

Abstract
Motivation
The CRISPR-Cas9 system is a Type II CRISPR system that has rapidly become the most versatile and widespread tool for genome engineering. It consists of two components, the Cas9 effector protein, and a single guide RNA that combines the spacer (for identifying the target) with the tracrRNA, a trans-activating small RNA required for both crRNA maturation and interference. While there are well-established methods for screening Cas effector proteins and CRISPR arrays, the detection of tracrRNA remains the bottleneck in detecting Class 2 CRISPR systems.
Results
We introduce a new pipeline CRISPRtracrRNA for screening and evaluation of tracrRNA candidates in genomes. This pipeline combines evidence from different components of the Cas9-sgRNA complex. The core is a newly developed structural model via covariance models from a sequence-structure alignment of experimentally validated tracrRNAs. As additional evidence, we determine the terminator signal (required for the tracrRNA transcription) and the RNA–RNA interaction between the CRISPR array repeat and the 5′-part of the tracrRNA. Repeats are detected via an ML-based approach (CRISPRidenify). Providing further evidence, we detect the cassette containing the Cas9 (Type II CRISPR systems) and Cas12 (Type V CRISPR systems) effector protein. Our tool is the first for detecting tracrRNA for Type V systems.
Availability and implementation
The implementation of the CRISPRtracrRNA is available on GitHub upon requesting the access permission, (https://github.com/BackofenLab/CRISPRtracrRNA). Data generated in this study can be obtained upon request to the corresponding person: Rolf Backofen (backofen@informatik.uni-freiburg.de).
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Clustered regularly interspaced short palindromic repeats (CRISPR) system is a widespread, prokaryotic acquired immune system to defend against invading phages or other genetic material. It consists of a CRISPR array made of repeats and spacers, which match foreign genetic material, and a set of CRISPR-associated (Cas) proteins. While there is a quite large variety of CRISPR systems found in nature, the system can be grouped into two classes and six major types (Makarova et al., 2020). Class 2 CRISPR systems, consisting of Types II, V and VI, have a single large effector protein, which makes them optimal for biotechnological applications. The CRISPR-Cas9 system is a Type II CRISPR system that has rapidly become the most versatile and widespread tool for genome engineering. It consists of three components, the Cas9 effector protein, the CRISPR array (i.e. the spacer-containing element for identifying targets) and the tracrRNA, which is a trans-activating small RNA required for both the maturation of the precursor crRNA (CRISPR RNA) as well as for the later interference. While there are well-established methods for screening Cas effector proteins and CRISPR arrays, the detection of tracrRNAs remains the bottleneck in detecting Class 2 CRISPR systems. Although different approaches have been developed to screen for tracrRNA, structural covariance models (CM) have mostly been used so far to compare clusters of screened putative tracrRNAs (Briner et al., 2014; Dooley et al., 2021; Fonfara et al., 2014). These CMs were based on a wide screen of tracrRNA candidates, combined with sequence-based clustering and subsequent sequence alignment in each cluster using MAFFT to generate the CM models. The found CMs covered the whole tracrRNA, including the anti-repeat part and the terminal hairpin, thus effectively using an evolutionary model for the anti-repeat part.
We developed a new approach for screening tracrRNA in this paper that supersedes existing approaches in several aspects. First, we use structural clustering on experimentally validated tracrRNAs, generating more specific CMs. Our models also exclude the anti-repeat part, as we can use here a more reliable information source by investigating the RNA–RNA interaction between the repeat and the tracrRNA. Thus, for screening tracrRNA, we first detect CRISPR arrays and the associated repeats in a genome, and then use the interaction between tracrRNA and a repeat as evidence for a tracrRNA candidate, instead of applying an evolutionary model of the anti-repeat. In addition, our models also represent information about which part of the structure is required for tracrRNA function. Second, we use improved methods to detect CRISPR arrays and CRISPR cassettes containing the relevant Cas proteins. And third, our system is not restricted to Type II systems but covers, for the first time, also Type V systems.
1.1 TracrRNA in Type II systems
Almost all Class II systems, including Type II-A, B, C1 and C2, rely on a small non-coding RNA called tracrRNA (trans-activating CRISPR RNA; Chylinski et al., 2013; Makarova et al., 2020) for both the maturation of crRNA and the later interference step. Concerning maturation, several studies were performed on crystal structures of Cas9 that help to understand how Cas9 specifically recognizes the crRNA: tracrRNA duplex (e.g. Anders et al., 2014; Huai et al., 2017; Jiang et al., 2015; Jinek et al., 2014). It is now clear that the tracrRNA consists of two parts, a 5′-‘anti-repeat’ part and a 3′-tail part called nexus. The crRNA: tracrRNA complex, which is recognized by Cas9, can be divided into a few domains: the anti-repeat interacting with repeat (stem and bulge), and the tail part containing two regular hairpin loops and one small hairpin. Anti-repeat sequences normally contain 24 nt, and the 3′-tail usually has a sequence length of ∼75 nt. Despite the tracrRNA from Type II-A (more specifically: from Streptococcus pyogenes) being well-investigated, there is a lot to be discovered about tracrRNA biology and biotechnological applications (see the recent review by Liao and Beisel, 2021). The critical part here is to screen and characterize the variety of tracrRNAs found in natural systems, which is the main application for our CRISPRtracrRNA tool.
