


Keywords: calcium, lung vascular endothelial cell, mechanotransduction, nonselective cation channel
Abstract
Mechanosensitive cation channels and Ca2+ influx through these channels play an important role in the regulation of endothelial cell functions. Transient receptor potential canonical channel 6 (TRPC6) is a diacylglycerol-sensitive nonselective cation channel that forms receptor-operated Ca2+ channels in a variety of cell types. Piezo1 is a mechanosensitive cation channel activated by membrane stretch and shear stress in lung endothelial cells. In this study, we report that TRPC6 and Piezo1 channels both contribute to membrane stretch-mediated cation currents and Ca2+ influx or increase in cytosolic-free Ca2+ concentration ([Ca2+]cyt) in human pulmonary arterial endothelial cells (PAECs). The membrane stretch-mediated cation currents and increase in [Ca2+]cyt in human PAECs were significantly decreased by GsMTX4, a blocker of Piezo1 channels, and by BI-749327, a selective blocker of TRPC6 channels. Extracellular application of 1-oleoyl-2-acetyl-sn-glycerol (OAG), a membrane permeable analog of diacylglycerol, rapidly induced whole cell cation currents and increased [Ca2+]cyt in human PAECs and human embryonic kidney (HEK)-cells transiently transfected with the human TRPC6 gene. Furthermore, membrane stretch with hypo-osmotic or hypotonic solution enhances the cation currents in TRPC6-transfected HEK cells. In HEK cells transfected with the Piezo1 gene, however, OAG had little effect on the cation currents, but membrane stretch significantly enhanced the cation currents. These data indicate that, while both TRPC6 and Piezo1 are involved in generating mechanosensitive cation currents and increases in [Ca2+]cyt in human PAECs undergoing mechanical stimulation, only TRPC6 (but not Piezo1) is sensitive to the second messenger diacylglycerol. Selective blockers of these channels may help develop novel therapies for mechanotransduction-associated pulmonary vascular remodeling in patients with pulmonary arterial hypertension.
INTRODUCTION
Lung vascular endothelial cells (ECs) form the pulmonary arterial and venous endothelium (or the intima) and the capillaries around alveoli. Similar to ECs in systemic vasculature, lung ECs are considered nonexcitable cells but abundantly express cation and anion channels in the plasma membrane. In addition to regulating membrane potential, ion flux through channels in ECs may directly contribute to the regulation of production and release of vasoactive substances (e.g., endothelium-derived relaxing and constricting factors), mitogenic and angiogenic factors, and inflammatory cytokines. In addition, ion channels (e.g., Ca2+-permeable cation channels) in lung ECs are involved in trans capillary transportation of macromolecules through regulation of endocytosis, trans cytosine, and exocytosis (1, 2).
Ca2+-permeable cation channels in lung vascular ECs play an important role in the regulation of intracellular Ca2+ homeostasis, membrane potential, and various vascular function (1, 2). An increase in cytosolic-free Ca2+ concentration ([Ca2+]cyt) in pulmonary arterial ECs (PAESs) is a trigger to activate endothelial nitric oxide (NO) synthase (eNOS) and stimulate endothelial NO production. By activating soluble guanylate cyclase to produce cGMP and/or activating various K+ channels to cause membrane hyperpolarization in smooth muscle cells (SMCs), NO is an important endothelium-derived relaxing factor (EDRF) that causes endothelium-dependent vasodilation (3–5). Persistent activation of eNOS due to EC calveolin-1 deficiency is, however, implicated in the development of pulmonary hypertension (PH) due to potentially eNOS uncoupling (6) and protein kinase G (PKG) nitration (7–10).
Vascular ECs including PAECs are nonexcitable cells that express both voltage-dependent Ca2+ channels (VDCC) (11, 12) and voltage-independent and/or receptor-operated cation channels (1). Changes of the activity, expression, and gating of these nonselective cation channels contribute to the regulation of EC function and vascular tone and structure (1, 13). Among the cation channels in ECs (including PAECs), the Ca2+-permeable nonselective cation channels [e.g., Transient receptor potential vanilloid 1 (TRPV1)] (14, 15) and Ca2+-activated K+ (KCa) channels (16, 17) are two important families of channels participating in the regulation of Ca2+ signaling and Ca2+-dependent translational and transcriptional cascades in vascular ECs. In addition to the rise in [Ca2+]cyt due to Ca2+ influx through nonselective cation channels, Na+ influx through these channels plays an important role in regulating EC migration and mobility (18).
More than 40% of cells in the lungs are arterial, arteriole, capillary, and venous ECs (19), although the lung vasculature or vascular wall is composed of ECs, smooth muscle cells (SMCs), and (myo)fibroblasts. In whole lung tissues and isolated pulmonary artery (PA) rings, we and others observed upregulation of transient receptor potential (TRP) channels in animals with experimental PH (20–22). In pulmonary arterial smooth muscle cells (PASMC) and PAEC from patients with pulmonary arterial hypertension (PAH), we and others found that transient receptor potential canonical channel 6 (TRPC6) and Piezo1 are upregulated and pathogenically involved in the development of sustained pulmonary vasoconstriction and concentric pulmonary vascular remodeling (23–25). TRPC6 is a canonical TRP channel subunit (with a single-channel conductance of 35 pS) that participates in forming receptor-operated Ca2+ channels in vascular SMCs and other types of cells including ECs (26–29) that are activated by ligand-mediated activation of G protein-coupled receptors and tyrosine kinase receptors (30–33) and by redox changes (34). In addition, TRPC6 is also a mechanosensitive cation channel that allows Ca2+ and Na+ to enter cell (35–37). Piezo1 is a newly identified mechanosensitive cation channel that is involved in the regulation of blood pressure, vascular reactivity, and angiogenesis (38–43), as well as cell proliferation (44–47).
In this study, we examine whether TRPC6 and Piezo1 channels are involved in receptor-operated Ca2+ influx (or cation currents) and mechanical stimulation-induced Ca2+ influx (or cation currents) in human PAECs. We compared the cation currents in TRPC6-transfected and Piezo1-transfected Human embryonic kidney (HEK)-293 cells with the native human PAECs. The data from this study demonstrate that both TRPC6 and Piezo1 channels contribute to the whole cell cation currents induced by mechanical stimulation or membrane stretch. TRPC6 also contributes to the whole cell cation currents induced by the second messenger, diacylglycerol, or its membrane-permeable analog 1-oleoyl-2-acetyl-sn-glycerol (OAG), but Piezo1-associated mechanosensitive currents are not sensitive to OAG. These results suggest that, in addition to other cation channels expressed in lung ECs, TRPC6 is an important OAG-sensitive receptor-operated or diacylglycerol-activated cation channel, whereas both TRPC6 and Piezo1 are important mechanosensitive cation channels in human PAECs.
MATERIALS AND METHODS
Cell Preparation and Culture
Normal human PAECs (Lonza, Rockville, MD) were cultured in VascuLife VEGF growth medium (Lifeline Cell Technologies, Frederick, MD) supplemented with 2% fetal bovine serum (FBS), 5 ng/mL human epidermal growth factor (EGF), 5 ng/mL human fibroblast growth factor (FGF), 50 µg/mL ascorbic acid, 1 µg/mL hydrocortisone hemisuccinate, 10 mM l-glutamine, 15 ng/mL human insulin-like growth factor (IGF-1), 5 ng/mL human vascular endothelial growth factor (VEGF), 0.75 U/mL heparin sulfate, 30 mg/mL gentamicin, and 15 µg/mL amphotericin B. HEK-293 cells were cultured in high-glucose DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen, Waltham, MA), 100 IU/mL penicillin and 100 μg/mL streptomycin (Sigma-Aldrich, St. Louis, MO). Cells were maintained in an incubator under a humidified environment at 37°C and 5% CO2. Upon reaching 70%–90% confluence, cells were detached with Trypsin-EDTA (0.05%) and plated on 25-mm cover slips for electrophysiological and fluorescent microscopy experiments.
Transfection
HEK-293 cells were transiently transfected with 2 µg of human TRPC6 construct (Plasmid No. 21084; Addgene) or mouse Piezo1 construct (Plasmid No. 80925, Addgene) using X-tremeGENE HP DNA Transfection Reagent (Millipore Sigma, Burlington, MA) for 4–6 h in Opti-MEM Reduced Serum Medium (Gibco, Grand Island, NY). Then, the Opti-MEM medium was replaced with 10% FBS DMEM and the cells were incubated in 10% FBS DMEM for 48–72 h before being used for electrophysiological and digital imaging fluorescence microscopy experiments. A GFP construct was used to visualize the transfected cells by co-transfecting the cells with the TRPC6 construct or the Piezo1 construct. In the pCMS-EGFP vector, the EGFP gene was expressed separately from the gene of interest and is used as a transfection marker; most of the cells would be co-transfected efficiently with the GFP and the TRPC6 or Piezo1 gene. Green fluorescence emitted at 507 nm was used to visualize the cells transfected with TRPC6 or Piezo1 and GFP constructs using an inverted Nikon microscope (Eclipse/TE200) with the TE-FM epifluorescence attachment. The cell images were acquired with an Image Intensifier Tube/Philips 1381 system (Stanford Photonics Electronic Imaging Technologies, Palo Alto, CA). We also used siRNA to specifically downregulate TRPC6 in human PAECs. TRPC6-siRNA (Ambion s14422) was introduced into human PAECs using RNA Max Lipofectamine reagent (Invitrogen, Waltham, MA) with a final concentration of 100 pM per well in 6-well Petri dishes. The cells were incubated in the siRNA-transfection media for 48–72 h before being used for the patch clamp and digital imaging fluorescence microscopy experiments.
Electrophysiological Recording
Whole cell cation currents were recorded with the patch-clamp technique using an Axopatch 200B amplifier and a DigiData1550B interface (Molecular Devices, Silicon Walley, CA). Patch pipettes (2–3 MΩ) were fabricated on an electrode puller (Sutter Instrument, Novato, CA) using borosilicate glass tubes and fire polished on a microforge (Narishige Scientific Instruments, Tokyo, Japan) to get a range of 4–9 MΩ series resistance. Command voltage protocols and data acquisition were performed using pCLAMP-10 software (Molecular Devices). The HEPES-buffered bath (extracellular) solution contained 137 mM NaCl, 5.9 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 14 mM glucose, and 10 mM HEPES (pH, 7.4, by NaOH). The pipette (intracellular) solution contained 120 mM CsOH, 120 mM aspartic acid, 0.6475 mM CaCl2, 4 mM MgCl2, 5 mM MgATP, 10 mM HEPES, and 10 mM EGTA (pCa = 8.0). The pH of the pipette solution was adjusted to 7.2 with 1 N CsOH. The hypotonic bath solution contained 90 mM NaCl, 5 mM KCl, 2.4 mM CaCl2, 1.3 mM MgCl2, 10 mM glucose, and 10 mM HEPES (pH = 7.4 with NaOH). The cell was clamped at a holding potential of 0 mV and an ascending ramp protocol from −100 to +100 mV for 1,000 ms was applied to the cell every 1.3 s. The step protocol has a 0-mV holding potential with 500-ms step in 200-mV increments from −100 to +100 mV. Currents were filtered at 1–2 kHz and digitized at 2–5 kHz. Electrophysiological recordings were performed at room temperature (24–25°C).
