

Abstract
Cancers are highly complex diseases that are characterized by not only the overgrowth of malignant cells but also an altered immune response. The inhibition and reprogramming of the immune system play critical roles in tumor initiation and progression. Immunotherapy aims to reactivate antitumor immune cells and overcome the immune escape mechanisms of tumors. Represented by immune checkpoint blockade and adoptive cell transfer, tumor immunotherapy has seen tremendous success in the clinic, with the capability to induce long-term regression of some tumors that are refractory to all other treatments. Among them, immune checkpoint blocking therapy, represented by PD-1/PD-L1 inhibitors (nivolumab) and CTLA-4 inhibitors (ipilimumab), has shown encouraging therapeutic effects in the treatment of various malignant tumors, such as non-small cell lung cancer (NSCLC) and melanoma. In addition, with the advent of CAR-T, CAR-M and other novel immunotherapy methods, immunotherapy has entered a new era. At present, evidence indicates that the combination of multiple immunotherapy methods may be one way to improve the therapeutic effect. However, the overall clinical response rate of tumor immunotherapy still needs improvement, which warrants the development of novel therapeutic designs as well as the discovery of biomarkers that can guide the prescription of these agents. Learning from the past success and failure of both clinical and basic research is critical for the rational design of studies in the future. In this article, we describe the efforts to manipulate the immune system against cancer and discuss different targets and cell types that can be exploited to promote the antitumor immune response.
Subject terms: Immunotherapy, Cancer
Introduction: The history of tumor immunotherapy
In 1891, William B Coley, an orthopedic surgeon at New York Memorial Hospital in the United States, injected bacteria into tumors to treat cancer.1,2 There were few developments in the use of tumor immunotherapy until specific immune cells and immune-regulating molecules were identified. In 1974, interleukin (IL)-2 was discovered to play an essential role in T-cell differentiation and growth, and its utilization on cancer patients by Steven Rosenberg and his team was a milestone of tumor immunotherapy in the modern era,3–5 which also led to many approaches in the 1980s involving the application of cytokines for stimulating immune responses in patients with cancer.6,7 However, direct application of cytokines to patients can result in significant side effects,8–10 which warrants the discovery of specific immune cells that mediate the antitumor response and can be precisely targeted.
Activation of T cells is a key event in both antiviral and antitumor adaptive immunity, which is mainly accomplished through dual signaling pathways. The first signal is an antigen-specific signal, which involves the specific binding of the T-cell surface receptor (TCR) to the antigenic peptide-major histocompatibility complex (MHC).11,12 The second signal is mediated by the communication of T cells with costimulatory molecules (CMs) on the surface of antigen-presenting cells (APCs).13 These “primed” T cells can produce perforin and granzyme, which lyse target cells, and can secrete cytokines and induce target cell apoptosis through the combination Fas-FasL interaction.14 Blocking the activation of T cells against malignant cells in cancer patients has been the central problem for tumor immunology research.15–17
After the identification of T-cell receptors (TCRs) that are responsible for antigen recognition,18,19 in 1986, scientists discovered the molecule CD28 expressed on activated T cells.20 Subsequently, it was found that T-cell activation requires both signals from the TCR and CD28, and CD28 was thereafter named a “costimulatory molecule”.21–23 Around the same time, Pierre Golstain’s team discovered a protein with a similar structure to CD28; it was named cytotoxic T lymphocyte-associated antigen 4 (CTLA-4)24 and hypothesized to be a potential T-cell activating molecule.25,26 The concept that CTLA-4 is a positive immune regulator was also shown in other studies22 but was later challenged by the teams of James Allison and Jeffery Bluestone, who independently discovered that blocking CTLA-4 enhanced the T-cell immune response.27,28 Consistently, disrupting the CTLA-4 gene was lethal in mice due to excessive immune activation, supporting the immunosuppressive function of CTLA-4.29,30 This discovery paved the way for Allison’s team to test whether blocking CTLA-4 can potentiate antitumor immunity and inhibit the growth of immunosuppressive tumors. At the end of 1994, Allison’s team developed an antagonistic CTLA-4 antibody to be evaluated in tumor-bearing mice and later reported the ground-breaking discovery that blocking CTLA-4 can increase the antitumor activity of T cells and inhibit tumor growth.31 Therefore, for the first time, it was demonstrated that inhibiting a negative immune regulator could suppress tumor progression; this approach was later named “immune checkpoint blockade” (ICB) by Allison.32 In 1997, Allison’s team suggested that both inducing T-cell costimulatory signals and reducing inhibitory signals can be potential approaches for cancer immunotherapy.33 In 2011, ipilimumab, the first antibody targeting CTLA-4, was approved for melanoma treatment and became the first immune checkpoint (IC) inhibitor.34,35
More than 20 years ago, the research group of Tasuku Honjo at Kyoto University discovered programmed cell death protein 1 (PD-1).36 PD-1 knockout led to autoimmune disease and abnormally activated immune cells in mice,37 suggesting its immune-suppressive role. In 1999, the research group of Lieping Chen at the Mayo Clinic discovered a molecule named B7-H1,38 which was later found to be expressed on tumor tissues such as melanoma and lung cancer and can promote the apoptosis of tumor-specific T cells, making them unable to attack cancer cells.39 In 2000, B7-H1 was identified as a ligand of PD-1, therefore acquiring its second name PD-L1.40 In 2002, PD-L2 was discovered, and the signaling pathway involving PD-1 was clarified.41,42 These discoveries demonstrated that PD-1 is another IC. Indeed, PD-1/L1 inhibitors are the most widely applied immunotherapy type to date, with 6 drugs that have been approved in the United States. In China, 4 PD-1 inhibitors have been approved for commercialization.43 The approved PD-1 inhibitors and PD-L1 inhibitors have changed the paradigm of cancer therapy.44–47
The concept of cellular immunotherapy arose from the observation of graft-versus-leukemia effects in allogeneic bone marrow transplantation.48 To support in vivo maintenance and tumor recognition, cell engineering technologies were integrated with adoptive transfer. The first chimeric antigen receptor (CAR) was generated in 1989 by the Zelig Eshhar group in Israel.49 These “first-generation” CARs are the variable regions of antibodies fused to the TCR signaling domain, which mediates T-cell activation against the targeted antigen but has shown limited in vivo expansion. In 2002, the Michel Sadelain group incorporated the costimulatory domain into the CAR construct.50 The resulting “second-generation” CARs have seen extraordinary clinical responses against malignancies of the B-cell lineages, including B-cell chronic lymphocytic leukemia (B-CLL) and B-cell acute lymphocytic leukemia (B-ALL).51 On 30 August 2017, the US FDA approved the first CAR-T product, Kymriah from Novartis, for the treatment of relapsed or refractory patients under the age of 25 with acute B-cell lymphoblastic leukemia (B-ALL). The commercialization of CAR-T-cell therapy soon followed with the approval of six other products targeting leukemia, lymphoma, and multiple myeloma.52 At present, ~1000 registered clinical studies are ongoing to evaluate CAR-T-cell immunotherapy against leukemia, lymphoma, melanoma, glioma and other malignant tumors.53–59
We summarized the major milestone breakthroughs in cancer immunotherapy over time (Fig. 1).
Fig. 1.

Historical landmarks in cancer immunotherapy development
Antibodies against CTLA-4 and PD-1, as well as CAR-T-cell therapy, represent promising approaches in which certain components of the immune system can be manipulated to reverse suppression and target tumors. However, not all patients respond to these therapies, indicating the complexity of tumor-induced immune alteration. In 2006, the concept of “cancer immunoediting” was introduced by Dr. Robert Schreiber, describing how malignant cells can respond to initial immune recognition and subsequently develop escape mechanisms and even “reprogram” the immune system to become protumorigenic.60,61 Such an immunoediting process can occur almost every time intratumoral or systemic immune cells are present, resulting in a highly suppressive tumor microenvironment (TME). The search for a synergistic approach for activating antitumor T-cell responses and targeting the suppressive TME has been the main focus of research in tumor immunotherapy.
New targets and drug candidates have been emerging for cancer immunotherapy, but most are still in the very early stage of development. Unfortunately, clinical studies have revealed that quite a few of these candidates may not exert satisfactory outcomes as monotherapies. In this article, we will discuss different strategies for cancer immunotherapy, including IC- and stimulatory molecule-targeted agents, cellular immunotherapy, and suppressive TME-targeting strategies. Furthermore, as ICB and CAR-T cells have been the most rigorously evaluated immunotherapy strategies in the clinic, we will also discuss biomarkers associated with the clinical efficacy of these two types of treatment.
ICs on T cells
ICB has revolutionized the field of cancer therapy and has become one of the most valuable methods in the treatment of many late-stage cancers.62–64 ICs are a class of immunosuppressive molecules that are expressed on immune cells and can suppress immune cell activation, therefore playing a key role in autoimmunity prevention (Fig. 2).65–68 In contrast, overexpression of ICs suppresses immune function and contributes to tumorigenesis.69–73 ICB therapy, therefore, inhibits tumor growth by blocking ICs and potentiating antitumor T-cell activity.74–77 The development of ICB was initiated by targeting two IC pathways, PD-1/PD-L1 and CTLA-4/B7-1/2,78–80 the blocking of which has made remarkable clinical progress, especially against non-small cell lung cancer, colon cancer, melanoma, and renal cell carcinoma.81–88 However, only 20–30% of patients achieve long-term survival following these ICB treatments, and one of the underlying mechanisms is the expression of other inhibitory molecules.88 Therefore, the continuous identification of new IC targets and the development of their corresponding ICBs have become critical. To date, several other IC molecules on T cells that mediate inhibitory signals through different mechanisms have been identified, with the potential to be exploited as targets for cancer immunotherapy.
Fig. 2.
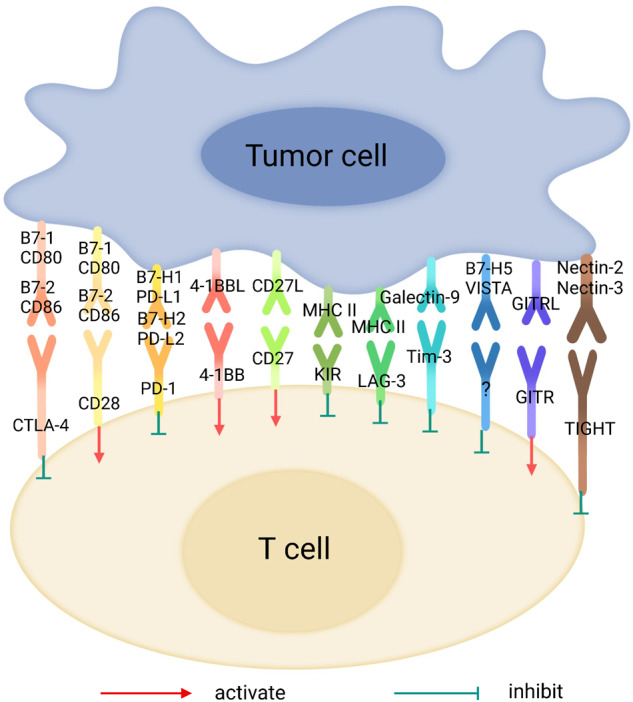
List of immune checkpoint inhibitors and their receptors
One of the key processes involved in cancer development is that cancer cells acquire immune escape by inducing and recruiting immunosuppressive cells, such as Treg cells, bone marrow-derived suppressor cells, and tumor-associated macrophages, as well as increasing the expression of various immunosuppressive molecules, such as PD-1 and PD-L1 (PD-L1). Blocking these immunosuppressive mechanisms can restore the underlying antitumor immune response. Cancer immunotherapy requires ICB, such as CTLA-4 and monoclonal antibodies against PD-1 or PD-L1, which restore the function of cytotoxic effector CD8+ T cells and kill cancer cells, leading to tumor suppression and a paradigm shift in cancer treatment for many cancer types. However, since more than half of treated patients do not respond to ICB even in combination with other therapies, the identification of biomarkers that predict clinical efficacy is an urgent issue.89
PD-1
PD-1 was found to inhibit the function of T lymphocytes, which is critical in controlling the autoimmune response.90–94 PD-L1 (initially identified as B7-H1), is highly expressed on multiple types of tumors and can bind to PD-1 and mediate tumor immune escape.39 Therefore, inhibition of PD-1 can reactivate T-cell function.95,96 Recent studies have also revealed that PD-1 is expressed not only on T cells but also on NK cells, B lymphocytes, macrophages and dendritic cells (DCs),97,98 suggesting that PD-1 may play a very effective role in remodeling the tumor immune microenvironment and even systemic antitumor immunity.46,99–102
PD-1 inhibitors can specifically bind to PD-1, thereby attenuating the immunosuppressive regulation of T lymphocytes and enabling T lymphocytes to participate in the killing of tumor cells (Fig. 3).84 Preclinical studies have shown that PD-1 inhibitors can inhibit the proliferation of cells and induce the programmed cell death (apoptosis) of various tumor cells.103 PD-1 antibodies can also enhance the apoptosis of tumor cells mediated by other cytotoxic agents, such as adriamycin.104,105
Fig. 3.

Schematic diagram of the working mechanism of PD-1 antibodies
The clinical efficacy of PD-1/L1 blocking antibodies was first observed against tumors with high PD-L1 expression, including melanoma, non-small cell lung cancer (NSCLC), and renal cell carcinoma (RCC).42,106,107 The PD-1 blocking antibody nivolumab (Opdivo) was approved in 2015 for advanced squamous cell lung cancer treatment, marking the first clinical application of anti-PD-1 therapy. Of note, the prescription criteria did not include the expression of PD-L1 on tumor cells.108,109 After that, pembrolizumab was the first immunotherapeutic drug approved by the FDA for first-line treatment of patients with metastatic NSCLC in 2016.110–112 Different from nivolumab, the prescription of pembrolizumab requires confirmed PD-L1 overexpression on tumors.113,114 Nivolumab and pembrolizumab (coreda) were later approved as single agents for the second-line treatment of NSCLC (non-small cell lung cancer).108,115–117 On the other hand, atezolizumab (Tecentriq) was approved in 2016 to treat patients with metastatic NSCLC and disease progression during or after first-line platinum chemotherapy.117,118 In addition, atezolizumab can also be used in patients with EGFR mutations or ALK rearrangements undergoing targeted therapy and disease progression.119–121 Another two new PD-L1 antibodies, durvalumab (Imfinzi) and avelumab (Bavencio), were approved in 2017.122,123 Furthermore, sindilizumab, a PD-1 antibody developed by Innovent Biologics in China, also achieved good results after two cycles of neoadjuvant administration,124 representing another candidate to target this pathway.125
However, more than 50% of patients with cancer do not respond to PD-1/L1 inhibitors. Of note, the objective response rate was only 45% with pembrolizumab for non-small cell lung cancer, even in patients with high expression of PD-L1.126 In addition, a small proportion of patients experience hyperprogressive disease (HPD),127–130 which may be a result of regulatory T-cell (Treg) outgrowth and subsequent inhibition of antitumor immunity.30,131–133 These results suggest that PD-1 blockade needs to be prescribed in a personalized manner to maximize its efficacy.
CTLA-4
CTLA-4, also known as CD152, is a transmembrane protein expressed in activated CD4+ and CD8+ T cells.134,135–138 While CD28 was found to be a T-cell costimulatory molecule,139 CTLA-4 was later discovered to mimic CD28 and act as a brake on T-cell activation.140,141 Under physiological conditions, CTLA-4 and CD80/CD86 binding can inhibit T-cell activation signals and prevent autoimmune disease.142,143 Blocking CTLA-4 can directly target inhibitory signals on effector T cells and reduce the inhibitory effect of Tregs,33,144–147 thus effectively enhancing the antitumor effect of T cells.
In 1996, James Allison found that blocking CTLA-4 caused tumor regression in mice.148 In subsequent human studies, the CTLA-4 antibody ipilimumab was the first-in-class ICB agent to be tested in clinical studies. Ipilimumab performed well and successfully inhibited disease progression in patients with refractory metastatic melanoma, which was a milestone of cancer immunotherapy.149 Intriguingly, CTLA-4 was particularly highly expressed on the surface of Treg cells infiltrated by melanoma, lung cancer and kidney cancer.150 Although it was later found that Treg depletion is not the main mechanism of the clinical antitumor efficacy of ipilimumab,144 these results suggest that the CTLA-4 antibody may also inhibit Treg cells in the TME under certain circumstances and contribute to immune activation.136,151,152
Although both are representative IC molecules, CTLA-4 and PD-1 regulate T-cell function in different manners (Fig. 4). While the inhibitory signal from CTLA-4 negatively regulates T-cell priming, PD-1 mainly mediates the subsequent activation and proliferation of primed T cells.153 In the context of tumors, it was found that ICB targeting PD-1 usually leads to the expansion and recruitment of existing antitumor T cells, while anti-CTLA-4 therapy generates new T-cell clones.153,154 Furthermore, anti-CTLA-4 therapy was found to induce a Th1-like CD4+ subset, which was not observed in anti-PD-1 therapy.155 Genetic models also revealed that CTLA-4 enforces boundaries on CD4+ T-cell phenotypes and that PD-1 subtly restrains CD8+ T-cell phenotypes.156 These results indicate that CTLA-4 and PD-1 may be simultaneously targeted for synergistic antitumor effects. The combinational therapy of CTLA-4 and PD-1 ICBs indeed resulted in superior clinical responses but led to more significant adverse effects than monotherapy.153
Fig. 4.

Illustration of CTLA-4 and PD-1
Tim-3
T-cell immunoglobulin domain and mucin domain-3 (Tim-3, CD366) is a T-cell surface inhibitory molecule that is mainly expressed on CD4+ T helper cell 1 (Th1) and CD8+ CTL cells157–160 and on a subset of Treg cells with enhanced inhibitory function.161,162 Tim-3, also known as HAVCR2, was later found to also be expressed on some innate immune cells, including dendritic cells, NK cells, monocytes, and macrophages.163 In IA/IB studies, the Tim-3 blocking antibody LY3321367 was well tolerated as a single agent or in combination with an anti-PD-L1 antibody.164 In addition, in one patient with extensive stage PD-L1-negative small cell lung cancer that was resistant to cisplatin/etoposide and PD-1/CTLA-4 antibodies, anti-TIM-3 monotherapy resulted in a partial response (PR). Therefore, preliminary antitumor activity of anti-TIM-3 therapy was observed in early clinical studies, but phase II and III studies are still needed to verify the efficacy in larger cohorts of patients.165
LAG-3
LAG-3 can be induced on CD4+ and CD8+ T cells under antigen stimulation. The inhibitory function of LAG-3 is closely related to its expression level on the cell surface, which is under stringent regulation during homeostasis.166–168 Long-term infection with viruses, bacteria and parasites causes continuous exposure to antigens, which leads to high levels and continuous expression of LAG-3 and subsequent reductions in cytokine release, cytolytic activity, and proliferation potential.169–172 Coexpression of LAG-3 and PD-1 on intratumor T cells has been observed in several mouse tumor models, and synergistic inhibition of tumor growth was observed when combining the blocking antibodies of these two molecules.173–176
LAG-3 has thus become one of the most critical new targets of cancer immunotherapy and is considered a major development direction after PD-1 with great application prospects.171,177,178 Relatlimab, the first inhibitor of LAG-3 to enter the clinic, blocks the interaction of LAG-3 with MHC II.179 RELATIVITY 047 (CA224-047), a phase II/III clinical study, was designed to evaluate a fixed-dose combination of relatlimab combined with nivolumab versus nivolumab monotherapy in patients with previously untreated metastatic or unresectable melanoma. The study resulted in a median progression-free survival (PFS) of 10.12 months (95% CI, 6.37– 15.74) in the combination group compared with 4.63 months (95% CI, 3.38–5.62) in the monotherapy group. In addition, the PFS rates at 12 months were 47.7% and 36.0%, respectively, supporting further development of anti-LAG-3 treatment.180
In 2019, Wang et al. identified fibrinogen-like protein 1 (FGL1) as the ligand for Lag-3.181 It was found to bind to Lag-3 to form a new PD-1/PD-L1-independent immune checkpoint pathway, leading to T-cell exhaustion, dysfunction, and tumor cell evasion of immune surveillance. Blocking FGL1 in addition to anti-PD-L1 has the potential to become another novel ICB strategy in clinical practice, especially in the targeted therapy of non-small cell lung cancer (NSCLC).182
NR2F6
Nuclear receptor subfamily 2 group F member 6 (NR2F6) was recently reported as an intracellular IC molecule, which is an orphan nuclear receptor inherent to lymphocytes.183,184 NR2F6 acts as a transcription factor regulating the activation, recruitment, proliferation, and homeostasis of cells associated with tumor antigen-specific T-cell responses. In NSCLC tissues, high expression of NR2F6 was detected in tumor-infiltrating lymphocytes (TILs), and upregulated NR2F6 expression was associated with impaired production of cytokines, including IL-2, TNF-α, and IFN-γ,185 suggesting that NR2F6 on TILs contributes to tumor immunosuppression. Moreover, the disruption of NR2F6 resulted in tumor suppression and enhanced the effect of PD-L1 blockade in tumor therapy, suggesting that NR2F6 inhibitors may become a new type of immunotherapy that can overcome resistance to existing ICB treatment.186,187
TIGIT
T-cell immunoglobulin and ITIM domain protein (TIGIT) is a type I transmembrane protein. TIGIT belongs to the immunoglobulin superfamily (IgSF) and can be expressed on T cells, regulatory T cells, memory T cells, and NK cells. TIGIT mediates the inhibitory effect on the activation of NK cells and T cells through its interaction with the ligands CD155 and CD112, which are expressed on antigen-presenting cells (APCs).188–191 In human tumors, TIGIT was found to be coexpressed with multiple IC molecules, including PD-1, TIM-3, and LAG-3.192 The coexpression of TIGIT, TIM-3 and PD-1 showed a correlation with poor survival in patients.193 In mouse models of malignant melanomas, it was found that the tumor growth rate was slowed down after TIGIT knockout, and survival was significantly prolonged.194 In human cancer models, simultaneous blockade of the TIGIT and PD-1 signaling pathways increased the expression of IFN-γ and TNF-α in tumor-specific CD8+ T cells, supporting the development of anti-TIGIT treatment. Two phase I clinical studies targeting TIGIT for cancer immunotherapy are currently ongoing.193
VISTA
V-set immunoregulatory receptor (VISTA), also known as PD-1H or DD1α, is an immunomodulatory protein that was discovered in recent years. It is mainly expressed in lymphoid organs and bone marrow cells, and its structure is similar to that of PD-L1.195,196 Studies have shown that VISTA-expressing APCs have inhibitory effects on CD4+ and CD8+ T cells; when this molecule is blocked, the immune function mediated by T cells is rescued, suggesting that VISTA is an IC molecule that inhibits T-cell responses.196 In T cells, the inhibitory effects of VISTA and PD-1 are independent of each other, and studies in mouse models of tumors also verified that the simultaneous application of anti-PD-1 and anti-VISTA antibodies can inhibit tumor growth and prolong survival.197
In gastric cancer patients, VISTA was found to be expressed in some tumor cells, as well as TILs. Furthermore, patients with VISTA-high oral squamous cell carcinoma have a poor prognosis. After treatment with ipilimumab in prostate cancer patients, the levels of VISTA+ TILs and macrophages were significantly upregulated, indicating that VISTA might contribute to acquired resistance to current ICB treatments, and the combined blockade of VISTA and CTLA-4 may exert better effects than blockade of either factor alone. The anti-VISTA antibody JNJ-61610588 is now being evaluated in a phase I clinical study for the treatment of solid tumors (NCT02671955).198
BTLA
B and T lymphocyte attenuator (BTLA) belongs to the immunoglobulin superfamily. It is expressed on T cells, resting B cells, macrophages, dendritic cells and NK cells and is similar in structure and function to PD-1 and CTLA-4. The ligand for BTLA is herpesvirus entry mediator (HVEM). When BTLA binds to HVEM, it generates inhibitory signals and inhibits T-cell activation. Anti-BTLA treatment can promote T-cell proliferation, and BTLA knockout mice show higher immune activity.199–201 In patients with malignant melanoma, tumor-specific T cells in both circulating lymph and metastatic lymph nodes expressed BTLA, and the expression of HVEM was also detected in the patient’s tumor tissue.202 The expression of BTLA was found to be significantly increased in pleural effusion samples from patients with lung cancer, which is an indicator of tumor aggressiveness.203 Therefore, BTLA, as an inhibitory molecule for immune regulation, has broad research prospects. At present, research on BTLA and HVEM inhibitors is still in the preclinical stage, and it is expected that related drugs will be launched as soon as possible and enter into clinical research verification.204–206
Immune stimulatory molecules on T cells
OX40
OX40, also known as CD134, is a member of the tumor necrosis factor receptor (TNFR) superfamily and is expressed 24–72 h after T-cell activation. Its ligand OX40L, also known as CD252, is mainly expressed on the surface of activated APCs. The OX40-OX40L interaction can initiate T-cell activation signals as well as the expression of cyclin A, Bcl-2 anti-apoptotic molecules, cytokines, and cytokine receptors.207 Mouse models have shown that specific antibodies that stimulate OX40 can reduce the number of Tregs, thereby maintaining the function of effector T cells and showing high antitumor activity.208–210
There are many clinical studies targeting the OX40-OX40L pathway, including those on single-agent application of specific antibodies that excite OX40 or combination with chemotherapy, radiotherapy, surgery, small molecule-targeted therapy, cytokines or other ICB drugs.211 The results of a study of the OX40 agonist MOXR0916 showed that as a monotherapy or in combination with atezolizumab, the treatment achieved PR in 2 out of 51 patients, and more phase I/II clinical studies are underway. However, there have been no clues about whether OX40 agonists should be used as a monotherapy or combined with other drugs. Future basic and clinical research is needed for an in-depth understanding of the mechanism by which OX40 regulates different T-cell subtypes in the TME.
