

Abstract
Background & aims
Excessive inflammatory responses and oxidative stress are considered the main characteristics of inflammatory bowel disease (IBD). Endogenous hydrogen sulfide (H2S) has been reported to show anti-inflammatory activity in IBD. The main aim of this study was to explore the role of 3-mercaptopyruvate sulfurtransferase (MPST), a key enzyme that regulates endogenous H2S biosynthesis, in IBD.
Methods
Colonic MPST expression was evaluated in mice and patients with IBD. Various approaches were used to explore the concrete mechanism underlying MPST regulation of the progression of colitis through in vivo and in vitro models.
Results
MPST expression was markedly decreased in colonic samples from patients with ulcerative colitis (UC) or Crohn's disease (CD) and from mice treated with DSS. MPST deficiency significantly aggravated the symptoms of murine colitis, exacerbated inflammatory responses and apoptosis, and inhibited epithelium stem cell-derived organoid formation in an H2S-independent manner. Consistently, when HT29 cells were treated with TNF-α, inhibition of MPST significantly increased the expression of proinflammatory cytokines, the amount of ROS and the prevalence of apoptosis, whereas overexpression of MPST markedly improved these effects. RNA-seq analysis showed that MPST might play a role in regulating apoptosis through AKT signaling. Mechanistically, MPST directly interacted with AKT and reduced the phosphorylation of AKT. Additionally, MPST expression was positively correlated with AKT expression in human IBD samples. In addition, overexpression of AKT rescued IEC apoptosis caused by MPST deficiency, while inhibition of AKT significantly aggravated it.
Conclusions
MPST protects the intestines from inflammation most likely by regulating the AKT/apoptosis axis in IECs. Our results may provide a novel therapeutic strategy for the treatment of colitis.
Keywords: 3-Mercaptopyruvate sulfurtransferase, Epithelial cell, Hydrogen sulfide, Anti-Apoptotic protein kinase
Abbreviations: IEC, intestinal epithelial cell
Graphical abstract
Abbreviations
- Akt
anti-apoptotic protein kinase
- CD
Crohn's disease
- CBS
cystathionine-β-synthase
- CSE
cystathionine -γ-synthase
- DAI
disease activity index
- DSS
dextran sulfate sodium
- H2S
hydrogen sulfide
- IBD
inflammatory bowel disease
- IEC
intestinal epithelial cell
- IL
interleukin
- MPST
3-mercaptopyruvate sulfurtransferase
- ROS
reactive oxygen species
- TNBS
2,4,6-trinitrobenzenesulfonic acid
- TNF
tumor necrosis factor
- UC
ulcerative colitis
- WT
wildtype
1. Introduction
Inflammatory bowel disease (IBD) is defined as a chronic inflammatory condition characterized by a relapsing and intermittent course comprising ulcerative colitis (UC) and Crohn's disease (CD) [1]. Chronic intestinal inflammation can lead to tumorigenesis, and a number of patients with IBD are at high risk of developing colorectal cancer [2,3]. Despite a continuous increase in the incidence of IBD worldwide, the complex pathogenesis remains incompletely understood [4]. Increasing efforts are being made to better understand the various aspects of this disease and to identify safer and more effective drugs, while satisfactory treatment is still lacking [5,6].
Under physiological conditions, the intestinal epithelium forms a physical and biochemical barrier between luminal bacteria and mucosal immune cells. Abnormal regulation of epithelial cell proliferation and cell death is considered to exacerbate intestinal damage [7]. Overactivated intestinal epithelial cell (IEC) apoptosis is frequently found in both UC and CD, leading to disruptions in intestinal barrier integrity that may allow the infiltration of bacteria from the lumen into the bowel wall and trigger an inflammatory cascade, including proinflammatory cytokine production. In murine models, bringing aggravated intestinal apoptosis to normal levels may be a potential therapeutic strategy for managing colitis [8,9]. Although most studies have linked apoptotic cell death to the pathogenesis of IBD, the physiological and pathophysiological roles of apoptosis in the gut have not been fully elucidated.
It has been well documented that exogenous hydrogen sulfide (H2S) and substrates that result in hydrogen sulfide production are anti-inflammatory and can protect mice from DSS colitis [[10], [11], [12]]. 3-Mercaptopyruvate sulfurtransferase (MPST), a key enzyme that regulates endogenous H2S biosynthesis, is a widely expressed protein in human tissues, and is more restricted in terms of tissue distribution. The highest MPST protein levels have been detected in the liver, kidney, and large intestine. Furthermore, MPST is the major source of H2S production in the rat colon in health and during colitis [13]. MPST activity is also known to be involved in tRNA thiolation, protein urmylation and cyanide detoxification [14]. Tissue-specific changes in MPST expression correlate with aging and the development of metabolic disease [14,15]. To date, whether MPST is involved in the regulation of inflammation and in the pathogenesis of UC remains unknown.