1.2 TracrRNA in Type V systems
Type V-K CRISPR-Cas system is a CRISPR-associated transposase system in Cyanobacteria, where the cas12k effector gene, its CRISPR array and the Tn7-like transposases TnsB, TnsC and TniQ form a shared transposon (Strecker et al., 2019). The CRISPR-Cas effector complex of this system is able to interact with the transposase complex to guide the transposon into its new location. This guidance is done by an interaction between Cas12k and the transposase TniQ as well as the DNA recognition by the crRNA (Park et al., 2021). CRISPR-associated transposase (CAST) systems are frequently found next to short protospacers of ∼17 nt corresponding to an intern spacer separated from the CRISPR array (Saito et al., 2021). This spacer is located downstream of a truncated Repeat sequence of ∼12 nt from the 3′-end (Saito et al., 2021). Different sRNA analyses of CAST systems also showed an expression of truncated crRNA from the CRISPR array itself with ∼17 nt Spacer and either 12 or 14 nt Repeat sequence length, even though the repeat sequence is persistently 37 nt long and the spacers usually reach over 30 nt (Saito et al., 2021; Strecker et al., 2019). The interaction between Cas12k and the crRNA is promoted by a tracrRNA located upstream of the CRISPR array. The binding between tracrRNA and crRNA seems to be facilitated by two different binding sites, one in the middle area (next to stem-loop 3) and one directly at the 3′-end of the tracrRNA (Querques et al., 2021; Xiao et al., 2021). The first one binds five nucleotides at the 3′-end of the repeat (5′-GAAAG-3′) and the second one binds nine nucleotides upstream of it (5′-YYYNYYYAA-3′). The fourth base of this second binding site is most of the time not corresponding to the tracrRNA and seems to be unbound in the single guide RNA (sgRNA) structure (Xiao et al., 2021). Interestingly, these positions correspond to the 12–14 nt processed 3′-end of the crRNA. The mechanism and the nuclease for this processing are still unknown, but this seems to indicate that the tracrRNA is part of it.
2 Materials and methods
As shown in Figure 1, our CRISPRtracrRNA investigates the different components relevant to CRISPR-Cas interference functionality, which in combination are tailored to robustly detect the trans-activating CRISPR RNA (tracrRNA) candidates in the provided input genomes. The core components (Component 1 + 2 in Fig. 1) are the determination of the tracrRNA structure motif by newly defined CMs, and the determination of the terminator hairpin, which is required for transcription. An additional part of the tracrRNA is the repeat/anti-repeat interaction (Component 3). We do not learn a motif here as we have more reliable genomic information that we can use. In more detail, we detect the first CRISPR arrays using the ML-based CRISPRidentify (Component 4), as an array is an evidence for the existence of a CRISPR system and it allows us to determine the repeat, which is subsequently used to determine the anti-repeat part of the tracrRNA via RNA–RNA interaction prediction. Additional evidence is acquired by the identification of cas9/cas12 genes (Component 5) using CRISPRcasIdentifier (Padilha et al., 2020), and computing the distance to the formed tracrRNA candidates. Each of the listed factors is provided with the corresponding certainty score. Our approach outputs the found candidate set in the readable format. The candidates are ranked according to their certainty scores with a clear indication of all the factors (pieces of evidence) that influenced their value. Moreover, the users can change the parameters for the respective search as well as assign different weight (importance) values to the factors they consider the most important (see Section 3.3.1). In the following description, we list the components in the order of usage in our pipeline. In each part, several scores are produced, which are then forwarded to a final combination step (see Section 3.3.1).
Fig. 1.
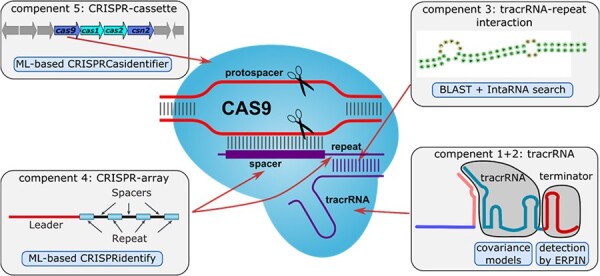
Components of the CRISPRtracrRNA tool. Components 1 and 2 comprise the structural model of tracrRNAs and are designed to robustly detect the tracrRNA tail location by comparing the candidate sequence with the existing model and searching for the terminator sequence. Component 3 uses RNA–RNA interaction prediction to determine the set of anti-repeat candidates. This component requires the set of repeat sequences, which are collected in Component 4. This step is designed to identify CRISPR arrays and the associated repeats. Component 5 uses a prediction of a whole CRISPR cassette (using CRISPRCasIndentifier) to reliably determine Cas9 or Cas12 effector proteins.
2.1 Anti-repeat search
The mature crRNA in Type II will contain one repeat-spacer unit, which is processed out of a tracrRNA: pre-crRNA duplex. After maturation, the mature crRNA remains in its interaction with the tracrRNA, forming a crRNA–tracrRNA duplex that is loaded into the Cas9 effector module (Chylinski et al., 2013). For that reason, a strong interaction between a tracrRNA and the repeat is a necessary requirement for crRNA maturation. Thus, we use RNA–RNA interaction prediction with the repeat sequence to screen for tracrRNA candidates. The repeat binding site on the tracrRNA is called anti-repeat and is located in the 5′-region. In order to perform such a search for a repeat: tracrRNA interaction, we first need to detect the CRISPR arrays to obtain the repeat candidates. With these candidates, we perform the coarse screening for the potential anti-repeat sequences. We then refine the set of anti-repeat candidates by using RNA–RNA interaction prediction to determine the repeat-anti-repeat duplex.
