

Keywords: amino acid transport, aristolochic acid, B0AT1, nephropathy, proximal tubule
Abstract
B0AT1 (Slc6a19) mediates absorption of neutral amino acids in the small intestine and in the kidneys, where it is primarily expressed in early proximal tubules (S1–S2). To determine the role of B0AT1 in nephropathy induced by aristolochic acid (AA), which targets the proximal tubule, littermate female B0AT1-deficient (Slc6a19−/−), heterozygous (Slc6a19+/−), and wild-type (WT) mice were administered AA (10 mg/kg ip) or vehicle every 3 days for 3 wk, and analyses were performed after the last injection or 3 wk later. Vehicle-treated mice lacking Slc6a19 showed normal body and kidney weight and plasma creatinine versus WT mice. The urinary glucose-to-creatinine ratio (UGCR) and urinary albumin-to-creatinine ratio (UACR) were two to four times higher in vehicle-treated Slc6a19−/− versus WT mice, associated with lesser expression of early proximal transporters Na+-glucose cotransporter 2 and megalin, respectively. AA caused tubular injury independently of B0AT1, including robust increases in cortical mRNA expression of p53, p21, and hepatitis A virus cellular receptor 1 (Havcr1), downregulation of related proximal tubule amino acid transporters B0AT2 (Slc6a15), B0AT3 (Slc6a18), and Slc7a9, and modest histological tubular damage and a rise in plasma creatinine. Absence of B0AT1, however, attenuated AA-induced cortical upregulation of mRNA markers of senescence (p16), inflammation [lipocalin 2 (Lcn2), C-C motif chemokine ligand 2 (Ccl2), and C-C motif chemokine receptor 2 (Ccr2)], and fibrosis [tissue inhibitor of metallopeptidase 1 (Timp1), transforming growth factor-β1 (Tgfb1), and collagen type I-α1 (Col1a1)], associated with lesser fibrosis staining, lesser suppression of proximal tubular organic anion transporter 1, restoration of Na+-glucose cotransporter 2 expression, and prevention of the AA-induced fivefold increase in the urinary albumin-to-creatinine ratio observed in WT mice. The data suggest that proximal tubular B0AT1 is important for the physiology of renal glucose and albumin retention but potentially deleterious for the kidney response following AA-induced kidney injury.
NEW & NOTEWORTHY Based on insights from studies manipulating glucose transport, the hypothesis has been proposed that inhibiting intestinal uptake or renal reabsorption of energy substrates has unique therapeutic potential to improve metabolic disease and kidney outcome in response to injury. The present study takes this idea to B0AT1, the major transporter for neutral amino acids in the intestine and kidney, and shows that its absence attenuates aristolochic acid-induced nephropathy.
INTRODUCTION
Chronic kidney disease (CKD) is one of the leading health problems. It affects ∼47 million people in the United States, which represents ∼15% of the total US adult population (1). Acute kidney injury (AKI) is associated with an increased risk of CKD (2, 3). For example, AKI episodes in patients with diabetes enhance the cumulative risk for developing advanced CKD, independently of other major risk factors of progression (4). CKD is an insidious disease with little symptoms over many years, and the factors involved in the progression of the disease are poorly understood. Therefore, studying and characterizing the molecular mechanisms underlying AKI and the transition to CKD is important to better understand the pathophysiology and develop potential novel therapies.
Meta-analyses and large clinical outcome trials have demonstrated that inhibitors of Na+-glucose cotransporter 2 (SGLT2), a Na+-dependent transporter that reabsorbs >90% of all filtered glucose in the early proximal tubule of the kidney, can reduce the risk of AKI and the development of CKD largely independent of their effect on blood glucose levels (5). Although the involved mechanisms of kidney protection remain to be fully delineated, the success of SGLT2 inhibitors may point to a particular deleterious role of Na+-coupled transport processes of energy substrates in the early proximal tubule.
Besides glucose, the proximal tubule avidly reabsorbs amino acids via Na+-dependent cotransport. This includes members of the SLC6 transporter family, such as B0AT1 (Slc6a19), B0AT2 (Slc6a15), and B0AT3 (Slc6a18 functional in mice but not in humans), which transport neutral amino acids by Na+-dependent transport (6–10). Among these, B0AT1, which is primarily expressed in the early proximal tubule (11, 12), plays a dominant role not only in renal but also in intestinal transport of neutral amino acids (13). Mutations in SLC6A19 lead to Hartnup disease, a metabolic disorder that is the consequence of reduced intestinal uptake of neutral amino acids combined with their urinary loss and that can manifest primarily in children with malnourishment (14). On the other hand, mice that lack Slc6a19 not only show the expected attenuated postprandial intestinal uptake of neutral amino acids and their continuous loss into the urine but also improved glycemic control, in part due to higher postprandial blood levels of glucagon-like peptide-1 (GLP-1) and greater liver release of the starvation hormone fibroblast growth factor 21 (FGF21) (15). Moreover, B0AT1 has a high affinity for the branched-chain amino acids (BCAA) valine, isoleucine, and leucine (8), the restriction of which is thought to improve metabolic health by inhibiting the mammalian target of rapamycin complex 1 and facilitating autophagy (16). Finally, genetic variants in SLC6A19 have been associated with reduced serum levels of creatinine, suggesting that loss of function of B0AT1 may potentially confer protection against CKD (17).
Aristolochic acid (AA) is a substance found in Chinese medicinal herb that can cause AKI, severe tubulointerstitial fibrosis, and CKD in both humans and mice (18). It is responsible for the so-called Balkan-endemic nephropathy caused by wheat contaminated with seeds of Aristolochia clematitis (18). Enzymatic nitroreduction of AA generates a reactive nitrenium intermediate that is capable of forming adducts with DNA (19), which induces a p53-mediated DNA damage response, thereby impairing the functionality and integrity of proximal tubular epithelial cells (20). As a consequence, AA enhances tubular markers of injury like kidney injury molecule-1 (KIM-1) as well as various proinflammatory and profibrotic cytokines including C-C motif chemokine ligand 2 (CCL2) and transforming growth factor-β1 (TGF-β1) (21). The susceptibility of proximal tubule segments to the deleterious effects of AA differs between species: in mice, the S2 segment of the proximal convoluted tubule appears to be the primary site of action (22), whereas the S3 segment seems most vulnerable in rats (23), with the human preference remaining unknown. Moreover, AA has a greater potency in male than female mice, including dose-dependent increases in serum creatinine and the urinary albumin-to-creatinine ratio (UACR) (24–26). Finally, suppression of proximal tubule BCAA catabolism has been linked to exacerbating kidney injury and fibrosis in response to AA (27), indicating a potential link to B0AT1.
In the present study, female mice with whole body gene targeting of Slc6a19 (Slc6a19−/−) (8, 15) were compared with littermate controls and heterozygous mice to better understand the functional contribution of this transporter to kidney physiology beyond amino acid transport and also to kidney pathophysiology. More specifically, this study aimed to determine whether the absence of B0AT1 can protect kidneys from injury, inflammation, or fibrosis when exposed to AA.
METHODS
Animals
Information provided was guided by the Animal Research: Reporting of In Vivo Experiments Guidelines checklist (28). All animal experiments were conducted in accordance with the National Institutes of Health (NIH) Guide for Care and Use of Laboratory Animals (NIH, Bethesda, MD) and were approved by the Veterans Affairs San Diego Healthcare System Institutional Animal Care and Use Committee. Experiments were performed on 10- to 17-wk-old female B0AT1-deficient (Slc6a19−/−) mice and littermate B0AT1 heterozygous (Slc6a19+/−) and wild-type (WT) mice, all on the C57BL/6 background. The generation of this mouse line has been previously described (29), and a license was purchased from Deltagen. All animals were housed in the same room with a 12:12-h light-dark cycle and free access to water and regular rodent chow (7001, Envigo, Indianapolis, IN). All mice that entered the study also completed the study.
Exposure of Mice to AA
After basal measurements, mice were randomized to intraperitoneal injection of 10 mg/kg of AA-I sodium salt (A9451, Sigma-Aldrich, St. Louis, MO) or vehicle (NaCl 0.9% Sodium Chloride Injection, Hospira) every 3 days for 3 wk and were then left for another 3 wk to recover, similarly to other AA models in female mice (21, 24, 27). Measurements were taken 3 days after the last injection to study the active phase and 3 wk after the last injection to study the recovery phase (Fig. 1).
Figure 1.

Experimental timeline. Slc6a19−/−, Slc6a19+/−, and wild-type female mice were given an intraperitoneal injection of 10 mg/kg aristolochic acid (AA) or vehicle (shown by arrowheads) every 3 days for 3 wk and were then left for another 3 wk to recover. Prior to the first injections, measurements were taken to obtain basal values (“Basal”). This was followed by measurements 3 days after the last injection to study the active phase (“After injections”) and 3 wk after the last injection when mice were euthanized and kidneys, hearts, and livers were harvested to study the recovery/chronic phase (“Harvest”). Urine and blood collections were paired with body weight to obtain basal values as well as after the injections and at harvest. Plasma creatinine concentration and urinary albumin-to-creatinine ratio levels were analyzed at the three time points. Kidney cortexes and medullas were analyzed by Masson’s trichrome staining, kidney cortexes used for quantitative RT-PCR analyses, and Western blot analysis was performed on membrane preparations of the whole kidney.
Urine and Blood Collection and Organ Harvest
Circadian changes affect kidney function. Therefore, we performed all AA injections and collected all samples at the same time of day for all of the groups, typically between 9:00 and 10:00 AM. Urine and blood collections were paired with body weight measurements the week before the first AA injection to obtain basal values and repeated 3 days and 3 wk after the last AA injection. Urine was obtained by gently grabbing the mice and inducing spontaneous urination, collected in a clean petri dish (Corning, Corning, NY). Samples were stored at −80°C for further analyses. The first two blood samples were collected under short isoflurane anesthesia (80 µL/collection), whereas the blood collection after 3 wk was performed under terminal anesthesia (160 µL/collection) before organ harvest, all by retroorbital plexus puncture. Capillaries were sealed and centrifuged to measure hematocrit, and plasma was stored at −80°C for further analyses. Three weeks after the last AA injection, terminal anesthesia was induced by injection of a ketamine (Henry Schein Animal Health, Dublin, OH) and xylazine (Bimeda-MTC Animal Health, Cambridge, ON, Canada) cocktail (100 mg/kg and 10 mg/kg, respectively, 10 µL/g ip). After confirmation of deep anesthesia, blood was collected by retroorbital puncture and the kidneys, heart, and liver were harvested, their weights were determined, and they were rapidly frozen in liquid nitrogen.
