

Abstract
Objectives
To analyse the clinical characteristics and prognosis of acute exacerbation (AE) in patients with idiopathic pulmonary fibrosis (IPF) and pulmonary emphysema.
Design
A multicentre retrospective cohort study
Setting
Two university hospitals in Japan
Participants
Patients admitted to hospitals due to AE of IPF diagnosed based on a multidisciplinary discussion.
Interventions
None
Primary and secondary outcome measures
90-day mortality rate
Methods
We retrospectively analysed consecutive patients with AE of IPF, with or without pulmonary emphysema, admitted to two university hospitals between 2007 and 2018.
Results
Among 62 patients (median age, 75 years; 48 men) admitted for AE of IPF, 29 patients (46%) presented with concomitant pulmonary emphysema. There was no significant difference in the arterial partial oxygen pressure/fraction of inhaled oxygen (P/F) ratio or other laboratory and radiographic data between patients with and without emphysema. The 90-day mortality rate was significantly lower in patients with emphysema than in those with IPF alone (23% vs 52%, p=0.03). The median survival time was significantly longer in patients with emphysema than in those with IPF alone (405 vs 242 days, p=0.02).
Conclusion
Patients with IPF and emphysema had better short-term survival after AE than those with non-emphysematous IPF.
Keywords: Interstitial lung disease, Emphysema, Adult intensive & critical care
Strengths and limitations of this study.
The studied population was patients with idiopathic pulmonary fibrosis (IPF) who developed acute exacerbations.
The diagnosis of IPF, emphysema and acute exacerbation was based on a multidisciplinary discussion by pulmonary physicians and a radiologist.
The number of patients studied was limited as a retrospective study in two clinical centres.
The incidence of acute exacerbation among patients with IPF with or without emphysema could not be analysed because of the retrospective design of this study.
Some data such as pulmonary function tests prior to acute exacerbation were not available.
Background
Idiopathic pulmonary fibrosis (IPF) is characterised by chronic, progressive, fibrosing interstitial pneumonia of unknown aetiology. The prognosis of IPF is poor, with a median survival time of 2–3 years. Acute exacerbation (AE) is the major cause of death in IPF patients, accounting for up to 40% of all deaths.1 AE–IPF is defined as the worsening of respiratory failure with acute or subacute onset that cannot be explained by cardiac failure or fluid overload, which parallels the Berlin criteria for acute respiratory distress syndrome.2
Pulmonary emphysema is common in lungs with IPF, as the estimated prevalence of emphysema ranges between 25% and 50%.3 Cottin et al proposed the term combined pulmonary fibrosis and emphysema (CPFE), which comprises upper lobe-dominant emphysema and lower lobe-dominant fibrosis.4 CPFE exhibits clinical characteristics, such as high prevalence in heavy smokers, relatively normal lung volumes accompanied by severely impaired gas exchange capacity, and a high risk for lung cancer and pulmonary arterial hypertension.3–5 Data on the prognosis of IPF with emphysema are inconsistent among reports although recent studies have reported that the prognosis of CPFE is as poor as that of IPF alone.6
The incidence of AEs in patients with CPFE has been reported to be 9.4% per year,7 which is compatible with the annual rate of AEs in patients with IPF.8 In contrast, AEs accounted for 31% of deaths in patients with IPF, but accounted for only 11.8% of deaths in patients with CPFE.9 Other causes, such as lung cancer and pulmonary artery hypertension, may play a major role in the mortality of patients with IPF and emphysema.3 4 Another possible explanation is that the prognosis of AE is better in patients with combined fibrosis and emphysema than in patients with IPF alone. Therefore, we analysed the clinical characteristics and prognoses of patients with AE of IPF and pulmonary emphysema.
Methods
Patients
Consecutive patients admitted to two university hospitals for AE–IPF between January 2007 and August 2018 were retrospectively analysed. IPF was diagnosed based on a multidisciplinary discussion by two pulmonologists and a radiologist using the patients’ clinical history, physical examination, laboratory test results and radiographic data from high-resolution CT.
