

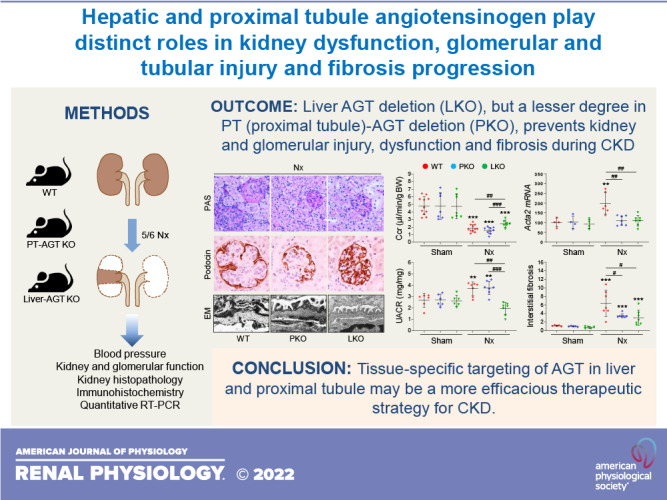
Keywords: angiotensinogen, chronic kidney disease, glomerulosclerosis, kidney fibrosis, kidney injury
Abstract
Components of the renin-angiotensin system, including angiotensinogen (AGT), are critical contributors to chronic kidney disease (CKD) development and progression. However, the specific role of tissue-derived AGTs in CKD has not been fully understood. To define the contribution of liver versus kidney AGT in the CKD development, we performed 5/6 nephrectomy (Nx), an established CKD model, in wild-type (WT), proximal tubule (PT)- or liver-specific AGT knockout (KO) mice. Nx significantly elevated intrarenal AGT expression and elevated blood pressure (BP) in WT mice. The increase of intrarenal AGT protein was completely blocked in liver-specific AGT KO mice with BP reduction, suggesting a crucial role for liver AGT in BP regulation during CKD. Nx-induced glomerular and kidney injury and dysfunction, as well as fibrosis, were all attenuated to a greater extent in liver-specific AGT KO mice compared with PT-specific AGT KO and WT mice. However, the suppression of interstitial fibrosis in PT- and liver-specific AGT KO mouse kidneys was comparable. Our findings demonstrate that liver AGT acts as a critical contributor in driving glomerular and tubular injury, renal dysfunction, and fibrosis progression, whereas the role of PT AGT was limited to interstitial fibrosis progression in chronic renal insufficiency. Our results provide new insights for the development of tissue-targeted renin-angiotensin system intervention in the treatment of CKD.
NEW & NOTEWORTHY Chronic kidney disease (CKD) is a major unmet medical need with no effective treatment. Current findings demonstrate that hepatic and proximal tubule angiotensinogen have distinct roles in tubular and glomerular injury, fibrogenesis, and renal dysfunction during CKD development. As renin-angiotensin system components, including angiotensinogen, are important targets for treating CKD in the clinic, the results from our study may be applied to developing better tissue-targeted treatment strategies for CKD and other fibroproliferative diseases.
INTRODUCTION
Chronic kidney disease (CKD) is globally increasing in incidence and prevalence. It remains a major health problem with no effective treatment apart from renal replacement therapy such as kidney transplant and dialysis. In addition, CKD is one of the major risk factors for triggering and accelerating cardiovascular diseases; thus, developing therapeutic strategies for CKD is urgently needed (1–3). CKD is characterized by persistent functional loss associated with tubular and glomerular structural damage, proteinuria, inflammation, and fibrosis progression (4, 5). The functional loss of nephrons, which develops and progresses by acute or chronic tubular and glomerular injury, is a fundamental cause of CKD (4). Hypertension and other metabolic diseases have been well recognized as critical factors for the development and progression of CKD (6–8). Currently, several drugs, such as angiotensin-converting enzyme (ACE) inhibitor and angiotensin II (ANG II) type 1 receptor (AT1R) inhibitor, are being used for not only controlling blood pressure (BP) but also slowing CKD progression (9). However, their efficacy in treating CKD is suboptimal with no or minor effect in many cases along with some significant deleterious effects (10–13). These limitations underscore the urgency of elucidating the precise molecular mechanisms of CKD progression to develop efficacious therapeutic strategies.
Angiotensinogen (AGT) is a precursor of angiotensin that is significantly elevated in kidney and urine during progression of CKD (14–17). The majority of AGT is derived from the liver, which is a major source of circulating AGT that is upregulated during CKD (17). The proximal tubule (PT) is another source of intrarenal AGT but is a lesser contributor than that of the liver (18, 19). Liver-derived circulating AGT as well as ANG II are absorbed in PTs through megalin-dependent endocytosis (18, 20–22). Previous reports have demonstrated that intrarenal AGT protein in normal mouse kidneys could originate from the liver and was increased under nephrotic conditions (18, 23, 24). However, little is known about whether and how tissue-specific AGT contribute to kidney injury, dysfunction, and fibrosis progression in CKD.
In the present study, to define the source-dependent effect of AGT in CKD development, we generated PT- and liver-specific AGT knockout (KO) mice and induced 5/6 nephrectomy (Nx), an established CKD mouse model. We demonstrated that both PT- and liver-specific AGT contributes to CKD development with distinct roles in tubular and glomerular damage and dysfunction as well as kidney fibrosis.
MATERIALS AND METHODS
Mice and Surgical Preparations
Mice were cared before and during the experimental procedures in accordance with the policies of the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All protocols had received prior approval from the Institutional Animal Care and Use Committee of the University of Nebraska Medical Center. Eight- to twelve-wk-old male AGT-floxed mice (B6 origin, Jackson Lab) were used. Phosphoenolpyruvate carboxykinase Cre recombinase-expressing mice (B6.129 origin, a kind gift from Dr. Volker Haase, Vanderbilt University) (25) and albumin Cre recombinase (B6 × DBA origin, Jackson Lab) were crossed with AGT-floxed mice to generate mice (mixed background) with PT- or liver-specific deletion of AGT. Cre recombinase-negative AGT-floxed mice were used for wild-type (WT) controls. 5/6 Nx was performed by a two-step procedure with a slight modification from previous report (26, 27); at day 0, each one-third of the upper and lower parts of the left kidney were removed through a left flank incision, and at day 7, the whole right kidney was removed through a right flank incision. Microfibrillar collagen hemostasis (Avitene, Davol, Warwick, RI) was used for bleeding control. The resected left kidney was weighed immediately to confirm whether the Nx model was accurately performed. Only those animals with appropriate resection for Nx were used for further experiments. After 16 wk of Nx or sham operation (sham), kidneys were either fixed in 4% formaldehyde for histological experiments or snap frozen in liquid nitrogen for biochemical experiments. Periodic acid-Schiff-stained sections were used for histopathological evaluation.
BP measurement.
Systolic BP, diastolic BP, and mean arterial BP (n = 6–12) were measured during daytime (1–4 PM) by a noninvasive tail-cuff system (CODA2, Kent Scientific, Torrington, CT) as previously described (28, 29). Mice were acclimated for 7 days before actual measurement. The BP value was an average of seven measurements taken in 15-s intervals per mouse.
RNA Extraction
Total RNA was extracted from kidneys using the TRIzol reagent (Invitrogen) and cleaned with the RNeasy mini kit (Qiagen, Hilden, Germany) as previously described (30). First-strand cDNA was prepared by reverse transcription using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA). To determine the conditions for the logarithmic phase during PCR amplification with target mRNA, aliquots (1 µg) were amplified using different numbers of cycles. A linear relationship between PCR product band visibility and the number of amplification cycles was observed for target mRNAs.
Quantitative RT-PCR
Quantitative real-time PCR (n = 4–9) was performed with 1 µL of the cDNA template added to 10 µL of 2× SYBR Premix Ex Taq (Applied Biosystems, Foster City, CA) and specific primers (10 pM each) as previously described (30). Real-time PCR (Bio-Rad) was carried out for 40 cycles of denaturation at 95°C for 15 s, annealing at 58°C for 15 s, and extension at 72°C for 15 s. Target gene expression was quantified relative to that of an internal control gene (GAPDH) based on comparison of the threshold cycle (CT) at constant fluorescence intensity. The amount of transcript was inversely related to the observed CT, and the CT value was expected to increase by 1 for every twofold dilution of the transcript. Relative expression was calculated using the following equation: relative expression = 2 − (ΔCT sample − ΔCT control). All data were normalized relative to GAPDH as well as to the respective controls. Primer sequences are provided in Supplemental Table S1.
Electron Microscopy
Kidneys were fixed in Trump’s fixative (American MasterTech Scientific, Lodi, CA) and processed on the Leica EM AMW Tissue Processor as previously described (31); kidneys (n = 3–5) were washed in distilled water, postfixed in 1% aqueous osmium tetroxide, and then dehydrated in a graded series using acetone. Tissue was infiltrated with and embedded in Poly/Bed 812 resin (Polysciences, Warrington, PA). Following an 800-nm survey section (Leica Ultracut R) stained with 0.5% toluidine blue in 0.5% sodium borate, ultrathin sections were cut by 80–90 nm, picked up onto copper grids, stained with saturated uranyl acetate in 50% ethyl alcohol and Reynold’s lead citrate, and imaged at 60 kV in a JEOL JEM 1230 transmission electron microscope. Digital images were captured with SIS iTem camera/software (Soft Imaging Systems).
Histological Analysis
PAS-stained sections were used for evaluating tubular injury and glomerular injury scoring. Tubular injury (n = 5–7) was calculated as we have previously described (30). The glomerulosclerosis index (n = 6–11) was evaluated in at least 30 randomly chosen 30 glomeruli per group, based on a modified semiquantitative scoring system: 0 = absence of glomerulosclerosis, 1 = 1–25%, 2 = 25–50%, 3 = 51–75%, and 3 = 76–100% (32). Glomerular volume was calculated from the cross-sectional area with the following formula: glomerular volume = β/k (AG)3/2, where β = 1.38 is the shape coefficient for a sphere and k = 1.1 is the size distribution coefficient (33). The relative mesangial area was expressed as a percentage of the mesangial-to-glomerular surface area as previously described (34). Tubular and glomerular diameters were evaluated as an actual length in at least 50 tubules or 30 glomeruli in randomly selected fields per kidney using ImageJ software (National Institutes of Health, Bethesda, MD).
