

Abstract
Bilirubin, a breakdown product of heme, is normally glucuronidated and excreted by the liver into bile. Failure of this system can lead to a buildup of conjugated bilirubin in the blood, resulting in jaundice.
Hyperbilirubinemia is an important clinical sign that needs to be investigated under a stepwise evaluation. Inherited non‐hemolytic conjugated hyperbilirubinemic conditions include Dubin‐Johnson syndrome (caused by mutations affecting ABCC2 gene) and Rotor syndrome (caused by the simultaneous presence of mutations in SLCO1B1 and SLCO1B3 genes). Although classically viewed as benign conditions requiring no treatment, they lately gained an increased interest since recent studies suggested that mutations in the responsible genes leading to hyperbilirubinemia, as well as minor genetic variants, may result in an increased susceptibility to drug toxicity.
This article provides a comprehensive review on the pathophysiology of Dubin‐Johnson and Rotor syndromes, presenting the current knowledge concerning the molecular details and basis of these conditions.
Keywords: ABCC2/MRP2, conjugated hyperbilirubinemia, Dubin‐Johnson syndrome, Rotor syndrome, SLCO1B1/OATP1B1, SLCO1B3/OATP1B3
INTRODUCTION
For each 1000 adults, 1.5 will present jaundice. 1 The differential diagnosis is extensive, reflecting the complex metabolism of bilirubin. Unconjugated or mixed hyperbilirubinemia translates increased production or decreased conjugation whilst conjugated hyperbilirubinemia translates disturbances in storing or biliary secretion, in both cases either acquired or inherited.
Two inherited disorders manifest by isolated conjugated hyperbilirubinemia: Dubin‐Johnson Syndrome (DJS) and Rotor Syndrome (RS). Although uncommon, their diagnosis is crucial since differentiation from harmful disorders can avoid unnecessary procedures and anxiety. Both are benign disorders, however they may result in increased susceptibility to drug toxicity.
We present a review on DJS and RS, focusing their pathophysiology, discussing the emerging genetic concepts, highlighting the need for evaluation in dedicated laboratories.
PHYSIOPATHOLOGY OF HEPATIC BILIRUBIN CLEARANCE
Bile secretion starts at the hepatocyte canaliculus, followed by modifications along the bile ducts. 2 Canalicular bile is formed by osmotic filtration of water and electrolytes in response to osmotic gradients generated by active transport systems for bile salts (bile salt‐dependent flow) and other organic anions such as reduced glutathione (GSH) and bicarbonate (HCO3‒) (bile salt‐independent flow) 2 (Figure 1). Bile secretion depends on: (1) active transmembrane transport systems generating osmotic gradients 3 , 4 ; (2) cytoskeleton apparatus allowing the movement of vesicles and bile canalicular contractions; (3) cell contacts sealing the canaliculi and maintaining cell polarity; and (4) signal transduction mechanisms regulating and coordinating bile formation.
FIGURE 1.
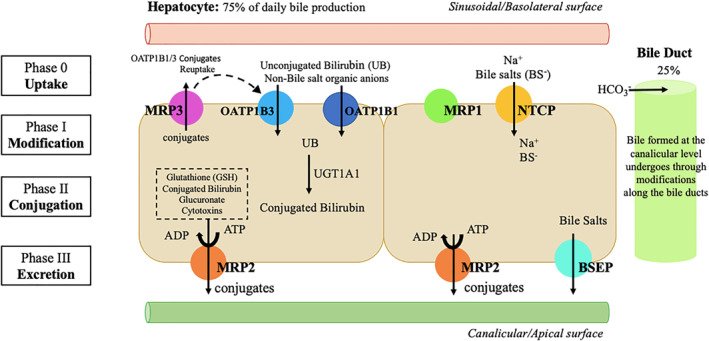
Mechanisms of bile formation and hepatobiliary transport systems in human liver. Bile formation begins in the hepatocyte, at the canalicular level (75% of daily bile production), followed by modifications along the bile ducts (25% of daily production). The uptake of various substances across the basolateral membrane is followed by conjugation reactions mediated by the enzyme UGT1A1 (Phase II). Posteriorly, excretion occurs and substances, including conjugated bilirubin, are secreted into the bile–this process is mediated by Multidrug Resistance‐associated Protein 2 (MRP2) with possible minor contribution of other transports at the canalicular (apical) membrane of hepatocytes. The hepatobiliary transport of bile salts is performed by a different system (their excretion occurs through bile salt export pump (BSEP) transport). In addition, even under physiologic conditions, a fraction of bilirubin conjugates is secreted by MRP3 across sinusoidal (basolateral) membrane into the blood, from where they can be subsequently reuptaken by sinusoidal membrane‐bound OATP1B1 and OATP1B3 transporters. MRP: Multidrug resistance‐associated protein; OATP: Organic anion transporting polypeptide; UGT: Uridine diphosphate glucuronosyltransferase; NTCP: Na+ taurocholate cotransporter; BSEP: Bile salt export pump; ATP: Adenosine triphosphate; ADP: Adenosine diphosphate; UB: unconjugated bilirubin; HCO3 −: Bicarbonate; Na+: Sodium
Hepatocytes are polarized epithelial cells with a polyhedric structure. Assuming a cuboidal structure, two surfaces face perisinusoidal space (basal domain), two face neighbor hepatocytes (lateral domain) and the remaining face a bile canaliculus (apical domain).
