

Abstract
The abundance of domesticated sheep varieties and phenotypes is largely the result of long-term natural and artificial selection. However, there is limited information regarding the genetic mechanisms underlying phenotypic variation induced by the domestication and improvement of sheep. In this study, to explore genomic diversity and selective regions at the genome level, we sequenced the genomes of 100 sheep across 10 breeds and combined these results with publicly available genomic data from 225 individuals, including improved breeds, Chinese indigenous breeds, African indigenous breeds, and their Asian mouflon ancestor. Based on population structure, the domesticated sheep formed a monophyletic group, while the Chinese indigenous sheep showed a clear geographical distribution trend. Comparative genomic analysis of domestication identified several selective signatures, including IFI44 and IFI44L genes and PANK2 and RNF24 genes, associated with immune response and visual function. Population genomic analysis of improvement demonstrated that candidate genes of selected regions were mainly associated with pigmentation, energy metabolism, and growth development. Furthermore, the IFI44 and IFI44L genes showed a common selection signature in the genomes of 30 domesticated sheep breeds. The IFI44 c. 54413058 C>G mutation was selected for genotyping and population genetic validation. Results showed that the IFI44 polymorphism was significantly associated with partial immune traits. Our findings identified the population genetic basis of domesticated sheep at the whole-genome level, providing theoretical insights into the molecular mechanism underlying breed characteristics and phenotypic changes during sheep domestication and improvement.
Keywords: Sheep, Whole-genome resequencing, Selection signature analysis, Immunity, IFI44 gene
INTRODUCTION
As one of the first domesticated herbivores (Chessa et al., 2009), sheep (Ovis aries) remain an essential source of meat, wool, and milk for humans (Jiang et al., 2014). Sheep were originally domesticated 8 000–11 000 years ago from wild sheep species found in the Fertile Crescent (Diamond, 2002; Zeder, 2008). After domestication, sheep distribution expanded to meet the needs of different human populations, thereby adapting to distinct climatic environments and forming varieties with diverse phenotypes (Scher, 2000). Thus, understanding the genetic differences and diversity of species and varieties is a focus of animal breeding research.
Publication of the sheep reference genome provides an opportunity to determine the genetic mechanisms involved in artificial and natural selection and the phenotypic differentiation of domesticated sheep from their wild ancestors. For example, Zhao et al. (2017) used population single-nucleotide polymorphisms (SNPs) to identify candidate genes associated with high fertility, coat color, tail type, and horn size and type in sheep. Based on pooled whole-genome resequencing data, Wang et al. (2019) identified several vision-associated genes with functional loci in Chinese indigenous sheep breeds. In addition, studies have identified candidate genes related to pigmentation, nervous system, sensory perception, litter size, tail fat deposition, immunity, wool fineness, and climatic adaptation using genome-wide analysis of selection signatures (Alberto et al., 2018; Cao et al., 2021; Chen et al., 2021; Hu et al., 2019; Li et al., 2020; Lv et al., 2022; Yang et al., 2016). Although previous research has focused on sheep domestication, few studies have explored the function and population genetic effects of candidate genes associated with phenotypic variation during domestication. Furthermore, only a handful of candidate loci have been identified in sheep compared to those associated with the domestication of dogs (Axelsson et al., 2013), pigs (Li et al., 2017), chicken (Wang et al., 2020), and cattle (Chen et al., 2018).
Here, we generated whole-genome sequencing data for 100 samples across 10 domesticated sheep breeds, combined with publicly available whole-genome resequencing data from 208 individuals representing 20 domesticated sheep breeds and 17 wild sheep (Asiatic mouflon, O. orientalis), to characterize population genetic structure and genomic diversity at the genome-wide level and to elucidate the genome-wide genetic mechanisms involved in the wild-to-domesticated process. Population genetic effects of important candidate genes were validated using phenotypic data of a Hu sheep population. Overall, we aimed to identify important genomic regions and molecular markers selected in sheep during domestication and improvement.
MATERIALS AND METHODS
Ethics statement
All applicable international, national, and/or institutional guidelines for the care and use of animals were strictly followed. All animal collection protocols complied with the current laws of China. The collection of blood samples and experimental protocols were approved by the Animal Care Committee of Lanzhou University (Permit No. 2010-1), in compliance with the recommendations of the Regulations for the Administration of Affairs Concerning Experimental Animals of China.
Sample collection and sequencing
Using the jugular vein method, we collected 100 blood samples from 10 different sheep breeds, including East Friesian milk sheep (EF), Dorper sheep (DP), Texel sheep (TK), South African mutton merino sheep (NM), Black Suffolk sheep (BS), Australian white sheep (AW), Mongolian sheep (MG), Lanzhou large-tailed sheep (LL), Altay sheep (AL), and large-tailed Han sheep (LH) (Supplementary Table S1). For each breed, 10 blood samples were obtained from five healthy females and five healthy males. Genomic DNA was extracted from whole blood of each individual using an EasyPure Blood Genomic DNA Kit (TransGen Biotech, China) according to the manufacturer’s recommended protocols. The A260/280 ratio and agarose gel electrophoresis were used to assess DNA quality and integrity, respectively. Paired-end sequencing libraries for each individual were constructed (mean insert size of 500 bp) using the Illumina NovaSeq 6000 platform (Illumina, USA). In addition, the genomic data of 225 individuals were downloaded from the NCBI database, including 34 African indigenous sheep, 61 improved sheep breeds, 113 Chinese indigenous sheep, and 17 Asiatic mouflons representing their wild ancestor (Supplementary Table S2).