AE–IPF was diagnosed based on the following criteria2: (1) previous or concurrent diagnosis of IPF; (2) acute worsening or development of dyspnoea typically of duration <1 month; (3) CT with new bilateral ground-glass opacity and/or consolidation superimposed on a background pattern consistent with the usual interstitial pneumonia (UIP) pattern and (4) deterioration not fully explained by cardiac failure or fluid overload. The patients with neoplasms undergoing active treatment, including radiation therapy, chemotherapy with cytotoxic or molecular-targeted drugs, or immunotherapy with immune checkpoint inhibitors, at the onset of AE were excluded.
AE–IPF was treated with an appropriate administration of oxygen and pharmacotherapy using prednisone (0.5–1 mg/kg per day) with or without preceding methylprednisolone pulse therapy (1 g/day for 3 days). The prednisone dose was tapered based on the response to therapy. In refractory cases, immunosuppressive therapy with intravenous cyclophosphamide (500 mg every 2–4 weeks) may have been added.
Clinical and laboratory parameters
The clinical and laboratory data within 12 months prior to and at the onset of AE were collected from medical charts. Spirometry was performed during stable disease using a Super Spiro DISCOM-21FX III spirometer (CHEST Corp., Tokyo, Japan) by trained clinical technicians. The predicted values of forced vital capacity (FVC) and forced expiratory volume in 1 s (FEV1) were calculated using a previously reported equation.10 In patients without arterial blood gas data at the time of admission, the partial pressure of arterial oxygen (PaO2) was calculated based on percutaneous arterial oxygen saturation (SpO2) using the Hill formula.11 The serum levels of Krebs von Lungen-6 (KL-6) were measured using an electrochemi-luminescence immunoassay and Lumipulse G1200 Analyzer (Rebio, Fuji, Japan). The surfactant protein D (SP-D) levels were measured using commercially available ELISA kits (RayBiotech, Norcross, GA, USA).
Radiographic evaluation of thoracic CT and diagnosis IPF
Thoracic CT with 1.5-mm-thick axial sections was obtained at 1 cm intervals throughout the entire thorax in the inspiratory phase. Emphysema was defined as demarcated areas of decreased attenuation compared with contiguous normal lung tissue and marginated by a very thin or no wall, with upper-zone predominance. The extent of emphysema was evaluated by low attenuation area (LAA) score in chest CT according to the method proposed by Goddard et al.12 The cases with LAA score>0 was classified in the emphysema group. The existence of emphysema and diagnostic categories of UIP based on CT patterns12 13 were diagnosed based on a discussion between two pulmonologists and a radiologist.
Statistical analysis
The data are expressed as mean±SD or median and IQR for continuous variables, and number and percentage for categorical data. Group comparisons were made using the Mann–Whitney U test for continuous variables and Fisher’s exact test for categorical variables. Kaplan–Meier survival curves and log-rank tests were used to evaluate the survival. Cox proportional hazards regression analysis of mortality within 90 days was performed using age, sex and other factors with p<0.1 in univariate analysis and data available in >90% of the cases. A statistical analysis was performed using IBM SPSS Statistics software V.21 (IBM, Chicago, IL, USA). The level of statistical significance was set at p<0.05.
Patient and public involvement
This was a retrospective cohort study with no direct patient and public engagement.
Results
Clinical characteristics prior to AE
Among 103 patients admitted to hospitals due to acute respiratory failure of idiopathic interstitial pneumonia during the study period, we identified 62 patients with AE–IPF (median age, 75 years; 48 men and 14 women, online supplemental figure 1). Notably, 29 patients (46.7%) had emphysema (IPF with emphysema group) and 33 patients had IPF without emphysema (IPF alone group).
bmjopen-2022-062236supp001.pdf (61.9KB, pdf)
The clinical characteristics at baseline (within 12 months before AE) are presented in table 1. All the patients with emphysema were smokers, whereas 19 patients (58%) with IPF alone were never smokers. The total exposure to cigarette smoke (pack-years) was greater in the IPF with emphysema group than in the IPF alone group (p<0.001). Age, sex, body mass index, proportion of patients with comorbidities, under long-term oxygen therapy or on pharmacotherapy with prednisolone or antifibrotic agents were not significantly different between the groups. The data of pulmonary function tests within 1 year prior to AE were available in 20 cases in the IPF with emphysema group and 17 cases in the IPF alone group. The FVC, %FVC of the predicted value and FEV1 were significantly higher in the IPF with emphysema group (p=0.045, 0.002 and 0.001, respectively), whereas the FEV1/FVC was not significantly different between the groups. The diagnostic categories of UIP based on CT patterns were not significantly different between the groups. One case, which was categorised as an alternative pattern, was diagnosed as IPF on autopsy.
