

Keywords: calcium, mitochondria, neutrophils, platelets, thrombosis
Abstract
Platelets play a key role in maintaining hemostasis. However, dysregulated platelet activation can lead to pathological thrombosis or bleeding. Once a platelet gets activated, it will either become an aggregatory platelet or eventually a procoagulant platelet with both types playing distinct roles in thrombosis and hemostasis. Although aggregatory platelets have been extensively studied, procoagulant platelets have only recently come into the spotlight. Procoagulant platelets are a subpopulation of highly activated platelets that express phosphatidylserine and P-selectin on their surface, allowing for coagulation factors to bind and thrombin to be generated. In recent years, novel roles for procoagulant platelets have been identified and they have increasingly been implicated in thromboinflammatory diseases. Here, we provide an up-to-date review on the mechanisms resulting in the formation of procoagulant platelets and how they contribute to hemostasis, thrombosis, and thromboinflammation.
PROCOAGULANT PLATELETS: WHAT IS IN A NAME?
Upon vessel wall trauma, platelets quickly adhere to the site of injury and become activated, resulting in the formation of aggregating and eventually procoagulant platelets. Together, these will cooperate in sealing of the injury site forming a fibrin-rich hemostatic plug. Although the platelet research community has uncovered the mechanisms of aggregating platelets in great detail, the mechanisms leading to and the function of procoagulant platelets remain a matter of debate.
Historically, procoagulant platelets were considered all platelets expressing high levels of phosphatidylserine (PS) on their surface. This allows for coagulation factors to bind and thrombin generation to initiate on the platelet surface (1, 2). There are two ways platelets can express PS on their surface; however, only one of them contributes to thrombosis and hemostasis. Similar to nucleated cells, dying platelets will express PS on their surface, marking them for clearance. This process is dependent on caspases and independent of Ca2+. Unique to platelets, strong platelet activation can also result in the expression of PS on the activated platelet surface independent of apoptosis and highly dependent on Ca2+ (3). In recent years, evidence emerged demonstrating apoptotic PS exposure has little or no role during hemostasis (4). Hence, the term procoagulant platelets is only used when discussing strongly activated PS-positive platelets. Moreover, procoagulant platelets and apoptotic platelets can easily be distinguished by the presence of high levels of P-selectin expression (or other α-granule proteins) exclusively on the procoagulant platelet surface (5).
MECHANISMS OF PROCOAGULANT PLATELET FORMATION
Quiescent platelets have a delicate plasma membrane lipid bilayer that has negatively charged PS on its inner layer. Homeostasis of the platelet phospholipid bilayer is maintained by ATP-dependent flippases, catalyzing the transport of phospholipids from the outer to the inner layer of the plasma membrane, and scramblases, responsible for the opposite (6, 7). For a platelet to become procoagulant, the balance between flippases and scramblases needs to be tipped in favor of scramblases. This will result in a PS-positive platelet. The key trigger for this is a high enough cytosolic calcium concentration as this will simultaneously activate scramblases (8) and inhibit flippases (9, 10). In general, it is assumed that the exposure of PS on the platelet surface is primarily driven by the scramblase transmembrane protein 16 F (TMEM16F), also known as Anocatamin 6 (Ano6) (11, 12).
Physiological Triggers for Procoagulant Platelet Formation
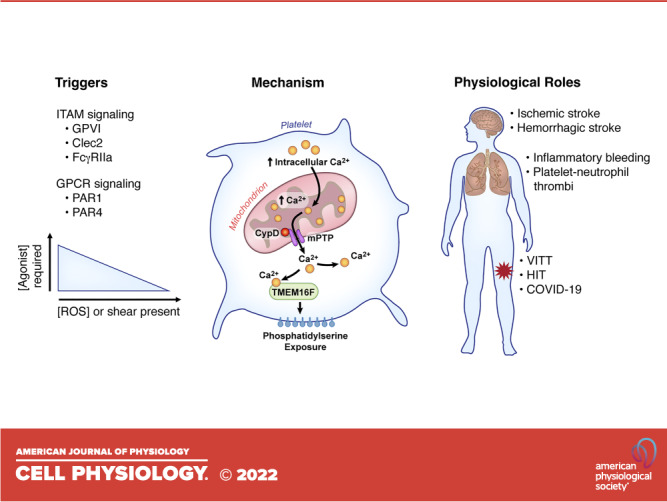
When procoagulant platelets were initially discovered, it was believed that for platelets to become procoagulant, both protease-activated receptors (PAR1 or PAR4) and immunoreceptor tyrosine-based activation motif (ITAM)-linked receptors [Glycoprotein VI (GPVI); Clec-2 or Fc Gamma RIIa (FcγRIIa)] needed to be activated simultaneously (Fig. 1) (12). However, this seems to mainly be the case in vitro. In vivo, the triggers for platelets to become procoagulant are less stringent and often times single agonist stimulation will suffice. From a signaling perspective, these various pathways will activate intracellular messengers that converge into the activation of distinct phospholipase C (PLC) isoforms (13). PARs signal through PLCβ, whereas ITAMs signal through PLCγ. Next, both PLC isoforms will cleave phosphatidylinositol-4,5-bisphosphate (PIP2) into inositol trisphosphate (IP3) and diacylglycerol (DAG). IP3 interacts with the IP3 receptor at the surface of the internal store of Ca2+, triggering the cytosolic release of Ca2+. Of note, these processes are not unique to procoagulant platelet formation and more research is needed to elucidate discrepancies in signaling events contributing to both subtypes of activated platelets.
Figure 1.

Overview of mechanisms of procoagulant platelet formation. Platelet stimulation through agonists that induce ITAM or GPCR signaling in combination with shear or ROS can induce procoagulant platelet formation. These triggers induce a rapid rise in intracellular calcium, which enters the mitochondria. Once excessive calcium enters the mitochondria, CypD acts to form the mPTP, resulting in decrease mitochondrial membrane stability and the release of calcium back into the cytoplasm. The release of calcium through the mPTP activates TMEM16F, a phosphatidylserine (PS) scramblase, resulting in PS exposure on the platelet surface. Pathological formation of procoagulant platelets can induce thrombotic complications such as stroke and pulmonary and venous thrombosis. CypD, cyclophilin D; FcγRIIa, Fc Gamma RIIa; GPCR, G protein-coupled receptor; GPVI, glycoprotein VI; HIT, heparin-induced thrombocytopenia; ITAM, immunoreceptor tyrosine-based activation motif; mPTP, mitochondrial permeability transition pore; PAR, protease-activated receptor; ROS, reactive oxygen species; TMEM16F, transmembrane protein 16F; VITT, vaccine-induced thrombotic thrombocytopenia.
