

Keywords: collecting duct, epithelial Na+ channel, Liddle syndrome, vasopressin, vasopressin-2 receptor
Abstract
The renin-angiotensin-aldosterone and arginine vasopressin-V2 receptor-aquaporin-2 (AQP2) systems converge on the epithelial Na+ channel (ENaC) to regulate blood pressure and plasma tonicity. Although it is established that V2 receptors initiate renal water reabsorption through AQP2, whether V2 receptors can also induce renal Na+ retention through ENaC and raise blood pressure remains an open question. We hypothesized that a specific increase in V2 receptor-mediated ENaC activity can lead to high blood pressure. Our approach was to test effects of chronic activation of V2 receptors in Liddle mice, a genetic mouse model of high ENaC activity, and compare differences in ENaC activity, urine Na+ excretion, and blood pressure with control mice. We found that ENaC activity was elevated in Liddle mice and could not be stimulated further by administration of desmopressin (dDAVP), a V2 receptor-specific agonist. In contrast, Liddle mice showed higher levels of expression of AQP2 and aquaporin-3, but they could still respond to dDAVP infusion by increasing phospho-AQP2 expression. With dDAVP infusion, Liddle mice excreted smaller urine volume and less urine Na+ and developed higher blood pressure compared with control mice; this hypertension was attenuated with administration of the ENaC inhibitor benzamil. We conclude that V2 receptors contribute to hypertension in the Liddle mouse model by promoting primary Na+ and concomitant water retention.
NEW & NOTEWORTHY Liddle syndrome is a classic model for hypertension from high epithelial Na+ channel (ENaC) activity. In the Liddle mouse model, vasopressin-2 receptors stimulate both ENaC and aquaporin-2, which increases Na+ and water retention to such an extent that hypertension ensues. Liddle mice will preserve plasma tonicity at the expense of a higher blood pressure; these data highlight the inherent limitation in which the kidney must use ENaC as a pathway to regulate both plasma tonicity and blood pressure.
INTRODUCTION
The epithelial Na+ channel (ENaC) sits at the crossroads between two feedback systems controlling water and Na+ balance. On the one hand, arginine vasopressin (AVP) activates the vasopressin-2 (V2) receptor to stimulate ENaC in principal cells of the cortical collecting duct (CCD) (1). During hypernatremia, activation of ENaC by AVP provides a driving force for isoosmotic water transport across the early distal nephron. Bulk absorption of isoosmotic fluid decreases the tubular fluid volume that reaches the inner medulla, where selective, but lower capacity, water transport takes place. The reduction in tubular fluid volume in the inner medulla ensures that parallel activation of aquaporin-2 (AQP2) and aquaporin-3 (AQP3) water channels by AVP will dilute plasma tonicity (1). On the other hand, the renin-angiotensin-aldosterone system (RAAS) stimulates ENaC during extracellular fluid (ECF) volume depletion. Activation of ENaC by aldosterone increases renal Na+ retention, expands ECF volume, and raises blood pressure. At the convergence of AVP and RAAS signaling, ENaC can regulate both water and Na+ balance.
This convergence on ENaC implies that there may be situations in which a conflict arises between regulation of plasma tonicity and blood pressure. Such situations could reveal how the kidney reconciles the competing requirements of the body to maintain water and Na+ balance. For example, AVP plays a critical role in stimulating ENaC activity and maintaining Na+ balance in adrenalectomized mice (2), which lack systemic mineralocorticoids. In adrenal insufficiency, and in situations where ECF volume status is threatened, AVP concentration is high and V2 receptors are activated so that ENaC activity and renal Na+ retention are at maximum levels. Administration of the V2 receptor antagonist tolvaptan to adrenalectomized mice decreases ENaC open probability (Po) and total cellular activity (NPo, where N is the number of channels in a patch) in freshly split-open murine connecting tubule (CNT)/CCD segments, demonstrating that ENaC activity is high in adrenalectomized mice because of elevations in AVP (2). However, a byproduct of this compensation in adrenal insufficiency is that the elevation in AVP triggers excess water reabsorption in the collecting duct and induces hyponatremia.
Whether V2 receptor activation can also lead to renal Na+ retention and high blood pressure remains an open question. Bankir et al. (3) have demonstrated in Sprague–Dawley rats that chronic V2 receptor activation can lead to high blood pressure. However, these rats were studied under conditions of high dietary Na+ intake, mineralocorticoid infusion, and uninephrectomy. Since a reduction in nephron number or mineralocorticoid infusion can independently reduce renal Na+ excretion, it remains unclear whether a specific increase in V2 receptor-mediated ENaC activity is sufficient to generate renal Na+ retention and high blood pressure. This question is clinically important because if an increase in V2 receptor-mediated ENaC activity can lead to high blood pressure, then there may be certain forms of hypertension that would be amenable to treatment with V2 receptor antagonists.
We therefore asked whether a specific increase in V2 receptor-mediated ENaC activity is sufficient to raise blood pressure. We infused desmopressin (dDAVP), a specific V2 receptor agonist, into Liddle mice, a genetic mouse model of high ENaC activity, and compared differences in ENaC activity, urine Na+ excretion, and blood pressure with dDAVP-infused control mice [wild-type littermates or pseudohypoaldosteronism type 1 (PHA1) mice]. Since Liddle mice do not exhibit blood pressure that is different from wild-type littermates on a regular Na+ diet, any increase in blood pressure with dDAVP infusion should reflect an increase in V2 receptor-mediated ENaC activity. We hypothesized the following: 1) Liddle mice develop high blood pressure when they receive dDAVP infusion, 2) blood pressure in Liddle mice receiving dDAVP infusion improves with ENaC inhibition with benzamil, and 3) hypertension in Liddle mice receiving dDAVP infusion associates with Na+ retention.