2.1.1. CRISPR array identification
Since the repeat sequence is required to perform the anti-repeat search, the initial step in order is to detect all potential CRISPR arrays in a genome. Here, CRISPRtracrRNA fully relies on CRISPRidentify (Mitrofanov et al., 2021) for CRISPR array detection since it can not only provide the detected CRISPR arrays but also utilizes Machine Learning approach to distinguish the true CRISPR arrays from repetitive genome structures that are similar to CRISPR arrays. In its final step, CRISPRidentify utilizes an extra-tree approach, which provides a certainty score (in fact a pseudo-class probability) for each array candidate. We use both Bona-fide (defined by a score higher than 0.8) and Possible candidates (defined by a score between 0.4 and 0.8) to form the candidate set for CRISPR arrays. If the user is only interested in the Bona-fide candidates they can use the corresponding input parameter. While the repeat sequence is extracted from the array, the certainty score is propagated to the last step of CRISPRtracrRNA where all the pieces of evidence are combined (see Section 3.3.1). On top of that CRISPRidentify utilizes CRISPRstrand (Alkhnbashi et al., 2014) to predict the array’s orientation. CRISPRstrand also provides a certainty score that gets also propagated to the last combination step.
2.1.2. Initial anti-repeat search
After the set of CRISPR arrays is determined, the initial search for anti-repeat sequences is possible. At the beginning of this step, the consensus sequence for each repeat is determined. Then, for each consensus repeat sequence, CRISPRtracrRNA utilizes BLAST (Altschul et al., 1990) to search for regions that show high similarity with the repeat sequence at hand. To avoid self-targeting within the CRISPR array, the hits within the CRISPR array regions are filtered out during this step. For the remaining set of candidates, the values for hit similarity and hit coverage are calculated. The set is then filtered again using thresholds for the listed values as well as the multiplication of both (i.e. square of the geometric mean). The threshold values can be set by the user. The default values used in the pipeline are 0.8 for both similarity and coverage, for the multiplication of similarity and coverage it is set to 0.7. This allows the approach to eliminate the large value of the false positive hits not related to the tracrRNA sequences. All three values represent the quality of the initial anti-repeat candidates. For each of them, all three values are also propagated to the last combination step.
2.1.3. Enhanced anti-repeat search
As the duplex formation between the tracrRNA and the repeat is based on RNA–RNA interaction, the BLAST search in the previous section can only determine initial candidates for repeat-anti-repeat duplexes. A more detailed investigation of the interaction between the candidate set of anti-repeat sequences obtained in the previous step and the corresponding repeat sequences using RNA–RNA interaction prediction tools is required. In the CRISPRtracrRNA pipeline, we utilize IntraRNA (Mann et al., 2017) to predict the most probable interaction. IntraRNA is used in the pipeline for two main reasons. First, it allows to determine the interaction site with greater precision than the previously used methods RNAhybrid and RNAcofold, as it takes accessibility of the interaction region and its structure into account. This step can therefore greatly improve the initial candidate for the anti-repeat region since the interaction between crRNA and tracrRNA is usually imperfect and often contains bulges, which can negatively influence the found hit regions if only the sequence similarity was taken into account. Second, it also provides the interaction energy as output, which indicates the strength of the interaction. Both the interaction interval as well as the interaction energy are then propagated to the last combination step.
2.2 Rho-independent termination search
A transcription terminator is a sequence that marks the end of a DNA operon during transcription. In prokaryotes two types of terminator sequences have been discovered: Rho-dependent terminators, where a large protein called Rho factor plays the crucial role in disrupting the mRNA–DNA–RNA polymerase transcriptional complex and Rho-independent terminators that form self-annealing hairpin structures, which results in the disruption of the mRNA–DNA–RNA polymease ternary complex. Bacterial small RNA transcripts are in general terminated by Rho-independent termination (Livny and Waldor, 2007). As the tracrRNA is a small RNA, it must also contain a rho-independent termination signal. In our CRISPRtracrRNA approach, we rely on the Erpin (Gautheret and Lambert, 2001) approach for the efficient search for the Rho-independent terminator sequence. In the pipeline, Erpin 5.5 is utilized with the default parameters. Erpin provides both the location of the predicted candidate, as well as a score. Both values are then used in the last combination step. The score is used directly as a piece of evidence for the presence of the terminator sequence. The interval location is used to check the consistency of all present evidence pieces.
2.3 Cas protein search
Since tracrRNA is used as a guide RNA in Type II CRISPR-Cas systems the presence of the Cas9 protein for Type II system and Cas12 protein for Type V system can be considered as a factor that is positively correlated with the found candidate being a true tracrRNA. Therefore, the search for cas genes is an important step in the CRISPRtracrRNA pipeline. In the CRISPRtracrRNA pipeline, we rely on CRISPRcasIdentifier version 1.1.0 with default parameters for the efficient search for cas genes. CRISPRcasIdentifier utilizes an ML approach for the robust identification and labeling of cas genes. Therefore, CRISPRcasIdentifier allows us to identify the locations of all found cas9/cas12 genes and determine the one closest to the tracrRNA candidate. The distance to the closest cas9/cas12 gene is negatively correlated with the probability of the candidate being a true tracrRNA since tracrRNAs tend to be distributed closely to their associated cas genes.
