cargo formulation | Study model | Antitumor parameters | Cardiotoxicity parameters | Ref |
|---|---|---|---|---|---|---|---|
| CSCs | EXOs | — | EXOs | In vivo (mice); in vitro (NRCMs) | NA | EF↑, FS↑, apoptosis↓, fibrosis↓, immune response↓ | [115] |
| MSCs | EXOs | DOX | EXOs-DOX | In vivo (MG63 cells, H9C2 cells) | MG63 cells: cell uptake rate↑, cell viability↓ | H9C2 cells: cell viability↑ | [122] |
| MDA-MB-231, STOSE, MDAMB-231 CD63-GFP and STOSE CD63-GFP cell lines | EXOs | DOX | EXOs-DOX | In vivo (mice); in vitro (human myocardial endothelia cells) | Maximum tolerated dose of DOX↑, tumor volume↓ | The ability to cross a reconstructed myocardial endothelial monolayer↓, vacuoles↓, and myofibril disorganization↓ in H&E staining | [124] |
| MDA-MB-231 and HCT-116 cell lines | EXOs | DOX | EXOs-DOX | In vivo (mice); in vitro (MDA-MB-231 cells) | Cell viability↓, tumor volume↓ | Distribution of DOX in the heart↓, no cardiac damage in H&E staining | [123] |
| HEK-293 cells | EXOs | miR-21a | EXOssiClathrin, followed by EXOsmiR-21a | In vivo (mice) | NA | miR-21a-5p expression in heart↑, EF↑, FS↑ | [120] |
| HEK-293 cells | EVs | Cx43, DOX | EVsCx43+-DOX | In vivo (mice); in vitro (4T1luc2 cells) | Cell viability↓, cell proliferation↓, cell motility↓, colony formation↓, tumor growth↓, apoptosis↑ | Fibrosis↓, histopathological changes↓, COX-2↓, HSP25↓ | [121] |
| NA | EXOs | miR-21 | UTMD+EXOsmiR-21 | In vivo (mice) | NA | miR-21 delivery efficiency↑, EF↑, E/A value↑ | [126] |
| LIM1215 cells | EXOs | DOX | A33Ab-US- EXOs-DOX | In vivo (mice); in vitro (LIM1215 cells) | Cell uptake rate↑, cell viability↓, half-maximal inhibitory concentrations↓, necrosis↑, apoptosis↑, tumor volume↓ | Apoptosis↓, cardiac damage↓ | [127] |
| Mouse immature dendritic cells | EXOs | DOX | iRGD-EXOs-DOX | In vitro (MDA-MB-231 cells); in vivo (mice) | DOX delivery efficiency↑, cell viability↓, tumor volume↓ | CK-MB↓, AST↓, no cardiac damage in H&E staining | [129] |
CSCs: cardiac stem cells; EXOs: exosomes; NRCMs: neonatal rat cardiomyocytes; MSCs: mesenchymal stem cells; Cx43: the gap junction protein connexin43; UTMD: ultrasound-targeted microbubble destruction; FS: shortening fraction; EF: ejection fraction; COX-2: cyclooxygenas-2; HSP25: heat shock protein 25; CK-MB: creatine kinase-MB; AST: aspartate transaminase; A33Ab-US-EXOs-DOX: DOX loaded in exosomes coated surface-carboxyl superparamagnetic iron oxide nanoparticles with A33 antibodies.
In acute DOX-induced DCM mice models, after being injected with human cardiac stem cell-derived EXOs (CSC-EXOs), the impaired heart function was improved, showing that both shortening fraction (FS) and ejection fraction (EF) were recovered. In addition, TUNEL staining showed that CSC-EXOs could inhibit cardiomyocyte apoptosis, Masson's trichrome staining revealed that CSC-EXOs could inhibit cardiomyocyte fibrosis, and H&E staining showed that CSC-EXOs could inhibit cardiac immune response [115].
miR-21a-5p was considered to be a potent cardiac protective miRNA in multiple studies. EXOs loaded with miR-21a-5p can be used to protect the heart from DOX damage [116–118]. However, the liver and spleen preferentially ingest EXOs and reduce their protective effects attributed to the mononuclear phagocyte system [114, 119]. Wan et al. demonstrated that clathrin played an important role in endocytosis by macrophages. Based on this, they invented a new two-step strategy to prevent DIC. Prior injection of EXOsblocking (EXOs encapsulated with siClathrin) reduced the localization of EXOs in the liver and spleen while improved the cardiomyocyte localization, therefore strengthening the beneficial effect of EXOstherapeutic (EXOs encapsulated with miR-21a-5p) in DIC model (5 mg/kg, IP; every week for 4 injections) which was characterized by higher EF and FS [120]. Martins-Marques et al. demonstrated that the gap junction protein connexin43 (Cx43) in EVs produced by HEK-293 cells was essential to reduce DIC because it formed a channel that accelerated the release of intravesical content into cardiomyocytes. Compared with EVsCx43--DOX treatment, EVsCx43+-DOX increased the transverse diameter of cardiomyocytes in HE staining, decreased fibrosis in Sirius staining, and downregulated oxidative stress indicators COX2 and HSP25 [121]. Wei et al. believed that MSC-EXOs as DOX carriers enhanced cellular uptake efficiency of DOX in osteosarcoma MG63 cells, while its effect on H9C2 cardiomyocytes exhibited opposite effects which were manifested by higher cell viability and IC50 [122]. One research has shown that DOX in EXOs originated from MDA-MB-231 and HCT-116 cell lines have a lower ability to cross human myocardial ECs than free DOX, which resulted in less DOX accumulation in the heart significantly [123, 124].