Measurement of Cytosolic Ca2+ Concentration ([Ca2+]cyt)
Briefly, human PAECs were grown to 60%–70% confluence on 25-mm round glass coverslips. The cells were loaded with fura-2 acetoxymethyl ester (fura-2/AM, 4 µM; Invitrogen/Molecular Probes, Eugene, OR) in the dark for 60 min at room temperature (22–24°C) in a normal physiological salt solution (PSS). The PSS contained 140 mM NaCl, 4.7 mM KCl, 1.8 mM CaCl2, 1.2 mM MgCl2, 10.0 mM glucose, and 10.0 mM HEPES. A coverslip containing fura-2/AM-loaded cells was placed in a recording chamber on the stage of the Nikon inverted fluorescence microscope (Eclipse Ti-E; Nikon, Tokyo, Japan). The excitation wavelengths were 340 nm and 380 nm, and the emission signal at 520 nm was detected using an EM-CCD camera (Evolve; Photometrics, Tucson, AZ) through a Nikon S-Plan Fluor ELWD ×20/0.45 objective lens; the fluorescence images were recorded and analyzed using the NIS Elements 3.2 software (Nikon, Tokyo, Japan). [Ca2+]cyt within the region of interest (5 × 5 μm) was positioned at the peripheral region of each cell and measured as the ratio of fluorescence intensities (F340/F380) and recorded every 2 s. [Ca2+]cyt was calculated by the ratiometric method using the following equation: [Ca2+]cyt = Kd × (Sf2/Sb2) × (R − Rmin)/(Rmax − R), where Kd (225 nM) is the dissociation constant of Fura-2 for Ca2+ and Sf2 and Sb2 are the fluorescent intensities at 380-nm excitation in Ca2+-free solution (with EGTA) and Ca2+-saturated solution (with ionomycin), respectively. R is the measured fluorescence ratio, while Rmin and Rmax are the minimum and maximum ratios that were determined in cells superfused with the Ca2+-free solution (plus 5 mM EGTA) and the Ca2+-containing solution (10 mM CaCl2), respectively, in the presence of ionomycin (2 µM). In Ca2+-free solution, CaCl2 was replaced by equimolar amounts of MgCl2, and EGTA was added to chelate residual Ca2+. All experiments for measurement of [Ca2+]cyt were performed at room temperature (22–24°C).
Drugs and Chemicals
All drugs and chemicals were obtained from Sigma Aldrich unless otherwise stated. A hydrophobic compound, 1-oleoyl-2-acetyl-sn-glycerol (OAG) which is a membrane-permeant analog of the second messenger, diacylglycerol (DAG), was dissolved in dimethyl sulfoxide (DMSO) at the concentration of 100 mM as a stock solution. BI-749327 (Cat. No. HY-111925, MedChemExpress, Monmouth, NJ) was prepared as an 11 mM stock solution in DMSO. GsMTx4 (Cat. No. HY-P1410, MedChemExpress, Monmouth, NJ) was dissolved into DMSO and prepared as a 500 mM stock solution. Yoda-1, 2-(5-{[(2,6-dichlorophenyl)methyl]sulfanyl}-1,3,4-thiadiazol-2-yl)pyrazine (Cat. No. 448947-81-7, Sigma-Aldrich, St. Louis, MO), was dissolved into DMSO and prepared as 30 mM stock solution. Aliquots of the stock solutions were then diluted to make a final concentration of OAG (Cat. No. 86390-77-4, Sigma-Aldrich, St. Louis, MO) at 100 µM, BI-749327 at 50 nM, and GsMTx4 at 5 µM in PSS on the day of the experiment.
Statistical Analysis
Pooled data are shown as means ± SE. The statistical significance between two groups was determined by Student’s t test. The statistical significance among groups was determined by one-way ANOVA analysis. A significant difference is expressed in the figures or figure legends by *P < 0.05, **P < 0.01, and ***P < 0.001.
RESULTS
We first conducted a series of patch clamp experiments to measure whole cell nonselective cation currents (ICation) in human PAECs induced by membrane stretch to demonstrate mechanosensitive cation currents. Lowering osmolality or decreasing osmotic pressure from 310 mosmol/kgH2O (Iso-Osm, iso-osmalality or iso-osmotic solution) to 220 mosmol/kgH2O (Hypo-Osm, hypo-osmolality or hypo-osmotic solution) in extracellular perfusate causes the movement of water molecules from the extracellular site into the cytosol and results in membrane stretch. The Hypo-Osm-mediated ICation is defined as the mechanosensitive cation current. OAG (a membrane-permeable analog of diacylglycerol) was used to induce diacylglycerol-sensitive cation currents in PAECs and the OAG-mediated ICation is defined as the receptor-operated cation current. Then, we used BI-749327, a selective blocker of TRPC6 (48), to characterize the pharmacological property of TRPC6 channels, and GsMTx4, a blocker of Piezo1 channels (49–51) and Yoda1, a selective activator of Piezo1 channels (52), to characterize Ca2+ influx through Piezo1 channels in human PAECs.
Membrane Stretch Induces Nonselective Currents in Human Pulmonary Artery Endothelial Cells
Membrane stretch by lowering extracellular osmolality from 310 mosmol/kgH2O (Iso-Osm) to 220 mosmol/kgH2O (Hypo-Osm) significantly and reversibly enhanced ICation in human PAECs (Fig. 1Aa). The membrane stretch-mediated ICation (Fig. 1Ab) was obtained by subtracting the current recorded when the cell was bathed in Iso-Osm solution from the current recorded when the cell was bathed in Hypo-Osm solution, which was reversed at ∼0 mV. The reversal potential of 0 mV indicated that these currents were nonselective cation currents. These data indicate that membrane stretch induces nonselective cation currents through, potentially, mechanosensitive cation channels in the PAEC plasma membrane. We then used a specific blocker of TRPC6 channels, BI-749327 (with an IC50 of 19 nM for TRPC6, which is 40–80 times more potent than on other TRPC channels) (48), to examine whether the membrane stretch-mediated ICation were due to, at least partially, cation influx through TRPC6 channels in PAECs.
Figure 1.

Membrane stretch and diacylglycerol (DAG) induce nonselective cation currents (ICation) in human pulmonary arterial endothelial cells (PAECs) and BI-749327, a selective blocker of TRPC6, inhibits the stretch- and DAG-mediated cation currents. A: representative whole cell ICation (a), elicited by a ramp protocol from −100 mV to +100 mV (for 1,000 ms) in a human PAEC before (iso-osmalality or iso-osmotic solution, Iso-Osm, 330 mosmol/kgH2O), during (hypo-osmolality or hypo-osmotic solution, Hypo-Osm), and after (Recovery) application of hypo-osmotic (Hypo-Osm; 220 mosmol/kgH2O) solution. The stretch-mediated ICation (b) was constructed by subtracting ICation recorded in Iso-Osm from ICation recorded in Hypo-Osm. Summarized data (means ± SE, n = 5 cells, c) showing the ICation amplitudes at −100 mV and +100 mV in cells before, during, and after application of Hypo-Osm solution. **P < 0.01, ***P < 0.001 vs. Iso-Osm and Recovery (one-way ANOVA). B: representative ICation (a), elicited by a ramp protocol in a PAEC bathed in Hypo-Osm solution before (Control), during (BI-749327), and after (Washout) application of BI-749327 (50 nM) in Hypo-Osm solution. The BI-749327-sensitive stretch-mediated ICation (b) was constructed by subtracting ICation recorded in Control + Hypo-Osm from ICation recorded in BI-749327 + Hypo-Osm. Summarized data (means ± SE, n = 5 cells, c) showing current amplitudes at −100 mV and +100 mV in PAECs in Hypo-Osm before, during, and after application of BI-749327. **P < 0.01, ***P < 0.001 vs. Control and Washout (one-way ANOVA). C: representative ICation (a) in a PAEC before (Control), during (1-oleoyl-2-acetyl-sn-glycerol, OAG), and after (Washout) application of the DAG analog OAG (100 µM). The OAG-mediated ICation (b) was constructed by subtracting the current recorded before application of OAG from the current recorded during application of OAG. Summarized data (means ± SE, n = 5 cells, c) showing the amplitudes of ICation at −100 mV and +100 mV in PAECs before, during, and after extracellular application of OAG (100 µM). **P < 0.01, ***P < 0.001 vs. Control and Washout (one-way ANOVA). D: representative ICation (a) in a PAEC bathed in OAG-containing Iso-Osm solution before, during, and after application of BI-749327 (50 nM). The BI-749327-sensitive OAG-mediated ICation (b) was constructed by subtracting ICation recorded in OAG from ICation recorded in OAG + BI-749327. Summarized data (means ± SE, n = 5 cells, c) showing the amplitudes of currents at −100 mV and +100 mV in PAECs in OAG solution before, during, and after application of BI-749327 (50 nM). ***P < 0.001 vs. Control (one-way ANOVA).
In the cells bathed in Hypo-Osm solution, the plasma membrane was stretched by hypo-osmolality (as a result of inward water transportation) and the mechanosensitive ICation were induced by the membrane stretch. Extracellular application of BI-749327 significantly and reversibly inhibited the stretch-mediated increases in whole cell ICation (Fig. 1Ba). The BI-749327-sensitive membrane stretch-mediated ICation (Fig. 1Bb) obtained by subtracting the stretch-induced current recorded in the absence of BI-749327 (Control) from the stretch-induced current recorded in the presence of 50 nM BI-749327 also reversed at 0 mV with a little inward rectification at the high voltage (+60 to +100 mV (Fig. 1Bb). BI-749327 resulted in an ∼60% inhibition of the stretch-induced ICation at −100 mV and +100 mV (Fig. 1Bc). These results indicate that, in human PAECs, membrane stretch activates nonselective cation channels, whereas BI-749327-sensitive TRPC6 channels contribute to the mechanosensitive ICation.
Diacylglycerol Analog Induces Nonselective Currents in Human PAECs
In addition to mechanosensitive stimulation (i.e., membrane stretch by extracellular hypo-osmolality), extracellular application of OAG (100 µM), a membrane-permeable analog of the second messenger diacylglycerol (DAG), significantly and reversibly enhanced the whole cell ICation in human PAECs (Fig. 1Ca). The reversal potential and current kinetics of the OAG-sensitive ICation (Fig. 1Cb), obtained by subtracting the current recorded before OAG treatment (Control) from the current recorded during OAG treatment, are similar to the stretch-mediated ICation (shown in Fig. 1Ab). In the presence of 100 µM of OAG, extracellular application of BI-749327 (50 nM), a selective blocker of TRPC6 channels, significantly and reversibly reduced the OAG-mediated ICation (Fig. 1Ca). The BI-749327-sensitive OAG-mediated ICation (Fig. 1Db), obtained by subtracting the OAG-induced ICation recorded in the absence of BI-749327 (Control) from the OAG-induced ICation recorded in the presence of 50 nM BI-749327, seems to have similar reversal potential (0 mV) and current kinetics to the BI-749327-sensitive stretch-mediated ICation (Fig. 1Bb). These data indicate that, in human PAECs, the second messenger DAG or the exogenous analog OAG activates nonselective cation channels, whereas BI-749327-sensitive TRPC6 channels contribute to the OAG-induced ICation.