ICOS
Inducible costimulatory molecule (ICOS), also known as CD278, is a member of the immunoglobulin superfamily. ICOS is expressed on the surface of activated T cells and regulates T-cell proliferation and function.212 The activation of ICOS is dependent on its ligand ICOS-L, which is mainly expressed in B cells and APCs. ICOS has been shown to be an important marker for ICB efficacy.212–214 When malignant melanomas were treated with anti-CLTA-4, the abundance of ICOS+ CD4+ T cells was found to be associated with better efficacy.155 In mouse models, ICOS agonist alone had difficulty eliciting sufficient antitumor responses against melanoma; however, there was a synergistic effect between ICOS and anti-CTLA-4; in addition, ICOS knockout mice responded poorly to anti-CTLA-4 treatment.215 Simultaneous use of ICOS agonists and anti-PD-1 and anti-CTLA-4 therapy can also enhance the antitumor effect against lung cancer in preclinical models.216 Currently, several phase I clinical studies targeting the ICOS pathway are ongoing.217
4-1BB
4-1BB, also known as CD137, is a member of the TNFR family.218 The function of 4-1BB on regulatory T cells is complex, and studies have led to contradictory results. However, importantly, 4-1BB gene knockout mice developed autoimmune diseases, suggesting that it plays an important role in immune balance and has the potential to be targeted to elicit tumor-specific immune recognition.219,220
Currently, clinical studies of two 4-1BB-specific agonistic antibodies, urelumab and PF-05082566, are ongoing, and the preliminary results support that 4-1BB agonism can promote the proliferation and activity of T cells and NK cells.221 Another clinical study used PF-05082566 combined with PD-1 antibody to treat NSCLC and renal cell carcinoma. With only 6 out of 23 patients achieving a complete response (CR) or PR, combinational therapy may need to be reinvestigated for better patient selection.219
CD27
Unlike other members of the TNFR family, CD27 is expressed only on the surface of lymphocytes, including naive and activated CD4+ and CD8+ T cells. When it interacts with its ligand CD70, CD27 induces the proliferation and differentiation of effector and memory T cells and enhances the activation of B cells and NK cells.222,223 Mouse models suggest that the induction of the CD27 signaling pathway can inhibit tumor growth.224 In addition, it was found that 1/3 of patients with Hodgkin's lymphoma and diffuse large B-cell lymphoma had germline depletion of CD27 or CD70, and most patients with diffuse large B-cell lymphoma and Burkitt's lymphoma had mutation or depletion of the CD70 gene, which further verified the antitumor immune effect of the CD27/CD70 signaling pathway.225–228
Varlilumab is a humanized monoclonal antibody against CD27 that promotes cytokine production and activation of T cells. In a phase I clinical study, varlilumab was well tolerated in patients with advanced solid tumors and has shown initial safety results: of 56 patients in the phase I study, only one patient developed grade 3 hyponatremia, and most treatment-related toxicities were grade 1–2.229 One patient with advanced renal cell carcinoma achieved a PR (tumor reduction of 78%), which lasted for 2.3 years. Eight patients had stable disease (SD) for more than 3 months, and one patient with advanced renal cell carcinoma had SD for more than 3.9 years.229 In addition to monotherapy, varlilumab has also been used in combination with anti-PD-L1 antibodies.230 The antitumor effect of this CD27 agonist still needs further, larger-scale investigation.227
ICs on NK cells
In the human body, NK cells are mainly characterized by a CD3-CD56+ lymphocyte population, and the CD16+CD56dim subtype is mainly found in the blood. As an important part of the natural immune system, NK cells play an important role in removing senescent cells and pathogenic microorganisms.231 NK cells do not recognize target cells through specific receptors like TCRs do; they recognize cells through receptors expressed by germline genes. The negative regulators of NK cells include KIRs (immunoglobulin-like receptors), CD94-NKG2 and MHC-I.232–234 In the context of tumor immunology, as tumor cells downregulate MHC expression to escape acquired immunity, they become more susceptible to NK-cell cytotoxicity. In addition, NK cells play an essential role in mediating antibody-induced cellular cytotoxicity (ADCC) in antibody therapy.235–237 NK cells can also directly exert an antitumor effect by secreting cytokines or mobilizing immune cells such as dendritic cells, macrophages, and T cells to participate in the process of removing tumor cells,238 making them attractive targets for cancer immunotherapy.
KIR
The killer cell immunoglobulin-like receptor (KIR) family is a class of highly polymorphic molecules mainly expressed on the surface of some NK cells and T cells, which can be divided into multiple subtypes. Among them, KIR2DTl1-3 and KIR3DL1 can exert inhibitory effects by binding MHC molecules (HLA-C/HLAB).239 Due to the characteristics of high gene polymorphism, the combination of multiple KIR genes and their ligands can cause a variety of diseases, including autoimmune diseases, especially the combination of some KIR genes and specific ligands, which can increase the risk of cancer.240 In mouse models, treatment targeting the activated NK-cell surface receptor KIR2DS2 showed significantly superior antitumor activity to treatment targeting conventional costimulatory molecules.241–243
The KIR inhibitor IPH2101 showed efficacy in preclinical models but not in phase I/II clinical studies,244 despite the observation that KIR inactivation was associated with prolonged survival in patients with colorectal cancer and glioblastoma.245,246 By further analyzing the blood sample of patients treated with IPH2101, Carlsten et al. found that IPH2101 binding to KIR resulted in NK-cell clearance via FcγR recognition from APCs.247 Lirilumab, another inhibitor of KIR, had an objective response rate of 24% in combination with nivolumab in advanced head and neck tumors, which may suggest less APC-mediated clearance using this antibody. A phase I/II clinical study of lirilumab is underway for advanced solid tumors and hematological tumors.
NKG2A
NK-cell lectin-like receptor subfamily C member 1 (NKG2A) is an “inhibitory” member of the NKG2 family and is mainly expressed in CD56hi NK cells, NKT cells and CD8+αβ T-cell subsets.248 It forms a heterodimeric receptor with CD94 and binds with its ligand, the nonclassical MHC I molecule HLA-E, which is expressed in most normal tissues. The interaction between NKG2A/CD94 and HLA-E can inhibit the activation of NK cells and T cells,249 indicating the potential to be targeted as an IC molecule.
Monalizumab (IPH2201), jointly developed by Innate Pharma and AstraZeneca, is an NKG2A monoclonal antagonistic antibody that can block the interaction between NKG2A and HLA-E and has shown therapeutic effects in a leukemia mouse model.250 Monalizumab has been used in phase II clinical trials against cancers of the female reproductive system and NSCLC.251 In these clinical studies, monalizumab was well tolerated but with limited therapeutic effect, and only showed short-term SD in some patients. However, monalizumab combined with cetuximab (EGFR blocking antibody) reached a 27.5% response rate against recurrent or metastatic head and neck squamous cell carcinoma, suggesting that combining monalizumab with targeting oncogenic pathways may enhance clinical antitumor efficacy.251
CD96
CD96 is a member of the immunoglobulin superfamily expressed on NK cells, recognizing the ligand CD155. It was found that CD96 expression on tumor-infiltrating NK cells was higher than that on NK cells in surrounding tissue.252 Higher levels of CD96 expression on NK cells in hepatocellular carcinoma samples predicted poor prognosis.253 Preclinical results in tumor models with implantable or spontaneous metastases have proven that CD96 inhibitors can reduce metastatic potential,187,254 which supports the future clinical application of agents with this target.
Cellular immunotherapy
Immune cells with cytotoxic potential, including T cells, NK cells and macrophages, recognize and eliminate infected or damaged cells under physiological conditions. The cytotoxic effect from T cells is distinct from others given its nature of antigen specificity. Cellular immunotherapy, also called adoptive cell transfer (ACT), exploits the killing capability of these types of immune cells for the treatment of cancers.255–258 Here, we discuss four major types of ACT that have achieved significant research and clinical progress: CAR T-cell therapy, TIL therapy, engineered TCR therapy, and NK cell therapy.
TIL therapy
TILs are heterogeneous lymphocytes that can be identified and purified from tumor tissues, and their abundance has been found to correlate with a better prognosis.259–261 Unfortunately, in most cancer patients, there are too few endogenous TILs to elicit a sufficient antitumor response. TILs were among the first set of cells exploited for ACT. These cells can be isolated from tumors, expanded in a laboratory environment in vitro, and reinjected in large numbers into cancer patients to eliminate tumor cells.262–264 TIL therapy has been tested rigorously in clinical studies, resulting in inspiring outcomes against certain types of tumors, with the longest reported survival of 11 years.265,266 TIL therapy has resulted in clinical remissions in some patients who have exhausted all other treatment options. One of the examples was a patient named Melinda Bachini, who was diagnosed with cholangiocarcinoma in 2009 and developed whole-body metastasis despite surgery and chemotherapy. This patient was then recruited into a clinical study of TILs. Only 1 month after TIL treatment, her whole-body tumor began to regress, and her physical strength recovered quickly. Now she is the first survivor of advanced bile duct cancer for more than 10 years. Another patient had metastatic adenocarcinoma that did not respond to chemoradiation and had metastases to retroperitoneal lymph nodes and to the surface of the liver. Before TIL treatment, tumor metastases were found in the retroperitoneum, abdominal wall, parahepatic and pelvic cavity. After treatment, a CR was declared in this patient with the regression of tumor sites detected.267 TILs have been considered to have the ability to accurately identify tumor antigens, which contributes to their tumor specificity, and this new therapy can be considered “tailored” to the patient.268 Below, we summarize the recent progress of TIL therapy against different types of tumors.
Melanoma
LN-144 (Lifileucel) is a TIL-based therapy against melanoma. A phase II clinical trial showed disease control rate of 80.3% and an objective response rate of 36.4%: two patients had a CR, 23 had a PR, and some patients' tumors completely disappeared after 2 years of treatment.268 More strikingly, some patients with PD-L1-negative tumors, who were likely not responsive to anti-PD-1 ICB, also responded to TIL therapy, suggesting that patients refractory to other types of immunotherapy can still benefit from TIL therapy. Indeed, another phase I clinical study used TILs to treat anti-PD-1-resistant tumors: two patients achieved a CR that lasted for more than 1.5 years,269,270 suggesting that for patients who have progressed after PD-1 therapy, TIL therapy is among the few other treatment options.
Lung cancer
A phase I clinical trial result was announced at the 2021 AACR meeting. In 12 evaluable patients with NSCLC, TIL therapy achieved a 25% overall remission rate. At a mean follow-up of 1.4 years, three patients were in remission, and two of these patients had durable complete for more than one year.271 Moreover, most of the patients had smaller tumor lesions after receiving TIL treatment. On the first CT scan after receiving treatment, the diameter of the tumor lesions was reduced by an average of 38%.
Cervical cancer
In a phase II clinical study of LN-145, a TIL therapy for advanced cervical cancer, most of the enrolled patients had refractory disease after 2–3 prior treatments.268 At a median follow-up time of 3.5 months post-infusion, the objective response rate of LN-145 treatment was 44%, and the disease control rate was 85%. Three patients' tumors completely disappeared, and nine patients' tumors shrank significantly. With a median follow-up of 3.5 months, 11 of the 12 patients had sustained responses, and no serious adverse events occurred. Based on the promising data of this clinical trial, the US FDA granted LN-145 with “breakthrough therapy” status for advanced cervical cancer, with approval entering the fast approval track.
Metastatic breast cancer
In 2018, a case report was published about a patient with refractory estrogen receptor-positive metastatic breast cancer who received TILs for four mutant proteins (SLC3A2, KIAA0368, CADPS2, and CTSB). Twenty-two months after the infusion, the tumor had completely disappeared, and 4 years later, there was no progression or recurrence.270 Larger-scale clinical studies of TIL therapy against breast cancer are currently ongoing.
Osteosarcoma
A clinical study was performed to determine the safety and efficacy of TILs and anti-PD-1 therapy for the treatment of osteosarcoma. In this study, 30 patients received anti-PD-1 monotherapy, and another 30 patients received TIL+anti-PD-1 combinational therapy. At the last follow-up assessment, no patients receiving monotherapy survived, with a mean overall survival of 6.6 months. In contrast, 10 of the 30 patients who received combinational therapy survived, with an objective response rate of 33.3%. Of note, two of the 10 patients experienced complete remission according to imaging examination. The mean overall survival was 15.2 months, which was more than doubled that of patients who received PD-1 monotherapy.272
Ovarian cancer
The combination of ICB with TIL therapy for the treatment of ovarian cancer has been tested in a phase I clinical study. The results showed that one patient achieved a PR, and the other five experienced SD for up to 12 months.273 The best strategy for the design of combinational treatment still requires further investigation.
From the clinical applications above, we can infer that the combination of ICB with TIL therapy can be an approach for the future design of immunotherapy. This combinational strategy has been proven for its safety against some types of tumors, with five potential advantages: (1) the killing ability of TILs can be improved by ICB;274 (2) TILs are believed to have specific clones that recognize tumor antigens, and advanced technologies have also allowed targeted screening processes to expand antigen-specific TILs and improve specificity;269 (3) there is potential for these treatments to be further combined with radiotherapy and chemotherapy to reduce tumor recurrence;275 (4) both TILs and ICB antibodies can be reinfused to maintain the antitumor response;276 and (5) the other host immune cells can be activated after ICB, which may form synergistic antitumor effects with TILs.54 TIL therapy has shown great potential for solid tumors, and new clinical studies have been conducted to expand the scenario of applying TILs to other cancers in the future.277
Engineered TCR T therapy
TCRs are specific receptors on the surface of T cells. By recognizing and binding to the antigens presented by MHC, they can activate the division and differentiation of T cells.278 However, not all patients have T cells that can recognize tumors. Therefore, TCR-T therapy involves taking T cells from patients and expanding these cells to equip patients with new TCRs that can recognize specific cancer antigens.
The design of engineered TCRs used for TCR-T therapy is highly dependent on the identification of specific tumor antigens. Some antigens, such as NY-ESO-1, are widely expressed in tumor tissues and can be exploited to develop TCRs to treat different types of tumors.279 However, TCRs can be identified and synthesized in a patient-specific manner. As TIL therapy exploits the difference between intratumoural versus systemic lymphocytes, the identification of specific mutations in a patient's tumor guides the generation and application of TCRs that can effectively target these mutations. These TCRs can then be isolated, cloned and expressed on T cells before these engineered T cells are expanded in vitro and reinfused into the patient.273,280 This is a highly personalized treatment approach that enhances the specificity of the therapy. TCR therapy has made breakthroughs in the treatment of melanoma and has also achieved certain results in the treatment of liver cancer, breast cancer, and ovarian cancer. However, TCR recognition of tumor antigens requires antigen expression by the MHC molecule, and tumor cells will escape T-cell killing by decreasing the expression of MHC.56,281
CAR-T cell therapy
CAR-T cell therapy is another type of ACT strategy.282 Sharing a similar principle with TCR T therapy, the patient's T cells are “equipped” with the synthetic CAR, expanded and reinfused into the patient to generate a tumor-specific immune response (Fig. 5).283–285 CARs are designed to recognize tumor-associated antigens (TAAs), which are independent of MHC presentation, therefore enabling T cells to recognize cancer cells in an MHC-nonrestricted manner.286–288 At present, ACT of CAR-T cells has become one of the main methods of tumor immunotherapy, providing new therapeutic solutions to many types of tumors.289–292 Rigorous clinical studies have also allowed researchers to understand the limitations and side effects of this type of treatment, fostering the development of future immunotherapies based on CAR-T cell refinement.
Fig. 5.

Structure of CAR-T cells
Principles and achievements of CAR-T cell therapy
CAR-T cells are generated by expressing tumor-specific CARs on the plasma membrane of T cells. The structure of CARs usually includes three parts: the extracellular antigen-binding domain, the linker/transmembrane domain, and the intracellular signaling domain.293 The extracellular antigen-binding region is designed utilizing the sequences of antibodies, ligands and peptides to specifically bind with TAAs. The transmembrane domain is responsible for connecting the extracellular binding domain with the intracellular signal domain and fixing it on the cell membrane.294,295 The intracellular signaling region, including the CD3-zeta domain and the costimulatory domain(s), transduces the signal of antigen recognition to the cell, mediating T-cell activation. The evolution of CAR-T cells has undergone four generations. For the first, second and third generation CAR designs, the intracellular signal transduction regions included zero, one and two costimulatory domains, respectively; the fourth generation usually refers to “armored” CAR-T cells with additional expression of immune-stimulatory factors or cytokines to further enhance T-cell activity.56,282
CAR-T cell therapy involves the integration of synthetic biology (CAR design), viral technology (CAR transduction) and cell manufacturing (CAR-T cell expansion) (Fig. 6).296 The introduction of the CAR generates tumor-specific activation potential in engineered T cells, while ex vivo culture and expansion allow the bypassing of tumor-induced immune suppression.297–299 As a result, large numbers of tumor-specific cells are infused back into the patient.300–302 CAR-T-cell therapy has shown promising clinical results against several types of cancers. At present, 6 CAR-T-cell therapy drugs have been approved for marketing worldwide, including four targeting CD19 in B-cell leukemia/lymphoma (Kymriah from Novartis; Yescarta and Tecartus from Kite/Gilead; Breyanzi from Bristol-Myers Squibb) and two targeting APRIL or BCMA in multiple myeloma (Abecma from Bristol-Myers Squibb and Cilta-Cel from J&J and Legend Biotech. Of the six products, two have also been approved by the Chinese FDA, including Yescarta (with the name of Achilles by Fosun Kite) and Breyanzi (with the name of Requilense by JW Therapeutic). In addition, there have been almost 1000 clinical trials registered for CAR-T cell treatment against various types of tumors.303
Fig. 6.

Workflow of CAR-T therapy
Challenges of CAR-T cell therapy
Despite the clinical successes listed above, the broader application of CAR-T cell therapy is still complicated by challenges from different aspects. First, when attacking tumor cells, CAR-T cells may cause severe side effects and toxicities that can be lethal. Second, the cytotoxicity of some CAR-T cells is not highly tumor-specific and may cause damage to normal tissue. Third, the manufacturing process of most CAR-T cell products is time consuming, which may result in further deterioration of some patients' tumors during the cell-producing window period. Furthermore, the long-term efficacy of CAR-T therapy against blood cancer still requires long-term follow-up observation, while CAR-T cell therapy application for solid tumors needs further study. These challenges will dictate the development of the entire field of T-cell engineering in the future.
Cytokine release syndrome (CRS), also known as a “cytokine storm”, is the most frequently observed adverse reaction with CAR-T treatment. After CAR-T cell infusions, the systemic inflammatory response can be elicited by the rapid rise of IL-6 and IL-13.304–308 The clinical manifestations mainly include fever, fatigue, headache, epilepsy, nausea, chills, and dyspnea. Patients with severe CRS may develop acute respiratory distress syndrome, hypotension, tachycardia, liver damage, renal failure and fulminant hemophagocytic lymphohistiocytosis (HLH), which can all become lethal.290,309 CRS usually occurs within a week after CAR-T infusion, with peaks occurring 1 to 2 weeks after infusion. It is worth noting that in the pathophysiological process of CRS, in addition to activated CAR-T cells, endogenous immune cells, such as monocytes, macrophages, and/or dendritic cells, are involved in the synthesis and release of various cytokines and clinical CRS symptoms.306,307
Immune effector cell-associated neurotoxicity syndrome (ICANS) refers to nervous system toxicity after CAR-T-cell infusions. The incidence of ICANS is closely correlated with CRS, with the rate differing between clinical studies but can be as high as 50%.310,311 The symptoms can manifest as mild behavioral abnormalities, unresponsiveness, aphasia, and epilepsy and are more common in patients with B-ALL than in those with other diseases. Mild ICANS is often reversible, but the etiology of ICANS is still unclear and may be related to various factors, such as cytokine release, infiltration of CAR-T cells into the central nervous system, and the dose of CAR-T-cell infusion.312,313
On-target, off-tumor toxicity is commonly observed in patients with B-cell malignancies after CAR-T therapy, as CAR-T cell targets (CD19, CD20 and/or CD22) are expressed in both normal and malignant B cells. The resulting B-cell aplasia can lead to hypoimmunoglobulinemia, and regular intravenous immunoglobulins can reduce the risk of opportunistic infections.314,315 However, the duration of B-cell aplasia is also an indicator of functional CAR-T-cell persistence and superior antitumor response. For CAR-T cells targeting other TAAs, the on-target, off-tumor toxicity needs additional attention. A clinical study using HER-2 CAR-T cells resulted in severe damage to the patients’ cardiac and respiratory systems.316 Due to the difficulty in finding TAAs that are exclusively expressed by tumor cells, CAR manipulation may be required to modify the activation potential against different antigen expression densities.317
Uncertain long-term efficacy is indicated in most clinical studies of CAR-T cells, even against leukemia, where the most promising CAR-T cell clinical response was observed. Several studies have shown that for B-ALL, although the clinical remission rate can reach more than 80%, the recurrence rate within one year can also reach more than 40%. This may be related to the inability of the engineered T cells to persist in the patients due to a variety of immune escape factors expressed by cancer cells, causing T-cell senescence and exhaustion.318,319 Furthermore, most clinical studies of CAR-T cell therapy against solid tumors have shown unsatisfactory results, despite some evidence of antitumor activity or even complete responses in some patients.320 CAR-T cells targeting solid tumors face challenges including vascular disorders that block T-cell infiltration, limited options for TAAs and tumor heterogeneity, which causes antigen escape. The application of CAR-T cells in solid tumors needs further exploration.319,321
Strategies of CAR-T cell refinement
Leveraging specificity with broad targeting
The long-term antitumor function of CAR-T cells has been complicated by tumor recurrence post-infusion. In view of the risk of antigen escape after CAR-T treatment, the design of a bispecific CAR-T that targets multiple TAAs can be adopted. These CAR-T cells are named “OR-gated”, meaning that the expression of either TAA on tumor cells can elicit CAR-T-cell activation. For B-ALL treated with CD19 CAR-T cells, recurrent malignant cells may downregulate CD19 but maintain CD22 expression. Therefore, “OR-gated” CARs targeting both CD19 and CD22 have the potential to reduce antigen escape.297 However, there are huge obstacles to applying “OR-gated” CAR-T-cell therapy in solid tumors.322,323 Given that solid tumor TAAs can hardly meet the criteria of stringent tumor specificity, off-target effects can be more significant if CAR-T cells require either of two TAAs for activation.324–326 In contrast, designing an “AND-gated” CAR construct, which needs both TAAs to activate T cells, can reduce the probability of on-target, off-tumor effects.327 The application of these CAR-T-cell designs will be highly dependent on the nature of the targeted tumors and clinical needs.
Enhancement of long-term antitumor effects
While tumor antigen escape accounts for many instances of postinfusion recurrence, quite a few relapsed cases still maintain the expression of the targeted antigen,328 indicating that CAR-T-cell dysfunction is an important contributor to treatment failure. Therefore, improving the fitness of CAR-T cells, including the activation potential, proliferation and survival capability, and prolonging the survival time of CAR-T cells in patients is one of the key directions to enhance clinical responses.329,330 Intriguingly, most strategies to optimize CAR-T-cell products are focused on preventing the overactivation and subsequent exhaustion and/or apoptosis of CAR-T cells.331 The approaches include modification of the manufacturing environment,332 pre-enrichment of memory T-cell subsets,333 CAR construct engineering to reduce signaling domains,334 and combination of small molecules to inhibit activation signals.335 Most of these methods have shown responses superior to those of traditional CAR-T therapy in preclinical models, which needs to be further validated for their safety and efficacy in clinical studies.
Reduction of manufacturing cost
To date, all approved CAR-T-cell therapies use autologous T cells to generate the therapeutic product. The production of this highly personalized therapy requires high cost.336 The CAR-T products approved by Novartis and Kite are priced at 475,000 USD and 373,000 USD, respectively, and the high prices limit their market potential. Novartis' CAR-T products yielded $12 million in revenue in the first quarter of 2018, only 30% of the expected revenue; Kite's CAR-T products also yielded less than expected within two months of approval. In addition to the high cost, the quality and stability of CAR-T-cell therapy have been major concerns. Autologous T cells are inconsistent in their quality and quantity, especially in patients who have been heavily pretreated with radiation and chemotherapy.337 The development of allogeneic “universal” CAR-T products aims to address these challenges. The technology has the potential to turn CAR-T cells into “off-the-shelf” drugs, with the advantages of large-scale production, lower cost and consistent characteristics. At present, although most general CAR-T cell therapies are still in the preclinical or early clinical stage, their attractive therapeutic potential is enough to serve as a strong driving force for continued research and development for the future benefit of more patients.338
Toxicity control
The toxicity and side effects exhibited by CAR-T cell therapy indicate that some control programs need to be developed to regulate the activity of CARs. A large number of methods have been used to control the safety of CAR-T cells; these include the rapid removal of infused cells by installing a suicide switch, which can be controlled by small molecules or antibodies. Commonly used suicide switches include inducible caspase-9 (iCasp9), thymidine kinase (HSV-TK) in herpes simplex virus, and suicide epitopes. However, such a suicide switch clears all therapeutic CAR-T cells, which compromises the antitumor response. Therefore, noncytotoxic reversible systems that do not clear CAR-T cells are under development and have the potential to maintain the balance between maintaining cytotoxicity and controlling toxic responses307 (Table 1).
Table 1.