METHODS
Animals
All mouse use and welfare adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals following protocols reviewed and approved by the Institutional Animal Care and Use Committees of the University of Texas Health Science Center (San Antonio, TX), Duke University School of Medicine and Durham Veterans Affairs Health Care System (Durham, NC), and Veterans Affairs Palo Alto Health Care System (Palo Alto, CA). Adult age- and weight-matched male and female mice on a C57BL/6J background were used for experiments. Liddle mice express a stop codon mutation inserted at R566 of β-ENaC, corresponding to a mutation found in the original kindred of Liddle syndrome (4). This gain-of-function mutation in β-ENaC increases ENaC activity in vivo, and Liddle mice develop salt-sensitive hypertension and largely reproduce the human form of Liddle syndrome. We used a mouse model for PHA1 as a control for Liddle mice. PHA1 mice express a disrupted β-ENaC locus that decreases ENaC activity in vivo, and these mice develop a PHA1 phenotype under salt restriction (5). Wild-type littermates of Liddle and PHA1 mice were used as additional controls. All mice were maintained with standard chow (0.39% Na+, Purina 5001) and ad libitum tap water.
Immunofluorescence
For immunofluorescence experiments, male and female mice (24–27 g), 7–9 wk of age, were administered dDAVP (Sigma, St. Louis, MO) via osmotic minipumps (model 1007 D, Alzet, Cupertino, CA) at a rate of 100 ng/h. In some mice, tolvaptan was administered directly into the stomach via an oral gavage at a concentration of 5 mg/kg/day for 3 days. Freshly isolated kidneys perfused with ice-cold PBS were fixed in 4% paraformaldehyde on ice. Kidney tissue was embedded in paraffin and sliced into 5-µm sections. Sections were deparaffinized, rehydrated, and stained with rabbit anti-AQP3 (1:1,000, PA5-36552, Invitrogen, Thermo Fisher Scientific, Waltham, MA) or goat anti-AQP2 (1:200, SC-9882, Santa Cruz Biotechnology, Dallas, TX) antibodies following standard protocols (2). After being washed, slices were incubated with the appropriate fluorophore-linked secondary antibodies (Alexa Fluor 568 anti-rabbit IgG or Alexa Fluor 488 anti-goat IgG, Invitrogen, Thermo Fisher Scientific).
Immunofluorescence images were collected on an Axiovert 200 M (Carl Zeiss Microscopy, Thornwood, NY) fluorescence microscope. Apical AQP2 and serosal AQP3 levels were quantified by holding illumination, gain, sampling period, and other parameters constant and below saturation during the acquisition of high-magnification immunofluorescent images. Regions of interest were selected by drawing lines across the apical and basolateral membranes of labeled tubular cells following a semiquantitative analysis protocol (6). ImageJ software (National Institutes of Health) was used to find peak intensities for the estimation of corresponding plasma membrane levels. For each group, we analyzed 5 sections from 4 different mice and quantified at least 20 cells per section at random for each labeled tubule. Secondary-only immunofluorescence images were acquired as controls.
Western Blots
The kidney cortex was isolated from male and female mice and homogenized in standard RIPA lysis buffer (No. 89901, Thermo Fisher Scientific) containing Pierce Protease and Phosphatase Inhibitors (A32956, Thermo Fisher Scientific). Lysates were sonicated and centrifuged at 14,000 g for 15 min at 4°C. Supernatant protein concentrations were determined with a Bio-Rad DC Protein Assay Kit (No. 5000001, Bio-Rad) with equal amounts of lysate protein loaded onto gels (10% Bis-Tris Midi Protein Gel, Thermo Fisher Scientific). Denatured protein samples were prepared in LDS Sample Buffer (LP0007, Thermo Fisher Scientific) and Sample Reducing Agent (NP0004, Thermo Fisher Scientific) and run in MOPS SDS Running Buffer (NP0001, Thermo Fisher Scientific) for protein electrophoresis. Protein blots were probed with anti-AQP2 (No. 3487, Cell Signaling), phospho-AQP2 (Ser256; No. 111346, Abcam), and β-actin (No. 8229, Abcam) antibodies. We imaged protein blots on a LI-COR Odyssey CLx Imaging System using IRDye 680RD anti-goat (No. 926–68074, LI-COR) and IRDye 800CW anti-rabbit (No. 926–32211, LI-COR) secondary antibodies. The intensities of immunoreactive bands were analyzed as the sum of pixel intensity minus background (Image Studio Acquisition Software, LI-COR). As typical (6), phospho-AQP2 levels are reported as the summated intensities of nonglycosylated and glycosylated species normalized to β-actin levels.
Patch-Clamp Electrophysiology in Isolated Split-Open Aldosterone-Sensitive Distal Nephrons
For patch-clamp analysis, 12- to 16-wk-old (∼30 g) male and female mice were used. We followed a previously published method for isolating aldosterone-sensitive distal nephrons containing CNT/CCD segments suitable for patch-clamp electrophysiology (2). The activity of ENaC in principal cells of murine aldosterone-sensitive distal nephrons was quantified in cell-attached patches of the apical membrane made under voltage-clamp conditions (negative pipette potential: −60 mV) using standard procedures (2). NPo was calculated. Bath and pipette solutions were (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 2 MgCl2, 5 glucose, and 10 HEPES (pH 7.4) and 140 LiCl, 2 MgCl2, and 10 HEPES (pH 7.4), respectively. For each experimental condition, aldosterone-sensitive distal nephrons from at least five different mice were assayed.