Downregulation of TRPC6 Inhibits Stretch-Induced Cation Currents in Human PAECs
To further confirm the contribution of TRPC6 to the mechanosensitive ICation, we treated human PAECs with siRNA specifically targeting TRPC6 (TRPC6-siRNA) and compared the stretch-mediated ICation with control cells. As shown in Fig. 2, Hypo-Osm solution resulted in a significant increase in whole cell ICation in control PAECs (Fig. 2, A and B, left). The averaged amplitude of mechanosensitive ICation in control PAECs was −144 pA at −100 mV and 214 pA at +100 mV (Fig. 2, B and C). Treatment of PAECs with TRPC6-siRNA (for 48 h) significantly reduced the Hypo-Osm-mediated ICation (Fig. 2, A and B, right); the averaged amplitude of mechanosensitive ICation in TRPC6-siRNA-treated PAECs was −60.8 pA at −100 mV and 33.4 pA at +100 mV (Fig. 2, B and C, left). The approximate 80% of inhibition of stretch-mediated ICation at −100 and +100 mV (Fig. 2C, right) indicate that cation currents through TRPC6 channels account for a large portion of mechanosensitive cation currents in human PAECs.
Figure 2.

Downregulation of transient receptor potential canonical channel 6 (TRPC6) with siRNA significantly inhibits membrane stretch-induced cation currents (ICation) in human pulmonary arterial endothelial cell (PAECs). A: representative whole cell ICation, elicited by a ramp protocol from −100 mV to + 100 mV (for 1,000 ms) before (iso-osmalality or iso-osmotic solution, Iso-Osm), during (hypo-osmolality or hypo-osmotic solution, Hypo-Osm), and after (Recovery) application of Hypo-Osm solution in a control PAEC (Control, left) and a PAEC treated with TRPC6-siRNA (100 pM/well for 48 h, right). B: summarized data (means ± SE, n = 5 cells showing ICation amplitudes at −100 mV and +100 mV before, during, and after application of Hypo-Osm solution in control PAECs (left) and TRPC6-siRNA-treated PAECs (right). ***P < 0.001 vs. Iso-Osm and Recovery (one-way ANOVA). C: the stretch-mediated ICation shown in the left panel was constructed by subtracting ICation recorded in Iso-Osm from ICation recorded in Hypo-Osm in control PAEC and TRPC6-siRNA-treated PAEC (A). Summarized data (means ± SE, n = 5, right) showing the percentage change of the amplitude of ICation (at −100 mV and +100 mV) in TRPC6-siRNA-treated PAEC compared to control cells.
TRPC6 Channels Are Sensitive to Membrane Stretch Due to Hypo-osmolality
The mechanosensitivity of TRPC6 has been demonstrated in different types of cells (35, 53–55). To further prove that TRPC6 channels are sensitive to membrane stretch, we conducted similar experiments in HEK-cells transiently transfected with the human TRPC6 gene. As shown in Fig. 3, membrane stretch, by superfusing hypo-osmotic solution (Hypo-Osm, for 10 min), significantly and reversibly increased whole cell nonselective cation currents through TRPC6 channels (ITRPC6) in TRPC6-transfected HEK cells (Fig. 3A, left). The membrane stretch-mediated ITRPC6 (Fig. 1A, right), obtained by subtracting the current recorded when the cell was bathed in Iso-Osm solution from the current recorded when the cell was bathed in Hypo-Osm solution, reversed at ∼0 mV. In the cells in which the currents were elicited by step-pulse, from a holding potential of 0 mV to +100 mV or to −100 mV, membrane stretch also significantly and reversibly enhanced the outward (at+100 mV) and inward (at −100 mV) currents (Fig. 3, B and C). These results indicate that membrane stretch of PAEC due to hypo-osmolality is sufficient to activate TRPC6 channels and that the stretch-mediated cation currents through TRPC6 are reversible upon restoration of extracellular osmolality.
Figure 3.

Membrane stretch and diacylglycerol (DAG) induce cation currents through transient receptor potential canonical channel 6 (TRPC6) channels (ITRPC6) in TRPC6-transfected human embryonic kidney (HEK)-293 cells and BI-749327 inhibits the stretch-and diacylglycerol-mediated ITRPC6. A: representative whole cell ITRPC6 (left), elicited by a ramp protocol from −100 mV to + 100 mV (for 1,000 ms) in a TRPC6-transfected HEK cell before (iso-osmalality or iso-osmotic solution, Iso-Osm), during (Hypo-osmolality or hypo-osmotic solution, Hypo-Osm), and after (Recovery) application of Hypo-Osm solution. The stretch-mediated ITRPC6 (right) was constructed by subtracting the current recorded in Iso-Osm from the current recorded in hypo-osmolality or hypo-osmotic solution (Hypo-Osm). B: representative whole cell ITRPC6, elicited by depolarization from a holding potential of 0 mV to +100 mV and −100 mV in a TRPC6-transfected HEK cell before, during, and after application of Hypo-Osm solution. C: summarized data (means ± SE, n = 5 cells) showing the amplitudes of ITRPC6 at −100 mV and +100 mV in TRPC6-transfected HEK cells before, during, and after application of Hypo-Osm solution. *P < 0.05, ***P < 0.001 vs. Iso-Osm and Iso-Osm Recovery (one-way ANOVA). D: representative ITRPC6 (left), elicited by a ramp protocol in a TRPC6-transfected HEK cell before (Control), during (1-oleoyl-2-acetyl-sn-glycerol, OAG) and after (Washout) application of the DAG analog OAG (100 µM). The OAG-mediated ITRPC6 (right) was constructed by subtracting the current recorded in Control and from the current recorded in OAG. E: representative ITRPC6, elicited by depolarization from a holding potential of 0 mV to +100 mV and −100 mV in a TRPC6-transfected HEK cell before, during, and after application of OAG (100 µM). F: summarized data (means ± SE, n = 5 cells) showing the amplitudes of ITRPC6 at −100 mV and +100 mV in TRPC6-transfected HEK cells before, during, and after application of OAG. ***P < 0.001 vs. Control and Washout (one-way ANOVA).
TRPC6 Channels Are Activated by the Diacylglycerol Analog
In addition to its mechanical sensitivity (53), TRPC6 is known as an important receptor-operated Ca2+ channel that is activated by ligands and the second messenger diacylglycerol (DAG) (26, 30, 36). Similar to human PAECs, extracellular application of the membrane-permeable DAG analog, OAG (100 µM), significantly and reversibly increased whole cell cation currents in TRPC6-transfected HEK cells (Fig. 3D, left). The OAG-sensitive currents, obtained by subtracting the currents recorded in the cell superfused with OAG-containing solution from the currents recorded in the cell bathed in control solution (Fig. 3D, right) indicate that the cation currents through TRPC6 channels (or predominantly through TRPC6 channels) seem to have a similar current-voltage (I-V) relationship as the native cation currents activated by OAG in human PAECs (Fig. 1C, a and b). In addition to the ramp protocol, we also used step protocol to elicit the currents by depolarizing the cell from a holding potential of 0 mV to +100 mV (to induce outward current) and −100 mV (to induce inward current). Extracellular application of 100 µM OAG resulted in a 162% increase of the inward current at −100 mV and a 97% increase of the outward currents at +100 mV in TRPC6-transfected HEK cells (Fig. 3, E and F). These results are in good agreement with other reports that the membrane-permeable DAG analog, OAG, can directly activate TRPC6 channels.
BI-749327 Significantly Reduces Cation Currents through TRPC6 Channels
In control HEK cells (Fig. 4A) and HEK cells treated with transfection agents (Mock, Fig. 4B), extracellular application of 50 nM BI-749237 had negligible effect on the “basal” cation currents elicited by either a ramp protocol (a and b) or a step protocol (c and d). Transient transfection of the human TRPC6 gene significantly enhanced the cation currents (Fig. 4C) compared with control HEK cells (Fig. 4A) and Mock HEK cells (Fig. 4B). In TRPC6-transfected HEK cells, extracellular application of BI-749327 (50 nM) significantly and reversibly inhibited the cation currents through TRPC6 channels (ITRPC6) (Fig. 4C) without membrane stretch or stimulation with OAG. The BI-749327-sensitive whole cell ITRPC6, obtained by subtracting the currents recorded in the cell superfused with solution containing 50 nM BI-749327 from the currents recorded in the cell bathed in control (C) solution (Fig. 5Cb) shows that the currents reversed at ∼0 mV and the I-V relationship is similar to the TRPC6 currents demonstrated by other investigators (56). Using both the ramp (Fig. 4C, a and b) and step (Fig. 4C, c and d) protocols, we showed that 50 nM BI-749327 resulted in a 62%–71% decrease of the inward ITRPC6 at −100 mV and an 81%–85% decrease of the outward ITRPC6 at +100 mV (Fig. 5C, c and d). The next set of experiments was designed to examine whether BI-749327-mediated inhibition of TRPC6 channels inhibits OAG-mediated increases in [Ca2+]cyt in TRPC6-transfected HEK cells. Consistent with the patch clamp experiments, extracellular application of 50 nM BI-749327 negligibly affected the resting [Ca2+]cyt, but significantly inhibited OAG-induced increases in [Ca2+]cyt (Fig. 5, A and B). These data indicate that BI-749327 is a potent inhibitor of TRPC6 channels, and BI-749327-mediated blockade of TRPC6 causes significant inhibition of receptor-operated or OAG-mediated cation currents and Ca2+ entry.
Figure 4.

BI-749327 inhibits cation currents through transient receptor potential canonical channel 6 (TRPC6) channels (ITRPC6) in TRPC6-transfected human embryonic kidney (HEK) cells. A and B: representative whole cell ICation (a), elicited by a ramp protocol from −100 mv to +100 mV (for 1,000 ms) in a control HEK cell (A) and a mock HEK cell (B, transfected with empty vector) before (C), during (BI), and after (W) application of BI-749327 (50 nM). The BI-749327-sensitive ICation (b) was constructed by subtracting the current in control (C) from the current recorded in BI-749327 (BI). Representative whole cell ICation (c), elicited by a step protocol from a holding potential of 0 mV to +100 mV and −100 mV in a control HEK cell (A) and a mock HEK cell (B) before (Control), during (BI-749327), and after (Washout) application of BI-749327. Summarized data (means ± SE, n = 5 cells, d) showing the amplitudes of ICation at −100 mV and +100 mV in control (A) and mock (B) HEK cells before, during, and after application of BI-749327. *P < 0.05 vs. Control (One-way ANOVA). C: representative whole cell ITRPC6 (a), elicited by a ramp protocol in TRPC6-transfected HEK cell before (C), during (BI), and after (W) application of BI-749327. The BI-749327-sensitive ITRPC6 (b) was constructed by subtracting ITRPC6 recorded before application of BI-749327 (C) from ITRPC6 recorded during application of BI-749327 (BI). Representative whole cell ITRPC6 (c), elicited by a step protocol from a holding potential of 0 mV to +100 mV and −100 mV in a TRPC6-transfected HEK cell before, during, and after application of BI-749327. Summarized data (means ± SE, n = 5 cells, d) showing the amplitude of ITRPC6 at −100 mV and +100 mV in TRPC6-transfected HEK cells before, during, and after application of BI-749327. ***P < 0.001 vs. Control (one-way ANOVA). Ication, nonselective cation currents.