List of CAR-T therapies available
| Product | Company | Approve times | Target | Indications | Price |
|---|---|---|---|---|---|
| Kymaiah | Novartis | 2017 | CD19 |
B-cell non-Hodgkin's lymphoma that failed first- or second-line therapy Acute lymphoblastic leukemia |
$475,000 |
| Tecartus | Gilead | 2020 | CD19 | $373,000 | |
| Yescarta | Kite | 2021 | CD19 | Large B-cell lymphoma or follicular lymphoma | $373,000 |
| Breyanzi | BMS | 2021 | CD19 | B-cell precursor acute lymphoblastic leukemia | $410,300 |
| Abecma | BMS and Bluebird Bio | 2021 | CD19-BCMA | Relapsed and refractory large B-cell lymphoma after second-line or above systemic therapy | $438,000 |
| Akilence | Fosunkite | 2021 | CD19 | Specific non-Hodgkin's lymphoma | ¥1,200,000 |
| JCAR014 | WuXi Junuo | 2017 | CD19 | Aggressive B-cell non-Hodgkin’s lymphoma (NHL) | – |
NK cell therapy
NK cells are another important type of immune cell that can mediate direct cytotoxicity. Mechanistically, NK cells play a key role in the first line of defense against cancer, mediating antitumor effects through two pathways: direct cytotoxicity through the release of post-perforin and granzyme or death receptors and the regulatory effect by secreting cytokines and chemokines that activate APCs and T cells.339–341 Therefore, in addition to drugs targeting the ICs on NK cells, which were discussed earlier in this article, ACT using NK cells is also under rapid development.342,343 There are many similarities between ACT strategies built around NK cells and T cells, despite the differences between the innate and acquired immune systems.
Similar to T cells, NK cells can also be transduced to express CARs. The development of CAR-NK cells followed the evolution of CAR-T cell therapy, and CAR-NK cells often directly adopt CAR-T cell designs. In 2020, the first CD19-targeted CAR-NK clinical study with confirmed safety and evidence of efficacy against B-cell malignancies was reported.344 Moreover, many preclinical studies have confirmed the antitumor activity of CAR-NK cells targeting other types of tumors.345,346
Advantages of NK cell therapy
The most important advantage of NK cell therapy relies on the nature of NK cells as part of the innate immune response. Compared with allogeneic T-cell products, allogeneic NK cells are significantly less concerning in terms of GvHD.347 Technological advances have also made it possible to expand NK cells in large numbers using feeder cells,238 providing a stable resource to manufacture “off-the-shelf” products with more controllable costs.
Because NK cytotoxicity is triggered by “missing-self” recognition, NK cells, in particular, have the capability of killing tumor cells with MHC downregulation. NK cells also have special killing ability to virus-infected cells, making them particularly suitable for the treatment of HPV- or EBV-associated tumors. Recent studies have also found that NK cells can inhibit the formation of tumor-associated blood vessels. In addition, NK cells are the most critical mediator of ADCC and have great potential to be combined with targeted antibody therapy. Furthermore, according to the clinical results of NK therapy, the incidence rates of CRS and ICANS are significantly reduced compared with those of CAR-T therapy,344 making it possible for this strategy to be applied in patients with less stringent limitations of age and prior treatment.
Challenges of NK cell therapy
Although allogeneic NK cells can provide a sufficient amount of starting material for ACT, freeze‒thaw cycles can significantly reduce NK-cell viability and cytotoxicity. Moreover, the in vivo expansion potential of NK cells is not as robust as that of CAR-T cells, which may lead to tumor recurrence early after infusion. Indeed, in a clinical study of CD19-CAR-NK cells, no correlation was found between the infusion doses and clinical outcome,344 indicating the major challenge of sustaining NK-cell activation against tumors.
For CAR-NK cell therapy, most current CARs have been directly adopted from CAR-T cells. The location of the CAR-binding epitope and its distance from the surface of CAR-NK cells may affect cytotoxicity in a T-cell-independent manner. The relatively high number of cells in infusions also makes NK approaches sensitive to insertional mutagenesis caused by viral CAR vectors. The Sleeping Beauty transposon system and mRNA transfection strategy, which have both been successfully applied to CAR-T cell production, remain to be evaluated as practical methods to generate CAR-NK cells.
Directions for the future development of NK cell therapy
NK cytotoxicity can be affected by multiple immunosuppressive mechanisms in the TME, including IL-10, indoleamine 2,3-dioxygenase, prostaglandin E2, transforming growth factor beta (TGF-β) and hypoxia.348,349 Enhancing NK-cell cytotoxicity and persistence in vivo is believed to be the major direction of advancing NK therapy.
Cytokines to support NK-cell maintenance
IL-15 has been identified as a key cytokine that enhances NK-cell activity. In syngeneic mouse models of cancers such as melanoma, colorectal cancer, lymphoma and lung cancer, injection of IL-15 was well tolerated and facilitated the expansion of NK cells. IL-15 can therefore be used as a monotherapy and as an adjuvant for NK-cell adoptive cell therapy. In a study targeting non-Hodgkin's lymphoma, medium and high concentrations of IL-15 effectively improved the survival rate of patients. IL-15 is also the main factor that induces NK-cell expansion in NK-cell culture in vitro.350 Therefore, multiple engineered NK-cell designs incorporate the expression of IL-15,344 and these designs are currently being tested in clinical studies.
Combination with NK checkpoint blockade
In 2011, the anti-KIR monoclonal antibody (lirilumab) was licensed to BMS for late-stage clinical development at a total price of $440 million and then tested in seven clinical trials. CD94/NKG2A-targeting monalizumab from AstraZeneca was soon developed as an NK ICB therapy. Moreover, NK-cell stimulatory receptors, including NKG2D, NCR, CD226, and CD16, provide targets for agonistic antibodies. Of note, most monotherapies targeting NK-cell checkpoints have failed to yield promising clinical responses; therefore, combination with NK-cell ACT might be a strategy for maximizing the stimulatory function of antibodies.
The “off-the-shelf” nature of NK-cell products makes their broad application possible. NK cells can specifically recognize and target cells with MHC downregulation, which can compensate for reduced antitumor T cell function. With more in-depth research on NK-cell activation and maintenance, future therapeutic methods must not only generate tumor-specific NK cells but must also increase their persistence in vivo to enhance their therapeutic potency.
CRISPR technology advances cellular immunotherapy
Recently, CRISPR/Cas9 technology has greatly improved our understanding of tumor genomics and contributed to cancer immunotherapy.351–353 Using this genome editing system, therapeutic immune cells can be further engineered to enhance tumor recognition and reduce exhaustion (Fig. 7).350,354–356 The first clinical study using CRISPR-engineered T cells was initiated in 2016 by Sichuan University in China. In 2020, a clinical study reported the use of PD-1 knockout T cells to treat patients with NSCLC that was refractory to radiotherapy and chemotherapy;357 it demonstrated that CRISPR engineering is safe in T cells, which paves the way for combining CRISPR technology with other T-cell modification approaches. NY-ESO-1 TCR-T cell therapy with CRISPR-mediated knockout of TCR and PD-1 represents the first tumor-specific T cells with further genetic modifications tested in the clinic.358 Moreover, the CRISPR gene editing system allows for broader application of allogeneic T-cell therapy.359 When allogeneic products are depleted of their endogenous TCRs and HLA molecules, they become less likely to be rejected and have a reduction in GvHD potential.360 Alternatively, CRISPR-mediated screening systems have been applied in multiple clinical studies. Such screening of tumor cells can identify targets that render tumors more sensitive to T-cell cytotoxicity.361 Moreover, screenings performed directly on therapeutic immune cells, such as CAR-T cells, have identified and validated critical factors that can be exploited in future research to further potentiate cellular immunotherapy.341,362–364
Fig. 7.

Workflow of CRISPR technology-based immunotherapy
Biomarkers for immunotherapy—lessons from ICB and CAR-T cells
Biomarkers for ICB
The overall impressive clinical effect of ICB has led to several approvals of related treatments. However, not all patients can benefit from ICB treatment, making it critical to identify biomarkers for efficacy prediction. For patients to receive accurate and effective treatment, biomarkers are responsible for screening and classifying patients, accurately identifying patients with drug response, and allowing them to receive the best treatment as soon as possible.365–367 Indeed, inappropriate application may even cause disease progression,368 illustrating the need for ICB to be prescribed in a personalized manner based on the analyses of certain biomarkers.
PD-L1 was used as the first biomarker for anti-PD-1 treatment, which was included in the prescription guide of pembrolizumab.369–372 However, PD-L1 can be induced by interferon and many other immunological signaling pathways during treatment,373 which undermines the utilization of PD-L1 as a predictive biomarker for ICB. The first reported study of acquired resistance to anti-PD-1 ICB identified mutations involved in interferon and antigen presentation pathways, which have become critical biomarkers to predict relapse post ICB.374 Further studies have identified additional mutations and immunosuppressive molecules that are associated with a poor prognosis in ICB-treated patients.375–377 In contrast, the T-cell inflammatory gene expression profile (GEP) and somatic copy number variation (SCNA) are correlated with a good prognosis in ICB-treated patients.378,379
At present, common or potential biomarkers related to immunotherapy efficacy have been reported mainly in the following categories based on their accessibility: (i) surface markers, including PD-L1 and some other inhibitory receptors, which can be examined by immunohistochemistry of tumor tissues; (ii) genetic biomarkers, such as tumor mutation load (TMB), mismatch repair system deficiency (dMMR), high microsatellite instability (MSI-H), neoantigens and mutations of the antigen presentation pathway, which all require genomic analyses of the tumor; and (iii) circulating tumor DNA (ctDNA), which is accessible by analyzing peripheral blood.380–382 Some of these biomarkers have been verified by phase III clinical trials and are widely used in the clinic.383–385 More biomarkers reflecting immune efficacy are still under continuous research and testing.386 One good example was the study from Sun Yat-sen University Cancer Hospital, which was a comprehensive analysis of genomic data identifying POLE and POLD1 gene mutations that can be used as independent biomarkers for predicting the efficacy of immunotherapy across cancers, which can provide more accurate guidance for the clinical application of immunotherapy384 (Table 2).
Table 2.
List of biomarkers for immunotherapy
| Biomarker | Origin | T cell source (Y/N) | Target of immunotherapy (Y/N) | Clinical acceptance |
|---|---|---|---|---|
| PD-L1 | Tumor tissue | N | Y | Broad |
| TMB | Tumor tissue | N | N | Broad |
| MSI-H | Tumor tissue | N | N | Limited |
| dMMR | Tumor tissue | N | N | Limited |
| cDNA | Plasma | N | N | Limited |
MSI-H and MMR
MSI-H refers to the variation in the length of short and repeated DNA sequences, which may include insertions, deletions or mutations caused by MMR functional defects.387–389 The MSI phenomenon was first found in colorectal cancer in 1993. According to the degree, it can be divided into the following: MSI-H, MSI-L and microsatellite stability.389,390 Mismatch repair (MMR) is a DNA damage repair mechanism against the wrong insertion, deletion and mismatch of bases that may occur in the process of DNA replication or recombination.391,392 The system consists of a series of specific DNA mismatch repair enzymes, which usually depend on four key genes: MLH1, PMS2, MSH2 and MSH6. Germline depletion of MMR genes is the “gold standard” for the diagnosis of Lynch syndrome.393 Due to the functional inactivation of MMR genes, patients with Lynch syndrome often simultaneously show MSI-H status and MMR defects (dMMR), which are also shared by some tumors.394
At present, it is recognized that dMMR/MSI-H is used as a prognostic factor for stage II colorectal cancer. For stage II colorectal cancer patients with the dMMR/MSI-H phenotype, grade 3/4 differentiation (low differentiation) is not considered a high-risk factor.393,395 Regarding ICB treatment, multiple clinical studies have shown that PD-1 antibodies can lead to survival benefits in patients with dMMR/MSI-H tumors.396,397 In May 2017, pembrolizumab was approved for solid tumor patients with MSI-H or dMMR who have progressed after previous treatment and have no satisfactory alternative treatment.386,396,398 In 2017 and 2018, the FDA successively approved the treatment of metastatic colorectal cancer patients with MSI-H or dMMR after treatment with fluorouracil, oxaliplatin and nivolumab alone or in combination with ipilimumab. Therefore, MSI-H and dMMR can be used as primary screening methods.399
TMB
TMB refers to the total number of mutations, base substitutions, and insertion or deletion errors detected per million bases.400,401 A number of clinical studies have confirmed that patients with high TMB tumors are more likely to benefit from ICB treatment.402–404 This correlation was found in tumors that generally have high immunogenicity, such as melanoma, urothelial cancer and NSCLC, as well as colorectal cancer, which has different degrees of immunogenicity between individuals.405 In the analyses of clinical and preclinical studies, TMB was also found to be associated with tumor T-cell infiltration and an “inflamed” TME and may be related to the high expression of immunoreactive neoantigens in these tumors.406–408
Neoantigens
Neoantigens are proteins that are specifically expressed only in tumor cells and can be recognized and killed by T cells of the immune system. During the development of tumor cells, nonsynonymous mutations change the amino acid coding sequence, causing tumor cells to express abnormal proteins in a tumor-specific manner. These proteins may also activate the immune system and lead to an attack by the immune system on tumor cells. These antigens from abnormal proteins that can be recognized by immune cells are neoantigens. Neoantigens have two major characteristics: first, they are unique to tumor cells and are not found in normal tissues or cells; second, these antigens should have corresponding TCRs that recognize them specifically.409
Neoantigens can be ideal biomarkers for ICB if clearly defined as “immunogenic neoepitopes”, which will reflect the extent of tumor immunogenicity with more accuracy than MSI status, MMR status and TMB level, so if neoantigens that bind with high affinity to MHC can be produced, the possibility of an immune response will be higher. However, it remains challenging to validate the “quality” of neoantigens, which refers to their capability to elicit immune recognition and activation.410 Currently, neoantigens are mostly used to support other biomarkers. For example, melanomas with TMB>10 often produce neoantigens with high frequency and are sensitive to PD-1 inhibitors; in contrast, melanomas with TMB<1 are unlikely to generate neoantigens and are insensitive to PD-1 inhibitors. The direct utilization of neoantigens as biomarkers would rely on the development of assays and algorithms that can precisely detect both the quantity and quality of neoantigens within a tumor.411
Circulating tumor DNA (ctDNA)
The DNA in the cell can sometimes dissociate into the blood, forming circulating DNA. ctDNAs are fragments derived from four sources: necrotic tumor cells, apoptotic tumor cells, circulating tumor cells, and exosomes secreted by tumor cells.402 By detecting gene mutations in blood ctDNA, we can understand the changes in tumor cells in the body in real time, thus providing a clinical basis for tumor treatment and prognosis.403,404 A research team from the Princess Margaret Cancer Center in Canada conducted a prospective phase II clinical trial in patients with 5 different types of advanced solid tumors who were treated with pembrolizumab. Analysis of the correlation between changes in ctDNA levels and immune efficacy after treatment revealed that ctDNA levels were associated with the clinical response to ICB.412 Another research group also proved that ctDNA could be a good biomarker for the immunotherapy response in different types of cancers. Therefore, ctDNA has the potential to become an easily accessible biomarker used for screening and prediction in a cost-effective manner.413,414
Biomarkers for CAR-T-cell therapy
Although the initial CR rate of CAR-T-cell therapy against B-cell leukemia can be as high as 90%, a significant proportion of patients develop tumor recurrence.328 Furthermore, not all patients with lymphoma or multiple myeloma achieved satisfactory response with CAR-T cell therapy, warranting the discovery of biomarkers that can help classify specific cohorts of patients who can benefit from this type of treatment. To date, no biomarkers have been utilized to guide the enrollment of patients, but some CAR-T cell intrinsic and extrinsic factors have shown intriguing correlations with the therapeutic response.415,416
Tumor antigen expression
The nature of CAR-T cells as a targeted therapy requires the expression of TAA on tumor cells to elicit T-cell activity, which is indeed the most critical biomarker for CAR-T cell efficacy. As tumor antigen escape is a major mechanism of tumor recurrence post CAR-T cell therapy, some studies have also revealed that downregulation, instead of complete loss of TAA, inhibits CAR-T cell function.417 While CAR-T cells can be designed to increase their sensitivity to low-level TAAs, the TAA expression density might become a predictive biomarker.417
Product characteristics
The inconsistent quality of therapeutic products has been a major challenge of autologous CAR-T cell therapy. The starting materials from patients can be significantly altered by the many lines of prior treatment. As a result, the composition of T-cell subsets showed more variation across patients with tumors compared with healthy donors. Therefore, the difference in product quality is an important contributor to clinical outcomes. In 2018, Kite Pharma evaluated the polyfunctionality of their CAR-T-cell products, which reflects the capability to produce multiple cytokines at the single-cell level and correlates with the therapeutic effect against lymphoma. Some other studies profiled the phenotypes and transcriptomes of the infusion products, identifying a memory-like population enriched in the products that ultimately lead to superior responses. With ongoing validations in larger-scale studies, these product characteristics can be exploited as useful biomarkers to predict the clinical response before infusion.317
Other types of immunotherapies
Tumor vaccines
Preventive tumor vaccines can prevent the development of certain cancers, including the HPV vaccine against cervical cancer, vaginal cancer, vulvar cancer, anal cancer and condyloma acuminatum and the HBV vaccine to prevent liver cancer.418–420 Therapeutic tumor vaccines involve the injection of tumor antigens in the form of free peptides or peptides loaded on APCs to activate immune cells to restore their autonomous antitumor ability. In preclinical models, therapeutic tumor vaccines have been confirmed to prevent cancer growth and metastasis and reduce relapse after the termination of other types of treatment.414,421,422 Tumor vaccines are mainly divided into the following four types: tumor whole-cell vaccines, genetically engineered vaccines, protein peptide vaccines, and dendritic cell vaccines.423–425
Dendritic cells (DCs) were first discovered by the Canadian scientist and 2011 Nobel Laureate Dr. Ralph M. Steinman. Dendritic cells are a heterogeneous group of innate immune cells with antigen-presenting functions and are considered the only immune cell type that can activate naive T cells.426 In the context of cancer, however, the number and vitality of DCs are not enough to trigger sufficient T-cell activation against malignant cells. Therefore, DCs can be isolated from cancer patients, primed and loaded with tumor antigens in vitro. The resulting dendritic cells express tumor antigens on their surface, and the activated DCs are then used to initiate an immune response, which is referred to as a “DC vaccine”.427,428 Unfortunately, DC isolation, priming, and antigen loading can be complex, time consuming, and labor intensive, which limits the capacity of DC vaccine application. However, the immune response elicited by tumor antigen-primed DCs is highly tumor-specific with limited side effects.416,429 Dr. Ralph M. Steinman also benefited from the DC vaccine, which extended his lifespan from an expected few months to four and a half years with refractory pancreatic cancer. On 29 April 2010, the US FDA approved a therapeutic tumor vaccine, sipuleucel-T from Dendreon, for treating advanced prostate cancer.430,431 With the continuous progress of science and technology, a variety of tumor vaccines have gradually entered the clinic.
Neoantigens and immunotherapy
Neoantigens are protein fragments present on cancer cells, offering a novel way to achieve cancer cell-specific targeting.432 Neoantigen vaccines are individualized based on a patient's specific tumor profile. Interest in the field has grown since the first human clinical trials using the neoantigen vaccine began in 2015.433,434 Despite subtle differences between platforms, the general steps of making neoantigen vaccines are mostly conserved; they include (1) tumor biopsy, in which tumor samples are taken from patients for genomic purification; (2) whole-exome sequencing of tumor cells and normal cells, which allows researchers to search for unique mutations in tumor cells; (3) prediction and selection of specific neoantigens as targets; and (4) development of personalized vaccines, which is based on predicted neoantigens and can be achieved using various approaches, including peptides, mRNA and DCs.435–440 The most critical and challenging step has been the identification of patient-specific neoantigens. While some platforms are focused on developing predictive algorithms to achieve greater accuracy, others use in silico prediction together with functional tests to ensure that the neoantigen indeed triggers immune cell activation.441 While the latter enables the validation of targets, it can be extremely time-consuming and expensive. Technical advances are therefore warranted before the application of neoantigen vaccines to larger-scale clinical studies.431
Oncolytic virus
Cancer patients with additional virus infection often experience worsening disease.442,443 However, viruses can also be modified to specifically target cancer cells. These “oncolytic viruses” are generated by genome editing and large-scale screenings, the readout of which includes the lysis ability against cancer cells while sparing normal cells. The resulting oncolytic virus candidates can replicate and subsequently lyse tumor cells, which releases more viral particles into the tumor sites.444,445 Therefore, a small dose of virus can be expanded in vivo. Talimogene laherparepvec (OncoVex, T-VEC) is an oncolytic virus agent approved by the FDA for use in melanoma in 2015.446,447 T-VEC is a type I herpes simplex virus and is an oncolytic immunotherapy preparation based on herpes simplex virus. Herpes simplex virus is genetically edited to help the virus evade the immune system, allowing the modified virus to replicate in cancer cells in a targeted manner. On the one hand, it can directly lyse cancer cells, and on the other hand, it can activate the human immune system by releasing antigens inside the tumor and priming “bystander” immune cells.448
Oncolytic virus therapy has demonstrated great potential in combination with other types of cancer immunotherapy. With their capability to mediate tumor antigen spread, oncolytic viruses can lead to an increase in lymphocytes infiltrating the tumors, which enhances the antitumor efficacy of ICB treatment. Another approach is to use oncolytic viruses as delivery vehicles in combination with cellular immunotherapy. With additional genetic engineering, the cytolytic function of the viruses can be suppressed while allowing for the expression of synthetic molecules (i.e., truncated CD19 as a CAR-T cell target). Combining these viruses with CD19-targeted CAR-T cell therapy can thus achieve homogenous expression of TAA and overcome the challenge of tumor antigen escape.449
Targeting the suppressive TME
Macrophages
These “soldiers” of the innate immune system remove damaged, senescent, and dangerous cells, but in cancer, macrophages facilitate their immune escape and have become an important field of drug development. While early researchers developed treatments by modulating the interactions between tumor cells and macrophages, this complex field of biology has been rewritten, and the application of macrophage therapy has slowed. The great progress of genetic engineering provides a greater possibility to use synthetic biology to redirect macrophages to fight tumors.450,451 Several researchers from Carisma Therapeutics in the United States and the University of Pennsylvania published a review titled “Macrophage-based approaches for cancer immunotherapy” in cancer research, outlining the progress made in macrophage immunotherapy and the impact of chimeric antigens. The rise of somatic macrophage therapy.452,453
Macrophages in cancer
Macrophages have a variety of functions, including removing cellular debris and pathogens and regulating inflammatory responses. Macrophages are also highly plastic cells that can switch from one phenotype to another depending on microenvironmental stimuli and signals.454 The activation state of macrophages is usually divided into two categories: M1-type macrophages and M2-type macrophages (Fig. 8).
Fig. 8.

Characteristics of M1 and M2 macrophages (Nature Reviews Immunology)
Certain M2 macrophage subsets are involved in promoting tumor progression and mediating immune suppression.455 Mechanistically, it has been found that tumors recruit monocytes and macrophages to the TME and polarize them to the M2-like phenotype. The central goal of macrophage-targeted cancer therapy is to reprogram tumor-associated macrophages (TAMs) into the proinflammatory (antitumor) subtype, which can be achieved in two ways: reducing the number of M2-like TAMs and/or restoring the antitumor function of TAMs within the TME.456 Advances in technology, such as single-cell sequencing, have allowed researchers to see different macrophage subsets with multiple complex biological functions in different TMEs and gain a deeper understanding of the relationship between macrophages and tumor immunotherapy.457
Inhibitory and stimulatory molecules on TAMs
The most established approach to target TAMs is the blockade of the colony-stimulating factor-1 (CSF-1, also known as the M-CSF)/CSF1R axis. This approach reduces the number of TAMs, which can also be associated with the repolarization of TAMs toward the M1 phenotype.458 However, the leading CSF1R inhibitor, cabiralizumab from Five Prime Therapeutics, did not show promising clinical responses in a series of clinical trials,459 indicating that the potency of CSF1R inhibition needs to be revisited.
The tumorigenic function of TAMs can also be mediated by TGF-β, an anti-inflammatory molecule normally expressed by macrophages during injury repair. Blockade of TGF-β and concurrent treatment with a STING agonist in a mouse model resulted in tumor regression by upregulating the expression of type I interferons.455,460
Toll-like receptors (TLRs) are involved in innate immune sensing. TLR agonists can increase monocyte recruitment/infiltration and induce macrophage repolarization toward the proinflammatory phenotype.461 TAMs also express CD40, and CD40 agonists can prevent tumor growth and attenuate drug resistance.462,463
These inhibitory (CSF1R, TGF-β) and stimulatory (TLRs, CD40) molecules can all be exploited as targets to restore the proinflammatory function of TAMs. However, the TME is composed of numerous immunosuppressive cells with functional redundancy, which may result in the clinical observation that targeting a single cell type will not lead to sufficient TME alterations to eradicate tumors.
CD47
CD47, also known as integrin-related protein, belongs to the immunoglobulin superfamily, which regulates cell proliferation, migration and apoptosis by binding to signal regulatory protein α (SIRPα) on the surface of macrophages or dendritic cells.464 CD47 is overexpressed on the surface of most tumor cells as a “do not eat me” signal that escapes phagocytosis by macrophages. Blocking the CD47/SIRPα pathway can induce the phagocytotic function of macrophages to target tumors, which was demonstrated in mouse xenograft models.465
Forty Seven was the company that developed the first-in-class CD47 antibodies. After its acquisition by Gilead Sciences, a phase Ib clinical study of the CD47 antibody magrolimab (Hu5F9-G4) was launched. The results showed that magrolimab combined with azacitidine in patients with myelodysplastic syndrome (MDS) reached an overall response rate of 92%.466 In addition, another clinical study showed that magrolimab combined with rituximab resulted in an overall response rate of 90% in lymphoma patients.467 These results have led to many additional drug candidates targeting the CD47/SIRPα pathway, such as TTI-621/622, ALX148, and OSE-172 (Table 3).
Table 3.