Plasma and Urine Analyses
Plasma creatinine concentration was determined by isotope dilution liquid chromatography-tandem mass spectrometry (LC-MS/MS) at the O’Brien Center for Acute Kidney Injury Research at the University of Alabama at Birmingham (Birmingham, AL). Concentrations of albumin and glucose in urine were measured using commercial assays (Bethyl Laboratories, Montgomery, TX, and Thermo Scientific, Louisville, CO), and urine creatinine was measured by the kinetics of the alkaline picrate Jaffe reaction (Infinity, Thermo Scientific). Urinary albumin/creatinine was used to evaluate albuminuria and urinary glucose/creatinine was used to assess glucosuria.
Reverse Transcription and Real-Time PCR
Kidney cortex RNA was isolated using the RNeasy Plus Mini Kit (Qiagen, Germantown, MD), and cDNA was prepared using the SuperScript IV First-Strand Synthesis System (Thermo Scientific). For quantification, TaqMan Universal PCR Master Mix (Applied Biosystems, Warrington, UK) and TaqMan Gene Expression Assay primers (Thermo Scientific) were used in a QuantStudio 6 Real-Time PCR System (2 min at 50°C and 10 min at 95°C with 40 cycles of 15 s at 95°C and 1 min at 60°C). Each experiment was performed in duplicate to determine the relative expression of specific genes (see Table 1). Expression levels were expressed as relative fold increases/decreases normalized to the housekeeping gene ribosomal protein L19 (Rpl19), which was not different between groups (not shown).
Table 1.
Real-time PCR primers used
| Target Gene | Assay ID |
|---|---|
| Ccl2 (MCP-1) | Mm00441242_m1 |
| Ccr2 | Mm04207877_m1 |
| Col1a1 | Mm00801666_m1 |
| Havcr1 (KIM-1) | Mm00506686_m1 |
| Lrp2 (megalin) | Mm01328171_m1 |
| Lcn2 (NGAL) | Mm01324470_m1 |
| p16 (Cdkn2a) | Mm00494449_m1 |
| p21 (Cdkn1a) | Mm01303209_m1 |
| p53 | Mm00441964_g1 |
| Rpl19 | Mm02601633_g1 |
| Slc6a19 (B0AT1) | Mm01352157_m1 |
| Slc6a15 (B0AT2) | Mm00558415_m1 |
| Slc6a18 (B0AT3) | Mm00496017_m1 |
| Slc7a9 | Mm00445269_m1 |
| Slc22a6 (OAT1) | Mm00456258_m1 |
| Slc22a8 (OAT3) | Mm00459534_m1 |
| Tgfb1 | Mm01178820_m1 |
| Timp1 | Mm01341361_m1 |
Ccl2, C-C motif chemokine ligand 2; Ccr2, C-C motif chemokine receptor 2; Col1a1, collagen type I-α1; Havcr1, hepatitis a virus cellular receptor 1; KIM-1, kidney injury molecule-1; MCP-1, monocyte chemoattractant protein-1; NGAL, neutrophil gelatinase-associated lipocalin; OAT1, organic anion transporter 1; OAT3, organic anion transporter 3; Tgfb1, transforming growth factor-β1; Timp1, tissue inhibitor of metallopeptidase 1.
Western Blot Analysis
Membrane fraction was extracted from whole kidneys as previously described (30). Protein concentration was determined using a DC Protein Assay kit (Bio-Rad Laboratories, Hercules, CA). Extracted proteins (15 μg) were loaded on TGX PAGE gels (Bio-Rad Laboratories). Gel proteins were transferred to PVDF membranes and immunoblotted with rabbit anti-SGLT2 antibody (1:1,000, BiCell Scientific, Maryland Heights, MO) and mouse anti-β-actin antibody (1:10,000, Sigma-Aldrich) followed by treatment with horseradish peroxidase-conjugated secondary antibody (1:3,000, Cell Signaling Technology, Danvers, MA). Protein expression levels were analyzed using ImageJ Software (NIH). Results were normalized to actin expression and the control WT group. Kidneys of mice lacking SGLT2 (30) were used to confirm specificity of the antibody.
Tubular Injury and Kidney Fibrosis Measurement by Histology
Kidney samples were fixed in formalin. The fixed tissues were embedded in paraffin and cut into 5-μm-thick sections. Masson’s trichrome staining was performed by Tissue Technology Shared Resource at the University of California-San Diego (CCSG Grant P30CA23100). For tubular injury semiquantification in the kidney cortex and outer medulla, slides were analyzed by a pathologist who was blinded to the experimental groups. For each animal, tubular injury was defined as tubular dilation, tubular atrophy, tubular cast formation, sloughing of tubular epithelial cells or loss of the brush border, and thickening of the tubular basement membrane using the following scoring system: no change (0), <25% (1+), 25%–50% (2+), 50%–75% (3+), and >75% (4+). To evaluate the fibrosis area in the scanned image, the ratio of the blue color area to the renal entire cortex area was measured using ImageJ software (v.1.53, NIH; https://imagej.nih.gov/ij) with the observer blinded to study groups.
Statistical Analyses
Data are presented as means ± SE and dots show individual values. Data were analyzed by two-way ANOVA to probe for a significant effect of AA treatment (PAA), Slc6a19 genotype (PSlc6a19), or the interaction between the two factors (Pinter). If the interaction was statistically significant, then a pair-wise multiple comparison procedure (Holm–Sidak method) identified the significant effects. For basal conditions, one-way ANOVA was performed to probe for a significant effect of genotypes. An overall significance level of P < 0.05 was considered as statistically significant.
RESULTS
AA Decreased Renal mRNA Expression of Proximal Tubule Amino Acid Transporters
Confirming the efficacy of the gene-targeting strategy, Slc6a19 mRNA expression was reduced by 50% in Slc6a19+/− versus WT mice and was not detectable in Slc6a19−/− mice in vehicle-treated groups (Fig. 2A).
Figure 2.
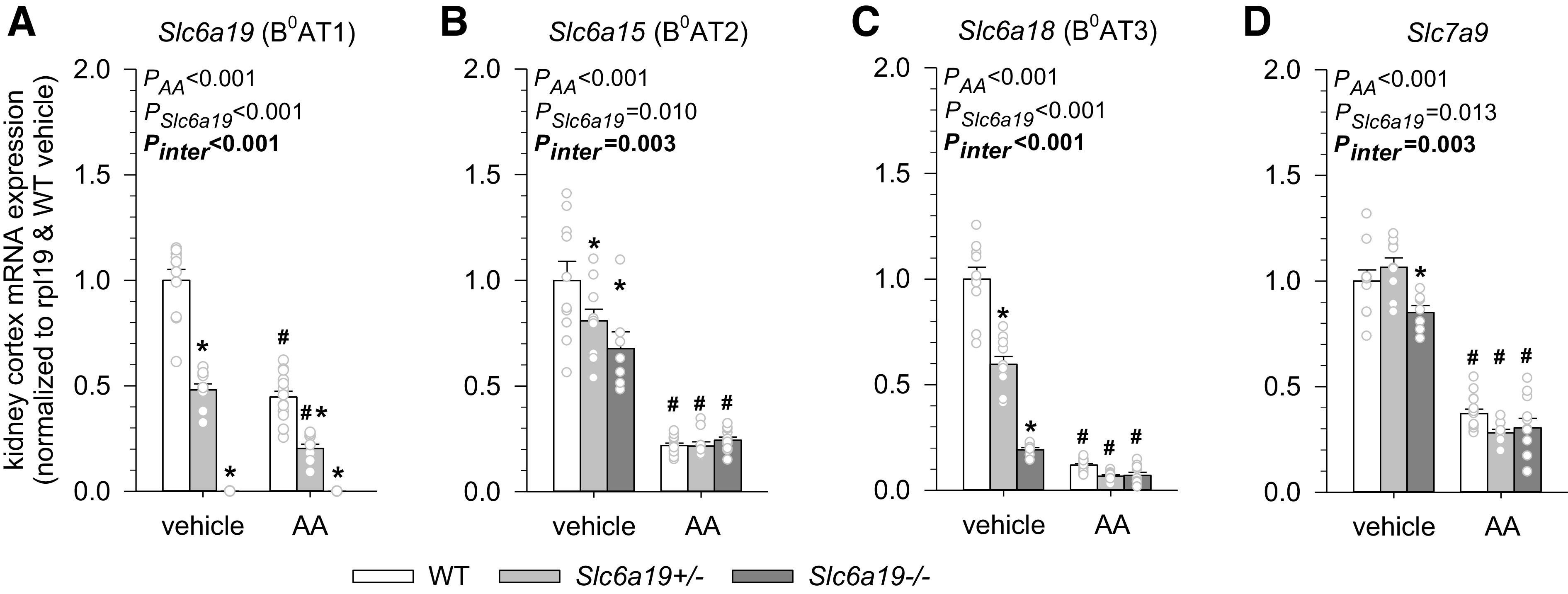
Aristolochic acid (AA) decreased renal mRNA expression of proximal tubule amino acid transporters. Kidney mRNA expression of Slc6a19 (B0AT1; A), Slc6a15 (B0AT2; B), Slc6a18 (B0AT3; C), and Slc7a9 (D) in mice treated with vehicle or AA. mRNA expression was normalized to the ribosomal protein L19 (Rpl19) gene. Values are means ± SE and dots show individual mouse data. Two-way ANOVA was performed to probe for a significant effect of treatment (PAA), genotypes (PSlc6a19), or the interaction between the two factors (Pinter). If the interaction was statistically significant, then a pair-wise multiple comparison procedure (Holm–Sidak method) identified the significant effects. *P < 0.05 vs. wild-type (WT) mice; #P < 0.05 vs. vehicle. n = 7–14 mice/group.
mRNA expression of Slc6a15 (B0AT2), Slc6a18 (B0AT3), and Slc7a9 was analyzed to determine the effect of B0AT1 knockout on related amino acid transporters in the brush border of the proximal tubule. B0AT2 (10) and B0AT3 (6) are Na+-dependent neutral amino acid transporters that are functionally and structurally related to B0AT1 and coexpressed in the proximal tubule. Slc7a9 is a Na+-independent transporter in the brush border of the proximal tubule that mainly transports positively charged amino acids in exchange for neutral amino acids and thereby is functionally coupled to B0AT1 (7). In vehicle-treated groups, mRNA expression of B0AT2 and B0AT3 was lower in Slc6a19+/− mice (20% and 40%, respectively) and even more so in Slc6a19−/− mice (30% and 80%, respectively) (Fig. 2, B and C), and the latter also had somewhat lower Slc7a9 mRNA expression (∼15%; Fig. 2D).