Table 1.
Baseline characteristics in patients with idiopathic pulmonary fibrosis with or without concomitant pulmonary emphysema
| Characteristic | IPF with emphysema (n=29) | IPF alone (n=33) |
P value |
| Age, years | 74±6 | 76±8 | 0.20 |
| Male, n (%) | 28 (97%) | 20 (60%) | |
| Body mass index | 21.9±3.2 (n=25) | 22.7±4.3 (n=29) | 0.40 |
| Smoking status | |||
| Smokers, n (%) | 29 (100%) | 14 (42%) | <0.001 |
| Pack-years | 59±68 | 12±20 | <0.001 |
| Comorbidity | |||
| Lung cancer, n (%) | 2 (7%) | 1 (3%) | 0.59 |
| Any cancer, n (%) | 5 (17%) | 3 (9%) | 0.33 |
| Diabetes mellitus, n (%) | 6 (21%) | 10 (30%) | 0.38 |
| Chronic heart failure, n (%) | 6 (21%) | 13 (39%) | 0.11 |
| Chronic renal failure, n (%) | 0 (0%) | 2 (6%) | 0.59 |
| Chronic respiratory infection, n (%) | 1 (3%) | 1 (3%) | 0.93 |
| Laboratory data | |||
| KL-6 in serum (U/mL) | 1266±697 (n=24) | 1255±817 (n=26) | 0.74 |
| Albumin in serum (g/mL) | 3.7±0.5 (n=20) | 3.7±0.3 (n=23) | 0.64 |
| Pulmonary functions | n=20 | n=17 | |
| FVC (L) | 2.26±0.68 | 1.54±0.61 | 0.045 |
| FVC, %predicted | 69.4±20.3 | 56.8±22.4 | 0.002 |
| FEV1 (L) | 1.9±0.5 | 1.3±0.4 | 0.001 |
| FEV1/FVC (%) | 85.9±7.6 | 89.3±9.2 | 0.11 |
| Thoracic CT | |||
| CT pattern (2018 IPF guideline) | |||
| Definite/probable/ indeterminate/alternative |
21/7/0/1 | 14/19/0/0 | 0.08 |
| Low-attenuation area score | 5.8±2.0 | 0.0±0.0 | < 0.001 |
| Treatment, n (%) | |||
| Prednisolone | 7 (24%) | 8 (24%) | 0.99 |
| Antifibrotic agents | 6 (20%) | 3 (9%) | 0.13 |
FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity; IPF, interstitial pulmonary fibrosis; KL-6, Krebs von Lungen-6.
Clinical characteristics on admission due to AE
The laboratory data on admission, such as leucocyte counts in peripheral blood and serum levels of lactate dehydrogenase, were not significantly different between the IPF with emphysema and IPF alone groups (table 2). In addition, the fraction of inhaled oxygen (FIO2) and the PaO2/FIO2 (P/F) ratio were not significantly different between the groups. The serum KL-6 and SP-D levels were higher in the IPF with emphysema group (p=0.05 and 0.025, respectively).
Table 2.