Sequencing data processing
The raw sequencing data downloaded from the NCBI Sequence Read Archive (SRA) were converted to fastq files using SRAToolkit (v2.9.2) (Kodama et al., 2012). All fastq files were then filtered using Trimmomatic (v0.36) to obtain clean reads. The following filter criteria were used to remove adapters and low-quality bases: reads with >10% unknown nucleotides (N), reads with >50% low-quality (Q-value<5) bases, and reads with >10 nucleotides aligned to the adaptor sequence with up to two mismatches. The resulting clean reads were mapped to the sheep Oar_v4.0 reference genome using BWA (Burrows-Wheeler Aligner) software (v0.7.8) with default parameters (Li & Durbin, 2009). Mapping files were converted to BAM files and sorted using SAMtools (v1.12) (Li et al., 2009). Picard software (v2.26.2) was used to mark potential polymerase chain reaction (PCR) duplicates for subsequent variant calling. SNP calling was performed using the Bayesian method in the GATK package (v3.4.0). VCFtools (v0.1.14) was used to correct the GATK results, with high-quality SNPs retained for further analysis based on the following criteria: (1) mean coverage depth ≥5, (2) missing rates <10% and minor allele frequencies (MAF) ≥5%, and (3) root mean squared (RMS) mapping quality ≥20. After filtering, all SNPs were functionally annotated based on the Oar_v.4.0 sheep reference genome using ANNOVAR (v2013-05-20) (Wang et al., 2010). From the genome annotation, the SNPs were classified as variations in intronic regions, upstream and downstream regions, splicing sites, and exonic regions (synonymous or non-synonymous SNPs), while mutations causing stop gain and stop loss were grouped as non-synonymous SNPs.
Population genetic structure analysis
To clarify the genetic relationships of domesticated sheep from a genome-wide perspective, a neighbor-joining (NJ) tree was constructed for the 325 sheep based on the matrix of pairwise genetic distances from the autosomal SNP data using TreeBeST v.1.9.2 (Vilella et al., 2009) and visualized using FigTree v.1.4.3. Population genetic structure was inferred using ADMIXTURE (v1.3.0) with default parameters (Alexander et al., 2009); the number of predefined ancestral clusters ranged from k=2 to k=15. Principal component analysis (PCA) was performed for the 325 individuals using GCTA software (v1.26.0), with the first and second principal components displayed using the R software (v3.5.2) (Yang et al., 2011).
Analysis of genome-wide selective sweep regions
We analyzed genome-wide selective sweeps during domestication and improvement based on fixation index (FST) analysis using VCFtools. The FST values were calculated using the sliding window approach, with 150 kb windows and 75 kb sliding steps according to previous study (Wang et al., 2019). For domestication analysis, we combined the 308 domesticated sheep (African indigenous breeds, improved breeds, and Chinese indigenous breeds) into a group and compared them with Asiatic mouflons (representing their wild ancestor). The selective sweeps of 30 domesticated sheep breeds were also detected by comparison with wild sheep. To establish the domestication and improvement process (i.e., wild sheep to indigenous breeds to improved breeds), we tested genomic selection signals for wild, indigenous, and improved sheep breeds. The formula used to calculate FST followed previous study (Wang et al., 2019). Windows with the top 5‰ of FST values were defined as the selective regions, and genes in the selective regions were identified as candidate genes.
Functional enrichment analysis of candidate genes
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment pathways and Human Phenotype Ontology (HPO) terms were analyzed to explore the most relevant functions of the protein-coding genes of selective regions using KOBAS-i (Bu et al., 2021) and g:Profiler, respectively. P<0.05 was used as the threshold for significantly enriched pathways and functions.
Genotyping of candidate SNP loci
To verify that the candidate genes selected from the 30 domesticated sheep breeds during the domestication process were associated with immune response, blood DNA from 904 individuals was extracted from our previously established Hu sheep population with accurate immune trait data records (Zhang et al., 2022). The candidate gene loci were genotyped using competitive allele-specific fluorescence resonance energy transfer (FRET)-based PCR (KBioscience competitive allele-specific PCR amplification of target sequences and endpoint fluorescence genotyping (KASPar™)) assays (LGC Genomics, UK) according to a previously published method (Zhang et al., 2021b). The primer pairs for SNPs designed for KASPar genotyping are listed in Supplementary Table S3.
Statistical analysis
A mixed linear model (MLM) in the lem4 package in R was used to test the SNP effect on hematological parameters. The animal model was defined as:
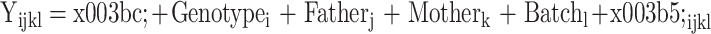 |
1 |
where Yijkl is the observed value of the hematological parameter indices, μ is the population mean, Genotypei is the effect of each genotype at the ith SNP locus, Batchl is the batch effect, Genotypei and Batchl are fixed effects, and Fatherj, Motherk, and εijkl are random residual effects. P<0.05 was considered as the criterion for statistical significance.
RESULTS
High-density sheep genomic variation map
In total, 100 domesticated sheep were collected for whole-genome resequencing, which yielded 16.97 billion clean reads, with a mean depth of 9.57× and an average genome coverage of 96.6% (Supplementary Table S1). The new genomic sequence data were combined with publicly available genomic data of 225 individuals from 21 breeds, for a total of 325 individuals (Figure 1A; Supplementary Table S2). After variant calling and filtration, we identified 39 331 475 high-quality SNPs for subsequent population analyses (Supplementary Table S4).
Figure 1.

Genomic diversity and population genetics of wild and domestic sheep
A: Geographic distribution of 31 breeds sampled. Each breed is indicated as a dot on the map. Population-based proportions of allele frequency changes are displayed using a pie chart. B: Neighbor-joining phylogenetic tree of 325 sheep. C: Proportion of allele frequency changes for each breed. D: Distribution of genetic differentiation (FST) across three groups. E, F: Population structure of 325 individuals using ADMIXTURE with k=2, 4. G–I: Principal component analysis (PCA) of 325 individuals, domesticated group, and Chinese indigenous breeds, respectively. Different subgroups or breeds are indicated by different colors and shapes in each figure; domesticated group includes African indigenous breeds, Chinese indigenous breeds, and improved breeds.
