

Abstract
Motivation
Selecting the splice variant that best represents a coding gene is a crucial first step in many experimental analyses, and vital for mapping clinically relevant variants. This study compares the longest isoforms, MANE Select transcripts, APPRIS principal isoforms, and expression data, and aims to determine which method is best for selecting biological important reference splice variants for large-scale analyses.
Results
Proteomics analyses and human genetic variation data suggest that most coding genes have a single main protein isoform. We show that APPRIS principal isoforms and MANE Select transcripts best describe these main cellular isoforms, and find that using the longest splice variant as the representative is a poor strategy. Exons unique to the longest splice isoforms are not under selective pressure, and so are unlikely to be functionally relevant. Expression data are also a poor means of selecting the main splice variant. APPRIS principal and MANE Select exons are under purifying selection, while exons specific to alternative transcripts are not. There are MANE and APPRIS representatives for almost 95% of genes, and where they agree they are particularly effective, coinciding with the main proteomics isoform for over 98.2% of genes.
Availability and implementation
APPRIS principal isoforms for human, mouse and other model species can be downloaded from the APPRIS database (https://appris.bioinfo.cnio.es), GENCODE genes (https://www.gencodegenes.org/) and the Ensembl website (https://www.ensembl.org). MANE Select transcripts for the human reference set are available from the Ensembl, GENCODE and RefSeq databases (https://www.ncbi.nlm.nih.gov/refseq/). Lists of splice variants where MANE and APPRIS coincide are available from the APPRIS database.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Most coding genes produce a range of alternatively spliced transcripts that can theoretically produce distinct protein isoforms. GENCODE v37 (Frankish et al., 2021) annotates more than 64 000 distinct translations for the 13 689 coding genes that are predicted to produce multiple protein isoforms, slightly more than four and a half distinct proteins per gene, and the complexity of the human coding gene set is growing.
There is overwhelming evidence that multi-exon coding genes can express a range of alternative transcripts (Reixachs-Solé and Eyras, 2022), and transcriptomics evidence suggests that many coding transcripts are important. However, not all splice variants are equal at the protein level. Most coding genes do have a main protein isoform (Ezkurdia et al., 2015a). While main splice variants have been predicted for coding genes at the transcript level (Gonzàlez-Porta et al., 2013), and methods have been developed to predict dominant transcripts using RNA-Seq data (Li et al., 2015; Olson and Ware, 2021), main splice variants are most obvious at the protein level. We showed that the main experimental isoforms determined from large-scale proteomics experiments were in agreement with two different sets of predicted reference isoforms over more than 96% of comparable genes (Ezkurdia et al., 2015a).
Evidence for the functional importance of alternative isoforms as a whole is less clear. Some alternative isoforms are clearly important—for example, at least 5% of alternative exons can be traced to a last common ancestor with fish (Martinez Gomez et al., 2021). However, genetic variation data show that alternative exons are not, in general, under purifying selection pressure (Liu and Lin, 2015; Tress et al., 2017), and there is much less evidence for alternative splicing at the protein level than would be expected (Abascal et al., 2015). What happens to the alternative variants that are not detected in proteomics experiments is one of the unanswered questions in genomics. For example, they might be translated in lower quantities or in restricted circumstances or they might be translated but have a shorter half-life than reference isoforms. Though, in each of these cases, the gene product is clearly an alternative isoform.
We have previously shown that APPRIS principal isoforms (Rodriguez et al., 2022), predicted from cross-species conservation and the preservation of protein features, are the best predictor of main protein isoforms (Ezkurdia et al., 2015a). Recently Ensembl (Cunningham et al., 2022) and RefSeq (Sayers et al., 2021) developed MANE Select (Morales et al., 2022), a new single reference splice variant for human coding genes. MANE Select transcripts are predicted from a range of experimental and computational data.
Here, we investigated the effectiveness of MANE Select transcripts for selecting main splice variants. We find that most coding genes have a single main splice isoform at the protein level and that these main splice isoforms almost always coincide with MANE Select transcripts and APPRIS principal isoforms. Exons exclusive to APPRIS principal isoforms and MANE Select transcripts are under selective pressure, while exons exclusive to alternative transcripts are not.
2 Materials and methods
2.1 Reference annotation
We downloaded the GENCODE v37 reference human gene set (Frankish et al., 2021), both the standard gtf file for the coordinates of the coding sequences (CDS) and the coding sequence translations. MANE Select transcripts are annotated in the GENCODE v37 reference set. We downloaded the APPRIS principal isoforms from the APPRIS website.