Under physiological conditions (i.e., in the presence of blood flow), platelets binding to collagen is sufficient to induce procoagulant platelets (14, 15). However, in the absence of flow, costimulation with PAR agonists is necessary to achieve similar procoagulant platelet responses. Nevertheless, platelets will almost always be subjected to some degree of shear stress in vivo. In fact, it has been reported that shear stress alone is sufficient to trigger procoagulant platelet formation and PS externalization increases with increasing shear stress (16). This could have implications for prosthetic cardiovascular devices that often induce very high shear stress. However, these results need validation, and the consensus is that rather than inducing procoagulant platelets, shear stress potentiates procoagulant platelet formation. This was elegantly demonstrated both in the absence and presence of platelet adhesion. Using a cone-plate rheometer, it was shown that arterial shear enabled low doses of a single agonist (thrombin or collagen) to induce full-scale procoagulant platelet activity (17, 18). Critically, in the setting of adherent immobilized platelets, low-shear flow conditions are sufficient to propagate procoagulant platelet formation. Mechanistically, this process requires platelet activation by von Willebrand factor (VWF) binding to platelet glycoprotein Ib (GPIb) in the presence of trace amounts of thrombin. Through a local enforcement loop, platelet PS will subsequently activate coagulation factors, which then generate sufficiently high amounts of thrombin to ensure focal fibrin formation. This was compellingly demonstrated by Cosemans et al. (19) using highly specific antibodies against the VWF-GPIb interaction, and plasma and blood from patients with von Willebrand’s disease or VWF-deficient mice. Of note, Nbeal2-deficient mice, lacking α-granules, also have strongly reduced procoagulant platelet responses (20). This indicates α-granules or components thereof (e.g., platelet-VWF) are critically required for platelet coagulant activity under shear stress.
Ca2+: Gatekeeper of Procoagulant Platelet Formation
A common feature of platelet activation is a rise in intracellular Ca2+ to initiate signaling events leading to either an aggregating or procoagulant platelet (21). Platelets take up Ca2+ from their surroundings mainly through Ca2+ channels ORAI1 and TRPC6. In conjunction with ORAI1, STIM1 will subsequently fill intracellular Ca2+ stores through a process known as store-operated calcium entry (SOCE). In parallel, sarco/endoplasmic reticulum Ca2+ ATPases (SERCA) will also fill the platelet Ca2+ stores. In platelets, three Ca2+ stores have been identified: the dense tubular system (equivalent to sarco/endoplasmic reticulum), lysosome-like acidic organelles, and mitochondria. From these stores, the dense tubular system is considered to be the major intracellular Ca2+ pool in platelets. In contrast, mitochondrial Ca2+ is considered to predominantly be involved in procoagulant platelet formation, since alterations in mitochondrial Ca2+ were reported to have little effect on classical platelet function (22–24). However, more studies are needed to investigate the contribution of mitochondrial Ca2+ to aggregatory platelet function.
Although both aggregating and procoagulant platelets require an increase in cytosolic Ca2+, procoagulant platelets need sustained levels of supramaximal Ca2+, whereas aggregating platelets only require transient increases in Ca2+ (24). The high and sustained increase in cytosolic calcium required for PS exposure is only achieved with strong platelet stimulation (combined ITAM and PAR signaling) and requires Ca2+ influx from the extracellular space. Early on during platelet activation, Ca2+ levels are considered oscillatory because platelets manage to handle excess Ca2+ through Ca2+ exporters [SERCA, the plasma membrane Ca2+ ATPases (PMCA), and the sodium calcium exchanger (NCX)] and mitochondrial Ca2+ uptake (21). However, in strongly stimulated platelets, over time, the NCX will reverse its direction and participate in sustaining high cytosolic Ca2+ levels (25). As a consequence, mitochondria will increasingly store more Ca2+ until a critical threshold is reached (24). Hereafter, the mitochondrial membranes will depolarize, initiating the process of platelet necrosis, resulting in the opening of the mitochondrial permeability transition pore (mPTP) (26, 27). Opening of the mPTP releases a significant amount of mitochondrial Ca2+ in the extracellular space. Critically, only platelets that have undergone mitochondrial depolarization will achieve the high and sustained Ca2+ levels required for activation of TMEM16F and subsequent PS exposure. In parallel, loss of the inner mitochondrial membrane potential will result in rapid ATP depletion, inhibiting flippases.
mPTP opening is dependent on cyclophilin D (CypD), as either pharmacological inhibition of CypD with cyclosporine A or genetic knockout of CypD strongly reduces procoagulant platelet formation (26, 27). Interestingly, in settings of oxidative stress, the Ca2+ threshold to activate CypD and mediate mPTP formation is lower, and platelets are more likely to become procoagulant (22, 28–30). However, oxidative stress alone has no effect on PS externalization, implying oxidative stress may play a role in priming platelets for procoagulant responses. As discussed later, this is of particular interest to several thrombotic diseases in which procoagulant platelets are known to be involved and where comorbidities greatly impact outcomes. Diabetes, for example, induces oxidative stress in platelets, increasing platelet reactive oxygen species and potentiating the formation of procoagulant platelets (28, 30).
Fluid Entry Influences Procoagulant Membrane Dynamics
Although Ca2+ is being mobilized to express PS on the platelet surface, fluids enter the procoagulant platelet inducing physical disruption of the membrane-cytoskeleton interaction, increasing internal hydrostatic pressure and resulting in irreversible membrane swelling (31). This membrane swelling is called ballooning and is required to support and localize thrombin generation on the platelet surface. In fact, the procoagulant quality of the platelet is dependent on the amount of coagulation factors they can bind on their enhanced surface area (32). Recently, it was shown that the transition to a balloon shape is dependent on a coordinated Na+, Cl–, and water entry and that inhibition of either of these processes is sufficient to block platelet ballooning and thrombin generation (31). Interestingly, ballooning could be inhibited by acetazolamide, an FDA-approved inhibitor of aquaporin water channels (AQP). In other cells, it has been shown that AQP1 is a critical regulator of cell volume and membrane dynamics (33). However, experiments with mice deficient in AQP1 highlight the complexity of ballooning and procoagulant platelet formation. Although procoagulant platelet responses were suppressed in AQP1 KO mice, procoagulant membrane ballooning was unaffected (34). This highlights the multifaceted role of aquaporins in procoagulant platelet responses and demonstrates the need for more research on this topic.