3 Results
3.1 Sequence structure model for Type II systems
The tracrRNA sequences in Type II systems have been closely studied. We took the available data of 41 identified tracrRNA sequences from the (Briner et al., 2014) publication. That study divided the dataset of tracrRNAs into three groups according to the structural similarity. In our study, we took the given sequences and removed the identified anti-repeat part. This step is necessary since the anti-repeat part of the tracrRNA strongly interacts with the corresponding crRNA in vivo but can affect the prediction of tracrRNA secondary structure with wrong interactions in silico when the structure is predicted without the crRNA attached. The truncated sequences are then subjected to the GraphClust2 (Miladi et al., 2019) pipeline via the European galaxy server. The GraphClust2 approach takes into account both sequence and structure similarities when forming clusters, which suited our approach perfectly. For each cluster, a CM is built using Infernal to screen for missing members of a cluster. These CMs constitute a sequence-structure model of that cluster, which can be used to screen genomes for further members of that model.
The GraphClust2 approach showed consistent results with the previous sequence-based clustering of the whole tracrRNA (consisting of anti-repeat and 3′-tail) as the same three distinctive clusters were determined (see Supplementary Fig. S2). We then enriched the dataset by adding 77 additional tracrRNA sequences from the (Gasiunas et al., 2020) publication. We used an iterative approach adding new sequences one by one, to be consistent with the original clustering. Thus, if the newly added sequence was able to fit the clustering it was added and discarded if it would break the current clustering scheme. In this way, we ensure that the sequence-structure models are preserved and refined to capture additional members. With this approach, we were able to preserve the clusters structures and obtain the model with 83 sequences.
We then further investigated the obtained classes. First, we built the phylogenetic tree of all combined tracrRNA sequences using MAFFT (Katoh and Standley, 2013) and marked the sequences that belong to the model according to the corresponding cluster (see Fig. 2A). The clusters showed consistent distribution over the tree as the cluster elements tend to stay close together.
Fig. 2.
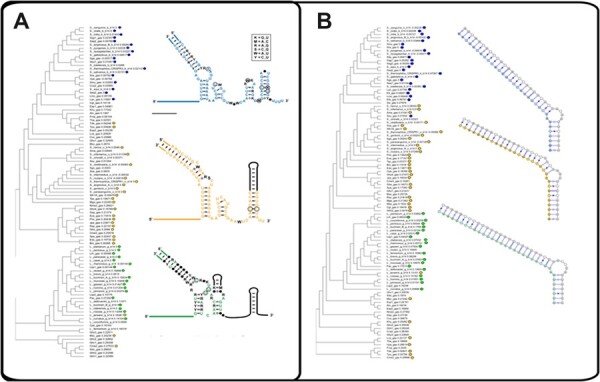
(A) Phylogenetic tree of the 3'-tail of experimentally validated tracrRNA used in the GraphClust2 analysis. The drawings of the three consensus structures were taken from the publication (Briner et al., 2014), license number 5287080399685, as they show additional information. They agree with the consensus models as predicted by the GraphClust2 pipeline (see Supplementary Fig. S1). The phylogenetic tree is based on sequence distance as GraphClust2 does not produce a tree but only clusters. The sequences that were finally used for our three CMs are indicated with blue, yellow and green dots. (B) MAFFT-generated phylogenetic tree of the anti-repeat part of the experimentally validated tracrRNA. It shows that the independent clustering of the anti-repeats gives consistent results, as nearly the same three main clusters are generated.
Similarly, we built the phylogenetic tree of the anti-repeat part of the tracrRNA sequences. Again, we could see the consistent results, the model clusters also form dense clusters on the formed phylogenetic tree. For each cluster, we used PETcofold (Seemann et al., 2011) to predict the consensus interaction between anti-repeat and repeat sequences (see Fig. 2B). The obtained interactions between anti-repeat and repeats were also consistent with the results from Briner et al. (2014).
For comparison with existing approaches, we also created a sequence model for the 83 tracrRNAs. The set of tracrRNAs was initially clustered with CD-hit (Li and Godzik, 2006). The corresponding clusters were then subjected to MAFFT to obtain the consensus cluster secondary structure. Finally, the HMM-build was used to create the model. As shown in detail in Section 3.5, these sequence models capture different properties of tracrRNAs.
In the CRISPRtracrRNA pipeline, we rely on Infernal to compare and score the candidate tracrRNA sequence with the pretrained model. In more detail, we extracted the three CMs generated by GraphClust2 and used Infernal’s cmscan, which not only produces the E-value of the hit and the hit score but also determines the hit interval. This approach allows CRISPRtracrRNA to also robustly predict the location of the tail part of the tracrRNA.
3.2 Sequence structure models for Type V systems
So far existing work on screening tracrRNA concentrated on Type II systems. To screen for tracrRNAs in Type V systems, we first performed computational searches for known Cas12k proteins in order to find CAST-associated DNA regions. We searched in these CAST regions for known components, like the transposon genes tnsB, tnsC and tniQ, CRISPR arrays and transposon insertion elements (see Supplementary Fig. S3). While doing so we also searched in the DNA regions for similarities to the known tracrRNAs from Anabaena sp. PCC7120 and Scytonema hofmanni (Reimann et al., 2020; Strecker et al., 2019), whose expression could be verified by small RNA-sequencing. We could identify multiple candidates for Type V-K tracrRNA, always downstream of the cas12k gene and if present, upstream of the CRISPR array. After that, these candidates were clustered by sequence similarity by CD-hit to align the sequences inside their respective clusters (Li and Godzik, 2006). To increase the significance and avoid wrong predictions of interaction between the anti-repeat and other areas of the tracrRNA, the sequences were aligned together with the upstream region until the second repeat of the CRISPR array. These alignments were then used for predicting conserved secondary structures, which was performed using the webtool shape studio from the University of Bielefeld (Janssen and Giegerich, 2015). When comparing the most prominent potential structures, all clusters showed four main loop areas inside the suspected tracrRNA areas. We could also detect three very conserved regions across all tracrRNA candidates. The first one was located upstream of the tracrRNAs and at least in Anabaena sp. PCC 7120 overlaps with the tracrRNA promoter (C1). The second one is in the center of the third main loop (C2) and the last one contains the fourth main loop as well as the following anti-repeat (AR) sequence (C3).