Population genetic structure
To understand the genetic relationships and divergences between all domesticated and wild sheep, we determined the population-based proportions of allele frequency changes for each geographic group of breeds compared with Asiatic mouflons (representing their wild ancestor). When the allele frequency changed by less than 25%, the proportions of African indigenous, Chinese indigenous, and improved sheep breeds were 80.17%, 78.71%, and 76.51%, respectively (Figure 1A). Data from 30 domesticated sheep breeds were also compared with data from wild sheep, which showed consistent results as the different groups mentioned above (Figure 1C; Supplementary Table S5). We also observed higher genetic differentiation (FST) between the Asiatic mouflons and improved breeds than between the Asiatic mouflons and indigenous (African and Chinese) breeds (Figure 1D). These results indicated that the differences between African indigenous breeds and wild sheep were the smallest, followed by Chinese indigenous breeds and improved breeds.
Using Asiatic mouflons as an outgroup, we characterized the genetic relationships among all individuals using the NJ tree, population structure analysis, and PCA. The NJ results suggested that the 325 individuals could be classified into four main groups (Figure 1B), representing wild sheep, African indigenous breeds, improved breeds, and Chinese indigenous breeds, respectively. Population structure analysis yielded similar results (Figure 1E, F). Based on PCA, the Asian mouflons were clustered together, while domesticated sheep were classified into African indigenous breeds, Chinese indigenous breeds, and improved breeds. The Chinese indigenous breeds were further divided into three groups, which showed strong geographical distribution characteristics. These results are consistent with the geographical and morphological classification of Chinese indigenous sheep as Mongolian, Kazakh, and Tibetan sheep (Figure 1G–I).
Selection signals during domestication in sheep
To explore the genome-wide selection signatures influenced by domestication, we combined the 308 domesticated sheep (African indigenous breeds, improved breeds, and Chinese indigenous breeds) as a group and compared them with Asiatic mouflons (representing their wild ancestor). The genome-wide FST value was calculated between the domesticated and wild sheep based on a 150 kb sliding window and 75 kb shift across the genome along the autosomes and sex chromosome. In total, 87 and two putatively selected genomic regions were identified on the autosomes and sex chromosome, respectively, with the top 5‰ of global FST values (spanning 100.915 Mb) accounting for 3.901% of the complete genome and harboring 328 genes (Figure 2A; Supplementary Table S6). We then carried out KEGG pathway and HPO category enrichment analyses of these genes influenced by domestication. KEGG analysis identified 32 significantly enriched pathways, 13 of which were associated with immune response, six of which were related to metabolic processes, and one of which was associated with longevity regulating pathway (Supplementary Table S7). HPO analysis of the candidate genes identified 23 significantly enriched HPO terms, including terms associated with visual function, such as iris coloboma, abnormal iris morphology, and abnormality of the eye (Supplementary Table S8).
Figure 2.

Genome-wide detection and annotations of regions selected during domestication
A: Selection signals in Asiatic mouflons compared with domestic sheep. B: Selection signals in Asiatic mouflons compared with 30 domestic sheep breeds, respectively. C: Fixation index (FST) values from Asiatic mouflons to four groups: i.e., domestic sheep, African indigenous breeds, Chinese indigenous breeds, and improved breeds; shadowed area indicates strong selection signatures. D: Haplotype differentiation patterns of two selected regions. E: Structures of IFI44L and IFI44 (left) and RFN24 and PANK2 (right) genes.
To investigate how many selective regions were shared among breeds, FST values were calculated for the following 30 comparisons: five African indigenous breeds, 15 Chinese indigenous breeds, and 10 improved breeds versus Asian mouflons, respectively (Figure 2B). In selective sweep analysis, windows with the top 5‰ of FST values for the autosomes and sex chromosome were defined as selective windows. Results identified 25 windows that overlapped in more than 25 domestic sheep breeds. Among them, two regions on chromosomes 1 and 13 (Chr1: 54.3-54.525 Mb and Chr13: 50.325-50.55 Mb) were identified from the above 30 pairwise comparisons (Table 1). These two regions contained genes associated with the immune response (IFI44 (encoding interferon induced protein 44 and IFI44L (encoding interferon induced protein 44 like) and visual function (PANK2 (encoding pantothenate kinase 2) and RNF24 (encoding ring finger protein 24)). Comparisons among the genomic regions adjacent to the four genes (IFI44, IFI44L, PANK2, and RNF24) revealed high genetic differentiation (FST) between the Asiatic mouflons and different groups of domestic sheep (Figure 2C). Furthermore, genotype pattern analysis of SNP loci revealed that the genotypes were significantly different between the Asiatic mouflons and domestic sheep (Figure 2D, E), suggesting the occurrence of an intensive selective sweep in the two regions. These results indicated that genes related to immunity and sensory ability were strongly selected during early domestication.
Table 1. Information on overlapping genomic regions on autosomes and X chromosome of more than 25 sheep breeds.