CONTRIBUTION OF PROCOAGULANT PLATELETS TO HEMOSTASIS AND THROMBOSIS
A reduced ability to form procoagulant platelets has been associated with mild bleeding disorders (35, 36). However, the most compelling evidence for a role of procoagulant platelets in hemostasis comes from patients with Scott syndrome. Patients with Scott syndrome patients lack the scramblase TMEM16F resulting in little PS expression on any surface and have a mild bleeding phenotype (37). Similarly, mice lacking TMEMF16 have a bleeding tendency (38). Critically, this bleeding phenotype was also observed in platelet-specific TMEM16F-deficient mice, implying a significant role for platelet PS in hemostasis (39). However, TMEM16F mediates both agonist-induced and apoptosis-induced PS scrambling, indicating the bleeding phenotype in these mice is not necessarily solely due to a lack of procoagulant platelet responses (8, 39). In contrast, inhibiting procoagulant platelet formation through deletion of platelet CypD had little or no effect on tail bleed times in mice (27). The discrepancy between bleeding associated with TMEMF16 deletion compared with CypD deficiency is currently unclear. However, differences in integrin αIIbβ3 inactivation between TMEMF16 and CypD deficient platelets may explain, in part, why increased bleeding is associated with TMEMF16 deletion. Furthermore, it is important to note that in either case, procoagulant platelet formation is not completely abolished (40). However, in TMEM16F-deficient animals and patients with Scott syndrome, platelet ballooning is averted (11). This phenotype has not been reported for CypD-deficient platelets. This raises the possibility that the combined reduction in PS and the inability for platelets to balloon could lead to a more pronounced hemostatic phenotype in the absence of TMEM16F. Taken together, these data suggest that in certain circumstances procoagulant platelet formation mediated though TMEM16F is critical for platelet hemostatic function, whereas CypD does not affect this. However, future exploration is needed to fully appreciate the role of procoagulant platelets on hemostasis.
General blocking of PS using either AnnexinA5 (41) or lactadherin (42) has been shown to block both arterial and venous thrombosis. However, the specific role of procoagulant platelets in thrombosis is less clear. Although deficiency of TMEM16F was associated with a reproducible reduction in arterial thrombosis(38, 39, 43), the role of CypD seems more model-dependent. Global and platelet-specific deletion of CypD results in accelerated times to occlusion in a photochemical arterial thrombosis model (27). The observed difference is independent of initial platelet adhesion and based on in vitro data, the acceleration in thrombosis is due largely to the inability of CypD-deficient platelets to inactivate αIIbβ3, resulting in a more proaggregatory platelet phenotype at the site of injury. However, in two different arterial thrombosis models, using FeCl3 or laser-induced injury, Hua et al. (44) in their study observed decreased platelet adhesion and fibrin formation as well as reduced procoagulant platelet formation in platelet-specific CypD knockout mice, indicating procoagulant platelet PS is important in thrombosis when thrombin and fibrin are generated quickly after injury. To add to the nuanced role of procoagulant platelets in thrombosis, platelet CypD-deficient mice have increased thrombus formation in a venous thrombosis model (45) and increased microvascular occlusion in a collagen-epinephrine pulmonary thrombosis model (46). Data from the venous and pulmonary thrombosis models support the concept that procoagulant platelets potentially play antithrombotic roles by limiting integrin activation. However, in instances where platelet-dependent thrombin generation mediates thrombotic complications, inhibition of procoagulant platelets may be beneficial. A limitation to these observations is that most studies have focused on CypD and mPTP formation to examine the in vivo role of procoagulant platelets. The generation of different models examining other molecular players in procoagulant platelet formation may shed additional light on their role in hemostasis and thrombosis.
THROMBOINFLAMMATORY FUNCTIONS OF PROCOAGULANT PLATELETS
It has become increasingly clear that besides their roles in thrombosis and hemostasis, platelets play a critical role in inflammation. Platelets help recruit and activate immune cells at sites of infection and subsequently aid in the resolution of inflammation. However, also in the absence of infection, platelets can interact with leukocytes mediating thromboinflammation. Thromboinflammation occurs when thrombotic and inflammatory pathways converge causing collateral tissue damage through neutrophil extracellular traps (NETs) and intravascular thrombosis (47). Interestingly, the receptors mediating thromboinflammation differ from those in hemostasis, making them attractive therapeutic targets (48). In particular, procoagulant platelets have emerged as mediators of thromboinflammation. In vitro, several studies have reported on a preferential interaction of procoagulant platelets with neutrophils (49–51).
Evidence from Animal Models
Several reports have linked procoagulant platelets to thrombotic complications during ischemia-reperfusion injury through their interactions with other cells. In the setting of inflammation, thrombotic complications can occur at distant locations compared with the site of injury. For example, gut ischemia followed by reperfusion induces thrombosis in the pulmonary circulation, which is dependent on procoagulant platelets mediating neutrophil recruitment. Interestingly, similar neutrophil-rich thrombi were found in the lungs of patients with human acute respiratory distress syndrome (50). Mechanistically, the formation of thrombi in the lungs after gut ischemia-reperfusion was both P-selectin dependent and independent. In particular, the rolling of neutrophils over PS-positive procoagulant platelet membranes was found as a key driver of pathological neutrophil macroaggregate formation. Importantly, deletion of platelet CypD reduces pulmonary thrombosis after gut ischemia-reperfusion. In contrast, mice deficient in Bak and Bax, both regulators of apoptotic PS expression, are not protected, providing a role for procoagulant platelets in reperfusion injury (50).