Variety of drugs targeting macrophage therapy
| Company | Candidate | Target | Status |
|---|---|---|---|
| Gilead (forty seven) | Magrolimab | CD47 | Phase II |
| Trillium | TTI-661; TTI-662 | CD47 | Phase I |
| ALX oncology | ALX148 | CD47 | Phase I |
| I-Mab | TJC-4 | CD47 | Phase I |
| Innovent biologics | IBI188 | CD47 | Phase I |
| Arch oncology | AO-176 | CD47 | Phase I |
| TG therapeutics/novlmmune | TG-1801 | CD47 | Phase I |
| BMS/celgene | CC-95251 | SIRPα | Phase I |
| OSE immunotherapeutic | OSE-172(BI-765063) | SIRPα | Phase I |
| Alector | AL008 | SIRPα | Preclinical |
| Gilead (forty seven) | FSI-189 | SIRPα | Preclinical |
TTI-621/622 are two CD47 inhibitors developed by Trillium Co.; they are SIRPa-Fc fusion proteins coupled with IgG1 and IgG4, respectively. IgG4 showed slightly weaker binding to Fc receptors on immune cells, which may compromise the potency but enhance protection against CD47-expressing nontumor cells.468 was developed by ALX Oncology and contains two SIRPα high-affinity CD47 binding domains linked to the inactive Fc domain of human immunoglobulins. Its Fc domain has been reengineered to inhibit Fcγ receptors. ALX148 is in phase I studies in patients with solid tumors and lymphomas in combination with a variety of chemotherapy agents.469 OSE-172 is a monoclonal antibody developed by OSE Immunotherapeutics that targets SIRP-α. In addition, it was designed not to bind with SIRP-γ, which plays a role in the migration of T cells in tissues. Therefore, this antibody will not inhibit the infiltration of T cells. OSE-172 is currently being tested in a phase I clinical study.470 In addition to single-drug applications, bifunctional antibodies with CD47/PD-L1, CD47/VEGF and other targets are under development, and we look forward to more studies to bring us more surprising drugs.471,472
Despite the clinical efficacy, the toxicity of CD47-targeted treatment cannot be ignored. CD47 is ubiquitously expressed on blood cells, which significantly compromises tumor specificity.463 The clinical study of magrolimab found that the number of red blood cells, hematocrit and hemoglobin decreased on the 2nd day of application of the drug and dropped to the lowest point on the 5th to 7th day. Although it was claimed that such a level of anemia can be relieved by supportive care and blood counts can return to normal within 2–3 weeks,473 prevention of anemia has been the key focus of toxicity management after anti-CD47 therapy. Magrolimab uses IgG4 for its Fc fragment to avoid excessive killing of red blood cells and has been applied in a low-dose induction manner during clinical application. When a low dose of antibody was given, mild anemia was induced, which subsequently accelerated hematopoiesis in the bone marrow, with the hope of inducing resistance to higher doses of medication.474 The clinical safety of this approach remains to be investigated in larger cohorts of patients.
Additional “do not eat me” signals may contribute to the acquired resistance of anti-CD47 therapy, and new targets for macrophages continue to emerge. CD24 is another “do not eat me” signaling molecule that is overexpressed in several types of human cancers, and its receptor Siglec-10 is expressed on TAMs.475 CD24 and other molecules with similar functions are emerging as new targets for reprogramming TAMs for immunotherapy.
Engineering macrophages
Macrophages have a long history of being exploited in ACT. In the late 1980s, Andreesen's group in Germany enrolled 15 patients. Monocytes were harvested from the patients, cultured in autologous serum for seven days, and “educated” with IFN-γ to differentiate into M1-like macrophages.476 Of the seven patients with peritoneal carcinoma, ascites disappeared in two of the seven patients who received intraperitoneal macrophage injection. Crucially, other than low-grade fever and abdominal discomfort after intraperitoneal injection, no other side effects were reported. These nonengineered, IFN-γ-activated macrophages have been considered a clinically safe agent but have limited efficacy. One of the main underlying reasons is that in the absence of tumor specificity, an IFN-γ-stimulated M1 phenotype can easily transform into the M2 phenotype in the TME. These challenges suggest that the development of macrophage therapy requires the addition of targeted activating receptors and a more durable approach to M1 macrophage polarization.476
To solve these problems, the use of genetic modification to enhance the antitumor ability of macrophages has gradually attracted more attention. One straightforward strategy is the depletion of inhibitory signals such as SIRPα. The antitumor efficacy of SIRPα-depleted macrophages has been preliminarily shown in combination with radiotherapy.477 Another approach is to engineer macrophages to express CARs (CAR-Ms) (Fig. 9). The first challenge of CAR-M generation is the difficulty of transducing macrophages. In 2016, it was reported that the chimeric adenoviral vector efficiently transduced macrophages.478 The resulting CAR-Ms, generated with the Ad5f35 vector, eliminated tumor cells more efficiently than macrophages with M1-oriented differentiation. CAR-Ms also induced proinflammatory features of the surrounding TME. The presence of M2 macrophages did not affect the tumor-killing ability of CAR-M cells, highlighting their resistance to TME immunosuppression. In addition, CAR-Ms exhibited a stronger T-cell-stimulating ability and were able to present antigens to T cells after phagocytosis, recruiting resting and activated T cells to tumors.479
Fig. 9.

Multipotent antitumor mechanisms of CAR-M therapy
As part of the innate immune response, macrophages can also be applied in an allogeneic manner; therefore, CAR-Ms have advantages as readily available products. Recent studies have also generated CAR-Ms from induced pluripotent stem cells, which can be further exploited to generate CAR-M banks as “off-the-shelf” products.480 CAR-Ms can also be combined with other macrophage-targeted therapies, such as anti-CD47.481,482
Targeting myeloid-derived suppressor cells (MDSCs)
MDSCs originate from hematopoietic stem cells (HSCs) as a result of altered myelopoiesis.483 This transient myelopoiesis is terminated after the stimulus is removed, and myeloid cell homeostasis is then restored.484 However, in chronic inflammation, cancer, and autoimmune diseases, persistent myelopoiesis may occur to prevent widespread tissue damage of the host, constantly generating IMCs. These cells have distinct characteristics, such as immature phenotype and morphology, relatively weak phagocytic function, and anti-inflammatory and immunosuppressive functions,485 matching their descriptive name.
In the 1970s, it was found that abnormal myeloid cells had inhibitory effects on other immune cells. After that, the surface markers Gr-1 and CD11b were adopted to define these immunosuppressive myeloid cells, especially in tumor-bearing mice.486 In humans, these myeloid cells are phenotypically characterized by the expression of CD34, CD14 and CD15 and functionally characterized by their ability to suppress T-cell activation.487 The term MDSCs indeed refers to a group of heterogeneous cells, which can be roughly divided into granulocytic (G-MDSCs or PMN-MDSCs) and monocytic (M-MDSCs) subtypes. Recent studies using single-cell profiling have uncovered the complexity of MDSC populations, which may differ between disease conditions and even individuals.485 Accumulating evidence indicates that the presence of MDSCs is one of the basic characteristics of tumor progression.488
MDSCs have been believed to be one of the most critical obstacles against effective cancer immunotherapy, which has also inspired research into therapeutic strategies targeting these cells. MDSCs exert multiple functions to influence T cells, Treg cells, DC cells and NK cells (Fig. 10). In the early 2000s, before the term MDSC was generated, vitamin D and all-trans retinoic acid (ATRA) were shown to reduce immature myeloid cells in patients with head and neck squamous cell carcinoma and metastatic renal cell carcinoma, respectively.489 Currently, MDSC-targeted therapy can be broadly classified into five types: (i) therapies that inhibit the expansion and recruitment of MDSCs; (ii) therapies that restore normal myeloid differentiation; (iii) therapies that target the IC molecules on MDSCs; (iv) therapies that block the inhibitory molecules secreted from MDSCs; and (v) therapies that directly deplete MDSCs.480,490 Since the enrichment and activation of MDSCs appear to be universal features of different malignancies, targeting these cells may have broader application potential. It is also intriguing to combine MDSC-targeted therapy with existing chemotherapy or immunotherapy agents, which have early evidence of clinical responses.490,481
Fig. 10.

Function of MDSCs
Targeting B cells
Tumor-associated B cells (TABs)
B cells, which are responsible for antibody production, have long been overlooked for their functions in tumor immunity. However, a recent study showed that similar to macrophages, B cells in the context of tumors, named TABs, display both pro- and anti-inflammatory functions, and the abundance of anti-inflammatory B cells is correlated with the resistance of melanoma to ICB treatment.491 However, there are not yet strategies to specifically deplete anti-inflammatory TABs. As proinflammatory TABs can attract effector T cells to tumors, depletion of all TABs has been believed to reduce, rather than induce, the antitumor immune response.492 TABs are enriched in tertiary lymphoid-tissue structures (TLSs) within tumor tissues, where they can be activated to recognize cancer cells. NSCLC patients with high levels of B cells within TLSs in their tumors were more likely to respond well to immunotherapy.493 It is still only partially understood how the numbers and characteristics of TABs differ across different tumor types. How B cells can be reprogrammed by tumors to inhibit T-cell activation is another topic that requires in-depth investigation.
Engineering B cells
As they have the capacity of antibody production, B cells can be upgraded into “biofactories”. Immusoft developed the Immune System Programming (ISP) platform, which can engineer B cells and differentiate them into plasma cells for ACT. The cells will efficiently secrete tens of thousands of personalized antibodies, enabling sustained delivery of therapeutic proteins.494,495 Walking Fish Therapeutics is another company focused on B cells, aiming to develop B-cell therapies for cancer, rare diseases, and autoimmune diseases, as well as regenerative therapies and recombinant antibodies.
Reversing the inhibitory signals from the TME
IDO
IDO is a tryptophan-metabolizing enzyme that can convert tryptophan to kynurenine and is overexpressed in multiple types of tumors.496 The tumor suppressor gene BIN1 negatively regulates the expression of IDO. In mouse models, the depletion of BIN1 has been confirmed to induce IDO expression and immune suppression in tumors.497 IDO can enhance the motility of cancer cells and inhibit the proliferation and function of tumor-targeting T cells.498 Mouse xenograft models show that treatment with IDO inhibitors can significantly increase the level of T cells and have a significant tumor-suppressive effect. IDO-targeted drugs have shown efficacy as monotherapies in preclinical models but can be more effective in combination with ICBs targeting CTLA-4 or PD-1/PD-L1.499–503
There are mainly four small molecule inhibitors targeting IDO that are currently undergoing clinical research: indoximod, navoximod, epacadostat and BMS-986205. In the reported phase I study of epacadostat, 52 patients with advanced solid tumors were enrolled. Unfortunately, no objective response was observed, but seven patients had stable disease for more than 16 weeks.502 In another phase I/II clinical study, epacadostat was combined with pembrolizumab, and a total of 54 patients with solid tumors were included. The overall response rate was 57%.493 However, an ECHO-301 clinical study suggested that epacadostat combined with pembrolizumab did not improve PFS in patients with advanced melanoma.504 Currently, a number of phase III clinical studies of epacadostat were closed without a satisfactory outcome, which also resulted in the termination of phase III clinical studies of BMS-986205 and indoximod. The enrolled patients were not stratified based on IDO1 expression levels, which may lead to the underestimation of efficacy. However, these terminated clinical studies indicate a lack of understanding of the underlying mechanism between IDO and various immune cells.504 IDO inhibitors have been combined with anti-CTLA-4, chemotherapy and other ICs. More types of drugs and larger clinical studies of inhibitors are underway, and the collection of more data will reveal the true antitumor effect of IDO-targeted drugs.505
IL-41
To explain the limited clinical efficacy of IDO inhibitors, some researchers hypothesized that there may be other activation pathways for aryl hydrocarbon receptors. From an analysis of 32 different types of tumors from the TCGA database, it was discovered that tumors with high expression of IDO1 or IDO2 also had high levels of aryl hydrocarbon receptors.506 A more in-depth study identified IL-41 with the highest occurrence rate among the aryl hydrocarbon receptor-related modules. IL-41 was later proven to enhance tumor aggressiveness and suppress antitumor immunity. Targeting IL-41 may be a new avenue of immunotherapy, as demonstrated in preclinical models,507 which needs to be validated in clinical studies.
Adenosine
Adenosine is an essential component of RNA synthesis. However, adenosine has also been shown to inhibit T-cell functions in the TME.505 CD39 is an enzyme involved in extracellular adenosine production and is highly expressed in various human tumors.506–508 Some tumor cells exhibited CD39 overexpression compared to normal cells. Moreover, multiple cell types with elevated CD39 expression levels in the TME are vascular endothelial cells, fibroblasts, and some immune cells.494,495,509,510 CD39 has been found to play an important role in a variety of immune cells. In macrophages, CD39 acts as a “molecular rheostat” that controls inflammation and regulates the balance between macrophage differentiation.511–513 In Treg cells, CD39 enhanced immunosuppressive activity. CD39 expression in MDSCs is positively correlated with NSCLC tumor stage, but its function remains unknown.511 Due to its important role in the TME, CD39 has become an emerging target for researchers to develop tumor immunotherapy.512,513 In vivo and in vitro data suggest that targeting CD39 sensitizes tumors to PD-1/PD-L1 ICB treatment. Therefore, CD39 therapy combination with ICB might become a new type of tumor immunotherapy.506,514
Conclusion and perspectives
Immunotherapy has become the “fourth bullet” of antitumor treatment following surgery, chemotherapy and radiotherapy. Here, we introduced the history as well as the different classifications of immunotherapy. Early studies to dissect the immune system have allowed the utilization of specific cells to achieve antitumor immunity. The clinical success of ICB and CAR-T cell therapy has revealed the potential that the “edited” immune system in patients with tumors can be “reprogrammed” to fight against malignant cells, which has been further highlighted by many ongoing preclinical and clinical studies targeting almost every type of immune cell.
Despite great success in the clinic, tumor immunotherapy is facing challenges including toxicity control and a low response rate. Although adverse events associated with immunotherapy may indicate the activation of the immune system, severe toxicity is sometimes lethal. Therefore, toxicity control of immunotherapy is not expected to compromise the therapeutic benefit, which is reminiscent of the efforts in controlling GvHD in allogeneic bone marrow transplantation while maintaining the graft-versus-leukemia effect. Moreover, immunotherapy is able to mediate long-term disease-free survival in some patients, but extending the therapeutic potency to more individuals is another barrier against its broader application.
With all emerging novel checkpoint inhibitors and different novel technologies, there is always one critical question for cancer immunotherapy: do patients respond or not respond? It is important to have good biomarkers to predict the response, and it is also important to seek a clinical strategy to improve the clinical response rate.
With a deeper understanding of the mechanisms underlying every type of therapeutic agent, there is also the potential of combinational treatment with chemotherapy, radiation or other types of immunotherapies. PD-1 (PD-L1). Only a small proportion of cancer patients have high TMB and other clinical biomarkers; therefore, the population for clinical application is very limited. Moreover, the therapeutic effect is sometimes not as good as expected. It has been confirmed that the response rate of immune drug monotherapy is low, only 10–30% in most solid tumors. Based on the response limitations, immune combination therapy has become a new research hot spot, and the combined use with other therapies can improve the efficacy and expand the beneficiary population. Combination therapy can expand the indications and applicable population of immunotherapy and effectively improve drug efficacy. To date, the FDA has approved four PD-1/PD-L1 drug combination therapies. An increasing number of combined immunotherapies are expected to be approved in the future, and combined immunotherapies are becoming conventional treatments in the clinic.
Another challenge of tumor immunotherapy is the lack of biomarkers to predict the immunotherapy response. Tumor immunotherapy has achieved remarkable success. However, it is very important to screen groups that may benefit from treatment, predict drug efficacy, and guide clinical treatment. The selection of biomarkers has become the key factor for immunotherapy treatment. The detection of molecular biomarkers such as PD-L1, TMB, MSI, and gene mutation has been widely used, but it is well known that these biomarkers are not good enough to guide clinical decision making, and factors such as cancer type, tumor heterogeneity, and dynamic changes in tumors will affect the accuracy and specificity. Some patients who may benefit from treatment will be missed by these methods, while some patients with high PD-L1 expression and TMB-H are not sensitive to immunotherapy at all. Therefore, a better understanding of the mechanism of cancer immunotherapy, improving existing biomarkers, and developing new tumor markers are important future directions of immunotherapy. Studies have shown that the TME is also an important factor affecting the effect of immunotherapy. According to the degree of infiltration, tumors can be divided into different types. Generally, tumors with a high level of immune cell infiltration (“hot” tumors) are more likely to respond to immunotherapy, while “cold” tumors generally need to be transformed into hot tumors through treatment. New biomarker systems integrating the results of immune profiling, clinical history and tumor biology are being developed with the hope of predicting therapeutic outcomes before the application of immunotherapy.
Novel technologies are emerging to solve these clinical problems. New strategies, such as CRISPR-mediated screening and cell engineering, enable the identification and targeting of specific genes that sensitize tumor cells to immunotherapy or potentiate immune cells for sustained tumor-targeting capability. Neoantigen identification and TCR-T cells have also brought new hope for immunotherapy of certain types of cancer that are not sensitive to checkpoint inhibitors and CAR-T cells. Other novel technologies, such as TIL therapy, DC-based therapy and CAR-NK cell therapy, are also making ground-breaking progress, and products could be expected in clinical application very soon.
Overall, a combination of solid research on tumor immunology and advanced technology for manipulating immune cells will shed light on the future development of cancer immunotherapy. The advancement of cancer immunotherapy calls for more integrated clinical and basic research programs, which will then allow for the analyses of unmet clinical needs in a comprehensive manner and subsequent guidance of research directions.
Supplementary information
Acknowledgements
This work was supported by the National Key R&D Program of China (2019YFC1315701 to Y.S.), sponsored by National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital & Shenzhen Hospital, Chinese Academic of Medical Sciences and Peking Union Medical College, Shenzhen (SZ2020ZD004, E010121002), supported by Sanming Project of Medicine in Shenzhen (No. SZSM201812062, No. SZSM201612097), Shenzhen Science and Technology Program (KCXFZ20201221173008022) and Shenzhen Key Medical Discipline Construction Fund (No.SZXK075). Some icons or graphic elements in all of our figures were adapted from BioRender.com (2022), and final schematic illustrations were created and integrated by our original design.
Author contributions
Y.-l.S. conceived and supervised the study and revised the paper. Y.-l.S., D.-r.W. and X.-l.W. formatted the paper and wrote different parts of the paper. Y.-l.S. and X.-l.W. organized the figures. All authors have read and approved the article.
Competing interests
The authors declare no competing interests.
Footnotes
These authors contributed equally: Dong-Rui Wang, Xian-Lin Wu
Contributor Information
Dong-Rui Wang, Email: dongrui-wang@zju.edu.cn.
Ying-Li Sun, Email: scaroll99@hotmail.com.
Supplementary information
The online version contains supplementary material available at 10.1038/s41392-022-01136-2.