Hepatic uptake of biliary constituents occurs at the sinusoidal/basal membrane, which is in contact with the sinusoidal blood via the space of Disse. A Na+‐dependent Na+/taurocholate cotransporter (NTCP) is the main uptake system for bile salts, 5 while the family of Na+‐independent organic anion transporting proteins transfers a large array of bile salts and non‐bile salt organic anions and cationic drugs 5 (Figure 1).
Canalicular bile excretion is the rate‐limiting step for bile formation. 6 The canalicular membrane contains both ATP‐dependent and independent transport systems. 4 ATP‐dependent transport systems, known as ATP‐binding cassette (ABC) proteins, transport biliary constituents against steep concentration gradients across the canalicular membrane. Most of ABC proteins belong to the multidrug resistance protein or multidrug resistance‐associated protein (MRP) gene superfamilies. 6 A bilirubin conjugate export pump (Multidrug Resistance‐associated Protein 2 (MRP2)), 7 also known as canalicular multispecific organic anion transporter (cMOAT), mediates the canalicular excretion of a broad range of organic anions, most of which are conjugates with glutathione, glucuronate and sulfate. 8
The MRP isoform MRP3 may serve as a compensatory mechanism when MRP2 function fails. MRP3 is located on the sinusoidal membrane of hepatocytes and its expression is regulated inversely to MRP2. MRP3 upregulation acts as an emergency export pump for conjugates that cannot be secreted into bile and therefore are released into blood for renal excretion. 9
In the ATP‐independent transport system, the canalicular bicarbonate secretion is mediated via Cl−/HCO3− anion exchanger localized to both bile canaliculi and ducts. Anion exchanger and MRP2 are the major driving forces for bile salt‐independent bile flow, while a bile salt export pump drives bile salt‐dependent flow 10 (Figure 1).
EPIDEMIOLOGY
Dubin‐Johnson Syndrome and RS are rare underdiagnosed hereditary disorders with autosomal recessive transmission. Dubin‐Johnson Syndrome is due to a defect on MRP2 (disturbing biliary bilirubin excretion) and RS to a deficiency of OATP1B1 and OATP1B3 (disturbing hepatic bilirubin uptake and storage).
These disorders have been described worldwide in all ethnic backgrounds, even though DJS is particularly prevalent (1:1300) among Iranian Jews. 11
Patients usually present in the second decade of life, however males tend to manifest earlier. 12
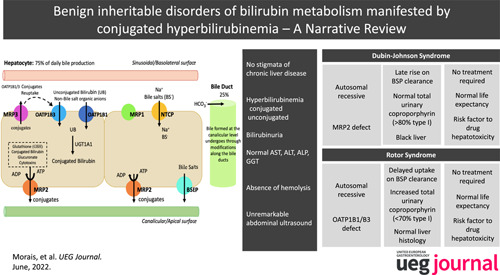
Rotor Syndrome is markedly less frequent than DJS, with an estimated prevalence under 1:1000000 12 , which may be explained by the need for the simultaneous defect of 2 proteins (OATPB1 and OAT1B3) (Table 1).
TABLE 1.
Comparison between Dubin‐Johnson Syndrome (DJS) and Rotor Syndrome (RS)
| Dubin‐Johnson Syndrome (DJS) | Rotor Syndrome (RS) | |
|---|---|---|
| Molecular mechanism | ABCC2 transporter affected; biliary transport deficiency of nonbile acid organic anions | OATP1B1 and OATP1B3 transporters affected; hepatic storage deficiency of conjugated bilirubin |
| Expected plasma concentration of conjugated bilirubin | 2.0–5.0 mg/dl, (possible up to 20–25 mg/dl) | 2.0–5.0 mg/dl (possible up to 20–25 mg/dl) |
| Histology | Gross pathology: Black liver; histology: Lysosomal pigment | Normal |
| Urine | Normal total urine coproporphyrin, but higher excretion of isomer I (>80%) | Increased excretion of total coproporphyrin |
| Aminotransferases | Normal | Normal |
| Hepatobiliary transport | Biphasic bromosulphthaleine clearance: Normal at 45 min; secondary peak at 90 min | Increased retention of bromosulphthaleine at 45 min; no 90 min peak |
| Pruritus | Absent | Absent |
| Prognosis | Benign | Benign |
| Precipitating factors | Pregnancy, intercurrent illness, drugs | Intercurrent illness, drugs |
CLINICAL CHARACTERISTICS
Laboratory findings
In DJS and RS, serum bilirubin concentration is typically between 2 and 5 mg/dl, although values as high as 20–25 mg/dl have been reported. 12 Serum bilirubin levels often fluctuate, may be normal occasionally and increase during intercurrent illness. More than half of serum bilirubin is direct‐reacting, and bilirubinuria is typically found. Other liver tests are normal and there is no evidence of hemolysis. 11 Bile acids are normal in the majority of DJS patients and increased in RS patients. 13
Bromosulphthaleine metabolism
Bromosulphthaleine (BSP) metabolism is characteristically abnormal, but different in DJS and RS.