Morphometry
As previously described (32), kidney or liver AGT- and collagen type IV- and α-smooth muscle actin (α-SMA)-positive areas were measured in five randomly chosen fields per section, and glomerular Wilms’ tumor 1 (WT1)-, podocin-, and α-SMA-positive areas or cell numbers were counted in at least 30 glomeruli per group using ImageJ software (National Institutes of Health).
Kidney and Glomerular Function
Plasma and urine creatinine and albuminuria were measured by the University of Alabama-University of California-San Diego O’Brien Center for Acute Kidney Injury Research. Blood and urine samples were collected from the vena cava and metabolic cages (Tecniplast, West Chester, PA), respectively. Creatinine clearance (n = 6–10) was calculated by the concentration of urine creatinine (mg/dL)/concentration of plasma creatinine (mg/dL) × urine volume (µL)/collection duration (min)/body weight (g), as previously described (35). Albuminuria (n = 6–7) was expressed as a ratio of albumin to creatinine (UACR).
Collagen Deposition
Collagen deposition was assessed by Sirius red staining as previously described (30). Interstitial or glomerular Sirius red-positive areas (n = 6–11) were expressed as the ratio of the Sirius red-positive area to total area in five randomly chosen fields or at least 30 glomeruli per group.
Immunohistochemistry
Immunohistochemical staining of the kidneys was performed on paraffin-embedded sections as previously described (36). Briefly, 4% paraformaldehyde-fixed kidney or liver sections (n ≥ 5) were rehydrated, retrieved in 10 mM sodium citrate by 10-min autoclave, and then labeled with antibodies against AGT (1:100, Immuno-Biological Laboratories, Minneapolis, MN) (18), WT1 (1:100, Santa Cruz Biotechnology, Santa Cruz, CA), platelet-derived growth factor receptor-β (PDGFRβ; 1:100), podocin (1:100, Abcam, Cambridge, MA), α-SMA (1:1,000, Sigma, St. Louis, MO), and collagen type IV (1:100, Southern Biotech, Birmingham, AL). Sections were then incubated with peroxidase-secondary antibodies (Vector Laboratories, Burlingame, CA). Meyer’s hematoxylin (Electron Microscopy Sciences, Hatfield, PA) was used as a counterstain.
Statistical Analyses
Data are expressed as means ± SD. Differences between two groups were assessed by a two-tailed unpaired Student’s t test. For multiple group comparison, one-way ANOVA with Bonferroni analysis was applied (GraphPrism 9.0 software). A P value of <0.05 was considered statistically significant.
RESULTS
Both PT- and Liver-Derived AGT Contribute to Intrarenal Expression of AGT in CKD
Both PTs and the liver are major sites that produce AGT, contributing to intrarenal AGT expression. To unveil the role of tissue-specific AGT in the development of CKD, we generated PT- and liver-specific AGT KO mice by crossing AGT-floxed mice with phosphoenolpyruvate carboxykinase- or albumin Cre recombinase-expressing mice, respectively (Supplemental Fig. S1 and Fig. 1A). We confirmed expression levels of AGT protein among WT, PT-specific AGT KO, and liver-specific AGT KO mouse kidneys in sham and after Nx. In sham mice, PT- and liver-specific AGT KO significantly downregulated AGT protein levels in the kidneys compared with WT mice, but the level was much lower or absent in liver-specific AGT KO mice (Fig. 1A). Nx markedly enhanced intrarenal AGT protein expression in WT mice. In a pattern similar to those of sham mice, PT-specific AGT KO partially, but liver-specific AGT KO almost completely, suppressed the AGT level in Nx kidneys (Fig. 1A). Next, to confirm whether AGT expression in the liver is changed in Nx mice, we performed immunohistochemistry for AGT in liver sections. AGT was expressed in hepatocytes, and that was upregulated in Nx mice compared with sham mice (Supplemental Fig. S2), suggesting a potential role in increased expression of intrarenal AGT during CKD. Megalin, which is the major receptor absorbing circulating AGT in the PT (18, 20), was increased in WT mouse kidneys with Nx, but PT- and liver-specific AGT KO suppressed its increase (Supplemental Fig. S3A). Similar to a previous report (18), in sham mouse kidneys, AGT mRNA was decreased in PT-specific AGT KO but increased in liver-specific AGT KO (Supplemental Fig. S3B). Nx in WT mice upregulated levels of AGT mRNA compared with that in sham kidneys. Unexpectedly, the level was increased in PT-specific AGT KO mice and was similar to those of WT and liver-specific AGT KO mice (Supplemental Fig. S3B), suggesting another intrarenal source of AGT mRNA during CKD.
Figure 1.
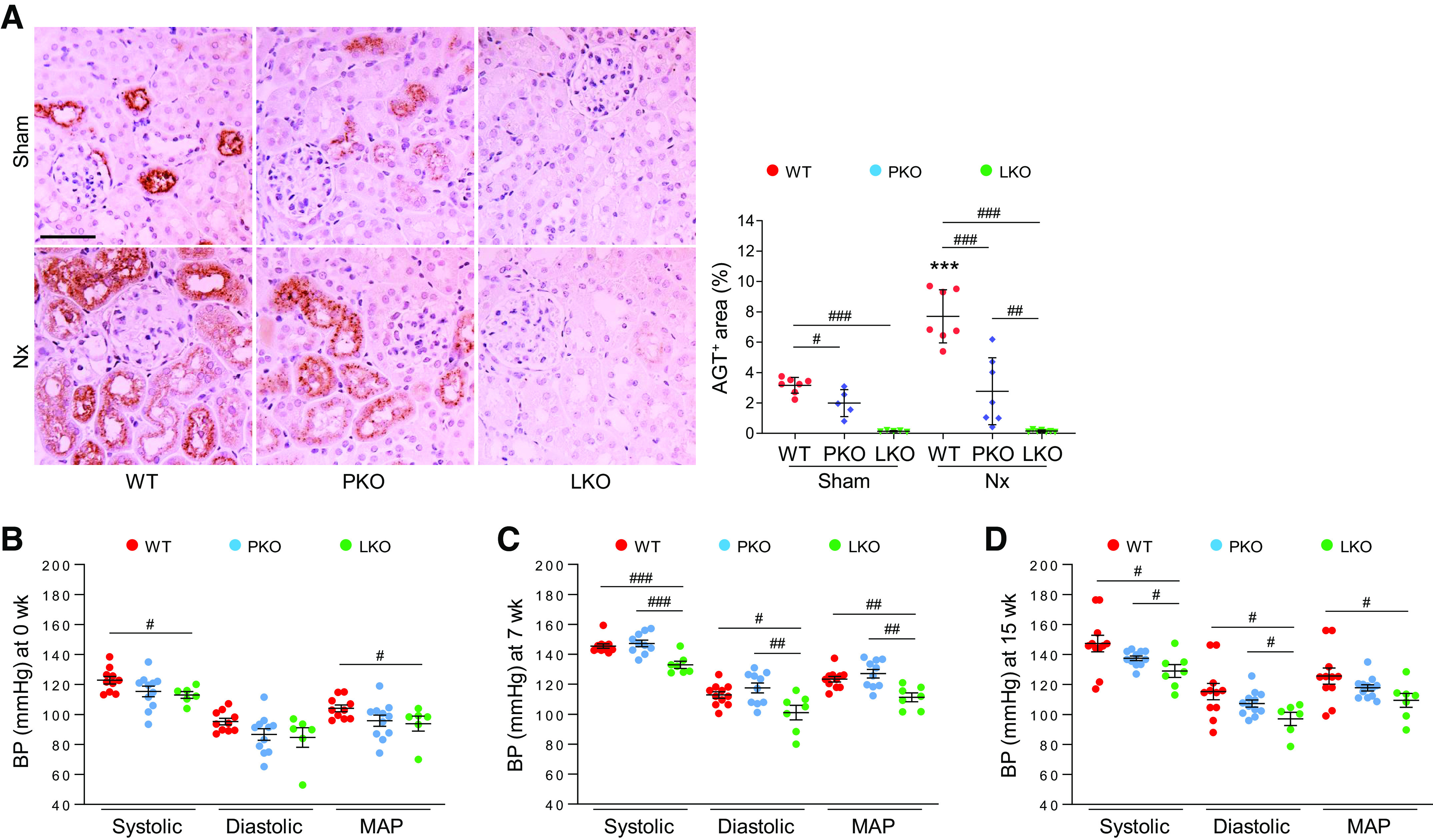
Contribution of tissue-specific angiotensinogen (AGT) in renal AGT expression and its role in blood pressure (BP) regulation during chronic kidney disease. 5/6 Nephrectomy (Nx) in wild-type (WT), proximal tubule-specific AGT knockout (PKO), and liver-specific AGT knockout (LKO) mice was induced by a two-step procedure as described in materials and methods. Mice were harvested 16 wk post-Nx. A: paraffin-embedded kidney sections were used to carry out immunohistochemistry for evaluating AGT. Data were quantified from five randomly chosen fields per kidney using ImageJ software (n = 5–7). Systolic BP, diastolic BP, and mean arterial pressure (MAP) at 0 wk (sham; B), 7 wk (C), and 15 wk (D) were measured using a noninvasive tail cuff system (CODA-2; n = 6–12). Scale bar = 50 µm. Data are expressed as means ± SD. ***P < 0.001 vs. sham; #P < 0.05; ##P < 0.01; and ###P < 0.001.
Liver-Derived AGT Augments BP and Impairs Glomerular and Kidney Function in Nx
CKD is often accompanied by hypertension, which is a critical contributor for the development and progression of CKD (37). To examine the specific effect of liver- or PT-derived AGT in BP regulation, we analyzed BP in liver- and PT-specific AGT KO mice with Nx. Liver-specific AGT KO, but not PT-specific AGT KO, suppressed BP in intact mice (Fig. 1, B–D). Nx significantly elevated BP in WT and PT-specific AGT KO mice, but the increase was significantly suppressed in liver-specific AGT KO mice (Fig. 1, B–D), suggesting a critical role for liver AGT in the increase of BP during CKD. Next, we evaluated glomerular and kidney function using UACR and creatinine clearance. Plasma creatinine levels were elevated in all Nx groups but were not different among groups (Fig. 2A), whereas creatinine clearance markedly declined in WT and PT-specific AGT KO mice but not in liver-specific AGT KO mice after Nx (Fig. 2B). Albuminuria significantly increased in WT and PT-specific AGT KO, but liver-specific AGT KO prevented the Nx-induced increase in albuminuria (Fig. 2C). These data suggest that kidney and glomerular dysfunction in CKD is associated with liver-derived AGT rather than that of the PT.