AA reduced renal mRNA expression of Slc6a19 (B0AT1), Slc6a15 (B0AT2), Slc6a18 (B0AT3), and Slc7a9, consistent with AA targeting the proximal tubule (Fig. 2). Notably, mRNA levels of Slc6a15 (B0AT2), Slc6a18 (B0AT3), and Slc7a9 were similar in all three AA-treated genotypes, suggesting that differences in these transporters may not explain differences in phenotypes after AA treatment.
Absence of B0AT1 Did Not Affect Markers of AA-Induced Kidney Injury
Amino acids are used as energy substrates and for tissue building. Hence, body and organ weight were analyzed to determine whether the absence of B0AT1 has an impact on body or organ mass. A previous study has reported that male and female Slc6a19−/− mice have a somewhat lower body weight than Slc6a19+/− and WT littermates (29). However, in the present study, no significant differences were observed in body weight or kidney, heart, and liver weights between vehicle-treated Slc6a19+/− or Slc6a19−/− mice and WT littermate mice (Fig. 3, A and B). Moreover, hematocrit (Fig. 3C) and plasma creatinine concentration (Fig. 3D), as an estimate of glomerular filtration rate, were not different between genotypes under basal conditions or following vehicle treatment.
Figure 3.
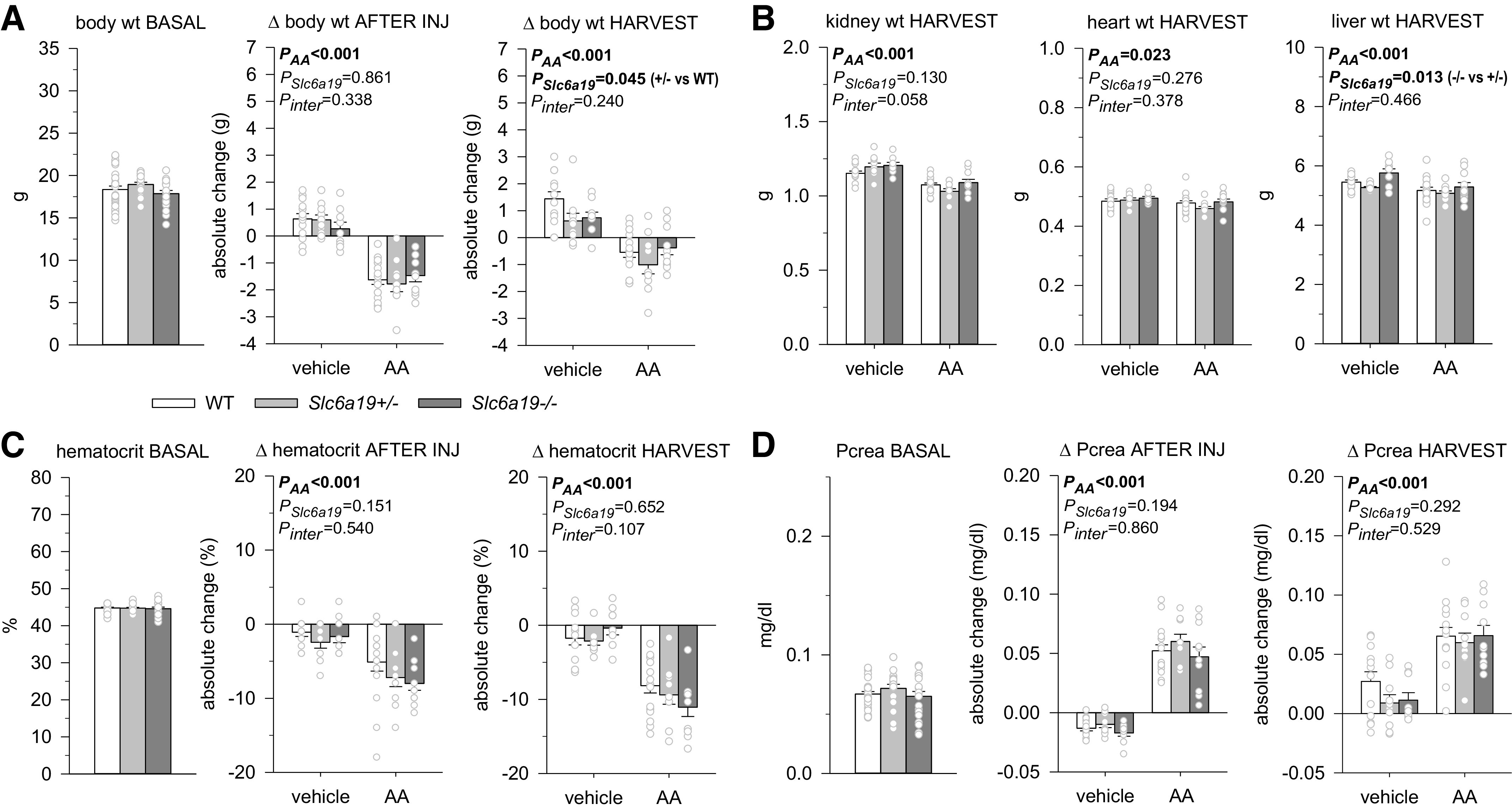
Absence of B0AT1 did not affect the aristolochic acid (AA)-induced reduction in body and organ weight, hematocrit, or an estimate of glomerular filtration rate. A: body weight under basal conditions (n = 18–30 mice/group, pooled groups before injection of vehicle or AA), after injections (INJ), and at harvest. B: kidney, heart, and liver weight at harvest. C: hematocrit under basal conditions (n = 19–28 mice/pooled group), after injections, and at harvest. D: plasma creatinine (Pcrea) under basal conditions (n = 19–30 mice/pooled group), after injections, and at harvest. Values are means ± SE and dots show individual mouse data. For basal conditions, one-way ANOVA was performed to probe for a significant effect of genotypes. For the other two time points, two-way ANOVA was performed to probe for a significant effect of AA treatment (PAA), genotypes (PSlc6a19), or the interaction between the two factors (Pinter). If the interaction was statistically significant, then a pair-wise multiple comparison procedure (Holm–Sidak method) identified the significant effects.
Due to similar basal body weight, absolute amounts of AA injected were not different between genotypes. Treatment with AA reduced body and organ weights and hematocrit independent of genotype (Fig. 3, A–C). Compared with basal values and independent of genotype, AA approximately doubled plasma creatinine concentrations when measured after the completion of injections or 3 wk later at the time of harvest (Fig. 3D).
AA treatment induced tubular injury independent of genotype as indicated by enhanced kidney cortex mRNA expression of the tubular injury marker KIM-1 (Fig. 4A) associated with histological evidence of tubular injury in the kidney cortex and outer medulla (Fig. 4, B and C), and similar kidney cortex mRNA upregulation of p53 and p21, which are part of the generic DNA damage response (Fig. 4D).
Figure 4.
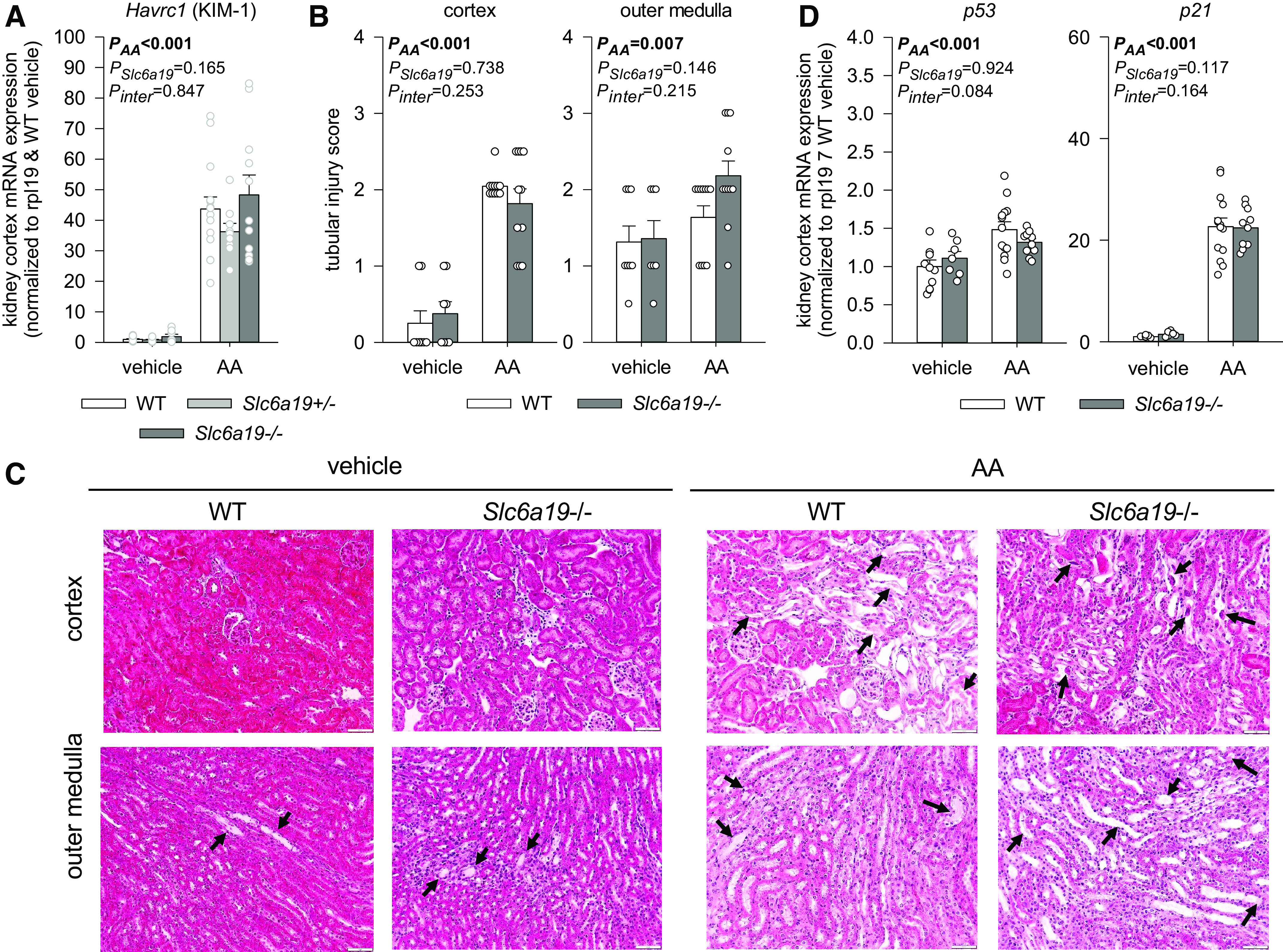
Absence of B0AT1 did not attenuate aristolochic acid (AA)-induced kidney injury. Kidney cortex mRNA expression of hepatitis A virus cellular receptor 1 [Havcr1); kidney injury molecule (KIM-1); A], histological assessment of tubular injury in the kidney cortex and medulla (B and C), and kidney cortex mRNA expression of p53 and p21 in mice treated with vehicle or AA (D). mRNA expression was normalized to the ribosomal protein L19 (Rpl19) gene. C: in both vehicle-treated genotypes, cortical histology appeared normal, and the outer medulla showed occasional dilated tubules with flattened epithelial cells and proteinous casts (black arrows). In both genotypes, AA induced a moderate degree of cortical tubular injury with a dilated lumen occasionally containing necrotic debris (black arrow) and flattened epithelial cells, normal-appearing glomeruli, and a mild to moderate degree of tubular injury in the outer medulla with dilated tubules, flattened epithelial cells, and proteinous casts (black arrows). Values are means ± SE and dots show individual mouse data. Two-way ANOVA was performed to probe for a significant effect of treatment (PAA), genotypes (PSlc6a19), or the interaction between the two factors (Pinter). If the interaction was statistically significant, then a pair-wise multiple comparison procedure (Holm–Sidak method) identified the significant effects. n = 7–11 mice/group for histology and n = 6–14 mice/group for quantitative PCR.