In DJS, following intravenous administration of BSP, initial plasma disappearance of the dye is normal, with either normal or mildly increased retention at 45 min. 14 However, there is a second (biphasic) peak with an increase in BSP concentration at 90 min, indicating altered transport, which results from the regurgitation of conjugated BSP from the hepatocyte into plasma, caused by a selective defect in biliary excretion of conjugate but not of free BSP. 14
In RS, following the administration of BSP, the initial plasma disappearance rate is markedly reduced with an elevated 45‐min retention. 15 There is no secondary rise in plasma BSP. Furthermore, during constant infusion studies, the maximum transport for BSP is only minimally decreased, while the relative storage capacity is greatly reduced, 15 suggesting a hepatic uptake and storage defect.
This test is no longer used in clinical practice due to its low specificity and potential for severe side effects, such as occasional anaphylaxis‐like shock. 16
Urinary coproporphyrin excretion
The pattern of urinary coproporphyrin excretion helps differentiating DJS and RS.
Approximately 75% of coproporphyrins excreted daily are eliminated by bile and 25% through urine. 12 Type I isomer is preferentially excreted in bile (comprising 65% of biliary coproporphyrins) and type III preferentially in urine (comprising 75% of urinary coproporphyrins). 17
During cholestasis, total urinary coproporphyrin levels are increased, by diversion into urine of substances normally excreted into bile, leading to an increase of isomer I proportion, albeit being almost always under 65%. 17
In DJS, total urinary coproporphyrin excretion is either normal or modestly increased, 18 but the proportion of urinary type I coproporphyrin is more than 80%. 19
In RS, the total urinary coproporphyrins are increased 2.5 to 5‐fold, and the ratio of isomer I to isomer III is high, but usually lower than in DJS and similar to other hepatobiliary diseases, typically <65% (though occasionally >80%). 20
Imaging studies
In patients with strong suspicion of DJS/RS, further diagnostic evaluation is usually not indicated, 21 albeit abdominal imaging may help excluding other conditions. Of note, in DJS, the CT scan may show a significantly increased hepatic attenuation.
Histopathology
Liver biopsy in DJS is characteristic for the accumulation of dark, coarsely granular pigment in centrilobular hepatocytes in an otherwise normal liver. 21 On electron microscopy, the pigment has lysosomal location. 22 Its nature is controversial, with some authors considering it a lipofuscin and others a melanin. 22
In RS, the liver is histologically normal, with no increase in pigmentation.
Importantly, suspicion of hereditary jaundice is not an indication for liver biopsy. 12
MOLECULAR BASIS
Dubin‐Johnson Syndrome
Canalicular multispecific organic anion transporter/Multidrug Resistance‐associated Protein 2
Dubin‐Johnson Syndrome is caused by mutations leading to the absence of functional MRP2 protein at the hepatocyte canalicular membrane.
Multidrug Resistance‐associated Protein 2 is an ABC transporter. Around 1100 ABC transporters have been described, 23 being ATP‐binding cassette C subfamily (ABCC) the largest subfamily, with 13 members. 24 The majority of those proteins are active transporters, with a wide array of endobiotics and xenobiotics substrates. In hepatocytes, 4 ABCC proteins are significantly expressed: ABCC3 (aka MRP3), ABCC4 (aka MRP4) and ABCC6 (aka MRP6) at the sinusoidal membrane, and ABCC2 (aka MRP2 or cMOAT) at the canalicular membrane. 25
ABCC2/MRP2 is a multispecific organic anion transporter expressed at enterocytes, proximal tubules, and hepatocytes. 26 ABCC2 plays the most critical role at the canalicular membrane of hepatocytes, where it functions as a biliary transporter. 27 Its substrate specificity comprises many organic anions, with the highest affinity for glucuronate and GSH conjugates of lipophilic substances. As such, MRP2 appears to play a role in eliminating endogenous metabolites (such as conjugated bilirubin), as well as xenobiotics.
Regarding structure, MRP2 has three membrane‐spanning domains (MSD‐0, MSD‐1 and MSD‐2), two nucleotide‐binding folds (NBF‐1 and NBF‐2), and the cytoplasmic loops (L‐0, L‐1 and within the C‐terminal (COOH)) (Figure 2). Multidrug Resistance‐associated Protein 2 shares a similar structure with other MRP proteins, but presents an extra membrane‐spanning domain (MSD‐0) and a specific sequence in the COOH region. Specific sequences within MSD‐0 and L‐0 regions are required for MRP2 activity and plasma membrane trafficking, respectively. 28 Its COOH region contains a putative protein domain (PDZ) binding motif that is involved in hepatocanalicular membrane targeting via its interaction with scaffolding proteins such as radixin. 29
FIGURE 2.