Figure 2.
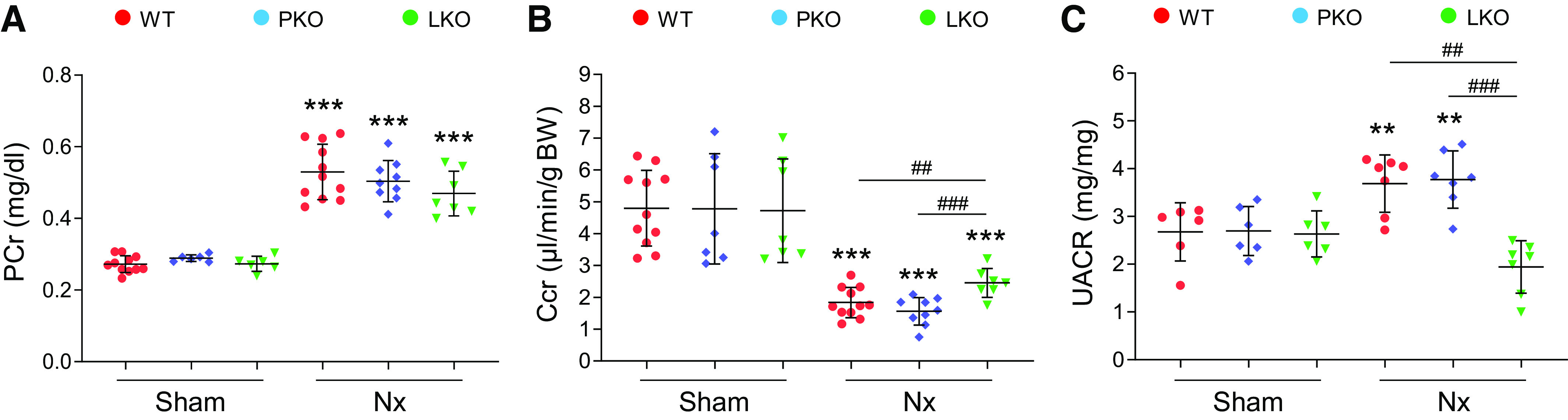
Role of tissue-specific angiotensinogen (AGT) in 5/6 nephrectomy (Nx)-induced kidney and glomerular dysfunction. Kidney and blood samples were collected at 16 wk post-Nx. Mouse urine was collected during 12 h using metabolic cages. Plasma creatinine (PCr; A) and creatinine clearance (Ccr; B), calculated with the formulas described in materials and methods, were used as an index of kidney function (n = 6–10). C: urine albumin-to-creatinine ratio (UACR) was evaluated using ELISA as described in materials and methods and used as an index of glomerular function (n = 6–7). Data are expressed as means ± SD. **P < 0.01 vs. the respective sham; ***P < 0.001 vs. the respective sham; ##P < 0.01; ###P < 0.001. LKO, liver-specific AGT knockout; PKO, proximal tubule-specific AGT knockout; WT, wild type.
Deletion of Liver AGT Primarily Prevents Tubular and Glomerular Injury in CKD
To delineate the role of tissue-derived AGT in glomerular and tubular injury as an initial cause of CKD, we assessed tubular and glomerular injury in WT, PT-specific AGT KO, or liver-specific AGT KO mice subjected to Nx. The severe loss of nephron number after Nx resulted in a large increase of glomerular volume and tubular diameter in WT mice compared with those of sham control mice (Fig. 3, A and B and Supplemental Fig. S4, A and B). Glomerular hypertrophy was decreased in the kidneys of both PT- and liver-specific AGT KO mice, and tubular hypertrophy was decreased in PT-specific AGT KO mice (Fig. 3, A and B and Supplemental Fig. S4, A and B). In addition, tubular atrophy, cast and brush border loss was seen in WT mouse kidneys after Nx (Fig. 3A and Supplemental Fig. S4, A and C). These characteristics were decreased to a greater extent in liver-specific AGT KO mice compared with PT-specific AGT KO mice (Fig. 3A and Supplemental Fig. S4). Most glomeruli in WT mouse kidneys with Nx were segmentally sclerotic and showed an increase in mesangial area, although globally sclerotic glomeruli were also seen (Fig. 3, A and C). After Nx, WT mouse kidneys showed a reduction in the number of WT1-positive podocyte and podocin-positive areas and podocin mRNA levels (Fig. 3, A and D–F). PT- and liver-specific AGT KO suppressed the podocyte loss and injury, resulting in decreased glomerular damage and dysfunction (Figs. 2C and 3, A and D–F). Liver-specific AGT KO was more effective in preventing podocyte injury compared with PT-specific AGT KO (Fig. 3, D–F). These data show that both PT and liver AGT deletion prevent glomerular and tubular damage during the development of CKD, but that PT AGT deletion contributes to a lesser extent.
Figure 3.
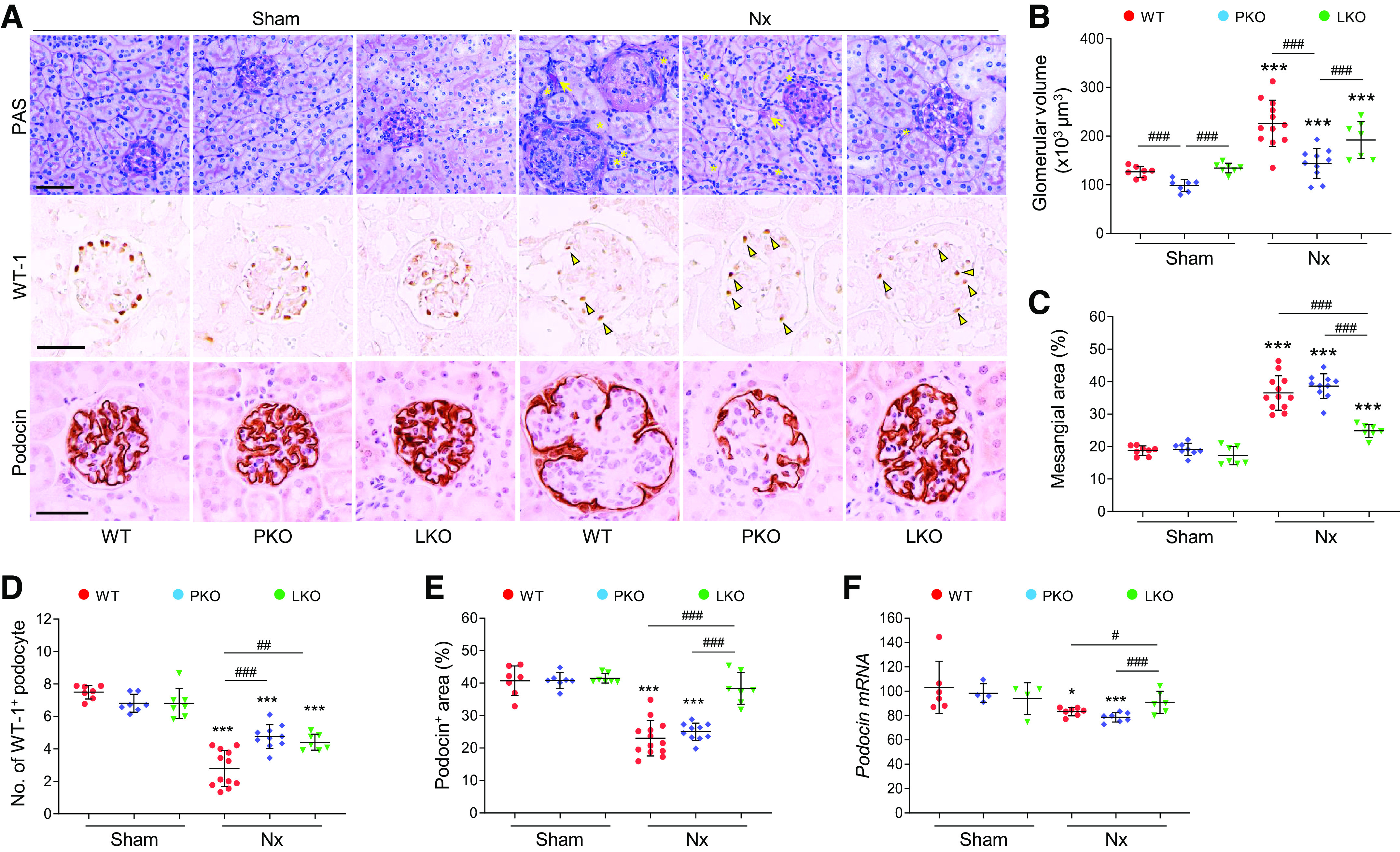
Role of tissue-specific angiotensinogen (AGT) in 5/6 nephrectomy (Nx)-induced glomerular injury. Kidney samples were collected at 16 wk post-Nx. A: paraffin-embedded kidney sections were used to carry out periodic acid-Schiff (PAS) staining and immunohistochemistry for evaluating the number of Wilms’ tumor-1 (WT1)-positive or podocin-positive podocytes. *Atrophied tubule. Arrows indicate tubular casts; arrowheads indicate WT1-positive podocytes. B: glomerular volume was calculated as described in materials and methods. C: mesangial area was expressed as a percentage of the mesangial-to-glomerular surface area. D and E: WT1-positive podocyte numbers and podocin-positive areas were counted from five randomly chosen fields or at least 30 glomeruli per kidney (n = 7–12). F: podocin mRNA levels were evaluated using quantitative RT-PCR and calculated using the formula described in MATERIALS AND METHODS (n = 4–7). Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.05 vs. the respective sham; ***P < 0.001 vs. the respective sham; #P < 0.05; ##P < 0.01; ###P < 0.001. LKO, liver-specific AGT knockout; PKO, proximal tubule-specific AGT knockout; WT, wild type.
Next, to examine the role of liver- versus PT-derived AGT in glomerular basement membrane (GBM) thickening and podocyte foot process effacement, we evaluated glomerular ultrastructure in Nx mouse kidneys using electron microscopy. Nx resulted in increased GBM thickness and podocyte foot process length in WT kidneys (Fig. 4 and Supplemental Fig. S5), indicating podocyte dysfunction. These increases were markedly suppressed in liver-specific AGT KO mice, but only to a marginal extent in PT-specific AGT KO mice (Fig. 4), demonstrating that liver-derived AGT primarily contributes to podocyte injury, a critical cause of albuminuria.