Similar observations were reported in a murine model of cerebral ischemia-reperfusion injury. After transient middle cerebral artery occlusion (tMCAO), circulating PS-positive platelets are increased systemically and were shown to preferentially interact with neutrophils (51). This results in increased neutrophil recruitment to the brain and platelet-mediated NET formation in the cerebral vasculature (52). Interestingly, platelet-specific CypD knockout mice are protected from ischemic stroke due to reduced PS-platelet-dependent neutrophil brain recruitment. The reduction of neutrophils to the brain after ischemia improves cerebral blood flow, resulting in reduced infarct injury and improved neurological and motor function (51). These studies indicate that procoagulant platelets are primed to interact with neutrophils to promote thrombosis during ischemia-reperfusion. Intriguingly, similar to the discrepancies in hemostasis, in cerebral ischemia-reperfusion injury, TMEM16F deficiency does not recapitulate the CypD knockout mice, highlighting the complexity of procoagulant platelets also in reperfusion injury (39).
In contrast to their detrimental role in sterile inflammation, procoagulant platelets were recently shown to maintain vascular integrity in the inflamed lung. Through intravital microscopy, Kaiser et al. (53) in their study demonstrated that procoagulant platelets binding to exposed collagen at sites of lung injury are crucial for the initiation of coagulation and the prevention of inflammatory bleeding in a mechanism that is CypD and TMEM16F dependent. Their study also identified that combined integrin-mediated outside-in-signaling and GPVI signaling were required to trigger procoagulant activity in the inflamed lung and that inhibition of both pathways was needed to breach vascular integrity. These results shed new light on the role of procoagulant platelets in the setting of inflammation. Depending on the setting, procoagulant platelets can either promote thrombosis or prevent bleeding. It is tempting to speculate that similar mechanisms that are preventing inflammatory bleeding in the lung are what is driving detrimental procoagulant platelet formation in the brain. Crucially, neither CypD (51) nor TMEM16F (39)-deficient mice showed signs of inflammatory brain bleeding when subjected to stroke, highlighting the tissue-specific role of platelets in maintaining vascular integrity in the setting of inflammation.
Clinical Implications for Procoagulant Platelets
In humans, procoagulant platelet formation is associated with clinical outcomes in certain diseases such as stroke, COVID-19, heparin-induced thrombocytopenia (HIT), and vaccine-induced thrombotic thrombocytopenia (VITT).
Similar to the murine stroke data, procoagulant platelet formation is associated with adverse outcomes in patients with ischemic stroke. In one study using patients with a previous lacunar stroke, procoagulant platelet formation predicted recurrent stroke within a year of the initial stroke (54). In a separate study, high levels of procoagulant platelet formation in patients with asymptomatic carotid artery stenosis at the time of enrollment were predictive of ipsilateral stroke or transient ischemic attack during a 40-mo follow-up period (55). In contrast, increased procoagulant platelet formation is associated with a reduced risk of hemorrhage after nonlacunar ischemic stroke (56). Likewise, lower levels of procoagulant platelets were linked to increased mortality after intracerebral hemorrhage (57).
Patients with COVID-19 are at increased risk for thrombotic complications. This is, in part, due to increased platelet reactivity (58). The role of procoagulant platelets in COVID-19 seems to depend on the disease severity and state. Procoagulant platelet formation of isolate platelets induced by dual agonists stimulation is significantly reduced in patients with moderate and severe COVID-19 compared with healthy donors (46, 59). In addition, mitochondrial depolarization is also significantly impaired in patients with COVID-19, suggesting COVID-19 induces mitochondrial dysfunction, which impairs the ability of platelets to become procoagulant in response to ex vivo stimulation (46). What the implications of these findings are, is currently not understood. However, although levels of procoagulant platelet potential are decreased in patients with COVID-19 early in the disease time course, they increase over time (46, 59). Furthermore, patients with COVID-19 with higher levels of procoagulant platelet potential were at increased risk for death (59). In agreement with this, baseline circulating levels of procoagulant platelets are significantly increased in severe COVID-19 compared with healthy donors as well as to other critically ill patient populations (60). Importantly, the level of circulating PS-positive platelets positively correlates with D-dimer levels and illness severity as well as associates with thrombotic complications (60). Interestingly, procoagulant platelet formation is mediated by COVID-19-induced antibody formation (60–62). Circulating immunoglobulins (IgGs) during COVID-19 activate platelets through FcγRIIa, altering cytoplasmic calcium and cyclic adenosine-monophosphate (cAMP) levels and activating AKT (61, 62). Therapeutically, inhibition of cAMP with IIoprost (62) or blocking AKT phosphorylation (61), inhibits COVID-19 IgG-induced procoagulant platelet formation.
HIT is an immune-mediated thrombocytopenia mediated by IgGs against platelet factor 4 (PF4)-heparin complexes, which in turn activates platelets through FcγRIIa (63). IgG-dependent platelet activation can result in aggregation and the release of procoagulant microparticle particles, which can participate in thrombin generation to promote a thrombotic environment (64). Interestingly, HIT antibodies in complex with PF4 are capable of inducing procoagulant platelet formation (65, 66). This is dependent on platelet FcγRIIa and the generation of monocyte-derived tissue factor and monocyte-supported thrombin generation (65, 67, 68). In a separate study, HIT plasma only induces procoagulant platelets in the presence of heparin and PAR stimulation (PAR1 or ADP) (65, 66). Mechanistically, HIT IgG binding to FcγRIIa induces procoagulant platelet formation, through robust calcium flux, resulting in calpain activation (64, 66). More recently, a similar procoagulant platelet phenotype was observed in patients who developed VITT after adenoviral-based COVID-19 vaccine administration (69, 70). Sera from patients with VITT contain high levels of anti-PF4 antibody and these antibodies significantly increase procoagulant platelet formation compared with healthy donors (69, 71). Similar to HIT, this is dependent on the presence of PF4 and heparin and the binding of antibody complexes to platelet FcγRIIa (69, 71). A similar finding of VITT-induced procoagulant platelet formation was also observed by Lee et al. (71), but also required PAR1 signaling. Although these studies suggest a role for procoagulant platelet formation in the pathophysiology of HIT and VITT, additional studies are necessary to mechanistically delineate their role in the disease process.