References
- 1.Bender E. Cancer Immunotherapy. Nature. 2017;552:S61. doi: 10.1038/d41586-017-08699-z. [DOI] [PubMed] [Google Scholar]
- 2.Feld E, Mitchell TC. Immunotherapy in melanoma. Immunotherapy. 2018;10:987–998. doi: 10.2217/imt-2017-0143. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA. Il-2: the first effective immunotherapy for human cancer. J. Immunol. 2014;192:5451–5458. doi: 10.4049/jimmunol.1490019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi L, et al. Combining Il-2-based immunotherapy with commensal probiotics produces enhanced antitumor immune response and tumor clearance. J. Immunother. Cancer. 2020;8:e000973. doi: 10.1136/jitc-2020-000973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang Y, Lundqvist A. Immunomodulatory effects of Il-2 and Il-15; implications for cancer immunotherapy. Cancers. 2020;12:3586. doi: 10.3390/cancers12123586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mansurov A, et al. Immunoengineering approaches for cytokine therapy. Am. J. Physiol. Cell Physiol. 2021;321:C369–C383. doi: 10.1152/ajpcell.00515.2020. [DOI] [PubMed] [Google Scholar]
- 7.Margolin K. Cytokine therapy in cancer. Expert Opin. Biol. Ther. 2008;8:1495–1505. doi: 10.1517/14712598.8.10.1495. [DOI] [PubMed] [Google Scholar]
- 8.Tamassia N, et al. Cytokine production by human neutrophils: revisiting the “dark side of the moon”. Eur. J. Clin. Invest. 2018;48(Suppl 2):e12952. doi: 10.1111/eci.12952. [DOI] [PubMed] [Google Scholar]
- 9.Vossen AC, et al. Fc receptor binding of anti-Cd3 monoclonal antibodies is not essential for immunosuppression, but triggers cytokine-related side effects. Eur. J. Immunol. 1995;25:1492–1496. doi: 10.1002/eji.1830250603. [DOI] [PubMed] [Google Scholar]
- 10.Mosinska P, Gabryelska A, Zasada M, Fichna J. Dual functional capability of dendritic cells - cytokine-induced killer cells in improving side effects of colorectal cancer therapy. Front. Pharm. 2017;8:126. doi: 10.3389/fphar.2017.00126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaud G, Lesourne R, Love PE. Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 2018;18:485–497. doi: 10.1038/s41577-018-0020-8. [DOI] [PubMed] [Google Scholar]
- 12.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu. Rev. Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klenerman P, Oxenius A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016;16:367–377. doi: 10.1038/nri.2016.38. [DOI] [PubMed] [Google Scholar]
- 15.Jiang S, Yan W. T-cell immunometabolism against cancer. Cancer Lett. 2016;382:255–258. doi: 10.1016/j.canlet.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Cheroutre H, Husain MM. Cd4 Ctl: living up to the challenge. Semin. Immunol. 2013;25:273–281. doi: 10.1016/j.smim.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanagi Y, et al. A human T cell-specific cdna clone encodes a protein having extensive homology to immunoglobulin chains. Nature. 1984;308:145–149. doi: 10.1038/308145a0. [DOI] [PubMed] [Google Scholar]
- 18.Allison JP, McIntyre BW, Bloch D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J. Immunol. 1982;129:2293–2300. [PubMed] [Google Scholar]
- 19.Martin PJ, et al. A 44 kilodalton cell surface homodimer regulates interleukin 2 production by activated human T lymphocytes. J. Immunol. 1986;136:3282–3287. [PubMed] [Google Scholar]
- 20.Allison JP. Cd28-B7 interactions in T-cell activation. Curr. Opin. Immunol. 1994;6:414–419. doi: 10.1016/0952-7915(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 21.Nandi D, Gross JA, Allison JP. Cd28-mediated costimulation is necessary for optimal proliferation of murine Nk cells. J. Immunol. 1994;152:3361–3369. [PubMed] [Google Scholar]
- 22.Krummel MF, Allison JP. Cd28 and Ctla-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Harper K, et al. Ctla-4 and Cd28 activated lymphocyte molecules are closely related in both mouse and human as to sequence, message expression, gene structure, and chromosomal location. J. Immunol. 1991;147:1037–1044. [PubMed] [Google Scholar]
- 24.Balzano C, Buonavista N, Rouvier E, Golstein P. Ctla-4 and Cd28: similar proteins, neighbouring genes. Int J. Cancer Suppl. 1992;7:28–32. [PubMed] [Google Scholar]
- 25.Buonavista N, et al. Molecular linkage of the human Ctla4 and Cd28 Ig-superfamily genes in yeast artificial chromosomes. Genomics. 1992;13:856–861. doi: 10.1016/0888-7543(92)90169-S. [DOI] [PubMed] [Google Scholar]
- 26.Linsley PS, et al. Coexpression and functional cooperation of Ctla-4 and Cd28 on activated T lymphocytes. J. Exp. Med. 1992;176:1595–1604. doi: 10.1084/jem.176.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tivol EA, et al. Loss of Ctla-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of Ctla-4. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 28.Walunas TL, Bluestone JA. Ctla-4 regulates tolerance induction and T cell differentiation in vivo. J. Immunol. 1998;160:3855–3860. [PubMed] [Google Scholar]
- 29.Waterhouse P, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–988. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 30.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by Ctla-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 31.Korman AJ, Peggs KS, Allison JP. Checkpoint blockade in cancer immunotherapy. Adv. Immunol. 2006;90:297–339. doi: 10.1016/S0065-2776(06)90008-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kwon ED, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc. Natl Acad. Sci. USA. 1997;94:8099–8103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodi FS, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hodi FS, et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (Checkmate 067): 4-Year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018;19:1480–1492. doi: 10.1016/S1470-2045(18)30700-9. [DOI] [PubMed] [Google Scholar]
- 35.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of Pd-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo J. 1992;11:3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishimura H, et al. Development of lupus-like autoimmune diseases by disruption of the Pd-1 gene encoding an itim motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/S1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 37.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-Stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 38.Dong H, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 39.Freeman GJ, et al. Engagement of the Pd-1 immunoinhibitory receptor by a novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okazaki T, Honjo T. Pd-1 and Pd-1 ligands: from discovery to clinical application. Int. Immunol. 2007;19:813–824. doi: 10.1093/intimm/dxm057. [DOI] [PubMed] [Google Scholar]
- 41.Latchman Y, et al. Pd-L2 is a second ligand for Pd-1 and inhibits T cell activation. Nat. Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 42.Smith KM, Desai J. Nivolumab for the Treatment of Colorectal Cancer. Expert Rev. Anticancer Ther. 2018;18:611–618. doi: 10.1080/14737140.2018.1480942. [DOI] [PubMed] [Google Scholar]
- 43.Khasraw M, Reardon DA, Weller M, Sampson JH. Pd-1 Inhibitors: do they have a future in the treatment of glioblastoma? Clin. Cancer Res. 2020;26:5287–5296. doi: 10.1158/1078-0432.CCR-20-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rossi C, Casasnovas RO. Pd-1 inhibitors in patients with hodgkin lymphoma. Eur. J. Cancer. 2022;164:114–116. doi: 10.1016/j.ejca.2021.06.059. [DOI] [PubMed] [Google Scholar]
- 45.Seki M, et al. Inflammatory myopathy associated with Pd-1 inhibitors. J. Autoimmun. 2019;100:105–113. doi: 10.1016/j.jaut.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 46.Henry L, et al. Pd-1 inhibitors in metastatic colorectal cancer. Anz. J. Surg. 2021;91:E758–E759. doi: 10.1111/ans.16878. [DOI] [PubMed] [Google Scholar]
- 47.Weiden PL, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N. Engl. J. Med. 1979;300:1068–1073. doi: 10.1056/NEJM197905103001902. [DOI] [PubMed] [Google Scholar]
- 48.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl Acad. Sci. USA. 1989;86:10024–10028. doi: 10.1073/pnas.86.24.10024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maher J, et al. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric Tcrzeta /Cd28 receptor. Nat. Biotechnol. 2002;20:70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 50.Porter DL, et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weber EW, Maus MV, Mackall CL. The emerging landscape of immune cell therapies. Cell. 2020;181:46–62. doi: 10.1016/j.cell.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He X, et al. Bispecific and split Car T cells targeting Cd13 and Tim3 eradicate acute myeloid leukemia. Blood. 2020;135:713–723. doi: 10.1182/blood.2019002779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Myers GD, Verneris MR, Goy A, Maziarz RT. Perspectives on outpatient administration of Car-T cell therapy in aggressive B-cell lymphoma and acute lymphoblastic leukemia. J. Immunother. Cancer. 2021;9:e002056. doi: 10.1136/jitc-2020-002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soltantoyeh T, et al. Chimeric antigen receptor (Car) T cell therapy for metastatic melanoma: challenges and road ahead. Cells. 2021;10:1450. doi: 10.3390/cells10061450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cobb DA, et al. Targeting of the alphav beta3 integrin complex by Car-T cells leads to rapid regression of diffuse intrinsic pontine glioma and glioblastoma. J. Immunother. Cancer. 2022;10:e003816. doi: 10.1136/jitc-2021-003816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Majzner RG, Mackall CL. Tumor antigen escape from Car T-cell therapy. Cancer Disco. 2018;8:1219–1226. doi: 10.1158/2159-8290.CD-18-0442. [DOI] [PubMed] [Google Scholar]
- 57.Parker KR, et al. Single-cell analyses identify brain mural cells expressing Cd19 as potential off-tumor targets for Car-T immunotherapies. Cell. 2020;183:126–142. doi: 10.1016/j.cell.2020.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brown CE, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006;6:836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 60.Xie W, et al. PD-1/PD-L1 Pathway and Its Blockade in Patients with Classic Hodgkin Lymphoma and Non-Hodgkin Large-Cell Lymphomas. Curr. Hematol. Malig. Rep. 2020;4:372–381. doi: 10.1007/s11899-020-00589-y. [DOI] [PubMed] [Google Scholar]
- 61.Yu H, et al. Efficacy and safety of Pd-L1 inhibitors versus Pd-1 inhibitors in first-line treatment with chemotherapy for extensive-stage small-cell lung cancer. Cancer Immunol. Immunother. 2022;71:637–644. doi: 10.1007/s00262-021-03017-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whooley, J. et al. Pd-1 Inhibitors in Esophageal Cancer: A Systematic Review of the Oncological Outcomes Associated with Pd-1 Blockade and the Evolving Therapeutic Paradigm. Dis Esophagus. 35, doab063 (2022). [DOI] [PubMed]
- 63.Li B, Chan HL, Chen P. Immune checkpoint inhibitors: basics and challenges. Curr. Med. Chem. 2019;26:3009–3025. doi: 10.2174/0929867324666170804143706. [DOI] [PubMed] [Google Scholar]
- 64.Xu F, Jin T, Zhu Y, Dai C. Immune checkpoint therapy in liver cancer. J. Exp. Clin. Cancer Res. 2018;37:110. doi: 10.1186/s13046-018-0777-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.He X, Xu C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020;30:660–669. doi: 10.1038/s41422-020-0343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Miguel M, Calvo E. Clinical challenges of immune checkpoint inhibitors. Cancer Cell. 2020;38:326–333. doi: 10.1016/j.ccell.2020.07.004. [DOI] [PubMed] [Google Scholar]
- 67.Johnson DB, Sullivan RJ, Menzies AM. Immune Checkpoint inhibitors in challenging populations. Cancer-Am. Cancer Soc. 2017;123:1904–1911. doi: 10.1002/cncr.30642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tian Y, Abu-Sbeih H, Wang Y. Immune checkpoint inhibitors-induced hepatitis. Adv. Exp. Med. Biol. 2018;995:159–164. doi: 10.1007/978-3-030-02505-2_8. [DOI] [PubMed] [Google Scholar]
- 69.Haanen JB, Robert C. Immune checkpoint inhibitors. Prog. Tumor Res. 2015;42:55–66. doi: 10.1159/000437178. [DOI] [PubMed] [Google Scholar]
- 70.Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015;33:1974–1982. doi: 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abril-Rodriguez G, Ribas A. Snapshot: immune checkpoint inhibitors. Cancer Cell. 2017;31:848. doi: 10.1016/j.ccell.2017.05.010. [DOI] [PubMed] [Google Scholar]
- 72.Venkatachalam S, McFarland TR, Agarwal N, Swami U. Immune checkpoint inhibitors in prostate cancer. Cancers. 2021;13:2187. doi: 10.3390/cancers13092187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kroemer G, Zitvogel L. Immune checkpoint inhibitors. J. Exp. Med. 2021;218:e20201979. doi: 10.1084/jem.20201979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hui E. Immune checkpoint inhibitors. J. Cell Biol. 2019;218:740–741. doi: 10.1083/jcb.201810035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Deczkowska A, Amit I, Schwartz M. Microglial immune checkpoint mechanisms. Nat. Neurosci. 2018;21:779–786. doi: 10.1038/s41593-018-0145-x. [DOI] [PubMed] [Google Scholar]
- 76.Qin S, et al. Novel immune checkpoint targets: moving beyond Pd-1 and Ctla-4. Mol. Cancer. 2019;18:155. doi: 10.1186/s12943-019-1091-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rotte A. Combination of Ctla-4 and Pd-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019;38:255. doi: 10.1186/s13046-019-1259-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Andrews LP, Yano H, Vignali D. Inhibitory receptors and ligands beyond Pd-1, Pd-L1 and Ctla-4: breakthroughs or backups. Nat. Immunol. 2019;20:1425–1434. doi: 10.1038/s41590-019-0512-0. [DOI] [PubMed] [Google Scholar]
- 79.Hong Y, Ding ZY. Pd-1 inhibitors in the advanced esophageal cancer. Front. Pharm. 2019;10:1418. doi: 10.3389/fphar.2019.01418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Annibali O, et al. Pd-1 /Pd-L1 checkpoint in hematological malignancies. Leuk. Res. 2018;67:45–55. doi: 10.1016/j.leukres.2018.01.014. [DOI] [PubMed] [Google Scholar]
- 81.Curran CS, Kopp JB. Pd-1 immunobiology in glomerulonephritis and renal cell carcinoma. Bmc Nephrol. 2021;22:80. doi: 10.1186/s12882-021-02257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dong Y, Sun Q, Zhang X. Pd-1 and its ligands are important immune checkpoints in cancer. Oncotarget. 2017;8:2171–2186. doi: 10.18632/oncotarget.13895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Olson DJ, et al. Pembrolizumab plus ipilimumab following anti-Pd-1/L1 failure in melanoma. J. Clin. Oncol. 2021;39:2647–2655. doi: 10.1200/JCO.21.00079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Verma V, et al. Pd-1 blockade in subprimed Cd8 cells induces dysfunctional Pd-1(+)Cd38(Hi) cells and anti-Pd-1 resistance. Nat. Immunol. 2019;20:1231–1243. doi: 10.1038/s41590-019-0441-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang S, Li W. Pd-1 inhibitors for urothelial cancer: combination or sequential therapy? Lancet. 2021;396:1977. doi: 10.1016/S0140-6736(20)32672-6. [DOI] [PubMed] [Google Scholar]
- 86.Raj N, et al. Pd-1 blockade in advanced adrenocortical carcinoma. J. Clin. Oncol. 2020;38:71–80. doi: 10.1200/JCO.19.01586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Koyama S, et al. Adaptive resistance to therapeutic Pd-1 blockade is associated with upregulation of alternative immune checkpoints. Nat. Commun. 2016;7:10501. doi: 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Xu-Monette ZY, Zhou J, Young KH. Pd-1 expression and clinical Pd-1 blockade in B-cell lymphomas. Blood. 2018;131:68–83. doi: 10.1182/blood-2017-07-740993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kumagai S, et al. The Pd-1 Expression Balance Between Effector and Regulatory T Cells Predicts the Clinical Efficacy of Pd-1 Blockade Therapies. Nat. Immunol. 2020;21:1346–1358. doi: 10.1038/s41590-020-0769-3. [DOI] [PubMed] [Google Scholar]
- 90.Chamoto K, Al-Habsi M, Honjo T. Role of Pd-1 in immunity and diseases. Curr. Top. Microbiol. Immunol. 2017;410:75–97. doi: 10.1007/82_2017_67. [DOI] [PubMed] [Google Scholar]
- 91.Jin HT, Ahmed R, Okazaki T. Role of Pd-1 in regulating T-cell immunity. Curr. Top. Microbiol. Immunol. 2011;350:17–37. doi: 10.1007/82_2010_116. [DOI] [PubMed] [Google Scholar]
- 92.Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to Pd-1 inhibition. N. Engl. J. Med. 2017;377:2500–2501. doi: 10.1056/NEJMc1713444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nie M, et al. Pd-1/Pd-L pathway potentially involved in Itp immunopathogenesis. Thromb. Haemost. 2019;119:758–765. doi: 10.1055/s-0039-1679909. [DOI] [PubMed] [Google Scholar]
- 94.Tobias J, Steinberger P, Drinic M, Wiedermann U. Emerging targets for anticancer vaccination: Pd-1. ESMO Open. 2021;6:100278. doi: 10.1016/j.esmoop.2021.100278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yu X, Gao R, Li Y, Zeng C. Regulation of Pd-1 in T cells for cancer immunotherapy. Eur. J. Pharmacol. 2020;881:173240. doi: 10.1016/j.ejphar.2020.173240. [DOI] [PubMed] [Google Scholar]
- 96.Wolchok JD. Pd-1 blockers. Cell. 2015;162:937. doi: 10.1016/j.cell.2015.07.045. [DOI] [PubMed] [Google Scholar]
- 97.Leake I. Pd-1 inhibitors for oesophageal cancer. Nat. Rev. Gastroenterol. Hepatol. 2019;16:706. doi: 10.1038/s41575-019-0237-4. [DOI] [PubMed] [Google Scholar]
- 98.Hill M, et al. The paradoxical roles of inflammation during Pd-1 blockade in cancer. Trends Immunol. 2020;41:982–993. doi: 10.1016/j.it.2020.09.003. [DOI] [PubMed] [Google Scholar]
- 99.Dong Y, et al. Pd-1 blockade prevents the progression of oral carcinogenesis. Carcinogenesis. 2021;42:891–902. doi: 10.1093/carcin/bgab035. [DOI] [PubMed] [Google Scholar]
- 100.Barroso-Sousa R, Ott PA. Pd-1 inhibitors in endometrial cancer. Oncotarget. 2017;8:106169–106170. doi: 10.18632/oncotarget.22583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vicki Brower. Programmed death protein 1 inhibitors making inroads in multiple cancers. J. Natl Cancer Inst. 2015;5:djv141. doi: 10.1093/jnci/djv141. [DOI] [PubMed] [Google Scholar]
- 102.Geng Y, et al. Effect of Pd-1 inhibitor combined with X-ray irradiation on the inflammatory microenvironment and lung tissue injury in mice. J. Inflamm. Res. 2022;15:545–556. doi: 10.2147/JIR.S350112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen R, Zhou L. Pd-1 signaling pathway in sepsis: does it have a future? Clin. Immunol. 2021;229:108742. doi: 10.1016/j.clim.2021.108742. [DOI] [PubMed] [Google Scholar]
- 104.Normann MC, et al. Early experiences with Pd-1 inhibitor treatment of platinum resistant epithelial ovarian cancer. J. Gynecol. Oncol. 2019;30:e56. doi: 10.3802/jgo.2019.30.e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Heine A, Kristiansen G, Schild HH, Brossart P. Successful treatment of refractory leiomyosarcoma with the Pd-1 inhibitor nivolumab. Ann. Oncol. 2016;27:1813–1814. doi: 10.1093/annonc/mdw243. [DOI] [PubMed] [Google Scholar]
- 106.Yin B, et al. Immune-related organizing pneumonitis in non-small cell lung cancer receiving Pd-1 inhibitor treatment: a case report and literature review. J. Cancer Res. Ther. 2020;16:1555–1559. doi: 10.4103/0973-1482.204842. [DOI] [PubMed] [Google Scholar]
- 107.Riaz N, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171:934–949. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Passiglia F, et al. Looking for the best immune-checkpoint inhibitor in pre-treated nsclc patients: an indirect comparison between nivolumab, pembrolizumab and atezolizumab. Int. J. Cancer. 2018;142:1277–1284. doi: 10.1002/ijc.31136. [DOI] [PubMed] [Google Scholar]
- 109.Halmos B, et al. Pembrolizumab+chemotherapy versus atezolizumab+chemotherapy+/-bevacizumab for the first-line treatment of non-squamous Nsclc: a matching-adjusted indirect comparison. Lung Cancer. 2021;155:175–182. doi: 10.1016/j.lungcan.2021.03.020. [DOI] [PubMed] [Google Scholar]
- 110.Rodriguez-Abreu D, et al. Pemetrexed plus platinum with or without pembrolizumab in patients with previously untreated metastatic nonsquamous nsclc: protocol-specified final analysis from keynote-189. Ann. Oncol. 2021;32:881–895. doi: 10.1016/j.annonc.2021.04.008. [DOI] [PubMed] [Google Scholar]
- 111.Herbst RS, et al. Five year survival update from keynote-010: pembrolizumab versus docetaxel for previously treated, programmed death-ligand 1-positive advanced Nsclc. J. Thorac. Oncol. 2021;16:1718–1732. doi: 10.1016/j.jtho.2021.05.001. [DOI] [PubMed] [Google Scholar]
- 112.Awad MM, et al. Long-term overall survival from keynote-021 cohort G: pemetrexed and carboplatin with or without pembrolizumab as first-line therapy for advanced nonsquamous Nsclc. J. Thorac. Oncol. 2021;16:162–168. doi: 10.1016/j.jtho.2020.09.015. [DOI] [PubMed] [Google Scholar]
- 113.Morgensztern D, Herbst RS. Nivolumab and pembrolizumab for non-small cell lung cancer. Clin. Cancer Res. 2016;22:3713–3717. doi: 10.1158/1078-0432.CCR-15-2998. [DOI] [PubMed] [Google Scholar]
- 114.Wong J, et al. Ipilimumab and nivolumab/pembrolizumab in advanced hepatocellular carcinoma refractory to prior immune checkpoint inhibitors. J. Immunother. Cancer. 2021;9:e001945. doi: 10.1136/jitc-2020-001945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lin SY, et al. Tumor Pd-L1 expression and clinical outcomes in advanced-stage non-small cell lung cancer patients treated with nivolumab or pembrolizumab: real-world data in Taiwan. J. Cancer. 2018;9:1813–1820. doi: 10.7150/jca.24985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang H, et al. Pdl1 high expression without Tp53, Keap1 and Epha5 mutations could better predict survival for patients with nsclc receiving atezolizumab. Lung Cancer. 2021;151:76–83. doi: 10.1016/j.lungcan.2020.11.006. [DOI] [PubMed] [Google Scholar]
- 117.Lam TC, et al. Combination atezolizumab, bevacizumab, pemetrexed and carboplatin for metastatic Egfr mutated Nsclc after Tki failure. Lung Cancer. 2021;159:18–26. doi: 10.1016/j.lungcan.2021.07.004. [DOI] [PubMed] [Google Scholar]
- 118.Gunjur A, et al. Occult gastrointestinal perforation in a patient with Egfr-mutant non-small-cell lung cancer receiving combination chemotherapy with atezolizumab and bevacizumab: brief report. Clin. Lung Cancer. 2020;21:e57–e60. doi: 10.1016/j.cllc.2019.11.014. [DOI] [PubMed] [Google Scholar]
- 119.Nogami N, et al. Impower150 final exploratory analyses for atezolizumab plus bevacizumab and chemotherapy in key nsclc patient subgroups with Egfr mutations or metastases in the liver or brain. J. Thorac. Oncol. 2022;17:309–323. doi: 10.1016/j.jtho.2021.09.014. [DOI] [PubMed] [Google Scholar]
- 120.Goldman JW, et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive-stage small-cell lung cancer (Caspian): updated results from a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2021;22:51–65. doi: 10.1016/S1470-2045(20)30539-8. [DOI] [PubMed] [Google Scholar]
- 121.Motzer RJ, et al. Avelumab plus axitinib versus sunitinib in advanced renal cell carcinoma: biomarker analysis of the phase 3 Javelin renal 101 trial. Nat. Med. 2020;26:1733–1741. doi: 10.1038/s41591-020-1044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Albuquerque AO, Da SJH, Sartori GR, Martins DSJ. Computationally-obtained structural insights into the molecular interactions between pidilizumab and binding partners Dll1 and Pd-1. J. Biomol. Struct. Dyn. 2021;9:1–13. doi: 10.1080/07391102.2021.1885492. [DOI] [PubMed] [Google Scholar]
- 123.Gao S, et al. Neoadjuvant Pd-1 inhibitor (Sintilimab) in Nsclc. J. Thorac. Oncol. 2020;15:816–826. doi: 10.1016/j.jtho.2020.01.017. [DOI] [PubMed] [Google Scholar]
- 124.Powles T, et al. Pembrolizumab plus axitinib versus sunitinib monotherapy as first-line treatment of advanced renal cell carcinoma (Keynote-426): extended follow-up from a randomised, open-label, phase 3 trial. Lancet Oncol. 2020;21:1563–1573. doi: 10.1016/S1470-2045(20)30436-8. [DOI] [PubMed] [Google Scholar]
- 125.Zhang H, et al. Hyperprogressive disease in patients receiving immune checkpoint inhibitors. Curr. Probl. Cancer. 2021;45:100688. doi: 10.1016/j.currproblcancer.2020.100688. [DOI] [PubMed] [Google Scholar]
- 126.Shen P, et al. Hyperprogressive disease in cancers treated with immune checkpoint inhibitors. Front. Pharm. 2021;12:678409. doi: 10.3389/fphar.2021.678409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Champiat S, et al. Hyperprogressive disease: recognizing a novel pattern to improve patient management. Nat. Rev. Clin. Oncol. 2018;15:748–762. doi: 10.1038/s41571-018-0111-2. [DOI] [PubMed] [Google Scholar]
- 128.Kim CG, et al. Hyperprogressive disease during Pd-1/Pd-L1 blockade in patients with non-small-cell lung cancer. Ann. Oncol. 2019;30:1104–1113. doi: 10.1093/annonc/mdz123. [DOI] [PubMed] [Google Scholar]
- 129.Wang X, et al. The biomarkers of hyperprogressive disease in Pd-1/Pd-L1 blockage therapy. Mol. Cancer. 2020;19:81. doi: 10.1186/s12943-020-01200-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim CG, et al. Hyperprogressive disease during Pd-1 blockade in patients with advanced hepatocellular carcinoma. J. Hepatol. 2021;74:350–359. doi: 10.1016/j.jhep.2020.08.010. [DOI] [PubMed] [Google Scholar]
- 131.Assi T, Mir O. Hyperprogressive disease in leiomyosarcoma: a threat to the use of single-agent anti-Pd-(L)1 therapy? Immunotherapy. 2022;14:271–274. doi: 10.2217/imt-2021-0297. [DOI] [PubMed] [Google Scholar]
- 132.Lo RG, et al. Hyperprogressive disease upon immune checkpoint blockade: focus on non-small cell lung cancer. Curr. Oncol. Rep. 2020;22:41. doi: 10.1007/s11912-020-00908-9. [DOI] [PubMed] [Google Scholar]
- 133.Camelliti S, et al. Mechanisms of hyperprogressive disease after immune checkpoint inhibitor therapy: what we (don't) know. J. Exp. Clin. Cancer Res. 2020;39:236. doi: 10.1186/s13046-020-01721-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zappasodi R, et al. Ctla-4 blockade drives loss of treg stability in glycolysis-low tumours. Nature. 2021;591:652–658. doi: 10.1038/s41586-021-03326-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Penter L, et al. Molecular and cellular features of ctla-4 blockade for relapsed myeloid malignancies after transplantation. Blood. 2021;137:3212–3217. doi: 10.1182/blood.2021010867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Formenti SC, et al. Radiotherapy induces responses of lung cancer to Ctla-4 blockade. Nat. Med. 2018;24:1845–1851. doi: 10.1038/s41591-018-0232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thompson RH, Allison JP, Kwon ED. Anti-cytotoxic T lymphocyte antigen-4 (Ctla-4) immunotherapy for the treatment of prostate cancer. Urol. Oncol. 2006;24:442–447. doi: 10.1016/j.urolonc.2005.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Egen JG, Kuhns MS, Allison JP. Ctla-4: new insights into its biological function and use in tumor immunotherapy. Nat. Immunol. 2002;3:611–618. doi: 10.1038/ni0702-611. [DOI] [PubMed] [Google Scholar]