After baseline measurements for ENaC activity were performed, we evaluated the response of ENaC to dDAVP and/or tolvaptan. For the dDAVP alone-treated group, mice received an infusion of dDAVP (100 ng/h) by miniosmotic pump for 7 days. ENaC activity was measured through the patch-clamp technique between days 4 and 7 of dDAVP infusion. For the dDAVP + tolvaptan-treated group, mice were infused with dDAVP for 7 days and given tolvaptan for 3 days (days 4−7). ENaC activity was measured on day 7. For the tolvaptan alone-treated group, mice received tolvaptan for 3 days, and ENaC activity was measured on day 3.
Blood Pressure Measurement in Conscious Mice
Blood pressure was measured by radiotelemetry in conscious, unrestrained 16- to 20-wk-old male mice, as previously described (7). Arterial blood pressure data were collected, stored, and analyzed using Dataquest A.R.T. software (v. 4.0, Data Sciences, St. Paul, MN). Telemetry data were collected continuously with sampling every 5 min for 10-s intervals. Baseline measurements were recorded for 7 consecutive days while mice were maintained on a normal diet (0.39% Na+, Purina 5001). On day 8, an osmotic minipump, which delivered dDAVP at a rate of 100 ng/h, was implanted under the skin of mice between the scapulae. Blood pressure measurements were continued until benzamil (1.4 mg/kg body wt) was administered via intraperitoneal injection once a day for 3 successive days (days 22−24).
Metabolic Cage Experiments
To evaluate whether changes in blood pressure were associated with changes in urine electrolyte excretion, we performed saline loading experiments in mice and housed them in metabolic cages (Hatteras Instruments, Cary, NC) to collect timed excretion of urine electrolytes. Male mice were fed a regular Na+ diet (0.39% Na+, Purina 5001) and acclimated in metabolic cages for 2 days. Mice then received chronic infusion of dDAVP at 100 ng/h via subcutaneous osmotic minipumps for 2 days. On the day of the experiment, mice were administered a saline load (8.4% NaCl in an intraperitoneal volume of 2.5% of body wt), and urine was collected over the ensuing 4 h. We performed saline loading after 2 days of dDAVP infusion so that we could associate changes in urine flow and electrolyte excretion with high blood pressure in Liddle mice.
Analysis of Hormones and Electrolytes
On euthanasia of mice, blood was collected by retroorbital puncture and electrolyte concentrations were quantified using an iSTAT portable analyzer (Abaxis, Union City, CA). A Siemens Dimension Xpand Chemistry Analyzer System or a Jenway (Staffordshire, UK) flame photometer was used to measure urine Na+ concentration. Urinary osmolality was measured with a VAPRO5520 osmometer (ELITechGroup, Puteaux, France). Plasma aldosterone concentration was measured with an aldosterone ELISA kit (IBL-America, Minneapolis, MN).
Statistical Analysis and Data Presentation
Data are reported as means ± SE. Statistical comparisons were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA). Comparisons were made using either one-way ANOVA, two-way ANOVA, or a Kruskal–Wallis test, as appropriate. Proportions were compared using a z test. The criterion for significance was two-sided P < 0.05. For the presentation of current traces, slow baseline drifts were corrected and data software filtered at 50 Hz.
RESULTS
V2 Receptors Maintain Maximal ENaC Activity in Liddle Mice
To test whether V2 receptors maintain maximal ENaC activity in Liddle mice, we measured ENaC activity in isolated CNT/CCD segments in the absence or presence of tolvaptan (Fig. 1A). At baseline, total ENaC NPo, N, and Po in isolated CNT/CCD segments from Liddle mice were higher than those from wild-type littermates (Fig. 1B). ENaC NPo, N, and Po in isolated CNT/CCD segments were also lower from PHA1 mice than those from wild-type littermates (Fig. 1C). Administration of tolvaptan via oral gavage decreased ENaC NPo and N in CNT/CCD segments from Liddle mice, indicating that V2 receptors maintain high ENaC activity in Liddle mice (Fig. 1D). However, chronic infusion of dDAVP, a V2 receptor-specific agonist, in Liddle mice produced no further significant increase in ENaC NPo (Fig. 1D). In contrast, chronic infusion of dDAVP in wild-type mice increased ENaC NPo, to a level that was commensurate to ENaC NPo in Liddle mice (Fig. 1D).
Figure 1.

V2 receptors maintain maximal epithelial Na+ channel (ENaC) activity in Liddle mice. A: experimental scheme. After mice were fed a regular chow diet, ENaC activity was measured in freshly isolated and split-open distal nephrons. B: summary graphs comparing mean (±SE) total ENaC activity (NPo), number of channels (N), and open probability (Po) in cortical collecting ducts of wild-type (WT) littermates (n = 10) and Liddle mice (n = 14). Male mice are indicated by closed circles; female mice are indicated by open circles. *P < 0.05 by one-way ANOVA with Tukey’s correction between the indicated groups. C: summary graphs comparing means (±SE) NPo, N, and Po in cortical collecting ducts of WT littermates (n = 9) and pseudohypoaldosteronism type 1 (PHA1) mice (n = 12). Female mice are indicated by open circles. *P < 0.05 by one-way ANOVA with Tukey’s correction between the indicated groups. D: summary graphs of means (±SE) ENaC NPo, N, and Po in cortical collecting ducts of WT or Liddle mice under the following conditions: 1) baseline (WT: n = 10 and Liddle: n = 14), 2) tolvaptan (30 mg/kg) for 3 days (WT: n = 10 and Liddle: n = 14), 3) desmopressin (dDAVP) infusion (100 ng/h) for 4 days (WT: n = 10 and Liddle: n = 21), and 4) dDAVP + tolvaptan (WT: n = 8 and Liddle: n = 12). Male mice are indicated by closed circles; female mice are indicated by open circles. *P < 0.05 by one-way ANOVA with Tukey correction between the indicated groups.