CONCLUSIONS AND PERSPECTIVES
Procoagulant platelets are increasingly recognized as mediators of thromboinflammation and therefore therapeutic targets for thromboinflammatory diseases. The discrepancy between procoagulant platelets and aggregating platelets and how they are distinctly formed raises the possibility that targeting procoagulant platelets could block pathological thrombosis while keeping hemostasis intact. However, there are currently no pharmacological inhibitors available that only block procoagulant platelet formation. Given the bleeding phenotype observed in patients with Scott syndrome (37), general scramblase blocking using TMEM16F inhibitors may not be ideal. However, drugs such as cyclosporin A (26) and acetazolamide (31) are FDA-approved drugs that, as a side effect, efficiently block procoagulant platelet formation without known bleeding consequences. In contrast, desmopressin has been shown to increase procoagulant platelet formation and reduce bleeding (36, 72). Nevertheless, a thorough characterization of these molecules in models of thrombosis, hemostasis, and thromboinflammation is needed to determine their safety and efficacy profile.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants K01AG059892 and R01HL163019 (to R.A.C.) and the American Heart Foundation Grant 2021Post830138 (to F.D).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
F.D. and R.A.C. prepared figures; F.D. and R.A.C. drafted manuscript; F.D. and R.A.C. edited and revised manuscript; F.D. and R.A.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Diana Lim for assistance with the figure preparation.
REFERENCES
- 1. Alberio L, Safa O, Clemetson KJ, Esmon CT, Dale GL. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood 95: 1694–1702, 2000. [PubMed] [Google Scholar]
- 2. Dale GL, Friese P, Batar P, Hamilton SF, Reed GL, Jackson KW, Clemetson KJ, Alberio L. Stimulated platelets use serotonin to enhance their retention of procoagulant proteins on the cell surface. Nature 415: 175–179, 2002. doi: 10.1038/415175a. [DOI] [PubMed] [Google Scholar]
- 3. Josefsson EC, Burnett DL, Lebois M, Debrincat MA, White MJ, Henley KJ, Lane RM, Moujalled D, Preston SP, O'Reilly LA, Pellegrini M, Metcalf D, Strasser A, Kile BT. Platelet production proceeds independently of the intrinsic and extrinsic apoptosis pathways. Nat Commun 5: 3455, 2014. doi: 10.1038/ncomms4455. [DOI] [PubMed] [Google Scholar]
- 4. Pleines I, Lebois M, Gangatirkar P, Au AE, Lane RM, Henley KJ, Kauppi M, Corbin J, Cannon P, Bernardini J, Alwis I, Jarman KE, Ellis S, Metcalf D, Jackson SP, Schoenwaelder SM, Kile BT, Josefsson EC. Intrinsic apoptosis circumvents the functional decline of circulating platelets but does not cause the storage lesion. Blood 132: 197–209, 2018. doi: 10.1182/blood-2017-11-816355. [DOI] [PubMed] [Google Scholar]
- 5. Pasalic L, Wing-Lun E, Lau JK, Campbell H, Pennings GJ, Lau E, Connor D, Liang HP, Muller D, Kritharides L, Hogg PJ, Chen VM. Novel assay demonstrates that coronary artery disease patients have heightened procoagulant platelet response. J Thromb Haemost 16: 1198–1210, 2018. doi: 10.1111/jth.14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lhermusier T, Chap H, Payrastre B. Platelet membrane phospholipid asymmetry: from the characterization of a scramblase activity to the identification of an essential protein mutated in Scott syndrome. J Thromb Haemost 9: 1883–1891, 2011. doi: 10.1111/j.1538-7836.2011.04478.x. [DOI] [PubMed] [Google Scholar]
- 7. Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood 130: 2171–2179, 2017. doi: 10.1182/blood-2017-05-787259. [DOI] [PubMed] [Google Scholar]
- 8. Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468: 834–838, 2010. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- 9. Takatsu H, Takayama M, Naito T, Takada N, Tsumagari K, Ishihama Y, Nakayama K, Shin H-W. Phospholipid flippase ATP11C is endocytosed and downregulated following Ca2+-mediated protein kinase C activation. Nat Commun 8: 1423, 2017. doi: 10.1038/s41467-017-01338-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Millington-Burgess SL, Harper MT. Maintaining flippase activity in procoagulant platelets is a novel approach to reducing thrombin generation. J Thromb Haemost 20: 989–995, 2022. doi: 10.1111/jth.15641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mattheij NJA, Braun A, Kruchten R, Castoldi E, Pircher J, Baaten CCFMJ, Wülling M, Kuijpers MJE, Köhler R, Poole AW, Schreiber R, Vortkamp A, Collins PW, Nieswandt B, Kunzelmann K, Cosemans JMEM, Heemskerk JWM. Survival protein anoctamin-6 controls multiple platelet responses including phospholipid scrambling, swelling, and protein cleavage. FASEB J 30: 727–737, 2016. doi: 10.1096/fj.15-280446. [DOI] [PubMed] [Google Scholar]
- 12. Veuthey L, Aliotta A, Bertaggia Calderara D, Pereira Portela C, Alberio L. Mechanisms underlying dichotomous procoagulant COAT platelet generation-a conceptual review summarizing current knowledge. IJMS 23: 2536, 2022. doi: 10.3390/ijms23052536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fernández DI, Kuijpers MJE, Heemskerk JWM. Platelet calcium signaling by G-protein coupled and ITAM-linked receptors regulating anoctamin-6 and procoagulant activity. Platelets 32: 863–871, 2021. doi: 10.1080/09537104.2020.1859103. [DOI] [PubMed] [Google Scholar]
- 14. Brouns SLN, van Geffen JP, Heemskerk JWM. High-throughput measurement of human platelet aggregation under flow: application in hemostasis and beyond. Platelets 29: 662–669, 2018. doi: 10.1080/09537104.2018.1447660. [DOI] [PubMed] [Google Scholar]