- 139.Cascone T, et al. Nodal immune flare mimics nodal disease progression following neoadjuvant immune checkpoint inhibitors in non-small cell lung cancer. Nat. Commun. 2021;12:5045. doi: 10.1038/s41467-021-25188-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Podlesnykh SV, et al. Peptide blocking Ctla-4 and B7-1 interaction. Molecules. 2021;26:253. doi: 10.3390/molecules26020253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Tiemann M, Atiakshin D, Samoilova V, Buchwalow I. Identification of Ctla-4-positive cells in the human tonsil. Cells. 2021;10:1027. doi: 10.3390/cells10051027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ren Z, et al. Ctla-4 limits anti-Cd20-mediated tumor regression. Clin. Cancer Res. 2017;23:193–203. doi: 10.1158/1078-0432.CCR-16-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Walker LSTreg. and Ctla-4: two intertwining pathways to immune tolerance. J. Autoimmun. 2013;45:49–57. doi: 10.1016/j.jaut.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu Y, Zheng P. How does an anti-Ctla-4 antibody promote cancer immunity? Trends Immunol. 2018;39:953–956. doi: 10.1016/j.it.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bengsch F, et al. Ctla-4/Cd80 pathway regulates T cell infiltration into pancreatic cancer. Cancer Immunol. Immunother. 2017;66:1609–1617. doi: 10.1007/s00262-017-2053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Pol J, Kroemer G. Anti-Ctla-4 immunotherapy: uncoupling toxicity and efficacy. Cell Res. 2018;28:501–502. doi: 10.1038/s41422-018-0031-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Gravitz L. Cancer immunotherapy. Nature. 2013;504:S1. doi: 10.1038/504S1a. [DOI] [PubMed] [Google Scholar]
- 148.Wing K, et al. Ctla-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 149.Sharma A, et al. Anti-Ctla-4 immunotherapy does not deplete Foxp3(+) regulatory T cells (Tregs) in human cancers. Clin. Cancer Res. 2019;25:1233–1238. doi: 10.1158/1078-0432.CCR-18-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Santoni G, et al. High Ctla-4 expression correlates with poor prognosis in thymoma patients. Oncotarget. 2018;9:16665–16677. doi: 10.18632/oncotarget.24645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Gaber T, et al. Ctla-4 mediates inhibitory function of mesenchymal stem/stromal cells. Int. J. Mol. Sci. 2018;19:2312. doi: 10.3390/ijms19082312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Disco. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 153.Wei SC, et al. Distinct cellular mechanisms underlie anti-Ctla-4 and anti-Pd-1 checkpoint blockade. Cell. 2017;170:1120–1133. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wei SC, et al. A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Disco. 2021;11:614–625. doi: 10.1158/2159-8290.CD-20-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Paz-Ares LG, et al. First-line nivolumab plus ipilimumab in advanced Nsclc: 4-year outcomes from the randomized, open-label, phase 3 checkmate 227 Part 1 trial. J. Thorac. Oncol. 2022;17:289–308. doi: 10.1016/j.jtho.2021.09.010. [DOI] [PubMed] [Google Scholar]
- 156.Wolchok JD, et al. Long-term outcomes with nivolumab plus ipilimumab or nivolumab alone versus ipilimumab in patients with advanced melanoma. J. Clin. Oncol. 2022;40:127–137. doi: 10.1200/JCO.21.02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Yasinska IM, et al. The Tim-3-Galectin-9 pathway and its regulatory mechanisms in human breast cancer. Front. Immunol. 2019;10:1594. doi: 10.3389/fimmu.2019.01594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Dixon KO, et al. Tim-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature. 2021;595:101–106. doi: 10.1038/s41586-021-03626-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 2017;276:97–111. doi: 10.1111/imr.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Acharya N, Sabatos-Peyton C, Anderson AC. Tim-3 finds its place in the cancer immunotherapy landscape. J. Immunother. Cancer. 2020;8:e000911. doi: 10.1136/jitc-2020-000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kim HS, et al. Glial Tim-3 modulates immune responses in the brain tumor microenvironment. Cancer Res. 2020;80:1833–1845. doi: 10.1158/0008-5472.CAN-19-2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.de Mingo PA, et al. Tim-3 regulates Cd103(+) dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell. 2018;33:60–74. doi: 10.1016/j.ccell.2017.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Harding JJ, et al. Blocking Tim-3 in treatment-refractory advanced solid tumors: a phase Ia/B study of Ly3321367 with or without an Anti-Pd-L1 antibody. Clin. Cancer Res. 2021;27:2168–2178. doi: 10.1158/1078-0432.CCR-20-4405. [DOI] [PubMed] [Google Scholar]
- 164.Suzuki K, et al. Immune-checkpoint profiles for T cells in bronchoalveolar lavage fluid of patients with immune-checkpoint inhibitor-related interstitial lung disease. Int. Immunol. 2020;32:547–557. doi: 10.1093/intimm/dxaa022. [DOI] [PubMed] [Google Scholar]
- 165.Girardi DM, et al. Cabozantinib plus nivolumab phase I expansion study in patients with metastatic urothelial carcinoma refractory to immune checkpoint inhibitor therapy. Clin. Cancer Res. 2022;28:1353–1362. doi: 10.1158/1078-0432.CCR-21-3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jones BE, et al. Fewer Lag-3(+) T cells in relapsing-remitting multiple sclerosis and type 1 diabetes. J. Immunol. 2022;208:594–602. doi: 10.4049/jimmunol.2100850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Huang CT, et al. Role of Lag-3 in regulatory T cells. Immunity. 2004;21:503–513. doi: 10.1016/j.immuni.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 168.Klumper N, et al. Lag3 (Lag-3, Cd223) Dna methylation correlates with Lag3 expression by tumor and immune cells, immune cell infiltration, and overall survival in clear cell renal cell carcinoma. J. Immunother. Cancer. 2020;8:e000552. doi: 10.1136/jitc-2020-000552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Robert CLag-3. and Pd-1 blockade raises the bar for melanoma. Nat. Cancer. 2021;2:1251–1253. doi: 10.1038/s43018-021-00276-8. [DOI] [PubMed] [Google Scholar]
- 170.Friedman LA, Ring KL, Mills AM. Lag-3 and Gal-3 in endometrial carcinoma: emerging candidates for immunotherapy. Int. J. Gynecol. Pathol. 2020;39:203–212. doi: 10.1097/PGP.0000000000000608. [DOI] [PubMed] [Google Scholar]
- 171.Sordo-Bahamonde C, et al. Lag-3 Blockade with relatlimab (Bms-986016) restores anti-leukemic responses in chronic lymphocytic leukemia. Cancers. 2021;13:2112. doi: 10.3390/cancers13092112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tu L, et al. Assessment of the expression of the immune checkpoint molecules Pd-1, Ctla4, Tim-3 and Lag-3 across different cancers in relation to treatment response, tumor-infiltrating immune cells and survival. Int. J. Cancer. 2020;147:423–439. doi: 10.1002/ijc.32785. [DOI] [PubMed] [Google Scholar]
- 173.Okagawa T, et al. Cooperation of Pd-1 and Lag-3 contributes to T-cell exhaustion in anaplasma marginale-infected cattle. Infect. Immun. 2016;84:2779–2790. doi: 10.1128/IAI.00278-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Tobin J, Bednarska K, Campbell A, Keane C. Pd-1 and Lag-3 checkpoint blockade: potential avenues for therapy in B-cell lymphoma. Cells. 2021;10:1152. doi: 10.3390/cells10051152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sobottka B, Moch H, Varga Z. Differential Pd-1/Lag-3 Expression and immune phenotypes in metastatic sites of breast cancer. Breast Cancer Res. 2021;23:4. doi: 10.1186/s13058-020-01380-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Schoffski P, et al. Phase I/Ii study of the Lag-3 inhibitor Ieramilimab (Lag525) +/- anti-Pd-1 spartalizumab (Pdr001) in patients with advanced malignancies. J. Immunother. Cancer. 2022;10:e003776. doi: 10.1136/jitc-2021-003776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Doe HT, et al. Expression of Pd-1/Lag-3 and cytokine production by Cd4(+) T cells during infection with plasmodium parasites. Microbiol. Immunol. 2016;60:121–131. doi: 10.1111/1348-0421.12354. [DOI] [PubMed] [Google Scholar]
- 178.Tawbi HA, et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 2022;386:24–34. doi: 10.1056/NEJMoa2109970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Wang J, et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of Lag-3. Cell. 2019;176:334–347. doi: 10.1016/j.cell.2018.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Qian W, Zhao M, Wang R, Li H. Fibrinogen-like protein 1 (Fgl1): the next immune checkpoint target. J. Hematol. Oncol. 2021;14:147. doi: 10.1186/s13045-021-01161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lv Z, et al. Fgl1 as a novel mediator and biomarker of malignant progression in clear cell renal cell carcinoma. Front. Oncol. 2021;11:756843. doi: 10.3389/fonc.2021.756843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.De Giglio A, et al. The Landscape of Immunotherapy in Advanced Nsclc: Driving Beyond Pd-1/Pd-L1 Inhibitors (Ctla-4, Lag3, Ido, Ox40, Tigit, Vaccines) Curr. Oncol. Rep. 2021;23:126. doi: 10.1007/s11912-021-01124-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Safe S, et al. Minireview: role of orphan nuclear receptors in cancer and potential as drug targets. Mol. Endocrinol. 2014;28:157–172. doi: 10.1210/me.2013-1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hermann-Kleiter N, et al. The nuclear orphan receptor Nr2F6 is a central checkpoint for cancer immune surveillance. Cell Rep. 2015;12:2072–2085. doi: 10.1016/j.celrep.2015.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Klepsch V, et al. Nuclear receptor Nr2F6 inhibition potentiates responses to Pd-L1/Pd-1 cancer immune checkpoint blockade. Nat. Commun. 2018;9:1538. doi: 10.1038/s41467-018-04004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Klepsch V, et al. Targeting the orphan nuclear receptor Nr2F6 in T cells primes tumors for immune checkpoint therapy. Cell Commun. Signal. 2020;18:8. doi: 10.1186/s12964-019-0454-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Dougall WC, Kurtulus S, Smyth MJ, Anderson AC. Tigit and Cd96: new checkpoint receptor targets for cancer immunotherapy. Immunol. Rev. 2017;276:112–120. doi: 10.1111/imr.12518. [DOI] [PubMed] [Google Scholar]
- 188.Harjunpaa H, Guillerey C. Tigit as an emerging immune checkpoint. Clin. Exp. Immunol. 2020;200:108–119. doi: 10.1111/cei.13407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chauvin JM, Zarour HM. Tigit in cancer immunotherapy. J. Immunother. Cancer. 2020;8:e000957. doi: 10.1136/jitc-2020-000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mullard A. Immuno-oncology target tigit attracts a new contender. Nat. Rev. Drug Discov. 2021;20:576. doi: 10.1038/d41573-021-00123-6. [DOI] [PubMed] [Google Scholar]
- 191.Chauvin JM, et al. Tigit and Pd-1 impair tumor antigen-specific Cd8(+) T cells in melanoma patients. J. Clin. Invest. 2015;125:2046–2058. doi: 10.1172/JCI80445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Deuss FA, Gully BS, Rossjohn J, Berry R. Recognition of nectin-2 by the natural killer cell receptor T cell immunoglobulin and itim domain (tigit) J. Biol. Chem. 2017;292:11413–11422. doi: 10.1074/jbc.M117.786483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Banta KL, et al. Mechanistic convergence of the tigit and Pd-1 inhibitory pathways necessitates Co-blockade to optimize anti-tumor Cd8(+) T cell responses. Immunity. 2022;55:512–526. doi: 10.1016/j.immuni.2022.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Wang L, et al. Vista, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011;208:577–592. doi: 10.1084/jem.20100619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Liu J, et al. Immune-checkpoint proteins vista and Pd-1 nonredundantly regulate murine T-cell responses. Proc. Natl Acad. Sci. USA. 2015;112:6682–6687. doi: 10.1073/pnas.1420370112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Boger C, Behrens HM, Kruger S, Rocken C. The Novel negative checkpoint regulator vista is expressed in gastric carcinoma and associated with Pd-L1/Pd-1: a future perspective for a combined gastric cancer therapy? Oncoimmunology. 2017;6:e1293215. doi: 10.1080/2162402X.2017.1293215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Wu L, et al. Expression of vista correlated with immunosuppression and synergized with Cd8 to predict survival in human oral squamous cell carcinoma. Cancer Immunol. Immunother. 2017;66:627–636. doi: 10.1007/s00262-017-1968-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Gao J, et al. Vista is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med. 2017;23:551–555. doi: 10.1038/nm.4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Derre L, et al. Btla mediates inhibition of human tumor-specific Cd8+ T cells that can be partially reversed by vaccination. J. Clin. Invest. 2010;120:157–167. doi: 10.1172/JCI40070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Demerle C, Gorvel L, Olive D. Btla-Hvem couple in health and diseases: insights for immunotherapy in lung cancer. Front. Oncol. 2021;11:682007. doi: 10.3389/fonc.2021.682007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Li X, et al. Btla expression in stage I-Iii non-small-cell lung cancer and its correlation with Pd-1/Pd-L1 and clinical outcomes. Onco Targets Ther. 2020;13:215–224. doi: 10.2147/OTT.S232234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Wang WD, et al. Up-regulation of Btla expression in myeloid dendritic cells associated with the treatment outcome of neonatal sepsis. Mol. Immunol. 2021;134:129–140. doi: 10.1016/j.molimm.2021.03.007. [DOI] [PubMed] [Google Scholar]
- 203.Hwang HJ, et al. The Btla and Pd-1 signaling pathways independently regulate the proliferation and cytotoxicity of human peripheral blood gammadelta T cells. Immun. Inflamm. Dis. 2021;9:274–287. doi: 10.1002/iid3.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Karabon L, et al. Abnormal expression of Btla and Ctla-4 immune checkpoint molecules in chronic lymphocytic leukemia patients. J. Immunol. Res. 2020;2020:6545921. doi: 10.1155/2020/6545921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Sordo-Bahamonde C, et al. Btla/Hvem axis induces Nk cell immunosuppression and poor outcome in chronic lymphocytic leukemia. Cancers. 2021;13:1766. doi: 10.3390/cancers13081766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Choi Y, et al. T-cell agonists in cancer immunotherapy. J. Immunother. Cancer. 2020;8:e000966. doi: 10.1136/jitc-2020-000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Fu Y, Lin Q, Zhang Z, Zhang L. Therapeutic strategies for the costimulatory molecule Ox40 in T-cell-mediated immunity. Acta Pharm. Sin. B. 2020;10:414–433. doi: 10.1016/j.apsb.2019.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Chen P, et al. Immune checkpoints Ox40 and Ox40L in small-cell lung cancer: predict prognosis and modulate immune microenvironment. Front. Oncol. 2021;11:713853. doi: 10.3389/fonc.2021.713853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yadav R, Redmond WL. Current clinical trial landscape of Ox40 agonists. Curr. Oncol. Rep. 2022;24:951–960. doi: 10.1007/s11912-022-01265-5. [DOI] [PubMed] [Google Scholar]
- 210.Kim TW, et al. First-in-human phase I study of the Ox40 agonist Moxr0916 in patients with advanced solid tumors. Clin. Cancer Res. 2022;14:4020. doi: 10.1158/1078-0432.CCR-21-4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Bulliard Y, et al. Ox40 engagement depletes intratumoral tregs via activating Fcgammars, leading to antitumor efficacy. Immunol. Cell Biol. 2014;92:475–480. doi: 10.1038/icb.2014.26. [DOI] [PubMed] [Google Scholar]
- 212.Fu T, He Q, Sharma P. The Icos/Icosl pathway is required for optimal antitumor responses mediated by anti-Ctla-4 therapy. Cancer Res. 2011;71:5445–5454. doi: 10.1158/0008-5472.CAN-11-1138. [DOI] [PubMed] [Google Scholar]
- 213.Fan X, et al. Engagement of the Icos pathway markedly enhances efficacy of Ctla-4 blockade in cancer immunotherapy. J. Exp. Med. 2014;211:715–725. doi: 10.1084/jem.20130590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Soldevilla MM, et al. Icos costimulation at the tumor site in combination with Ctla-4 blockade therapy elicits strong tumor immunity. Mol. Ther. 2019;27:1878–1891. doi: 10.1016/j.ymthe.2019.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shi LZ, et al. Blockade of Ctla-4 and Pd-1 enhances adoptive T-cell therapy efficacy in an Icos-mediated manner. Cancer Immunol. Res. 2019;7:1803–1812. doi: 10.1158/2326-6066.CIR-18-0873. [DOI] [PubMed] [Google Scholar]
- 216.Wang Y, et al. Combined 4-1Bb and Icos co-stimulation improves anti-tumor efficacy and persistence of dual anti-Cd19/Cd20 chimeric antigen receptor T cells. Cytotherapy. 2021;23:715–723. doi: 10.1016/j.jcyt.2021.02.117. [DOI] [PubMed] [Google Scholar]
- 217.Etxeberria I, Glez-Vaz J, Teijeira A, Melero I. New emerging targets in cancer immunotherapy: Cd137/4-1Bb costimulatory axis. ESMO Open. 2020;4:e733. doi: 10.1136/esmoopen-2020-000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Chester C, Sanmamed MF, Wang J, Melero I. Immunotherapy targeting 4-1Bb: mechanistic rationale, clinical results, and future strategies. Blood. 2018;131:49–57. doi: 10.1182/blood-2017-06-741041. [DOI] [PubMed] [Google Scholar]
- 219.Segal NH, et al. Phase I study of single-agent utomilumab (Pf-05082566), a 4-1Bb/Cd137 agonist, in patients with advanced cancer. Clin. Cancer Res. 2018;24:1816–1823. doi: 10.1158/1078-0432.CCR-17-1922. [DOI] [PubMed] [Google Scholar]
- 220.Claus C, et al. Tumor-targeted 4-1Bb agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019;11:eaav5989. doi: 10.1126/scitranslmed.aav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Vinay DS, Kwon BS. Immunotherapy of cancer with 4-1Bb. Mol. Cancer Ther. 2012;11:1062–1070. doi: 10.1158/1535-7163.MCT-11-0677. [DOI] [PubMed] [Google Scholar]
- 222.Van de Ven K, Borst J. Targeting the T-cell co-stimulatory Cd27/Cd70 pathway in cancer immunotherapy: rationale and potential. Immunotherapy. 2015;7:655–667. doi: 10.2217/imt.15.32. [DOI] [PubMed] [Google Scholar]
- 223.Starzer AM, Berghoff AS. New emerging targets in cancer immunotherapy: Cd27 (Tnfrsf7) ESMO Open. 2020;4:e629. doi: 10.1136/esmoopen-2019-000629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Latorre I, et al. Study of Cd27 and Ccr4 markers on specific Cd4(+) T-cells as immune tools for active and latent tuberculosis management. Front. Immunol. 2018;9:3094. doi: 10.3389/fimmu.2018.03094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Chen H, et al. Cd27 enhances the killing effect of Car T cells targeting trophoblast cell surface antigen 2 in the treatment of solid tumors. Cancer Immunol. Immunother. 2021;70:2059–2071. doi: 10.1007/s00262-020-02838-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Muth S, et al. Cd27 expression on Treg cells limits immune responses against tumors. J. Mol. Med. 2022;100:439–449. doi: 10.1007/s00109-021-02116-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Burris HA, et al. Safety and activity of varlilumab, a novel and first-in-class agonist anti-Cd27 antibody, in patients with advanced solid tumors. J. Clin. Oncol. 2017;35:2028–2036. doi: 10.1200/JCO.2016.70.1508. [DOI] [PubMed] [Google Scholar]
- 228.Buchan SL, et al. Pd-1 blockade and Cd27 stimulation activate distinct transcriptional programs that synergize for Cd8(+) T-cell-driven antitumor immunity. Clin. Cancer Res. 2018;24:2383–2394. doi: 10.1158/1078-0432.CCR-17-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Ramakrishna V, et al. Characterization of the human T cell response to in vitro Cd27 costimulation with varlilumab. J. Immunother. Cancer. 2015;3:37. doi: 10.1186/s40425-015-0080-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Ansell SM, et al. Safety and activity of varlilumab, a novel and first-in-class agonist Anti-Cd27 antibody, for hematologic malignancies. Blood Adv. 2020;4:1917–1926. doi: 10.1182/bloodadvances.2019001079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Mehta RS, Randolph B, Daher M, Rezvani K. Nk cell therapy for hematologic malignancies. Int. J. Hematol. 2018;107:262–270. doi: 10.1007/s12185-018-2407-5. [DOI] [PubMed] [Google Scholar]
- 232.Lachota M, et al. Prospects for Nk cell therapy of sarcoma. Cancers. 2020;12:3719. doi: 10.3390/cancers12123719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lee J, et al. An antibody designed to improve adoptive Nk-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol. Res. 2019;7:219–229. doi: 10.1158/2326-6066.CIR-18-0317. [DOI] [PubMed] [Google Scholar]
- 234.Masuyama J, Chaiyasit K, Sanphasitvong W, Wiwanitkit V. Nk cell therapy for end-stage cancerous patient: a case study. South Asian J. Cancer. 2014;3:143. doi: 10.4103/2278-330X.130480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Farhan S, Lee DA, Champlin RE, Ciurea SO. Nk cell therapy: targeting disease relapse after hematopoietic stem cell transplantation. Immunotherapy. 2012;4:305–313. doi: 10.2217/imt.11.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Fang F, Xiao W, Tian Z. Nk cell-based immunotherapy for cancer. Semin. Immunol. 2017;31:37–54. doi: 10.1016/j.smim.2017.07.009. [DOI] [PubMed] [Google Scholar]
- 237.Shimasaki N, Jain A, Campana D. Nk cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020;19:200–218. doi: 10.1038/s41573-019-0052-1. [DOI] [PubMed] [Google Scholar]
- 238.Myers JA, Miller JS. Exploring the Nk cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021;18:85–100. doi: 10.1038/s41571-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Colucci F. The role of Kir and Hla interactions in pregnancy complications. Immunogenetics. 2017;69:557–565. doi: 10.1007/s00251-017-1003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Naumova E, Mihaylova A, Ivanova M, Mihailova S. Impact of Kir/Hla ligand combinations on immune responses in malignant melanoma. Cancer Immunol. Immunother. 2007;56:95–100. doi: 10.1007/s00262-006-0151-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Agrawal S, Prakash S. Significance of Kir like natural killer cell receptors in autoimmune disorders. Clin. Immunol. 2020;216:108449. doi: 10.1016/j.clim.2020.108449. [DOI] [PubMed] [Google Scholar]
- 242.Benson DJ, et al. A phase I trial of the anti-Kir antibody Iph2101 and lenalidomide in patients with relapsed/refractory multiple myeloma. Clin. Cancer Res. 2015;21:4055–4061. doi: 10.1158/1078-0432.CCR-15-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Manzanares-Martin B, et al. Improving selection of patients with metastatic colorectal cancer to benefit from cetuximab based on Kir genotypes. J. Immunother. Cancer. 2021;9:e001705. doi: 10.1136/jitc-2020-001705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Sarac ME, et al. The evaluation of killer cell immunoglobulin-like receptor gene polymorphism in glioblastoma patients. Turk. Neurosurg. 2019;29:570–575. doi: 10.5137/1019-5149.JTN.24329-18.3. [DOI] [PubMed] [Google Scholar]
- 245.Carlsten M, et al. Checkpoint inhibition of Kir2D with the monoclonal antibody Iph2101 induces contraction and hyporesponsiveness of Nk cells in patients with myeloma. Clin. Cancer Res. 2016;22:5211–5222. doi: 10.1158/1078-0432.CCR-16-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Betser-Cohen G, et al. Reduced Kir2Dl1 recognition of Mhc class I molecules presenting phosphorylated peptides. J. Immunol. 2006;176:6762–6769. doi: 10.4049/jimmunol.176.11.6762. [DOI] [PubMed] [Google Scholar]
- 247.Gooneratne SL, Center RJ, Kent SJ, Parsons MS. Functional advantage of educated Kir2Dl1(+) natural killer cells for anti-Hiv-1 antibody-dependent activation. Clin. Exp. Immunol. 2016;184:101–109. doi: 10.1111/cei.12752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Van Hall T, et al. Monalizumab: inhibiting the novel immune checkpoint Nkg2a. J. Immunother. Cancer. 2019;7:263. doi: 10.1186/s40425-019-0761-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Herbst RS, et al. Coast: an open-label, phase ii, multidrug platform study of durvalumab alone or in combination with oleclumab or monalizumab in patients with unresectable, stage iii non-small-cell lung cancer. J. Clin. Oncol. 2022;22:O2200227. doi: 10.1200/JCO.22.00227. [DOI] [PubMed] [Google Scholar]
- 250.Tinker AV, et al. Dose-ranging and cohort-expansion study of monalizumab (Iph2201) in patients with advanced gynecologic malignancies: a trial of the Canadian Cancer Trials Group (Cctg): Ind221. Clin. Cancer Res. 2019;25:6052–6060. doi: 10.1158/1078-0432.CCR-19-0298. [DOI] [PubMed] [Google Scholar]
- 251.Galot R, et al. A phase Ii study of monalizumab in patients with recurrent/metastatic squamous cell carcinoma of the head and neck: the I1 cohort of the Eortc-Hncg-1559 upstream trial. Eur. J. Cancer. 2021;158:17–26. doi: 10.1016/j.ejca.2021.09.003. [DOI] [PubMed] [Google Scholar]
- 252.Mittal D, et al. Cd96 is an immune checkpoint that regulates Cd8(+) T-cell antitumor function. Cancer Immunol. Res. 2019;7:559–571. doi: 10.1158/2326-6066.CIR-18-0637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Georgiev H, Ravens I, Papadogianni G, Bernhardt G. Coming of age: Cd96 emerges as modulator of immune responses. Front. Immunol. 2018;9:1072. doi: 10.3389/fimmu.2018.01072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Singh AK, McGuirk JP. Car T cells: continuation in a revolution of immunotherapy. Lancet Oncol. 2020;21:e168–e178. doi: 10.1016/S1470-2045(19)30823-X. [DOI] [PubMed] [Google Scholar]
- 255.Miliotou AN, Papadopoulou LC. Car T-cell therapy: a new era in cancer immunotherapy. Curr. Pharm. Biotechnol. 2018;19:5–18. doi: 10.2174/1389201019666180418095526. [DOI] [PubMed] [Google Scholar]
- 256.Rodriguez-Garcia A, et al. Car-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021;12:877. doi: 10.1038/s41467-021-20893-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Shi J, Li M, Yang R. Tumor-infiltrating lymphocytes as a feasible adjuvant immunotherapy for osteosarcoma with a poor response to neoadjuvant chemotherapy. Immunotherapy. 2020;12:641–652. doi: 10.2217/imt-2020-0107. [DOI] [PubMed] [Google Scholar]
- 258.Siddiqui I, et al. Intratumoral Tcf1(+)Pd-1(+)Cd8(+) T cells with stem-like properties promote tumor control in response to vaccination and checkpoint blockade immunotherapy. Immunity. 2019;50:195–211. doi: 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 259.Ti D, et al. Adaptive T cell immunotherapy in cancer. Sci. China Life Sci. 2021;64:363–371. doi: 10.1007/s11427-020-1713-9. [DOI] [PubMed] [Google Scholar]
- 260.Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021;18:842–859. doi: 10.1038/s41423-020-00565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Luen SJ, et al. Tumour-infiltrating lymphocytes and the emerging role of immunotherapy in breast cancer. Pathology. 2017;49:141–155. doi: 10.1016/j.pathol.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 262.Lee N, Zakka LR, Mihm MJ, Schatton T. Tumour-infiltrating lymphocytes in melanoma prognosis and cancer immunotherapy. Pathology. 2016;48:177–187. doi: 10.1016/j.pathol.2015.12.006. [DOI] [PubMed] [Google Scholar]
- 263.Romano F, et al. Preoperative Il-2 immunotherapy enhances tumor infiltrating lymphocytes (Tils) in gastric cancer patients. Hepatogastroenterology. 2006;53:634–638. [PubMed] [Google Scholar]