2.2 APPRIS principal isoforms
APPRIS principal isoforms are determined in several steps. The most reliable predictions (PRINCIPAL:1, P:1) are predicted from core modules that map protein structure (Burley et al., 2017; Jumper et al., 2021) and functional (Lopez et al., 2011; Mistry et al., 2021) features to isoforms and calculate cross-species conservation.
Where it is not possible to select a P:1 isoform, APPRIS uses TRIFID, a machine-learning tool trained on APPRIS data (Pozo et al., 2021) and proteomics data to select principal isoforms (Rodriguez et al., 2022). TRIFID is used in Steps 2 and 4 to generate PRINCIPAL:2 (P:2) and PRINCIPAL:4 (P:4) isoforms. APPRIS uses proteomics data in Step 3 to generate PRINCIPAL:3 (P:3) isoforms. For the proteomics comparison in this analysis, we could only use P:1 and P:2 isoforms. All APPRIS isoforms were used in the germline variation analysis.
2.3 MANE Select transcripts
MANE Select transcripts from the Matched Annotation from NCBI and EMBL-EBI collaboration (Morales et al., 2022) are representative transcripts that have exactly the same exonic structure in the RefSeq and Ensembl human gene sets. To generate these transcripts, RefSeq and Ensembl developed independent pipelines to identify representative transcripts and compared the outputs to determine the final MANE Select transcripts. The two pipelines have a range of inputs, based on conservation, expression and protein length. In addition, the APPRIS principal isoform is part of the input to the Ensembl pipeline, but not the RefSeq pipeline.
2.4 Transcript expression
We used a score developed in a previous study (Pozo et al., 2021) to determine dominant transcripts from RNA-seq data. The score calculates a score per transcript from splice junction reads. We downloaded RNA-seq data from the large-scale Human Protein Atlas RNA-seq experiments (Uhlén et al., 2015), a collection of samples from 36 different tissues. We aligned the reads to GENCODE v37 using STAR 2.6 (Dobin et al., 2013). We used default parameters, except that we set the maximum number of multiple alignments allowed to 50 and forced end-to-end read alignments to avoid unwanted alignments to repetitive regions.
We calculated a score per transcript from either the exon or CDS splice junction reads depending on whether we restricted the analysis to coding transcripts. For each splice junction, the number of reads was the number of junction spanning reads in the tissue with most reads. From these values, we calculated a mean splice junction value for each gene and the final score for each transcript was based on the least supported splice junction. The score for each transcript was calculated as the number of reads that supported the lowest scoring splice junction divided by the average read count of all the junctions in the gene.
2.5 Proteomics analysis
In order to distinguish dominant proteomics isoforms for as many genes as possible, we re-analysed five large-scale proteomics analyses (Bekker-Jensen et al., 2017; Carlyle et al., 2017; Kim et al., 2014; Schiza et al., 2019; Wang et al., 2019). The spectra from experiments PXD000561, PXD004452, PXD005445, PXD008333 and PXD010154 were downloaded from ProteomeXchange (Deutsch et al., 2017). These experiments were carried out using a range of tissue types (52 distinct tissue types in total) and one cell line. To optimize reproducibility, we set aside experiments that used proteolytic enzymes other than trypsin.
We searched against the GENCODE v37 human reference set with read-through transcripts eliminated (Abascal et al., 2018). We mapped spectra to the gene set using Comet (Eng et al., 2013) with default parameters, allowing only fully tryptic peptides, a maximum of one missed cleavage and the oxidation of methionines. Allowing missed cleavages is an important step, because trypsin is not 100% efficient and means we can detect splice events that might otherwise be undetectable (Wang et al., 2018). We post-processed the peptide spectrum matches (PSMs) with Percolator (The et al., 2016) and Percolator posterior error probabilities (PEPs) values were used to identify correct PSMs. We considered as valid those PSMs with PEP values <0.001. This PEP threshold approximates to q-values of 0.0001, so it is highly conservative. Moonlighting peptides, those that mapped to more than one gene, were discarded.
To reduce the false positive identifications that will inevitably occur when combining results from many different experiments (Ezkurdia et al., 2015b), peptides identified in each of the five analyses had to be validated by PSMs from at least two of the individual experiments that made up each proteomics analysis. All large-scale analyses contained replicate experiments, so this rule did not exclude tissue-specific peptides.