We compared these findings to the cryo-electron microscopic structures of sgRNAs from S. hofmanni by (Querques et al., 2021) and (Xiao et al., 2021) and looked for common or similar features in all three models. Our previously predicted four main loops were comparable to the eight stem-loop (S1–8) model from Xiao et al. (2021). The first and last main loops were overlapping with stem-loops S1 and S8. The second main loop included the stem-loops S2 and S3, while the third main loop consisted of S4, S5, S6 and S7. The conserved sequence inside main loop three overlaps with part of a pseudoknot and S5. The model of (Querques et al., 2021) showed a triplex formation of the repeat binding area (AR1) that corresponds to two highly conserved short sequences (5′-TTT-3′ and 5′-CTTTC-3′) inside main Loop 2. The corresponding binding region on the repeat is highly conserved as well, which suggests a conserved anti-repeat binding region in this location. Out of this comparison, we identified local palindromic structures and looked for sequence similarities in order to map the eight stem-loop model onto the individual tracrRNAs. The already identified conserved areas (C1–3) and the less specific main loop model were used as fix points for this identification. We could locate S2, S3, S4 and S8 stem-loops as well as both Anti-Repeat sequences in all tracrRNA candidates, while S1, S5, S6 and S7 could be identified in most candidates (99%, 92%, 90% and 95%).
In order to obtain the best sequence-structure model for screening tracrRNAs in the Type V systems, we again utilized the GrapthClust2 approach. Unlike in the sequence/structure model generation for Type II systems, where we had experimentally verified tracrRNAs and known structural classes, we needed to determine a fitting structural model by testing the performance of a model in a different fashion. We took the available 91 tracrRNA candidate sequences and split them into the pairs of train validation set in a 5-fold fashion. We then trained a model on the training set and checked the recall potential on the validation set. For each train test split, we could see that the dataset is very consistent. Namely, each sequence from the validation set consistently got 16–18 hits from different sequences in the training dataset for each of the five runs (see Supplementary Fig. S4).
3.3 Usage of CRISPRtracrRNA
3.3.1. Combining evidence and ranking candidates
The ultimate goal of the CRISPRtracrRNA pipeline is to detect all the potential tracrRNA candidates in the given genome. For user convenience, such a list should be filtered and sorted according to the trust level for each candidate. For that purpose, each tracrRNA candidate is transformed into an n-dimensional feature vector.
The first five dimensions of this vector are related to the anti-repeat part of the tracrRNA. The first value is the certainty score of the corresponding CRISPR array obtained by the CRISPRidentify. The next two values are the hit similarity and coverage between the anti-repeat part of the tracrRNA and the corresponding repeat. We also introduced the multiplication (i.e. square of the geometric mean) of these two values as a balanced estimation of the overall similarity between the repeat and anti-repeat parts. The last value of the anti-repeat component of the vector is the interaction energy of the repeat-anti-repeat interaction.
The next value in the vector representation of the candidate is the sequence/structure similarity score of the candidate to the pre-trained model.
The rest of the values are related support information and consistency scores between observations. The support scoring consists of the three values: The first support evidence score is the certainty score of the orientation of the CRISPR array predicted with CRISPRstrand. The second support evidence score is the evidence score obtained from the terminator sequence search. The last support score is the score of the Cas9 protein.
On top of the direct support scores, we also introduced the consistency scores. Consistency scores were created to indicate that the pieces of evidence we consider to be important in the tracrRNA identification are internally consistent. All the consistency scores are binary, i.e. the value of each score can only take values 0 and 1. Value 1 means that the predictions are consistent and the value 0 means that they contradict each other. The first score indicates the consistency between the interval that represents the anti-repeat and the part of the tracrRNA that forms a secondary structure accepted by one of the CMs (using Infernal’s cmscan). In our approach, we call the results consistent if the overlap and or gap between the predicted intervals does not exceed 10 nucleotides. The second consistency value is related to the secondary structure part of the tracrRNA and the terminator sequence. Similar to the previous criterion, we call the observations consistent if the overlap or the gap does not exceed the value of 10 nucleotides. The last consistency value indicates the consistency of the predicted strand. If the terminator sequence and the strand prediction of the CRISPR array lead to the same strand for the tracrRNA candidate the observations are called consistent.
The user is given the option of ranking the identified candidates. The hierarchy of the candidates can be achieved by providing the weights (float values between 0 and 1) to the corresponding evidence factor. That is, if the user is interested in the tracrRNA candidates where the terminator sequence was identified by Erpin and the tail part of the tracrRNA candidate was identified with the provided CMs, they can put 1 for these values. And if they expect a low similarity between repeat and anti-repeat they can put 0 as the corresponding weight value. The default values are set to 1 for all anti-repeat evidence-related values, identification of the tail with the model and presence of the anti-repeat. The rest of the values are set to 0. This allows the researcher to pick candidates that fit prior information and are most suitable for further investigations.
3.3.2. Different ways for screening tracrRNAs with CRISPRtracrRNA
Our CRISPRtracrRNA tool incorporates two different approaches of searching the tracrRNA candidates. The first approach is based on the assumption that there should be a clear anti-repeat tail present in a tracrRNA candidate. When choosing this mode, CRISPRtracrRNA first searches for the CRISPR arrays, extracts the corresponding repeat sequences and performs a search for anti-repeat sequences (see Material and Methods). The initial set of tracrRNA candidates is formed based on the anti-repeat sequences. Subsequently, each of the candidates is subjected to the sequence/structure similarity search with the pretrained CMs.