AQP2/3 Expression Is High in Liddle Mice
To determine the status of V2 receptor signaling in the collecting ducts of Liddle mice, we compared the level of AQP2 and AQP3 expression in response to V2 receptor activation and inhibition in Liddle and wild-type mice (Fig. 2). Expression of AQP2 in the apical membrane and AQP3 in the serosal membrane of collecting ducts of Liddle mice was higher than those in wild-type mice, to such an extent that the levels of expression of AQP2 and AQP3 in untreated Liddle mice were similar to those in dDAVP-treated wild-type mice (Fig. 2, B and C). Since ENaC activity was elevated and sensitive to inhibition by tolvaptan in Liddle mice, we asked whether the high level of AQP2 and AQP3 would also decrease with tolvaptan. In Liddle and wild-type mice, administration of tolvaptan decreased expression of AQP2 and AQP3 in the collecting duct (Fig. 2, B and C). Since ENaC activity in Liddle mice was high and not further stimulated with dDAVP, we tested whether expression of phospho-AQP2 (Ser256), a surrogate for AQP2 activity, would similarly be saturated in Liddle mice. Even though Liddle mice showed higher levels of expression of AQP2 and AQP3 compared with wild-type mice, Liddle mice could still respond to dDAVP infusion by increasing expression of phospho-AQP2 (Ser256) in kidney lysates, to a similar degree as observed in wild-type mice (Fig. 2D).
Figure 2.

Expression of aquaporin-2 (AQP2) and aquaporin-3 (AQP3) in the collecting duct is high in Liddle mice. A: representative kidney sections of wild-type (WT) and Liddle male mice. Red fluorescence indicates AQP2 expression; green fluorescence indicates AQP3 expression. Expression of AQP2 was in the apical membrane and expression of AQP3 was in the serosal membrane of collecting ducts of WT and Liddle mice. Mice were infused with desmopressin (dDAVP) in the absence or presence of the V2 receptor antagonist tolvaptan, as indicated. B: apical expression of AQP2 was significantly higher in principal cells of the collecting ducts of Liddle mice compared with WT mice. Means (±SE) apical AQP2 intensity was from n = 20 random cells for each group, analyzed from 5 kidney sections from 4 different mice. *P < 0.05 by two-way ANOVA with Tukey’s correction between the indicated groups. C: serosal expression of AQP3 was significantly higher in principal cells of the collecting ducts of Liddle mice compared with WT mice. Means (±SE) serosal AQP3 intensity was from n = 20 random cells for each group, analyzed from 5 kidney sections from 4 different mice. *P < 0.05 by two-way ANOVA with Tukey’s correction between the indicated groups. D: Liddle mice responded to V2 receptor stimulation and inhibition to a similar extent as WT mice. Male or female mice were treated with vehicle control, dDAVP, and dDAVP with tolvaptan. Immunoblots of kidney lysates were probed with antibodies directed against phospho-AQP2 (Ser256; pAQP2) and β-actin. Means (±SE) densitometric analysis for expression of pAQP2 was normalized to the expression of β-actin in kidney lysates from WT and Liddle mice. *P < 0.05 by two-way ANOVA with Tukey’s correction between dDAVP and vehicle control. **P < 0.05 between dDAVP and dDAVP + tolvaptan. n = 6−11 mice for each group as indicated.
Chronic Stimulation of V2 Receptors Raises Blood Pressure in Liddle and Wild-Type Mice
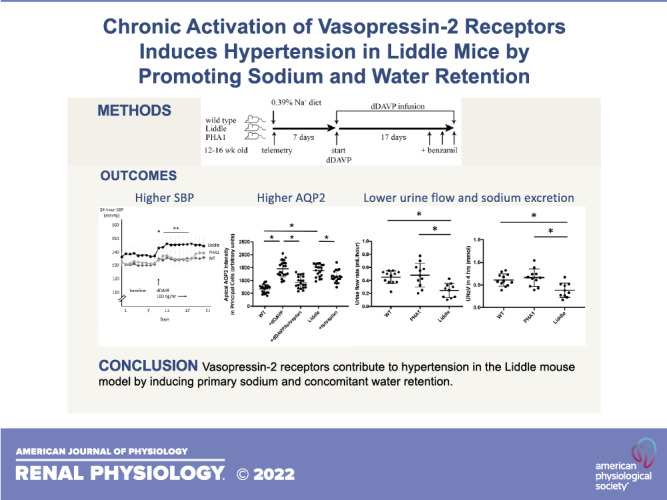
To test whether V2 receptors contribute to the pathogenesis of hypertension in Liddle mice, we compared blood pressure responses to dDAVP infusion in Liddle, wild-type, and PHA1 mice (Fig. 3A). Baseline blood pressure was similar for all genotypes of mice when they were maintained on a regular Na+ diet (Fig. 3B), similar to findings from the study by Pradervand et al. (4). Liddle and wild-type mice developed significantly higher blood pressure with dDAVP infusion (Fig. 3, C and D): infusion of dDAVP into Liddle mice increased systolic blood pressure (SBP) by 7.3 ± 1.2 mmHg (P < 0.05) and mean arterial pressure (MAP) by 7.0 ± 1.6 mmHg (P < 0.05), and infusion of dDAVP into wild-type mice increased SBP by 4.3 ± 0.5 mmHg (P < 0.05) and MAP by 3.7 ± 0.4 mmHg (P < 0.05). In contrast, PHA1 mice were relatively protected from hypertension and did not respond with a statistically significant increase in SBP or MAP with dDAVP infusion (Fig. 3, C and D). Blood pressure was highest in dDAVP-infused Liddle mice compared with dDAVP-infused wild-type or PHA1 mice; for example, during a 1-wk period of dDAVP infusion (days 11−17 in Fig. 3B), Liddle mice had an average SBP of 146 ± 6 mmHg and MAP of 128 ± 4 mmHg compared with wild-type mice with an average SBP of 135 ± 1 mmHg and MAP of 119 ± 1 mmHg or with PHA1 mice with an average SBP of 135 ± 5 mmHg and MAP of 119 ± 4 mmHg (P < 0.05 for higher SBP and higher MAP for Liddle compared with wild-type or PHA1 genotypes).