Figure 4.
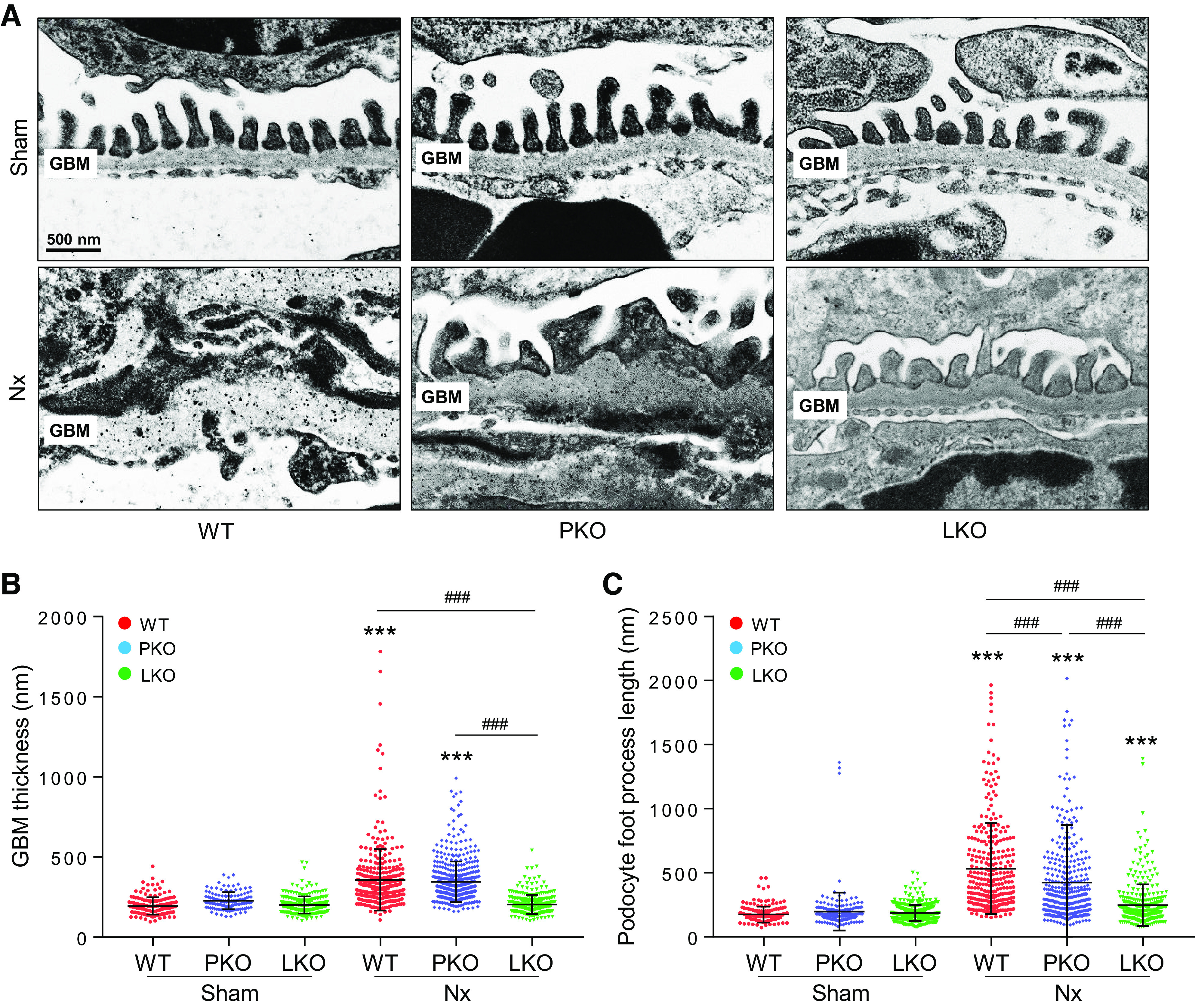
Effect of tissue-specific angiotensinogen (AGT) in glomerular basement membrane (GBM) thickening and foot process effacement in post-5/6 nephrectomy (Nx) kidneys. Kidney samples were collected at 16 wk post-Nx. A: electron microscopy was used to evaluate GBM thickening and foot process length. GBM thickness (B) and podocyte foot process length (C) were measured in at least 50 randomly chosen areas per kidney (n = 3–5). Scale bars = 500 nm. Data are expressed as means ± SD. ***P < 0.001 vs. the respective sham; ###P < 0.001. LKO, liver-specific AGT knockout; PKO, proximal tubule-specific AGT knockout; WT, wild type.
Blockade of PT- and Liver-Derived AGT Suppresses Glomerular and Interstitial Fibrosis in Nx Mouse Kidneys
Kidney fibrosis is a common characteristic of CKD and has been shown to serve as a main cause and effect of CKD (38). To elucidate the roles of PT and liver AGT in fibrosis progression in CKD, we checked both glomerular and interstitial fibrosis using a glomerulosclerosis index, Sirius red stain, and immunohistochemistry against α-SMA, a marker of myofibroblasts, and quantitative evaluation of fibrotic cytokines. The glomerulosclerosis index was highly increased in WT mice with Nx, but both liver- and PT-specific AGT KO diminished the injury level. The decrease was greater in liver-specific AGT KO than PT-specific AGT KO (Figs. 3A and 5, A and B). In sham mice, both PT- and liver-specific AGT KO did not affect collagen deposition in glomeruli, as evaluated by Sirius red staining (Fig. 5, A and C). After Nx, collagen deposition in glomerular regions was highly increased in WT mouse kidneys (Fig. 5, A and C). The deposition was suppressed in liver-specific AGT KO compared with WT and PT-specific AGT KO (Fig. 5, A and C). The Nx-induced increase of α-SMA-positive areas was mitigated in glomeruli of both PT- and liver-specific AGT KO mouse kidneys, but the effect was significantly greater in liver-specific AGT KO mouse kidneys (Fig. 5, A and D), indicating a major contribution of liver-derived AGT in glomerulosclerosis.
Figure 5.
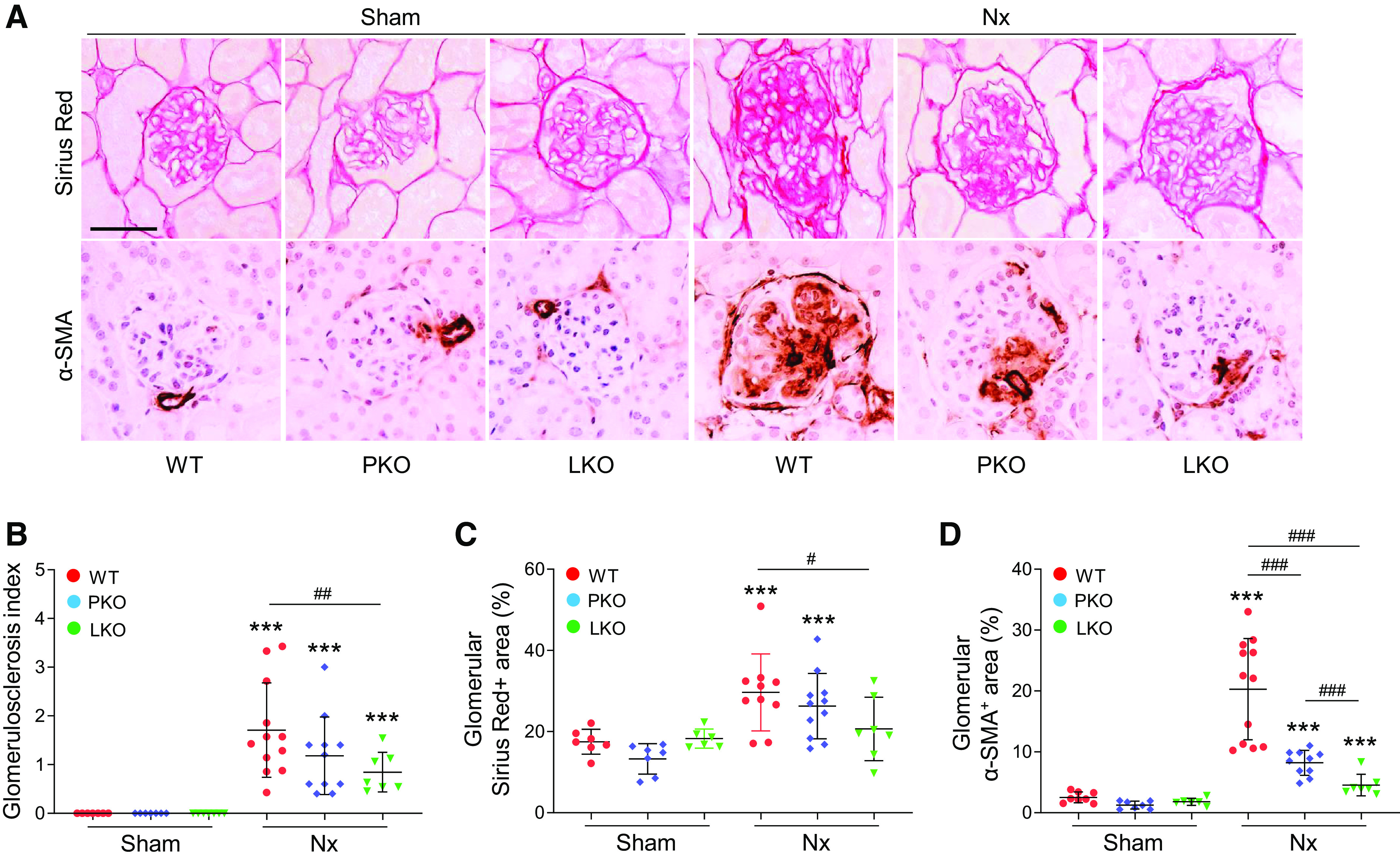
Role of tissue-specific angiotensinogen (AGT) in 5/6 nephrectomy (Nx)-induced glomerulosclerosis. Kidney samples were collected at 16 wk post-Nx. A: paraffin-embedded kidney sections were used to carry out periodic acid-Schiff staining, Sirius red staining, and immunohistochemistry for evaluating the α-smooth muscle actin (α-SMA)-positive area. B: the glomerulosclerosis index was evaluated in at least 30 randomly chosen glomeruli per group, based on a modified semiquantitative scoring system as described in materials and methods. C and D: Sirius red- and α-SMA-positive areas were measured in at least 30 randomly chosen areas per kidney (n = 6–11). Scale bars = 50 µm. Data are expressed as means ± SD. ***P < 0.001 vs. the respective sham; #P < 0.05; ##P < 0.01; and ###P < 0.001. LKO, liver-specific AGT knockout; PKO, proximal tubule-specific AGT knockout; WT, wild type.