We determined the main proteomics isoform by counting up the PSMs that mapped to each annotated protein isoform (Ezkurdia et al., 2015a). The isoform with the largest number of PSM over the five analyses was determined to be the main experimental isoform for each gene (Fig. 1). Main isoforms had to have at least two more PSM than any other isoform (ties between distinct isoforms were not possible) and be supported by a minimum of four PSM.
Fig. 1.
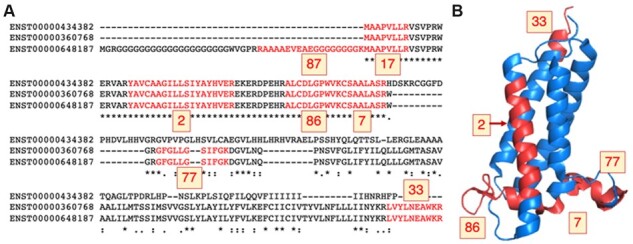
Peptides mapping to isoforms from gene VKORC1L1. (A) The alignment between the three protein sequence unique isoforms of gene VKORC1L1 (Vitamin K epoxide reductase complex subunit 1-like protein 1) with the peptides detected in the five large-scale proteomics analyses in red. The number of PSM detected for each peptide is highlighted in yellow boxes. The protein coded by transcript ENST00000648187 was chosen as the main experimental isoform because 309 PSM map to this isoform (against just 189 and 222 PSM for the other two isoforms). (B) VKORC1L1 peptides mapped to the structure of human VKOR (PDB: 6WVH, Burley et al., 2017). Peptides are marked in red on the structure and the number of PSM detected is in yellow boxes. The protein is membrane-bound and no peptides were found in membrane-spanning regions, except for peptide YAVCAAGIIISIYAYHVER (2 PSM). The N-terminal peptides (87 and 17 PSM) do not map to the structure; this region is likely to be disordered. Peptide mapping was carried out using HHPRED (Gabler et al., 2020). The image was generated with PyMol
2.6 Analysis of germline variants
We analysed the distribution of variants from 2504 individuals in the 1000 Genomes Project, phase 3 (1000 Genomes Project Consortium, 2012). We mapped the variants from GRCh37 to GRCh38 using dbSNP v149 (Sherry et al., 2001) with a success rate of greater than 99% (Abascal et al., 2018). We discarded all genes that had a single coding transcript and all genes that did not have a MANE Select transcript.
We generated seven sets of exons for the remaining genes based on the APPRIS principal isoforms, the MANE Select transcripts and the longest isoforms. The first, and largest, set contained exons from genes where APPRIS principal isoforms, MANE Select transcripts and longest isoforms coincided. Then we created sets of non-overlapping exons that were part of a reference transcript according to one method, but in part of an alternative transcript according to another method. There were therefore six further sets of exons.
Finally, we compared exons from reference transcripts selected using RNA-Seq reads that did not overlap exons from APPRIS principal transcripts, against exons from APPRIS alternative transcripts that did not overlap exons in RNA-Seq reference transcripts.
For each analysis, we removed all exons that were present in both reference and alternative transcripts and any alternative exons that overlapped reference exons. We tagged all exons that belonged to the reference splice variants in each set as reference exons. All other exons were alternatives. Non-synonymous-to-synonymous ratios for both rare and common allele frequencies were calculated for all sets of exons. We used an allele frequency cut-off of 0.005 to separate rare (<0.005) and common (>0.005) alleles.
3 Results
The study aimed to determine whether the principal isoforms selected by APPRIS, the MANE Select transcripts, the longest isoform or the isoforms chosen from transcript level data were a better choice as the representative splice variant. We compared the predicted representative isoforms to the main cellular isoform derived from proteomics experiments, and used human germline variation data to analyse selective pressure in exons unique to each method.
3.1 Principal isoforms and MANE Select transcripts coincide with the main proteomics isoform
In total, we detected 337 737 distinct peptides mapping to 15 137 coding genes across the five large-scale proteomics analyses. We chose a main proteomics isoform by counting the PSM that supported each annotated splice isoform in GENCODE v37 (Fig. 1). There were 7697 genes for which we could determine a main proteomics isoform (50.8% of coding genes for which we detected peptides). The 7440 genes left out of the analysis were either annotated with a single coding transcript or did not have enough peptides to determine a main isoform.