| No. | Combined region (Mb) | Chr | Position | Gene name | Breed No. | |
| Start | End | |||||
| –: Not available. | ||||||
| 1 | Chr1: 54.3–54.525 | 1 | 54300001 | 54450000 | LOC105613526, PTGFR, LOC101103103, LOC101110201, IFI44, IFI44L | 30 |
| 1 | 54375001 | 54525000 | IFI44L, IFI44 | 30 | ||
| 2 | Chr2: 122.625–122.85 | 2 | 122625001 | 122775000 | LOC105608888 | 27 |
| 2 | 122700001 | 122850000 | – | 27 | ||
| 3 | Chr3: 94.425–94.575 | 3 | 94425001 | 94575000 | EXOC6B, LOC106991063, LOC105614905 | 26 |
| 4 | Chr5: 106.95–107.1 | 5 | 106950001 | 107100000 | LOC105615337 | 25 |
| 5 | Chr6: 81.525–81.675 | 6 | 81525001 | 81675000 | – | 25 |
| 6 | Chr6: 115.875–116.025 | 6 | 115875001 | 116025000 | LETM1, LOC106991222, WHSC1 | 27 |
| 7 | Chr9:30.825–30.975 | 9 | 30825001 | 30975000 | LOC101110602, LOC101110341 | 25 |
| 8 | Chr13: 50.325–50.55 | 13 | 50325001 | 50475000 | RNF24, LOC105606258 | 30 |
| 13 | 50400001 | 50550000 | PANK2, RNF24 | 30 | ||
| 9 | Chr14: 34.275–34.575 | 14 | 34275001 | 34425000 | HSD11B2, ATP6V0D1, AGRP, LOC105606370, FAM65A, LOC106991595, CTCF | 26 |
| 14 | 34350001 | 34500000 | FAM65A, LOC106991595, CTCF, RLTPR, ACD, PARD6A, ACTB, C14H16orf86, GFOD2 | 29 | ||
| 14 | 34425001 | 34575000 | CTCF, RLTPR, ACD, PARD6A, ACTB, C14H16orf86, GFOD2, RANBP10, TRNAG-CCC, TSNAXIP1 | 28 | ||
| 10 | Chr15: 0.225–0.375 | 15 | 225001 | 375000 | LOC106990761, LOC105614369, LOC105614142 | 25 |
| 11 | Chr24: 34.575–34.725 | 24 | 34575001 | 34725000 | CUX1, LOC105604781, TRNAE-UUC, TRNAG-CCC, LOC105604780, LOC106991890 | 26 |
| 12 | ChrX: 24.6–24.825 | X | 24600001 | 24750000 | – | 26 |
| X | 24675001 | 24825000 | – | 25 | ||
| 13 | ChrX: 76.125–76.5 | X | 76125001 | 76275000 | VAMP7 | 29 |
| X | 76200001 | 76350000 | LOC106990663, LOC101106825 | 29 | ||
| X | 76275001 | 76425000 | LOC101106825 | 28 | ||
| X | 76350001 | 76500000 | LOC101116810 | 28 | ||
| 14 | ChrX: 94.125–94.275 | X | 94125001 | 94275000 | LOC101105554, LOC101117996 | 25 |
| 15 | ChrX:121.5–121.725 | X | 121500001 | 121650000 | CXHXorf57 | 26 |
| X | 121575001 | 121725000 | LOC101116890, LOC101117144, LOC105601901, NGFRAP1, WBP5 | 26 | ||
Selective imprints during domestication and breeding
To further determine the significance of genomic divergence in sheep domestication and breeding history, we analyzed and compared the genomes of wild and indigenous groups, as well as indigenous and improved groups, to identify potential selection imprints that occurred during this process (Figure 3A). The global FST values for each comparison were calculated using 150 kb sliding windows with 75 kb steps across the genome. The top 5‰ of windows were defined as selective windows. After the adjacent selective windows were merged, a total of 90 domestication regions and 72 improvement regions were identified, comprising 327 and 326 candidate genes, respectively (Figure 3B, F; Supplementary Tables S9, S10). The FSIP2 (encoding fibrous sheath interacting protein 2) gene, which is associated with reproduction traits on chromosome 2, was identified during domestication. The genotype pattern of FSIP2 showed significant differences between the Asiatic mouflons and indigenous sheep, and linkage disequilibrium analysis indicated strong linkage of SNPs in this region (Figure 3C–E). To explore the strongly selected regions of chromosomes 10 and 19 during the breeding process, we analyzed genomic architecture by calculating allele frequency at non-synonymous SNPs in two genes (FGF9 (encoding fibroblast growth factor 9) and MITF (encoding melanocyte inducing transcription factor)) (Figure 3G–I). Two variant alleles (c. 35570188 A>G, c. 35570233 T>C) located in the exon region of FGF9 showed higher frequencies in the improved breeds, but lower frequencies in the indigenous breeds. Similarly, one variant allele (c. 31605530 C>T) located in the second exon of MITF differed between white sheep and non-white sheep. Annotation of candidate genes for domestication indicated that they were mainly related to immune processes (RAP1 signaling pathway, lysosome, and NOD-like receptor signaling pathway) and the thyroid hormone signaling pathway (Figure 4A; Supplementary Table S11). Furthermore, genes selected during breeding and improvement were mainly association with melanoma, pathways in cancer, notch signaling pathway, and starch and sucrose metabolism. The most significantly enriched pathway was melanoma (Figure 4B; Supplementary Table S12). Notably, seven candidate genomic regions overlapped in both domestication and improvement (Supplementary Table S13), suggesting that several important domestication loci may have undergone a second round of artificial selection for continued improvement of vital economic traits. Furthermore, candidate genes of the overlapping genomic regions were significantly enriched in cholinergic synapse, glycosylphosphatidylinositol (GPI)-anchor biosynthesis, and phototransduction pathways (Figure 4C; Supplementary Table S14).
Figure 3.

Genome-wide distribution of selective sweeps during domestication and breeding
A: Schematics of sheep evolution during domestication and breeding, values between pairs indicate population differentiation. B, F: Selection signals in Asiatic mouflons compared with indigenous breeds and indigenous breeds compared with improved breeds. C: Plot of FST windows of chr2 region (119.9–129.8 Mb) based on Asiatic mouflons compared with indigenous breeds. D: Haplotype diversity of a local region of FSIP2 (chromosome 2: 122325000-122475000). E: Linkage disequilibrium analysis of SNPs in FSIP2 region. G: Plot of FST windows of two regions: chr10 (30.15–40.05 Mb) and chr19 (30.15–36.15 Mb) based on indigenous breeds compared with improved breeds. H: Structure of FGF9 and MITF genes. I: Allele frequencies of FGF9 and MITF genes.