Similar to glomerular fibrosis progression, interstitial fibrosis, as evaluated by interstitial collagen deposition and immunostaining for interstitial fibrosis-related factors (38), was significantly upregulated in WT mouse kidneys with Nx (Fig. 6 and Supplemental Fig. S6). The fibrosis progression was mitigated by PT- and liver-specific AGT KO and the beneficial effect was comparable (Fig. 6). In addition, the interstitial α-SMA-positive area and its quantification, as well as fibrogenic molecules, connective tissue growth factor (CTGF) and transforming growth factor (TGF)-β, were markedly increased in the kidney after Nx (Fig. 6). PT- and liver-specific AGT KO suppressed interstitial fibrosis in the kidneys of Nx mice compared with kidneys of WT mice. The protective effect was similar in both groups, but some of the fibrogenic factor Ctgf was lower in liver-specific AGT KO than PT-specific AGT KO (Fig. 6), suggesting that PT- and liver-derived AGT can similarly contribute to interstitial fibrosis progression. Expression of PDGFRβ, which is expressed in mesangial cells, glomerular parietal cells, and interstitial fibroblasts/pericytes (38, 39), and collagen type IV, which is expressed in glomerular and tubular basement membranes (40), showed a similar pattern with the fibrosis-related markers (Fig. 6, A, C, and D). Together, these results indicate that PT- and liver-derived AGT distinctly contribute to region-specific fibrosis progression in CKD.
Figure 6.
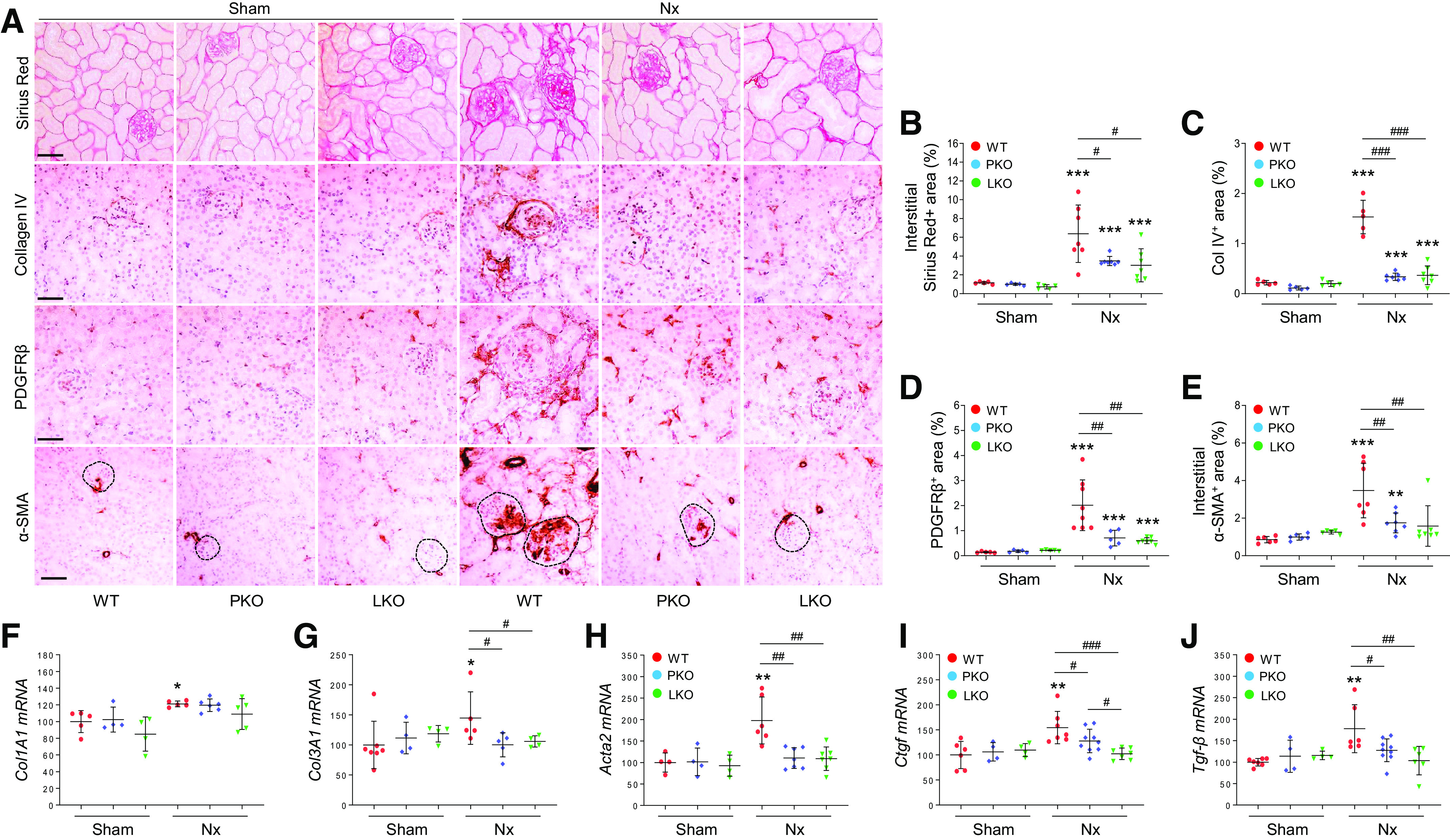
Role of tissue-specific angiotensinogen (AGT) in 5/6 nephrectomy (Nx)-induced interstitial fibrosis progression. Kidney samples were collected at 16 wk post-Nx. A: paraffin-embedded kidney sections were used to carry out Sirius red staining and immunohistochemistry for evaluating collagen type IV (Col IV)-, platelet-derived growth factor receptor-β (PDGFRβ)-, and α-smooth muscle actin (α-SMA)-positive areas. Circles with dashed lines indicate glomeruli. B–E: data quantified from five randomly chosen fields per kidney (n = 5–7). Collagen type I-α1 (Col1A1; F), collagen type III-α1 (Col3A1; G), α-SMA (Acta2; H), connective tissue growth factor (Ctgf; I), and transforming growth factor-β (Tgf-β; J) mRNA levels were evaluated using quantitative RT-PCR and calculated using the formula described in materials and methods (n = 4–9). Scale bars = 50 µm. Data are expressed as means ± SD. *P < 0.01 vs. the respective sham; **P < 0.01 vs. the respective sham; ***P < 0.001 vs. the respective sham; #P < 0.05; ##P < 0.01; and ###P < 0.001. LKO, liver-specific AGT knockout; PKO, proximal tubule-specific AGT knockout; WT, wild type.
DISCUSSION
Our present study demonstrates that 1) liver AGT is a primary contributor of augmented intrarenal AGT protein and hypertension in CKD; 2) liver-specific AGT KO suppresses kidney and glomerular injury and dysfunction, GBM thickening, and podocyte foot process effacement during CKD; 3) hepatic AGT KO, but to a lesser degree than PT-specific AGT KO, blunts glomerulosclerosis in CKD mice; and 4) both PT- and liver-specific AGT KO ameliorate interstitial fibrosis progression in mouse kidneys with Nx. These findings provide insights into the development of renin-angiotensin system (RAS)-based treatment for CKD.
The RAS is an important target for treating chronic diseases, including cardiovascular diseases and CKD. A number of experimental and clinical studies have shown that inhibition of RAS components, such as ACE and AT1R inhibitors, is beneficial to treat CKD (9). Most inhibitors of RAS components have a BP-lowering effect, but the effect on glomerular and tubular fibrosis and dysfunction can vary depending on the type and stage of CKD (41–43). However, the molecular mechanism of how RAS components contribute to CKD development and progression is not fully understood. Matsusaka et al. (18, 24) demonstrated that intrarenal AGT protein originates from the liver in normal mouse kidneys and that protein accumulation increases after podocyte injury. In the present study, we sought to identify the tissue-specific role of AGT in CKD progression. It was shown that liver AGT-targeted siRNA and antisense oligonucleotide (ASO) prevent hypertension and renal injury (44–47). However, unconjugated siRNA and ASO could downregulate AGT in other organs, including the kidney and adipocyte (48), and thus the distinct tissue-specific effects of AGT on hypertension or renal injury were not clarified. The use of tissue-specific AGT deletion has a benefit to overcome this limitation. In the present study, we showed that liver-specific AGT KO inhibits the increase of BP and intrarenal AGT expression and prevents kidney and glomerular injury, dysfunction, and fibrogenesis in the kidney after Nx. On the other hand, PT-specific AGT KO did not affect the Nx-induced increase of BP and partially suppressed AGT expression. Interestingly, PT-specific AGT KO suppresses only interstitial fibrosis progression, to a similar level to liver-specific AGT KO. Given that AGT deletion in the kidney does not affect the plasma AGT level (18), renal-derived AGT may contribute to the progression through presumably diffusion into the interstitial space and not via circulation (18, 48, 49). These results indicate that regardless of the origin, AGT can contribute to interstitial fibrosis progression during CKD. Although the mechanism by which liver-derived AGT can induce both glomerular and interstitial injury while PT-derived AGT induces only interstitial fibrosis is not defined, we propose that the circulating AGT, mainly derived from the liver, will get deposited in the glomeruli during filtration and contribute to glomerular injury and dysfunction during CKD.