Absence of B0AT1 Is Associated With Basal Albuminuria but Prevented AA-Induced Increases
Urinary albumin-to-creatinine ratios (UACRs) under basal conditions or following vehicle treatment were similar in WT and Slc6a19+/− mice but two to three times higher in Slc6a19−/− mice (Fig. 5A). To gain further insights, renal mRNA expression of megalin (Fig. 5B) was analyzed as a determinant of tubular handling of albumin. Expression of megalin was lower in Slc6a19−/− compared with WT mice, indicating a potential role of tubular processes to explain the modestly higher albuminuria in Slc6a19−/− mice.
Figure 5.

Absence of B0AT1 associated with basal albuminuria but mitigated aristolochic acid (AA)-induced albuminuria. A: the urinary albumin-to-creatinine ratio (UACR) under basal conditions (n = 18–28 mice/pooled groups before injection of vehicle or AA), after injections (INJ), and at harvest. Kidney mRNA expression of Lrp2 (megalin; B), and organic anion transporters Slc22a6 [organic anion transporter 1 (OAT1)] and Slc22a8 [organic anion transporter 3 (OAT3)] (C) in mice treated with vehicle or AA. mRNA expression was normalized to the ribosomal protein L19 (Rpl19) gene. Values are means ± SE and dots show individual mouse data. For basal conditions, one-way ANOVA was performed to probe for a significant effect of genotypes. For the other two time points and the mRNA expression data, two-way ANOVA was performed to probe for a significant effect of treatment (PAA), genotypes (PSlc6a19), or the interaction between the two factors (Pinter). If the interaction was statistically significant, then a pair-wise multiple comparison procedure (Holm–Sidak method) identified the significant effects. *P < 0.05 vs. wild-type (WT) mice; #P < 0.05 vs. vehicle. n = 6–14 mice/group.
AA significantly increased UACR in WT and Slc6a19+/− mice, as observed after completion of injections and at harvest (Fig. 5A); this response to AA was completely prevented in Slc6a19−/− mice, and UACR values were lower in Slc6a19−/− versus WT mice after AA treatment, suggesting a potential protective effect of lacking B0AT1. AA reduced megalin expression to similar levels in Slc6a19−/− and WT mice, indicating that the differences in renal megalin mRNA expression do not explain differences in the UACR response.
AA enters proximal tubular cells through uptake across the basolateral membrane by organic anion transporter 1 (OAT1; SLC22A6) and organic anion transporter 3 (OAT3; SLC22A8) (22, 31). Renal mRNA expression of OAT1 and OAT3 did not differ between vehicle-treated Slc6a19−/− and WT mice (Fig. 5C), suggesting similar proximal tubular uptake of AA in Slc6a19−/− and WT mice. AA reduced mRNA expression of both OAT1 and OAT3 consistent with targeting the proximal tubule. The suppression of OAT1 mRNA expression by AA, however, was significantly less in Slc6a19−/− versus WT mice, suggesting better recovery or preservation of OAT1 expression in the absence of B0AT1, which may reflect proximal tubule protection.
Absence of B0AT1 Reduced Renal SGLT2 Protein Expression Associated With Low-Level Glucosuria
SGLT2 mediates glucose reabsorption in the S1/S2 segments of the proximal tubule (30, 32). Urinary glucose-to-creatinine ratios (UGCRs) were two to four times higher in vehicle-treated Slc6a19−/− versus WT mice (Fig. 6A). After the specificity of the SGLT2 antibody was confirmed using mice that lack the targeted protein as a negative control (Fig. 6B), the present study showed, for the first time, that the low-level glucosuria in vehicle-treated Slc6a19−/− mice is associated with and potentially explained by reduced renal SGLT2 protein expression (Fig. 6C).
Figure 6.

Absence of B0AT1 reduced renal Na+-glucose transporter 2 (SGLT2) protein expression associated with low-level glucosuria. A: the urinary glucose-to-creatinine ratio after injections (INJ) of vehicle or aristolochic acid (AA) and at harvest. B: Western blot confirming the specificity of the SGLT2 antibody using mice lacking Slc5a2. C: Western blots of the whole kidney membrane fraction using the same SGLT2 antibody and an antibody for the housekeeping protein β-actin in mice treated with vehicle or AA. D: quantification of SGLT2 protein expression normalized to β-actin. β-Actin showed some variability but was not statistically different between groups by two-way ANOVA. Values are means ± SE and dots show individual mouse data. Two-way ANOVA was performed to probe for a significant effect of treatment (PAA), genotypes (PSlc6a19), or the interaction between the two factors (Pinter). If the interaction was statistically significant, then a pair-wise multiple comparison procedure (Holm–Sidak method) identified the significant effects. *P < 0.05 vs. wild-type (WT) mice; #P < 0.05 vs. vehicle. n = 6–14 mice/group.
UGCR was not affected after the last AA injection in any genotype or in vehicle-treated WT and Slc6a19−/− mice when reanalyzed 3 wk later (Fig. 6A). In comparison, at harvest, UGCR was no longer higher in AA-treated Slc6a19−/− versus WT mice. This was associated with and potentially explained by an upregulation of renal SGLT2 protein expression in Slc6a19−/− mice in response to AA (Fig. 6C), which may likewise suggest better preservation of proximal tubular function in response to AA by the absence of B0AT1.
Absence of B0AT1 Attenuated AA-Induced Markers of Senescence and Inflammation in the Kidney Cortex
As analyzed 3 wk after the last vehicle or AA injection, no significant differences were found between genotypes in vehicle-treated mice for kidney cortex mRNA expression of markers of senescence (p16) and inflammation [neutrophil gelatinase-associated lipocalin (NGAL; a marker of inflammation and injury) (33), monocyte chemoattractant protein-1 (MCP-1), and C-C motif chemokine receptor 2 (CCR2); Fig. 7, A–C]. The AA-induced increases in mRNA expression of p16, NGAL, MCP-1, and CCR2, however, were significantly attenuated in Slc6a19−/− versus WT mice (Fig. 7, A–C).
Figure 7.
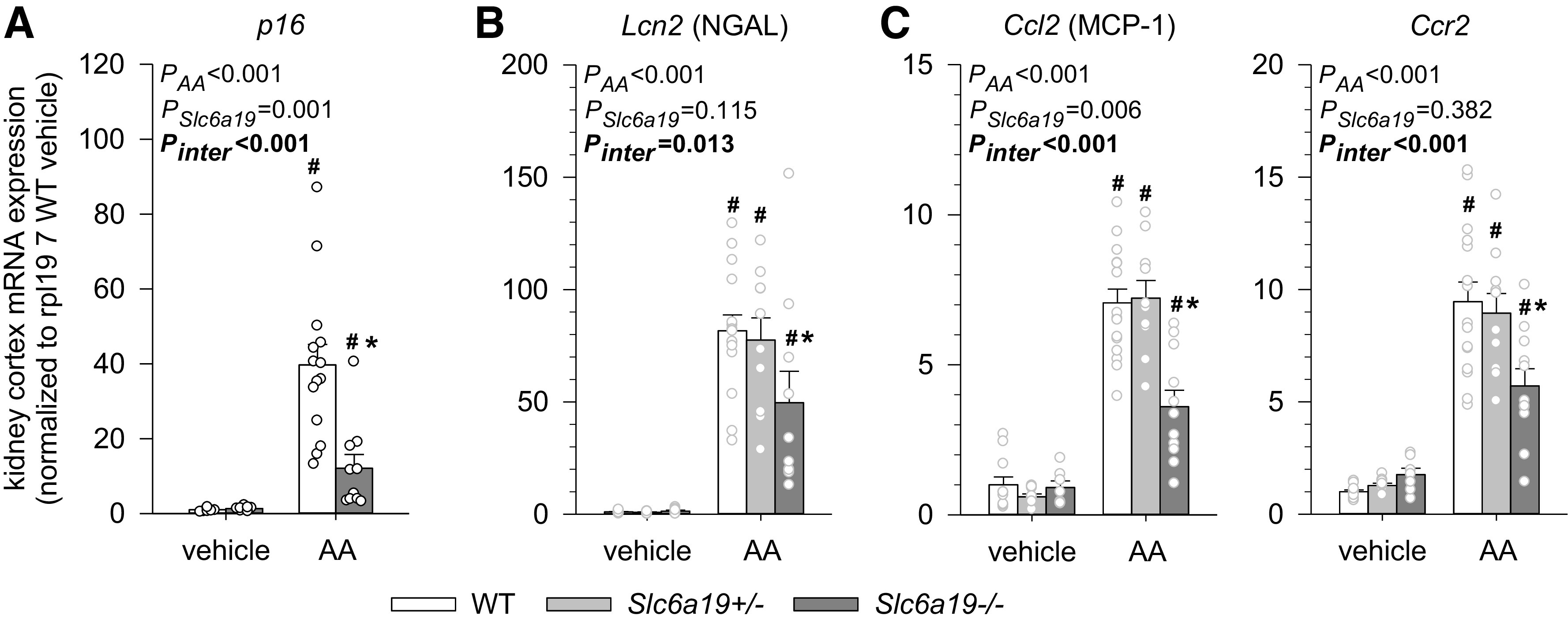
Absence of B0AT1 attenuated aristolochic acid (AA)-induced markers of senescence and inflammation in the kidney cortex. Kidney mRNA expression of p21 (A), Lcn2 [neutrophil gelatinase-associated lipocalin (NGAL); B], and C-C motif chemokine ligand 2 [Ccl2; monocyte chemoattractant protein-1 (MCP-1)] and C-C motif chemokine receptor 2 (Ccr2) (C) in mice treated with vehicle or AA. mRNA expression was normalized to the ribosomal protein L19 (Rpl19) gene. Values are means ± SE and dots show individual mouse data. Two-way ANOVA was performed to probe for a significant effect of treatment (PAA), genotypes (PSlc6a19), or the interaction between the two factors (Pinter). If the interaction was statistically significant, then a pair-wise multiple comparison procedure (Holm–Sidak method) identified the significant effects. *P < 0.05 vs. wild-type (WT) mice; #P < 0.05 vs. vehicle. n = 6–14 mice/group.