To further enhance the anticancer effect of EXOs-DOX, some medical technologies have been applied to EXOs. miR-21 in EXOs alleviated DIC [120, 125]. Sun et al. isolated EXOs from mouse plasma by centrifugation and then loaded miR-21 into EXOs by electroporation to alleviate DIC. By using ultrasound targeted microbubble destruction (UTMD), the delivery efficiency of miR-21 in EXOs to the heart was significantly enhanced, as manifested by the increased EF and E/A ratio [126]. Li et al. demonstrated EXOs coated with surface-carboxyl superparamagnetic iron oxide nanoparticles (US) with A33 antibodies (A33Ab-US), also known as A33Ab-US-EXOs-DOX, enhanced the targeting ability of DOX to A33-positive colorectal cancer cells. Compared with free DOX and EXOs-DOX, A33Ab-US-EXOs-DOX was more effective against cancer both in vivo and in vitro, manifested by higher cell uptake rates, lower cell viability, lower half-maximal inhibitory concentrations, more necrosis and apoptosis of cancer cells, and smaller tumor volumes. More importantly, H&E staining showed no significant cardiotoxicity [127]. Quah and O'Neill transfected immature dendritic cells which lacked immunostimulatory markers on their surface (such as CD40, CD86, MHC-I, and MHC-II) [128] via iRGD-lamp2b to obtain EXOs containing lamp2b, which loaded DOX (iRGD-EXOs-DOX) targeted metastatic breast cancer tissues, achieving more precise and effective anticancer effects both in vivo and in vitro. Besides, iRGD-EXOs-DOX had fewer cardiac side effects than free DOX and empty EXOs loaded with DOX, manifesting as lower levels of CK-MB and AST, and H&E staining showed no significant pathological damage [129].
As nanoscale biofilm structures, therapeutic EXOs can be produced by patients themselves without immune responses. In addition, the special structure of EXOs enables them to be engineered to be loaded with therapeutic miRNAs, proteins, or DOX, making them an ideal drug carrier for the prevention and treatment of DIC. In addition, despite the great promise of EXOs for the treatment of DIC, there are some challenges. Different cell-derived EXOs have different characteristics and functions, and the relationship between EXOs' subsets and DIC efficacy needs to be further explored. In addition, the efficient isolation of EXOs, the standardization of EXO preparations, and the determination of the effective dose of therapeutic EXOs are also key issues that need to be solved urgently in the process of clinical transformation of EXOs [10, 18, 19].
7. Conclusions and Perspectives
In conclusion, this review highlights and summarizes current studies regarding the role of EXOs in DIC, with a focus on cardioprotection. Different cell-derived EXOs can inhibit DOX-induced oxidative stress, inflammation, senescence, apoptosis, and pyroptosis. Besides, some components in EXOs play an important role in the occurrence and development of DIC. By detecting these components, DIC can be diagnosed sensitively and specifically; that is, EXOs have the potential to serve as DIC biomarkers. As a natural low-immunogenic DOX delivery carrier, EXOs can improve the loading rate of DOX, increase the uptake rate of DOX by tumor cells, exert a stronger antitumor effect, and reduce the dosage of DOX, thus indirectly reducing DIC. In addition, EXOs-DOX can reduce the uptake of DOX by cardiomyocytes and the distribution of DOX in cardiomyocytes, thereby directly reducing cardiotoxicity. It is conceivable that EXOs hold the excellent prospect for diagnosis and treatment soon with further research.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number 82174349), the CACMS Innovation Fund (grant number CI2021A00919), and the National Key R&D Program of China (grant numbers 2018YFC1704901 and 2018YFC1704900). We would like to thank Biorender (http://biorender.com) for providing an easy tool to make figures.
Contributor Information
Yuchen Jiang, Email: jiangyuchenxxn@163.com.
Yanwei Xing, Email: xingyanwei12345@163.com.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Authors' Contributions
XYW and JYC designed this study; ZGX wrote the first draft of this manuscript and created figures; YXY searched the literature; SX and AN participated in discussions and improved pictures related to the manuscript; YF edited the language and revised the manuscript; LXY critically revised and approved the final manuscript. All authors contributed to the article and approved the submitted version.
References
- 1.Curigliano G., Lenihan D., Fradley M., et al. Management of cardiac disease in cancer patients throughout oncological treatment: ESMO consensus recommendations. Annals of Oncology . 2020;31(2):171–190. doi: 10.1016/j.annonc.2019.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cardinale D., Colombo A., Lamantia G., et al. Cardio-oncology: a new medical issue. Ecancermedicalscience . 2008;2:p. 126. doi: 10.3332/ecancer.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ewer M. S., Ewer S. M. Erratum: Cardiotoxicity of anticancer treatments. Nature Reviews. Cardiology . 2015;12(11):p. 620. doi: 10.1038/nrcardio.2015.133. [DOI] [PubMed] [Google Scholar]
- 4.Zamorano J. L., Lancellotti P., Rodriguez Muñoz D., et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC) European Heart Journal . 2016;37(36):2768–2801. doi: 10.1093/eurheartj/ehw211. [DOI] [PubMed] [Google Scholar]
- 5.Swain S. M., Whaley F. S., Ewer M. S. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer . 2003;97(11):2869–2879. doi: 10.1002/cncr.11407. [DOI] [PubMed] [Google Scholar]