It is suggested that podocyte injury, depletion, and subsequent dysfunction are the main causes of glomerulosclerosis and proteinuria in diabetic and nondiabetic CKD. Furthermore, the podocyte injury is renal ANG II-dependent and can be attenuated by AT1R blockade (50–52). As liver-derived AGT is a determinant of the intrarenal ANG II level, its targeting results in a reduction of the intrarenal ANG II level (18, 24, 45, 47). Our data indicate that liver AGT, but not PT AGT, is critical for podocyte injury and subsequent glomerular dysfunction and fibrosis progression. It has been shown that liver-derived AGT activates podocytes and mesangial cells for both cell death and fibrotic signaling via an ANG II-dependent mechanism (18, 24, 53–55), which, in turn, leads to damage in the podocyte ultrastructure, such as podocyte foot process effacement. In this regard, liver-specific AGT KO may result in a suppression of UACR in Nx mice through the mitigation of albumin excretion into the urine, despite similar levels of plasma creatinine among groups. Our data also suggest that since PT-specific AGT KO did not affect the Nx-induced BP increase, PT-derived AGT has a BP-independent role in suppressing interstitial fibrosis during CKD progression, including downregulation of fibrogenic factors CTGF and TGF-β that are secreted by injured tubules (56). However, we could not rule out the BP-independent effect of liver AGT, because RAS blockers, such as AT1R blocker, have additional effects beyond improving hypertension on albuminuria during chronic heart failure (57). Our data would suggest the notion that regulation of intrarenal AGT, regardless of its origin, would be critical for better outcomes in CKD.
It has been shown that the increase of proteinuria/albuminuria by podocyte injury, which can activate profibrotic signaling in the PT or cortical collecting duct, results in the progression of interstitial fibrosis (58–62). Conversely, PT injury can trigger both glomerulosclerosis and interstitial fibrosis progression. Targeted PT injury by diphtheria toxin results in glomerular pathology, including glomerulosclerosis and atubular glomeruli, which is a cause of tubular atrophy and interstitial fibrosis (63–65). Prolonged tubular injury and its consequences, including interstitial fibrosis, are likely to cause interstitial capillary rarefaction that can lead to secondary glomerulosclerosis, thus ultimately progressing to end-stage kidney disease (63, 66, 67). This may explain why our present data show that PT-specific AGT KO mildly ameliorated glomerular collagen deposition and fibroblast activation and podocyte damage compared with WT. Furthermore, it is possible that increased protein uptake by PTs causes progressive renal damage through modified tubular cell death mechanism, phenotypic change, and fibrogenic pathway activation (68–70). Moreover, there seems to be a reciprocal communication between injured podocytes and PTs as simultaneous injury worsens podocyte injury, proteinuria, and glomerulosclerosis compared with injury on podocytes only (64).
AGT that is expressed in podocytes and mesangial cells, which is derived from circulating AGT, is upregulated in glomerular diseases and CKD (17, 71, 72). ANG II that is newly formed in glomeruli from circulating AGT causes podocyte damage as well as mesangial cell proliferation and activation to secrete collagen and extracellular matrix through signaling cascades, e.g., MAPK signaling and transient receptor potential cation channel subfamily C member 6 (51, 72–74). However, ANG II in tubules and the interstitium is primarily involved in tubular atrophy and secretion of proinflammatory and fibrotic factors (75). The ANG II-dependent mechanism would be depending on its receptor signaling; ANG II signaling via AT1R has procell death, proinflammatory, and profibrotic roles, whereas that via the ANG II type 2 receptor has opposing roles in CKD (75). Further study would be necessary to define the molecular mechanism of accumulated intrarenal AGT and the cell type-specific role of RAS components that triggers kidney injury or repair in CKD as well as the distinct molecular mechanism between liver and PT AGT in glomerular and interstitial injury and fibrosis progression.
Our study has certain limitations. In line with Matsusaka et al.’s report (18), we found that liver-specific AGT KO mice are polyuric and hypotensive as well as have mild hyperplasia in intrarenal arteries at baseline but no major developmental issues, including growth retardation or changes in body weight and kidney size in both liver- and PT-specific AGT KO. We could not rule out an effect of the first-line differences in 5/6 Nx CKD. However, based on the literature (44–47) and our present findings, targeting liver AGT by specific gene KO or a siRNA approach shows that liver-derived AGT can contribute to both BP-dependent and -independent kidney and glomerular dysfunction and fibrosis progression. Another limitation is that PT-specific AGT KO demonstrated an increase of AGT mRNA in the kidneys of Nx mice. It may be associated with other tubular segments, such as the medullary thick ascending limb, glomerular epithelial cells, and mesangial cells, which express AGT mRNA that can be upregulated in diseased kidneys, including diabetic nephropathy (53, 76). In the absence of AGT from PTs, it is possible that other tubular segments might compensate for the downregulation in PTs. However, based on our findings, it would not be a major contributor in glomerular and kidney dysfunction and fibrosis progression in CKD. PT-specific AGT KO also showed decreased glomerular and tubular hypertrophy in the kidneys of Nx mice but seemed not to be associated with a protective effect on CKD progression, as PT-specific AGT KO resulted in a greater mesangial area and/or interstitial fibrosis compared with WT and/or liver-specific AGT KO, suggesting a distinct mechanism between WT and PT-specific AGT KO in CKD pathogenesis. Further study would be required to dissect the detailed mechanism.
Taken together, our findings demonstrate distinct roles of tissue-specific AGT in glomerular and kidney dysfunction and fibrosis progression during CKD; liver-derived AGT, but not PT-derived AGT, via activation of an ANG II-dependent mechanism, induces podocyte loss and damage, e.g., foot process effacement, and mesangial cell expansion, resulting in glomerular dysfunction and glomerulosclerosis. However, PT-derived AGT induces interstitial fibrosis that is comparable with liver-derived AGT. In addition, the data regarding intrarenal AGT accumulation during CKD progression may provide a rationale for developing an AGT-based biomarker for diagnosing CKD progression. Our data would provide not only clues to overcome limitations of efficacy of current RAS-based drugs for CKD treatment but also new avenues to develop more effective therapeutics for RAS-dependent CKD.
Perspectives and Significance
Our data demonstrate that hepatic and PT AGT have distinct roles in kidney and glomerular injury and dysfunction and fibrogenesis, suggesting that tissue-specific targeting of AGT in the liver and PT may be a more efficacious therapeutic strategy for CKD. Our findings may be extrapolated to understand mechanisms of the systemic versus local RAS in diseases, which could be affected by both BP-dependent and -independent effects, including cardiovascular diseases and fibroproliferative disorders in other tissues.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S6 and Supplemental Table S1: https://doi.org/10.6084/m9.figshare.19134965.v4.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK116987, DK120533, and DK120846 (to B.J.P.) and by American Heart Association Postdoctoral Fellowship 15POST25130003 (to H.S.J.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H-S.J. and B.J.P. conceived and designed research; H-S.J. and M.R.N. performed experiments; H-S.J., and M.R.N. analyzed data; H-S.J., T.P., K.L., J.C.H., and B.J.P. interpreted results of experiments; H-S.J. prepared figures; H-S.J. drafted manuscript; H-S.J., F.A.F., and B.J.P. edited and revised manuscript; H-S.J., M.R.N., T.P., K.L., J.C.H., F.A.F., and B.J.P. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Volker H. Haase (Vanderbilt University) for providing the transgenic mouse with the phosphoenolpyruvate carboxykinase-Cre recombinase and the University of Alabama at Birmingham-University of California-San Diego O’Brien Center for Acute Kidney Injury Research for measuring urine creatinine and albumin.
REFERENCES
- 1. Gansevoort RT, Correa-Rotter R, Hemmelgarn BR, Jafar TH, Heerspink HJ, Mann JF, Matsushita K, Wen CP. Chronic kidney disease and cardiovascular risk: epidemiology, mechanisms, and prevention. Lancet 382: 339–352, 2013. doi: 10.1016/S0140-6736(13)60595-4. [DOI] [PubMed] [Google Scholar]
- 2. Di Lullo L, House A, Gorini A, Santoboni A, Russo D, Ronco C. Chronic kidney disease and cardiovascular complications. Heart Fail Rev 20: 259–272, 2015. doi: 10.1007/s10741-014-9460-9. [DOI] [PubMed] [Google Scholar]
- 3. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 351: 1296–1305, 2004. [Erratum in N Engl J Med 18: 4, 2008]. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 4. Romagnani P, Remuzzi G, Glassock R, Levin A, Jager KJ, Tonelli M, Massy Z, Wanner C, Anders HJ. Chronic kidney disease. Nat Rev Dis Primers 3: 17088, 2017. doi: 10.1038/nrdp.2017.88. [DOI] [PubMed] [Google Scholar]
- 5. Webster AC, Nagler EV, Morton RL, Masson P. Chronic kidney disease. Lancet 389: 1238–1252, 2017. doi: 10.1016/S0140-6736(16)32064-5. [DOI] [PubMed] [Google Scholar]
- 6. Locatelli F, Pozzoni P, Del Vecchio L. Renal manifestations in the metabolic syndrome. J Am Soc Nephrol 17: S81–S85, 2006. doi: 10.1681/ASN.2005121332. [DOI] [PubMed] [Google Scholar]