- 15. Nagy M, Perrella G, Dalby A, Becerra MF, Garcia Quintanilla L, Pike JA, Morgan N. V, Gardiner EE, Heemskerk JWM, Azócar L, Miquel JF, Mezzano D, Watson SP. Flow studies on human GPVI-deficient blood under coagulating and noncoagulating conditions. Blood Adv 4: 2953–2961, 2020. doi: 10.1182/bloodadvances.2020001761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roka-Moiia Y, Walk R, Palomares DE, Ammann KR, Dimasi A, Italiano JE, Sheriff J, Bluestein D, Slepian MJ. Platelet activation via shear stress exposure induces a differing pattern of biomarkers of activation versus biochemical agonists. Thromb Haemost 120: 776–792, 2020. doi: 10.1055/s-0040-1709524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Delaney MK, Liu J, Kim K, Shen B, Stojanovic-Terpo A, Zheng Y, Cho J, Du X. Agonist-induced platelet procoagulant activity requires shear and a Rac1-dependent signaling mechanism. Blood 124: 1957–1967, 2014. doi: 10.1182/blood-2014-03-560821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pang A, Cui Y, Chen Y, Cheng N, Delaney MK, Gu M, Stojanovic-Terpo A, Zhu C, Du X. Shear-induced integrin signaling in platelet phosphatidylserine exposure, microvesicle release, and coagulation. Blood 132: 533–543, 2018. doi: 10.1182/blood-2017-05-785253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cosemans JMEM, Schols SEM, Stefanini L, de Witt S, Feijge MAH, Hamulyák K, Deckmyn H, Bergmeier W, Heemskerk JWM. Key role of glycoprotein Ib/V/IX and von Willebrand factor in platelet activation-dependent fibrin formation at low shear flow. Blood 117: 651–660, 2011. doi: 10.1182/blood-2010-01-262683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Deppermann C, Cherpokova D, Nurden P, Schulz J-N, Thielmann I, Kraft P, Vögtle T, Kleinschnitz C, Dütting S, Krohne G, Eming SA, Nurden AT, Eckes B, Stoll G, Stegner D, Nieswandt B. Gray platelet syndrome and defective thrombo-inflammation in Nbeal2-deficient mice. J Clin Invest 123: 3331–3342, 2013. doi: 10.1172/JCI69210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mammadova-Bach E, Nagy M, Heemskerk JWM, Nieswandt B, Braun A. Store-operated calcium entry in thrombosis and thrombo-inflammation. Cell Calcium 77: 39–48, 2019. doi: 10.1016/j.ceca.2018.11.005. [DOI] [PubMed] [Google Scholar]
- 22. Choo H-J, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol 32: 2946–2955, 2012. doi: 10.1161/ATVBAHA.112.300433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kholmukhamedov A, Janecke R, Choo H-J, Jobe SM. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J Thromb Haemost 16: 2315–2321, 2018. doi: 10.1111/jth.14284. [DOI] [PubMed] [Google Scholar]
- 24. Abbasian N, Millington-Burgess SL, Chabra S, Malcor J-D, Harper MT. Supramaximal calcium signaling triggers procoagulant platelet formation. Blood Adv 4: 154–164, 2020. doi: 10.1182/bloodadvances.2019000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aliotta A, Bertaggia Calderara D, Zermatten MG, Alberio L. Sodium-calcium exchanger reverse mode sustains dichotomous ion fluxes required for procoagulant COAT platelet formation. Thromb Haemost 121: 309–321, 2021. doi: 10.1055/s-0040-171670. [DOI] [PubMed] [Google Scholar]
- 26. Remenyi G, Szasz R, Friese P, Dale GL. Role of mitochondrial permeability transition pore in coated-platelet formation. Arterioscler Thromb Vasc Biol 25: 467–471, 2005. doi: 10.1161/01.ATV.0000152726.49229.bf. [DOI] [PubMed] [Google Scholar]
- 27. Jobe SM, Wilson KM, Leo L, Raimondi A, Molkentin JD, Lentz SR, di Paola J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 111: 1257–1265, 2008. doi: 10.1182/blood-2007-05-092684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang WH, Stitham J, Jin Y, Liu R, Lee SH, Du J, Atteya G, Gleim S, Spollett G, Martin K, Hwa J. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation 129: 1598–1609, 2014. doi: 10.1161/CIRCULATIONAHA.113.005224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang M, Kholmukhamedov A, Schulte ML, Cooley BC, Scoggins NO, Wood JP, Cameron SJ, Morrell CN, Jobe SM, Silverstein RL. Platelet CD36 signaling through ERK5 promotes caspase-dependent procoagulant activity and fibrin deposition in vivo. Blood Adv 2: 2848–2861, 2018. doi: 10.1182/bloodadvances.2018025411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Denorme F, Portier I, Kosaka Y, Campbell RA. Hyperglycemia exacerbates ischemic stroke outcome independent of platelet glucose uptake. J Thromb Haemost 19: 536–546, 2021. doi: 10.1111/jth.15154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Agbani EO, van den Bosch MTJ, Brown E, Williams CM, Mattheij NJA, Cosemans JMEM, Collins PW, Heemskerk JWM, Hers I, Poole AW. Coordinated membrane ballooning and procoagulant spreading in human platelets. Circulation 132: 1414–1424, 2015. doi: 10.1161/CIRCULATIONAHA.114.015036. [DOI] [PubMed] [Google Scholar]
- 32. Heemskerk JW, Vuist WM, Feijge MA, Reutelingsperger CP, Lindhout T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood 90: 2615–2625, 1997. [PubMed] [Google Scholar]
- 33. Zhang W, Zitron E, Hömme M, Kihm L, Morath C, Scherer D, Hegge S, Thomas D, Schmitt CP, Zeier M, Katus H, Karle C, Schwenger V. Aquaporin-1 channel function is positively regulated by protein kinase C. J Biol Chem 282: 20933–20940, 2007. doi: 10.1074/jbc.M703858200. [DOI] [PubMed] [Google Scholar]
- 34. Agbani EO, Williams CM, Li Y, van den Bosch MT, Moore SF, Mauroux A, Hodgson L, Verkman AS, Hers I, Poole AW. Aquaporin-1 regulates platelet procoagulant membrane dynamics and in vivo thrombosis. JCI Insight 3, 2018. doi: 10.1172/jci.insight.99062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Daskalakis M, Colucci G, Keller P, Rochat S, Silzle T, Biasiutti FD, Barizzi G, Alberio L. Decreased generation of procoagulant platelets detected by flow cytometric analysis in patients with bleeding diathesis. Cytometry B Clin Cytom 86: 397–409, 2014. doi: 10.1002/cyto.b.21157. [DOI] [PubMed] [Google Scholar]