Absence of B0AT1 Attenuated AA-Induced Kidney Fibrosis
In vehicle-treated mice, renal mRNA expression of the profibrotic marker genes tissue inhibitor of metallopeptidase 1 (Timp1), transforming growth factor-β1 (Tgfb1), and collagen type I-α1 (Col1a1) were similar between genotypes (Fig. 8A). AA treatment caused robust and similar increases in the expression of Timp1, Tgfb1, and Col1a1 in WT and Slc6a19+/− mice, but mRNA levels for all three genes were significantly lower in Slc6a19−/− versus WT mice (Fig. 8A). As explored in WT and Slc6a19−/− mice and consistent with mRNA expression data of profibrotic genes, Masson’s trichrome staining revealed a robust AA treatment-induced increase in collagen staining in the kidney cortex and medulla of WT mice, with significantly reduced collagen staining in the absence of B0AT1 (Fig. 8, B and C).
Figure 8.
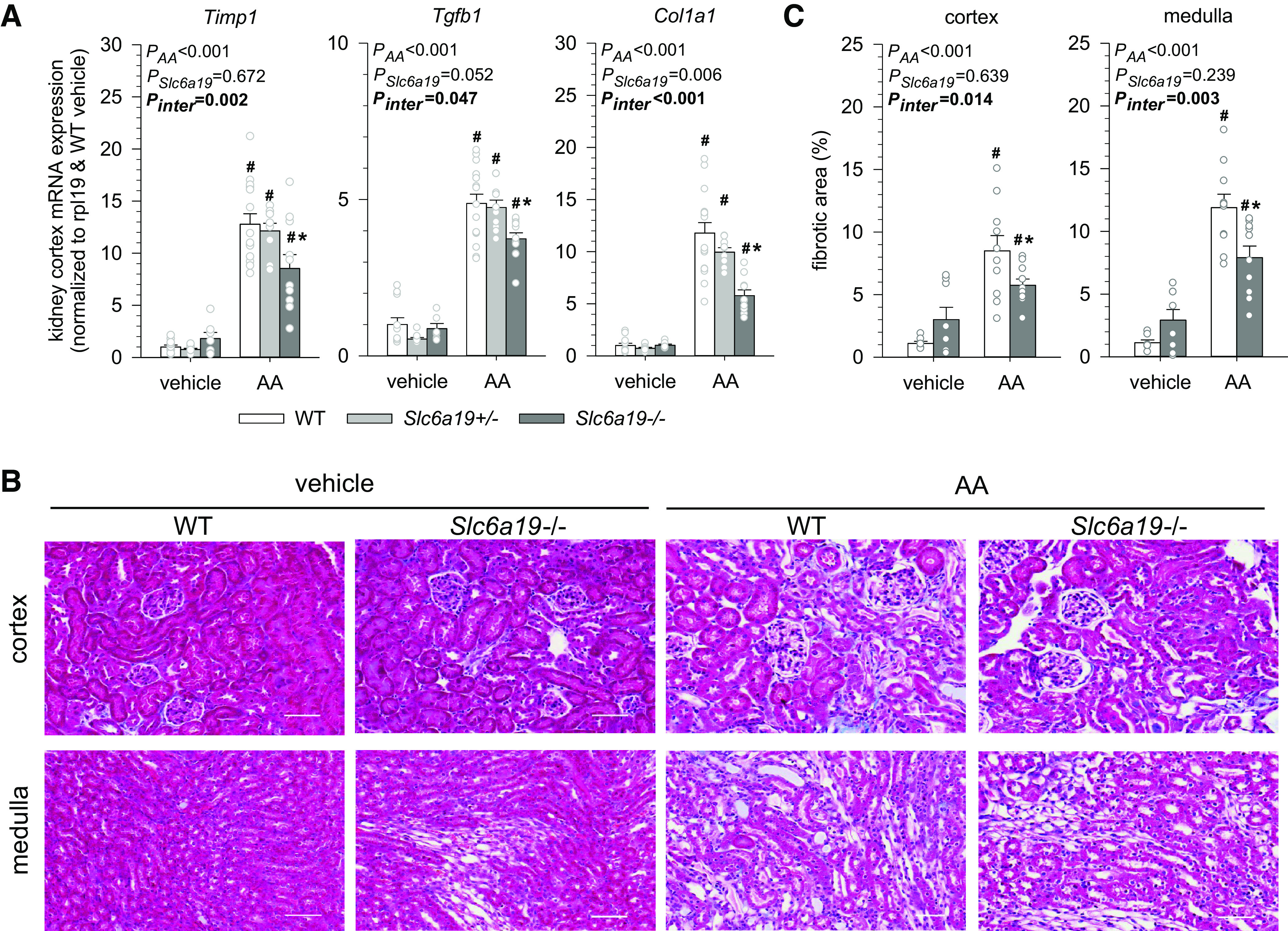
Absence of B0AT1 attenuates aristolochic acid (AA)-induced kidney fibrosis. A: kidney mRNA expression of tissue inhibitor of metallopeptidase 1 (Timp1), transforming growth factor-β1 (Tgfb1), and collagen type 1-α1 (Col1a1) in mice treated with vehicle or AA. mRNA expression was normalized to the ribosomal protein L19 (Rpl19) gene. B: Masson’s trichrome staining of the cortex and medulla from mice treated with vehicle or AA. Scale bars = 50 µm. C: quantification of the fibrotic area according to Masson’s trichrome staining. Values are means ± SE and dots show individual mouse data. Two-way ANOVA was performed to probe for a significant effect of treatment (PAA), genotypes (PSlc6a19), or the interaction between the two factors (Pinter). If the interaction was statistically significant, then a pairwise multiple comparison procedure (Holm–Sidak method) identified the significant effects. *P < 0.05 vs. wild-type (WT) mice; #P < 0.05 vs. vehicle. n = 6–14 mice/group.
DISCUSSION
B0AT1 (Slc6a19) is the major epithelial transporter for neutral amino acids in the intestine and kidney proximal tubule (7). As a consequence, mice lacking B0AT1 show reduced uptake of neutral amino acids in the intestine and loss of neutral amino acids in the urine (29). Moreover, the overload of amino acids in the lumen of the intestine results in higher postprandial levels of GLP-1, whereas the reduced amounts of amino acid presented to the liver enhance the release of the starvation hormone FGF21 (15). In mice lacking B0AT1, the combined upregulation of these hormones makes removal of glucose more efficient, particularly by the heart, reduces adipose tissue mass, enhances browning of subcutaneous white adipose tissue and production of ketone bodies, and reduces hepatic glucose output (15). Thus, absence of B0AT1 improves glucose homeostasis, and this metabolic phenotype is remarkably similar to the inhibition of intestinal absorption of glucose via SGLT1, which also increases GLP-1 (34), and/or inhibition of renal retention of glucose via SGLT2, which raises FGF21 (35). Little is known, however, about the physiology of B0AT1 in the kidney beyond amino acid retention or its role in kidney pathophysiology. The present study shows that absence of B0AT1 in female mice does not affect basal body and kidney weight or plasma creatinine values but modestly impairs the physiological retention of glucose and albumin in the kidney. Moreover, absence of B0AT1 attenuated the pathological upregulation of albuminuria as well as of markers of kidney senescence, inflammation, and fibrosis in response to AA-induced nephropathy, suggesting that absence of B0AT1 can have kidney protective effects.
Previous studies in Slc6a19−/− mice found no changes in the expression of the ancillary proteins collectrin and angiotensin-converting enzyme 2, which upregulate membrane expression of B0AT1 in the kidney and intestine, respectively, as well as normal dipeptide uptake as tested in the intestine (29). The present study shows that Slc6a19−/− mice not only lack renal mRNA expression of Slc6a19 but also have reduced renal mRNA expression of the related proximal tubular amino acid transporters Slc6a15 (B0AT2), Slc6a18 (B0AT3), and Slc7a9. B0AT1 and B0AT3 share the same chromosomal location (36), and thus the knockout strategy may explain the reduced B0AT3 mRNA expression levels in Slc6a19−/− mice. Alternatively, suppression of B0AT1, which is primarily expressed in the early proximal tubule in rats and mice (S1/S2 segments) (11, 12, 37), enhances amino acid delivery and most likely compensatory transport in downstream S3 and further distal segments. In these segments, which are not used to handle large quantities of amino acids, Slc6a15 (B0AT2) and Slc6a18 (B0AT3) may be downregulated by enhanced intracellular amino acid concentrations to prevent excessive transport work or amino acid loading (Fig. 9); indeed, robust mRNA expression has been detected for both genes along the entire proximal tubule and descending thin limbs in mice (12). The downregulation of B0AT2 and B0AT3 needs to be confirmed at the protein level, but specific antibodies verified in knockout tissue are sparse. Nevertheless, this observation is reminiscent of the downregulation of Na+-glucose cotransporter 1 (SGLT1) in the late proximal tubule when upstream SGLT2 is inhibited by genetic or pharmacological means and glucose load and net uptake by SGLT1 in the S3 segment is enhanced (30, 38, 39).