Our findings identified the protective role of MPST in intestinal homeostasis. In this study, we first reported that MPST is mainly expressed by IECs, where it protects the intestine from inflammation by regulating apoptosis via the AKT pathway but not the H2S-dependent pathway. These results enable the identification of a novel pathway of intestinal inflammation that may be targeted to treat patients with IBD.
2. Material and methods
2.1. Tissue samples
Human paraffin-embedded colon sections from IBD patients and healthy control subjects were obtained from the Department of Gastroenterology, The First Affiliated Hospital, Zhejiang University. The use of colon tissue samples from patients in this study was approved by the Ethics Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University, and the studies abided by the Declaration of Helsinki principles.
2.2. Mice
Heterozygous MPST-deficient (MPST+/−) mice on a C57BL/6 background were generated through frame shift mutation by the TAL effector nuclease system by Beijing View-Solid Biotechnology (Beijing, China) as previously described [16]. Animals, which were maintained under specific pathogen-free (SPF) conditions, were bred and maintained at 23 ± 2 °C on a 12 h light/12 h dark cycle at the Medical Science Institution of Zhejiang Province (Hangzhou, China). All animals were given mouse chow and water ad libitum (5 mice per cage). Six-to 8-week-old male mice weighing 20–22 g were used for all experiments. All animal experiments were performed according to guidelines approved by the Animal Care and Use Committee of the First Affiliated Hospital, College of Medicine, Zhejiang University.
2.3. Induction of DSS-induced and TNBS-induced colitis and treatment
For colitis induction, mice were administered 3.5% DSS (MP Biomedicals, Solon, OH) through the drinking water for 6 or 7 days. For TNBS treatment, mice were presensitized by application of 1% TNBS (Sigma‒Aldrich, St. Louis, MO) to the skin for 1 week. After fasting for 24 h, the mice received 100 μl of 2.5% TNBS by rectal injection with a 1-mL syringe fitted with a catheter, as described by Stefan [17]. To elevate H2S level, NaSH was administered by intraperitoneal injection at a dosage of 4 mg/kg every day along with DSS administration.
2.4. Molecular analysis from publicly available datasets from IBD patients
The data used is from GEO database (GSE59071) (https://www.ncbi.nlm.nih.gov/geo/), including 97 UC patients, 8 CD patients and 11 controls. The box plot is implemented by the R (version 4.1.1) software package ggplot2; the correlation analysis is conducted by the R (version 4.1.1) software package ggstatsplot.
2.5. Epithelial isolation and dissociation
IECs were isolated as previously reported [18]. Briefly, colon tissue from the rectum to the cecum (approximately 6 cm) was excised from control mice and DSS-treated mice, opened, cut longitudinally and washed in cold Dulbecco's phosphate-buffered saline (DPBS). The tissue was first placed in ice-cold dissociation reagent #1 (47 mL of DPBS, 3 mL of 0.5 M EDTA (Sigma), 75 μL of 1 M DTT) for 30 min, then in reagent #2 (47 mL of DPBS, 3 mL of 0.5 M EDTA) at 37 °C for 10 min, and shaken for 30 s to release the epithelium from the basement membrane. After being removed from the remaining intestinal tissue, the cells were washed in 10 mL DPBS with 10% FBS, dissociated with 8 mg dispase for 10 min at 37 °C in a water bath and stopped by adding 10% FBS and 50 μL of 10 mg/mL DNase to the cell solution. The cells were then sifted through 40-μm filters and used for determining IEC MPST protein expression.
2.6. Organoid culture
Mouse colon stem cells were cultured using IntestiCult Organoid Growth Media according to the manufacturer's instructions (06005, STEMCELL Technologies) [19]. Small intestines 20 cm in length nearly to the stomach were removed from untreated WT,MPST+/- and MPST−/− mice and rinsed with cold PBS. The colon was cut into 2 mm segments and washed 15 times with cold PBS. Colonic segments were incubated in Gentle Cell Dissociation Reagent (07174, STEMCELL Technologies) and rotated at 350 g for 15 min at room temperature, followed by resuspension in 0.1% BSA (A6003, Sigma) in PBS. Dissociated colonic crypts were filtered through 40μm strainers. Dissociated colonic crypts were resuspended in DMEM/F12 medium with 15 mM HEPES (36254, STEMCELL Technologies), counted and resuspended in IntestiCult Organoid Growth Media and Matrigel (356230, Corning) in a 1:1 ratio. Cells were plated in 24-well culture plates (3738, Corning). IntestiCult Organoid Growth Media was added to the cell culture plates to immerse the matrix composed of IntestiCult Organoid Growth Media and Matrigel. The culture medium was changed every other day, and the organoids were observed daily. After 5–7 days, the organoids were stimulated with 100 ng/mL mouse recombinant TNF-α (PeproTech, China) for 24 h.