Figure 5.

BI-749327 (BI) reversibly inhibits 1-oleoyl-2-acetyl-sn-glycerol (OAG)-mediated increases in [Ca2+]cyt in human pulmonary arterial endothelial cell (PAECs). A: representative record of changes in [Ca2+]cyt before and during application of OAG (100 µM) in single PAECs (individual, a) and in group of PAECs (averaged, b) on the same cover slip bathed in physiological salt solution (PSS) (Control), PSS with BI-749327 (50 nM), and PSS with vehicle (Vehicle). B: summarized data (means ± SE, n = 29–32 cells) showing the basal [Ca2+]cyt (left) and the OAG-induced increases in cytosolic-free Ca2+ concentration ([Ca2+]cyt) (right), calculated from individual PAECs, in cells bathed in control PSS, PSS with BI-749327, and PSS with vehicle (DMSO). ***P < 0.001 vs. Control and Vehicle (one-way ANOVA).
The results from these experiments shown in Figs. 1, 2, 3, 4, and 5 indicate that the BI-749327-inhibited and OAG-activated TRPC6 channels are an important receptor-operated or diacylglycerol-activated cation channel in human PAECs that can be activated by mechanical stimulation. The sensitivity of TRPC6 channels in lung ECs to membrane stretch and second messenger make the channel as an important Ca2+ influx pathway for increasing [Ca2+]cyt. Under normal conditions, the agonist- and stretch-mediated Ca2+ influx through TRPC6 channels in PAECs exerts vasodilative effect on the pulmonary vasculature by activating eNOS and increasing endothelial NO release. Under pathological conditions, the upregulated TRPC6 and enhanced Ca2+ influx through TRPC6 channels may be an important pathogenic mechanism for stretch- and mitogen-mediated pulmonary vascular remodeling.
The next set of experiments was designed to investigate whether Piezo1 channels are involved in generating mechanosensitive cation currents and increases in [Ca2+]cyt in human PAECs.
GsMTx4 Inhibits Stretch-Mediated Nonselective Cation Currents in Human PAECs
In the cells bathed in Hypo-Osm solution or when membrane was stretched by hypo-osmolality, extracellular application of GsMTx4 significantly and reversibly inhibited the stretch-mediated increases in whole cell ICation (Fig. 6A, left). The GsMTx4-sensitive membrane stretch-mediated ICation (Fig. 6A, right), obtained by subtracting the stretch-induced current recorded in the absence of GsMTx4 (Control) from the stretch-induced current recorded in the presence of 5 µM GsMTx4, reversed at −20 mV (Fig. 6A, right). GsMTx4 resulted in an 40%–60% inhibition of the stretch-induced ICation at −100 mV and +100 mV (Fig. 6B). These results indicate that, in human PAECs, membrane stretch activates nonselective cation channels, whereas GsMTx4-sensitive Piezo1 channels also contribute to the mechanosensitive ICation.
Figure 6.

Membrane stretch induces cation currents (ICation) in human pulmonary arterial endothelial cell (PAECs) and GsMTx4 (a blocker of Piezo1) inhibits the stretch-mediated (ICation). A: representative whole cell ICation (left), elicited by a ramp protocol in a PAEC bathed in hypo-osmolality or hypo-osmotic solution (Hypo-Osm) solution before (Control), during (GsMTx4), and after (Washout) application of GsMTx4 (5 µM). The GsMTx4-sensitive stretch-mediated ICation (right) was constructed by subtracting ICation recorded in Control + Hypo-Osm from ICation recorded in GsMTx4 + Hypo-Osm. B: summarized data (means ± SE, n = 5 cells) showing the amplitudes of currents at −100 mV and +100 mV in PAECs bathed in Hypo-Osm before, during, and after application of GsMTx4. ***P < 0.001 vs. Control (one-way ANOVA).
We also compared the effects of GsMTx3 (a Piezo1 blocker) and BI-749327 (a TRPC6 blocker) on the increases in [Ca2+]cyt induced by membrane stretch (Hypo-Osm) or Yoda-1, an allosteric activator of Piezo1 in human PAECs. Membrane stretch with Hypo-Osm solution induced a transient increase in [Ca2+]cyt in PAECs (Fig. 7, A and B), the Hypo-Osm-mediated increase in [Ca2+]cyt was significantly inhibited by the Piezo1 blocker GsMTx4 (GsM) (49) (Fig. 7C) and by the selective TRPC6 blocker BI-749327 (Fig. 7C). Extracellular application of Yoda-1 induced a transient increase in [Ca2+]cyt followed by a sustained or plateaued increase in PAECs (Fig. 7, D and E). GsMTx4 significantly inhibited the Yoda-1-mediated increase in [Ca2+]cyt, whereas BI-749327 had no effect on Yoda-1-mediated increase in [Ca2+]cyt (Fig. 7F). These data imply that both GsMTx4-sensitive Piezo1 and BI-749327-sensitive TRPC6 contribute to the stretch-mediated increase in [Ca2+]cyt in human PAECs, whereas only GsMTx4-sensitive Piezo1 contributes to Yoda-1-mediated increase in [Ca2+]cyt.
Figure 7.
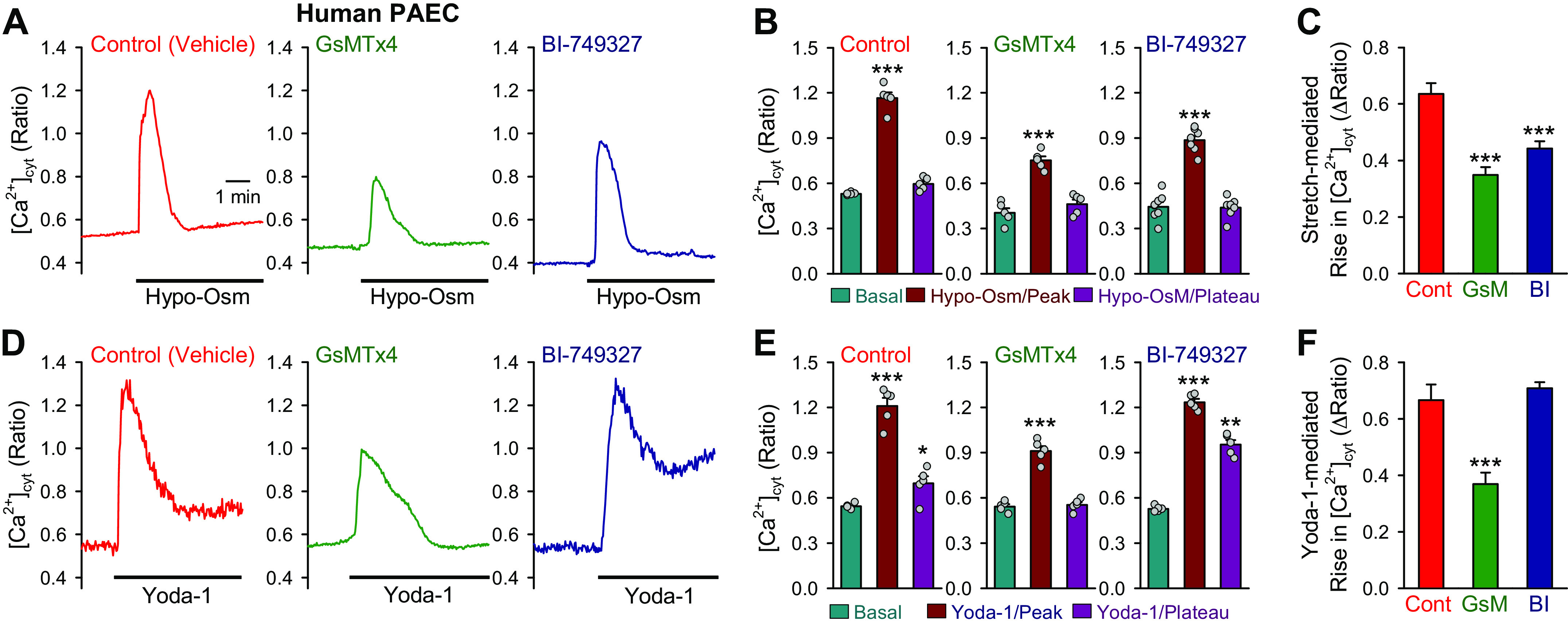
Effects of GsMTx4 (a Piezo1 blocker) and BI-749327 (a TRPC6 blocker) on membrane stretch- and Yoda-1-mediated increases in cytosolic-free Ca2+ concentration ([Ca2+]cyt) in human Pulmonary arterial endothelial cell (PAECs). A: representative record showing [Ca2+]cyt before and after membrane stretch induced by Hypo-Osm solution in the absence (Control, with vehicle) and presence of GxMTx4 (5 µM) or BI-749327 (50 nM). B: summarized data (means ± SE, n = 5) showing the basal level (Basal) of [Ca2+]cyt and the peak and plateau levels of [Ca2+]cyt during membrane stretch with Hypo-Osm solution in the absence and presence of GxMTx4 or BI-749327. ***P < 0.001 vs. Basal (one-way ANOVA). C: summarized data (means ± SE, n = 5) showing the transient (peak) increases of [Ca2+]cyt induced by membrane stretch in the absence (Cont) and presence of GxMTx4 (GsM) or BI-749327 (BI). ***P < 0.001 vs. Cont (one-way ANOVA). D: representative record showing [Ca2+]cyt before and after application of Yoda-1 (5 µM) in the absence and presence of GxMTx4 or BI-749327. E: summarized data (means ± SE, n = 5) showing the basal level (Basal) of [Ca2+]cyt and the peak and plateau levels of [Ca2+]cyt during application of Yoda-1 in the absence and presence of GxMTx4 or BI-749327. *P < 0.05, **P < 0.01, ***P < 0.001 vs. Basal (one-way ANOVA). F: summarized data (means ± SE, n = 5) showing the transient (peak) increases of [Ca2+]cyt induced by Yoda-1 in the absence and presence of GxMTx4 or BI-749327. ***P < 0.001 vs. Cont (one-way ANOVA).