Figure 4.

Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of candidate genes during domestication and breeding
A: Top 20 enriched signaling pathways of candidate genes during domestication. B: Top 20 enriched signaling pathways of candidate genes during breeding. C: Top 20 enriched signaling pathways of candidate genes from overlapping regions during domestication and breeding.
SNP genotyping and association analysis
To further explore the effects of key genes in significant candidate regions on immunity traits, a novel mutation (c. 54413058 C>G) in the IFI44 gene was genotyped using KASPar technology (Supplementary Figure S1, blue, green, and red dots indicate three different genotypes, respectively). We performed association analysis of hematological parameter indices based on our previously established Hu sheep population with accurate phenotypic data records (Zhang et al., 2022). Results indicated that the IFI44 c. 54413058 C>G polymorphism was significantly associated with white blood cells (WBCs), neutrophils, and monocytes (P<0.05). Furthermore, the WBC phenotype value in Hu lambs with the CC and GC genotypes was significantly higher than in those with the GG genotype, while the difference between CC and GC lambs was not significant (Table 2). These results indicated that CC was the dominant genotype associated with WBC count.
Table 2. Association results between genotypes of ovine IFI44 gene and hematological parameters.
| Item | IFI44 c. 54413058 C>G | P-value | ||
| CC | GC | GG | ||
| Phenotype values are indicated as mean±standard error (SE). Letters with different genotypes in same trait (a, b) indicate significant difference at P<0.05. RBC: Red blood cell count; HGB: Hemoglobin concentration; HCT: Hematocrit; MCV: Mean erythrocyte volume; MCH: Mean erythrocyte hemoglobin content; MCHC: Mean erythrocyte hemoglobin concentration; RDW_SD: Red blood cell distribution-standard deviation; RDW_CV: Red blood cell distribution-coefficient of variation; PLT: Platelet count; MPV: Mean platelet volume; WBC: White blood cell count; NEUT: Neutrophils; LYMPH: Lymphocytes; MONO: Monocytes; EO: Eosinophils; BASO: Basophils. | ||||
| No. | 673 | 181 | 31 | |
| RBC (M/μL) | 14.169±0.053 | 14.281±0.102 | 14.452±0.247 | 0.676 |
| HGB (g/dL) | 13.186±0.041 | 13.286±0.079 | 13.313±0.191 | 0.256 |
| HCT (%) | 30.865±0.138 | 30.774±0.266 | 28.235±0.643 | 0.149 |
| MCV (fL) | 21.923±0.112 | 21.702±0.216 | 19.826±0.521 | 0.86 |
| MCH (pg) | 9.34±0.025 | 9.334±0.049 | 9.061±0.117 | 0.053 |
| MCHC (g/dL) | 43.126±0.187 | 43.597±0.36 | 46.358±0.871 | 0.060 |
| RDW-SD (fL) | 26.718±0.124 | 27.078±0.24 | 26.948±0.579 | 0.397 |
| RDW-CV (%) | 42.434±0.098 | 42.787±0.189 | 42.677±0.456 | 0.256 |
| PLT (K/μL) | 556.318±4.621 | 551.276±8.911 | 533.806±21.533 | 0.905 |
| MPV (fL) | 8.337±0.023 | 8.316±0.044 | 8.161±0.106 | 0.541 |
| WBC (K/μL) | 12.076±0.106a | 12.517±0.205a | 11.138±0.496b | 0.043 |
| NEUT (K/μL) | 4.615±0.075ab | 5.021±0.145a | 4.092±0.35b | 0.017 |
| LYMPH (K/μL) | 5.373±0.054 | 5.519±0.104 | 5.286±0.251 | 0.422 |
| MONO (K/μL) | 1.894±0.029a | 1.748±0.056ab | 1.563±0.136b | 0.032 |
| EO (K/μL) | 0.134±0.009 | 0.17±0.017 | 0.138±0.041 | 0.608 |
| BASO (K/μL) | 0.06±0.002 | 0.059±0.004 | 0.058±0.009 | 0.840 |
DISCUSSION
Understanding the molecular basis underlying genetic variation and phenotypic changes during animal domestication and subsequent selection could contribute to animal breeding and help determine how phenotypes are influenced by genotypic changes. Whole-genome sequencing has been widely applied to reveal the genomic variants under selection in domestic animals (e.g., horse, pig, cattle, and sheep) (Chen et al., 2021; Daetwyler et al., 2014; Liu et al., 2019b; Xu et al., 2020). Here, we collected whole-genome data from 325 sheep for comprehensive population genetic and selective signaling analysis at the genome level.
Elucidating population structure of sheep at genome level
Analysis of population genetic structure can enhance our understanding of the domestication process of certain species or breeds. Genome-level analysis of allele frequency changes showed that differences were the smallest between African indigenous breeds and wild sheep, followed by differences between Chinese indigenous breeds and improved breeds (Figure 1A, C). Based on population structure analysis, the 325 individuals could be grouped into two clusters, i.e., domestic sheep and wild sheep, suggesting that all domestic sheep originated from a single domestication event (Figure 1G). Domestic sheep were further clustered into three groups, i.e., African indigenous, Chinese indigenous, and improved sheep breeds (Figure 1H). The Chinese indigenous breeds were also grouped into three clusters, i.e., Mongolian, Kazakh, and Tibetan sheep, which showed strong geographical distribution and characteristic morphological patterns. These results are in accordance with previous study using the Illumina Ovine SNP 50 K Bead Chip assay (Wei et al., 2015).