Figure 3.

Chronic infusion of desmopressin (dDAVP) increases blood pressure in wild-type (WT) and Liddle mice. A: experimental scheme. After mice were fed a regular chow diet for 7 days, they received infusion of dDAVP (100 ng/h) for 14 days. Benzamil was administered to mice on the last 3 days of the experiment. B: radiotelemetric measurement of means (±SE) systolic blood pressure (SBP) or mean arterial pressure (MAP) before and after chronic infusion of dDAVP in male WT, Liddle, and pseudohypoaldosteronism type 1 (PHA1) mice. *P < 0.05 by Kruskal–Wallis test for SBP or MAP between baseline and dDAVP for Liddle and WT mice. **P < 0.05 by Kruskal–Wallis test for SBP or MAP during days 11−17 (during days 2−8 of dDAVP infusion) for dDAVP-infused Liddle mice vs. SBP or MAP in other groups of dDAVP-infused mice. C: SBP response to chronic dDAVP infusion. Baseline represents the average of SBP of mice from days 1−7; dDAVP represents the average SBP of mice from days 11−21. *P < 0.05 vs. baseline by a Kruskal–Wallis test. n = 7 WT mice, 8 Liddle mice, and 7 PHA1 mice. D: MAP response to chronic dDAVP infusion. Baseline represents the average of MAP of mice from days 1−7; dDAVP represents the average MAP of mice from days 11−21. *P < 0.05 vs. baseline by a Kruskal–Wallis test. n = 7 WT mice, 8 Liddle mice, and 7 PHA1 mice.
To ensure that differences in blood pressure among dDAVP-infused Liddle, wild-type, and PHA1 mice were not due to differences in RAAS signaling, we compared plasma aldosterone concentrations for all genotypes of mice. Liddle mice had lower plasma aldosterone concentrations compared with wild-type or PHA1 mice (Liddle: 0.34 ± 0.03 nM vs. wild type: 1.21 ± 0.11 nM or PHA1: 1.15 ± 0.15 nM, P < 0.05; Fig. 4A); infusion of dDAVP had no effect on plasma aldosterone concentrations for all genotypes (Fig. 4A). We also compared plasma electrolyte parameters, such as plasma Na+, K+, Cl−, total CO2, and urea nitrogen, in untreated or dDAVP-infused Liddle, wild-type, and PHA1 mice. At baseline, Liddle mice had significantly lower plasma K+ and Cl− concentrations and higher total CO2 concentrations compared with wild-type and PHA1 mice (Fig. 4B). With chronic dDAVP infusion, Liddle mice had significantly higher plasma K+ concentrations compared with other genotypes (Liddle: 4.9 ± 0.1 mM vs. wild type: 4.4 ± 0.1 mM or PHA1: 4.4 ± 0.2 mM, P < 0.05; Fig. 4B).
Figure 4.

Plasma measurements in wild-type (WT), Liddle, and pseudohypoaldosteronism type 1 (PHA1) mice at baseline and with chronic infusion of desmopressin (dDAVP). A: plasma aldosterone concentrations were not different with dDAVP infusion. Means (±SE) plasma aldosterone concentration was suppressed in Liddle mice with infusion of vehicle or dDAVP (100 ng/h) for 5 days. P < 0.05 by two-way ANOVA with Tukey’s correction for the effect of genotype. n = 6 WT mice, 6 Liddle mice, and 6 PHA1 mice with vehicle treatment; n = 7 WT mice, 6 Liddle mice, and 5 PHA1 mice with dDAVP treatment. B: means (±SE) plasma measurements in WT, Liddle, and PHA1 mice at baseline and with chronic infusion of dDAVP. Male mice were fed a regular chow diet or also administered dDAVP at 100 ng/h for 5 days. *P < 0.05 by two-way ANOVA with Tukey’s correction from other groups of mice. tCO2, total CO2.
Hypertension in Liddle Mice Is Sensitive to ENaC Inhibition
To test whether ENaC accounts for hypertension in dDAVP-infused Liddle mice, we treated dDAVP-infused mice with benzamil for 3 successive days and compared subsequent blood pressure in the three genotypes of mice. Administration of benzamil significantly reduced blood pressure in dDAVP-infused Liddle mice (SBP decrease by 4.9 ± 1.3 mmHg, P < 0.05; MAP decrease by 4.9 ± 1.3 mmHg, P < 0.05). In contrast, hypertension in dDAVP-infused wild-type mice was not sensitive to benzamil inhibition (SBP increase by 0.2 ± 0.6 mmHg, not significant; MAP decrease by 0.5 ± 0.5 mmHg, not significant; Fig. 5, A and B).