GsMTx4 Reduces Stretch-Induced Cation Currents but Not OAG-Induced Cation Currents in Piezo1-Transfected HEK Cells
In HEK cells treated with transfection agents (Mock, Fig. 8A), Hypo-Osm solution induced a small increase of cation currents elicited by either a ramp protocol (a and b). Transient transfection of the Piezo1 gene significantly enhanced not only the basal cation currents in HEK cells bathed in Iso-Osm solution (without membrane stretch or stimulation with OAG), but also the stretch-mediated cation currents in HEK cells bathed in Hypo-Osm solution (Fig. 8B, left). In Piezo1-transfected HEK cells bathed in Hypo-Osm solution, extracellular application of 5 µM GsMTx4 significantly inhibited the cation currents through Piezo1 channels (IPiezo1) (Fig. 8B, right). The GsMTx4-sensitive whole cell IPiezo1, obtained by subtracting the currents recorded in the cell superfused with solution containing 5 µM GsMTx4 from the currents recorded in the cell bathed in control (Control) Hypo-Osm solution (Fig. 8Bb, left) shows that the reversal potential of the currents is close to 0 mV and that the I-V relationship shows more outward rectification. GsMTx4 (5 µM) resulted in 55% decrease of the inward IPiezo1 at −100 mV and 64% reduction of the outward IPiezo1 at +100 mV (Fig. 8Bc, right) in Piezo1-transfected HEK cells. However, extracellular application of OAG had negligible effect on the whole cell IPiezo1 in Piezo1-transfected HEK cells (Fig. 8C). The stretch-mediated enhancement of IPiezo1 in Piezo1-transfected HEK cells are consistent with the Hypo-Osm-mediated increase in [Ca2+]cyt. Membrane stretch by hypo-osmolality (or Hypo-Osm solution) induced a transient increase in [Ca2+]cyt followed by a sustained increase in Piezo1-transfected HEK cells (Fig. 9A). By directly activating Piezo1 channels, extracellular application of Yoda-1 (5 µM) also resulted in a transient increase in [Ca2+]cyt in Piezo1-transfected HEK cells (Fig. 9B). The pattern of Yoda1-mediated increase in [Ca2+]cyt via Ca2+ influx through Piezo1 channels is similar to those reported by other investigators (52).
Figure 8.

GsMTX4 inhibits stretch-mediated cation currents through Piezo1 channels (IPiezo1) in Piezo1-transfected human embryonic kidney (HEK) cells. A: representative whole cell ICation (a), elicited by a ramp protocol in a mock HEK cell (transfected with empty vector) before (iso-osmalality or iso-osmotic solution, Iso-Osm), during (hypo-osmolality or hypo-osmotic solution, Hypo-Osm), and after (Recovery) application of Hypo-Osm solution. The stretch-mediated ICation (b) was constructed by subtracting ICation recorded in Iso-Osm from ICation recorded in Hypo-Osm. Summarized data (means ± SE, n = 5 cells, c) showing the amplitude of ICation at −100 mV and +100 mV in mock HEK cells before, during, and after application of Hypo-Osm solution. ***P < 0.001 vs. Iso-Osm (one-way ANOVA). B: representative whole cell IPiezo1 (a, left), elicited by a ramp protocol in a Piezo1-transfected HEK cell before, during, and after application of Hypo-Osm solution. The stretch-mediated IPiezo1 (b, left) was constructed by subtracting IPiezo1 recorded in Iso-Osm from IPiezo1 recorded in Hypo-Osm. Summarized data (means ± SE, n = 5 cells, c, left) showing the amplitude of IPiezo1 at −100 mV and +100 mV in mock HEK cells before, during, and after application of Hypo-Osm solution. ***P < 0.001 vs. Iso-Osm (one-way ANOVA). B, right: representative IPiezo1 (a, right) in a Piezo1-transfected HEK cell bathed in Hypo-Osm solution before (Control), during (GsMTx4), and after (Washout) application of GsMTx4 (5 µM) in Hypo-Osm solution. The GsMTx4-sensitive stretch-mediated IPiezo1 (b, right) was constructed by subtracting IPiezo1 recorded in Control from IPiezo1 recorded in GsMTx4. Summarized data (means ± SE, n = 5 cells, c, right) showing the amplitudes of currents at −100 mV and +100 mV in Piezo1-transfected HEK cells bathed in Hypo-Osm solution before, during, and after application of GsMTx4. ***P < 0.001 vs. Control (one-way ANOVA). C: representative IPiezo1 (a) in a Piezo1-transfected HEK cell before (Control), during (1-oleoyl-2-acetyl-sn-glycerol, OAG), and after (Washout) application of the diacylglycerol analog OAG (100 µM). The OAG-mediated IPiezo1 (b) was constructed by subtracting the current recorded in Control from the current recorded in OAG. Summarized data (means ± SE, n = 5 cells, c) showing the amplitudes of IPiezo1 at −100 mV and +100 mV in Piezo1-transfected HEK cells before, during, and after application of OAG. Ication, nonselective cation currents.
Figure 9.

Membrane stretch and Yoda-1 induce Ca2+ influx by activating Piezo1 channels. A: representative record (left) of changes in cytosolic-free Ca2+ concentration ([Ca2+]cyt) in a Piezo1-transfected human embryonic kidney (HEK) cell before and during application of hypo-osmolality (Hypo-Osm) solution. Summarized data (means ± SE, n = 5 individual experiments) showing the basal [Ca2+]cyt before Hypo-Osm and the peak and plateau increases in [Ca2+]cyt during Hypo-Osm solution (right). ***P < 0.001 vs. Basal (one-way ANOVA). B: representative record (left) of changes in [Ca2+]cyt in a Piezo1-transfected HEK cell before and during application of Yoda-1 (5 µM). Summarized data (means ± SE, n = 5 individual experiments) showing the basal [Ca2+]cyt before Yoda-1 application and the peak and plateau increases in [Ca2+]cyt during Yoda-1 application (right). **P < 0.01 vs. Basal (one-way ANOVA).
These data indicate that GsMTx4 is an inhibitor of Piezo1 channels and GsMTx-mediated blockade of Piezo1 causes significant inhibition of stretch-mediated cation currents through Piezo1 channels, whereas OAG had negligible effect on the cation currents through Piezo1 channels. Taken together, the data indicate that TRPC6 in human PAECs contributes to cation currents that are sensitive to the second messenger diacylglycerol (or OAG) and membrane stretch, whereas Piezo1 is a mechanosensitive cation channel in PAECs that is not directly activated by diacylglycerol (or OAG).
DISCUSSION
In this study, we characterized whole cell nonselective cation currents (ICation) in human PAECs that are induced or enhanced by membrane stretch and diacylglycerol. The observations showed that the currents generated by Ca2+ or cation flux through TRPC6 and Piezo1 channels contributed to the mechanosensitive cation currents in human PAECs (Fig. 10). Downregulation of TRPC6 with siRNA or blockade of TRPC6 with BI-749327, a potent and selective blocker of TRPC6 (48), significantly inhibited the stretch-mediated cation currents and the stretch-mediated increase in [Ca2+]cyt in human PAECs. In HEK-293 cells transiently transfected with TRPC6 gene, OAG (a diacylglycerol analog) and membrane stretch (via superfusion of hypo-osmotic solution) both enhanced the cation currents through TRPC6 channels (ITRPC6). Inhibition of Piezo1 channels with GsMTx4, a blocker of Piezo1 channels (49–51), also significantly reduced the stretch-mediated ICation and the stretch-mediated increase in [Ca2+]cyt in human PAECs. In HEK-293 cells transiently transfected with the Piezo1 gene, membrane stretch enhanced the cation currents through Piezo1 channels (IPiezo1) and GsMTx4 significantly reduced the stretch-mediated IPiezo1, whereas OAG had negligible effect on IPiezo1. The data from this study indicate that TRPC6 and Piezo1 are major cation channels involved in generating mechanosensitive cation currents in human PAECs. TRPC6 is not only a diacylglycerol-sensitive cation channel, but also a mechanosensitive cation channel in human PAECs. Piezo1, on the other hand, is a mechanosensitive channel that is insensitive to diacylglycerol (Fig. 10).
Figure 10.

Simplified schematic diagram illustrating the potential role of mechanosensitive cation channels of transient receptor potential canonical channel 6 (TRPC6) and Piezo1 in human pulmonary arterial endothelial cell (PAEC). Membrane stretch and/or flow shear stress activate both TRPC6 and Piezo1, as mechanosensitive cation channels, on the surface membrane of PAEC, enhance Ca2+ influx and raise cytosolic-free Ca2+ concentration ([Ca2+]cyt). The stretch-mediated increase in [Ca2+]cyt activates endothelial nitric oxide (NO) synthase (eNOS), increases NO synthesis and release, and causes endothelium-dependent pulmonary vasodilation. Increased [Ca2+]cyt can also activate CaM/CaMK and AKT/mTOR signaling cascades to stimulate cell proliferation and gene expression, contributing to pulmonary vascular remodeling. Vasoconstrictor or mitogenic factor (e.g., endothelin-1, TXA2) also opens TRPC6, as a receptor-operated cation channel, through diacylglycerol (DAG), to induce Ca2+ influx and contribute to regulating endothelium-dependent vasodilation and vascular remodeling. mTOR, mammalian target of rapamycin.
In the lungs, more than 40% of cells are endothelial cells (19) and the pulmonary vasculature or the pulmonary vascular endothelium is under constant stretch due to respiration and cardiac output. Under normal conditions, the stretch-mediated Ca2+ influx through mechanosensitive channels and agonist-induced Ca2+ influx through diacylglycerol-activated Ca2+ channels serve as an important mechanism for inducing the endothelium-derived relaxation by activating eNOS activity and NO production. In addition to its vasodilatative effect through activation of eNOS, the enhanced Ca2+ influx through mechanosensitive and receptor-operated cation channels (e.g., TRPC6 and Piezo1) in PAECs may contribute to the development and progression of PH by causing eNOS uncoupling (7, 57), activating the AKT/mammalian target of rapamycin (mTOR) signaling (25), and enhancing endothelial-to-mesenchymal transition (EndMT) (58, 59).
Mechanosensitive Ca2+ influx through Piezo1 channels in the endothelium is implicated in pulmonary vasodilation using isolated intrapulmonary arterial vessels (42). In the isolated and perfused/ventilated whole lung preparation, however, activation of Piezo1 channels with Yoda-1 caused an increase in pulmonary arterial pressure (PAP), whereas inhibition of eNOS with nitro-l-arginine methyl ester (l-NAME) significantly enhanced Yoda-1-mediated increase in PAP (data not shown). In addition, agonist (such as acetylcholine/ACh), by inducing Ca2+ influx through receptor-operated cation channels in the endothelium or ECs, causes pulmonary vasodilation, whereas functional removal (or injury) of endothelium from isolated pulmonary arterial rings converts the ACh-mediated pulmonary vasodilation to ACh-mediated vasoconstriction (60, 61) and significantly enhances vasoconstriction induced by other agonists (60, 62, 63). These data indicate that mechanosensitive and receptor-operated cation channels in ECs or PAECs are responsible for Ca2+ influx that activates eNOS and causes vasodilation. However, upregulation of the cation channels and persistent increases in [Ca2+]cyt in ECs (due to Ca2+ influx through the upregulated mechanosensitive and receptor-operated cation channels) would activate other Ca2+- or Ca2+/CaM-sensitive signaling cascades (e.g., AKT/mTOR) to induce EndMT and EC proliferation, and ultimately contribute to the development of pulmonary vascular remodeling in patients with PAH and animals with experimental PH (25). The data from this study suggest that TRPC6 and Piezo1 channels are two important cation channels involved in raising [Ca2+]cyt in human PAECs in response to mechanical stretch and flow shear stress reinforced by respiration and cardiac output.