- 6.Cardinale D., Colombo A., Bacchiani G., et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation . 2015;131(22):1981–1988. doi: 10.1161/CIRCULATIONAHA.114.013777. [DOI] [PubMed] [Google Scholar]
- 7.Kitakata H., Endo J., Ikura H., et al. Therapeutic targets for DOX-induced cardiomyopathy: role of apoptosis vs. ferroptosis. International journal of molecular sciences . 2022;23(3):p. 1414. doi: 10.3390/ijms23031414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rocca C., Pasqua T., Cerra M. C., Angelone T. Cardiac damage in anthracyclines therapy: focus on oxidative stress and inflammation. Antioxidants & Redox Signaling . 2020;32(15):1081–1097. doi: 10.1089/ars.2020.8016. [DOI] [PubMed] [Google Scholar]
- 9.Vejpongsa P., Yeh E. T. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. Journal of the American College of Cardiology . 2014;64(9):938–945. doi: 10.1016/j.jacc.2014.06.1167. [DOI] [PubMed] [Google Scholar]
- 10.Tian C., Yang Y., Bai B., et al. Potential of exosomes as diagnostic biomarkers and therapeutic carriers for doxorubicin-induced cardiotoxicity. International Journal of Biological Sciences . 2021;17(5):1328–1338. doi: 10.7150/ijbs.58786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hensley M. L., Hagerty K. L., Kewalramani T., et al. American Society of Clinical Oncology 2008 clinical practice guideline update: use of chemotherapy and radiation therapy protectants. Journal of Clinical Oncology . 2009;27(1):127–145. doi: 10.1200/JCO.2008.17.2627. [DOI] [PubMed] [Google Scholar]
- 12.Li J., Chang H. M., Banchs J., et al. Detection of subclinical cardiotoxicity in sarcoma patients receiving continuous doxorubicin infusion or pre-treatment with dexrazoxane before bolus doxorubicin. Cardiooncology . 2020;6(1):p. 1. doi: 10.1186/s40959-019-0056-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kojima R., Bojar D., Rizzi G., et al. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nature communications . 2018;9(1):1–10. doi: 10.1038/s41467-018-03733-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Q., Piao H., Wang Y., Zheng D., Wang W. Circulating exosomes in cardiovascular disease: novel carriers of biological information. Biomedicine & Pharmacotherapy . 2021;135, article 111148 doi: 10.1016/j.biopha.2020.111148. [DOI] [PubMed] [Google Scholar]
- 15.Zamani P., Fereydouni N., Butler A. E., Navashenaq J. G., Sahebkar A. The therapeutic and diagnostic role of exosomes in cardiovascular diseases. Trends in Cardiovascular Medicine . 2019;29(6):313–323. doi: 10.1016/j.tcm.2018.10.010. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y., Hu Y. W., Zheng L., Wang Q. Characteristics and roles of exosomes in cardiovascular disease. DNA and Cell Biology . 2017;36(3):202–211. doi: 10.1089/dna.2016.3496. [DOI] [PubMed] [Google Scholar]
- 17.Théry C., Witwer K. W., Aikawa E., et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of extracellular vesicles . 2018;7(1, article 1535750) doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng L., Hill A. F. Therapeutically harnessing extracellular vesicles. Nature Reviews. Drug Discovery . 2022;21(5):379–399. doi: 10.1038/s41573-022-00410-w. [DOI] [PubMed] [Google Scholar]
- 19.Kalluri R., LeBleu V. S. The biology, function, and biomedical applications of exosomes. Science . 2020;367(6478) doi: 10.1126/science.aau6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Niel G., D'Angelo G., Raposo G. Shedding light on the cell biology of extracellular vesicles. Nature Reviews. Molecular Cell Biology . 2018;19(4):213–228. doi: 10.1038/nrm.2017.125. [DOI] [PubMed] [Google Scholar]
- 21.Yang B., Chen Y., Shi J. Exosome biochemistry and advanced nanotechnology for next-generation theranostic platforms. Advanced Materials . 2019;31(2, article e1802896) doi: 10.1002/adma.201802896. [DOI] [PubMed] [Google Scholar]
- 22.Barile L., Vassalli G. Exosomes: therapy delivery tools and biomarkers of diseases. Pharmacology & Therapeutics . 2017;174:63–78. doi: 10.1016/j.pharmthera.2017.02.020. [DOI] [PubMed] [Google Scholar]
- 23.Jiang L., Gu Y., Du Y., Liu J. Exosomes: diagnostic biomarkers and therapeutic delivery vehicles for cancer. Molecular Pharmaceutics . 2019;16(8):3333–3349. doi: 10.1021/acs.molpharmaceut.9b00409. [DOI] [PubMed] [Google Scholar]
- 24.Wani T. U., Mohi-Ud-Din R., Mir R. H., et al. Exosomes harnessed as nanocarriers for cancer therapy - current status and potential for future clinical applications. Current Molecular Medicine . 2021;21(9):707–723. doi: 10.2174/1566524020666200915111618. [DOI] [PubMed] [Google Scholar]
- 25.de Freitas R. C. C., Hirata R. D. C., Hirata M. H., Aikawa E. Circulating extracellular vesicles as biomarkers and drug delivery vehicles in cardiovascular diseases. Biomolecules . 2021;11(3):p. 388. doi: 10.3390/biom11030388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rawat P. S., Jaiswal A., Khurana A., Bhatti J. S., Navik U. Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomedicine & Pharmacotherapy . 2021;139, article 111708 doi: 10.1016/j.biopha.2021.111708. [DOI] [PubMed] [Google Scholar]
- 27.Šimůnek T., Štěrba M., Popelová O., Adamcová M., Hrdina R., Geršl V. Anthracycline-induced cardiotoxicity: overview of studies examining the roles of oxidative stress and free cellular iron. Pharmacological Reports . 2009;61(1):154–171. doi: 10.1016/S1734-1140(09)70018-0. [DOI] [PubMed] [Google Scholar]