Genomic regions related to domestication and improvement in sheep
First, for selection signature analysis, we focused on the whole domestication process in domestic sheep. Based on genome-wide comparisons of wild sheep and 308 domesticated sheep from 30 breeds, we identified several genes associated with immune response, metabolic processes, longevity-regulating pathway, and visual function (Supplementary Tables S7, S8). Vision plays a vital role in the survival and evolution of animals, such as predator avoidance, mate selection, and foraging (Yokoyama, 2002). Second, we detected selective sweeps in the 30 domesticated sheep breeds in comparison to wild sheep. Results identified two overlapping genomic regions for the 30 pairwise comparisons, which contained genes associated with immune response (IFI44 and IFI44L) and visual function (PANK2 and RNF24). The IFI44 and IFI44L genes belong to the type I interferon-inducible gene family and are located on the same chromosome. These two genes play important roles in regulating autoimmune disorders, inflammation, and immune response, and inhibit respiratory syncytial virus infection (Busse et al., 2020). Previous studies have also reported that variants of the PANK2 gene are related to pantothenate kinase-associated neurodegeneration (e.g., optic atrophy), and genetic variants of RNF24 and PANK2 are associated with optic disc morphology (Axenovich et al., 2011). Thus, these two genes may be associated with the evolution of vision during sheep domestication. Vision plays a vital role in animal survival, and many studies have demonstrated that visual acuity is weaker in domestic animals (e.g., chickens, dogs, and ducks) compared to their wild ancestors (Henderson et al., 2000; Peichl, 1992; Wang et al., 2016). We hypothesized that sensory ability and immunity may have been important targets of selection during domestication.
We also analyzed the potential selection signatures of wild-to-indigenous sheep domestication and identified candidate genes associated with immune response (IFI44L, IFI44), reproductive traits (SPAG16 (encoding sperm associated antigen 16) and FSIP2), and visual function (PDE6B (encoding phosphodiesterase 6B), FOXC1 (encoding forkhead box C1), and GMDS (GDP-mannose 4,6-dehydratase)) during domestication. FSIP2 is a protein-coding gene and plays an important role in spermatogenesis (Fang et al., 2021). Homozygous loss-of-function mutations in FSIP2 can lead to male infertility (Liu et al., 2019a). In the present study, FSIP2 was located in a significant selective region, and the genotype pattern differed between the wild and indigenous sheep breeds, suggesting that FSIP2 may play an important role in sheep fertility.
In the process of breeding and improvement, we identified several candidate genes associated with pigmentation, including several identified in previous research (e.g., ASIP (encoding agouti signaling protein) and MITF) (Li et al., 2014). Mutations in MITF can decrease pigmentation in dogs (Karlsson et al., 2007), pigs (Chen et al., 2016), ducks (Zhou et al., 2018), and quails (Minvielle et al., 2010). MITF also encodes a protein in the melanoma pathway. We identified a SNP in MITF that differed between white and non-white sheep. Moreover, two non-synonymous mutations of FGF9 were found at higher frequencies in the improved breeds than in the indigenous breeds. FGF9 is a member of the fibroblast growth factor family, and is involved in multiple biological processes, such as cartilage development, cell growth, and embryonic development (Zhang et al., 2021a, 2021c). These results imply that important economic traits (e.g., coat color, body size) are the preferred targets during breeding improvement. In addition, seven genomic regions were continuously selected at the two stages, indicating that some candidate loci may have undergone a second round of artificial selection for continued improvement of important economics traits.
Functional exploration of candidate SNPs in IFI44 gene
To further verify the function of the IFI44 gene, an IFI44 SNP (c. 54413058 C>G) was genotyped and subjected to association analysis in a Hu sheep population. Results indicated that the SNP at IFI44 c. 54413058 C>G was significantly associated with WBCs, neutrophils, and monocytes (Table 2). WBCs play an important role in the immune system. A feature of the inflammatory response is an increase in WBC count following bacterial or viral infection, and neutrophils, lymphocytes, and monocytes also play critical roles in innate and adaptive immunity (Parish, 2006). Thus, we concluded that the polymorphic sites in IFI44 may be important molecular markers of overcoming immune deficiency in animals. Nevertheless, the immune function of the IFI44 gene needs to be verified at the cellular and protein levels.
DATA AVAILABILITY
Raw data were deposited in the National Center for Biotechnology Information database under BioProjectID PRJNA795904 and PRJNA777695, in the Genome Sequence Archive under Accession No. CRA007173, and in the Science Data Bank under DOI: 10.57760/sciencedb.01846.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online.
COMPETING INTERESTS
The authors declare that they have no competing interests.
AUTHORS’ CONTRIBUTIONS
W.M.W. and F.D.L. designed the project. D.Y.Z., X.X.Z., F.D.L., and W.M.W. contributed to blood sample collection. D.Y.Z., L.F.Y., W.M.W., X.L.L., Y.K.Z., and Y.Z. analyzed the data. D.Y.Z., X.X.Z., L.M.Z., J.H.W., D.X., J.B.C., X.B.Y., W.X.L., C.C.L., and B.B.Z. contributed to data collection for the validation of experimental populations. D.Y.Z., X.X.Z., X.L.L., and X.B.Y. participated in DNA extraction. D.Y.Z. wrote the paper. W.M.W., X.X.Z., and D.Y.Z. reviewed and edited the manuscript. All authors read and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We would like to thank the staff at our laboratory for their ongoing assistance.