We first ran a control experiment in which we selected isoforms randomly from each of the 7697 genes and measured the agreement with the main isoform. Over 100 simulations, randomly selected isoforms agreed with the main isoform just 29.97% of the time.
We then analysed the agreement between the main proteomics isoforms and other means of selecting a reference splice variant. The agreement with the main isoform is shown in Figure 2. RNA-Seq data often support non-coding transcripts from coding genes as the reference transcript. When we ignored reads for non-coding transcripts, untranslated exons and unfinished coding transcripts, the agreement between the most expressed transcript and the main proteomics isoform was 70.05% over 6434 genes.
Fig. 2.
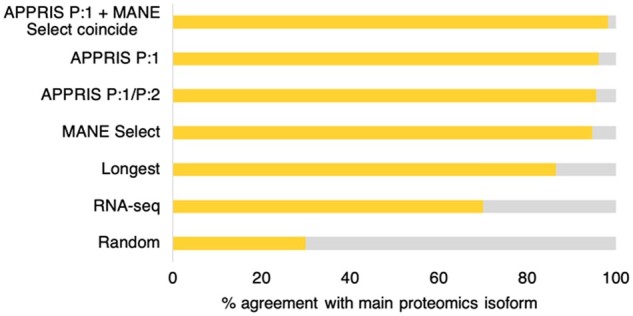
Agreement between main protein isoforms and reference predictions. The percentage of genes in which predicted reference isoforms coincided with proteomics main isoforms (yellow). Predicted reference isoforms were the longest isoform, dominant RNA-seq transcripts, the MANE Select transcripts and APPRIS principal isoforms
The agreement between MANE Select transcripts and the main proteomics isoform was 94.58% (over the 6995 genes with MANE Select transcripts), while APPRIS P:1 principal isoforms coincided with the main proteomics isoform 96.13% of the time over 6285 genes (P:1 isoforms are chosen by the core APPRIS methods). The agreement between P:1 and P:2 isoforms and the main cellular isoform was 95.52% over 6847 genes. P:2 isoforms are chosen using the TRIFID functional isoform predictor (Pozo et al., 2021), recently incorporated into APPRIS. APPRIS P:3 isoforms are chosen based on proteomics evidence, so cannot be compared.
For the 5888 genes in which the APPRIS P:1 isoform and MANE Select transcript coincided, the agreement with the main proteomics isoform was 98.2%. Therefore, APPRIS P:1 isoforms and MANE Select transcripts are particularly good predictors of the main cellular isoform when they agree.
The agreement between main proteomics isoforms and longest isoforms was just 86.4%, even though in this analysis the longest isoforms have an inbuilt advantage—the longer the isoform, the higher the probability that peptides map to the isoform. This is clear in the example in Figure 1, VKORC1L1. The first and third isoforms have unique sequences, so they have unique peptides and can be the main proteomics isoform. The second isoform (translated from ENST00000360768) technically cannot be the main proteomics isoform because its sequence is identical (but shorter) to that of the third (longest) isoform, produced from an upstream ATG. Incidentally, this second isoform is highly conserved and is the one selected by APPRIS and MANE for this gene.
The gene RAB7A can serve as an example of the differences between predictors. RAB7A produces a small GTPase (Rab-7a) that plays a central role in endosome–lysosome transport via protein–protein interaction cascades (Wu et al., 2005). RAB7A is strikingly conserved across vertebrates and invertebrates; for example, human and bagworm moth Rab-7a sequences are 83% sequence identical. MANE and APPRIS both select the highly conserved five exon transcript as the most important splice variant (Fig. 3), while the variant chosen from the RNA-Seq data is missing the first coding exon with respect to the APPRIS/MANE transcript.
Fig. 3.