- 28.Octavia Y., Tocchetti C. G., Gabrielson K. L., Janssens S., Crijns H. J., Moens A. L. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. Journal of Molecular and Cellular Cardiology . 2012;52(6):1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 29.Finn N. A., Findley H. W., Kemp M. L. A switching mechanism in doxorubicin bioactivation can be exploited to control doxorubicin toxicity. PLoS Computational Biology . 2011;7(9, article e1002151) doi: 10.1371/journal.pcbi.1002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu M. F., Tang P. L., Qian Z. M., Ashraf M. Effects by doxorubicin on the myocardium are mediated by oxygen free radicals. Life Sciences . 2001;68(8):889–901. doi: 10.1016/S0024-3205(00)00990-5. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Y., McLaughlin D., Robinson E., et al. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with doxorubicin chemotherapy. Cancer Research . 2010;70(22):9287–9297. doi: 10.1158/0008-5472.CAN-10-2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Angsutararux P., Luanpitpong S., Issaragrisil S. Chemotherapy-induced cardiotoxicity: overview of the roles of oxidative stress. Oxidative Medicine and Cellular Longevity . 2015;2015:13. doi: 10.1155/2015/795602.795602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vedam K., Nishijima Y., Druhan L. J., et al. Role of heat shock factor-1 activation in the doxorubicin-induced heart failure in mice. American Journal of Physiology. Heart and Circulatory Physiology . 2010;298(6):H1832–H1841. doi: 10.1152/ajpheart.01047.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ueno M., Kakinuma Y., Yuhki K., et al. Doxorubicin induces apoptosis by activation of caspase-3 in cultured cardiomyocytes in vitro and rat cardiac ventricles in vivo. Journal of Pharmacological Sciences . 2006;101(2):151–158. doi: 10.1254/jphs.FP0050980. [DOI] [PubMed] [Google Scholar]
- 35.Konorev E. A., Vanamala S., Kalyanaraman B. Differences in doxorubicin-induced apoptotic signaling in adult and immature cardiomyocytes. Free Radical Biology & Medicine . 2008;45(12):1723–1728. doi: 10.1016/j.freeradbiomed.2008.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim D. S., Woo E. R., Chae S. W., et al. Plantainoside D protects adriamycin-induced apoptosis in H9c2 cardiac muscle cells via the inhibition of ROS generation and NF-κB activation. Life Sciences . 2007;80(4):314–323. doi: 10.1016/j.lfs.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 37.Li H., Gu H., Sun B. Protective effects of pyrrolidine dithiocarbamate on myocardium apoptosis induced by adriamycin in rats. International Journal of Cardiology . 2007;114(2):159–165. doi: 10.1016/j.ijcard.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 38.Wang S., Kotamraju S., Konorev E., Kalivendi S., Joseph J., Kalyanaraman B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. The Biochemical Journal . 2002;367(3):729–740. doi: 10.1042/bj20020752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao L., Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Scientific Reports . 2017;7(1):p. 44735. doi: 10.1038/srep44735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An J., Li P., Li J., Dietz R., Donath S. ARC is a critical cardiomyocyte survival switch in doxorubicin cardiotoxicity. Journal of Molecular Medicine . 2009;87(4):401–410. doi: 10.1007/s00109-008-0434-z. [DOI] [PubMed] [Google Scholar]
- 41.Guo R., Wu K., Chen J., et al. Exogenous hydrogen sulfide protects against doxorubicin-induced inflammation and cytotoxicity by inhibiting p38MAPK/NFκB pathway in H9c2 cardiac cells. Cellular Physiology and Biochemistry . 2013;32(6):1668–1680. doi: 10.1159/000356602. [DOI] [PubMed] [Google Scholar]
- 42.Hadi N., Yousif N. G., Al-Amran F. G., Huntei N. K., Mohammad B. I., Ali S. J. Vitamin E and telmisartan attenuates doxorubicin induced cardiac injury in rat through down regulation of inflammatory response. BMC Cardiovascular Disorders . 2012;12(1):p. 63. doi: 10.1186/1471-2261-12-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi W., Boliang W., Xiaoxi T., Guoqiang F., Jianbo X., Gang W. Cardamonin protects against doxorubicin-induced cardiotoxicity in mice by restraining oxidative stress and inflammation associated with Nrf2 signaling. Biomedicine & Pharmacotherapy . 2020;122, article 109547 doi: 10.1016/j.biopha.2019.109547. [DOI] [PubMed] [Google Scholar]
- 44.Wang L., Chen Q., Qi H., et al. Doxorubicin-induced systemic inflammation is driven by upregulation of Toll-like receptor TLR4 and endotoxin leakage. Cancer Research . 2016;76(22):6631–6642. doi: 10.1158/0008-5472.CAN-15-3034. [DOI] [PubMed] [Google Scholar]
- 45.Zhai J., Tao L., Zhang S., et al. Calycosin ameliorates doxorubicin-induced cardiotoxicity by suppressing oxidative stress and inflammation via the sirtuin 1-NOD-like receptor protein 3 pathway. Phytotherapy Research . 2020;34(3):649–659. doi: 10.1002/ptr.6557. [DOI] [PubMed] [Google Scholar]
- 46.Meng L., Lin H., Zhang J., et al. Doxorubicin induces cardiomyocyte pyroptosis via the TINCR-mediated posttranscriptional stabilization of NLR family pyrin domain containing 3. Journal of Molecular and Cellular Cardiology . 2019;136:15–26. doi: 10.1016/j.yjmcc.2019.08.009. [DOI] [PubMed] [Google Scholar]
- 47.Christidi E., Brunham L. R. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death & Disease . 2021;12(4):p. 339. doi: 10.1038/s41419-021-03614-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng X., Zhong T., Ma Y., et al. Bnip3 mediates doxorubicin-induced cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sciences . 2020;242, article 117186 doi: 10.1016/j.lfs.2019.117186. [DOI] [PubMed] [Google Scholar]