2.7. Determination of DAI scores
DAI scores were calculated as previously described [20]. Body weight, bleeding and stool consistency were monitored every day during the colitis experiments. All scores were relative to the scores on Day 0, which were set to 0. For body weight, no loss was scored as 0, 1–5% weight loss was scored as 1, 6–10% weight loss was scored as 2, 11–20% weight loss was scored as 3, and higher than 20% was scored as 4. Bleeding scores were determined as follows: 0, no blood as examined by Hemoccult (Beckman Coulter) analysis; 2, positive Hemoccult test; and 4, gross bleeding. For stool consistency, 0 indicated well-formed stool pellets, 2 indicated pasty and semiformed stools that did not adhere to the anus, and 4 indicated liquid stools that adhered to the anus.
2.8. Analysis of histological scores
Paraffin-embedded sections (4 mm thickness) were subjected to hematoxylin and eosin (H&E) staining for histological analysis. Histological scoring was performed as previously described [21]. Briefly, the scores for inflammatory cell infiltration (score 0–3) and tissue damage (score 0–3) were assessed. For inflammatory cell infiltration, the presence of occasional inflammatory cells in the lamina propria was scored as 0, the presence of increased numbers of inflammatory cells in the lamina propria was scored as 1, a confluence of inflammatory cells extending into the submucosa was scored as 2, and transmural extension of the infiltrate was scored as 3. For tissue damage, no mucosal damage was scored as 0, the presence of lymphoepithelial lesions was scored as 1, the presence of surface mucosal erosion or focal ulceration was scored as 2, and extensive mucosal damage with extension into the deeper structures of the bowel wall was scored as 3. The combined histological score ranged from 0 (no changes) to 6 (extensive cell infiltration and tissue damage).
2.9. Cell culture
HT29 cells and HEK293T cells were purchased from the Chinese Academy of Sciences (Shanghai, China). To establish a cell apoptosis model, HT29 cells were treated with 100 ng/mL TNF-α for 24 h. HT29 cells were transfected with MPST-specific siRNA (siMPST forward: 5′-GCCAUCUGUUCCAGGAGAATT-3'; reverse: 5′-UUCUCCUGGAACAGAUGGCTT-3′) or corresponding scrambled siRNA as a negative control (forward: 5′-UUCUCCGAACGUGUCACGUTT-3′, reverse: 5′-ACGUGACACGUUCGGAGAATT-3′) using Lipofectamine 3000 (Invitrogen, St Louis, MO). An overexpression plasmid containing the full-length MPST DNA sequence was transfected into HT29 cells using Lipofectamine 3000 according to the manufacturer's instructions. Cells were transfected with siRNA or plasmid DNA for 24 h and then incubated for another 24 h with TNF-α before harvesting. To elevate the H2S level, NaSH (1 mM, Sigma) or GYY4137 (400 μM, Sigma) was added to the culture medium. To inhibit PP2A enzymatic activity, LB100 (1 μM, Topscience) was added to the culture medium.
2.10. Bone marrow chimeras
Bone marrow chimeras were established as previously reported [22]. Briefly, WT recipient mice received a sublethal dose of γ-ray irradiation (7.5 Gy) to kill the bone marrow cells, and at 6 h post-irradiation, the recipients received 100 μl of fresh MPST+/+, MPST+/- or MPST−/− bone marrow cells at a concentration of 2 × 107/mL. Four weeks after bone marrow transplantation, blood was collected and evaluated with MPST genotyping analysis to exclude mice with failed reconstitution. Then, the mice were fed 3.5% DSS for 6 days to induce colitis.
2.11. Immunohistochemistry
Immunohistochemistry was performed as described previously [16]. Colon sections were deparaffinized with xylene and rehydrated. Slides were incubated in 3% hydrogen peroxide. Heat epitope retrieval was performed for 20 min in target-retrieval solution at pH 7.5. The sections were preblocked with 10% normal goat serum (ZSGB-BIO, Beijing, China) and then incubated with antibody at a dilution of 1:100 overnight at 4 °C. Tissue sections were stained with HPR secondary antibody (dilution: 1:1000, ZSGB-BIO, Beijing, China) for 1 h at 37 °C in an incubator. Immunoreactivity was detected using a DAB kit (ZSGB-BIO, Beijing, China) and visualized as brown staining. Slides were counterstained with hematoxylin. The antibodies used for IHC are listed as follows: MPST (Genetex; #GTX108274).