Lung vascular injury associated with inflammation and oxidative stress is implicated as one of the initial triggers for pulmonary vasoconstriction and vascular remodeling in patients with PAH and animals with experimental PH. Loss of vasodilative effect of the mechanosensitive Ca2+ influx through TRPC6 and Piezo1 in PAECs, when endothelial injury and inflammation occur, would enhance vasoconstrictive effect of the mechanosensitive Ca2+ influx through TRPC6 and Piezo1 in PASMCs, thereby causing PH. TRPC6 and Piezo1 channels are nonselective cation channels, although the Ca2+ permeability (PCa) is greater than the Na+ permeability (PNa) for both channels. The Na+ influx through TRPC6 and Piezo can also increase local Na+ concentration ([Na+]), which is a major driving force to induce inward transportation of Ca2+ through Na+/Ca2+ exchangers (64–68); the late phase of Ca2+ inward transportation via Na+/Ca2+ may contribute to the maintaining of high levels of [Ca2+]cyt in mechanically stimulated PAECs and PASMCs to induce vasodilation and vasoconstriction, respectively.
Functional TRPC6 channels are homo- or heterotetramers composed of either TRPC6 subunits (homotetrameric channel) or TRPC6 with other TRPC subunits (e.g., TRPC1, TRPC3, and TRPC7) (heterotetrameric channel) (69). Furthermore, TRPC6/TRPC1 dimers can also form functional tetramers with TRPC4 and TRPC5 (70). Topology of TRPC6 shows that there are six transmembrane domains (TM) with the predicted pore region between TM5 and TM6. The cytoplasmic N- and C-termini contain multiple phosphorylation sites (71), two glycosylation sites, Ca2+/CaM and inositol (1,4,5)-trisphosphate (IP3) receptor phosphoinositide-binding site (CIRPIB), and a diacylglycerol-sensitive lipid trafficking domain (26, 30, 37, 69). As a nonselective cation channel, TRPC6 is more permeable to Ca2+ than to other cations (e.g., PCa/PNa = 5; PNa/PCs = 0.7) (26). As an important subunit for forming the receptor-operated Ca2+ channel, TRPC6 can be activated by diacylglycerol (26, 31, 72–74), as shown in this study in TRPC6-transfected HEK cells and human PAECs, and by IP3 receptor (37, 75) and tyrosine phosphorylation (30, 33, 36). In addition to its sensitivity to receptor-operated mechanisms, TRPC6 can be activated by mechanical stimulation (53, 73), as shown in this study in TRPC6-transfected HEK cells and human PAECs.
The pulmonary vascular endothelium is always exposed to high level of ligands (e.g., vasoactive substances, mitogenic and angiogenic factors, and inflammatory cytokines) in the blood (or locally released from ECs and infiltrated inflammatory cells). Synergistic activation of TRPC6 in lung ECs by receptor-mediated second messengers (e.g., diacylglycerol) and mechanical stimulation enables the channel to simultaneously sense chemical and mechanical stimuli and regulate pulmonary vascular function. TRPC6 channels can be blocked by 2-APB (2-aminoethoxydiphenyl borate), a nonselective inhibitor of many subtypes of TRP channels (76), and by BI-749327, a selective small-molecule inhibitor of TRPC6 (48). The IC50 of BI-749327 for human and animal TRPC6 is 13–19 nM, which is ∼85-fold less than the IC50 for TRPC3 and 42-fold less than the IC50 for TRPC7 (48). BI-749327 is thus much more selective for TRPC6 than other TRPC channels that can form heterotetrameric channels with TRPC6.
As shown in this study, OAG, a membrane-permeable analog of diacylglycerol, significantly enhanced the cation currents in human PASMCs (ICation) (Fig. 1C) and TRPC6-transfected HEK cells (ITRPC6) (Fig. 3D); BI-749327 (50 nM) reversibly reduced ICation in human PASMCs (Fig. 1D) and ITRPC6 in TRPC6-transfected HEK cells (Fig. 4). BI-749327 at 50 nM was also sufficient to inhibit OAG-mediated increases in [Ca2+]cyt in human PAECs (Fig. 5). These data indicate that, in human PAECs, TRPC6 is an important channel contributing to the whole cell cation currents responsible for agonist- and stretch-induced increase in [Ca2+]cyt in human PAECs. It is, however, unknown whether TRPC6 channels are mainly present in human lung ECs in homotetrameric channels or heterotetrameric channels (with other TRPC channel subunits).
Piezo1 is a newly discovered mechanosensitive channel (38, 43, 77). Functional Piezo1 channels exhibit a unique propeller-shaped, three-bladed homotrimer structure; each subunit contains 38 transmembrane (TM) helices, a C-terminal extracellular domain (CED), an anchor between TM36 and TM37, and a long (9 nm) intracellular beam between TM28 and TM29 (78–80). Single-channel conductance of Piezo1 is reported to be 41.3 ± 0.9. pS at negative potential (−120 to −40 mV) and 27.1 ± 1.2 pS at positive potential (+20 to +110 mV), which indicates a significant outward rectification at positive potential (81). Whole cell outward currents at positive potential are also much greater than the inward currents at negative potential. The outward rectification of Piezo1 channels is also shown in this study (Fig. 8). In addition to mechanical forces, Yoda1 is selectively activated by the small-molecule allosteric activator, Yoda1 (52, 82, 83). The allosteric binding pocket of Yoda1 is located in the mechanosensory domain of Piezo1 nearby (4-nm away) the central pore. Yoda1 functions as a molecular wedge facilitating force-induced conformational changes to lower Piezo1’s mechanical threshold for activation (52). In human PAECs, 5 µM Yoda1 induced an increase in [Ca2+]cyt that was similar to membrane stretch-mediated increase in [Ca2+]cyt (Fig. 9) indicating that Piezo1 is an important channel in sensing mechanical force during vascular extension and recruitment.
There is no selective blocker for Piezo1 channels although many cation channel blockers can inhibit cation currents through mechanosensitive channels. GxMTx4 is a spider venom peptide that has been shown to block Piezo1 channels (49) and inhibit stretch-activated cation channels in astrocytes, cardiomyocytes, smooth muscle cells, and skeletal muscle cells. In this study, we found that 5 µM GsMTx4 resulted in an 50%–55% inhibition of membrane stretch-mediated increases in [Ca2+]cyt (54.9 ± 9.5%) and Yoda-1-mediated increases in [Ca2+]cyt (55.3 ± 11.0%) in human PAECs (Fig. 7). GsMTx4 (5 µM) also significantly inhibited whole cell cation currents in human PAECs bathed in Hypo-Osm solution; extracellular application of GsMTx4 resulted in 35.8 ± 10.7% inhibition of the inward currents at −100 mV and 62/2 ± 7.0% inhibition of the outward currents at +100 mV (Fig. 6). In Piezo1-transfected HEK cells, we found that OAG, which significantly enhanced cation currents through TRPC6 channels (ITRPC6) in TRPC6-transfected HEK cells, had no effect on cation currents through Piezo1 channels (IPiezo1) (Fig. 8C). These results indicate that GsMTx4-sensitive cation currents through Piezo1 channels are important mechanosensitive currents that are insensitive to OAG in human PAECs.
In summary, mechanosensitive cation channels in ECs play a significant role in sensing mechanical forces including flow-mediated shear stretch and pressure-induced stretch in the lung vasculature. This study indicates that TRPC6 and Piezo1 are two important mechanosensitive cation channels present in human PAECs (Fig. 10). TRPC6, but not Piezo1, is also sensitive to diacylglycerol, an important second messenger that mediates receptor-operated signal transduction in many types of cells. It is thus critical to conduct further studies to define potential pathogenic roles of these two channels in human PAECs and whether and how to target these channels for developing future therapeutic approaches for pulmonary vascular disease.
GRANTS
This work is supported in part by the National Heart, Lung, and Blood Institute of the National Institutes of Health Grants HL135807 and HL146764.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.M. and J.X.-J.Y. conceived and designed research; T.Z. performed experiments; T.Z., S.P., Z.S., M.W., L.Y., P.A.T., A.M., and J.X.-J.Y. analyzed data; T.Z., S.P., Z.S., M.W., L.Y., P.A.T., A.M., and J.X.-J.Y. interpreted results of experiments; T.Z., S.P., Z.S., M.W., L.Y., P.A.T., A.M., and J.X.-J.Y. prepared figures; A.M. and J.X.-J.Y. drafted manuscript; T.Z., S.P., Z.S., M.W., L.Y., P.A.T., A.M., and J.X.J.Y. edited and revised manuscript; T.Z., S.P., Z.S., M.W., L.Y., P.A.T., A.M., and J.X.-J.Y. approved final version of manuscript.