The second approach of running CRISPRtracrRNA concentrates on the structural models we defined for tracrRNAs. This mode starts with the sequence-structure similarity search between the provided sequence and the trained models using cmscan. This mode is especially suitable for large datasets since it is less computationally demanding, or in cases where the anti-repeat part of the certain tracrRNA sequences is relatively small and thus will not produce a high complementarity score with the corresponding repeat sequence. In such a scenario the naive search for anti-repeat candidates based solely on the sequence similarity would naturally fail as it would report an overwhelming number of false-positive hits. In order to overcome this challenge, CRISPRtracrRNA utilizes the sequence structure search first in this mode. Then the user has an option to search for anti-repeat sequences in the flanking regions of the identified hits.
Both of the approaches form the final candidate set by complementing the candidate set with the support information see Support Information.
CRISPRtracrRNA is implemented in python and available as a standalone command line interface. It can be downloaded from our GitHub repository (https://github.com/BackofenLab/CRISPRtracrRNA). The initial setup of the CRISPRtracrRNA is done via creation of a conda environment and can be done with a single command. All the components of the CRISPRtracrRNA are either included in the GitHub repository or integrated in the conda environment which eliminates the need for additional download or setup steps. The algorithm can be executed with the command line. The parameters are fully described in the help option as well as in the GitHub page.
The algorithm output is written in an easy-to-read CSV file. The tracrRNA candidates are ranked according to the corresponding certainty score and the important factors specified by the user. The user can find the flanking regions for each candidate as well as the candidate location. On top of that, each evidence factor is listed in the corresponding column.
The user is allowed to assign the importance weights to the evidence factors of their desire or use the default ones (See Table S1 in Supplementary Materials for the default weight distribution).
3.4 Comprehensive annotation of tracrRNA
We utilized CRISPRtracrRNA to identify potential tracrRNA candidates on the comprehensive dataset of Type II organisms. During the first step of our research, we ran the ‘sequence/structure mode’ search on the given dataset (see previous Section 3.3.2). With this approach, we were able to identify potential tracrRNA sequences in 18 227 genomes. We then ran the ‘sequence’ based search mode and were able to identify tracrRNA in 12 651 organisms with the overlap with the sequence/structure model in 11 797 of the candidates. The rest 22 335 were subjected to the CRISPRtracrRNA with the anti-repeat mode. The search yielded 71 353 potential candidates from 11 882 organisms.
We also conducted the taxonomy analysis of the dataset checking the performance of our models. For that purpose, we downloaded the GenBank files for the corresponding genomes and extracted the origin taxonomy.
We then checked the distribution of the organisms by phylum. Three major phyla in the dataset are Firmicutes, Proteobacteria and Bacteroidetes with a cumulative sum of 38 983 elements.
We then investigated how well the sequence/structure model and the sequence-only model agree on the corresponding phyla. For Firmicutes the models showed the same recall ability and both were able to detect potential tracrRNA in 10 662 candidates out of 11 602. In Proteobacteria we were able to see the superior performance of the sequence/structure model. It was able to provide 5113 candidates while the sequence-only model detected six candidates and only in Gammaproteobacteria. The most interesting results were obtained in the Bacteroidetes phylum where sequence/structure and sequence-only models produced large sets of unique predictions for each of the classes: Bacteroidia, Flavobacteria, Chitinophagia, Sphingobacteriia and Cytophagia. The sequence/structure model reported tracrRNA candidates in 235 unique organisms while the sequence-only model found candidates in 442 unique organisms. In 528 organisms candidates were found both by sequence/structure and sequence-only models (see Fig. 3). To summarize, we found tracrRNA candidates in 17 443 organisms. In 514 of them, the candidates were found only with the sequence-based model while in 5683 the candidates were reported uniquely by the sequence/structure model. In 11 246 organisms, the candidates were identified with both models.
Fig. 3.
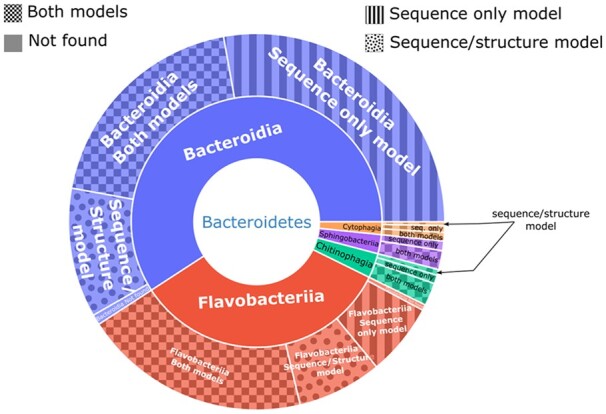
The distribution of organisms of the Bacteroidetes phylum by Class (inner circle of the pie chart) and by model coverage (outer circle of the pie chart). It can be seen that sequence/structure and sequence-only models found unique candidates for the vast majority of the represented classes.