Funding Statement
This work was supported by the National Key R&D Program of China (2021YFD1300901), National Natural Science Foundation of China (31960653), West Light Foundation of the Chinese Academy of Sciences, and National Joint Research on Improved Breeds of Livestock and Poultry (19210365)
References
- 1.Alberto FJ, Boyer F, Orozco-Terwengel P, Streeter I, Servin B, de Villemereuil P, et al Convergent genomic signatures of domestication in sheep and goats. Nature Communications. 2018;9(1):813. doi: 10.1038/s41467-018-03206-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander DH, Novembre J, Lange K Fast model-based estimation of ancestry in unrelated individuals. Genome Research. 2009;19(9):1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Axelsson E, Ratnakumar A, Arendt ML, Maqbool K, Webster MT, Perloski M, et al The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495(7441):360–364. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- 4.Axenovich T, Zorkoltseva I, Belonogova N, van Koolwijk LME, Borodin P, Kirichenko A, et al Linkage and association analyses of glaucoma related traits in a large pedigree from a Dutch genetically isolated population. Journal of Medical Genetics. 2011;48(12):802–809. doi: 10.1136/jmedgenet-2011-100436. [DOI] [PubMed] [Google Scholar]
- 5.Bu DC, Luo HT, Huo PP, Wang ZH, Zhang S, He ZH, et al KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Research. 2021;49(W1):W317–W325. doi: 10.1093/nar/gkab447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busse DC, Habgood-Coote D, Clare S, Brandt C, Bassano I, Kaforou M, et al Interferon-induced protein 44 and interferon-induced protein 44-like restrict replication of respiratory syncytial virus. Journal of Virology. 2020;94(18):e00297–20. doi: 10.1128/JVI.00297-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao YH, Xu SS, Shen M, Chen ZH, Gao L, Lv FH, et al Historical introgression from wild relatives enhanced climatic adaptation and resistance to pneumonia in sheep. Molecular Biology and Evolution. 2021;38(3):838–855. doi: 10.1093/molbev/msaa236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, Guo WW, Ren LL, Yang MY, Zhao YF, Guo ZY, et al A de novo silencer causes elimination of MITF-M expression and profound hearing loss in pigs. BMC Biology. 2016;14:52. doi: 10.1186/s12915-016-0273-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen NB, Cai YD, Chen QM, Li R, Wang K, Huang YZ, et al Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nature Communications. 2018;9(1):2337. doi: 10.1038/s41467-018-04737-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen ZH, Xu YX, Xie XL, Wang DF, Aguilar-Gómez D, Liu GJ, et al Whole-genome sequence analysis unveils different origins of European and Asiatic mouflon and domestication-related genes in sheep. Communications Biology. 2021;4(1):1307. doi: 10.1038/s42003-021-02817-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chessa B, Pereira F, Arnaud F, Amorim A, Goyache F, Mainland I, et al Revealing the history of sheep domestication using retrovirus integrations. Science. 2009;324(5926):532–536. doi: 10.1126/science.1170587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daetwyler HD, Capitan A, Pausch H, Stothard P, van Binsbergen R, Brøndum RF, et al Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nature Genetics. 2014;46(8):858–865. doi: 10.1038/ng.3034. [DOI] [PubMed] [Google Scholar]
- 13.Diamond J Evolution, consequences and future of plant and animal domestication. Nature. 2002;418(6898):700–707. doi: 10.1038/nature01019. [DOI] [PubMed] [Google Scholar]
- 14.Fang X, Gamallat Y, Chen ZH, Mai HR, Zhou P, Sun CB, et al Hypomorphic and hypermorphic mouse models of Fsip2 indicate its dosage-dependent roles in sperm tail and acrosome formation. Development. 2021;148(11):dev199216. doi: 10.1242/dev.199216. [DOI] [PubMed] [Google Scholar]
- 15.Henderson JV, Wathes CM, Nicol CJ, White RP, Lines JA Threat assessment by domestic ducklings using visual signals: implications for animal-machine interactions. Applied Animal Behaviour Science. 2000;69(3):241–253. doi: 10.1016/S0168-1591(00)00132-5. [DOI] [PubMed] [Google Scholar]
- 16.Hu XJ, Yang J, Xie XL, Lv FH, Cao YH, Li WR, et al The genome landscape of tibetan sheep reveals adaptive introgression from argali and the history of early human settlements on the Qinghai-Tibetan Plateau. Molecular Biology and Evolution. 2019;36(2):283–303. doi: 10.1093/molbev/msy208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Y, Xie M, Chen WB, Talbot R, Maddox JF, Faraut T, et al The sheep genome illuminates biology of the rumen and lipid metabolism. Science. 2014;344(6188):1168–1173. doi: 10.1126/science.1252806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlsson EK, Baranowska I, Wade CM, Hillbertz NHCS, Zody MC, Anderson N, et al Efficient mapping of mendelian traits in dogs through genome-wide association. Nature Genetics. 2007;39(11):1321–1328. doi: 10.1038/ng.2007.10. [DOI] [PubMed] [Google Scholar]
- 19.Kodama Y, Shumway M, Leinonen R The sequence read archive: explosive growth of sequencing data. Nucleic Acids Research. 2012;40(D1):D54–D56. doi: 10.1093/nar/gkr854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li MH, Tiirikka T, Kantanen J A genome-wide scan study identifies a single nucleotide substitution in ASIP associated with white versus non-white coat-colour variation in sheep (Ovis aries) Heredity. 2014;112(2):122–131. doi: 10.1038/hdy.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li MZ, Chen L, Tian SL, Lin Y, Tang QZ, Zhou XM, et al Comprehensive variation discovery and recovery of missing sequence in the pig genome using multiple de novo assemblies. Genome Research. 2017;27(5):865–874. doi: 10.1101/gr.207456.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Yang J, Shen M, Xie XL, Liu GJ, Xu YX, et al Whole-genome resequencing of wild and domestic sheep identifies genes associated with morphological and agronomic traits. Nature Communications. 2020;11(1):2815. doi: 10.1038/s41467-020-16485-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu WJ, Wu H, Wang L, Yang XY, Liu CY, He XJ, et al Homozygous loss-of-function mutations in FSIP2 cause male infertility with asthenoteratospermia. Journal of Genetics and Genomics. 2019a;46(1):53–56. doi: 10.1016/j.jgg.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Liu XX, Zhang YL, Li YF, Pan JF, Wang DD, Chen WH, et al EPAS1 gain-of-function mutation contributes to high-altitude adaptation in Tibetan horses. Molecular Biology and Evolution. 2019b;36(11):2591–2603. doi: 10.1093/molbev/msz158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lv FH, Cao YH, Liu GJ, Luo LY, Lu R, Liu MJ, et al Whole-genome resequencing of worldwide wild and domestic sheep elucidates genetic diversity, introgression, and agronomically important loci. Molecular Biology and Evolution. 2022;39(2):msab353. doi: 10.1093/molbev/msab353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minvielle F, Bed'hom B, Coville JL, Ito S, Inoue-Murayama M, Gourichon D The "silver" Japanese quail and the MITF gene: causal mutation, associated traits and homology with the "blue" chicken plumage. BMC Genetics. 2010;11:15. doi: 10.1186/1471-2156-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parish CR The role of heparan sulphate in inflammation. Nature Reviews Immunology. 2006;6(9):633–643. doi: 10.1038/nri1918. [DOI] [PubMed] [Google Scholar]
- 30.Peichlcu L Topography of ganglion cells in the dog and wolf retina. The Journal of Comparative Neurology. 1992;324(4):603–620. doi: 10.1002/cne.903240412. [DOI] [PubMed] [Google Scholar]
- 31.Scher BD. 2000. World Watch List for Domestic Animal Diversity. 3rd ed. Rome: Food and Agriculture Organization of the United Nations.