REFERENCES
- 1. Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev 81: 1415–1459, 2001. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 2. Santos-Gomes J, Le Ribeuz H, Brás-Silva C, Antigny F, Adão R. Role of ion channel remodeling in endothelial dysfunction induced by pulmonary arterial hypertension. Biomolecules 12: 484, 2022. doi: 10.3390/biom12040484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fleming I, Busse R. NO: the primary EDRF. J Mol Cell Cardiol 31: 5–14, 1999. doi: 10.1006/jmcc.1998.0839. [DOI] [PubMed] [Google Scholar]
- 4. Ignarro LJ, Byrns RE, Buga GM, Wood KS. Endothelium-derived relaxing factor from pulmonary artery and vein possesses pharmacologic and chemical properties identical to those of nitric oxide radical. Circ Res 61: 866–879, 1987. doi: 10.1161/01.res.61.6.866. [DOI] [PubMed] [Google Scholar]
- 5. Cremona G, Dinh Xuan AT, Higenbottam TW. Endothelium-derived relaxing factor and the pulmonary circulation. Lung 169: 185–202, 1991. doi: 10.1007/BF02714154. [DOI] [PubMed] [Google Scholar]
- 6. Aggarwal S, Gross CM, Sharma S, Fineman JR, Black SM. Reactive oxygen species in pulmonary vascular remodeling. Compr Physiol 3: 1011–1034, 2013. doi: 10.1002/cphy.c120024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Black SM, Sanchez LS, Mata-Greenwood E, Bekker JM, Steinhorn RH, Fineman JR. sGC and PDE5 are elevated in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 281: L1051–L1057, 2001. doi: 10.1152/ajplung.2001.281.5.L1051. [DOI] [PubMed] [Google Scholar]
- 8. Zhao YY, Zhao YD, Mirza MK, Huang JH, Potula HH, Vogel SM, Brovkovych V, Yuan JX, Wharton J, Malik AB. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. J Clin Invest 119: 2009–2018, 2009. doi: 10.1172/JCI33338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhao YD, Cai L, Mirza MK, Huang X, Geenen DL, Hofmann F, Yuan JX, Zhao YY. Protein kinase G-I deficiency induces pulmonary hypertension through Rho A/Rho kinase activation. Am J Pathol 180: 2268–2275, 2012. doi: 10.1016/j.ajpath.2012.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee GH, Kim CY, Zheng C, Jin SW, Kim JY, Lee SY, Kim MY, Han EH, Hwang YP, Jeong HG. Rutaecarpine increases nitric oxide synthesis via eNOS phosphorylation by TRPV1-dependent CaMKII and CaMKK0/AMPK signaling pathway in human endothelial cells. Int J Mol Sci 22: 9407, 2021. doi: 10.3390/ijms22179407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu S, Haynes J Jr, Taylor JT, Obiako BO, Stubbs JR, Li M, Stevens T. Cav3.1 (a1G) T-type Ca2+ channels mediate vaso-occlusion of sickled erythrocytes in lung microcirculation. Circ Res 93: 346–353, 2003. doi: 10.1161/01.RES.0000087148.75363.8F. [DOI] [PubMed] [Google Scholar]
- 12. Yakubu MA, Leffler CW. L-type voltage-dependent Ca2+ channels in cerebral microvascular endothelial cells and ET-1 biosynthesis. Am J Physiol Cell Physiol 283: C1687–C1695, 2002. doi: 10.1152/ajpcell.00071.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev 87: 165–217, 2007. doi: 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 14. Lu Q, Zemskov EA, Sun X, Wang H, Yegambaram M, Wu X, Garcia-Flores A, Song S, Tang H, Kangath A, Cabanillas GZ, Yuan JX, Wang T, Fineman JR, Black SM. Activation of the mechanosensitive Ca2+ channel TRPV4 induces endothelial barrier permeability via the disruption of mitochondrial bioenergetics. Redox Biol 38: 101785, 2021. doi: 10.1016/j.redox.2020.101785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Otto M, Bucher C, Liu W, Müller M, Schmidt T, Kardell M, Driessen MN, Rossaint J, Gross ER, Wagner NM. 12(S)-HETE mediates diabetes-induced endothelial dysfunction by activating intracellular endothelial cell TRPV1. J Clin Invest 130: 4999–5010, 2020. doi: 10.1172/JCI136621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feletou M. Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol 156: 545–562, 2009. doi: 10.1111/j.1476-5381.2009.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ledoux J, Bonev AD, Nelson MT. Ca2+-activated K+ channels in murine endothelial cells: block by intracellular calcium and magnesium. J Gen Physiol 131: 125–135, 2008. doi: 10.1085/jgp.200709875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu N, Ayers L, Pastukh V, Alexeyev M, Stevens T, Tambe DT. Impact of Na+ permeation on collective migration of pulmonary arterial endothelial cells. PLoS One 16: e0250095, 2021. doi: 10.1371/journal.pone.0250095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Townsley MI. Structure and composition of pulmonary arteries, capillaries, and veins. Compr Physiol 2: 675–709, 2012. doi: 10.1002/cphy.c100081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang J, Shimoda LA, Weigand L, Wang W, Sun D, Sylvester JT. Acute hypoxia increases intracellular [Ca2+] in pulmonary arterial smooth muscle by enhancing capacitative Ca2+ entry. Am J Physiol Lung Cell Mol Physiol 288: L1059–L1069, 2005. doi: 10.1152/ajplung.00448.2004. [DOI] [PubMed] [Google Scholar]
- 21. Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006. doi: 10.1161/01.RES.0000227551.68124.98. [DOI] [PubMed] [Google Scholar]
- 22. Smith KA, Voiriot G, Tang H, Fraidenburg DR, Song S, Yamamura H, Yamamura A, Guo Q, Wan J, Pohl NM, Tauseef M, Bodmer R, Ocorr K, Thistlethwaite PA, Haddad GG, Powell FL, Makino A, Mehta D, Yuan JX. Notch activation of Ca2+ signaling in the development of hypoxic pulmonary vasoconstriction and pulmonary hypertension. Am J Respir Cell Mol Biol 53: 355–367, 2015. doi: 10.1165/rcmb.2014-0235OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101: 13861–13866, 2004. doi: 10.1073/pnas.0405908101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu Y, Keller SH, Remillard CV, Safrina O, Nicholson A, Zhang SL, Jiang W, Vangala N, Landsberg JW, Wang JY, Thistlethwaite PA, Channick RN, Robbins IM, Loyd JE, Ghofrani HA, Grimminger F, Schermuly RT, Cahalan MD, Rubin LJ, Yuan JX. A functional single-nucleotide polymorphism in the TRPC6 gene promoter associated with idiopathic pulmonary arterial hypertension. Circulation 119: 2313–2322, 2009. doi: 10.1161/CIRCULATIONAHA.108.782458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Z, Chen J, Babicheva A, Jain PP, Rodriguez M, Ayon RJ, Ravellette KS, Wu L, Balistrieri F, Tang H, Wu X, Zhao T, Black SM, Desai AA, Garcia JGN, Sun X, Shyy JY, Valdez-Jasso D, Thistlethwaite PA, Makino A, Wang J, Yuan JX. Endothelial upregulation of mechanosensitive channel Piezo1 in pulmonary hypertension. Am J Physiol Cell Physiol 321: C1010–C1027, 2021. doi: 10.1152/ajpcell.00147.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263, 1999. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 27. Ichikawa J, Inoue R. TRPC6 regulates cell cycle progression by modulating membrane potential in bone marrow stromal cells. Br J Pharmacol 171: 5280–5294, 2014. doi: 10.1111/bph.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Inoue R, Jensen LJ, Shi J, Morita H, Nishida M, Honda A, Ito Y. Transient receptor potential channels in cardiovascular function and disease. Circ Res 99: 119–131, 2006. doi: 10.1161/01.RES.0000233356.10630.8a. [DOI] [PubMed] [Google Scholar]
- 29. Weber EW, Han F, Tauseef M, Birnbaumer L, Mehta D, Muller WA. TRPC6 is the endothelial calcium channel that regulates leukocyte transendothelial migration during the inflammatory response. J Exp Med 212: 1883–1899, 2015. doi: 10.1084/jem.20150353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hisatsune C, Kuroda Y, Nakamura K, Inoue T, Nakamura T, Michikawa T, Mizutani A, Mikoshiba K. Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem 279: 18887–18894, 2004. doi: 10.1074/jbc.M311274200. [DOI] [PubMed] [Google Scholar]
- 31. Imai Y, Itsuki K, Okamura Y, Inoue R, Mori MX. A self-limiting regulation of vasoconstrictor-activated TRPC3/C6/C7 channels coupled to PI(4,5)P(2)-diacylglycerol signalling. J Physiol 590: 1101–1119, 2012. doi: 10.1113/jphysiol.2011.221358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular α1-adrenoceptor-activated Ca2+-permeable cation channel. Circ Res 88: 325–332, 2001. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- 33. Kanda S, Harita Y, Shibagaki Y, Sekine T, Igarashi T, Inoue T, Hattori S. Tyrosine phosphorylation-dependent activation of TRPC6 regulated by PLC-g1 and nephrin: effect of mutations associated with focal segmental glomerulosclerosis. Mol Biol Cell 22: 1824–1835, 2011. doi: 10.1091/mbc.E10-12-0929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Takahashi S, Lin H, Geshi N, Mori Y, Kawarabayashi Y, Takami N, Mori MX, Honda A, Inoue R. Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J Physiol 586: 4209–4223, 2008. doi: 10.1113/jphysiol.2008.156083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gottlieb P, Folgering J, Maroto R, Raso A, Wood TG, Kurosky A, Bowman C, Bichet D, Patel A, Sachs F, Martinac B, Hamill OP, Honoré E. Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch 455: 1097–1103, 2008. doi: 10.1007/s00424-007-0359-3. [DOI] [PubMed] [Google Scholar]
- 36. Itsuki K, Imai Y, Hase H, Okamura Y, Inoue R, Mori MX. PLC-mediated PI(4,5)P2 hydrolysis regulates activation and inactivation of TRPC6/7 channels. J Gen Physiol 143: 183–201, 2014. doi: 10.1085/jgp.201311033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dionisio N, Albarran L, Berna-Erro A, Hernandez-Cruz JM, Salido GM, Rosado JA. Functional role of the calmodulin- and inositol 1,4,5-trisphosphate receptor-binding (CIRB) site of TRPC6 in human platelet activation. Cell Signal 23: 1850–1856, 2011. doi: 10.1016/j.cellsig.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 38. Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330: 55–60, 2010. doi: 10.1126/science.1193270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Douguet D, Patel A, Xu A, Vanhoutte PM, Honoré E. Piezo ion channels in cardiovascular mechanobiology. Trends Pharmacol Sci 40: 956–970, 2019. doi: 10.1016/j.tips.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 40. John L, Ko NL, Gokin A, Gokina N, Mandalà M, Osol G. The Piezo1 cation channel mediates uterine artery shear stress mechanotransduction and vasodilation during rat pregnancy. Am J Physiol Heart Circ Physiol 315: H1019–H1026, 2018. doi: 10.1152/ajpheart.00103.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kang H, Hong Z, Zhong M, Klomp J, Bayless KJ, Mehta D, Karginov AV, Hu G, Malik AB. Piezo1 mediates angiogenesis through activation of MT1-MMP signaling. Am J Physiol Cell Physiol 316: C92–C103, 2019. doi: 10.1152/ajpcell.00346.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lhomme A, Gilbert G, Pele T, Deweirdt J, Henrion D, Baudrimont I, Campagnac M, Marthan R, Guibert C, Ducret T, Savineau JP, Quignard JF. Stretch-activated Piezo1 channel in endothelial cells relaxes mouse intrapulmonary arteries. Am J Respir Cell Mol Biol 60: 650–658, 2019. doi: 10.1165/rcmb.2018-0197OC. [DOI] [PubMed] [Google Scholar]