- 49.Tavakoli Dargani Z., Singla D. K. Embryonic stem cell-derived exosomes inhibit doxorubicin-induced TLR4-NLRP3-mediated cell death-pyroptosis. American Journal of Physiology. Heart and Circulatory Physiology . 2019;317(2):H460–h471. doi: 10.1152/ajpheart.00056.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sun Z., Lu W., Lin N., et al. Dihydromyricetin alleviates doxorubicin-induced cardiotoxicity by inhibiting NLRP3 inflammasome through activation of SIRT1. Biochemical Pharmacology . 2020;175, article 113888 doi: 10.1016/j.bcp.2020.113888. [DOI] [PubMed] [Google Scholar]
- 51.Abdelgawad I. Y., Sadak K. T., Lone D. W., Dabour M. S., Niedernhofer L. J., Zordoky B. N. Molecular mechanisms and cardiovascular implications of cancer therapy-induced senescence. Pharmacology & Therapeutics . 2021;221, article 107751 doi: 10.1016/j.pharmthera.2020.107751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altieri P., Spallarossa P., Barisione C., et al. Inhibition of doxorubicin-induced senescence by PPARδ activation agonists in cardiac muscle cells: cooperation between PPARδ and Bcl6. PLoS One . 2012;7(9, article e46126) doi: 10.1371/journal.pone.0046126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bielak-Zmijewska A., Wnuk M., Przybylska D., et al. A comparison of replicative senescence and doxorubicin-induced premature senescence of vascular smooth muscle cells isolated from human aorta. Biogerontology . 2014;15(1):47–64. doi: 10.1007/s10522-013-9477-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maejima Y., Adachi S., Ito H., Hirao K., Isobe M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell . 2008;7(2):125–136. doi: 10.1111/j.1474-9726.2007.00358.x. [DOI] [PubMed] [Google Scholar]
- 55.Spallarossa P., Altieri P., Aloi C., et al. Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. American Journal of Physiology. Heart and Circulatory Physiology . 2009;297(6):H2169–H2181. doi: 10.1152/ajpheart.00068.2009. [DOI] [PubMed] [Google Scholar]
- 56.Hodjat M., Haller H., Dumler I., Kiyan Y. Urokinase receptor mediates doxorubicin-induced vascular smooth muscle cell senescence via proteasomal degradation of TRF2. Journal of Vascular Research . 2013;50(2):109–123. doi: 10.1159/000343000. [DOI] [PubMed] [Google Scholar]
- 57.Przybylska D., Janiszewska D., Goździk A., et al. NOX4 downregulation leads to senescence of human vascular smooth muscle cells. Oncotarget . 2016;7(41):66429–66443. doi: 10.18632/oncotarget.12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spallarossa P., Altieri P., Barisione C., et al. p38 MAPK and JNK antagonistically control senescence and cytoplasmic p16INK4A expression in doxorubicin-treated endothelial progenitor cells. PLoS One . 2010;5(12, article e15583) doi: 10.1371/journal.pone.0015583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heo J. I., Kim W., Choi K. J., Bae S., Jeong J. H., Kim K. S. XIAP-associating factor 1, a transcriptional target of BRD7, contributes to endothelial cell senescence. Oncotarget . 2016;7(5):5118–5130. doi: 10.18632/oncotarget.6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Piegari E., De Angelis A., Cappetta D., et al. Doxorubicin induces senescence and impairs function of human cardiac progenitor cells. Basic Research in Cardiology . 2013;108(2):p. 334. doi: 10.1007/s00395-013-0334-4. [DOI] [PubMed] [Google Scholar]
- 61.Dixon S. J., Lemberg K. M., Lamprecht M. R., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell . 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fang X., Wang H., Han D., et al. Ferroptosis as a target for protection against cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America . 2019;116(7):2672–2680. doi: 10.1073/pnas.1821022116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Panjrath G. S., Patel V., Valdiviezo C. I., Narula N., Narula J., Jain D. Potentiation of doxorubicin cardiotoxicity by iron loading in a rodent model. Journal of the American College of Cardiology . 2007;49(25):2457–2464. doi: 10.1016/j.jacc.2007.02.060. [DOI] [PubMed] [Google Scholar]
- 64.Kotamraju S., Chitambar C. R., Kalivendi S. V., Joseph J., Kalyanaraman B. Transferrin receptor-dependent iron uptake is responsible for doxorubicin-mediated apoptosis in endothelial cells: role of oxidant-induced iron signaling in apoptosis. The Journal of Biological Chemistry . 2002;277(19):17179–17187. doi: 10.1074/jbc.M111604200. [DOI] [PubMed] [Google Scholar]
- 65.Zhuang S., Ma Y., Zeng Y., et al. METTL14 promotes doxorubicin-induced cardiomyocyte ferroptosis by regulating the KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biology and Toxicology . 2021 doi: 10.1007/s10565-021-09660-7. [DOI] [PubMed] [Google Scholar]
- 66.Minotti G., Ronchi R., Salvatorelli E., Menna P., Cairo G. Doxorubicin irreversibly inactivates iron regulatory proteins 1 and 2 in cardiomyocytes: evidence for distinct metabolic pathways and implications for iron-mediated cardiotoxicity of antitumor therapy. Cancer Research . 2001;61(23):8422–8428. [PubMed] [Google Scholar]
- 67.Canzoneri J. C., Oyelere A. K. Interaction of anthracyclines with iron responsive element mRNAs. Nucleic Acids Research . 2008;36(21):6825–6834. doi: 10.1093/nar/gkn774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Corna G., Galy B., Hentze M. W., Cairo G. IRP1-independent alterations of cardiac iron metabolism in doxorubicin-treated mice. Journal of Molecular Medicine . 2006;84(7):551–560. doi: 10.1007/s00109-006-0068-y. [DOI] [PubMed] [Google Scholar]