Figure 5.

The epithelial Na+ channel inhibitor benzamil lowers blood pressure in desmopressin (dDAVP)-infused Liddle mice. Successive administration of benzamil (1.4 mg/kg body wt) for 3 days lowered systolic blood pressure (SBP; A) and mean arterial pressure (MAP; B) in dDAVP-infused (100 ng/h) Liddle mice but not in dDAVP-infused wild-type (WT) or pseudohypoaldosteronism type 1 (PHA1) mice. *P < 0.05 by a Kruskal–Wallis test before and after benzamil treatment. n = 7 WT mice, 8 Liddle mice, and 7 PHA1 mice. NS, not significant.
Hypertension in Liddle Mice Is Associated With Na+ and Water Retention
To test whether dDAVP-infused Liddle mice have impaired natriuresis associated with hypertension, we administered a saline load to dDAVP-infused Liddle, wild-type, and PHA1 mice and compared rates of urine flow and Na+ excretion from a 4-h timed collection (Fig. 6A). In response to a saline load, Liddle mice that received dDAVP infusion excreted a smaller urine volume compared with wild-type or PHA1 mice that received the same dDAVP infusion (Liddle: 0.25 ± 0.03 mL urine/h, wild type: 0.45 ± 0.03 mL urine/h, and PHA1: 0.48 ± 0.05 mL urine/h, P < 0.05; Fig. 6B), but urine Na+ concentration and urine osmolality were similar for all groups of dDAVP-infused mice (Fig. 6B). Liddle mice that received dDAVP infusion also excreted less urine Na+ and osmoles over a 4-h period compared with similarly treated wild-type or PHA1 mice (Fig. 6C).
Figure 6.

Liddle mice excrete smaller urine volume and less urine Na+ in response to a saline load compared with wild-type (WT) or pseudohypoaldosteronism type 1 (PHA1) mice. A: experimental scheme. After male mice were fed a regular chow diet, they received infusion of desmopressin (dDAVP; 100 ng/h) for 2 days. Mice were then administered a saline load (8.4% NaCl in an intraperitoneal volume of 2.5% of body wt), and urine was collected over the ensuing 4 h. B: summary graphs of a timed (4-h) urine collection for means (±SE) urine flow rate, urine Na+ concentration (UNa+), and urine osmolality (urine osM) in WT, PHA1, and Liddle mice. C: summary graphs of a timed (4-h) urine collection for means (±SE) urine Na+ excretion (UNaV) and urine osmolar excretion (urine osM V) in WT, PHA1, and Liddle mice. *P < 0.05 by two-way ANOVA with Tukey correction from other groups of mice. n = 12 WT mice, 11 PHA1 mice, and 10 Liddle mice.
DISCUSSION
Our findings demonstrate that a specific increase in V2 receptor-mediated ENaC activity in Liddle mice, and in wild-type littermates, is sufficient to raise blood pressure. In Liddle mice, we found that ENaC activity was elevated and could not be stimulated further with dDAVP. However, ENaC activity could still be inhibited by tolvaptan, indicating that ENaC activity is maximal in Liddle mice with respect to V2 receptor stimulation. Liddle mice also showed higher levels of expression of AQP2 and AQP3 under basal conditions, with levels of AQP2 and AQP3 expression similar to those found in dDAVP-treated wild-type mice. Yet Liddle mice could still respond to dDAVP by increasing phospho-AQP2 expression, demonstrating that ENaC and AQP2 respond differently to V2 receptors in Liddle mice. When Liddle mice received chronic infusion of dDAVP, blood pressure increased, and this hypertension was blunted by benzamil. The hypertension in dDAVP-infused Liddle mice was associated with a limitation in urine Na+ and water excretion. We conclude that V2 receptors contribute to hypertension in Liddle mice by inducing primary Na+ and concomitant (secondary) water retention.
The regulation of ENaC activity in Liddle mice has recently been revisited. Nesterov and colleagues (8) compared the effects of dietary salt on ENaC activity in the CNT/CCD and late distal convoluted tubule (DCT2)/CNT segment of wild-type and Liddle mice. ENaC activity was higher in both nephron segments of Liddle mice compared with wild-type mice, but ENaC responded differently to dietary salt depending on its location along the nephron. In the CNT/CCD segment, ENaC could respond to dietary salt intake in Liddle mice such that ENaC activity increased as dietary salt decreased. In the DCT2/CNT segment, ENaC activity was fixed in Liddle mice and did not change with dietary salt. In our study, we found that ENaC activity in the CNT/CCD is higher in Liddle mice and that V2 receptors help to maintain this higher level of ENaC activity, adding to the list of factors such as low dietary Na+ (8, 9), aldosterone infusion (9), or high dietary K+ (9) that can stimulate ENaC activity in the CNT/CCD of Liddle mice. Since we did not evaluate ENaC activity in the DCT2/CNT, it remains to be determined if ENaC in this nephron segment is either maintained by V2 receptors or can also respond to V2 receptor stimulation.
Our findings prompt the question of how V2 receptors raise benzamil-sensitive blood pressure without increasing ENaC activity in Liddle mice. The combination of high ENaC activity and AQP2/AQP3 expression in Liddle mice may facilitate ENaC-dependent isoosmotic water reabsorption in the early segments of the distal nephron. Water retention may directly increase ECF volume in the context of maximal ENaC activity. This physiology becomes apparent when dDAVP-infused Liddle mice were challenged with a saline load: they excreted a smaller urine volume and less urine Na+ compared with dDAVP-infused control mice. It has been established in rodents and humans that a Na+ load cannot be excreted easily when the urine Na+ concentration becomes maximal and the urine volume becomes limiting (10, 11). In our study, the urine Na+ concentration and urine osmolality were similar and at maximal concentration for all groups of dDAVP-infused mice but urine flow rate was significantly lower in dDAVP-infused Liddle mice, reflecting higher Na+ reabsorption through ENaC and possibly other Na+-coupled transporters in proximal segments of the nephron.