2.12. Western blot (WB) analysis
WB analysis was performed as described previously [16]. RIPA buffer (Beyotime, Jiangsu, China) was used to extract proteins supplemented with protease and phosphatase inhibitors (Invitrogen). Protein concentrations were measured using the BCA Protein Assay Kit (Beyotime). A total of 20 μg of protein from each group was separated by 10% SDS‒PAGE and then electrophoretically transferred to PVDF membranes (Millipore). After the membranes were blocked with 5% nonfat milk (Sangon, Shanghai, China) in a Tris-buffered saline Tween solution (TBST) at room temperature for 1 h, they were incubated overnight with primary antibodies at the dilutions specified by the manufacturers. The membranes were then incubated with the corresponding HRP-conjugated secondary antibody at a 1:5000 dilution for 1 h. After three washes with TBST, specific bands were visualized using an ECL detection kit (Beyotime) and imaged with a ChemiScope 6000 Pro Touch (Clinx Science Instruments Co., Ltd., Shanghai, China). The Western blot assays were repeated at least 3 times in the cell models, and representative blots (n = 2) are shown in the figures. The antibodies used for WB are listed as follows: MPST (Genetex; #GTX108274); p-AKT (Ser 473; CST, #4060); CSE (Proteintech; #12217-1-AP); CBS (Proteintech; # 14787-1-AP); p-P65 (Ser 536; CST; #3039 for mice; Senta Cruz # 166748 for human); AKT (CST; #4685); p-Iκbα (Ser32; CST; #9246); p-P38 (Thr180/Tyr182; CST; #4155); iNOS (Abcam; # ab202427); Cleaved Caspase 3 (CST; #9661); Cleaved Caspase 7 (CST; #8438); Cleaved Caspase 8 (CST; #9694); BCL-XL (Proteintech; #26967-1-AP); PP2A (Abcam; #32104); PTEN (Abcam; #170941); PI3K (Genetex; #GTX100462); GST (Abcam; #111947); Flag (Invitrogen; #MA1-91878); HA (Invitrogen; #26183); GAPDH (CST; #5174); Tubulin (CST; #2148).
2.13. Immunoprecipitation
HEK293T cells were plated in 10 cm2 dishes and then transfected with Flag-tagged full-length MPST DNA, HA-tagged full-length AKT, HA-tagged AKT (1–149), HA-tagged AKT (150–408) or HA-tagged AKT (150–480) for 48 h before being lysed with 1 mL of ice-cold IP lysis buffer (Pierce, St Louis, MO) and a protease inhibitor cocktail (Invitrogen). The cell lysates were immunoprecipitated with 18 μl of anti-FLAG M2 Affinity gel (Sigma) and incubated with rocking at 4 °C for 3 h (7.5 circles/min). Then, the agarose was washed four times with lysis buffer and boiled in SDS sample buffer. For recombinant protein IP, recombinant-His-MPST (Abcam), recombinant-GST-AKT (Biotechne) and anti-GST antibody (Abcam) were incubated with rocking at 4 °C overnight, then A/G agarose was added and the sample was rocked at 4 °C for another 1 h. Then, the agarose was washed four times with lysis buffer and boiled in SDS sample buffer. The proteins were then separated using SDS‒PAGE and subjected to WB analysis with anti-AKT or anti-MPST antibodies.
2.14. Analysis of apoptosis
To identify apoptotic cells in vitro, caspase 3/7 activity (Promega, Fitchburg, USA) was measured according to the manufacturer's instructions.
2.15. Real-time PCR
Total RNA was isolated from colon tissue samples or cell lysates using the standard TRIzol (TaKaRa, Otsu, Japan) method according to the manufacturer's instructions. The isolated RNA was reverse transcribed into cDNA using the PrimeScript® RT reagent Kit (TaKaRa). Quantitative real-time PCR was performed using the ABI 7500 real-time PCR System (Applied Biosystems, Carlsbad, CA) with SYBR Green (TaKaRa). The relative expression levels of target genes were normalized to the expression level of GAPDH, which was used as an internal control, and calculated by the 2−ΔΔCT method. The primer sequences are listed in Table S1.
2.16. RNA-seq analysis
MPST was knocked down in HT29 cells, and then the cells were stimulated with 100 ng/mL TNF-α for 24 h. Vials containing snap-frozen cell samples were sent on dry ice to Novogene (China), where RNA was extracted from samples using standard extraction protocols (TRIzol). RNA was quality-checked with electropherograms, gel images and RNA integrity number (RIN); RNA with an RIN of greater than six was deemed of sufficient quality for gene expression profiling experiments. The detailed results can be found in SAR (PRJNA725951). The different gene results can be found in the supplementary data (Supplementary Table 2).