3.5 Comparison with the existing approaches on Cas9-tracrRNA (Type II)
Despite certain similarities, our approach significantly differs from the published methods. In their approach, Dooley et al. (2021) relied on the anti-repeats and the presence of the RTS sequences to identify the tracrRNA boundaries. In contrast, CRISPRtracrRNA represents a multiple evidence level approach and utilizes ML models that were trained on the verified tracrRNA sequences. On top of that instead of Minced and PilerCR for the CRISPR array identification, CRISPRtracrRNA relies on CRISPRidentify, which is a Machine Learning based tool that showed a robust approach for the CRISPR array detection. Second, we used CRISPRcasIdentifier, which also utilizes a state-of-the-art ML approach for cas gene detection. Finally, in the CRISPRtracrRNA approach, we utilize HMM search based on both sequence and structure similarities. Our model targets the tail of the tracrRNA candidate while the model by Dooley et al. tries to predict the whole tracrRNA sequence.
For that reason, we decided to compare how specific the models are in their prediction. In order to do that we first calibrated our sequence/structure model for Type II systems with Infernal cmcalibrate. This step sets exponential tail parameters for E-value determination by generating random sequences, searching them with the CM and collecting the scores of the resulting hits. The model from Dooley et al. did not need an additional calibration since that step was already done. We then submitted the tracrRNA tail sequences with the known structure (see Sequence structure model for Type II systems) with Infernal cmscan.
When using the truncated tracrRNA tail sequences, we could immediately see that our CRISPRtracrRNA model is more specific and shows much lower E-values for the identified hits (see Fig. 4). As the Dooley et al. CM models cover the whole tracrRNA and our CM models on the tracrRNA tail sequence, we cannot exclude that some of the difference is due the fact that the sequence was truncated for the Dooley et al. CM models, but complete sequences for our models. We thus further investigated the model by Dooley et al. and submitted the anti-repeats sequences, to check whether they constitute a significant part of the information captured by the Dooley et al. CM models. We can see that that model is also consistently showing strong hits on the anti-repeat part, showing that a large portion of the model describes anti-repeat properties as the Dooley et al. CM model was trained on the complete tracrRNA sequences. While this approach improves the specificity of the Dooley et al. CM model, modeling the anti-repeat with CM models has also negative effects as it (i) ignores the direct evidence of repeat/anti-repeat interactions in the genome, and (ii) might tend to overfit to known repeat sequences. In contrast to this approach, our CRISPRtracrRNA predicts the anti-repeat part and the tail part separately, which can improve the false positive rate of the reported candidates.
Fig. 4.
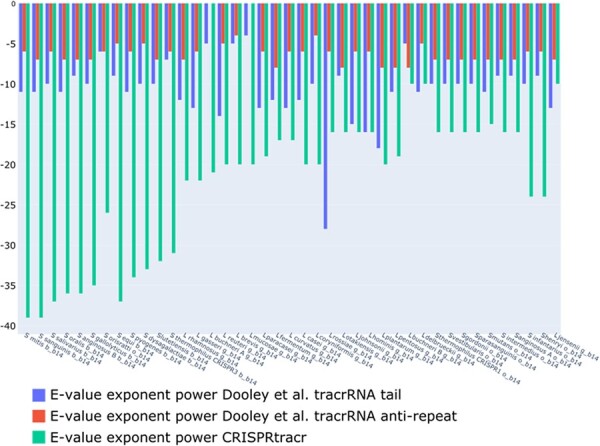
Comparison of the E-value exponents between CMs of Dooley et al. and CRISPRtracrRNA on the Type II tracrRNA sequences. CRISPRtracrRNA shows much higher specificity. The model by Dooley et al. also heavily relies on the anti-repeat part of the tracrRNA candidate while in CRISPRtracrRNA the CM is used to search for the tail part.
4 Conclusion
We developed CRISPRtracrRNA: an end-to-end standalone bioinformatic tool for tracrRNA predictions. In order to robustly predict the new tracrRNA candidates, CRISPRtracrRNA combines different sources of evidence. It separately performs a search for the anti-repeat and tail of the tracrRNA, detects the signal of the terminator sequence and complements the result with the information about the cas genes. For that reason, CRISPRtracrRNA utilizes Machine Learning based start of the art tools such as CRISPRidentify and CRISPRcasIdentier. For the tracrRNA tail detection, CRISPRtracrRNA uses the sequence-structure similarity search based on Infernal and data-driven pre-trained models. In our approach, we obtained models not only for Type II systems but also, for the first time, for CRISPR Type V systems. In order to build the model for Type V systems, we constructed a novel dataset of tracrRNA candidates.
We then compared the specificity of our Type II model with the existing CMs. We noticed that our model is more specific and in contrast to the other method targets only the structural part of the tracrRNA. Finally, we compared the performance of our sequence/structure and sequence-only models on the comprehensive dataset of Type II organisms and saw that despite the large overlap (11 246 organisms the candidates were identified with both models) both sequence and sequence/structure models also reported unique candidates (514 were uniquely reported by sequence model and 5683 by sequence/structure model), thus capturing different properties of tracrRNA. As tracrRNA is a small, structured RNA, we believe that the sequence/structure models are more appropriate to determine tracrRNA candidates.
CRISPRtracrRNA is implemented in python and can be easily set up using the conda environment. It can be executed in two modes: the complete evidence search, where all the evidence factors are computed and the fast search where the candidates are formed based on the sequence/structure model. The latter is suitable for large datasets. We plan to integrate CRISPRtracrRNA into the CRISPRloci (Alkhnbashi et al., 2021) web server in order to provide users with a more in-depth CRISPR-Cas analysis.
Supplementary Material
Acknowledgements
The authors also thank Milad Miladi for his help with the GraphClust2 tool.