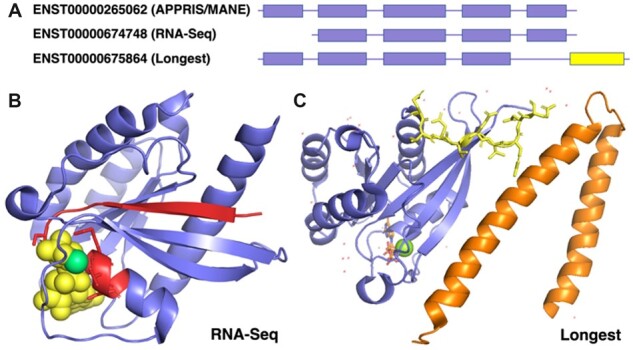
RNA-Seq dominant transcripts and longest isoforms for RAB7A. (A) The reference transcripts selected by different methods for RAB7A. APPRIS and MANE chose the same transcript. The RNA-Seq transcript (ENST00000674748) is missing the first coding exon; the longest isoform has a different 3′ CDS (in yellow). (B) The predicted effect of the transcript chosen by RNA-Seq data, mapped to the resolved structure of Rab-7a (PDB: 1T91). The region in red would be missing from this isoform of Rab-7a. The GTP molecule is shown in spacefill in yellow and the magnesium ion in green. The lost GTP binding motif is shown as red sticks. (C) The predicted changes brought about by the longest isoform, mapped to the resolved structure of Rab-7a (PDB: 1YHN). Rab-7a is shown in blue and the RILP structure in orange. Residues that would be swapped for unrelated residues are shown as yellow sticks. Tyrosine 183, the residue closest to RILP, nestles in a hydrophobic pocket in RILP (Wu et al., 2005). It would be lost in the longest isoform
Isoforms chosen by RNA-Seq data tend to have fewer exons. In RAB7A, the first exon codes for a vital strand in the Rab-7a structure and the loop that harbours the conserved GTP-binding PM/G motif is vital for all Ras proteins (Valencia et al., 1991). The loss of the strand would compromise the folding of Rab-7a and the loss of the binding motif would mean that this splice isoform would not bind GTP or magnesium (Fig. 3). GTP binding is necessary for protein–protein interactions; an isoform lacking this motif is unlikely to have a functional role.
The longest isoform annotated for RAB7A is produced from a distinct fifth exon. The fifth exon in RAB7A is the least conserved (human and Malaysian fruit fly share just 48% of residues), but the conserved hydrophobic residues at the start of the exon are important for the interaction between Rab-7a- and Rab7-interacting lysosomal protein (Wu et al., 2005, RILP), so the loss of this exon would abolish this interaction (and the conserved C-terminal double-cysteine prenylation motif involved in binding to the endosome membrane).
3.2 MANE Select and APPRIS principal exons are under purifying selection
We calculated non-synonymous-to-synonymous ratios for exons from reference coding transcripts and exons from non-reference (alternative) transcripts using MANE Select transcripts, APPRIS principal isoforms, longest isoforms and the most expressed transcripts. As a control, we looked at exons from genes where MANE Select transcripts, APPRIS principal isoforms and the longest isoforms agreed on the reference splice variant. Then we analysed exons from genes in which the APPRIS principal isoform did not coincide with the longest isoform, where MANE Select transcripts did not coincide with the longest isoforms, where longest isoforms did not overlap with the principal isoform, and where the most expressed transcript did not coincide the principal isoform. In each set, we calculated non-synonymous-to-synonymous ratios for exons unique to reference transcripts and for exons unique to alternative transcripts.
Where all three methods agreed, the results were clear (see Fig. 4 and Supplementary Fig. S1). The non-synonymous-to-synonymous ratio for the common variants of the reference exons was half that of the rarer variants, indicating clear evidence of purifying selection (Fig. 4A). The non-synonymous-to-synonymous ratios for common and rare variants of exons alternative to these transcripts were indistinguishable, which suggests that most of these alternative exons are evolving neutrally.
Fig. 4.