2.17. FITC-dextran permeability assay
Intestinal permeability was assessed by oral administration of FITC-labeled dextran (MW 4000; Sigma-Aldrich) as previously described [23]. After the withdrawal of food overnight, all mice were gavaged with FITC-dextran (60 mg/100 g of body weight) 4 h before sacrifice. The serum was then collected, and the FITC-dextran level in the serum was measured with a fluorescence spectrophotometer with emission and excitation wavelengths of 488 nm and 520 nm, respectively. The serum FITC-dextran concentration was calculated from standard curves generated from serial dilutions of FITC-dextran in PBS.
2.18. Reactive oxygen species (ROS) measurement
The levels of mitochondrial ROS generation were determined using MitoSOX assay via flow cytometry. Briefly, approximate 2 × 106 cells of each cell line were harvested, resuspended in PBS supplemented with 5 μM MitoSOX™ (Invitrogen, USA) reagents and then incubated at 37 °C for 10 min.
2.19. Statistical analysis
Statistical analysis was performed using GraphPad Prism. All data are shown as the mean ± SD. For comparisons among three or more groups, one-way ANOVA followed by Bonferroni analysis for multiple comparisons (for data meeting homogeneity of variance criteria) or Tamhane's T2 analysis (for data demonstrating heteroscedasticity) was performed. The differences in values between two groups were evaluated using Student's t-test. A difference was considered statistically significant when P < 0.05. All authors had access to the study data and reviewed and approved the final manuscript.
3. Results
3.1. Intestinal MPST expression was downregulated in IBD
H2S is regarded as a protective factor for IBD [24,25], yet how MPST, an endogenous enzyme regulating H2S synthesis, influences intestinal homeostasis and inflammation remains poorly understood. To this end, we evaluated the expression of MPST in human IBD, and colonic biopsy specimens were supplied for subsequent analysis. Initially, as shown in Fig. 1A, we found that MPST, which is mainly expressed in intestinal epithelial cells (IECs) in the human colon, was substantially decreased in the samples from both CD patients and UC patients compared with those from healthy control subjects. We also analyzed mucosal MPST protein expression in public gene datasets (GSE59071) of CD and UC samples. The results indicate that MPST was downregulated in these samples compared with samples from healthy controls (Fig. 1B). Western blot analysis also confirmed the reduced MPST protein levels in UC patients (Fig. 1C). Then, we established a DSS-induced experimental mouse model of acute colitis, in which mice were exposed to DSS (3.5%) in drinking water for 7 days. Consistent with the results of the human samples, the MPST protein level was also decreased in murine colitis (Fig. 1D). In addition, downregulated MPST mRNA levels were found in the 3.5% DSS-treated mice compared with the controls (Fig. 1E). As MPST was mainly located in IECs, we isolated IECs from DSS-treated mouse colons and found that MPST protein expression was decreased in the DSS group compared with the control group (Fig. 1F). In vitro, we stimulated HT29 cells with TNF-α (100 ng/mL) for 24 h to establish a cellular model of intestinal inflammation and found that TNF-α treatment markedly downregulated MPST mRNA and protein expression levels (Fig. 1G and H). Furthermore, colonic epithelial stem cells harvested from WT mice developed into organoids and were stimulated with TNF-α (100 ng/mL) for 24 h to establish a cellular model of colonocyte inflammation. We found that TNF-α treatment markedly downregulated the epithelial MPST protein level in the organoids, too (Fig. 1I).
Fig. 1.

Reduced MPST expression was found in the intestine of patients with IBD and in chemically induced mouse acute experimental colitis. (A) Representative images of immunohistochemical staining of intestinal sections from the controls and the patients with UC and CD. (B) Boxplot of MPST expression levels in endoscopic-derived intestinal mucosal biopsies from patients with CD, patients with UC and healthy controls (using dataset GSE59071; control, n = 11; CD, n = 8; UC, n = 97). (C) The expression of MPST in intestinal tissues from the controls and the patients with UC was assessed via Western blots, and densitometry analysis is shown. (D) Western blotting and densitometry analysis of MPST protein expression in colons from mice treated with or without 3.5% DSS for 7 days. (E) Intestinal MPST mRNA expression in normal mice (n = 4) and mice treated with 3.5% DSS (n = 5) for 7 days was determined by quantitative PCR. (F) Western blotting and densitometry analysis of MPST protein in IECs isolated from the controls and the 3.5% DSS-treated mice. (G and H) MPST mRNA (G) and protein expression (H) in HT29 cells treated with or without TNF-α. (I) Intestinal stem cells were harvested from untreated WT mice, and the organoids were stimulated with mouse recombinant TNF-α for 24 h starting on Day 6. Western blotting and densitometry analysis of MPST protein in organoids are shown. Fig. 1C, D, Fig. 1F, H and I were repeated three times on three different experimental days. Three to four replicates were used in Fig. 1C, D and Fig. 1, Fig. 2 replicates were used in Fig. 1H and I. Values are expressed as the mean ± S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Taken together, these results suggested that the downregulation of MPST expression was present in IBD.