- 36. Segot A, Adler M, Aliotta A, Matthey-Guirao E, Nagler M, Bertaggia Calderara D, Grandoni F, Gomez FJ, Alberio L. Low COAT platelets are frequent in patients with bleeding disorders of unknown cause (BDUC) and can be enhanced by DDAVP. J Thromb Haemost 20: 1271–1274, 2022. doi: 10.1111/jth.15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Toti F, Satta N, Fressinaud E, Meyer D, Freyssinet JM. Scott syndrome, characterized by impaired transmembrane migration of procoagulant phosphatidylserine and hemorrhagic complications, is an inherited disorder. Blood 87: 1409–1415, 1996. [PubMed] [Google Scholar]
- 38. Yang H, Kim A, David T, Palmer D, Jin T, Tien J, Huang F, Cheng T, Coughlin SR, Jan YN, Jan LY. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 151: 111–122, 2012. doi: 10.1016/j.cell.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baig AA, Haining EJ, Geuss E, Beck S, Swieringa F, Wanitchakool P, Schuhmann MK, Stegner D, Kunzelmann K, Kleinschnitz C, Heemskerk JWM, Braun A, Nieswandt B. TMEM16F-mediated platelet membrane phospholipid scrambling is critical for hemostasis and thrombosis but not thromboinflammation in mice-brief report. Arterioscler Thromb Vasc Biol 36: 2152–2157, 2016. doi: 10.1161/ATVBAHA.116.307727. [DOI] [PubMed] [Google Scholar]
- 40. van Kruchten R, Mattheij NJA, Saunders C, Feijge MAH, Swieringa F, Wolfs JLN, Collins PW, Heemskerk JWM, Bevers EM. Both TMEM16F-dependent and TMEM16F-independent pathways contribute to phosphatidylserine exposure in platelet apoptosis and platelet activation. Blood 121: 1850–1857, 2013. doi: 10.1182/blood-2012-09-454314. [DOI] [PubMed] [Google Scholar]
- 41. Kuijpers MJE, Munnix ICA, Cosemans JMEM, Vlijmen B. V, Reutelingsperger CPM, Egbrink MO, Heemskerk JWM. Key role of platelet procoagulant activity in tissue factor-and collagen-dependent thrombus formation in arterioles and venules in vivo differential sensitivity to thrombin inhibition. Microcirculation 15: 269–282, 2008. doi: 10.1080/10739680701653517. [DOI] [PubMed] [Google Scholar]
- 42. Shi J, Pipe SW, Rasmussen JT, Heegaard CW, Gilbert GE. Lactadherin blocks thrombosis and hemostasis in vivo: correlation with platelet phosphatidylserine exposure. J Thromb Haemost 6: 1167–1174, 2008. doi: 10.1111/j.1538-7836.2008.03010.x. [DOI] [PubMed] [Google Scholar]
- 43. Fujii T, Sakata A, Nishimura S, Eto K, Nagata S. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc Natl Acad Sci USA 112: 12800–12805, 2015. doi: 10.1073/pnas.1516594112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hua VM, Abeynaike L, Glaros E, Campbell H, Pasalic L, Hogg PJ, Chen VMY. Necrotic platelets provide a procoagulant surface during thrombosis. Blood 126: 2852–2862, 2015. doi: 10.1182/blood-2015-08-663005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Skaer C, Kholmukhamedov A, Liu F, Jobe SM. Platelet cyclophilin D-dependent events limit venous thrombotic occlusion and platelet accretion. Blood 130: 454, 2017.doi: 10.1182/blood.V130.Suppl_1.454.454. [DOI] [Google Scholar]
- 46. Denorme F, Manne BK, Portier I, Petrey AC, Middleton EA, Kile BT, Rondina MT, Campbell RA. COVID‐19 patients exhibit reduced procoagulant platelet responses. J Thromb Haemost 18: 3067–3073, 2020. doi: 10.1111/jth.15107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo L, Rondina MT. The era of thromboinflammation: platelets are dynamic sensors and effector cells during infectious diseases. Front Immunol 10: 2204, 2019. doi: 10.3389/fimmu.2019.02204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Denorme F, Rustad JL, Campbell RA. Brothers in arms: platelets and neutrophils in ischemic stroke. Curr Opin Hematol 28: 301–307, 2021. doi: 10.1097/MOH.0000000000000665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kulkarni S, Woollard KJ, Thomas S, Oxley D, Jackson SP. Conversion of platelets from a proaggregatory to a proinflammatory adhesive phenotype: role of PAF in spatially regulating neutrophil adhesion and spreading. Blood 110: 1879–1886, 2007. doi: 10.1182/blood-2006-08-040980. [DOI] [PubMed] [Google Scholar]
- 50. Yuan Y, Alwis I, Wu MCL, Kaplan Z, Ashworth K, Bark D, Pham A, Mcfadyen J, Schoenwaelder SM, Josefsson EC, Kile BT, Jackson SP. Neutrophil macroaggregates promote widespread pulmonary thrombosis after gut ischemia. Sci Transl Med 9: eaam5861, 2017. doi: 10.1126/scitranslmed.aam5861. [DOI] [PubMed] [Google Scholar]
- 51. Denorme F, Manne BK, Portier I, Eustes AS, Kosaka Y, Kile BT, Rondina MT, Campbell RA. Platelet necrosis mediates ischemic stroke outcome in mice. Blood 135: 429–440, 2020. doi: 10.1182/blood.2019002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Denorme F, Portier I, Rustad JL, Cody MJ, de Araujo C. V, Hoki C, Alexander MD, Grandhi R, Dyer MR, Neal MD, Majersik JJ, Yost CC, Campbell RA. Neutrophil extracellular traps regulate ischemic stroke brain injury. J Clin Invest 132: e154225, 2022. doi: 10.1172/JCI154225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kaiser R, Escaig R, Kranich J, Hoffknecht M-L, Anjum A, Polewka V, Mader M, Hu W, Belz L, Gold C, Titova A, Lorenz M, Pekayvaz K, Kääb S, Gaertner F, Stark K, Brocker T, Massberg S, Nicolai L. Procoagulant platelet sentinels prevent inflammatory bleeding through GPIIBIIIA and GPVI. Blood 140: 121–139, 2022. doi: 10.1182/blood.2021014914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kirkpatrick AC, Vincent AS, Dale GL, Prodan CI. Increased platelet procoagulant potential predicts recurrent stroke and TIA after lacunar infarction. J Thromb Haemost 18: 660–668, 2020. doi: 10.1111/jth.14714. [DOI] [PubMed] [Google Scholar]