- 264.Hua JM, Zheng ZG, Xu GD. A clinical study on adoptive immunotherapy of bone metastatic neoplasm with tumor infiltrating lymphocytes. Zhonghua Zhong Liu Za Zhi. 1994;16:203–206. [PubMed] [Google Scholar]
- 265.Creelan BC, et al. Tumor-infiltrating lymphocyte treatment for anti-Pd-1-resistant metastatic lung cancer: a phase 1 trial. Nat. Med. 2021;27:1410–1418. doi: 10.1038/s41591-021-01462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Wang X, et al. Assessment of immune status of laryngeal squamous cell carcinoma can predict prognosis and guide treatment. Cancer Immunol. Immunother. 2021;71:1199–1220. doi: 10.1007/s00262-021-03071-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Losurdo A, et al. Insights for the application of Tils and Ar in the treatment of Tnbc in routine clinical practice. Sci. Rep. 2020;10:20100. doi: 10.1038/s41598-020-77043-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wang Y, et al. Targeting Cd96 overcomes Pd-1 blockade resistance by enhancing Cd8+ Til function in cervical cancer. J. Immunother. Cancer. 2022;10:e003667. doi: 10.1136/jitc-2021-003667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Sarnaik AA, et al. Lifileucel, a tumor-infiltrating lymphocyte therapy, in metastatic melanoma. J. Clin. Oncol. 2021;39:2656–2666. doi: 10.1200/JCO.21.00612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Savas P, Loi S. Metastatic breast cancer: til it is too late. Clin. Cancer Res. 2020;26:526–528. doi: 10.1158/1078-0432.CCR-19-3490. [DOI] [PubMed] [Google Scholar]
- 271.Hashemi S, et al. Surprising impact of stromal Til's on immunotherapy efficacy in a real-world. Lung Cancer Study Lung Cancer. 2021;153:81–89. doi: 10.1016/j.lungcan.2021.01.013. [DOI] [PubMed] [Google Scholar]
- 272.Zhou X, Wu J, Duan C, Liu Y. Retrospective Analysis of adoptive Til therapy plus anti-Pd1 therapy in patients with chemotherapy-resistant metastatic osteosarcoma. J. Immunol. Res. 2020;2020:7890985. doi: 10.1155/2020/7890985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Wang Z, Wu Z, Liu Y, Han W. New development in Car-T cell therapy. J. Hematol. Oncol. 2017;10:53. doi: 10.1186/s13045-017-0423-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Han G, et al. 9P21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat. Commun. 2021;12:5606. doi: 10.1038/s41467-021-25894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Qin SS, Melucci AD, Chacon AC, Prieto P. A. adoptive T cell therapy for solid tumors: pathway to personalized standard of care. Cells. 2021;10:808. doi: 10.3390/cells10040808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Watanabe K, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. 2018;3:e99573. doi: 10.1172/jci.insight.99573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Merhavi-Shoham E, et al. Adoptive cell therapy for metastatic melanoma. Cancer J. 2017;23:48–53. doi: 10.1097/PPO.0000000000000240. [DOI] [PubMed] [Google Scholar]
- 278.Delgoffe GM, et al. The role of exhaustion in Car T cell therapy. Cancer Cell. 2021;39:885–888. doi: 10.1016/j.ccell.2021.06.012. [DOI] [PubMed] [Google Scholar]
- 279.Sadelain M. Cd19 Car T cells. Cell. 2017;171:1471. doi: 10.1016/j.cell.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 280.Tallantyre EC, et al. Neurological updates: neurological complications of Car-T therapy. J. Neurol. 2021;268:1544–1554. doi: 10.1007/s00415-020-10237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Poorebrahim M, et al. Counteracting Car T cell dysfunction. Oncogene. 2021;40:421–435. doi: 10.1038/s41388-020-01501-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Hamieh M, et al. Car T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568:112–116. doi: 10.1038/s41586-019-1054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Bao C, et al. The application of nanobody in Car-T therapy. Biomolecules. 2021;11:238. doi: 10.3390/biom11020238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Sheih A, et al. Clonal kinetics and single-cell transcriptional profiling of Car-T cells in patients undergoing Cd19 Car-T immunotherapy. Nat. Commun. 2020;11:219. doi: 10.1038/s41467-019-13880-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Wagner DL, et al. Immunogenicity of Car T cells in cancer therapy. Nat. Rev. Clin. Oncol. 2021;18:379–393. doi: 10.1038/s41571-021-00476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Wei J, Han X, Bo J, Han W. Target selection for Car-T therapy. J. Hematol. Oncol. 2019;12:62. doi: 10.1186/s13045-019-0758-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Bielamowicz K, et al. Trivalent Car T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018;20:506–518. doi: 10.1093/neuonc/nox182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Melenhorst JJ, et al. Decade-long leukaemia remissions with persistence of Cd4(+) Car T cells. Nature. 2022;602:503–509. doi: 10.1038/s41586-021-04390-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Nakajima M, et al. Improved survival of chimeric antigen receptor-engineered T (Car-T) and tumor-specific T cells caused by anti-programmed cell death protein 1 single-chain variable fragment-producing Car-T cells. Cancer Sci. 2019;110:3079–3088. doi: 10.1111/cas.14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Curran KJ, et al. Toxicity and response after Cd19-specific Car T-cell therapy in pediatric/young adult relapsed/refractory B-All. Blood. 2019;134:2361–2368. doi: 10.1182/blood.2019001641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Xie G, et al. Car-T cells targeting a nucleophosmin neoepitope exhibit potent specific activity in mouse models of acute myeloid leukaemia. Nat. Biomed. Eng. 2021;5:399–413. doi: 10.1038/s41551-020-00625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Yan L, et al. Sequential Cd19 and Bcma-specific Car T-cell treatment elicits sustained remission of relapsed and/or refractory myeloma. Cancer Med. 2021;10:563–574. doi: 10.1002/cam4.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Liu S, et al. Cd19-specific Car-T cell therapy for relapsed/refractory Non-B-cell acute leukaemia with Cd19 antigen expression. Eur. J. Cancer. 2021;153:1–4. doi: 10.1016/j.ejca.2021.04.042. [DOI] [PubMed] [Google Scholar]
- 294.Zhu G, Zhang Q, Zhang J, Liu F. Targeting tumor-associated antigen: a promising Car-T therapeutic strategy for glioblastoma treatment. Front. Pharm. 2021;12:661606. doi: 10.3389/fphar.2021.661606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Qin VM, D'Souza C, Neeson PJ, Zhu JJ. Chimeric antigen receptor beyond Car-T cells. Cancers. 2021;13:404. doi: 10.3390/cancers13030404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Adeel K, et al. Efficacy and safety of Cd22 chimeric antigen receptor (Car) T cell therapy in patients with B cell malignancies: a protocol for a systematic review and meta-analysis. Syst. Rev. 2021;10:35. doi: 10.1186/s13643-021-01588-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Strati P, Neelapu SS. Car-T failure: beyond antigen loss and T cells. Blood. 2021;137:2567–2568. doi: 10.1182/blood.2020010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Salinas RD, Durgin JS, O'Rourke DM. Potential of glioblastoma-targeted chimeric antigen receptor (Car) T-cell therapy. Cns Drugs. 2020;34:127–145. doi: 10.1007/s40263-019-00687-3. [DOI] [PubMed] [Google Scholar]
- 299.Xue G, et al. Adoptive cell therapy with tumor-specific Th9 cells induces viral mimicry to eliminate antigen-loss-variant tumor cells. Cancer Cell. 2021;39:1610–1622. doi: 10.1016/j.ccell.2021.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Jatiani SS, et al. Myeloma Car-T Crs management with Il-1R antagonist anakinra. Clin. Lymphoma Myeloma Leuk. 2020;20:632–636. doi: 10.1016/j.clml.2020.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Pochon C, et al. Complications other than infections, Crs and Icans following Car T-cells therapy: recommendations of the Francophone Society of bone marrow transplantation and cell therapy (Sfgm-Tc) Bull. Cancer. 2021;108:S98–S103. doi: 10.1016/j.bulcan.2021.10.004. [DOI] [PubMed] [Google Scholar]
- 302.Jiang H, et al. Improving the safety of Car-T cell therapy by controlling Crs-related coagulopathy. Ann. Hematol. 2019;98:1721–1732. doi: 10.1007/s00277-019-03685-z. [DOI] [PubMed] [Google Scholar]
- 303.Hao Z, et al. Macrophage, the Potential Key Mediator in Car-T Related Crs. Exp. Hematol. Oncol. 2020;9:15. doi: 10.1186/s40164-020-00171-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Pan J, et al. Ruxolitinib mitigates steroid-refractory Crs during Car T therapy. J. Cell. Mol. Med. 2021;25:1089–1099. doi: 10.1111/jcmm.16176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Sandler RD, et al. Diagnosis and management of secondary Hlh/Mas following Hsct and Car-T cell therapy in adults; a review of the literature and a survey of practice within Ebmt Centres on behalf of the autoimmune diseases working party (Adwp) and transplant complications working party (Tcwp) Front. Immunol. 2020;11:524. doi: 10.3389/fimmu.2020.00524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Lichtenstein DA, et al. Characterization of Hlh-like manifestations as a Crs variant in patients receiving Cd22 Car T cells. Blood. 2021;138:2469–2484. doi: 10.1182/blood.2021011898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Schubert ML, et al. Side-effect management of chimeric antigen receptor (Car) T-cell therapy. Ann. Oncol. 2021;32:34–48. doi: 10.1016/j.annonc.2020.10.478. [DOI] [PubMed] [Google Scholar]
- 308.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in Car T-cell therapy. Mol. Ther. Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Fu W, et al. Car exosomes derived from effector Car-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019;10:4355. doi: 10.1038/s41467-019-12321-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Huang X, Yang Y. Driving an improved CAR for cancer immunotherapy. J. Clin. Invest. 2016;126:2795–2798. doi: 10.1172/JCI88959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Kasakovski D, Xu L, Li Y. T cell senescence and Car-T cell exhaustion in hematological malignancies. J. Hematol. Oncol. 2018;11:91. doi: 10.1186/s13045-018-0629-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Morgan RA, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing Erbb2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Majzner RG, et al. Tuning the antigen density requirement for Car T-cell activity. Cancer Disco. 2020;10:702–723. doi: 10.1158/2159-8290.CD-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Wang L, et al. Chimeric antigen receptor (Car)-modified Nk cells against cancer: opportunities and challenges. Int. Immunopharmacol. 2019;74:105695. doi: 10.1016/j.intimp.2019.105695. [DOI] [PubMed] [Google Scholar]
- 315.Edeline J, Houot R, Marabelle A, Alcantara M. Car-T cells and bites in solid tumors: challenges and perspectives. J. Hematol. Oncol. 2021;14:65. doi: 10.1186/s13045-021-01067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Brown CE, Mackall CL. Car T cell therapy: inroads to response and resistance. Nat. Rev. Immunol. 2019;19:73–74. doi: 10.1038/s41577-018-0119-y. [DOI] [PubMed] [Google Scholar]
- 317.Martinez BD, Dutoit V, Migliorini D. Allogeneic Car T cells: an alternative to overcome challenges of Car T cell therapy in glioblastoma. Front. Immunol. 2021;12:640082. doi: 10.3389/fimmu.2021.640082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Zhao Z, et al. The application of Car-T cell therapy in hematological malignancies: advantages and challenges. Acta Pharm. Sin. B. 2018;8:539–551. doi: 10.1016/j.apsb.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Spiegel JY, et al. Car T cells with dual targeting of Cd19 and Cd22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat. Med. 2021;27:1419–1431. doi: 10.1038/s41591-021-01436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Jia H, et al. Haploidentical Cd19/Cd22 bispecific Car-T cells induced Mrd-negative remission in a patient with relapsed and refractory adult B-All after haploidentical hematopoietic stem cell transplantation. J. Hematol. Oncol. 2019;12:57. doi: 10.1186/s13045-019-0741-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Bashiri DA, et al. Nk cells armed with chimeric antigen receptors (Car): roadblocks to successful development. Cells. 2021;10:3390. doi: 10.3390/cells10123390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Sutherland AR, Owens MN, Geyer CR. Modular chimeric antigen receptor systems for universal Car T cell retargeting. Int. J. Mol. Sci. 2020;21:7222. doi: 10.3390/ijms21197222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Morsut L, et al. Engineering Customized. Cell Sens. Response Behav. Using Synth. Notch Receptors. Cell. 2016;164:780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Shah NN, Fry TJ. Mechanisms of resistance to Car T cell therapy. Nat. Rev. Clin. Oncol. 2019;16:372–385. doi: 10.1038/s41571-019-0184-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Blaeschke F, et al. Augmenting anti-Cd19 and anti-Cd22 Car T-cell function using Pd-1-Cd28 checkpoint fusion proteins. Blood Cancer J. 2021;11:108. doi: 10.1038/s41408-021-00499-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Baird JH, et al. Cd22-directed Car T-cell therapy induces complete remissions in Cd19-directed Car-refractory large B-cell lymphoma. Blood. 2021;137:2321–2325. doi: 10.1182/blood.2020009432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Weber EW, et al. Transient rest restores functionality in exhausted Car-T cells through epigenetic remodeling. Science. 2021;372:eaba1786. doi: 10.1126/science.aba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Alizadeh D, et al. Il15 enhances Car-T cell antitumor activity by reducing Mtorc1 activity and preserving their stem cell memory phenotype. Cancer Immunol. Res. 2019;7:759–772. doi: 10.1158/2326-6066.CIR-18-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Wang X, et al. Phenotypic and functional attributes of lentivirus-modified Cd19-specific human Cd8+ central memory T cells manufactured at clinical scale. J. Immunother. 2012;35:689–701. doi: 10.1097/CJI.0b013e318270dec7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Feucht J, et al. Calibration of Car activation potential directs alternative T cell fates and therapeutic potency. Nat. Med. 2019;25:82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Shao M, et al. Inhibition of calcium signaling prevents exhaustion and enhances anti-leukemia efficacy of Car-T cells via soce-calcineurin-Nfat and glycolysis pathways. Adv. Sci. 2022;9:e2103508. doi: 10.1002/advs.202103508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Jayaraman K. Cut-price Car-T cell therapies top India's biotech agenda. Nat. Biotechnol. 2019;37:1388–1389. doi: 10.1038/s41587-019-0346-1. [DOI] [PubMed] [Google Scholar]
- 333.Kron F, Franz J, Kron A, Hallek M. Economics and management of Car T-cell therapy: status quo and future perspectives. Internist. 2021;62:620–626. doi: 10.1007/s00108-021-01042-9. [DOI] [PubMed] [Google Scholar]
- 334.Fathi E, et al. A general view of Cd33(+) leukemic stem cells and Car-T cells as interesting targets in acute myeloblatsic leukemia therapy. Blood Res. 2020;55:10–16. doi: 10.5045/br.2020.55.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Zoine JT, et al. Thrombopoietin-based Car-T cells demonstrate in vitro and in vivo cytotoxicity to mpl positive acute myelogenous leukemia and hematopoietic stem cells. Gene Ther. 2021;29:1–12. doi: 10.1038/s41434-021-00283-5. [DOI] [PubMed] [Google Scholar]
- 336.El KN, et al. Demethylating therapy increases anti-Cd123 Car T cell cytotoxicity against acute myeloid leukemia. Nat. Commun. 2021;12:6436. doi: 10.1038/s41467-021-26683-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Kiesgen S, et al. Comparative analysis of assays to measure Car T-cell-mediated cytotoxicity. Nat. Protoc. 2021;16:1331–1342. doi: 10.1038/s41596-020-00467-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Thomas R, Yang X. Nk-Dc crosstalk in immunity to microbial infection. J. Immunol. Res. 2016;2016:6374379. doi: 10.1155/2016/6374379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Marofi F, et al. Car-Nk cell: a new paradigm in tumor immunotherapy. Front. Oncol. 2021;11:673276. doi: 10.3389/fonc.2021.673276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 340.Chen S, et al. Genome-wide Crispr screen in a mouse model of tumor growth and metastasis. Cell. 2015;160:1246–1260. doi: 10.1016/j.cell.2015.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Gautron A, et al. Crispr screens identify tumor-promoting genes conferring melanoma cell plasticity and resistance. Embo Mol. Med. 2021;13:e13466. doi: 10.15252/emmm.202013466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.King CCar. Nk cell therapy for T follicular helper cells. Cell Rep. Med. 2020;1:100009. doi: 10.1016/j.xcrm.2020.100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Marofi F, et al. Car-Nk cell in cancer immunotherapy; a promising frontier. Cancer Sci. 2021;112:3427–3436. doi: 10.1111/cas.14993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Liu E, et al. Cord blood Nk cells engineered to express Il-15 and a Cd19-targeted car show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–531. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Becker PS, et al. Selection and expansion of natural killer cells for Nk cell-based immunotherapy. Cancer Immunol. Immunother. 2016;65:477–484. doi: 10.1007/s00262-016-1792-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Eguizabal C, et al. Natural killer cells for cancer immunotherapy: pluripotent stem cells-derived Nk cells as an immunotherapeutic perspective. Front. Immunol. 2014;5:439. doi: 10.3389/fimmu.2014.00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Qin Z, et al. Effect of Nk cell immunotherapy on immune function in patients with hepatic carcinoma: a preliminary clinical study. Cancer Biol. Ther. 2017;18:323–330. doi: 10.1080/15384047.2017.1310346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Zhang M, et al. Il-15 enhanced antibody-dependent cellular cytotoxicity mediated by Nk cells and macrophages. Proc. Natl Acad. Sci. USA. 2018;115:E10915–E10924. doi: 10.1073/pnas.1811615115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Waldmann TA, Dubois S, Miljkovic MD, Conlon KC. Il-15 in the combination immunotherapy of cancer. Front. Immunol. 2020;11:868. doi: 10.3389/fimmu.2020.00868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Wellhausen N, et al. Better living through chemistry: Crispr/Cas engineered T cells for cancer immunotherapy. Curr. Opin. Immunol. 2022;74:76–84. doi: 10.1016/j.coi.2021.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Deng S, et al. Codelivery of Crispr-Cas9 and chlorin E6 for spatially controlled tumor-specific gene editing with synergistic drug effects. Sci. Adv. 2020;6:b4005. doi: 10.1126/sciadv.abb4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Azangou-Khyavy M, et al. Crispr/Cas: from tumor gene editing to T cell-based immunotherapy of cancer. Front. Immunol. 2020;11:2062. doi: 10.3389/fimmu.2020.02062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Chen P, You L, Lu Y. Applications of Crispr-Cas9 technology in translational research on solid-tumor cancers. CRISPR J. 2018;1:47–54. doi: 10.1089/crispr.2017.0001. [DOI] [PubMed] [Google Scholar]
- 354.Lu Y, et al. Safety and feasibility of Crispr-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020;26:732–740. doi: 10.1038/s41591-020-0840-5. [DOI] [PubMed] [Google Scholar]
- 355.Stadtmauer, E. A. et al. Crispr-engineered T cells in patients with refractory cancer. Science367, eaba7365 (2020). [DOI] [PMC free article] [PubMed]
- 356.Hu Y, et al. Crispr/Cas9-engineered universal Cd19/Cd22 dual-targeted Car-T cell therapy for relapsed/refractory B-cell acute lymphoblastic leukemia. Clin. Cancer Res. 2021;27:2764–2772. doi: 10.1158/1078-0432.CCR-20-3863. [DOI] [PubMed] [Google Scholar]
- 357.Singh N, et al. Impaired death receptor signaling in leukemia causes antigen-independent resistance by inducing Car T-cell dysfunction. Cancer Disco. 2020;10:552–567. doi: 10.1158/2159-8290.CD-19-0813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Wang D, et al. Crispr screening of Car T cells and cancer stem cells reveals critical dependencies for cell-based therapies. Cancer Disco. 2021;11:1192–1211. doi: 10.1158/2159-8290.CD-20-1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Agarwal S, Wellhausen N, Levine BL, June CH. Production of human Crispr-engineered Car-T cells. J. Vis. Exp. 2021;15:e62299. doi: 10.3791/62299. [DOI] [PubMed] [Google Scholar]
- 360.Razeghian E, et al. A deep insight into Crispr/Cas9 application in Car-T cell-based tumor immunotherapies. Stem Cell Res. Ther. 2021;12:428. doi: 10.1186/s13287-021-02510-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Mollanoori H, Shahraki H, Rahmati Y, Teimourian S. Crispr/Cas9 and Car-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum. Immunol. 2018;79:876–882. doi: 10.1016/j.humimm.2018.09.007. [DOI] [PubMed] [Google Scholar]
- 362.Artegiani B, et al. Probing the tumor suppressor function of Bap1 in Crispr-engineered human liver organoids. Cell Stem Cell. 2019;24:927–943. doi: 10.1016/j.stem.2019.04.017. [DOI] [PubMed] [Google Scholar]
- 363.Stenger D, et al. Endogenous Tcr promotes in vivo persistence of Cd19-Car-T cells compared to a Crispr/Cas9-mediated Tcr knockout Car. Blood. 2020;136:1407–1418. doi: 10.1182/blood.2020005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Borcoman E, et al. Patterns of response and progression to immunotherapy. Am. Soc. Clin. Oncol. Educ. Book. 2018;38:169–178. doi: 10.1200/EDBK_200643. [DOI] [PubMed] [Google Scholar]
- 365.Dumoulin DW, et al. Renal toxicity from pemetrexed and pembrolizumab in the era of combination therapy in patients with metastatic nonsquamous cell Nsclc. J. Thorac. Oncol. 2020;15:1472–1483. doi: 10.1016/j.jtho.2020.04.021. [DOI] [PubMed] [Google Scholar]
- 366.Goldberg SB, et al. Pembrolizumab for management of patients with nsclc and brain metastases: long-term results and biomarker analysis from a non-randomised, open-label, phase 2 trial. Lancet Oncol. 2020;21:655–663. doi: 10.1016/S1470-2045(20)30111-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Powell, S. F. et al. Outcomes with pembrolizumab plus platinum-based chemotherapy for patients with Nsclc and stable brain metastases: pooled analysis of keynote-021, -189, and -407. J. Thorac. Oncol. 16, 1883–1892 (2021). [DOI] [PubMed]
- 368.Paz-Ares L, et al. A randomized, placebo-controlled trial of pembrolizumab plus chemotherapy in patients with metastatic squamous Nsclc: protocol-specified final analysis of keynote-407. J. Thorac. Oncol. 2020;15:1657–1669. doi: 10.1016/j.jtho.2020.06.015. [DOI] [PubMed] [Google Scholar]
- 369.Garcia-Diaz A, et al. Interferon receptor signaling pathways regulating Pd-L1 and Pd-L2 expression. Cell Rep. 2019;29:3766. doi: 10.1016/j.celrep.2019.11.113. [DOI] [PubMed] [Google Scholar]
- 370.Zaretsky JM, et al. Mutations associated with acquired resistance to Pd-1 blockade in melanoma. N. Engl. J. Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Costa SF, et al. Plasma thymidine kinase activity as a novel biomarker in metastatic melanoma patients treated with immune checkpoint inhibitors. Cancers. 2022;14:702. doi: 10.3390/cancers14030702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Powles T, Morrison L. Biomarker challenges for immune checkpoint inhibitors in urothelial carcinoma. Nat. Rev. Urol. 2018;15:585–587. doi: 10.1038/s41585-018-0056-3. [DOI] [PubMed] [Google Scholar]
- 373.Doroshow DB, et al. Pd-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021;18:345–362. doi: 10.1038/s41571-021-00473-5. [DOI] [PubMed] [Google Scholar]
- 374.Hirashima T, et al. The levels of interferon-gamma release as a biomarker for non-small-cell lung cancer patients receiving immune checkpoint inhibitors. Anticancer Res. 2019;39:6231–6240. doi: 10.21873/anticanres.13832. [DOI] [PubMed] [Google Scholar]
- 375.Lyu Q, et al. Alterations in Tp53 are a potential biomarker of bladder cancer patients who benefit from immune checkpoint inhibition. Cancer Control. 2020;27:1148411929. doi: 10.1177/1073274820976665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Wang F, et al. Evaluation of pole and Pold1 mutations as biomarkers for immunotherapy outcomes across multiple cancer types. JAMA Oncol. 2019;5:1504–1506. doi: 10.1001/jamaoncol.2019.2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Rizvi NA, et al. Cancer immunology. Mutational landscape determines sensitivity to Pd-1 blockade in non-small cell lung cancer. Science. 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Domingo E, et al. Somatic pole proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol. Hepatol. 2016;1:207–216. doi: 10.1016/S2468-1253(16)30014-0. [DOI] [PubMed] [Google Scholar]
- 379.Lopez-Beltran A, et al. Immune checkpoint inhibitors in urothelial carcinoma: recommendations for practical approaches to Pd-L1 and other potential predictive biomarker testing. Cancers. 2021;13:1424. doi: 10.3390/cancers13061424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Chaput L, Jordheim LP. Current landscape of biomarker development for immune checkpoint inhibitors targeting Pd-1/Pd-L1 pathway in oncology. Therapie. 2021;76:597–615. doi: 10.1016/j.therap.2021.06.005. [DOI] [PubMed] [Google Scholar]
- 381.Zouein J, Kesrouani C, Kourie HR. Pd-L1 expression as a predictive biomarker for immune checkpoint inhibitors: between a dream and a nightmare. Immunotherapy. 2021;13:1053–1065. doi: 10.2217/imt-2020-0336. [DOI] [PubMed] [Google Scholar]
- 382.Yamauchi T, et al. T-cell Cx3Cr1 expression as a dynamic blood-based biomarker of response to immune checkpoint inhibitors. Nat. Commun. 2021;12:1402. doi: 10.1038/s41467-021-21619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Liu H, et al. Metabolic molecule Pla2G2D is a potential prognostic biomarker correlating with immune cell infiltration and the expression of immune checkpoint genes in cervical squamous cell carcinoma. Front. Oncol. 2021;11:755668. doi: 10.3389/fonc.2021.755668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Gong J, et al. Response to Pd-1 blockade in microsatellite stable metastatic colorectal cancer harboring a pole mutation. J. Natl Compr. Canc. Netw. 2017;15:142–147. doi: 10.6004/jnccn.2017.0016. [DOI] [PubMed] [Google Scholar]