Clinical characteristics on admission due to acute exacerbation
| IPF with emphysema (n=29) | IPF alone (n=33) | P value | |
| Respiratory rate (/min) | 23±5 | 25±6 | 0.18 |
| Laboratory data | |||
| Leukocytes (×103/ µL) | 10.1±3.1 | 11.0±5.1 | 0.27 |
| Lactate dehydrogenase (U/L) | 377±164 | 372±103 | 0.60 |
| KL-6 (U/mL) | 2109±1249 | 1644±1193 | 0.049 |
| SP-D (U/mL) (n=46) | 599±380 (n=24) | 388±305 (n=22) | 0.025 |
| BNP (pg/mL) (n=47) | 116±149 (n=24) | 166±194 (n=23) | 0.25 |
| FIO2 | 0.38±0.24 | 0.33±0.20 | 0.94 |
| P/F ratio | 233±105 | 221±80 | 0.51 |
| Treatment, n (%) | |||
| Drug | |||
| High dose corticosteroids | 29 (100%) | 32 (97%) | – |
| Methylprednisolone pulse therapy | 17 (58%) | 22 (66%) | 0.51 |
| Immunosuppressive therapy | 3 (10%) | 4 (12%) | 0.45 |
| Oxygen therapy | |||
| High flow nasal cannula | 4 (13%) | 8 (24%) | 0.29 |
| NIPPV | 0 (0%) | 2 (6%) | 0.49 |
| IPPV | 0 (0%) | 2 (6%) | 0.49 |
| Do-not-resuscitation order, n (%) | 29 (100%) | 31 (94%) | 0.92 |
AE, acute exacerbation; IPF, interstitial pulmonary fibrosis; IPPV, intermittent positive pressure ventilation; KL-6, Krebs von Lungen-6; NIPPV, non-invasive intermittent positive pressure ventilation; P/F ratio, PaO2/FIO2 ratio; SIRS, systemic inflammatory response syndrome; SP-D, surfactant protein D.
All the patients, except for one patient who was not treated with prednisolone, were treated with high-dose prednisolone and oxygen therapy. There was no difference in the proportion of patients treated with methylprednisolone pulse therapy and/or immunosuppressive therapy between the groups. Four patients with IPF and emphysema and 12 patients with IPF alone required a high-flow nasal cannula or positive-pressure ventilation for the treatment of respiratory failure.
Short-term prognosis within 90 days after admission
Nine (15%), 15 (24%) and 23 (38%) patients died within 7, 30 and 90 days of admission due to AE–IPF, respectively. The survivors were more likely to have emphysema (56%) than the deceased (30%), with an OR of 0.27 (95% CI, 0.07 to 0.88; p=0.02). FVC and P/F ratio were lower (p=0.02 and 0.06, respectively) in the deceased patients (table 3). The survival rate was significantly higher in patients with emphysema than in those without emphysema (p=0.03), and the 90-days survival rate was higher in patients with emphysema (76% and 50%, respectively; figure 1). The age, sex, smoking history (pack-years), serum KL-6 levels, comorbidity of lung cancer, long-term oxygen therapy and the proportion of patients using prednisolone and/or antifibrotic agents before AE were not significantly different between patients who died and those who survived for the first 90 days (table 3). A Cox proportional hazards regression analysis revealed that the presence of emphysema (HR 0.33 (95% CI 0.14 to 0.82), p=0.01) and P/F ratio (HR 0.99 (95% CI 0.98 to 0.99), p=0.01) were predictors of mortality within 90 days after adjustment for age and sex (table 4).
Table 3.