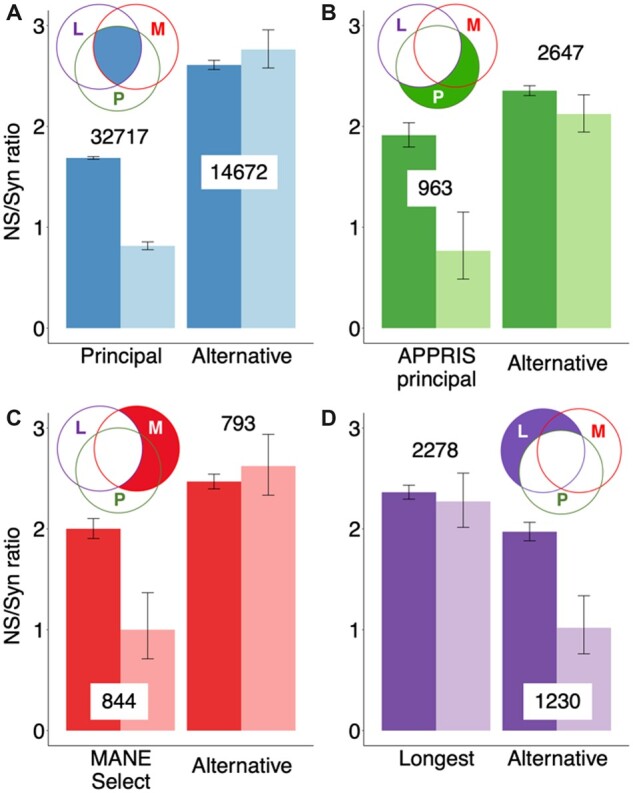
Non-synonymous-to-synonymous ratios for reference and alternative exons. Non-synonymous-to-synonymous ratios for rare (dark bars) and common (light bars) variants, along with error bars showing 95% confidence intervals. The Venn diagrams indicate the subset of exons in each plot; exons ‘L’ are those that produce the longest isoform, ‘M’ are present in MANE Select transcripts and ‘P’ produce principal isoforms. The number of exons in each set is indicated. (A) The non-synonymous-to-synonymous ratios for reference and alternative exons from genes where the longest splice variant, the APPRIS principal isoform and the MANE Select transcript all agree. (B) Ratios for exons that generate APPRIS principal isoforms and not alternative isoforms (APPRIS principal), and for exons that produce alternative isoforms only (Alternative). (C) Ratios for exons from MANE Select transcripts and not in alternative transcripts (MANE Select), and for exons that do not overlap MANE Select transcripts (Alternative). (D) Ratios for exons that produce the longest isoform and not any other isoform (Longest), and for exons that produce isoforms other than the longest (Alternative)
Exons that produced APPRIS principal isoforms but not longest isoforms followed a similar pattern. The non-synonymous-to-synonymous ratio in common alleles was less than half that of rare alleles and this difference was significant despite the smaller sample size (Fig. 4B). Non-synonymous-to-synonymous ratios for common and rare alleles were similar (not significantly different) for exons that are alternative in APPRIS, but that produce the longest isoforms.
The non-synonymous-to-synonymous ratio in common alleles for MANE Select exons that did not generate the longest isoform was also less than half that of rare alleles, and again the difference was significant (Fig. 4C). Non-synonymous-to-synonymous ratios for alternative exons to MANE Select transcripts were actually higher for rare variants, but again this was not significant.
By way of contrast, non-synonymous-to-synonymous ratios for exons that produce the longest isoform but not the APPRIS principal isoform were not significantly lower in common alleles than in rare alleles (Fig. 4D). Alternative exons unique to shorter splice variants did have a substantially lower ratio for common variants, however, and this difference was clearly significant, suggesting that they are under selective pressure. Non-synonymous-to-synonymous ratios for common and rare alleles were also similar when we compared exons that produced the longest isoform with those of the MANE Select transcript (Supplementary Table S1).
The non-synonymous-to-synonymous ratios show that exons unique to APPRIS principal isoforms and MANE Select transcripts are both under purifying selection, while there is no evidence for purifying selection for exons that produce the longest isoforms, exons unique to APPRIS alternative isoforms or MANE alternative transcripts. Indeed, all these exons seem mostly to be under neutral selective pressure.
We also compared exons that generated MANE Select transcripts but not APPRIS principal isoforms with those that produced APPRIS principal isoforms but not MANE Select transcripts. We found that non-synonymous-to-synonymous ratios for these exons were lower in common alleles than in rare alleles, both for exons that would produce the reference splice variants and for those that would produce alternative splice variants (see Supplementary Table S1). However, the differences between common alleles and rare alleles were less marked in all sets, and not always significant, in part because there were fewer exons in these comparisons because of the greater overlap between APPRIS principal and MANE Select splice variants.
Finally, reference transcripts predicted from transcript expression had relatively few unique exons (as we have seen transcript expression levels tend to favour shorter transcripts), but exons unique to the most expressed transcripts had a lower non-synonymous-to-synonymous ratio among common variants, suggesting that they are under selection pressure (Supplementary Fig. S1). For these genes at least there may be evidence for the functional importance of more than one isoform, because exons alternative to the most expressed transcripts were clearly under selection pressure too. One in 11 exons from the most expressed transcripts came from tandem duplications (Martinez Gomez et al., 2021).
4 Conclusions
Both peptide evidence and germline variation analysis support the hypothesis that most coding genes have a single main protein isoform. We show that this main cellular isoform is best described by MANE Select transcripts and APPRIS principal isoforms. The two methods are particularly powerful when the predictions agree, and they agreed over 94.2% of coding genes in GENCODE v37.