3.2. MPST deficiency aggravated DSS-induced experimental colitis
To investigate the functional role and the underlying molecular mechanisms of MPST in IBD, we generated MPST knockout mice. At baseline conditions, 6- to 8-week MPST-deficient (MPST−/−) and partly MPST-deficient (MPST+/−) mice did not exhibit colitis compared to MPST+/+ mice, as indicated by HE staining (Fig. S1). However, MPST−/− mice, MPST +/- mice and MPST+/+ littermates were treated with 3.5% DSS in the drinking water for 7 days to establish an acute experimental colitis model, but the MPST−/− mice showed poor health and thus sacrificed on day 6.The MPST−/− and MPST +/- mice exhibited more severe clinical symptoms, such as loss of body weight (Fig. 2A), higher DAI scores (Fig. 2B) and shorter colon length (Fig. 2C), indicating a greater extent of intestinal tissue damage in the MPST-deficient mice. Additionally, we measured intestinal permeability by assessing FITC-dextran levels. Following DSS treatment, the intestinal barrier of the MPST−/− and MPST +/- mice was further disrupted, leading to the presence of 2-fold more FITC-dextran in their bloodstream than in that of the WT mice (Fig. 2D). Moreover, MPST−/− and MPST +/- mice had a more severe intestinal inflammatory response and epithelial injury than MPST+/+ mice. Notably, H&E staining showed that the colons of the MPST−/− and MPST+/- mice following DSS treatment exhibited more severe inflammation in the mucosa, muscularis propria and submucosa, with the entire loss of the crypts and partial loss of the surface epithelia, than those of the MPST+/+ mice (Fig. 2E). The histological examination results of colon sections with murine colitis corresponded with the clinical observations (Fig. 2F).
Fig. 2.

MPST deficiency markedly aggravates murine colitis. (A–H) MPST+/+ (n = 7), MPST+/− (n = 7), and MPST−/− (n = 7) mice were treated with 3.5% DSS for 6 days. (A–C) Body weight (A) and the disease activity index (B) calculated from weight loss, stool consistency, and bleeding were scored daily. Mice were sacrificed on day 6, and colon length (C) was measured. (D) Mice were fed FITC-dextran (60 mg/100 g) 4 h before sacrifice on day 6, and the serum level of FITC-dextran was detected with a microplate reader. (E) Histological changes in colon tissue samples from DSS-treated MPST+/+ mice (MPST+/+ DSS), DSS-treated MPST +/− mice (MPST +/− DSS) and DSS-treated MPST−/− mice (MPST−/− DSS) were examined by hematoxylin and eosin staining (original magnification, × 50). (F) Semiquantitative scoring of histopathology. (G) The relative mRNA levels of cytokines and chemokines in the colon were calculated (TNF-α, IL-1β, IL-6 and CCL3). (H) Western blotting was used to analyze iNOS, p-Iκbα (Ser32), p-p65 (Ser 536) and MPST protein expression in colons harvested from mice after DSS administration. Values are expressed as the mean ± S.D. in this figure. The WB was repeated three times on three different experimental days. Three to four replicates were used in Fig. 2H.
Consistently, MPST−/− and MPST+/- mice exhibited higher expression of cytokines and chemokines (TNF-α, IL-6, IL-1β and CCL3) in the colon than WT mice did in the DSS-induced colitis model, as determined by mRNA expression (Fig. 2G). Furthermore, as determined by Western blotting, we found that the NF-κb signaling pathway, the most relevant inflammatory signaling pathway, was more activated in the MPST−/− and MPST +/- mice compared with the WT mice (Fig. 2H).
Overall, MPST played an important role in colitis, and knockdown of this molecule increased the levels of inflammatory cytokines, thereby aggravating the symptoms of colitis.