- 32.Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, Birney E EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Research. 2009;19(2):327–335. doi: 10.1101/gr.073585.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang K, Li MY, Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Research. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang MS, Thakur M, Peng MS, Jiang Y, Frantz LAF, Li M, et al 863 genomes reveal the origin and domestication of chicken. Cell Research. 2020;30(8):693–701. doi: 10.1038/s41422-020-0349-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang MS, Zhang RW, Su LY, Li Y, Peng MS, Liu HQ, et al Positive selection rather than relaxation of functional constraint drives the evolution of vision during chicken domestication. Cell Research. 2016;26(5):556–573. doi: 10.1038/cr.2016.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang WM, Zhang XX, Zhou X, Zhang YZ, La YF, Zhang Y, et al Deep genome resequencing reveals artificial and natural selection for visual deterioration, plateau adaptability and high prolificacy in Chinese domestic sheep. Frontiers in Genetics. 2019;10:300. doi: 10.3389/fgene.2019.00300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wei CH, Wang HH, Liu G, Wu MH, Cao JX, Liu Z, et al Genome-wide analysis reveals population structure and selection in Chinese indigenous sheep breeds. BMC Genomics. 2015;16(1):194. doi: 10.1186/s12864-015-1384-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu JY, Fu YH, Hu Y, Yin LL, Tang ZS, Yin D, et al Whole genome variants across 57 pig breeds enable comprehensive identification of genetic signatures that underlie breed features. Journal of Animal Science and Biotechnology. 2020;11(1):115. doi: 10.1186/s40104-020-00520-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Li WR, Lv FH, He SG, Tian SL, Peng WF, et al Whole-genome sequencing of native sheep provides insights into rapid adaptations to extreme environments. Molecular Biology and Evolution. 2016;33(10):2576–2592. doi: 10.1093/molbev/msw129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yokoyama S Molecular evolution of color vision in vertebrates. Gene. 2002;300(1-2):69–78. doi: 10.1016/S0378-1119(02)00845-4. [DOI] [PubMed] [Google Scholar]
- 41.Yang J, Lee SH, Goddard ME, Visscher PM. 2011. GCTA: a tool for genome-wide complex trait analysis. The American Journal of Human Genetics, 88(1): 76−82.
- 42.Zeder MA Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(33):11597–11604. doi: 10.1073/pnas.0801317105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang DL, Huang JL, Sun XD, Chen HG, Huang S, Yang J, et al Targeting local lymphatics to ameliorate heterotopic ossification via FGFR3-BMPR1a pathway. Nature Communications. 2021a;12(1):4391. doi: 10.1038/s41467-021-24643-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang DY, Zhang XX, Li FD, Yuan LF, Zhang YK, Li XL, et al Polymorphisms in ovine ME1 and CA1 genes and their association with feed efficiency in Hu sheep. Journal of Animal Breeding and Genetics. 2021b;138(5):589–599. doi: 10.1111/jbg.12541. [DOI] [PubMed] [Google Scholar]
- 45.Zhang DY, Zhang XX, Li FD, Zhao Y, Li XL, Wang JH, et al Expression profiles of the ovine IL18 gene and association of its polymorphism with hematologic parameters in Hu lambs. Frontiers in Veterinary Science. 2022;9:925928. doi: 10.3389/fvets.2022.925928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang XY, Weng MJ, Chen ZQ Fibroblast growth factor 9 (FGF9) negatively regulates the early stage of chondrogenic differentiation. PLoS One. 2021c;16(2):e0241281. doi: 10.1371/journal.pone.0241281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao YX, Yang J, Lv FH, Hu XJ, Xie XL, Zhang M, et al Genomic reconstruction of the history of native sheep reveals the peopling patterns of nomads and the expansion of early pastoralism in East Asia. Molecular Biology and Evolution. 2017;34(9):2380–2395. doi: 10.1093/molbev/msx181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou ZK, Li M, Cheng H, Fan WL, Yuan ZR, Gao Q, et al An intercross population study reveals genes associated with body size and plumage color in ducks. Nature Communications. 2018;9(1):2648. doi: 10.1038/s41467-018-04868-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data to this article can be found online.
Data Availability Statement
Raw data were deposited in the National Center for Biotechnology Information database under BioProjectID PRJNA795904 and PRJNA777695, in the Genome Sequence Archive under Accession No. CRA007173, and in the Science Data Bank under DOI: 10.57760/sciencedb.01846.