Clinical characteristics of non-survivors and survivors at day 90
| Non-survivors at day 90 (n=23) |
Survivors at day 90 (n=36) |
P value | |
| Baseline clinical characteristics | |||
| Age, years | 77±6 | 73±7 | 0.25 |
| male, n (%) | 17 (74%) | 32 (89%) | 0.18 |
| Smoking status | |||
| Smokers, n (%) | 12 (52%) | 26 (72%) | 0.11 |
| Pack-years | 22±27 | 42.3±33.1 | 0.13 |
| Pulmonary functions | n=13 | n=23 | |
| FVC (L) | 1.56±0.72 | 2.23±0.67 | 0.02 |
| FVC, %predicted | 56.4±27.3 | 70.6±17.1 | 0.12 |
| FEV1/FVC (%) | 89.5±9.5 | 86.3±7.6 | 0.24 |
| Thoracic CT | |||
| Emphysema, n (%) | 7 (30%) | 20 (56%) | 0.02 |
| CT pattern (2018 IPF guideline) | |||
| Definite/probable/indeterminate/alternative | 13/10/0/0 | 22/13/0/1 | 0.35 |
| Treatment, n (%) | |||
| Long-term oxygen therapy | 5 (21%) | 7 (19%) | 0.83 |
| Prednisolone | 6 (26%) | 8 (22%) | 0.73 |
| Antifibrotic agents | 2 (7%) | 6 (16%) | 0.22 |
| Clinical data on admission | |||
| Respiratory rate (/min) | 26±5 | 23±6 | 0.18 |
| Laboratory data | |||
| Leukocytes (×103/ µL) | 11.2±3.5 | 11.0±5.1 | 0.34 |
| Lactate dehydrogenase (U/L) | 380±106 | 372±103 | 0.42 |
| KL-6 (U/mL) | 1926±1642 | 1644±1193 | 0.63 |
| SP-D (U/mL) | 352±147 (n=15) | 388±305 (n=22) | 0.23 |
| FIO2 | 0.40±0.29 | 0.34±0.20 | 0.30 |
| P/F ratio | 194±94 | 241±90 | 0.06 |
| Treatments, n (%) | |||
| Drugs | |||
| Methylprednisolone pulse therapy | 17 (74%) | 22 (61%) | 0.60 |
| Immunosuppressive therapy | 3 (13%) | 4 (11%) | 0.14 |
| Oxygen therapy | |||
| High flow nasal cannula | 7 (30%) | 5 (14%) | 0.11 |
| NPPV | 2 (9%) | 0 (0%) | 0.16 |
| IPPV | 1 (4%) | 1 (3%) | 1.00 |
FEV1, forced expiratory volume in one second; FVC, forced vital capacity; IPPV, intermittent positive pressure ventilation; KL-6, Krebs von Lungen-6; NIPPV, non-invasive intermittent positive pressure ventilation; P/F ratio, PaO2/FIO2 ratio; SIRS, systemic inflammatory response syndrome; SP-D, surfactant protein D.
Figure 1.
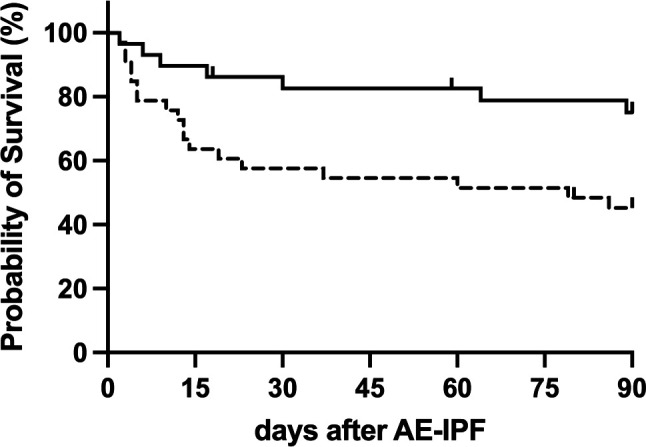
Kaplan–Meier survival curves for patients admitted to the hospital with AE–IPF with pulmonary emphysema (solid line, n=29) and without emphysema (dashed line, n=33). AE, acute exacerbation; IPF, idiopathic pulmonary fibrosis.
Table 4.
Cox proportional hazards regression analysis of mortality within 90 days
| HR | 95% CI | P value* | |
| Pulmonary emphysema | 0.33 | 0.14 to 0.82 | 0.01 |
| PaO2/FIO2 ratio | 0.99 | 0.98 to 0.99 | 0.01 |
*Adjusted for age and sex.
PaO2/FIO2, partial pressure of arterial oxygen/fraction of inhaled oxygen.
In the multivariate analysis, FVC was not included because of the high rate of missing values. Therefore, we performed a subanalysis by dividing the patients into four subgroups according to the presence or absence of emphysema and baseline FVC (≥60% or <60% of the predicted value). A Kaplan–Meier estimate according to this subgrouping revealed that the presence of emphysema tended to have a good prognosis regardless of the %FVC (online supplemental figure 2). A Cox proportional hazards regression analysis including the %FVC revealed that the presence of emphysema remained a predictor of mortality within 90 days (HR, 0.30 (95% CI 0.09 to 0.98), p=0.04) (online supplemental table 1).