3.3. MPST-deficient mice were susceptible to TNBS-induced experimental colitis
In addition to DSS-induced colitis, trinitrobenzene sulfonic acid (TNBS)-induced experimental colitis is characterized by a predominant Th1/Th17-mediated immune response and mucosal inflammation that closely resembles important immunological and histopathological aspects of CD [17]. In the control group, MPST+/+, MPST +/- and MPST−/− mice were treated with ethanol, and the body weight, colon length and histological analysis showed no difference in the baseline condition (Figs. S2A–2C). Consistent with previous data, following TNBS treatment, the MPST-deficient mice displayed much worse colonic damage, as demonstrated by worse megascopic viewing, decreased body weight, shorter colonic length and poorer survival than the controls (Fig. 3A–D). In addition, histological analysis revealed a high degree of crypt architecture disruption in MPST-deficient mice (Fig. 3E and F), indicating accelerated colitis progression. Furthermore, colonic inducible IL-6, CCL3, MCP-1 and S100A8 mRNA expression was upregulated, and MPST deficiency significantly aggravated these elevated mRNA expression levels (Fig. 3G). Together, these results indicate that MPST is an important factor that facilitates the development of IBD.
Fig. 3.

MPST deficiency aggravated TNBS-induced experimental colitis. (A–D) MPST+/+ (n = 8), MPST+/− (n = 6), and MPST−/− (n = 5) mice were pre-sensitized by application of 1% TNBS to the skin for 1 week and received 2.5% TNBS by rectal injection for 48 h. Megascopic viewing (A), body weight (B), survival analysis (C) and colonic length (D) of the MPST +/+, MPST−/− and MPST +/- mice treated with TNBS. (E) Representative images of H&E staining of colonic samples from the above mice. (F) Histological scores of murine colitis colon sections from the above mice. (G) mRNA expression of cytokines and chemokines (IL-6, CCL3, MCP-1 and S100A8).
3.4. MPST expression in hematopoietic cells is essential for colitis development in the DSS-induced colitis model
To determine the cell populations that contribute to MPST-mediated protection against colitis, we generated 3 groups of MPST bone marrow chimeras. The WT recipient mice underwent a sublethal dose of γ-ray irradiation (7.5 Gly) to kill the bone marrow cells. Bone marrow isolated from MPST+/+, MPST +/- and MPST−/− mice was injected into WT recipients (termed “MPST+/+ to WT”, “MPST+/- to WT”, and “MPST−/− to WT” mice, respectively). Four weeks after bone marrow reconstitution, genetic information in the bone marrow was identified (Fig. S3), and the mice were subjected to DSS. MPST+/+ to WT mice, in which MPST was intact in all cells, had significantly lower body weight (Fig. S4A) and shorter colon length (Fig. S4B) compared with MPST +/- or MPST−/− to WT mice, in which MPST was partly or completely disrupted only in myeloid cells. Histological examination of colon sections corresponded with the clinical observations (Figs. S4C–D). Moreover, increased proinflammatory cytokines (IL-1β) were observed in the MPST+/+ to WT group compared with the other two groups (Fig. S4E), suggesting that MPST in immune cells exacerbated colitis.
Collectively, these data indicated that a lack of MPST in the epithelium contributed to colitis development, while its role in immune cells may be reversed.
3.5. MPST deficiency in IECs aggravated inflammation and oxidative stress in vitro
The data described above suggested that MPST in the epithelium is critical to improve colitis, and MPST-deficient IECs were further used in this study. We found that MPST was hardly detected in MPST+/- and MPST−/− organoids compared with MPST+/+ organoids by WB (Fig. 4A) and IHC (Fig. 4B). Colonic epithelial stem cells harvested from MPST +/- and MPST−/− mice developed into organoids more slowly in ex vitro culture than those from MPST+/+ mice (Fig. 4C). Moreover, when stimulated with TNF-α for 24 h, the organoids derived from MPST +/- and MPST−/− mice showed an abnormal cellular morphology compared with those from MPST+/+ mice (Fig. 4D).
Fig. 4.

MPST deficiency inhibited stem-cell-derived organoid formation (A–B) Western blotting and IHC were used to analyze MPST protein expression in organoids harvested from MPST+/+, MPST +/- and MPST−/− mice. (C) Intestinal stem cells were harvested from untreated MPST+/+, MPST +/- and MPST−/− mice, and the organoids were observed daily. (D) Organoids were stimulated with mouse recombinant TNF-α for 24 h on Day 7.