The large-scale proteomics experiments allowed us to determine a main cellular isoform for more than a third of coding genes. This isoform coincided much more often with the APPRIS principal isoforms and MANE Select variants than with the longest isoform, or with the variant selected from transcript expression data. Where MANE Select and APPRIS principal isoforms coincided, the agreement with the main cellular isoforms was 98.2%. Since main isoforms from proteomics experiments, APPRIS principal isoforms and MANE Select transcripts are three orthogonal means of determining a reference splice variant, the three-way agreement over such a high proportion of genes is remarkably strong evidence for a single main isoform per gene.
The distribution of germline variants also provided strong support for the importance of APPRIS principal isoforms and MANE Select transcripts. Non-synonymous-to-synonymous ratios showed that exons unique to principal and MANE Select exons are subject to purifying selection and that exons unique to alternative transcripts were not under selective pressure. For the longest isoform in each coding gene, often the default representative in databases and large-scale studies, the opposite was true. Exons unique to the longest isoforms appear to be evolving neutrally.
Both proteomics data and germline variations suggest that most coding genes have one functionally important protein isoform. Most alternative exons are not under selective pressure, suggesting that any functional role is not vital to the cell and alternative isoforms not detected in proteomics experiments are missing either because they are translated in low quantities or restricted circumstances, or because they have shorter half-lives. Alternative isoforms are less important than principal isoforms in most coding genes.
The fact that most coding genes have one functionally important protein isoform does not mean that alternative isoforms have no cellular role. Literature studies suggest functional differences for alternative isoforms (Kelemen et al., 2013; Bhuiyan et al., 2018), though the exact cellular purpose is often not well defined. In addition, while most alternative exons are not conserved (Martinez Gomez et al., 2021; Rodriguez et al., 2020), approximately 5% of alternative exons are ancient (Martinez Gomez et al., 2021) and some alternative isoforms have clear evidence of tissue specificity at the protein level (Rodriguez et al., 2020). The likely functional importance of splice isoforms can be predicted (Pozo et al., 2021) and these predictions are included in APPRIS.
We have shown that MANE Select transcripts and APPRIS principal isoforms are important tools for determining the biological relevance of splice isoforms and predict the main cellular isoform with a high degree of reliability. Researchers ought to use these two sets of reference transcripts, rather than the longest isoform, in all clinical and biomedical research.
Supplementary Material
Acknowledgements
The authors would like to thank Thomas A. Walsh for the proteomics analysis.
Funding
This paper was published as part of a special issue financially supported by ECCB2022. This work was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U24HG007234. This work was also supported by the following grants: PGC2018-097019-B-I00/Ministry of Science, Innovation and Universities; IPT17/0019/Carlos III Institute of Health-Fondo de Investigación Sanitaria and HR17-00247/‘la Caixa’ Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflict of Interest: none declared.
Contributor Information
Fernando Pozo, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), 28029 Madrid, Spain.
José Manuel Rodriguez, Cardiovascular Proteomics Laboratory, Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), 28029 Madrid, Spain.
Laura Martínez Gómez, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), 28029 Madrid, Spain.
Jesús Vázquez, Cardiovascular Proteomics Laboratory, Centro Nacional de Investigaciones Cardiovasculares Carlos III (CNIC), 28029 Madrid, Spain; CIBER de Investigaciones Cardiovasculares (CIBERCV), 28029 Madrid, Spain.
Michael L Tress, Bioinformatics Unit, Spanish National Cancer Research Centre (CNIO), 28029 Madrid, Spain.
Data Availability
There is no new data associated with this article. All data is already available from the public sources listed in the abstract and the methods section. The analysis of this public data will be shared on reasonable request to the corresponding author.