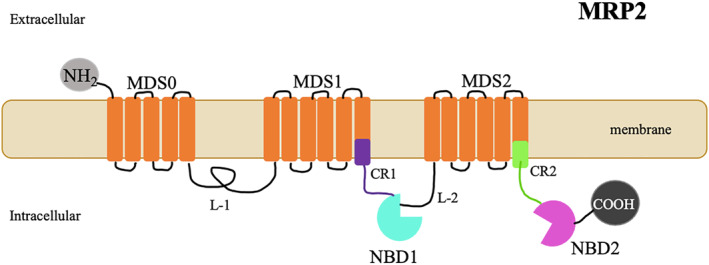
Schematic representation of the Multidrug Resistance‐associated Protein 2 (MRP2) protein. MRP2 is a multispecific organic anion transporter, exclusively located on the apical membrane of polarized cells, especially in the hepatocyte. A bidimensional schematic representation of its proposed structural topology is shown–MRP2 contains an extra membrane spanning domain (MSD0) of 5 transmembrane helices in the NH2 terminal region, along with two other membrane‐spanning domains (MSD‐0) (MSD 1 and MSD2) of six transmembrane helices and two nucleotide binding domains (NBD1 and NBD2). Cytoplasmic loops (L‐1 and L‐2); Connecting Regions (CR1, CR2); N‐terminal (NH2) and C‐terminal (COOH)
Genetic variants in Multidrug Resistance‐associated Protein 2
Human ABCC2 gene localizes in chromosome 10q23‐q24 30 and contains 32 exons. 31
Over 200 sequence variants have been identified in exons, introns, and at the 5′‐ and 3′‐flanking regions of ABCC2 gene. Many of these variants are single‐nucleotide polymorphisms (SNPs) not resulting in amino‐acid changes and without functional consequences. 19 Accordingly, healthy subjects may present many known ABCC2 polymorphisms. 19
DJS‐associated genetic variants resulting in the absence of functional ABCC2 protein at the hepatocyte canalicular membrane include: splice‐site mutations, 32 missense mutations resulting in amino‐acids substitutions, 32 a deletion mutation leading to the loss of two amino‐acids from the second nucleotide‐binding domain, a deletion/in‐frame insertion mutation, 33 and nonsense mutations leading to premature stop‐codons. 34
Premature stop codons may cause rapid degradation of the mutated ABCC2 mRNA by a mechanism termed nonsense mediated decay. 35 Other variants result in deficient maturation, impaired housing of ABCC2 protein to the canalicular membrane, or non‐functional ABCC2 protein. 36 Non‐functional MRP2 protein may result from point mutations and small deletions in NBDs and other cytoplasmic regions. 37 SNPs in the promoter/enhancer and/or in the DNA‐binding proteins may result in different MRP2 expression levels. 36
The regulation of MRP2 function occurs at three levels: (1) transcription, (2) translation, and (3) endocytic retrieval from the canalicular membrane. 38 ABCC2 is constitutively expressed, but its expression is regulated at the transcriptional and posttranscriptional levels in response to many endogenous and xenobiotic substances and to different disease states. Multidrug Resistance‐associated Protein 2 transcription is regulated by intracellular bile acids and nuclear hormone receptors (Nuclear Receptors) such as pregnane X receptor, constitutive androstane receptor, peroxisome proliferator‐activated receptor‐α and retinoic acid receptor‐α.
Posttranscriptional regulation of ABCC2 can occur through the interchange between its intracellular and apical membrane pools. For example, translocation of Abcc2 into the bile canalicular membranes was reduced in mice deficient in radixin (a protein that crosslinks actin filaments to integral membrane proteins), resulting in a DJS‐like phenotype. 29 In rodents, targeting Abcc2 to the canalicular membrane also requires lipid kinase phosphoinositide 3‐kinase, protein kinase C, and cyclic adenosine monophosphate. 39
Lastly, a balance between MRP2 synthesis and degradation rates modulates the canalicular concentration of MRP2. During cholestasis, Abcc2 degradation in periportal hepatocytes increases.
Heterozygous carriers of MRP2/ABCC2 mutations present normal bilirubin concentrations, but their urinary levels of coproporphyrin isomer I may be elevated, 40 suggesting some disturbances in metabolite disposition and elimination. Furthermore, MRP2 loss of function also impacts the pharmacokinetics of a wide range of drugs, toxicants and endobiotics, leading to changes in their absorption, organ distribution and clearance, with an increased potential for drug toxicity and interactions. Indeed, genetic variations in MRP2 have been associated with altered sensitivity to several drugs including anticancer agents, antiepileptic drugs, and antibiotics 41 (Table 2). Nevertheless, and, despite of intense efforts, most attempts to determine a correlation between MRP2 expression and its clinical impact have met little success and therefore the functional consequences of these genetic variants are still largely unknown.
TABLE 2.
Drugs with potential pharmacokinetic/pharmacodynamics modulation by Multidrug Resistance‐associated Protein 2 (MRP2) dysfunction
| Class of drugs | Examples |
|---|---|
| Anticancer agents | Methotrexate, tamoxifen, docetaxel, vinblastine, irinotecan |
| Antiepileptic drugs | Carbamazepine, valproic acid |
| Antibiotics | Ampicillin, ceftriaxone, rifampin |
| NSAIDs | Salicylates, ibuprofen, naproxen |
| HIV drugs | Tenofovir |
Abbreviations: HIV, Human Immunodeficiency Virus; SAIDs, non‐steroidal anti‐inflammatory drugs.
Rotor Syndrome
OATP1B1 and OATP1B3
OATPs are ubiquitous membrane influx Na+‐independent transporters that regulate cellular uptake of several endogenous and xenobiotic compounds. A total of 52 members comprising 12 families have been identified across species. 42 OATPs are encoded by genes of the SLCO‐family (previously SLC21).
OATP1B1 and 1B3 are the major OATPs expressed on the hepatocyte sinusoidal membrane. 43 They share several structural features, including similar 12 transmembrane domain (TMs) with intracellularly located N‐ and C‐termini. 44 A highly conserved amino‐acid signature sequence at the TM interface stabilizes the proteins within the membrane 45 (Figure 3). Substrates include bilirubin and its glucuronide conjugates, bile acids, peptides, eicosanoids, hormones and their conjugates, toxins, and drugs. There is important overlap in substrate specificity between the two OATP1B subfamily members, being the sequence of residues within TMs critical for substrate binding and processing of the transporter proteins. 46
FIGURE 3.