- 7. Prasad GV. Metabolic syndrome and chronic kidney disease: current status and future directions. World J Nephrol 3: 210–219, 2014. doi: 10.5527/wjn.v3.i4.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas G, Sehgal AR, Kashyap SR, Srinivas TR, Kirwan JP, Navaneethan SD. Metabolic syndrome and kidney disease: a systematic review and meta-analysis. Clin J Am Soc Nephrol 6: 2364–2373, 2011. doi: 10.2215/CJN.02180311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ruggenenti P, Cravedi P, Remuzzi G. Mechanisms and treatment of CKD. J Am Soc Nephrol 23: 1917–1928, 2012. doi: 10.1681/ASN.2012040390. [DOI] [PubMed] [Google Scholar]
- 10. Appel LJ, Wright JT Jr, Greene T, Agodoa LY, Astor BC, Bakris GL, Cleveland WH, Charleston J, Contreras G, Faulkner ML, Gabbai FB, Gassman JJ, Hebert LA, Jamerson KA, Kopple JD, Kusek JW, Lash JP, Lea JP, Lewis JB, Lipkowitz MS, Massry SG, Miller ER, Norris K, Phillips RA, Pogue VA, Randall OS, Rostand SG, Smogorzewski MJ, Toto RD, Wang X; for the AASK Collaborative Research Group. Intensive blood-pressure control in hypertensive chronic kidney disease. N Engl J Med 363: 918–929, 2010. doi: 10.1056/NEJMoa0910975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muntner P, Anderson A, Charleston J, Chen Z, Ford V, Makos G, O'Connor A, Perumal K, Rahman M, Steigerwalt S, Teal V, Townsend R, Weir M, Wright JT Jr; Chronic Renal Insufficiency Cohort Study Investigators. Hypertension awareness, treatment, and control in adults with CKD: results from the Chronic Renal Insufficiency Cohort (CRIC) Study. Am J Kidney Dis 55: 441–451, 2010. doi: 10.1053/j.ajkd.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sharma P, Blackburn RC, Parke CL, McCullough K, Marks A, Black C. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers for adults with early (stage 1 to 3) non-diabetic chronic kidney disease. Cochrane Database Syst Rev 10: CD007751, 2011.doi: 10.1002/14651858.CD007751.pub2. [DOI] [PubMed] [Google Scholar]
- 13. Sinha AD, Agarwal R. Clinical pharmacology of antihypertensive therapy for the treatment of hypertension in CKD. Clin J Am Soc Nephrol 14: 757–764, 2019. doi: 10.2215/CJN.04330418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mills KT, Kobori H, Hamm LL, Alper AB, Khan IE, Rahman M, Navar LG, Liu Y, Browne GM, Batuman V, He J, Chen J. Increased urinary excretion of angiotensinogen is associated with risk of chronic kidney disease. Nephrol Dial Transplant 27: 3176–3181, 2012. doi: 10.1093/ndt/gfs011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kobori H, Harrison-Bernard LM, Navar LG. Expression of angiotensinogen mRNA and protein in angiotensin II-dependent hypertension. J Am Soc Nephrol 12: 431–439, 2001. doi: 10.1681/ASN.V123431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kobori H, Alper AB Jr, Shenava R, Katsurada A, Saito T, Ohashi N, Urushihara M, Miyata K, Satou R, Hamm LL, Navar LG. Urinary angiotensinogen as a novel biomarker of the intrarenal renin-angiotensin system status in hypertensive patients. Hypertension 53: 344–350, 2009. doi: 10.1161/HYPERTENSIONAHA.108.123802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yamamoto T, Nakagawa T, Suzuki H, Ohashi N, Fukasawa H, Fujigaki Y, Kato A, Nakamura Y, Suzuki F, Hishida A. Urinary angiotensinogen as a marker of intrarenal angiotensin II activity associated with deterioration of renal function in patients with chronic kidney disease. J Am Soc Nephrol 18: 1558–1565, 2007. doi: 10.1681/ASN.2006060554. [DOI] [PubMed] [Google Scholar]
- 18. Matsusaka T, Niimura F, Shimizu A, Pastan I, Saito A, Kobori H, Nishiyama A, Ichikawa I. Liver angiotensinogen is the primary source of renal angiotensin II. J Am Soc Nephrol 23: 1181–1189, 2012. doi: 10.1681/ASN.2011121159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Navar LG. Translational studies on augmentation of intratubular renin–angiotensin system in hypertension. Kidney Int Suppl (2011) 3: 321–325, 2013. doi: 10.1038/kisup.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pohl M, Kaminski H, Castrop H, Bader M, Himmerkus N, Bleich M, Bachmann S, Theilig F. Intrarenal renin angiotensin system revisited: role of megalin-dependent endocytosis along the proximal nephron. J Biol Chem 285: 41935–41946, 2010. doi: 10.1074/jbc.M110.150284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nielsen R, Christensen EI, Birn H. Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int 89: 58–67, 2016. doi: 10.1016/j.kint.2015.11.007. [DOI] [PubMed] [Google Scholar]
- 22. Gonzalez-Villalobos R, Klassen RB, Allen PL, Navar LG, Hammond TG. Megalin binds and internalizes angiotensin II. Am J Physiol Renal Physiol 288: F420–F427, 2005. doi: 10.1152/ajprenal.00243.2004. [DOI] [PubMed] [Google Scholar]
- 23. Koizumi M, Ueda K, Niimura F, Nishiyama A, Yanagita M, Saito A, Pastan I, Fujita T, Fukagawa M, Matsusaka T. Podocyte injury augments intrarenal angiotensin II generation and sodium retention in a megalin-dependent manner. Hypertension 74: 509–517, 2019. doi: 10.1161/HYPERTENSIONAHA.118.12352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsusaka T, Niimura F, Pastan I, Shintani A, Nishiyama A, Ichikawa I. Podocyte injury enhances filtration of liver-derived angiotensinogen and renal angiotensin II generation. Kidney Int 85: 1068–1077, 2014. doi: 10.1038/ki.2013.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rankin EB, Tomaszewski JE, Haase VH. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res 66: 2576–2583, 2006. doi: 10.1158/0008-5472.CAN-05-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Souza AC, Tsuji T, Baranova IN, Bocharov AV, Wilkins KJ, Street JM, Alvarez-Prats A, Hu X, Eggerman T, Yuen PS, Star RA. TLR4 mutant mice are protected from renal fibrosis and chronic kidney disease progression. Physiol Rep 3: e12558, 2015. doi: 10.14814/phy2.12558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang X, Chaudhry MA, Nie Y, Xie Z, Shapiro JI, Liu J. A mouse 5/6th nephrectomy model that induces experimental uremic cardiomyopathy. J Vis Exp 55825, 2017. doi: 10.3791/55825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jang HS, Kim JI, Kim J, Na YK, Park JW, Park KM. Bone marrow derived cells and reactive oxygen species in hypertrophy of contralateral kidney of transient unilateral renal ischemia-induced mouse. Free Radic Res 46: 903–911, 2012. doi: 10.3109/10715762.2012.686664. [DOI] [PubMed] [Google Scholar]
- 29. Noh MR, Kong MJ, Han SJ, Kim JI, Park KM. Isocitrate dehydrogenase 2 deficiency aggravates prolonged high-fat diet intake-induced hypertension. Redox Biol 34: 101548, 2020. doi: 10.1016/j.redox.2020.101548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jang HS, Padanilam BJ. Simultaneous deletion of Bax and Bak is required to prevent apoptosis and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 309: F540–F550, 2015. doi: 10.1152/ajprenal.00170.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jang HS, Noh MR, Jung EM, Kim WY, Southekal S, Guda C, Foster KW, Oupicky D, Ferrer FA, Padanilam BJ. Proximal tubule cyclophilin D regulates fatty acid oxidation in cisplatin-induced acute kidney injury. Kidney Int 97: 327–339, 2020. doi: 10.1016/j.kint.2019.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jang HS, Noh MR, Ha L, Kim J, Padanilam BJ. Proximal tubule cyclophilin D mediates kidney fibrogenesis in obstructive nephropathy. Am J Physiol Renal Physiol 321: F431–F442, 2021. doi: 10.1152/ajprenal.00171.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, Shankland SJ, D'Agati VD, Duffield JS. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol 26: 1040–1052, 2015. doi: 10.1681/ASN.2013111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hong Q, Zhang L, Das B, Li Z, Liu B, Cai G, Chen X, Chuang PY, He JC, Lee K. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int 93: 1330–1343, 2018. doi: 10.1016/j.kint.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim J, Padanilam BJ. Renal denervation prevents long-term sequelae of ischemic renal injury. Kidney Int 87: 350–358, 2015. doi: 10.1038/ki.2014.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jang HS, Kim JI, Noh M, Rhee MH, Park KM. Regulator of G protein signaling 2 (RGS2) deficiency accelerates the progression of kidney fibrosis. Biochim Biophys Acta 1842: 1733–1741, 2014. doi: 10.1016/j.bbadis.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 37. Horowitz B, Miskulin D, Zager P. Epidemiology of hypertension in CKD. Adv Chronic Kidney Dis 22: 88–95, 2015. doi: 10.1053/j.ackd.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 38. Ruiz-Ortega M, Rayego-Mateos S, Lamas S, Ortiz A, Rodrigues-Diez RR. Targeting the progression of chronic kidney disease. Nat Rev Nephrol 16: 269–288, 2020. doi: 10.1038/s41581-019-0248-y. [DOI] [PubMed] [Google Scholar]
- 39. Buhl EM, Djudjaj S, Klinkhammer BM, Ermert K, Puelles VG, Lindenmeyer MT, Cohen CD, He C, Borkham-Kamphorst E, Weiskirchen R, Denecke B, Trairatphisan P, Saez-Rodriguez J, Huber TB, Olson LE, Floege J, Boor P. Dysregulated mesenchymal PDGFR-beta drives kidney fibrosis. EMBO Mol Med 12: e11021, 2020. doi: 10.15252/emmm.201911021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bulow RD, Boor P. Extracellular matrix in kidney fibrosis: more than just a Scaffold. J Histochem Cytochem 67: 643–661, 2019. doi: 10.1369/0022155419849388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie X, Liu Y, Perkovic V, Li X, Ninomiya T, Hou W, Zhao N, Liu L, Lv J, Zhang H, Wang H. Renin–angiotensin system inhibitors and kidney and cardiovascular outcomes in patients with CKD: a Bayesian network meta-analysis of randomized clinical trials. Am J Kidney Dis 67: 728–741, 2016. doi: 10.1053/j.ajkd.2015.10.011. [DOI] [PubMed] [Google Scholar]