Our findings highlight the inherent limitation of how terrestrial organisms must use Na+ as an osmotic solute to absorb water from renal tubular fluid. ENaC likely plays a broader, and less recognized, role in controlling both Na+ conservation and the urine concentrating mechanism. Bankir and colleagues (3) have postulated that V2 receptors might activate ENaC to prioritize conservation of body water over Na+. Although ENaC activation by V2 receptors may be adaptive for water conservation, it can become pathological for Na+ balance and blood pressure. Whether activation of V2 receptors can lead to Na+ retention and high blood pressure has remained an open question. Chronic activation of V2 receptors in Sprague-Dawley rats induces high blood pressure (12). However, these rats were studied under combined conditions of high dietary Na+ intake, mineralocorticoid infusion, and uninephrectomy (DS-Nx model). Since a reduction in nephron number or mineralocorticoid infusion can independently induce Na+ retention, factors apart from V2 receptors could have accounted for high blood pressure in this model. Our findings demonstrate that, even in the absence of high dietary Na+ intake, mineralocorticoid excess, or nephrectomy, chronic activation of V2 receptors in Liddle mice is sufficient to raise blood pressure by inducing ENaC-mediated Na+ retention, which, in turn, drives water retention.
There are several limitations in our study. First, since chronic activation of V2 receptors in Liddle mice increased blood pressure, we also tested whether treatment of dDAVP-infused Liddle mice with tolvaptan could decrease blood pressure; however, treatment of dDAVP-infused Liddle mice with tolvaptan (for 1 day) induced no decrease in blood pressure (data not shown). It remains to be determined if more chronic treatment of dDAVP-infused Liddle mice with tolvaptan will lower blood pressure. Indeed, in prior studies that have examined effects of V2 receptor antagonists on lowering blood pressure, in a mineralocorticoid high-salt or DS-Nx model, only long-term treatment with a V2 receptor antagonist, consisting of at least 2-wk duration, decreased blood pressure (12–14). Second, we did not design our study to examine sex differences in the contribution of V2 receptors to hypertension in the Liddle mouse model. Although both female and male mice were used for expression and electrophysiological experiments, only male mice were used for blood pressure experiments. The use of both sexes of mice for expression experiments could have introduced variability in our results, and future studies will be needed to study the possibility of sex differences in the blood pressure response to V2 receptor activation in Liddle mice. Third, blood pressure readings for PHA1 mice generally increased with dDAVP infusion, but statistical significance was not reached because some PHA1 mice showed a decrease in blood pressure. It is possible that the response profile for PHA1 mice would be different with a larger sample size. Fourth, we did not measure all components of the water concentrating mechanism in Liddle mice, such as plasma AVP concentration or renal V2 receptor expression, which may be dysregulated at baseline. Finally, we only tested the responses of ENaC and AQP2 to chronic dDAVP infusion in Liddle and PHA1 mice; we did not examine whether chronic dDAVP infusion can stimulate phosphorylation of the NaCl cotransporter (NCC) in the distal convoluted tubule or Na+-K+-2Cl− cotransporter isoform 2 (NKCC2) in the thick ascending limb of these mice. Saritas et al. (15) have demonstrated that chronic infusion of dDAVP can stimulate phosphorylation of NCC and NKCC2 through a STE20/SPS1-related proline/alanine-rich kinase-dependent mechanism. Future studies will be needed to delineate the specific contribution of NCC or NKCC2 to AVP/V2 receptor-induced Na+ retention and hypertension in wild-type or Liddle mice.
Perspectives and Significance
In conclusion, our findings demonstrate that V2 receptors contribute to high blood pressure in the Liddle mouse model by stimulating ENaC, which induces not only renal Na+ but also water retention, thereby raising blood pressure. These findings indicate that V2 receptors contribute to the pathophysiology of Liddle syndrome by preserving plasma tonicity at the expense of a higher blood pressure that results from Na+ and water retention. We suggest that a subset of human hypertension may be driven by high levels of ENaC and/or V2 receptor activity; if present, this form of hypertension may respond to V2 receptor antagonists or prescription of high fluid intake to suppress V2 receptors.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK103758 (to J.D.S. and A.C.P.), DK098141 (to J.A.M.), and DK096493 (to S.B.G.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.D.S., E.V.M., H.X., J.A.M., S.B.G., and A.C.P. conceived and designed research; J.D.S., E.V.M., H.X., A.G.S., J.C., J.A.M., and A.C.P. performed experiments; J.D.S., E.V.M., H.X., A.G.S., J.C., J.A.M., S.B.G., and A.C.P. analyzed data; J.D.S., E.V.M., H.X., A.G.S., J.C., J.A.M., S.B.G., and A.C.P. interpreted results of experiments; J.D.S., E.V.M., H.X., A.G.S., J.C., J.A.M., and A.C.P. prepared figures; J.D.S., E.V.M., J.A.M., and A.C.P. drafted manuscript; J.D.S., E.V.M., H.X., J.A.M., S.B.G., and A.C.P. edited and revised manuscript; J.D.S., E.V.M., H.X., J.A.M., and A.C.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Edith Hummler for sharing Liddle and PHA1 mice and to Dr. Lise Bankir for helpful discussion and critical evaluation of the experiments in the report.