- 385.Mehnert JM, et al. Immune activation and response to pembrolizumab in pole-mutant endometrial cancer. J. Clin. Invest. 2016;126:2334–2340. doi: 10.1172/JCI84940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Rizzo A, Ricci AD, Brandi G. Pd-L1, Tmb, Msi, and other predictors of response to immune checkpoint inhibitors in biliary tract cancer. Cancers. 2021;13:553. doi: 10.3390/cancers13030553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Lobo J, et al. Detailed characterization of immune cell infiltrate and expression of immune checkpoint molecules Pd-L1/Ctla-4 and Mmr proteins in testicular germ cell tumors disclose novel disease biomarkers. Cancers. 2019;11:1535. doi: 10.3390/cancers11101535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Guo X, et al. Case Report: complete response to antiangiogenesis and immune checkpoint blockade in an unresectable mmr-deficient leiomyosarcoma harboring biallelic loss of Pten. Front. Oncol. 2022;12:802074. doi: 10.3389/fonc.2022.802074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Sahin IH, et al. Mismatch repair (Mmr) gene alteration and Braf V600E mutation are potential predictive biomarkers of immune checkpoint inhibitors in mmr-deficient colorectal cancer. Oncologist. 2021;26:668–675. doi: 10.1002/onco.13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Roudko V, et al. Lynch syndrome and Msi-H cancers: from mechanisms to “off-the-shelf” cancer vaccines. Front. Immunol. 2021;12:757804. doi: 10.3389/fimmu.2021.757804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Oliveira AF, Bretes L, Furtado I. Review of Pd-1/Pd-L1 inhibitors in metastatic Dmmr/Msi-H colorectal cancer. Front. Oncol. 2019;9:396. doi: 10.3389/fonc.2019.00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Zhou C, et al. Good tumor response to chemoradioimmunotherapy in Dmmr/Msi-H advanced colorectal cancer: a case series. Front. Immunol. 2021;12:784336. doi: 10.3389/fimmu.2021.784336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Oaknin A, et al. Safety and Antitumor Activity of Dostarlimab in Patients with Advanced Or Recurrent Dna Mismatch Repair Deficient/Microsatellite Instability-High (Dmmr/Msi-H) Or Proficient/Stable (Mmrp/Mss) Endometrial Cancer: Interim Results From Garnet-a Phase I, Single-Arm Study. J. Immunother. Cancer. 2022;10:e003777. doi: 10.1136/jitc-2021-003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Overman MJ, et al. Nivolumab in patients with metastatic dna mismatch repair-deficient or microsatellite instability-high colorectal cancer (checkmate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18:1182–1191. doi: 10.1016/S1470-2045(17)30422-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Sclafani F. Pd-1 inhibition in metastatic Dmmr/Msi-H colorectal cancer. Lancet Oncol. 2017;18:1141–1142. doi: 10.1016/S1470-2045(17)30512-0. [DOI] [PubMed] [Google Scholar]
- 396.Sahin IH, et al. Immune Checkpoint Inhibitors for the Treatment of Msi-H/Mmr-D Colorectal Cancer and a Perspective On Resistance Mechanisms. Br. J. Cancer. 2019;121:809–818. doi: 10.1038/s41416-019-0599-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Olivares-Hernandez A, et al. Influence of Dna mismatch repair (Mmr) system in survival and response to immune checkpoint inhibitors (Icis) in non-small cell lung cancer (Nsclc): retrospective analysis. Biomedicines. 2022;10:360. doi: 10.3390/biomedicines10020360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Fuca G, et al. Ascites and resistance to immune checkpoint inhibition in Dmmr/Msi-H metastatic colorectal and gastric cancers. J. Immunother. Cancer. 2022;10:e004001. doi: 10.1136/jitc-2021-004001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Mohamed A, et al. High-risk features are prognostic in Dmmr/Msi-H stage Ii colon cancer. Front. Oncol. 2021;11:755113. doi: 10.3389/fonc.2021.755113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Foote MB, et al. Tmb cut-offs fail to predict benefit of Pd-1 blockade in gastroesophageal adenocarcinoma in keynote-061. Ann. Oncol. 2021;32:1188–1189. doi: 10.1016/j.annonc.2021.06.006. [DOI] [PubMed] [Google Scholar]
- 401.Liu L, et al. Combination of Tmb and Cna stratifies prognostic and predictive responses to immunotherapy across metastatic cancer. Clin. Cancer Res. 2019;25:7413–7423. doi: 10.1158/1078-0432.CCR-19-0558. [DOI] [PubMed] [Google Scholar]
- 402.Weber S, et al. Dynamic changes of circulating tumor dna predict clinical outcome in patients with advanced non-small-cell lung cancer treated with immune checkpoint inhibitors. JCO Precis Oncol. 2021;5:1540–1553. doi: 10.1200/PO.21.00182. [DOI] [PubMed] [Google Scholar]
- 403.Powles T, et al. Ctdna guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;595:432–437. doi: 10.1038/s41586-021-03642-9. [DOI] [PubMed] [Google Scholar]
- 404.Fenner AUsing. Ctdna to guide immunotherapy for urothelial cancer. Nat. Rev. Urol. 2021;18:443. doi: 10.1038/s41585-021-00503-y. [DOI] [PubMed] [Google Scholar]
- 405.Furness AJ, Quezada SA, Peggs KS. Neoantigen heterogeneity: a key driver of immune response and sensitivity to immune checkpoint blockade? Immunotherapy. 2016;8:763–766. doi: 10.2217/imt-2016-0064. [DOI] [PubMed] [Google Scholar]
- 406.Luksza M, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551:517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.McGranahan, N. & Swanton, C. Neoantigen quality, not quantity. Sci. Transl. Med. 11, aax4905 (2019). [DOI] [PubMed]
- 408.Eckardt, J. et al. Tmb and Braf mutation status are independent predictive factors in high-risk melanoma patients with adjuvant anti-Pd-1 therapy. J. Cancer Res. Clin. Oncol. Epub ahead of print (2022). [DOI] [PMC free article] [PubMed]
- 409.Anagnostou V, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Disco. 2017;7:264–276. doi: 10.1158/2159-8290.CD-16-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Wan J, et al. Liquid biopsies come of age: towards implementation of circulating tumour Dna. Nat. Rev. Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 411.Lo AA, et al. Indication-Specific Tumor Evolution and its Impact On Neoantigen Targeting and Biomarkers for Individualized Cancer Immunotherapies. J. Immunother. Cancer. 2021;9:e003001. doi: 10.1136/jitc-2021-003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Fry TJ, et al. Cd22-targeted Car T cells induce remission in B-All that is naive or resistant to Cd19-targeted Car immunotherapy. Nat. Med. 2018;24:20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Thomas P, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;595:432–437. doi: 10.1038/d41586-021-01958-0. [DOI] [PubMed] [Google Scholar]
- 414.Takamura-Ishii M, Nakaya T, Hagiwara K. Regulation of constitutive interferon-stimulated genes (Isgs) in tumor cells contributes to enhanced antitumor response of newcastle disease virus-infected tumor vaccines. Cancers. 2018;10:186. doi: 10.3390/cancers10060186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Rossi J, et al. Preinfusion polyfunctional anti-Cd19 chimeric antigen receptor T cells are associated with clinical outcomes in Nhl. Blood. 2018;132:804–814. doi: 10.1182/blood-2018-01-828343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Goyvaerts C, Breckpot K. Pros and cons of antigen-presenting cell targeted tumor vaccines. J. Immunol. Res. 2015;2015:785634. doi: 10.1155/2015/785634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Fraietta JA, et al. Determinants of response and resistance to Cd19 chimeric antigen receptor (Car) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018;24:563–571. doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Accolla RS, et al. Editorial: novel strategies for anti-tumor vaccines. Front. Immunol. 2019;10:3117. doi: 10.3389/fimmu.2019.03117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Melenhorst JJ, Barrett AJ. Tumor vaccines and beyond. Cytotherapy. 2011;13:8–18. doi: 10.3109/14653249.2010.530649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Kaiser J. Personalized tumor vaccines keep cancer in check. Science. 2017;356:122. doi: 10.1126/science.356.6334.122. [DOI] [PubMed] [Google Scholar]
- 421.Menez-Jamet J, Gallou C, Rougeot A, Kosmatopoulos K. Optimized tumor cryptic peptides: the basis for universal neo-antigen-like tumor vaccines. Ann. Transl. Med. 2016;4:266. doi: 10.21037/atm.2016.05.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Cassell DJ, Schwartz RH. A quantitative analysis of antigen-presenting cell function: activated B cells stimulate naive Cd4 T cells but are inferior to dendritic cells in providing costimulation. J. Exp. Med. 1994;180:1829–1840. doi: 10.1084/jem.180.5.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Liu L, et al. Synergistic killing effects of Pd-L1-Car T cells and colorectal cancer stem cell-dendritic cell vaccine-sensitized T cells in Aldh1-positive colorectal cancer stem cells. J. Cancer. 2021;12:6629–6639. doi: 10.7150/jca.62123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Subklewe M, et al. New generation dendritic cell vaccine for immunotherapy of acute myeloid leukemia. Cancer Immunol. Immunother. 2014;63:1093–1103. doi: 10.1007/s00262-014-1600-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Carreno BM, et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348:803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Shi W, et al. A new Pd-1-specific nanobody enhances the antitumor activity of T-cells in synergy with dendritic cell vaccine. Cancer Lett. 2021;522:184–197. doi: 10.1016/j.canlet.2021.09.028. [DOI] [PubMed] [Google Scholar]
- 427.Liu Y, et al. The adjuvant of alpha-galactosylceramide presented by gold nanoparticles enhances antitumor immune responses of Muc1 antigen-based tumor vaccines. Int J. Nanomed. 2021;16:403–420. doi: 10.2147/IJN.S273883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Phua KK. Towards targeted delivery systems: ligand conjugation strategies for Mrna nanoparticle tumor vaccines. J. Immunol. Res. 2015;2015:680620. doi: 10.1155/2015/680620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 429.Pappalardo F, et al. Induction of T-cell memory by a dendritic cell vaccine: a computational model. Bioinformatics. 2014;30:1884–1891. doi: 10.1093/bioinformatics/btu059. [DOI] [PubMed] [Google Scholar]
- 430.Van de Loosdrecht AA, et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol. Immunother. 2018;67:1505–1518. doi: 10.1007/s00262-018-2198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Ott PA, et al. Aneoantigen vaccine plus anti-Pd-1 elicits antitumor T-cell responses. Cancer Disco. 2020;10:1787. [Google Scholar]
- 432.Ott PA, et al. Corrigendum: An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2018;555:402. doi: 10.1038/nature25145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Keskin DB, et al. Neoantigen vaccine generates intratumoral T cell responses in phase ib glioblastoma trial. Nature. 2019;565:234–239. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Ott PA, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Guo Y, Lei K, Tang L. Neoantigen vaccine delivery for personalized anticancer immunotherapy. Front. Immunol. 2018;9:1499. doi: 10.3389/fimmu.2018.01499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Liu CJ, et al. Treatment of an aggressive orthotopic murine glioblastoma model with combination checkpoint blockade and a multivalent neoantigen vaccine. Neuro Oncol. 2020;22:1276–1288. doi: 10.1093/neuonc/noaa050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Joglekar AV, et al. T cell antigen discovery via signaling and antigen-presenting bifunctional receptors. Nat. Methods. 2019;16:191–198. doi: 10.1038/s41592-018-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Corrigan PA, Beaulieu C, Patel RB, Lowe DK. Talimogene laherparepvec: an oncolytic virus therapy for melanoma. Ann. Pharmacother. 2017;51:675–681. doi: 10.1177/1060028017702654. [DOI] [PubMed] [Google Scholar]
- 439.Gourd E. Oncolytic virus therapy in advanced melanoma. Lancet Oncol. 2017;18:e649. doi: 10.1016/S1470-2045(17)30782-9. [DOI] [PubMed] [Google Scholar]
- 440.Fukuhara H, Ino Y, Todo T. Oncolytic virus therapy: a new era of cancer treatment at dawn. Cancer Sci. 2016;107:1373–1379. doi: 10.1111/cas.13027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Patel MR, Kratzke RA. Oncolytic virus therapy for cancer: the first wave of translational clinical trials. Transl. Res. 2013;161:355–364. doi: 10.1016/j.trsl.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 442.Taguchi S, Fukuhara H, Todo T. Oncolytic virus therapy in Japan: progress in clinical trials and future perspectives. Jpn. J. Clin. Oncol. 2019;49:201–209. doi: 10.1093/jjco/hyy170. [DOI] [PubMed] [Google Scholar]
- 443.Godlewski J, et al. Oncolytic virus therapy alters the secretome of targeted glioblastoma cells. Cancers. 2021;13:1287. doi: 10.3390/cancers13061287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Kaufman HL, Maciorowski D. Advancing oncolytic virus therapy by understanding the biology. Nat. Rev. Clin. Oncol. 2021;18:197–198. doi: 10.1038/s41571-021-00490-4. [DOI] [PubMed] [Google Scholar]
- 445.Li J, et al. Gospel of malignant glioma: oncolytic virus therapy. Gene. 2022;818:146217. doi: 10.1016/j.gene.2022.146217. [DOI] [PubMed] [Google Scholar]
- 446.Cheema TA, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc. Natl Acad. Sci. USA. 2013;110:12006–12011. doi: 10.1073/pnas.1307935110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Ribas A, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-Pd-1 immunotherapy. Cell. 2017;170:1109–1119. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Park AK, et al. Effective combination immunotherapy using oncolytic viruses to deliver car targets to solid tumors. Sci. Transl. Med. 2020;12:eaaz1863. doi: 10.1126/scitranslmed.aaz1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449.Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer J. 2012;18:69–81. doi: 10.1097/PPO.0b013e31824671c9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Fidler IJ. Macrophage Therapy of Cancer Metastasis. Ciba Found. Symp. 1988;141:211–222. doi: 10.1002/9780470513736.ch12. [DOI] [PubMed] [Google Scholar]
- 451.Petty AJ, Yang Y. Tumor-associated macrophages: implications in cancer immunotherapy. Immunotherapy. 2017;9:289–302. doi: 10.2217/imt-2016-0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40:310–327. doi: 10.1016/j.it.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 453.Ngambenjawong C, Gustafson HH, Pun SH. Progress in tumor-associated macrophage (Tam)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017;114:206–221. doi: 10.1016/j.addr.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Italiani P, Boraschi D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front. Immunol. 2014;5:514. doi: 10.3389/fimmu.2014.00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Rothlin CV, Carrera-Silva EA, Bosurgi L, Ghosh S. Tam receptor signaling in immune homeostasis. Annu. Rev. Immunol. 2015;33:355–391. doi: 10.1146/annurev-immunol-032414-112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Lian G, et al. Colon cancer cell secretes Egf to promote M2 polarization of Tam through Egfr/Pi3K/Akt/Mtor pathway. Technol. Cancer Res. Treat. 2019;18:1078116716. doi: 10.1177/1533033819849068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Ramesh A, Kumar S, Nandi D, Kulkarni A. Csf1R- and Shp2-inhibitor-loaded nanoparticles enhance cytotoxic activity and phagocytosis in tumor-associated macrophages. Adv. Mater. 2019;31:e1904364. doi: 10.1002/adma.201904364. [DOI] [PubMed] [Google Scholar]
- 458.Vonderheide RH. Cd40 agonist antibodies in cancer immunotherapy. Annu. Rev. Med. 2020;71:47–58. doi: 10.1146/annurev-med-062518-045435. [DOI] [PubMed] [Google Scholar]
- 459.Aehnlich P, et al. Tam receptor inhibition-implications for cancer and the immune system. Cancers. 2021;13:1195. doi: 10.3390/cancers13061195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Xiao Z, et al. Antibody mediated therapy targeting Cd47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett. 2015;360:302–309. doi: 10.1016/j.canlet.2015.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Bewersdorf JP, Zeidan AM. Risk-adapted, individualized treatment strategies of myelodysplastic syndromes (Mds) and chronic myelomonocytic leukemia (Cmml) Cancers. 2021;13:1610. doi: 10.3390/cancers13071610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462.Lin F, et al. A novel blockade Cd47 antibody with therapeutic potential for cancer. Front. Oncol. 2020;10:615534. doi: 10.3389/fonc.2020.615534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Barkal AA, et al. Cd24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature. 2019;572:392–396. doi: 10.1038/s41586-019-1456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Sun J, et al. Cd47-targeting antibodies as a novel therapeutic strategy in hematologic malignancies. Leuk. Res. Rep. 2021;16:100268. doi: 10.1016/j.lrr.2021.100268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Lakhani NJ, et al. Evorpacept alone and in combination with pembrolizumab or trastuzumab in patients with advanced solid tumours (Aspen-01): a first-in-human, open-label, multicentre, phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2021;22:1740–1751. doi: 10.1016/S1470-2045(21)00584-2. [DOI] [PubMed] [Google Scholar]
- 466.Petrova PS, et al. Tti-621 (Sirpalphafc): a Cd47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin. Cancer Res. 2017;23:1068–1079. doi: 10.1158/1078-0432.CCR-16-1700. [DOI] [PubMed] [Google Scholar]
- 467.Chao MP, et al. Anti-Cd47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Wang J, Zhang H, Yin X, Bian Y. Anti-Cd47 antibody synergizes with cisplatin against laryngeal cancer by enhancing phagocytic ability of macrophages. Clin. Exp. Immunol. 2021;205:333–342. doi: 10.1111/cei.13618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Zhu S, et al. Tumor-associated macrophages: role in tumorigenesis and immunotherapy implications. J. Cancer. 2021;12:54–64. doi: 10.7150/jca.49692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Gholamin S, et al. Disrupting the Cd47-sirpalpha anti-phagocytic axis by a humanized anti-Cd47 antibody is an efficacious treatment for malignant pediatric brain tumors. Sci. Transl. Med. 2017;9:eaaf2968. doi: 10.1126/scitranslmed.aaf2968. [DOI] [PubMed] [Google Scholar]
- 471.Daver N, et al. New directions for emerging therapies in acute myeloid leukemia: the next chapter. Blood Cancer J. 2020;10:107. doi: 10.1038/s41408-020-00376-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Ni YH, Zhao X, Wang W. Cd24, a review of its role in tumor diagnosis, progression and therapy. Curr. Gene Ther. 2020;20:109–126. doi: 10.2174/1566523220666200623170738. [DOI] [PubMed] [Google Scholar]
- 473.Huang Y, et al. Engineered macrophages as near-infrared light activated drug vectors for chemo-photodynamic therapy of primary and bone metastatic breast cancer. Nat. Commun. 2021;12:4310. doi: 10.1038/s41467-021-24564-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.Bian Z, et al. Intratumoral sirpalpha-deficient macrophages activate tumor antigen-specific cytotoxic T cells under radiotherapy. Nat. Commun. 2021;12:3229. doi: 10.1038/s41467-021-23442-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Klichinsky M, et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020;38:947–953. doi: 10.1038/s41587-020-0462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Anderson NR, Minutolo NG, Gill S, Klichinsky M. Macrophage-based approaches for cancer immunotherapy. Cancer Res. 2021;81:1201–1208. doi: 10.1158/0008-5472.CAN-20-2990. [DOI] [PubMed] [Google Scholar]
- 477.Yang H, et al. Engineering macrophages to phagocytose cancer cells by blocking the Cd47/Sirpa axis. Cancer Med. 2019;8:4245–4253. doi: 10.1002/cam4.2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Fricker J. Engineered macrophages: a new weapon in the war on cancer? Mol. Med. Today. 2000;6:181–182. doi: 10.1016/S1357-4310(00)01700-7. [DOI] [PubMed] [Google Scholar]
- 479.Moyes KW, et al. Genetically engineered macrophages: a potential platform for cancer immunotherapy. Hum. Gene Ther. 2017;28:200–215. doi: 10.1089/hum.2016.060. [DOI] [PubMed] [Google Scholar]
- 480.Weiskopf K, et al. Myeloid cell origins, differentiation, and clinical implications. Microbiol. Spectr. 2016;4:1128. doi: 10.1128/microbiolspec.MCHD-0031-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37:208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 482.Hegde S, Leader AM, Merad M. Mdsc: markers, development, states, and unaddressed complexity. Immunity. 2021;54:875–884. doi: 10.1016/j.immuni.2021.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Gao X, et al. Immunotherapy targeting myeloid-derived suppressor cells (Mdscs) in tumor microenvironment. Front. Immunol. 2020;11:585214. doi: 10.3389/fimmu.2020.585214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Li Q, Xiang M. Metabolic reprograming of Mdscs within tumor microenvironment and targeting for cancer immunotherapy. Acta Pharmacol. Sin. 2021;43:1337–1348. doi: 10.1038/s41401-021-00776-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Almand B, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J. Immunol. 2001;166:678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- 486.Yang F, et al. The effect of immunosuppressive drugs on Mdscs in transplantation. J. Immunol. Res. 2018;2018:5414808. doi: 10.1155/2018/5414808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Cioccarelli C, Molon B. Mdscs and T cells in solid tumors and non-hodgkin lymphomas: an immunosuppressive speech. Clin. Exp. Immunol. 2022;208:147–157. doi: 10.1093/cei/uxac025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Xu X, et al. Carbohydrate-based adjuvants activate tumor-specific Th1 and Cd8(+) T-cell responses and reduce the immunosuppressive activity of Mdscs. Cancer Lett. 2019;440–441:94–105. doi: 10.1016/j.canlet.2018.10.013. [DOI] [PubMed] [Google Scholar]
- 489.Hofer F, et al. A complex metabolic network confers immunosuppressive functions to myeloid-derived suppressor cells (Mdscs) within the tumour microenvironment. Cells. 2021;10:2700. doi: 10.3390/cells10102700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Davis RJ, Van Waes C, Allen CT. Overcoming Barriers to effective immunotherapy: mdscs, tams, and tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral. Oncol. 2016;58:59–70. doi: 10.1016/j.oraloncology.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Cho SF, Anderson KC, Tai YT. Targeting B cell maturation antigen (Bcma) in multiple myeloma: potential uses of bcma-based immunotherapy. Front. Immunol. 2018;9:1821. doi: 10.3389/fimmu.2018.01821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Qin M, Jin Y, Pan LY. Tertiary lymphoid structure and B-cell-related pathways: a potential target in tumor immunotherapy. Oncol. Lett. 2021;22:836. doi: 10.3892/ol.2021.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Xue P, Fu J, Zhou Y. The aryl hydrocarbon receptor and tumor immunity. Front. Immunol. 2018;9:286. doi: 10.3389/fimmu.2018.00286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Vijayan D, Young A, Teng M, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer. 2017;17:709–724. doi: 10.1038/nrc.2017.86. [DOI] [PubMed] [Google Scholar]
- 495.Tallon DLP, et al. Cd39(+)Pd-1(+)Cd8(+) T cells mediate metastatic dormancy in breast cancer. Nat. Commun. 2021;12:769. doi: 10.1038/s41467-021-21045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Zheng Q, et al. Targeting the Ido-Bcl2a1-cytochrome C pathway promotes apoptosis in oral squamous cell carcinoma. Onco Targets Ther. 2021;14:1673–1687. doi: 10.2147/OTT.S288692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Liu Y, et al. Blockade of Ido-Kynurenine-Ahr metabolic circuitry abrogates Ifn-gamma-induced immunologic dormancy of tumor-repopulating cells. Nat. Commun. 2017;8:15207. doi: 10.1038/ncomms15207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Munn DHBlocking. Ido activity to enhance anti-tumor. Immun. Front. Biosci. 2012;4:734–745. doi: 10.2741/e414. [DOI] [PubMed] [Google Scholar]
- 499.Popp FC, et al. Expression of immune checkpoint regulators Ido, Vista, Lag3, and Tim3 in resected pancreatic ductal adenocarcinoma. Cancers. 2021;13:2689. doi: 10.3390/cancers13112689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Beatty GL, et al. First-in-human phase I study of the oral inhibitor of indoleamine 2,3-dioxygenase-1 epacadostat (Incb024360) in patients with advanced solid malignancies. Clin. Cancer Res. 2017;23:3269–3276. doi: 10.1158/1078-0432.CCR-16-2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Mitchell TC, et al. Epacadostat plus pembrolizumab in patients with advanced solid tumors: phase I results from a multicenter, open-label phase I/Ii trial (Echo-202/Keynote-037) J. Clin. Oncol. 2018;36:3223–3230. doi: 10.1200/JCO.2018.78.9602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Long GV, et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (Echo-301/Keynote-252): a phase 3, randomised, double-blind study. Lancet Oncol. 2019;20:1083–1097. doi: 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- 503.Li F, Zhang R, Li S, Liu J. Ido1: an important immunotherapy target in cancer treatment. Int. Immunopharmacol. 2017;47:70–77. doi: 10.1016/j.intimp.2017.03.024. [DOI] [PubMed] [Google Scholar]
- 504.Liu M, et al. Targeting the Ido1 pathway in cancer: from bench to bedside. J. Hematol. Oncol. 2018;11:100. doi: 10.1186/s13045-018-0644-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Chen S, et al. Cd39: the potential target in small cell lung cancer. Transl. Lung Cancer Res. 2020;9:1483–1495. doi: 10.21037/tlcr-20-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Kashyap AS, et al. Antisense oligonucleotide targeting Cd39 improves anti-tumor T cell immunity. J. Immunother. Cancer. 2019;7:67. doi: 10.1186/s40425-019-0545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Sonigo G, et al. Involvement of the Cd39/Cd73/adenosine pathway on T cell proliferation and Nk cell-mediated Adcc in sezary syndrome. Blood. 2022;139:2712–2716. doi: 10.1182/blood.2021014782. [DOI] [PubMed] [Google Scholar]
- 508.Yan J, et al. Control of metastases via myeloid Cd39 and Nk cell effector function. Cancer Immunol. Res. 2020;8:356–367. doi: 10.1158/2326-6066.CIR-19-0749. [DOI] [PubMed] [Google Scholar]
- 509.Timperi E, Barnaba V. Cd39 regulation and functions in T cells. Int. J. Mol. Sci. 2021;22:8068. doi: 10.3390/ijms22158068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Moesta AK, Li XY, Smyth MJ. Targeting Cd39 in cancer. Nat. Rev. Immunol. 2020;20:739–755. doi: 10.1038/s41577-020-0376-4. [DOI] [PubMed] [Google Scholar]
- 511.Koh J, et al. Mdsc subtypes and Cd39 expression on Cd8(+) T cells predict the efficacy of anti-Pd-1 immunotherapy in patients with advanced Nsclc. Eur. J. Immunol. 2020;50:1810–1819. doi: 10.1002/eji.202048534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Takenaka MC, Robson S, Quintana FJ. Regulation of the T cell response by Cd39. Trends Immunol. 2016;37:427–439. doi: 10.1016/j.it.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Allard D, Allard B, Stagg J. On the mechanism of anti-Cd39 immune checkpoint therapy. J. Immunother. Cancer. 2020;8:e000186. doi: 10.1136/jitc-2019-000186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Leem G, et al. 4-1Bb co-stimulation further enhances anti-Pd-1-mediated reinvigoration of exhausted Cd39(+) Cd8 T cells from primary and metastatic sites of epithelial ovarian cancers. J. Immunother. Cancer. 2020;8:e001650. doi: 10.1136/jitc-2020-001650. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.