References
- 1000 Genomes Project Consortium. (2012) An integrated map of genetic variation from 1,092 human genomes. Nature, 491, 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abascal F. et al. (2015) Alternatively spliced homologous exons have ancient origins and are highly expressed at the protein level. PLoS Comp. Biol., 11, 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abascal F. et al. (2018) Loose ends: almost one in five human genes still have unresolved coding status. Nucleic Acids Res., 46, 7070–7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker-Jensen D.B. et al. (2017) An optimized shotgun strategy for the rapid generation of comprehensive human proteomes. Cell Syst., 4, 587–599.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuiyan S.A. et al. (2018) Systematic evaluation of isoform function in literature reports of alternative splicing. BMC Genomics, 19, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley S.K. et al. (2017) Protein data bank (PDB): the single global macromolecular structure archive. Methods Mol. Biol., 1607, 627–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlyle B.C. et al. (2017) A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci., 20, 1787–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham F. et al. (2022) Ensembl 2022. Nucleic Acids Res., 50, D988–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch E.W. et al. (2017) The ProteomeXchange consortium in 2017: supporting the cultural change in proteomics public data deposition. Nucleic Acids Res., 45, D1100–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A. et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng J.K. et al. (2013) Comet: an open-source MS/MS sequence database search tool. Proteomics, 13, 22–24. [DOI] [PubMed] [Google Scholar]
- Ezkurdia I. et al. (2015a) Most highly expressed protein-coding genes have a single dominant isoform. J. Proteome Res., 14, 1880–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezkurdia I. et al. (2015b) The potential clinical impact of the release of two drafts of the human proteome. Exp. Rev. Proteomics, 12, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankish A. et al. (2021) Gencode 2021. Nucleic Acids Res., 49, D916–D923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabler F. et al. (2020) Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinformatics, 72, e108. [DOI] [PubMed] [Google Scholar]
- Gonzàlez-Porta M. et al. (2013) Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome Biol., 14, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J. et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature, 596, 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelemen O. et al. (2013) Function of alternative splicing. Gene, 514, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.S. et al. (2014) A draft map of the human proteome. Nature, 509, 575–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.D. et al. (2015) Functional networks of highest-connected splice isoforms: from the chromosome 17 human proteome project. J Proteome Res, 14, 3484–3491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T., Lin K. (2015) The distribution pattern of genetic variation in the transcript isoforms of the alternatively spliced protein-coding genes in the human genome. Mol. Biosyst., 11, 1378–1388. [DOI] [PubMed] [Google Scholar]
- Lopez G. et al. (2011) Firestar—advances in the prediction of functionally important residues. Nucleic Acids Res., 39, W235–W241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Gomez L. et al. (2021) The clinical importance of tandem exon duplication-derived substitutions. Nucleic Acids Res., 49, 8232–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry J. et al. (2021) Pfam: the protein families database in 2021. Nucleic Acids Res., 49, D412–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales J. et al. (2022) A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature, 604, 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson A.J., Ware D. (2021) Ranked choice voting for representative transcripts with TRaCE. Bioinformatics, 38, 261–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozo F. et al. (2021) Assessing the functional relevance of splice isoforms. NAR Genom. Bioinformatics, 3, lqab044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reixachs-Solé M., Eyras E. (2022) Uncovering the impacts of alternative splicing on the proteome with current omics techniques. Wiley Interdiscip. Rev., e1707. 10.1002/wrna.1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J.M. et al. (2020) An analysis of tissue-specific alternative splicing at the protein level. PLoS Comput. Biol., 16, e1008287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J.M. et al. (2022) APPRIS: selecting functionally important isoforms. Nucleic Acids Res., 50, D54–D59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers E.W. et al. (2021) Database resources of the national center for biotechnology information. Nucleic Acids Res., 49, D10–D17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiza C. et al. (2019) Identification of TEX101-associated proteins through proteomic measurement of human spermatozoa homozygous for the missense variant rs35033974. Mol. Cell. Proteomics, 18, 338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S.T. et al. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The M. et al. (2016) Fast and accurate protein false discovery rates on large-scale proteomics data sets with percolator 3.0. J. Am. Soc. Mass Spectrom., 27, 1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tress M.L. et al. (2017) Alternative splicing may not be the key to proteome complexity. Trends Biochem. Sci., 42, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M. et al. (2015) Proteomics. Tissue-based map of the human proteome. Science, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Valencia A. et al. (1991) The ras protein family: evolutionary tree and role of conserved amino acids. Biochemistry, 30, 4637–4648. [DOI] [PubMed] [Google Scholar]
- Wang D. et al. (2019) A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol., 15, e8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. et al. (2018) Detection of proteome diversity resulted from alternative splicing is limited by trypsin cleavage specificity. Mol. Cell. Proteomics, 17, 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M. et al. (2005) Structural basis for recruitment of RILP by small GTPase Rab7. EMBO J., 24, 1491–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
There is no new data associated with this article. All data is already available from the public sources listed in the abstract and the methods section. The analysis of this public data will be shared on reasonable request to the corresponding author.