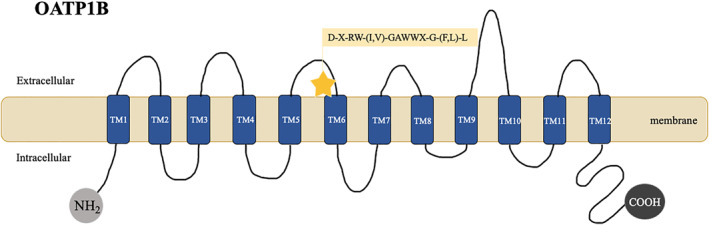
Schematic representation of the OATP1B protein. OATP1B encompass a subfamily of membrane transport proteins. OATP1B1/1B3 are expressed on the basolateral membrane of hepatocytes. A bidimensional schematic representation of its core structure is shown–OATP1B proteins include 12 transmembrane domains (TM1–TM12) and a conserved signature sequence of (D‐X‐RW‐(I,V)‐GAWWX‐G‐(F,L)‐L). N‐terminal (NH2) and C‐terminal (COOH)
Genetic variants in OATP1B1/OATP1B3
The molecular basis of RS is the deficiency of two organic anion transporters OATP1B1 and OATP1B3, 47 encoded respectively by genes SLCO1B1 and SLCO1B3, which lie very close together on chromosome 12 (SLCO1B1 12p12.1 and SLCO1B3 12p.12.2). 48 RS is an obligate two‐gene disorder, 47 and the presence of a single functional allele of either SLCO1B1 or SLCO1B3 prevents the development of conjugated hyperbilirubinemia.
Animal models suggest that MPR3 and OATP1B1/OATP1B3 may form a liver‐blood shuttling loop for bilirubin glucuronide: MRP3 spills out bilirubin glucuronides from hepatocytes to the sinusoidal blood and OATP1B1/3 mediates its reuptake by hepatocytes. 47 In RS, this reuptake is hampered, causing increased plasma bilirubin glucuronide levels and jaundice.
The OATP1B1/3 abnormality also explains other diagnostic traits of RS: decrease plasma clearance of anionic diagnostic dyes such as BSP (also a substrate of these proteins), reduced visualization of the liver by anionic cholescintigraphic radiotracers such as 99mTc‐HIDA, as well as increased urinary coproporphyrin excretion (resulting from the interaction of several porphyrins with OATP1B1). 43
Recently, there has been a particular effort to identify SNPs in SLCO1B1 and SLCO1B3, determine their frequency in different populations, and establish their functional consequences. 48 Two common SNPs are 388A > G (130Asn > Asp, rs2306283) and 521T > C (174Val > Ala, rs4149056). 48 In these genetic variants an alternative pathway becomes responsible for most of the hepatobiliary elimination of organic anions, including anionic drugs. 42 However, and despite moderate phenotypic abnormalities, even reduced‐activity OATP1B1/3 polymorphisms can result in drug toxicities and such risks are likely increased in RS subjects. OATP1B1/3 variations play a role in the pharmacokinetics and pharmacodynamics of several drugs, such as HMG‐CoA reductase inhibitors, antidiabetic agents, cyclosporine, and rifampicin 48 (Table 3). For example, SLCO1B1 variants are a risk factor for HMG‐CoA reductase inhibitors adverse reactions such as myopathy or rhabdomyolysis that depend on their plasma concentrations.
TABLE 3.
OATP1B1 and OATP1B3 substrates associated with Drug‐Drug Interactions
| OATP1B1 | OATP1B3 |
|---|---|
| Cyclosporin A | Cyclosporin A |
| Atorvastatin | Pravastatin |
| Pravastatin | Rifampin |
| Simvastatin | Taurocholate |
| Rifampin | Troglitazone sulfate |
| Gemfibrozil | |
| HIV drugs |
Abbreviation: HIV, Human Immunodeficiency Virus.
NATURAL HISTORY OF DISEASE AND CLINICAL MANAGEMENT
Dubin‐Johnson Syndrome and RS are benign disorders associated with a normal life expectancy, 49 characteristically presenting during the second decade of life. Some factors, such as intercurrent illness, oral contraception, and pregnancy may trigger jaundice. 49 Importantly, these conditions do not have an impact on maternal or fetus prognosis. 49
Phenobarbital has been used, in DJS, as an attempt to reduce hyperbilirubinemia, with highly variable results. 50 Some patients reported amelioration of mild unspecific constitutional and abdominal complains, irrespective of hyperbilirubinemia improvement. Importantly, chronic phenobarbital administration is not recommended. 50
Dubin‐Johnson Syndrome or RS do not induce liver toxicity neither progression to fibrosis/cirrhosis, 12 and therefore do not need specific treatment. 15 Patients should be reassured regarding its benign nature.