- 69.Zhao L., Qi Y., Xu L., et al. MicroRNA-140-5p aggravates doxorubicin-induced cardiotoxicity by promoting myocardial oxidative stress via targeting Nrf2 and Sirt2. Redox Biology . 2018;15:284–296. doi: 10.1016/j.redox.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sunitha M. C., Dhanyakrishnan R., PrakashKumar B., Nevin K. G. p-Coumaric acid mediated protection of H9c2 cells from doxorubicin-induced cardiotoxicity: involvement of augmented Nrf2 and autophagy. Biomedicine & Pharmacotherapy . 2018;102:823–832. doi: 10.1016/j.biopha.2018.03.089. [DOI] [PubMed] [Google Scholar]
- 71.Ichikawa Y., Ghanefar M., Bayeva M., et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. The Journal of Clinical Investigation . 2014;124(2):617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fox C. A., Ryan R. O. Dye binding assay reveals doxorubicin preference for DNA versus cardiolipin. Analytical Biochemistry . 2020;594, article 113617 doi: 10.1016/j.ab.2020.113617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pereira G. C., Pereira S. P., Tavares L. C., et al. Cardiac cytochrome c and cardiolipin depletion during anthracycline-induced chronic depression of mitochondrial function. Mitochondrion . 2016;30:95–104. doi: 10.1016/j.mito.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 74.Maccarinelli F., Gammella E., Asperti M., et al. Mice lacking mitochondrial ferritin are more sensitive to doxorubicin-mediated cardiotoxicity. Journal of Molecular Medicine . 2014;92(8):859–869. doi: 10.1007/s00109-014-1147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ichikawa Y., Bayeva M., Ghanefar M., et al. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proceedings of the National Academy of Sciences of the United States of America . 2012;109(11):4152–4157. doi: 10.1073/pnas.1119338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mouli S., Nanayakkara G., AlAlasmari A., et al. The role of frataxin in doxorubicin-mediated cardiac hypertrophy. American Journal of Physiology. Heart and Circulatory Physiology . 2015;309(5):H844–H859. doi: 10.1152/ajpheart.00182.2015. [DOI] [PubMed] [Google Scholar]
- 77.Tadokoro T., Ikeda M., Ide T., et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. Insight . 2020;5(9) doi: 10.1172/jci.insight.132747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Keung E. C., Toll L., Ellis M., Jensen R. A. L-type cardiac calcium channels in doxorubicin cardiomyopathy in rats morphological, biochemical, and functional correlations. The Journal of Clinical Investigation . 1991;87(6):2108–2113. doi: 10.1172/JCI115241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim D. H., Landry A. B., Lee Y. S., Katz A. M. Doxorubicin-induced calcium release from cardiac sarcoplasmic reticulum vesicles. Journal of Molecular and Cellular Cardiology . 1989;21(5):433–436. doi: 10.1016/0022-2828(89)90782-7. [DOI] [PubMed] [Google Scholar]
- 80.Saeki K., Obi I., Ogiku N., Shigekawa M., Imagawa T., Matsumoto T. Doxorubicin directly binds to the cardiac-type ryanodine receptor. Life Sciences . 2002;70(20):2377–2389. doi: 10.1016/S0024-3205(02)01524-2. [DOI] [PubMed] [Google Scholar]
- 81.Hanna A. D., Lam A., Tham S., Dulhunty A. F., Beard N. A. Adverse effects of doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Molecular Pharmacology . 2014;86(4):438–449. doi: 10.1124/mol.114.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sag C. M., Köhler A. C., Anderson M. E., Backs J., Maier L. S. CaMKII-dependent SR Ca leak contributes to doxorubicin-induced impaired Ca handling in isolated cardiac myocytes. Journal of Molecular and Cellular Cardiology . 2011;51(5):749–759. doi: 10.1016/j.yjmcc.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lim C. C., Zuppinger C., Guo X., et al. Anthracyclines Induce Calpain-dependent Titin Proteolysis and Necrosis in Cardiomyocytes∗. The Journal of Biological Chemistry . 2004;279(9):8290–8299. doi: 10.1074/jbc.M308033200. [DOI] [PubMed] [Google Scholar]
- 84.Kalivendi S. V., Konorev E. A., Cunningham S., et al. Doxorubicin activates nuclear factor of activated T-lymphocytes and Fas ligand transcription: role of mitochondrial reactive oxygen species and calcium. The Biochemical Journal . 2005;389(2):527–539. doi: 10.1042/BJ20050285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tscheschner H., Meinhardt E., Schlegel P., et al. CaMKII activation participates in doxorubicin cardiotoxicity and is attenuated by moderate GRP78 overexpression. PLoS One . 2019;14(4, article e0215992) doi: 10.1371/journal.pone.0215992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ranganath S. H., Levy O., Inamdar M. S., Karp J. M. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell Stem Cell . 2012;10(3):244–258. doi: 10.1016/j.stem.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ni J., Liu Y., Wang K., et al. Trophoblast stem-cell-derived exosomes improve doxorubicin-induced dilated cardiomyopathy by modulating the let-7i/YAP pathway. Molecular Therapy-Nucleic Acids . 2020;22:948–956. doi: 10.1016/j.omtn.2020.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ni J., Liu Y., Kang L., et al. Human trophoblast-derived exosomes attenuate doxorubicin-induced cardiac injury by regulating miR-200b and downstream Zeb1. Journal of Nanobiotechnology . 2020;18(1):p. 171. doi: 10.1186/s12951-020-00733-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zeringer E., Barta T., Li M., Vlassov A. V. Strategies for isolation of exosomes. Cold Spring Harbor Protocols . 2015;2015(4):319–323. doi: 10.1101/pdb.top074476. [DOI] [PubMed] [Google Scholar]