- 43. Li J, Hou B, Tumova S, Muraki K, Bruns A, Ludlow MJ, Sedo A, Hyman AJ, McKeown L, Young RS, Yuldasheva NY, Majeed Y, Wilson LA, Rode B, Bailey MA, Kim HR, Fu Z, Carter DA, Bilton J, Imrie H, Ajuh P, Dear TN, Cubbon RM, Kearney MT, Prasad RK, Evans PC, Ainscough JF, Beech DJ. Piezo1 integration of vascular architecture with physiological force. Nature 515: 279–282, 2014. doi: 10.1038/nature13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gudipaty SA, Lindblom J, Loftus PD, Redd MJ, Edes K, Davey CF, Krishnegowda V, Rosenblatt J. Mechanical stretch triggers rapid epithelial cell division through Piezo1. Nature 543: 118–121, 2017. doi: 10.1038/nature21407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hasegawa K, Fujii S, Matsumoto S, Tajiri Y, Kikuchi A, Kiyoshima T. YAP signaling induces PIEZO1 to promote oral squamous cell carcinoma cell proliferation. J Pathol 253: 80–93, 2021. doi: 10.1002/path.5553. [DOI] [PubMed] [Google Scholar]
- 46. Nonomura K, Lukacs V, Sweet DT, Goddard LM, Kanie A, Whitwam T, Ranade SS, Fujimori T, Kahn ML, Patapoutian A. Mechanically activated ion channel PIEZO1 is required for lymphatic valve formation. Proc Natl Acad Sci USA 115: 12817–12822, 2018. doi: 10.1073/pnas.1817070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pathak MM, Nourse JL, Tran T, Hwe J, Arulmoli J, Le DT, Bernardis E, Flanagan LA, Tombola F. Stretch-activated ion channel Piezo1 directs lineage choice in human neural stem cells. Proc Natl Acad Sci USA 111: 16148–16153, 2014. doi: 10.1073/pnas.1409802111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin BL, Matera D, Doerner JF, Zheng N, Del Camino D, Mishra S, Bian H, Zeveleva S, Zhen X, Blair NT, Chong JA, Hessler DP, Bedja D, Zhu G, Muller GK, Ranek MJ, Pantages L, McFarland M, Netherton MR, Berry A, Wong D, Rast G, Qian HS, Weldon SM, Kuo JJ, Sauer A, Sarko C, Moran MM, Kass DA, Pullen SS. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc Natl Acad Sci USA 116: 10156–10161, 2019. doi: 10.1073/pnas.1815354116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bae C, Sachs F, Gottlieb PA. The mechanosensitive ion channel Piezo1 is inhibited by the peptide GsMTx4. Biochemistry 50: 6295–6300, 2011. doi: 10.1021/bi200770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Copp SW, Kim JS, Ruiz-Velasco V, Kaufman MP. The mechano-gated channel inhibitor GsMTx4 reduces the exercise pressor reflex in decerebrate rats. J Physiol 594: 641–655, 2016. doi: 10.1113/JP271714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gnanasambandam R, Ghatak C, Yasmann A, Nishizawa K, Sachs F, Ladokhin AS, Sukharev SI, Suchyna TM. GsMTx4: mechanism of inhibiting mechanosensitive ion channels. Biophys J 112: 31–45, 2017. doi: 10.1016/j.bpj.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Botello-Smith WM, Jiang W, Zhang H, Ozkan AD, Lin YC, Pham CN, Lacroix JJ, Luo Y. A mechanism for the activation of the mechanosensitive Piezo1 channel by the small molecule Yoda1. Nat Commun 10: 4503, 2019. doi: 10.1038/s41467-019-12501-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spassova MA, Hewavitharana T, Xu W, Soboloff J, Gill DL. A common mechanism underlies stretch activation and receptor activation of TRPC6 channels. Proc Natl Acad Sci USA 103: 16586–16591, 2006. doi: 10.1073/pnas.0606894103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tang Q, Guo W, Zheng L, Wu JX, Liu M, Zhou X, Zhang X, Chen L. Structure of the receptor-activated human TRPC6 and TRPC3 ion channels. Cell Res 28: 746–755, 2018. doi: 10.1038/s41422-018-0038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yamaguchi Y, Iribe G, Nishida M, Naruse K. Role of TRPC3 and TRPC6 channels in the myocardial response to stretch: linking physiology and pathophysiology. Prog Biophys Mol Biol 130: 264–272, 2017. doi: 10.1016/j.pbiomolbio.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 56. Chen X, Sooch G, Demaree IS, White FA, Obukhov AG. Transient receptor potential canonical (TRPC) channels: then and now. Cells 9: 1983, 2020. doi: 10.3390/cells9091983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Black SM, Fineman JR, Steinhorn RH, Bristow J, Soifer SJ. Increased endothelial NOS in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Heart Circ Physiol 275: H1643–H1651, 1998. doi: 10.1152/ajpheart.1998.275.5.H1643. [DOI] [PubMed] [Google Scholar]
- 58. Babicheva A, Song S, Carr S, Makino A, Yuan JX. TGF-β1-induced endothelial-to-mesenchymal transition in normal human lung vascular endothelial cells is Ca2+-dependent event (Abstract). Am J Respir Crit Care Med 197: A3735, 2018. [Google Scholar]
- 59. Gorelova A, Berman M, Al Ghouleh I. Endothelial-to-mesenchymal transition in pulmonary arterial hypertension. Antioxid Redox Signal 34: 891–914, 2021. doi: 10.1089/ars.2020.8169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Conraads VM, Bosmans JM, Claeys MJ, Vrints CJ, Snoeck JP, De Clerck L, Vermeire PA. Paradoxic pulmonary vasoconstriction in response to acetylcholine in patients with primary pulmonary hypertension. Chest 106: 385–390, 1994. doi: 10.1378/chest.106.2.385. [DOI] [PubMed] [Google Scholar]
- 61. Altiere RJ, Kiritsy-Roy JA, Catravas JD. Acetylcholine-induced contractions in isolated rabbit pulmonary arteries: role of thromboxane A2. J Pharmacol Exp Ther 236: 535–541, 1986. [PubMed] [Google Scholar]
- 62. Xu M, Platoshyn O, Makino A, Dillmann WH, Akassoglou K, Remillard CV, Yuan JX. Characterization of agonist-induced vasoconstriction in mouse pulmonary artery. Am J Physiol Heart Circ Physiol 294: H220–H228, 2008. doi: 10.1152/ajpheart.00968.2007. [DOI] [PubMed] [Google Scholar]
- 63. Ludmer PL, Selwyn AP, Shook TL, Wayne RR, Mudge GH, Alexander RW, Ganz P. Paradoxical vasoconstriction induced by acetylcholine in atherosclerotic coronary arteries. N Engl J Med 315: 1046–1051, 1986. doi: 10.1056/NEJM198610233151702. [DOI] [PubMed] [Google Scholar]
- 64. Blaustein MP. The pump, the exchanger, and the holy spirit: origins and 40-year evolution of ideas about the ouabain-Na+ pump endocrine system. Am J Physiol Cell Physiol 314: C3–C26, 2018. doi: 10.1152/ajpcell.00196.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev 79: 763–854, 1999. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 66. Zhang S, Dong H, Rubin LJ, Yuan JX. Upregulation of Na+/Ca2+ exchanger contributes to the enhanced Ca2+ entry in pulmonary artery smooth muscle cells from patients with idiopathic pulmonary arterial hypertension. Am J Physiol Cell Physiol 292: C2297–C2305, 2007. doi: 10.1152/ajpcell.00383.2006. [DOI] [PubMed] [Google Scholar]
- 67. Zhang S, Yuan JX, Barrett KE, Dong H. Role of Na+/Ca2+ exchange in regulating cytosolic Ca2+ in cultured human pulmonary artery smooth muscle cells. Am J Physiol Cell Physiol 288: C245–C252, 2005. doi: 10.1152/ajpcell.00411.2004. [DOI] [PubMed] [Google Scholar]
- 68. Poburko D, Liao CH, Lemos VS, Lin E, Maruyama Y, Cole WC, van Breemen C. Transient receptor potential channel 6-mediated, localized cytosolic [Na+] transients drive Na+/Ca2+ exchanger-mediated Ca2+ entry in purinergically stimulated aorta smooth muscle cells. Circ Res 101: 1030–1038, 2007. doi: 10.1161/CIRCRESAHA.107.155531. [DOI] [PubMed] [Google Scholar]
- 69. Azumaya CM, Sierra-Valdez F, Cordero-Morales JF, Nakagawa T. Cryo-EM structure of the cytoplasmic domain of murine transient receptor potential cation channel subfamily C member 6 (TRPC6). J Biol Chem 293: 10381–10391, 2018. doi: 10.1074/jbc.RA118.003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bröker-Lai J, Kollewe A, Schindeldecker B, Pohle J, Nguyen Chi V, Mathar I, Guzman R, Schwarz Y, Lai A, Weißgerber P, Schwegler H, Dietrich A, Both M, Sprengel R, Draguhn A, Köhr G, Fakler B, Flockerzi V, Bruns D, Freichel M. Heteromeric channels formed by TRPC1, TRPC4 and TRPC5 define hippocampal synaptic transmission and working memory. EMBO J 36: 2770–2789, 2017. doi: 10.15252/embj.201696369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu X, Yao X, Tsang SY. Post-translational modification and natural mutation of TRPC channels. Cells 9: 135, 2020. doi: 10.3390/cells9010135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fuchs B, Rupp M, Ghofrani HA, Schermuly RT, Seeger W, Grimminger F, Gudermann T, Dietrich A, Weissmann N. Diacylglycerol regulates acute hypoxic pulmonary vasoconstriction via TRPC6. Respir Res 12: 20, 2011. doi: 10.1186/1465-9921-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Inoue R, Jensen LJ, Jian Z, Shi J, Hai L, Lurie AI, Henriksen FH, Salomonsson M, Morita H, Kawarabayashi Y, Mori M, Mori Y, Ito Y. Synergistic activation of vascular TRPC6 channel by receptor and mechanical stimulation via phospholipase C/diacylglycerol and phospholipase A2/w-hydroxylase/20-HETE pathways. Circ Res 104: 1399–1409, 2009. doi: 10.1161/CIRCRESAHA.108.193227. [DOI] [PubMed] [Google Scholar]
- 74. Shi J, Takahashi S, Jin XH, Li YQ, Ito Y, Mori Y, Inoue R. Myosin light chain kinase-independent inhibition by ML-9 of murine TRPC6 channels expressed in HEK293 cells. Br J Pharmacol 152: 122–131, 2007. doi: 10.1038/sj.bjp.0707368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Song T, Hao Q, Zheng YM, Liu QH, Wang YX. Inositol 1,4,5-trisphosphate activates TRPC3 channels to cause extracellular Ca2+ influx in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 309: L1455–L1466, 2015. doi: 10.1152/ajplung.00148.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xu SZ, Zeng F, Boulay G, Grimm C, Harteneck C, Beech DJ. Block of TRPC5 channels by 2-aminoethoxydiphenyl borate: a differential, extracellular and voltage-dependent effect. Br J Pharmacol 145: 405–414, 2005. doi: 10.1038/sj.bjp.0706197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ranade SS, Qiu Z, Woo SH, Hur SS, Murthy SE, Cahalan SM, Xu J, Mathur J, Bandell M, Coste B, Li YS, Chien S, Patapoutian A. Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc Natl Acad Sci USA 111: 10347–10352, 2014. doi: 10.1073/pnas.1409233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Tang H, Zeng R, He E, Zhang I, Ding C, Zhang A. Piezo-type mechanosensitive ion channel component 1 (Piezo1): a promising therapeutic target and its modulators. J Med Chem 65: 6441–6453, 2022. doi: 10.1021/acs.jmedchem.2c00085. [DOI] [PubMed] [Google Scholar]
- 79. Saotome K, Murthy SE, Kefauver JM, Whitwam T, Patapoutian A, Ward AB. Structure of the mechanically activated ion channel Piezo1. Nature 554: 481–486, 2018. doi: 10.1038/nature25453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Fang XZ, Zhou T, Xu JQ, Wang YX, Sun MM, He YJ, Pan SW, Xiong W, Peng ZK, Gao XH, Shang Y. Structure, kinetic properties and biological function of mechanosensitive Piezo channels. Cell Biosci 11: 13, 2021. doi: 10.1186/s13578-020-00522-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Moroni M, Servin-Vences MR, Fleischer R, Sánchez-Carranza O, Lewin GR. Voltage gating of mechanosensitive PIEZO channels. Nat Commun 9: 1096, 2018. doi: 10.1038/s41467-018-03502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Evans EL, Cuthbertson K, Endesh N, Rode B, Blythe NM, Hyman AJ, Hall SJ, Gaunt HJ, Ludlow MJ, Foster R, Beech DJ. Yoda1 analogue (Dooku1) which antagonizes Yoda1-evoked activation of Piezo1 and aortic relaxation. Br J Pharmacol 175: 1744–1759, 2018. doi: 10.1111/bph.14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Dela Paz NG, Frangos JA. Yoda1-induced phosphorylation of Akt and ERK1/2 does not require Piezo1 activation. Biochem Biophys Res Commun 497: 220–225, 2018. doi: 10.1016/j.bbrc.2018.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]