Of note, these conditions may be a risk factor to drug hepatotoxicity. Indeed, drug elimination and drug‐drug interaction is modulated by the interplay of hepatocyte transporters that act coordinately and influence mutually their activity. Patients with DJS and RS present an altered hepatobiliary metabolism that predispose them to drug toxicity. As such, physicians should careful monitor these patients when prescribing anticancer, antiepileptic, antibiotics and statis, as enumerated in Tables 2 and 3. 24 , 41 Beyond inducing hyperbilirubinemia, genetic variations of MRP2 and OATP1B1/3 may be contributing factors towards inter‐individual variability in drug disposition and response and are important issues that require consideration for the discovery and development of drugs with safer profiles.
CONCLUSION
Hereditary causes of hyperbilirubinemia encompass a broad array of clinical conditions, from benign to potentially life‐threatening liver damage. Among benign forms, DJS and RS course with predominantly conjugated hyperbilirubinemia. They are rare entities with an unspecific clinical presentation but a distinguishable pattern of urinary coproporphyrin excretion.
The most important issue in the management of these patients is an accurate differential diagnosis with other hepatobiliary disorders associated with hepatic injury, as no treatment is required. Importantly, these conditions modulate the pharmacokinetic behavior of many substrate drugs, as well as drug‐drug interactions, which may confer an increased susceptibility for drug toxicity.
AUTHOR CONTRIBUTIONS
Mariana B. Morais wrote the manuscript. Mariana Verdelho Machado revised the manuscript.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
ETHICS STATEMENT
Ethical approval is not required for this study in accordance with local and/or national guidelines.
ACKNOWLEDGMENT
The author declares that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Morais MB, Machado MV. Benign inheritable disorders of bilirubin metabolism manifested by conjugated hyperbilirubinemia–A narrative review. United European Gastroenterol J. 2022;10(7):747–55. 10.1002/ueg2.12279
DATA AVAILABILITY STATEMENT
All data analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.
REFERENCES
- 1. Taylor A, Stapley S, Hamilton W. Jaundice in primary care: a cohort study of adults aged >45 years using electronic medical records. Fam Pract. 2012;29(4):416–20. 10.1093/fampra/cmr118 [DOI] [PubMed] [Google Scholar]
- 2. Ferenci P, Zollner G, Trauner M. Hepatic transport systems. J Gastroenterol Hepatol. 2002;17(Suppl 1):S105–12. 10.1046/j.1440-1746.17.s1.15.x [DOI] [PubMed] [Google Scholar]
- 3. Baiocchi L, Lesage G, Glaser S, Alpini G. Regulation of cholangiocyte bile secretion. J Hepatol. 1999;31(1):179–91. 10.1016/S0168-8278(99)80180-9 [DOI] [PubMed] [Google Scholar]
- 4. Müller M, Jansen PLM. Molecular aspects of hepatobiliary transport. Am J Physiol Gastrointest Liver Physiol. 1997;272(6 35‐6):G1285–303. 10.1152/ajpgi.1997.272.6.g1285 [DOI] [PubMed] [Google Scholar]
- 5. Hagenbuch B, Meier PJ. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest. 1994;93(3):1326–31. 10.1172/JCI117091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Keppler D, Arias IM. Introduction: transport across the hepatocyte canalicular membrane. Faseb J. 1997;11(1):15–18. 10.1096/fasebj.11.1.9034161 [DOI] [PubMed] [Google Scholar]
- 7. Büchler M, König J, Brom M, Kartenbeck J, Spring H, Horie T, et al. cDNA cloning of the hepatocyte canalicular isoform of the multidrug resistance protein, cMrp, reveals a novel conjugate export pump deficient in hyperbilirubinemic mutant rats. J Biol Chem. 1996;271(25):15091–8. 10.1074/jbc.271.25.15091 [DOI] [PubMed] [Google Scholar]
- 8. Oude Elferink RPJ, Jansen PLM. The role of the canalicular multispecific organic anion transporter in the disposal of endo‐and xenobiotics. Pharmacol Ther. 1994;64(1):77–97. 10.1016/0163-7258(94)90034-5 [DOI] [PubMed] [Google Scholar]
- 9. Kool M, Van Der Linden M, De Haas M, Scheffer GL, de Vree JML, Smith AJ, et al. MRP3, an organic anion transporter able to transport anti‐cancer drugs. Proc Natl Acad Sci U S A. 1999;96(12):6914–19. 10.1073/pnas.96.12.6914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, et al. The sister of P‐glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273(16):10046–50. 10.1074/jbc.273.16.10046 [DOI] [PubMed] [Google Scholar]
- 11. Shani M, Seligsohn U, Gilon E, Sheba C, Adam A. Dubin‐Johnson syndrome in Israel. I. Clinical, laboratory, and genetic aspects of 101 cases. Q J Med. 1970;39(156):549–67. [PubMed] [Google Scholar]