- 90.Montecalvo A., Larregina A. T., Shufesky W. J., et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood . 2012;119(3):756–766. doi: 10.1182/blood-2011-02-338004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee J. Y., Chung J., Byun Y., Kim K. H., An S. H., Kwon K. Mesenchymal stem cell-derived small extracellular vesicles protect cardiomyocytes from doxorubicin-induced cardiomyopathy by upregulating survivin expression via the miR-199a-3p-Akt-Sp1/p53 signaling pathway. International Journal of Molecular Sciences . 2021;22(13):p. 7102. doi: 10.3390/ijms22137102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhong Z., Tian Y., Luo X., Zou J., Wu L., Tian J. Extracellular vesicles derived from human umbilical cord mesenchymal stem cells protect against DOX-induced heart failure through the miR-100-5p/NOX4 pathway. Frontiers in Bioengineering and Biotechnology . 2021;9, article 703241 doi: 10.3389/fbioe.2021.703241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xia W., Chen H., Xie C., Hou M. Long-noncoding RNA MALAT1 sponges microRNA-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting ATG4a. Aging . 2020;12(9):8241–8260. doi: 10.18632/aging.103136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu Y., Liu Z., Xie Y., Zhao C., Xu J. Serum extracellular vesicles retard H9C2 cell senescence by suppressing miR-34a expression. Journal of Cardiovascular Translational Research . 2019;12(1):45–50. doi: 10.1007/s12265-018-9847-4. [DOI] [PubMed] [Google Scholar]
- 95.Zhuang L., Xia W., Chen D., et al. Exosomal LncRNA-NEAT1 derived from MIF-treated mesenchymal stem cells protected against doxorubicin-induced cardiac senescence through sponging miR-221-3p. Journal of Nanobiotechnology . 2020;18(1):p. 157. doi: 10.1186/s12951-020-00716-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Milano G., Biemmi V., Lazzarini E., et al. Intravenous administration of cardiac progenitor cell-derived exosomes protects against doxorubicin/trastuzumab-induced cardiac toxicity. Cardiovascular Research . 2020;116(2):383–392. doi: 10.1093/cvr/cvz108. [DOI] [PubMed] [Google Scholar]
- 97.Lei B., Wu X., Xia K., Sun H., Wang J. Exosomal micro-RNA-96 derived from bone marrow mesenchymal stem cells inhibits doxorubicin-induced myocardial toxicity by inhibiting the Rac1/nuclear factor-κB signaling pathway. Journal of the American Heart Association . 2021;10(17, article e020589) doi: 10.1161/JAHA.120.020589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun X., Shan A., Wei Z., Xu B. Intravenous mesenchymal stem cell-derived exosomes ameliorate myocardial inflammation in the dilated cardiomyopathy. Biochemical and Biophysical Research Communications . 2018;503(4):2611–2618. doi: 10.1016/j.bbrc.2018.08.012. [DOI] [PubMed] [Google Scholar]
- 99.Zhang W., Chen Z., Qiao S., et al. The effects of extracellular vesicles derived from Krüppel-like factor 2 overexpressing endothelial cells on the regulation of cardiac inflammation in the dilated cardiomyopathy. Journal of Nanobiotechnology . 2022;20(1):p. 76. doi: 10.1186/s12951-022-01284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Singla D. K., Johnson T. A., Tavakoli D. Z. Exosome treatment enhances anti-inflammatory M2 macrophages and reduces inflammation-induced pyroptosis in doxorubicin-induced cardiomyopathy. Cell . 2019;8(10):p. 1224. doi: 10.3390/cells8101224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bhagat A., Kleinerman E. S. Anthracycline-induced cardiotoxicity: causes, mechanisms, and prevention. Advances in Experimental Medicine and Biology . 2020;1257:181–192. doi: 10.1007/978-3-030-43032-0_15. [DOI] [PubMed] [Google Scholar]
- 102.Songbo M., Lang H., Xinyong C., Bin X., Ping Z., Liang S. Oxidative stress injury in doxorubicin-induced cardiotoxicity. Toxicology Letters . 2019;307:41–48. doi: 10.1016/j.toxlet.2019.02.013. [DOI] [PubMed] [Google Scholar]
- 103.Cardinale D., Colombo A., Lamantia G., et al. Anthracycline-induced cardiomyopathy: clinical relevance and response to pharmacologic therapy. Journal of the American College of Cardiology . 2010;55(3):213–220. doi: 10.1016/j.jacc.2009.03.095. [DOI] [PubMed] [Google Scholar]
- 104.Pudil R., Mueller C., Čelutkienė J., et al. Role of serum biomarkers in cancer patients receiving cardiotoxic cancer therapies: a position statement from the Cardio-Oncology Study Group of the Heart Failure Association and the Cardio-Oncology Council of the European Society of Cardiology. European Journal of Heart Failure . 2020;22(11):1966–1983. doi: 10.1002/ejhf.2017. [DOI] [PubMed] [Google Scholar]
- 105.Tan L. L., Lyon A. R. Role of biomarkers in prediction of cardiotoxicity during cancer treatment. Current Treatment Options in Cardiovascular Medicine . 2018;20(7):p. 55. doi: 10.1007/s11936-018-0641-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Marini M. G., Cardillo M. T., Caroli A., Sonnino C., Biasucci L. M. Increasing specificity of high-sensitivity troponin: new approaches and perspectives in the diagnosis of acute coronary syndromes. Journal of Cardiology . 2013;62(4):205–209. doi: 10.1016/j.jjcc.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 107.Yarana C., Carroll D., Chen J., et al. Extracellular vesicles released by cardiomyocytes in a doxorubicin-induced cardiac injury mouse model contain protein biomarkers of early cardiac injury. Clinical Cancer Research . 2018;24(7):1644–1653. doi: 10.1158/1078-0432.CCR-17-2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhu Y., Gius D. Glycogen phosphorylase: a novel biomarker in doxorubicin-induced cardiac injury. Clinical Cancer Research . 2018;24(7):1516–1517. doi: 10.1158/1078-0432.CCR-17-3276. [DOI] [PubMed] [Google Scholar]