Long-term prognosis
Notably, 48 patients, including 21 patients in the IPF with emphysema group and 27 patients in the IPF alone group, died during the observation period (median: 325 days, range: 2–1721 days). Although there was no difference in the total mortality between the groups, the median survival time was significantly longer in the IPF with emphysema group than in the IPF alone group (405 days vs 254 days, p=0.02, table 5).
Table 5.
Prognosis after acute exacerbation of IPF
| IPF with emphysema | IPF alone | P value | |
| All patients | n=29 | n=33 | |
| Median survival time, days | 405 (2–1544) | 254 (2–1721) | 0.02 |
| Survivors at day 90 | n=22 | n=16 | |
| Median survival time, days | 573 (110–1544) | 565 (106–1721) | 0.96 |
| Cause of death | n=14 | n=11 | |
| Acute exacerbation, n (%) | 4 (28%) | 6 (55%) | 0.24 |
| Chronic respiratory failure, n (%) | 3 (22%) | 4 (36%) | 0.66 |
| Pneumonia, n (%) | 3 (22%) | 0 (0%) | 0.23 |
| Unknown, n (%) | 4 (28%) | 1 (9%) | – |
IPF, idiopathic pulmonary fibrosis
There were 38 patients who survived 90 days or more after admission due to AE–IPF, including 22 patients with emphysema and 16 patients without emphysema (table 5). In these acute phase survivors, the long-term prognosis and cause of death was not significantly different between patients with and without emphysema (p=0.8) (online supplemental figure 3, table 5). The median survival times for the IPF with emphysema and IPF alone groups were 573 and 565 days, respectively (p=0.96). The leading cause of death in survivors was the exacerbation of IPF, regardless of the presence of emphysema (table 5).
Discussion
We retrospectively analysed 62 consecutive patients admitted for AE–IPF in two hospitals. Approximately half of the patients with AE–IPF exhibited pulmonary emphysema, who showed higher exposure to cigarette smoking, better FVC before AE and higher levels of KL-6 and SP-D at AE. The patients with emphysema had better short-term survival than those without emphysema, although the long-term survival of the survivors was equivalent.
Several possibilities may explain why the presence of emphysema was associated with better short-term prognosis in patients with IPF. First, the higher baseline FVC observed in the IPF with emphysema group may have contributed to a better survival rate. However, several studies have demonstrated that higher FVC values were not associated with better prognosis in patients with AE of chronic fibrosing interstitial pneumonia (CFIP).14 15 Furthermore, the copresence of emphysema may have masked the decrease in FVC in the IPF with emphysema group and greater FVC does not suggest a better capacity for alveolar gas exchange, which is essential for favourable oxygenation. In fact, there was no difference in the P/F ratio at exacerbation, an essential factor associated with the poor prognosis of AE–IPF, between patients with and without emphysema. Ikuyama et al also reported that patients with CPFE had better baseline %FVC and better prognosis after exacerbation than those with IPF alone;16 however, there was no difference in the % diffusing capacity of lung for carbon monoxide of the predicited or gender-age-physiology (GAP) score between the groups. Therefore, the higher baseline FVC observed in the IPF with emphysema group is unlikely to be the cause of the better short-term prognosis in these patients. Although substantial FVC data were lacking in the present study, pulmonary emphysema was an independent factor in determining the short-term prognosis even after adjustment for FVC values.
Ikuyama et al reported that patients with CPFE exhibited significantly lower serum KL-6 levels on admission and postulated that lung damage was less extensive in these patients.16 Another recent study also reported that higher serum KL-6 levels on admission were associated with a poorer prognosis in patients with AE-IPF.17 However, in our study, the serum KL-6 levels were higher in patients with IPF and emphysema. Therefore, patients with IPF and emphysema had better short-term survival, regardless of their serum KL-6 levels on admission.