- 12. Syndrome D, Features C. The familial conjugated hyperbilirubinemias. Semin Liver Dis. 1994;14(4):386–94. 10.1055/s-2007-1007329 [DOI] [PubMed] [Google Scholar]
- 13. Kimura A, Kagawa T, Takei H, Maruo Y, Sakugawa H, Sasaki T, et al. Rotor syndrome: glucuronidated bile acidemia from defective reuptake by hepatocytes. Hepatol Commun. 2021;5(4):629–33. 10.1002/hep4.1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Paulusma CC, Oude Elferink RPJ. The canalicular multispecific organic anion transporter and conjugated hyperbilirubinemia in rat and man. J Mol Med. 1997;75(6):420–8. 10.1007/s001090050127 [DOI] [PubMed] [Google Scholar]
- 15. Zimniak P. Dubin‐Johnson and Rotor syndromes: molecular basis and pathogenesis. Semin Liver Dis. 1993;13(3):248–60. 10.1055/s-2007-1007353 [DOI] [PubMed] [Google Scholar]
- 16. Elder GH. Enzymatic defects in porphyria: an overview. Semin Liver Dis. 1982;2(2):87–99. 10.1055/s-2008-1040699 [DOI] [PubMed] [Google Scholar]
- 17. Kaplowitz N, Javitt N, Kappas A. Coproporphyrin I and 3 excretion in bile and urine. J Clin Invest. 1972;51(11):2895–9. 10.1172/JCI107113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Frank M, Doss M, De Carvalho DG. Diagnostic and pathogenetic implications of urinary coproporphyrin excretion in the Dublin‐Johnson Syndrome. Hepato‐Gastroenterology. 1990;37(1):147–51. [PubMed] [Google Scholar]
- 19. Ito S, Ieiri I, Tanabe M, Suzuki A, Higuchi S, Otsubo K. Polymorphism of the ABC transporter genes, MDR1, MRP1 and MRP2/cMOAT, in healthy Japanese subjects. Pharmacogenetics. 2001;11(2):175–84. 10.1097/00008571-200103000-00008 [DOI] [PubMed] [Google Scholar]
- 20. Neuvonen M, Tornio A, Hirvensalo P, Backman JT, Niemi M. Performance of plasma coproporphyrin I and III as OATP1B1 biomarkers in humans. Clin Pharmacol Ther. 2021;110(6):1622–32. 10.1002/cpt.2429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sprinz H, Nelson RS. Persistent non‐hemolytic hyperbilirubinemia associated with lipochrome‐like pigment in liver cells: report of four cases. Ann Intern Med. 1954;41(5):952–62. 10.7326/0003-4819-41-5-952 [DOI] [PubMed] [Google Scholar]
- 22. Muscatello U, Mussini I, Agnolucci MT. The Dubin‐Johnson syndrome: an electron microscopic study of the liver cell. Acta Hepato‐Splenol. 1967;14(3):162–70. [PubMed] [Google Scholar]
- 23. Gottesman MM, Ambudkar SV. Overview: ABC transporters and human disease. J Bioenerg Biomembr. 2001;33(6):453–8. 10.1023/A:1012866803188 [DOI] [PubMed] [Google Scholar]
- 24. Kruh GD, Belinsky MG. The MRP family of drug efflux pumps. Oncogene. 2003;22(47 REV. ISS. 6):7537–52. 10.1038/sj.onc.1206953 [DOI] [PubMed] [Google Scholar]
- 25. Keppler D. Uptake and efflux transporters for conjugates in human hepatocytes. Methods Enzymol. 2005;400(05):531–42. 10.1016/S0076-6879(05)00029-7 [DOI] [PubMed] [Google Scholar]
- 26. Jemnitz K, Heredi‐Szabo K, Janossy J, Ioja E, Vereczkey L, Krajcsi P. ABCC2/Abcc2: a multispecific transporter with dominant excretory functions. Drug Metab Rev. 2010;42(3):402–36. 10.3109/03602530903491741 [DOI] [PubMed] [Google Scholar]
- 27. Paulusma CC, Van Geer MA, Evers R, Heijn M, Ottenhoff R, Borst P, et al. Canalicular multispecific organic anion transporter/multidrug resistance protein 2 mediates low‐affinity transport of reduced glutathione. Biochem J. 1999;338(2):393–401. 10.1042/0264-6021:3380393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bandler PE, Westlake CJ, Grant CE, Cole SPC, Deeley RG. Identification of regions required for apical membrane localization of human multidrug resistance protein 2. Mol Pharmacol. 2008;74(1):9–19. 10.1124/mol.108.045674 [DOI] [PubMed] [Google Scholar]
- 29. Wang W, Soroka CJ, Mennone A, Rahner C, Harry K, Pypaert M, et al. Radixin is required to maintain apical canalicular membrane structure and function in rat hepatocytes. Gastroenterology. 2006;131(3):878–84. 10.1053/j.gastro.2006.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cancer CH, Lines C, Kagotani K, Okumura K, Kawakami M. A human canalicular multispecific organic anion transporter (cMOAT). Gene. 1996:4124–9. [PubMed] [Google Scholar]
- 31. Tsujii H, Konig J, Rost D, Stockel B, Leuschner U, Keppler D. Exon‐intron organization of the human multidrug‐resistance protein 2 (MRP2) gene mutated in Dubin‐Johnson syndrome. Gastroenterology. 1999;117(3):653–60. 10.1016/S0016-5085(99)70459-2 [DOI] [PubMed] [Google Scholar]
- 32. Kajihara S, Hisatomi A, Mizuta T, Hara T, Ozaki I, Wada I, et al. A splice mutation in the human canalicular multispecific organic anion transporter gene causes Dubin‐Johnson syndrome. Biochem Biophys Res Commun. 1998;253(2):454–7. 10.1006/bbrc.1998.9780 [DOI] [PubMed] [Google Scholar]