- 55. Kirkpatrick AC, Tafur AJ, Vincent AS, Dale GL, Prodan CI. Coated-platelets improve prediction of stroke and transient ischemic attack in asymptomatic internal carotid artery stenosis. Stroke 45: 2995–3001, 2014. doi: 10.1161/STROKEAHA.114.006492. [DOI] [PubMed] [Google Scholar]
- 56. Prodan CI, Stoner JA, Dale GL. Acute hemorrhagic complications are associated with lower coated-platelet levels in non-lacunar brain infarction. J Thromb Haemost 13: 2233–2239, 2015. doi: 10.1111/jth.13160. [DOI] [PubMed] [Google Scholar]
- 57. Prodan CI, Stoner JA, Dale GL. Lower coated-platelet levels are associated with increased mortality after spontaneous intracerebral hemorrhage. Stroke 46: 1819–1825, 2015. doi: 10.1161/STROKEAHA.115.009068. [DOI] [PubMed] [Google Scholar]
- 58. Portier I, Campbell RA, Denorme F. Mechanisms of immunothrombosis in COVID-19. Curr Opin Hematol 28: 445–453, 2021. doi: 10.1097/MOH.0000000000000666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Khattab MH, Prodan CI, Vincent AS, Xu C, Jones KR, Thind S, Rabadi M, Mithilesh S, Mathews E, Guthery L, Dale GL, Kirkpatrick AC. Increased procoagulant platelet levels are predictive of death in COVID-19. Geroscience 43: 2055–2065, 2021. doi: 10.1007/s11357-021-00385-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Althaus K, Marini I, Zlamal J, Pelzl L, Singh A, Häberle H, Mehrländer M, Hammer S, Schulze H, Bitzer M, Malek N, Rath D, Bösmüller H, Nieswandt B, Gawaz M, Bakchoul T, Rosenberger P. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood 137: 1061–1071, 2021. doi: 10.1182/blood.2020008762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pelzl L, Singh A, Funk J, Witzemann A, Marini I, Zlamal J, Weich K, Abou-Khalel W, Hammer S, Uzun G, Althaus K, Bakchoul T. Antibody-mediated procoagulant platelet formation in COVID-19 is AKT dependent. J Thromb Haemost 20: 387–398, 2022. doi: 10.1111/jth.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zlamal J, Althaus K, Jaffal H, Häberle H, Pelzl L, Singh A, Witzemann A, Weich K, Bitzer M, Malek N, Göpel S, Bösmüller H, Gawaz M, Mirakaj V, Rosenberger P, Bakchoul T. Upregulation of cAMP prevents antibody-mediated thrombus formation in COVID-19. Blood Adv 6: 248–258, 2022. doi: 10.1182/bloodadvances.2021005210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Arepally GM, Padmanabhan A. Heparin-induced thrombocytopenia: a focus on thrombosis. Arterioscler Thromb Vasc Biol 41: 141–152, 2021. doi: 10.1161/ATVBAHA.120.315445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nevzorova TA, Mordakhanova ER, Daminova AG, Ponomareva AA, Andrianova IA, Le Minh G, Rauova L, Litvinov RI, Weisel JW. Platelet factor 4-containing immune complexes induce platelet activation followed by calpain-dependent platelet death. Cell Death Discov 5: 106, 2019. doi: 10.1038/s41420-019-0188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tutwiler V, Madeeva D, Ahn HS, Andrianova I, Hayes V, Zheng XL, Cines DB, McKenzie SE, Poncz M, Rauova L. Platelet transactivation by monocytes promotes thrombosis in heparin-induced thrombocytopenia. Blood 127: 464–472, 2016. doi: 10.1182/blood-2013-11-539262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee CSM, Selvadurai M. V, Pasalic L, Yeung J, Konda M, Kershaw GW, Favaloro EJ, Chen VM. Measurement of procoagulant platelets provides mechanistic insight and diagnostic potential in heparin-induced thrombocytopenia. J Thromb Haemost 20: 975–988, 2022. doi: 10.1111/jth.15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kasthuri RS, Glover SL, Jonas W, McEachron T, Pawlinski R, Arepally GM, Key NS, Mackman N. PF4/heparin-antibody complex induces monocyte tissue factor expression and release of tissue factor positive microparticles by activation of FcγRI. Blood 119: 5285–5293, 2012. doi: 10.1182/blood-2011-06-359430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pouplard C, Iochmann S, Renard B, Herault O, Colombat P, Amiral J, Gruel Y. Induction of monocyte tissue factor expression by antibodies to heparin-platelet factor 4 complexes developed in heparin-induced thrombocytopenia. Blood 97: 3300–3302, 2001. doi: 10.1182/blood.v97.10.3300. [DOI] [PubMed] [Google Scholar]
- 69. Althaus K, Möller P, Uzun G, Singh A, Beck A, Bettag M, Bösmüller H, Guthoff M, Dorn F, Petzold GC, Henkes H, Heyne N, Jumaa H, Kreiser K, Limpach C, Luz B, Maschke M, Müller JA, Münch J, Nagel S, Pötzsch B, Müller J, Schlegel C, Viardot A, Bäzner H, Wolf M, Pelzl L, Warm V, Willinek WA, Steiner J, Schneiderhan-Marra N, Vollherbst D, Sachs UJ, Fend F, Bakchoul T. Antibody-mediated procoagulant platelets in SARS-CoV-2-vaccination associated immune thrombotic thrombocytopenia. Haematologica 106: 2170–2179, 2021. doi: 10.3324/haematol.2021.279000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Huynh A, Kelton JG, Arnold DM, Daka M, Nazy I. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopaenia. Nature 596: 565–569, 2021. doi: 10.1038/s41586-021-03744-4. [DOI] [PubMed] [Google Scholar]
- 71. Lee CSM, Liang HPH, Connor DE, Dey A, Tohidi-Esfahani I, Campbell H, Whittaker S, Capraro D, Favaloro EJ, Donikian D, Kondo M, Hicks SM, Choi PY-I, Gardiner EE, Clarke LJ, Tran H, Passam FH, Brighton TA, Chen VM. A novel flow cytometry procoagulant assay for diagnosis of vaccine-induced immune thrombotic thrombocytopenia. Blood Adv 6: 3494–3506, 2022. doi: 10.1182/bloodadvances.2021006698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Colucci G, Stutz M, Rochat S, Conte T, Pavicic M, Reusser M, Giabbani E, Huynh A, Thürlemann C, Keller P, Alberio L. The effect of desmopressin on platelet function: a selective enhancement of procoagulant COAT platelets in patients with primary platelet function defects. Blood 123: 1905–1916, 2014. doi: 10.1182/blood-2013-04-497123. [DOI] [PubMed] [Google Scholar]