REFERENCES
- 1. Mironova E, Chen Y, Pao AC, Roos KP, Kohan DE, Bugaj V, Stockand JD. Activation of ENaC by AVP contributes to the urinary concentrating mechanism and dilution of plasma. Am J Physiol Renal Physiol 308: F237–F243, 2015. doi: 10.1152/ajprenal.00246.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mironova E, Bugaj V, Roos KP, Kohan DE, Stockand JD. Aldosterone-independent regulation of the epithelial Na+ channel (ENaC) by vasopressin in adrenalectomized mice. Proc Natl Acad Sci USA 109: 10095–10100, 2012. doi: 10.1073/pnas.1201978109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bankir L, Bichet DG, Bouby N. Vasopressin V2 receptors, ENaC, and sodium reabsorption: a risk factor for hypertension? Am J Physiol Renal Physiol 299: F917–F928, 2010. doi: 10.1152/ajprenal.00413.2010. [DOI] [PubMed] [Google Scholar]
- 4. Pradervand S, Wang Q, Burnier M, Beermann F, Horisberger JD, Hummler E, Rossier BC. A mouse model for Liddle's syndrome. J Am Soc Nephrol 10: 2527–2533, 1999. doi: 10.1681/ASN.V10122527. [DOI] [PubMed] [Google Scholar]
- 5. Pradervand S, Barker PM, Wang Q, Ernst SA, Beermann F, Grubb BR, Burnier M, Schmidt A, Bindels RJ, Gatzy JT, Rossier BC, Hummler E. Salt restriction induces pseudohypoaldosteronism type 1 in mice expressing low levels of the beta-subunit of the amiloride-sensitive epithelial sodium channel. Proc Natl Acad Sci USA 96: 1732–1737, 1999. doi: 10.1073/pnas.96.4.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cheema MU, Irsik DL, Wang Y, Miller-Little W, Hyndman KA, Marks ES, Frøkiær J, Boesen EI, Norregaard R. Estradiol regulates AQP2 expression in the collecting duct: a novel inhibitory role for estrogen receptor α. Am J Physiol Renal Physiol 309: F305–F317, 2015. doi: 10.1152/ajprenal.00685.2014. [DOI] [PubMed] [Google Scholar]
- 7. Gurley SB, Riquier-Brison ADM, Schnermann J, Sparks MA, Allen AM, Haase VH, Snouwaert JN, Le TH, McDonough AA, Koller BH, Coffman TM. AT1A angiotensin receptors in the renal proximal tubule regulate blood pressure. Cell Metab 13: 469–475, 2011. doi: 10.1016/j.cmet.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nesterov V, Krueger B, Bertog M, Dahlmann A, Palmisano R, Korbmacher C. In Liddle syndrome, epithelial sodium channel is hyperactive mainly in the early part of the aldosterone-sensitive distal nephron. Hypertension 67: 1256–1262, 2016. doi: 10.1161/HYPERTENSIONAHA.115.07061. [DOI] [PubMed] [Google Scholar]
- 9. Dahlmann A, Pradervand S, Hummler E, Rossier BC, Frindt G, Palmer LG. Mineralocorticoid regulation of epithelial Na+ channels is maintained in a mouse model of Liddle’s syndrome. Am J Physiol Renal Physiol 285: F310–F318, 2003. doi: 10.1152/ajprenal.00016.2003. [DOI] [PubMed] [Google Scholar]
- 10. Bankir L, Perucca J, Norsk P, Bouby N, Damgaard M. Relationship between sodium intake and water intake: the false and the true. Ann Nutr Metab 70, Suppl 1: 51–61, 2017. doi: 10.1159/000463831. [DOI] [PubMed] [Google Scholar]
- 11. Choukroun G, Schmitt F, Martinez F, Drüeke TB, Bankir L. Low urine flow reduces the capacity to excrete a sodium load in humans. Am J Physiol Regul Integr Comp Physiol 273: R1726–R1733, 1997. doi: 10.1152/ajpregu.1997.273.5.R1726. [DOI] [PubMed] [Google Scholar]
- 12. Fernandes S, Bruneval P, Hagege A, Heudes D, Ghostine S, Bouby N. Chronic V2 vasopressin receptor stimulation increases basal blood pressure and exacerbates deoxycorticosterone acetate-salt hypertension. Endocrinology 143: 2759–2766, 2002. doi: 10.1210/endo.143.7.8918. [DOI] [PubMed] [Google Scholar]
- 13. Hashimoto J, Imai Y, Minami N, Munakata M, Abe K. Effects of vasopressin V1 and V2 receptor antagonists on the development of salt-induced hypertension in Dahl rats. J Cardiovasc Pharmacol 26: 548–554, 1995. doi: 10.1097/00005344-199510000-00007. [DOI] [PubMed] [Google Scholar]
- 14. Okada H, Suzuki H, Kanno Y, Saruta T. Effect of nonpeptide vasopressin receptor antagonists on developing, and established DOCA-salt hypertension in rats. Clin Exp Hypertens 17: 469–483, 1995. doi: 10.3109/10641969509037419. [DOI] [PubMed] [Google Scholar]
- 15. Saritas T, Borschewski A, McCormick JA, Paliege A, Dathe C, Uchida S, Terker A, Himmerkus N, Bleich M, Demaretz S, Laghmani K, Delpire E, Ellison DH, Bachmann S, Mutig K. SPAK differentially mediates vasopressin effects on sodium cotransporters. J Am Soc Nephrol 24: 407–418, 2013. doi: 10.1681/ASN.2012040404. [DOI] [PMC free article] [PubMed] [Google Scholar]