Figure 9.
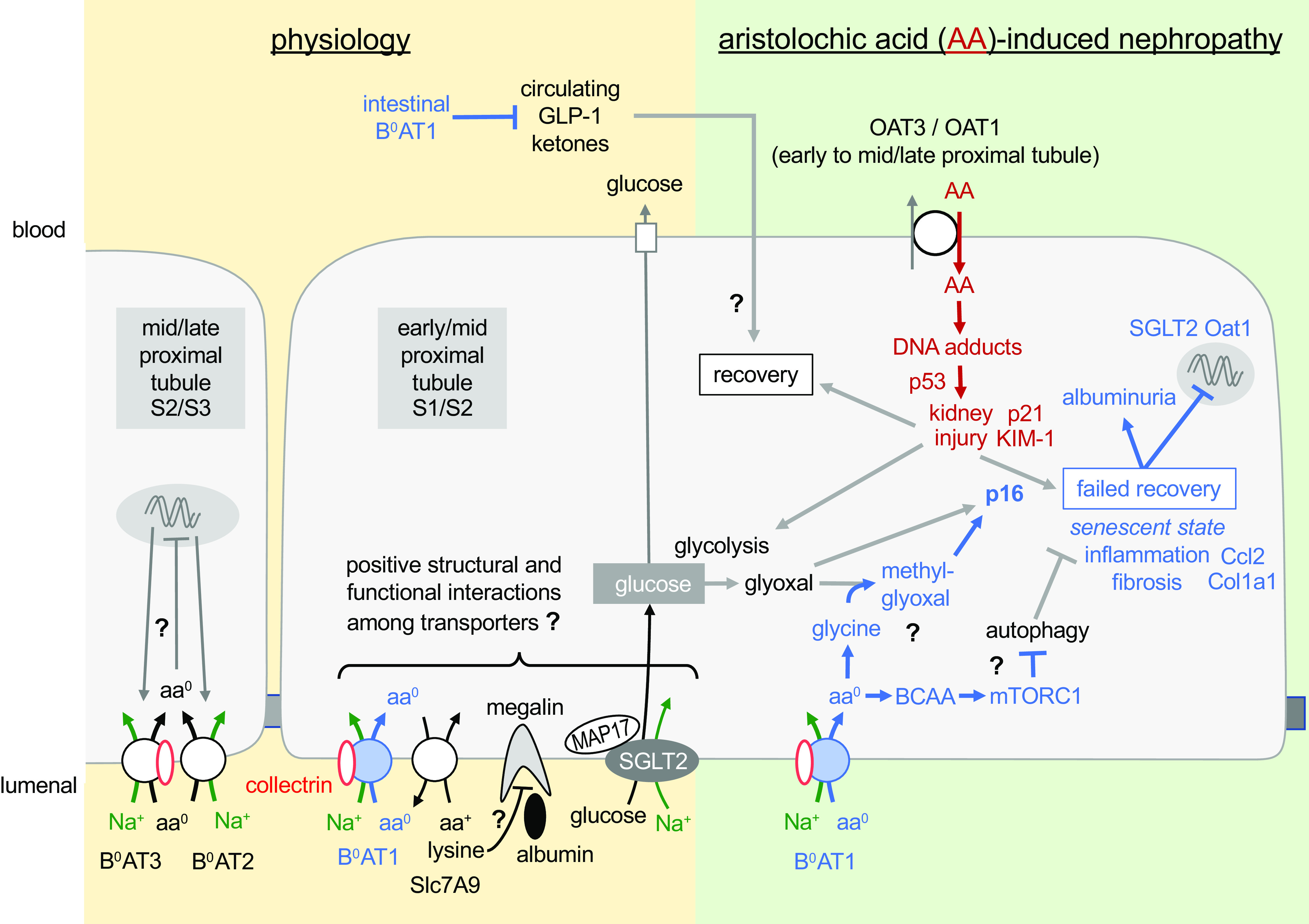
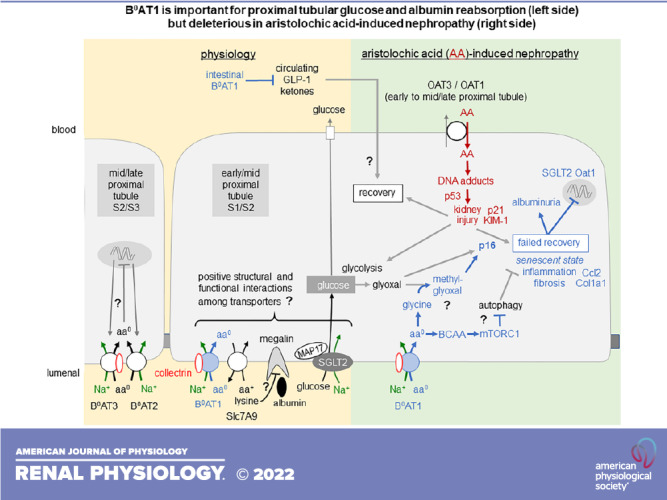
Proximal tubular B0AT1 is important for the physiology of renal glucose and albumin retention (left) but potentially deleterious in aristolochic acid (AA)-induced nephropathy (right). The former may be due to structural or functional interactions of transport proteins colocalized to the brush border of the early proximal tubule. The latter may involve promotion of p16-mediated senescence that is facilitated by the uptake of neutral amino acids (aa0) via B0AT1, including glycin and branched-chain amino acids (BCAAs). Blue text and arrows refer to effects related to B0AT1 function. “?” indicates hypothetical changes/pathways. See discussion for more details. aa+, cationic amino acid; Ccl2, C-C motif chemokine ligand 2; Col1a1, collagen type I-α1; GLP-1, glucagon-like peptide-1; KIM-1, kidney injury molecule-1; MAP17, 17-kDa membrane-associated protein; mTORC1, mammalian target of rapamycin complex 1; OAT, organic anion transporter; SGLT2, Na+-glucose cotransporter 2.
The present study revealed low-level glucosuria in Slc6a19−/− mice that is associated with and probably due to a robust reduction in the expression of SGLT2 protein, likely in the proximal tubule brush border since the analysis was based on kidney membrane fractions. This is in accordance with a previous study showing that kidney brush-border membrane vesicles of mice lacking B0AT1 not only had impaired amino acid transport but also a 50% reduction in glucose transport (29). Why absence of B0AT1 should suppress SGLT2 expression remains unknown. Both Na+-dependent transporters are expressed in the early proximal tubule and may functionally interact as proposed for other early proximal tubular transporters, like SGLT2 and Na+/H+ exchanger 3 (NHE3), where the inhibition of one inhibits the other (40–42). Positive interactions among apical transporters involves the scaffolding protein 17-kDa membrane-associated protein (MAP17) (42, 43), and makes physiological sense because it facilitates postprandial glomerulotubular balance when a rise in glomerular filtration rate increases not only the filtered amounts of Na+ but also of glucose, bicarbonate, and amino acids, which are all reabsorbed in the early proximal tubule (35). Further studies are needed to probe for interactions between B0AT1, SGLT2, and MAP17 (Fig. 9).
Slc6a19−/− mice show a modest increase in albumin excretion under basal conditions. Like the reuptake of glucose, tubular reabsorption of albumin predominantly takes place in early proximal tubules. Some studies have observed a doubling in albuminuria in normal mice that received a SGLT2 inhibitor (44), potentially due to functional interactions between SGLT2 and NHE3, with the latter being involved in megalin-mediated albumin reabsorption (45). Slc6a19−/− mice showed not only lower SGLT2 expression but also reduced levels of kidney megalin mRNA, which may indicate an early proximal tubular cause for the modest basal albuminuria. The application of large amounts of lysine can attenuate megalin-mediated albumin reabsorption in the proximal tubule (46). B0AT1 functionally interacts in the proximal tubule with SLC7A9 to reabsorb lysine, and Slc6a19−/− mice excrete more lysine (29). Whether altered handling of lysine contributes to the modest basal albuminuria in the absence of B0AT1 remains unclear (Fig. 9).
Due to the clinical relevance, the targeting of the proximal tubule, and good reproducibility of the model, AA-induced nephropathy was used as a mouse model of AKI/CKD. In the mouse kidney, AA affects the entire proximal tubule, but the S2 segment seems to be the primary site of its deleterious actions (22). This may explain in part the strong downregulation (by 80%–90%) in WT mice of mRNA expression of B0AT2 and B0AT3, which in mice are strongest expressed in S2 and S3 segments (12), whereas B0AT1 and Slc7a9, which are highest expressed in the S1 segment (12), were also reduced by AA, but somewhat less (by 50%–60%; Fig. 2).
AA enters the targeted proximal tubular cells primarily through basolateral OAT1 and OAT3 (Fig. 9) (31), which in mice have their highest mRNA expression in S2 and S1 segments, respectively (12). The observed similar renal mRNA expression for OAT1 and OAT3 in vehicle-treated Slc6a19−/− versus WT mice argued against a general downregulation of proximal tubular transporters in the absence of B0AT1 and for a similar proximal tubular uptake of AA in both genotypes. In accordance with this assumption, absence of B0AT1 did not affect the AA-induced reduction in kidney and body weight, the doubling in plasma creatinine values, histological evidence of tubular injury, or the increase in kidney cortical mRNA expression of the tubular injury marker KIM-1 observed in WT mice. Moreover, kidney cortex mRNA expression of p53 and p21 was similarly induced by AA in both genotypes. p21 is part of the generic DNA damage response and mainly regulated by direct transactivation via p53 (47), and p53 activation has been implicated in AA-induced tubular injury (20).