Funding
This paper was published as part of a special issue financially supported by ECCB2022. This work was supported by German Research Foundation (DFG) [BA 2168/23-1/2 and HE 2544/14-2 SPP 2141]; Much more than Defence: the Multiple Functions and Facets of CRISPR–Cas; Baden-Wuerttemberg Ministry of Science, Research and Art; University of Freiburg. Funding for open access charge. Probabilistic Structures in Evolution [BA 2168/13-1 SPP 1590]. The authors acknowledge the support of the Freiburg Galaxy Team: Rolf Backofen and Björn Grüning, Bioinformatics, University of Freiburg (Germany) funded by the Collaborative Research Centre 992 Medical Epigenetics (DFG grant SFB 992/1 2012) and the German Federal Ministry of Education and Research BMBF grant 031 A538A de. NBI-RBC.
Conflict of Interest: none declared.
Contributor Information
Alexander Mitrofanov, Chair of Bioinformatics, University of Freiburg, Freiburg, Germany.
Marcus Ziemann, Faculty of Biology, Genetics and Experimental Bioinformatics, University of Freiburg, Freiburg, Germany.
Omer S Alkhnbashi, Information and Computer Science Department, King Fahd University of Petroleum and Minerals, Dhahran, Saudi Arabia.
Wolfgang R Hess, Faculty of Biology, Genetics and Experimental Bioinformatics, University of Freiburg, Freiburg, Germany.
Rolf Backofen, Chair of Bioinformatics, University of Freiburg, Freiburg, Germany; Signalling Research Centres BIOSS and CIBSS, University of Freiburg, Freiburg, Germany.
Data Availability
Finally, our method and the corresponding materials are available as an open source tool on GitHub (https://github.com/BackofenLab/CRISPRtracrRNA).
References
- Alkhnbashi O.S. et al. (2021) CRISPRloci: comprehensive and accurate annotation of CRISPR–cas systems. Nucleic Acids Res., 49, W125–W130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhnbashi O.S. et al. (2014) CRISPRstrand: predicting repeat orientations to determine the crRNA-encoding strand at CRISPR loci. Bioinformatics, 30, i489–i496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F. et al. (1990) Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Anders C. et al. (2014) Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature, 513, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briner A.E. et al. (2014) Guide RNA functional modules direct Cas9 activity and orthogonality. Mol. Cell., 56, 333–339. [DOI] [PubMed] [Google Scholar]
- Chylinski K. et al. (2013) The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol., 10, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dooley S.K. et al. (2021) Identification and evolution of Cas9 tracrRNAs. Crispr J., 4, 438–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfara I. et al. (2014) Phylogeny of Cas9 determines functional exchangeability of dual-RNA and Cas9 among orthologous type II CRISPR-Cas systems. Nucleic Acids Res., 42, 2577–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas G. et al. (2020) A catalogue of biochemically diverse CRISPR-Cas9 orthologs. Nat. Commun., 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautheret D., Lambert A. (2001) Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol., 313, 1003–1011. [DOI] [PubMed] [Google Scholar]
- Huai C. et al. (2017) Structural insights into DNA cleavage activation of CRISPR-Cas9 system. Nat. Commun., 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen S., Giegerich R. (2015) The RNA shapes studio. Bioinformatics, 31, 423–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F. et al. (2015) A Cas9–guide RNA complex preorganized for target DNA recognition. Science, 348, 1477–1481. [DOI] [PubMed] [Google Scholar]
- Jinek M. et al. (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science, 343, 1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol., 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Godzik A. (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics, 22, 1658–1659. [DOI] [PubMed] [Google Scholar]
- Liao C., Beisel C.L. (2021) The tracrRNA in CRISPR biology and technologies. Annu. Rev. Genet., 55, 161–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livny J., Waldor M.K. (2007) Identification of small RNAs in diverse bacterial species. Curr. Opin. Microbiol., 10, 96–101. [DOI] [PubMed] [Google Scholar]
- Makarova K.S. et al. (2020) Evolutionary classification of CRISPR–cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol., 18, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M. et al. (2017) IntaRNA 2.0: enhanced and customizable prediction of RNA–RNA interactions. Nucleic Acids Res., 45, W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miladi M. et al. (2019) GraphClust2: annotation and discovery of structured RNAs with scalable and accessible integrative clustering. GigaScience, 8, giz150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrofanov A. et al. (2021) CRISPRidentify: identification of CRISPR arrays using machine learning approach. Nucleic Acids Res., 49, e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilha V.A. et al. (2020) CRISPRcasIdentifier: machine learning for accurate identification and classification of CRISPR-Cas systems. GigaScience, 9, giaa062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.-U. et al. (2021) Structural basis for target site selection in RNA-guided DNA transposition systems. Science, 373, 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querques I. et al. (2021) Target site selection and remodelling by type V CRISPR-transposon systems. Nature, 599, 497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann V. et al. (2020) Specificities and functional coordination between the two Cas6 maturation endonucleases in anabaena sp. PCC 7120 assign orphan CRISPR arrays to three groups. RNA Biol., 17, 1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M. et al. (2021) Dual modes of CRISPR-associated transposon homing. Cell, 184, 2441–2453.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann S.E. et al. (2011) PETcofold: predicting conserved interactions and structures of two multiple alignments of RNA sequences. Bioinformatics, 27, 211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J. et al. (2019) RNA-guided DNA insertion with CRISPR-associated transposases. Science, 365, 48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao R. et al. (2021) Structural basis of target DNA recognition by CRISPR-Cas12k for RNA-guided DNA transposition. Mol. Cell., 81, 4457–4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Finally, our method and the corresponding materials are available as an open source tool on GitHub (https://github.com/BackofenLab/CRISPRtracrRNA).