- 33. Shoda J, Suzuki H, Suzuki H, Sugiyama Y, Hirouchi M, Utsunomiya H, et al. Novel mutations identified in the human multidrug resistance‐associated protein 2 (MRP2/ABCC2) gene in a Japanese patient with Dubin‐Johnson syndrome. Hepatol Res. 2003;27(4):323–6. 10.1016/s1386-6346(03)00267-5 [DOI] [PubMed] [Google Scholar]
- 34. Ito K, Suzuki H, Hirohashi T, Kume K, Shimizu T, Sugiyama Y. Molecular cloning of canalicular multispecific organic anion transporter defective in EHBR. Am J Physiol Gastrointest Liver Physiol. 1997;272(1 35‐1):G16–22. 10.1152/ajpgi.1997.272.1.g16 [DOI] [PubMed] [Google Scholar]
- 35. Thermann R, Neu‐Yilik G, Deters A, Frede U, Weht K, Hagemeier C, et al. Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J. 1998;17(12):3484–94. 10.1093/emboj/17.12.3484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Suzuki H, Sugiyama Y. Single nucleotide polymorphisms in multidrug resistance associated protein 2 (MRP2/ABCC2): its impact on drug disposition. Adv Drug Deliv Rev. 2002;54(10):1311–31. 10.1016/S0169-409X(02)00075-3 [DOI] [PubMed] [Google Scholar]
- 37. Conseil G, Deeley RG, Cole SPC. Polymorphisms of MRP1 (ABCC1) and related ATP‐dependent drug transporters. Pharmacogenet Genomics. 2005;15(8):523–33. 10.1097/01.fpc.0000167333.38528.ec [DOI] [PubMed] [Google Scholar]
- 38. Jedlitschky G, Hoffmann U, Kroemer HK. Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expet Opin Drug Metabol Toxicol. 2006;2(3):351–66. 10.1517/17425255.2.3.351 [DOI] [PubMed] [Google Scholar]
- 39. Roelofsen H, Soroka CJ, Keppler D, Boyer JL. Cyclic AMP stimulates sorting of the canalicular organic anion transporter (Mrp2/cMoat) to the apical domain in hepatocyte couplets. J Cell Sci. 1998;111(8):1137–45. 10.1242/jcs.111.8.1137 [DOI] [PubMed] [Google Scholar]
- 40. Toh S, Wada M, Uchiumi T, Inokuchi A, Makino Y, Horie Y, et al. Genomic structure of the canalicular multispecific organic anion‐transporter gene (MRP2/cMOAT) and mutations in the ATP‐ binding‐cassette region in Dubin‐Johnson syndrome. Am J Hum Genet. 1999;64(3):739–46. 10.1086/302292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Van Der Schoor LWE, Verkade HJ, Kuipers F, Jonker JW. New insights in the biology of ABC transporters ABCC2 and ABCC3: impact on drug disposition. Expet Opin Drug Metabol Toxicol. 2015;11(2):273–93. 10.1517/17425255.2015.981152 [DOI] [PubMed] [Google Scholar]
- 42. Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158(3):693–705. 10.1111/j.1476-5381.2009.00430.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smith NF, Figg WD, Sparreboom A. Role of the liver‐specific transporters OATP1B1 and OATP1B3 in governing drug elimination. Expet Opin Drug Metabol Toxicol. 2005;1(3):429–45. 10.1517/17425255.1.3.429 [DOI] [PubMed] [Google Scholar]
- 44. Hong M. Critical domains within the sequence of human organic anion transporting polypeptides. Curr Drug Metabol. 2014;15(3):265–70. 10.2174/1389200214666131229111118 [DOI] [PubMed] [Google Scholar]
- 45. Taylor‐Wells J, Meredith D. The signature sequence region of the human drug transporter organic anion transporting polypeptide 1B1 is important for protein surface expression. J Drug Deliv. 2014;2014:1–10. 10.1155/2014/129849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. König J, Cui Y, Nies AT, Keppler D. A novel human organic anion transporting polypeptide localized to the basolateral hepatocyte membrane. Am J Physiol Gastrointest Liver Physiol. 2000;278(1 41‐1):156–64. 10.1152/ajpgi.2000.278.1.g156 [DOI] [PubMed] [Google Scholar]
- 47. Erlinger S, Arias IM, Dhumeaux D. Inherited disorders of bilirubin transport and conjugation: new insights into molecular mechanisms and consequences. Gastroenterology. 2014;146(7):1625–38. 10.1053/j.gastro.2014.03.047 [DOI] [PubMed] [Google Scholar]
- 48. Ieiri I, Higuchi S, Sugiyama Y. Genetic polymorphisms of uptake (OATP1B1, 1B3) and efflux (MRP2, BCRP) transporters: implications for inter‐individual differences in the pharmacokinetics and pharmacodynamics of statins and other clinically relevant drugs. Expet Opin Drug Metabol Toxicol. 2009;5(7):703–29. 10.1517/17425250902976854 [DOI] [PubMed] [Google Scholar]
- 49. Memon N, Weinberger BI, Hegyi T, Aleksunes LM. Inherited disorders of bilirubin clearance. Pediatr Res. 2016;79(3):378–86. 10.1038/pr.2015.247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Merdier C, Burke M, Shani M, Felner S, Lurie I. The effect of phénobarbital on patients with Dubin‐Johnson syndrome. Digestion. 1976;399(5‐6):394–9. 10.1159/000197962 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data analyzed during this study are included in this article. Further enquiries can be directed to the corresponding author.