Absence of B0AT1, however, prevented the AA-induced increase in UACR observed in WT mice after the last injection or 3 wk later at kidney harvest, suggesting a potential deleterious role of B0AT1 in the active and recovery phase of AA nephropathy. Differences in kidney megalin mRNA expression cannot explain the blunted UACR response to AA in the absence of B0AT1. The latter may have better preserved the integrity of early proximal tubules in response to AA. This may be reflected by the observed unexpected increase in kidney SGLT2 protein expression in response to AA in Slc6a19−/− mice as well as better preservation of OAT1 mRNA expression in Slc6a19−/− versus WT mice.
In WT mice and as expected, AA robustly enhanced mRNA expression of the kidney injury/inflammation marker NGAL, proinflammatory markers CCL2 and CCR2, and profibrotic markers TIMP1, TGF-β1, and COL1A1, associated with enhanced renal fibrosis staining. All these effects were significantly attenuated in Slc6a19−/− mice compared with WT mice, consistent with the observed attenuation of AA-induced fibrosis in the cortex and medulla in the absence of B0AT1.
The differences in renal mRNA expression of B0AT2, B0AT3, and SLC7A9 detected in vehicle-treated Slc6a19−/− versus WT mice were not observed after AA treatment, indicating that lower mRNA expression in these genes cannot explain the blunted UACR responses or other protective effects between genotypes.
The data indicate that the absence of B0AT1 did not affect AA-induced tubular injury but attenuated the proinflammatory and profibrotic responses (Fig. 9). Mouse models show that kidney tubules have an intrinsic regenerative potential; however, on repeated injury, cells can be diverted to a nonproductive senescent state that is linked to maladaptive failed repair and profibrotic/inflammation (including the mRNA markers Ccl2 and Col1a1) (48). Although p21 is upregulated by different senescence-inducing stimuli (49), it paradoxically is also necessary for cell cycle progression (50), which makes it difficult to use as a unique senescence marker (47). In comparison, cyclin-dependent kinase inhibitor p16 is often used as a unique and specific marker for senescence, and its transcriptional activation has been used extensively to report the presence of senescent cells in vivo (47). The observed blunted rise in kidney cortex p16 as well as Ccl2 and Col1a1 mRNA expression in response to AA in Slc6a19−/− mice compared with WT mice may indicate a deleterious role of B0AT1-mediated amino acid transport in facilitating tubular senescence and repair following AA-induced tubular injury (Fig. 9). Due to its high plasma concentrations, the neutral amino acid glycine is one of the primary substrates of SLC6A19. Moreover, enhanced glycolysis and excess cytosolic glycine due to impaired cellular metabolism promote the formation of glyoxal and methylglyoxal (51). These two compounds are precursors of advanced glycation end products, whose presence marks normal and pathological aging and can trigger a senescence phenotype maintained by p16 (52). Further studies are needed to follow up on this hypothesis at the cellular level of the proximal tubule and its subsegments to explore a potential role of changes in cellular metabolism, autophagy, or BCAAs (7, 16, 27) and to probe for potential secondary effects of B0AT1 inhibition by affecting systemic metabolism, e.g., increasing GLP-1 and ketone bodies (Fig. 9) (15). Moreover, can these effects be extended to other models of AKI and CKD, how does genetic deletion of Slc6a19 compare with the pharmacological inhibition of B0AT1, and is B0AT1 inhibition complementing standard-of-care therapies, like SGLT2 inhibition, which may also attenuate the glyoxal/methylglyoxal/p16 pathway (Fig. 9)?
In summary, the data suggest that proximal tubular B0AT1 is important for the physiology of renal glucose and albumin retention but potentially deleterious in AA-induced nephropathy. The findings further support the hypothesis, as proposed for glucose (34, 53), that targeting intestinal uptake or renal reabsorption of energy substrates has unique therapeutic potential to improve metabolic disease and kidney outcome in response to injury.
GRANTS
This work was supported by National Institutes of Health (NIH) Grants R01DK112042 and RF1AG061296 (to V.V.), University of Alabama at Birmingham/University of California-San Diego O’Brien Center of Acute Kidney Injury NIH Grant P30DK079337 (to V.V. and P.W.S.), and the Department of Veterans Affairs. Y.O. was supported by a fellowship of the Manpei Suzuki Diabetes Foundation.
DISCLOSURES
Over the past 36 mo, V.V. has served as a consultant and received honoraria from Astra-Zeneca, Boehringer Ingelheim, Lexicon, and Retrophin and received grant support for investigator-initiated research from Astra-Zeneca, Gilead, Janssen Pharmaceutical, Kyowa-Kirin, Merck, and Novo-Nordisk. S.B. is a consultant for Axcella Health Inc. and Maze Therapeutics. None of the other authors have any conflicts of interest, financial or otherwise, to disclose.
AUTHOR CONTRIBUTIONS
A.N.G., Y.C.K., and V.V. conceived and designed research; A.N.G., Y.C.K., Y.O., H.Z., M.C-M., H.A.G., and S.K. performed experiments; A.N.G., Y.C.K., Y.O., H.Z., M.C-M., H.A.G., S.K., and V.V. analyzed data; A.N.G., Y.C.K., Y.O., H.Z., M.C-M., H.A.G., S.K., and V.V. interpreted results of experiments; A.N.G. and V.V. prepared figures; A.N.G. and V.V. drafted manuscript; A.N.G., Y.C.K., Y.O., H.Z., M.C-M., H.A.G., S.K., P.W.S., S.B., and V.V. edited and revised manuscript; A.N.G., Y.C.K., Y.O., H.Z., H.A.G., S.K., P.W.S., S.B., and V.V. approved final version of manuscript.
ACKNOWLEDGMENTS
Part of these data was presented at the Experimental Biology 2021 Meeting and published in abstract form in The FASEB Journal (54).
REFERENCES
- 1. Saran R, Robinson B, Abbott KC, Agodoa LY, Albertus P, Ayanian J, et al. US renal data system 2016 annual data report: epidemiology of kidney disease in the United States. Am J Kidney Dis 69: A7–A8, 2017. [Erratum in Am J Kidney Dis 69: 712, 2017]. doi: 10.1053/j.ajkd.2016.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. He L, Wei Q, Liu J, Yi M, Liu Y, Liu H, Sun L, Peng Y, Liu F, Venkatachalam MA, Dong Z. AKI on CKD: heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int 92: 1071–1083, 2017. doi: 10.1016/j.kint.2017.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vallon V. Do tubular changes in the diabetic kidney affect the susceptibility to acute kidney injury? Nephron Clin Pract 127: 133–138, 2014. doi: 10.1159/000363554. [DOI] [PubMed] [Google Scholar]
- 4. Thakar CV, Christianson A, Himmelfarb J, Leonard AC. Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. Clin J Am Soc Nephrol 6: 2567–2572, 2011. doi: 10.2215/CJN.01120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vallon V, Verma S. Effects of SGLT2 inhibitors on kidney and cardiovascular function. Annu Rev Physiol 83: 503–528, 2021. doi: 10.1146/annurev-physiol-031620-095920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Singer D, Camargo SM, Huggel K, Romeo E, Danilczyk U, Kuba K, Chesnov S, Caron MG, Penninger JM, Verrey F. Orphan transporter SLC6A18 is renal neutral amino acid transporter B0AT3. J Biol Chem 284: 19953–19960, 2009. doi: 10.1074/jbc.M109.011171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Broer S. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev 88: 249–286, 2008. doi: 10.1152/physrev.00018.2006. [DOI] [PubMed] [Google Scholar]
- 8. Bohmer C, Broer A, Munzinger M, Kowalczuk S, Rasko JE, Lang F, Broer S. Characterization of mouse amino acid transporter B0AT1 (slc6a19). Biochem J 389: 745–751, 2005. doi: 10.1042/BJ20050083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Broer A, Klingel K, Kowalczuk S, Rasko JE, Cavanaugh J, Broer S. Molecular cloning of mouse amino acid transport system B0, a neutral amino acid transporter related to Hartnup disorder. J Biol Chem 279: 24467–24476, 2004. doi: 10.1074/jbc.M400904200. [DOI] [PubMed] [Google Scholar]
- 10. Broer A, Tietze N, Kowalczuk S, Chubb S, Munzinger M, Bak LK, Broer S. The orphan transporter v7-3 (slc6a15) is a Na+-dependent neutral amino acid transporter (B0AT2). Biochem J 393: 421–430, 2006. doi: 10.1042/BJ20051273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Limbutara K, Chou CL, Knepper MA. Quantitative proteomics of all 14 renal tubule segments in rat. J Am Soc Nephrol 31: 1255–1266, 2020. doi: 10.1681/ASN.2020010071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen L, Chou CL, Knepper MA. A comprehensive map of mRNAs and their isoforms across all 14 renal tubule segments of mouse. J Am Soc Nephrol 32: 897–912, 2021. doi: 10.1681/ASN.2020101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Broer S. The role of the neutral amino acid transporter B0AT1 (SLC6A19) in Hartnup disorder and protein nutrition. IUBMB Life 61: 591–599, 2009. doi: 10.1002/iub.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Broer S. Diseases associated with general amino acid transporters of the solute carrier 6 family (SLC6). Curr Mol Pharmacol 6: 74–87, 2013. doi: 10.2174/18744672113069990034. [DOI] [PubMed] [Google Scholar]
- 15. Jiang Y, Rose AJ, Sijmonsma TP, Broer A, Pfenninger A, Herzig S, Schmoll D, Broer S. Mice lacking neutral amino acid transporter B0AT1 (Slc6a19) have elevated levels of FGF21 and GLP-1 and improved glycaemic control. Mol Metab 4: 406–417, 2015. doi: 10.1016/j.molmet.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Holecek M. Branched-chain amino acids in health and disease: metabolism, alterations in blood plasma, and as supplements. Nutr Metab (Lond) 15: 33, 2018. doi: 10.1186/s12986-018-0271-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sveinbjornsson G, Mikaelsdottir E, Palsson R, Indridason OS, Holm H, Jonasdottir A, Helgason A, Sigurdsson S, Jonasdottir A, Sigurdsson A, Eyjolfsson GI, Sigurdardottir O, Magnusson OT, Kong A, Masson G, Sulem P, Olafsson I, Thorsteinsdottir U, Gudbjartsson DF, Stefansson K. Rare mutations associating with serum creatinine and chronic kidney disease. Hum Mol Genet 23: 6935–6943, 2014. doi: 10.1093/hmg/ddu399. [DOI] [PubMed] [Google Scholar]