The heterogeneity of prognosis in patients with AE-IPF may be due to different pathological changes in the lungs during AE. Diffuse alveolar damage is the major pathological finding in the lungs with AE-IPF in autopsy cases, accounting for approximately 80% of cases, followed by alveolar haemorrhage.18 In contrast, lung specimens obtained by surgical biopsy often show organising pneumonia. Churg et al19 reported that 6 of 12 patients with AE–CFIP presented with organising pneumonia and survived the acute phase, whereas half of the patients presenting with diffuse alveolar damage died. Currently, there is no histopathological information explaining the different prognoses of AE-IPF due to emphysema, and further studies are required.
The Kaplan–Meier estimate revealed significantly better short-term survival in the IPF with emphysema group than in the IPF alone group. However, in the survivors of acute phase, long-term prognosis was not significantly different between patients with and without emphysema. The major cause of death was AE–IPF, observed in 33 cases, comprising 11 patients (52%) with emphysema and 22 patients (81%) without emphysema. This observation may be consistent with a previous report which stated that patients with IPF who never smoked and developed AE had poorer prognoses than those with IPF who smoked.17
This study has several limitations. First, this was a retrospective study of cases in two clinical centres; therefore, the number of patients studied was limited. In addition, we could not estimate the incidence of AEs in patients with IPF and pulmonary emphysema because of the retrospective design of this study. Prospective multicentre studies, such as the Japan Idiopathic Interstitial Pneumonias Registry are currently ongoing in Japan and are warranted. Second, due to the real-world nature of the study, some clinical data, especially data on the diffusion capacity and/or the 6 min walk test, which are used to calculate GAP scores to evaluate the IPF severity,20 21 were missing in most cases. However, we propose that the presence or absence of emphysema on thoracic CT may be associated with the prognosis of AE, which is a leading cause of death in patients with IPF.
Conclusion
Patients with AE–IPF and concomitant pulmonary emphysema had better short-term prognoses than those without emphysema. Further studies are required to identify the histopathological characteristics of AE–IPF lungs with emphysema associated with better prognosis.
Supplementary Material
Footnotes
Contributors: YH and KA contributed to the conception and design of the study, analysed and interpreted the data and wrote the first version of the draft. TT contributed to the conception and design of the study, analysed the data (radiological images) and wrote the first version of the draft. KE contributed to the acquisition of data, analysed the data (radiological images) and revised the manuscript critically for important intellectual content. NN analysed the data (radiological images) and revised the manuscript critically for important intellectual content. FT, JT, KT, KN, ST, NH, YI and TO contributed to the acquisition of data and revised the manuscript critically for important intellectual content. All authors read and approved the final version of the draft, and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. KA is responsible for the overall content as the guarantor.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient and public involvement: Patients and/or the public were not involved in the design, or conduct, or reporting or dissemination plans of this research.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request. The data set used in this study is available from the corresponding author upon request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study involves human participants and was approved by the Institutional Review Board of Tokai University Hospital (17R-198) the Institutional Review Board of Tokai University Oiso Hospital (18R-241). Informed consent was obtained from the Tokai University Hospital website as an opt-out.