- 42. Chung EY, Ruospo M, Natale P, Bolignano D, Navaneethan SD, Palmer SC, Strippoli GF. Aldosterone antagonists in addition to renin angiotensin system antagonists for preventing the progression of chronic kidney disease. Cochrane Database Syst Rev 10: CD007004, 2020. doi: 10.1002/14651858.CD007004.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mishima E, Haruna Y, Arima H. Renin–angiotensin system inhibitors in hypertensive adults with non-diabetic CKD with or without proteinuria: a systematic review and meta-analysis of randomized trials. Hypertens Res 42: 469–482, 2019. doi: 10.1038/s41440-018-0116-3. [DOI] [PubMed] [Google Scholar]
- 44. Mullick AE, Yeh ST, Graham MJ, Engelhardt JA, Prakash TP, Crooke RM. Blood pressure lowering and safety improvements with liver angiotensinogen inhibition in models of hypertension and kidney injury. Hypertension 70: 566–576, 2017. doi: 10.1161/HYPERTENSIONAHA.117.09755. [DOI] [PubMed] [Google Scholar]
- 45. Uijl E, Mirabito Colafella KM, Sun Y, Ren L, van Veghel R, Garrelds IM, de Vries R, Poglitsch M, Zlatev I, Kim JB, Hoorn EJ, Foster D, Danser AHJ. Strong and sustained antihypertensive effect of small interfering RNA targeting liver angiotensinogen. Hypertension 73: 1249–1257, 2019. doi: 10.1161/HYPERTENSIONAHA.119.12703. [DOI] [PubMed] [Google Scholar]
- 46. Olearczyk J, Gao S, Eybye M, Yendluri S, Andrews L, Bartz S, Cully D, Tadin-Strapps M. Targeting of hepatic angiotensinogen using chemically modified siRNAs results in significant and sustained blood pressure lowering in a rat model of hypertension. Hypertens Res 37: 405–412, 2014. doi: 10.1038/hr.2013.155. [DOI] [PubMed] [Google Scholar]
- 47. Bovee DM, Ren L, Uijl E, Clahsen-van Groningen MC, van Veghel R, Garrelds IM, Domenig O, Poglitsch M, Zlatev I, Kim JB, Huang S, Melton L, Lu X, Hoorn EJ, Foster D, Danser AHJ. Renoprotective effects of small interfering RNA targeting liver angiotensinogen in experimental chronic kidney disease. Hypertension 77: 1600–1612, 2021. doi: 10.1161/HYPERTENSIONAHA.120.16876. [DOI] [PubMed] [Google Scholar]
- 48. Arendse LB, Danser AHJ, Poglitsch M, Touyz RM, Burnett JC, Jr., Llorens-Cortes C, Ehlers MR, Sturrock ED. Novel therapeutic approaches targeting the renin–angiotensin system and associated peptides in hypertension and heart failure. Pharmacol Rev 71: 539–570, 2019. doi: 10.1124/pr.118.017129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Lannoy LM, Danser AH, van Kats JP, Schoemaker RG, Saxena PR, Schalekamp MA. Renin-angiotensin system components in the interstitial fluid of the isolated perfused rat heart. Local production of angiotensin I. Hypertension 29: 1240–1251, 1997. doi: 10.1161/01.HYP.29.6.1240. [DOI] [PubMed] [Google Scholar]
- 50. Fukuda A, Wickman LT, Venkatareddy MP, Sato Y, Chowdhury MA, Wang SQ, Shedden KA, Dysko RC, Wiggins JE, Wiggins RC. Angiotensin II-dependent persistent podocyte loss from destabilized glomeruli causes progression of end stage kidney disease. Kidney Int 81: 40–55, 2012. doi: 10.1038/ki.2011.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nijenhuis T, Sloan AJ, Hoenderop JG, Flesche J, van Goor H, Kistler AD, Bakker M, Bindels RJ, de Boer RA, Moller CC, Hamming I, Navis G, Wetzels JF, Berden JH, Reiser J, Faul C, van der Vlag J. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol 179: 1719–1732, 2011. doi: 10.1016/j.ajpath.2011.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kang JS, Lee SJ, Lee JH, Kim JH, Son SS, Cha SK, Lee ES, Chung CH, Lee EY. Angiotensin II-mediated MYH9 downregulation causes structural and functional podocyte injury in diabetic kidney disease. Sci Rep 9: 7679, 2019. doi: 10.1038/s41598-019-44194-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lai KN, Leung JC, Lai KB, To WY, Yeung VT, Lai FM. Gene expression of the renin–angiotensin system in human kidney. J Hypertens 16: 91–102, 1998. doi: 10.1097/00004872-199816010-00014. [DOI] [PubMed] [Google Scholar]
- 54. Fern RJ, Yesko CM, Thornhill BA, Kim HS, Smithies O, Chevalier RL. Reduced angiotensinogen expression attenuates renal interstitial fibrosis in obstructive nephropathy in mice. J Clin Invest 103: 39–46, 1999. doi: 10.1172/JCI4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Arai M, Wada A, Isaka Y, Akagi Y, Sugiura T, Miyazaki M, Moriyama T, Kaneda Y, Naruse K, Naruse M. and In vivo transfection of genes for renin and angiotensinogen into the glomerular cells induced phenotypic change of the mesangial cells and glomerular sclerosis. Biochem Biophys Res Commun 206: 525–532, 1995. doi: 10.1006/bbrc.1995.1075. [DOI] [PubMed] [Google Scholar]
- 56. Yang L, Besschetnova TY, Brooks CR, Shah JV, Bonventre JV. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rafiq K, Noma T, Fujisawa Y, Ishihara Y, Arai Y, Nabi AH, Suzuki F, Nagai Y, Nakano D, Hitomi H, Kitada K, Urushihara M, Kobori H, Kohno M, Nishiyama A. Renal sympathetic denervation suppresses de novo podocyte injury and albuminuria in rats with aortic regurgitation. Circulation 125: 1402–1413, 2012. doi: 10.1161/CIRCULATIONAHA.111.064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Abbate M, Zoja C, Remuzzi G. How does proteinuria cause progressive renal damage? J Am Soc Nephrol 17: 2974–2984, 2006. doi: 10.1681/ASN.2006040377. [DOI] [PubMed] [Google Scholar]
- 59. Dizin E, Hasler U, Nlandu-Khodo S, Fila M, Roth I, Ernandez T, Doucet A, Martin PY, Feraille E, de Seigneux S. Albuminuria induces a proinflammatory and profibrotic response in cortical collecting ducts via the 24p3 receptor. Am J Physiol Renal Physiol 305: F1053–F1063, 2013. doi: 10.1152/ajprenal.00006.2013. [DOI] [PubMed] [Google Scholar]
- 60. Sharma S, Smyth B. From proteinuria to fibrosis: an update on pathophysiology and treatment options. Kidney Blood Press Res 46: 411–420, 2021. doi: 10.1159/000516911. [DOI] [PubMed] [Google Scholar]
- 61. Figueroa SM, Araos P, Reyes J, Gravez B, Barrera-Chimal J, Amador CA. Oxidized albumin as a mediator of kidney disease. Antioxidants (Basel) 10: 404, 2021. doi: 10.3390/antiox10030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. de Zeeuw D, Remuzzi G, Parving HH, Keane WF, Zhang Z, Shahinfar S, Snapinn S, Cooper ME, Mitch WE, Brenner BM. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int 65: 2309–2320, 2004. doi: 10.1111/j.1523-1755.2004.00653.x. [DOI] [PubMed] [Google Scholar]
- 63. Grgic I, Campanholle G, Bijol V, Wang C, Sabbisetti VS, Ichimura T, Humphreys BD, Bonventre JV. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int 82: 172–183, 2012. doi: 10.1038/ki.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lim BJ, Yang JW, Zou J, Zhong J, Matsusaka T, Pastan I, Zhang MZ, Harris RC, Yang HC, Fogo AB. Tubulointerstitial fibrosis can sensitize the kidney to subsequent glomerular injury. Kidney Int 92: 1395–1403, 2017. doi: 10.1016/j.kint.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Takaori K, Nakamura J, Yamamoto S, Nakata H, Sato Y, Takase M, Nameta M, Yamamoto T, Economides AN, Kohno K, Haga H, Sharma K, Yanagita M. Severity and frequency of proximal tubule injury determines renal prognosis. J Am Soc Nephrol 27: 2393–2406, 2016. doi: 10.1681/ASN.2015060647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mori Y, Ajay AK, Chang JH, Mou S, Zhao H, Kishi S, Li J, Brooks CR, Xiao S, Woo HM, Sabbisetti VS, Palmer SC, Galichon P, Li L, Henderson JM, Kuchroo VK, Hawkins J, Ichimura T, Bonventre JK. 1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab 33: 1042–1061, 2021. doi: 10.1016/j.cmet.2021.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bonventre JV. Primary proximal tubule injury leads to epithelial cell cycle arrest, fibrosis, vascular rarefaction, and glomerulosclerosis. Kidney Int Suppl 4: 39–44, 2014. doi: 10.1038/kisup.2014.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nolin AC, Mulhern RM, Panchenko MV, Pisarek-Horowitz A, Wang Z, Shirihai O, Borkan SC, Havasi A. Proteinuria causes dysfunctional autophagy in the proximal tubule. Am J Physiol Renal Physiol 311: F1271–F1279, 2016. doi: 10.1152/ajprenal.00125.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zandi-Nejad K, Eddy AA, Glassock RJ, Brenner BM. Why is proteinuria an ominous biomarker of progressive kidney disease? Kidney Int Suppl 66: S76–S89, 2004. doi: 10.1111/j.1523-1755.2004.09220.x. [DOI] [PubMed] [Google Scholar]
- 70. Zoja C, Morigi M, Remuzzi G. Proteinuria and phenotypic change of proximal tubular cells. J Am Soc Nephrol 14 Suppl 1: S36–S41, 2003. doi: 10.1097/01.asn.0000068626.23485.e0. [DOI] [PubMed] [Google Scholar]
- 71. Takamatsu M, Urushihara M, Kondo S, Shimizu M, Morioka T, Oite T, Kobori H, Kagami S. Glomerular angiotensinogen protein is enhanced in pediatric IgA nephropathy. Pediatr Nephrol 23: 1257–1267, 2008. doi: 10.1007/s00467-008-0801-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ohashi N, Urushihara M, Satou R, Kobori H. Glomerular angiotensinogen is induced in mesangial cells in diabetic rats via reactive oxygen species–ERK/JNK pathways. Hypertens Res 33: 1174–1181, 2010. doi: 10.1038/hr.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Che G, Gao H, Hu Q, Xie H, Zhang Y. Angiotensin II promotes podocyte injury by activating Arf6-Erk1/2-Nox4 signaling pathway. PLoS One 15: e0229747, 2020. doi: 10.1371/journal.pone.0229747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Osborne MJ, Droz B, Meyer P, Morel F. Angiotensin II: renal localization in glomerular mesangial cells by autoradiography. Kidney Int 8: 245–254, 1975. doi: 10.1038/ki.1975.108. [DOI] [PubMed] [Google Scholar]
- 75. Kobori H, Nangaku M, Navar LG, Nishiyama A. The intrarenal renin–angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacol Rev 59: 251–287, 2007. doi: 10.1124/pr.59.3.3. [DOI] [PubMed] [Google Scholar]
- 76. Reinhold SW, Kruger B, Barner C, Zoicas F, Kammerl MC, Hoffmann U, Bergler T, Banas B, Kramer BK. Nephron-specific expression of components of the renin-angiotensin-aldosterone system in the mouse kidney. J Renin Angiotensin Aldosterone Syst 13: 46–55, 2012. doi: 10.1177/1470320311432184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S6 and Supplemental Table S1: https://doi.org/10.6084/m9.figshare.19134965.v4.