- 109.Beaumier A., Robinson S. R., Robinson N., et al. Extracellular vesicular microRNAs as potential biomarker for early detection of doxorubicin-induced cardiotoxicity. Journal of Veterinary Internal Medicine . 2020;34(3):1260–1271. doi: 10.1111/jvim.15762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li B., Cai X., Wang Y., et al. Circ-SKA3 enhances doxorubicin toxicity in AC16 cells through miR-1303/TLR4 Axis. International Heart Journal . 2021;62(5):1112–1123. doi: 10.1536/ihj.20-809. [DOI] [PubMed] [Google Scholar]
- 111.Shafei A., El-Bakly W., Sobhy A., et al. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomedicine & Pharmacotherapy . 2017;95:1209–1218. doi: 10.1016/j.biopha.2017.09.059. [DOI] [PubMed] [Google Scholar]
- 112.Goh W. J., Zou S., Lee C. K., et al. EXOPLEXs: chimeric drug delivery platform from the fusion of cell-derived nanovesicles and liposomes. Biomacromolecules . 2018;19(1):22–30. doi: 10.1021/acs.biomac.7b01176. [DOI] [PubMed] [Google Scholar]
- 113.Schindler C., Collinson A., Matthews C., et al. Exosomal delivery of doxorubicin enables rapid cell entry and enhanced in vitro potency. PLoS One . 2019;14(3, article e0214545) doi: 10.1371/journal.pone.0214545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Patras L., Ionescu A. E., Munteanu C., et al. Trojan horse treatment based on PEG-coated extracellular vesicles to deliver doxorubicin to melanomain vitroandin vivo. Cancer Biology & Therapy . 2022;23(1):1–16. doi: 10.1080/15384047.2021.2003656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vandergriff A. C., de Andrade J. B., Tang J., et al. Intravenous cardiac stem cell-derived exosomes ameliorate cardiac dysfunction in doxorubicin induced dilated cardiomyopathy. Stem Cells International . 2015;2015:8. doi: 10.1155/2015/960926.960926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Song Y., Zhang C., Zhang J., et al. Localized injection of miRNA-21-enriched extracellular vesicles effectively restores cardiac function after myocardial infarction. Theranostics . 2019;9(8):2346–2360. doi: 10.7150/thno.29945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Luther K. M., Haar L., McGuinness M., et al. Exosomal miR-21a-5p mediates cardioprotection by mesenchymal stem cells. Journal of Molecular and Cellular Cardiology . 2018;119:125–137. doi: 10.1016/j.yjmcc.2018.04.012. [DOI] [PubMed] [Google Scholar]
- 118.Bejerano T., Etzion S., Elyagon S., Etzion Y., Cohen S. Nanoparticle delivery of miRNA-21 mimic to cardiac macrophages improves myocardial remodeling after myocardial infarction. Nano Letters . 2018;18(9):5885–5891. doi: 10.1021/acs.nanolett.8b02578. [DOI] [PubMed] [Google Scholar]
- 119.Smyth T., Kullberg M., Malik N., Smith-Jones P., Graner M. W., Anchordoquy T. J. Biodistribution and delivery efficiency of unmodified tumor-derived exosomes. Journal of Controlled Release . 2015;199:145–155. doi: 10.1016/j.jconrel.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wan Z., Zhao L., Lu F., et al. Mononuclear phagocyte system blockade improves therapeutic exosome delivery to the myocardium. Theranostics . 2020;10(1):218–230. doi: 10.7150/thno.38198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Martins-Marques T., Pinho M. J., Zuzarte M., et al. Presence of Cx43 in extracellular vesicles reduces the cardiotoxicity of the anti-tumour therapeutic approach with doxorubicin. Journal of Extracellular Vesicles . 2016;5(1):p. 32538. doi: 10.3402/jev.v5.32538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wei H., Chen J., Wang S., et al. A nanodrug consisting of doxorubicin and exosome derived from mesenchymal stem cells for osteosarcoma treatment in vitro. International Journal of Nanomedicine . 2019;14:8603–8610. doi: 10.2147/IJN.S218988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Toffoli G., Hadla M., Corona G., et al. Exosomal doxorubicin reduces the cardiac toxicity of doxorubicin. Nanomedicine . 2015;10(19):2963–2971. doi: 10.2217/nnm.15.118. [DOI] [PubMed] [Google Scholar]
- 124.Hadla M., Palazzolo S., Corona G., et al. Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models. Nanomedicine . 2016;11(18):2431–2441. doi: 10.2217/nnm-2016-0154. [DOI] [PubMed] [Google Scholar]
- 125.Tong Z., Jiang B., Wu Y., et al. MiR-21 protected cardiomyocytes against doxorubicin-induced apoptosis by targeting BTG2. International Journal of Molecular Sciences . 2015;16(12):14511–14525. doi: 10.3390/ijms160714511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sun W., Zhao P., Zhou Y., et al. Ultrasound targeted microbubble destruction assisted exosomal delivery of miR-21 protects the heart from chemotherapy associated cardiotoxicity. Biochemical and Biophysical Research Communications . 2020;532(1):60–67. doi: 10.1016/j.bbrc.2020.05.044. [DOI] [PubMed] [Google Scholar]
- 127.Li Y., Gao Y., Gong C., et al. A33 antibody-functionalized exosomes for targeted delivery of doxorubicin against colorectal cancer. Nanomedicine . 2018;14(7):1973–1985. doi: 10.1016/j.nano.2018.05.020. [DOI] [PubMed] [Google Scholar]
- 128.Quah B. J., O'Neill H. C. The immunogenicity of dendritic cell-derived exosomes. Blood Cells, Molecules & Diseases . 2005;35(2):94–110. doi: 10.1016/j.bcmd.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 129.Tian Y., Li S., Song J., et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials . 2014;35(7):2383–2390. doi: 10.1016/j.biomaterials.2013.11.083. [DOI] [PubMed] [Google Scholar]