- 18. Debelle FD, Vanherweghem JL, Nortier JL. Aristolochic acid nephropathy: a worldwide problem. Kidney Int 74: 158–169, 2008. doi: 10.1038/ki.2008.129. [DOI] [PubMed] [Google Scholar]
- 19. Shibutani S, Dong H, Suzuki N, Ueda S, Miller F, Grollman AP. Selective toxicity of aristolochic acids I and II. Drug Metab Dispos 35: 1217–1222, 2007. doi: 10.1124/dmd.107.014688. [DOI] [PubMed] [Google Scholar]
- 20. Zhou L, Fu P, Huang XR, Liu F, Lai KN, Lan HY. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J Am Soc Nephrol 21: 31–41, 2010. doi: 10.1681/ASN.2008111133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Honarpisheh M, Foresto-Neto O, Steiger S, Kraft F, Koehler P, von Rauchhaupt E, Potempa J, Adamowicz K, Koziel J, Lech M. Aristolochic acid I determine the phenotype and activation of macrophages in acute and chronic kidney disease. Sci Rep 8: 12169, 2018. doi: 10.1038/s41598-018-30628-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dickman KG, Sweet DH, Bonala R, Ray T, Wu A. Physiological and molecular characterization of aristolochic acid transport by the kidney. J Pharmacol Exp Ther 338: 588–597, 2011. doi: 10.1124/jpet.111.180984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lebeau C, Debelle FD, Arlt VM, Pozdzik A, De Prez EG, Phillips DH, Deschodt-Lanckman MM, Vanherweghem JL, Nortier JL. Early proximal tubule injury in experimental aristolochic acid nephropathy: functional and histological studies. Nephrol Dial Transplant 20: 2321–2332, 2005. doi: 10.1093/ndt/gfi042. [DOI] [PubMed] [Google Scholar]
- 24. Huang L, Scarpellini A, Funck M, Verderio EA, Johnson TS. Development of a chronic kidney disease model in C57BL/6 mice with relevance to human pathology. Nephron Extra 3: 12–29, 2013. doi: 10.1159/000346180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sato N, Takahashi D, Chen SM, Tsuchiya R, Mukoyama T, Yamagata S, Ogawa M, Yoshida M, Kondo S, Satoh N, Ueda S. Acute nephrotoxicity of aristolochic acids in mice. J Pharm Pharmacol 56: 221–229, 2004. doi: 10.1211/0022357023051. [DOI] [PubMed] [Google Scholar]
- 26. Shi M, Ma L, Zhou L, Fu P. Renal protective effects of 17β-estradiol on mice with acute aristolochic acid nephropathy. Molecules 21: 1391, 2016. doi: 10.3390/molecules21101391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Piret SE, Guo Y, Attallah AA, Horne SJ, Zollman A, Owusu D, Henein J, Sidorenko VS, Revelo MP, Hato T, Ma'ayan A, He JC, Mallipattu SK. Kruppel-like factor 6-mediated loss of BCAA catabolism contributes to kidney injury in mice and humans. Proc Natl Acad Sci USA 118: e2024414118, 2021. doi: 10.1073/pnas.2024414118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Percie Du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M, Browne WJ, Clark A, Cuthill IC, Dirnagl U, Emerson M, Garner P, Holgate ST, Howells DW, Karp NA, Lazic SE, Lidster K, MacCallum CJ, Macleod M, Pearl EJ, Petersen OH, Rawle F, Reynolds P, Rooney K, Sena ES, Silberberg SD, Steckler T, Wurbel H. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol 18: e3000410, 2020. doi: 10.1371/journal.pbio.3000410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broer A, Juelich T, Vanslambrouck JM, Tietze N, Solomon PS, Holst J, Bailey CG, Rasko JE, Broer S. Impaired nutrient signaling and body weight control in a Na+ neutral amino acid cotransporter (Slc6a19)-deficient mouse. J Biol Chem 286: 26638–26651, 2011. doi: 10.1074/jbc.M111.241323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li C, Wang X, Bi Y, Yu H, Wei J, Zhang Y, Han L, Zhang Y. Potent inhibitors of organic anion transporters 1 and 3 from natural compounds and their protective effect on aristolochic acid nephropathy. Toxicol Sci 175: 279–291, 2020. doi: 10.1093/toxsci/kfaa033. [DOI] [PubMed] [Google Scholar]
- 32. Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 91: 733–794, 2011. doi: 10.1152/physrev.00055.2009. [DOI] [PubMed] [Google Scholar]
- 33. Martensson J, Bellomo R. The rise and fall of NGAL in acute kidney injury. Blood Purif 37: 304–310, 2014. doi: 10.1159/000364937. [DOI] [PubMed] [Google Scholar]
- 34. Song P, Onishi A, Koepsell H, Vallon V. Sodium glucose cotransporter SGLT1 as a therapeutic target in diabetes mellitus. Expert Opin Ther Targets 20: 1109–1125, 2016. doi: 10.1517/14728222.2016.1168808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vallon V. Glucose transporters in the kidney in health and disease. Pflugers Arch 472: 1345–1370, 2020. doi: 10.1007/s00424-020-02361-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seow HF, Broer S, Broer A, Bailey CG, Potter SJ, Cavanaugh JA, Rasko JE. Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat Genet 36: 1003–1007, 2004. doi: 10.1038/ng1406. [DOI] [PubMed] [Google Scholar]
- 37. Lee JW, Chou CL, Knepper MA. Deep sequencing in microdissected renal tubules identifies nephron segment-specific transcriptomes. J Am Soc Nephrol 26: 2669–2677, 2015. doi: 10.1681/ASN.2014111067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vallon V, Gerasimova M, Rose MA, Masuda T, Satriano J, Mayoux E, Koepsell H, Thomson SC, Rieg T. SGLT2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol 306: F194–F204, 2014. doi: 10.1152/ajprenal.00520.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, Cunard R, Sharma K, Thomson SC, Rieg T. Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304: F156–F167, 2013. doi: 10.1152/ajprenal.00409.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Onishi A, Fu Y, Patel R, Darshi M, Crespo-Masip M, Huang W, Song P, Freeman B, Kim YC, Soleimani M, Sharma K, Thomson SC, Vallon V. A role for tubular Na+/H+ exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am J Physiol Renal Physiol 319: F712–F728, 2020. doi: 10.1152/ajprenal.00264.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Onishi A, Fu Y, Darshi M, Crespo-Masip M, Huang W, Song P, Patel R, Kim YC, Nespoux J, Freeman B, Soleimani M, Thomson SC, Sharma K, Vallon V. Effect of renal tubule-specific knockdown of the Na+/H+ exchanger NHE3 in Akita diabetic mice. Am J Physiol Renal Physiol 317: F419–F434, 2019. doi: 10.1152/ajprenal.00497.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Coady MJ, El TA, Santer R, Bissonnette P, Sasseville LJ, Calado J, Lussier Y, Dumayne C, Bichet DG, Lapointe JY. MAP17 is a necessary activator of renal Na+/glucose cotransporter SGLT2. J Am Soc Nephrol 28: 85–93, 2017. doi: 10.1681/ASN.2015111282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Calado J, Santos AR, Aires I, Lebre F, Nolasco F, Rueff J, Ramalho J. The Na+ -coupled glucose transporter SGLT2 interacts with its accessory unit MAP17 in vitro and their expressions overlap in the renal proximal tubule. FEBS Lett 592: 3317–3326, 2018. doi: 10.1002/1873-3468.13233. [DOI] [PubMed] [Google Scholar]
- 44. Song P, Huang W, Onishi A, Patel R, Kim YC, van Ginkel C, Fu Y, Freeman B, Koepsell H, Thomson S, Liu R, Vallon V. Knockout of Na+-glucose cotransporter SGLT1 mitigates diabetes-induced upregulation of nitric oxide synthase NOS1 in the macula densa and glomerular hyperfiltration. Am J Physiol Renal Physiol 317: F207–F217, 2019. doi: 10.1152/ajprenal.00120.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gekle M, Volker K, Mildenberger S, Freudinger R, Shull GE, Wiemann M. NHE3 Na+/H+ exchanger supports proximal tubular protein reabsorption in vivo. Am J Physiol Renal Physiol 287: F469–F473, 2004. doi: 10.1152/ajprenal.00059.2004. [DOI] [PubMed] [Google Scholar]
- 46. Tojo A, Kinugasa S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int J Nephrol 2012: 481520, 2012. doi: 10.1155/2012/481520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol 28: 436–453, 2018. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 48. Naved BA, Bonventre JV, Hubbell JA, Hukriede NA, Humphreys BD, Kesselman C, Valerius MT, McMahon AP, Shankland SJ, Wertheim JA, White MJV, de Caestecker MP, Drummond IA. Kidney repair and regeneration: perspectives of the NIDDK (Re)Building a Kidney consortium. Kidney Int 101: 845–853, 2022. doi: 10.1016/j.kint.2022.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol 27: 2652–2660.e4, 2017. doi: 10.1016/j.cub.2017.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal 22: 1003–1012, 2010. doi: 10.1016/j.cellsig.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kang PJ, Zheng J, Lee G, Son D, Kim IY, Song G, Park G, You S. Glycine decarboxylase regulates the maintenance and induction of pluripotency via metabolic control. Metab Eng 53: 35–47, 2019. doi: 10.1016/j.ymben.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 52. Halkoum R, Salnot V, Capallere C, Plaza C, L'Honore A, Pays K, Friguet B, Nizard C, Petropoulos I. Glyoxal induces senescence in human keratinocytes through oxidative stress and activation of the protein kinase B/FOXO3a/p27(KIP1) pathway. J Invest Dermatol 142: 2068–2078.e7, 2021. doi: 10.1016/j.jid.2021.12.022. [DOI] [PubMed] [Google Scholar]
- 53. Vallon V, Thomson SC. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia 60: 215–225, 2017. doi: 10.1007/s00125-016-4157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Navarro-Garrido A, Kim YC, Goodluck H, Kanoo S, Crespo-Masip M, Bröer S, Vallon V. Basal renal phenotype and response to induction of aristolochic acid nephropathy in mice lacking the neutral amino acid transporter B0AT1 (SLC6A19). FASEB J 35: issue S1, Abstract, 2021. doi: 10.1096/fasebj.2021.35.S1.04004. [DOI] [PMC free article] [PubMed] [Google Scholar]