References
- 1.Natsuizaka M, Chiba H, Kuronuma K, et al. Epidemiologic survey of Japanese patients with idiopathic pulmonary fibrosis and investigation of ethnic differences. Am J Respir Crit Care Med 2014;190:773–9. 10.1164/rccm.201403-0566OC [DOI] [PubMed] [Google Scholar]
- 2.Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2007;176:636–43. 10.1164/rccm.200703-463PP [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mejía M, Carrillo G, Rojas-Serrano J, et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest 2009;136:10–15. 10.1378/chest.08-2306 [DOI] [PubMed] [Google Scholar]
- 4.Cottin V, Nunes H, Brillet P-Y, et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J 2005;26:586–93. 10.1183/09031936.05.00021005 [DOI] [PubMed] [Google Scholar]
- 5.Usui K, Tanai C, Tanaka Y, et al. The prevalence of pulmonary fibrosis combined with emphysema in patients with lung cancer. Respirology 2011;16:326–31. 10.1111/j.1440-1843.2010.01907.x [DOI] [PubMed] [Google Scholar]
- 6.Jiang C-G, Fu Q, Zheng C-M. Prognosis of combined pulmonary fibrosis and emphysema: comparison with idiopathic pulmonary fibrosis alone. Ther Adv Respir Dis 2019;13:1753466619888119. 10.1177/1753466619888119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kishaba T, Shimaoka Y, Fukuyama H, et al. A cohort study of mortality predictors and characteristics of patients with combined pulmonary fibrosis and emphysema. BMJ Open 2012;2. doi: 10.1136/bmjopen-2012-000988. [Epub ahead of print: 15 05 2012]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song JW, Hong S-B, Lim C-M, et al. Acute exacerbation of idiopathic pulmonary fibrosis: incidence, risk factors and outcome. Eur Respir J 2011;37:356–63. 10.1183/09031936.00159709 [DOI] [PubMed] [Google Scholar]
- 9.Kurashima K, Takayanagi N, Tsuchiya N, et al. The effect of emphysema on lung function and survival in patients with idiopathic pulmonary fibrosis. Respirology 2010;15:843–8. 10.1111/j.1440-1843.2010.01778.x [DOI] [PubMed] [Google Scholar]
- 10.Kubota M, Kobayashi H, Quanjer PH, et al. Reference values for spirometry, including vital capacity, in Japanese adults calculated with the LMS method and compared with previous values. Respir Investig 2014;52:242–50. 10.1016/j.resinv.2014.03.003 [DOI] [PubMed] [Google Scholar]
- 11.Hill A. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J Physiol 1910;40:4–7. [Google Scholar]
- 12.Goddard PR, Nicholson EM, Laszlo G, et al. Computed tomography in pulmonary emphysema. Clin Radiol 1982;33:379–87. 10.1016/S0009-9260(82)80301-2 [DOI] [PubMed] [Google Scholar]
- 13.Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society white paper. Lancet Respir Med 2018;6:138–53. 10.1016/S2213-2600(17)30433-2 [DOI] [PubMed] [Google Scholar]
- 14.Akira M, Kozuka T, Yamamoto S, et al. Computed tomography findings in acute exacerbation of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2008;178:372–8. 10.1164/rccm.200709-1365OC [DOI] [PubMed] [Google Scholar]
- 15.Oishi K, Aoe K, Mimura Y, et al. Survival from an acute exacerbation of idiopathic pulmonary fibrosis with or without direct hemoperfusion with a polymyxin B-immobilized fiber column: a retrospective analysis. Intern Med 2016;55:3551–9. 10.2169/internalmedicine.55.6056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikuyama Y, Ushiki A, Kosaka M, et al. Prognosis of patients with acute exacerbation of combined pulmonary fibrosis and emphysema: a retrospective single-centre study. BMC Pulm Med 2020;20:144. 10.1186/s12890-020-01185-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kishaba T, Nagano H, Nei Y, et al. Clinical characteristics of idiopathic pulmonary fibrosis patients according to their smoking status. J Thorac Dis 2016;8:1112–20. 10.21037/jtd.2016.03.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oda K, Ishimoto H, Yamada S, et al. Autopsy analyses in acute exacerbation of idiopathic pulmonary fibrosis. Respir Res 2014;15:109. 10.1186/s12931-014-0109-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Churg A, Müller NL, Silva CIS, et al. Acute exacerbation (acute lung injury of unknown cause) in UIP and other forms of fibrotic interstitial pneumonias. Am J Surg Pathol 2007;31:277–84. 10.1097/01.pas.0000213341.70852.9d [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi H, Omori S, Nakashima K, et al. Modified gap index for prediction of acute exacerbation of idiopathic pulmonary fibrosis in non-small cell lung cancer. Respirology 2017;22:1379–85. 10.1111/resp.13075 [DOI] [PubMed] [Google Scholar]
- 21.Ryerson CJ, Vittinghoff E, Ley B, et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest 2014;145:723–8. 10.1378/chest.13-1474 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjopen-2022-062236supp001.pdf (61.9KB, pdf)
Data Availability Statement
Data are available upon reasonable request. The data set used in this study is available from the corresponding author upon request.