We also used the HT29 cell line exposed to TNF-α to establish cell models to mimic gut inflammation in IECs. MPST expression was knocked down by MPST small interfering RNA (siRNA) in cultured HT29 cells (Fig. S5A), and following TNF-α treatment, the knockdown significantly aggravated the levels of proinflammatory cytokines and chemokines (TNF-α, ICAM, CXCL5 and CX3CL1) (Fig. 5A). The levels of inflammation-associated cytokines and chemokines (TNF-α, ICAM, CXCL5 and CX3CL1) were significantly decreased when MPST was overexpressed (Fig. S5B) via plasmids (Fig. 5B). To further confirm the anti-inflammation phenotypes induced by MPST in the cell model, we searched UniProt (https://www.uniprot.org/uniprot/P25325) for a potential MPST enzymatic active site, and found site 248 (cysteine persulfide intermediate). The MPST plasmid or mutated MPST (C248A) plasmid was transfected into MPST-knockdown HT29 cells, and the increased cytokines (TNF-α and ICAM) by MPST deficiency could be rescued by overexpression of MPST, showing a decreasing trend but no significant difference in the mutated MPST (C248A) plasmid in HT29 cells (Fig. S5C).
Fig. 5.

MPST deficiency markedly aggravates cellular inflammation (A) mRNA expression of proinflammatory cytokines and chemokines (TNF-α, ICAM, CXCL5 and CX3CL1) after MPST knockdown in TNF-α-treated HT29 cells (n = 3/per group). (B) mRNA expression of proinflammatory cytokines and chemokines (TNF-α, ICAM, CXCL5 and CX3CL1) after MPST overexpression in TNF-α-treated HT29 cells (n = 3/per group). (C) ROS levels after MPST overexpression in TNF-α-treated HT29 cells (n = 6/per group). (D) ROS levels after MPST knockdown in TNF-α-treated HT29 cells (n = 3/per group). (E) Western blotting was used to analyze NF-κb protein expression in MPST-deficient HT29 cells. Values are expressed as the mean ± S.D. in this figure. This WB was repeated three times (2 replicates were used each) on three different experimental days. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Currently, it is generally believed that aggravated inflammation can induce reactive oxygen species (ROS) in the pathogenesis of IBD, further enhancing colonic damage [26]. Thus, we assessed ROS activity by assessing superoxide in the mitochondria and found that MPST knockdown markedly increased ROS levels (Fig. 5D) and that MPST overexpression significantly decreased ROS levels (Fig. 5C). Furthermore, MPST knockdown activated the NF-κB pathway compared with that of the controls (Fig. 5E).
3.6. MPST deficiency contributed to IEC apoptosis
To systematically evaluate the role of MPST in the pathogenesis of IBD at the transcriptomic level, we performed RNA-seq analysis on HT29 cells with and without MPST knockdown in a TNF-α-induced model. The different genes between the siMPST and siNC groups are shown in Supplemental Table 2. KEGG analysis revealed that pathways such as TNF signaling, apoptosis, and IL-17 signaling were markedly enriched in the MPST knockdown group (Fig. 6A). By analyzing the detailed apoptosis-related gene expression, we also found that proapoptotic genes (for example, Bax, Box, and AIFM1) were active, whereas antiapoptotic genes (for example, Bcl-2, API5, and BIRC8) were inhibited in the MPST-deficient group (Fig. 6B). These data indicate that MPST deficiency may exacerbate cell apoptosis. Indeed, the knockdown of MPST in HT29 cells significantly increased the prevalence of apoptosis, as confirmed by flow cytometry analysis (Fig. 6C and D). As shown in Fig. 6E, the response to TNF-α was greater in the MPST-silenced cells than in the control cells in terms of caspase 3/7 activity. Furthermore, the knockdown of MPST significantly activated the expression of cleaved caspase 3 and cleaved caspase 8 and suppressed the expression of BCL-XL (Fig. 6F). In contrast, the overexpression of MPST via the transfection of the pcDNA3.1-MPST plasmid decreased cleaved caspase 3 protein levels (Fig. 6G) in HT29 cells. Thus, MPST deficiency contributed to IEC apoptosis.
Fig. 6.

MPST deficiency contributed to IEC apoptosis. (A–F) MPST was knocked down in HT29 cells, and then the cells were stimulated with 100 ng/mL TNF-α for 24 h. (A) Heatmap showing the pathways enriched by KEGG in HT29 cells after MPST knockdown (siNC, n = 3; siMPST, n = 3; siNC + TNF-α, n = 3; siMPST + TNF-α, n = 3). (B) Heatmap showing apoptosis-related genes that are differentially expressed between HT29 cells after MPST knockdown. (C) Apoptotic cells were analyzed by PI and Annexin V double staining. (D) Quantitative results of flow cytometric analysis (siNC, n = 3; siMPST, n = 3; siNC + TNF-α, n = 3; siMPST + TNF-α, n = 3). (E) The activities of cleaved caspase 3/7 were measured as described in the experimental procedures, and the results were normalized to the control group (n = 4–8 per group). (F) The protein levels of cleaved caspase 8, caspase 3 and BCL-XL were assessed by Western blots in vitro. (G) MPST was overexpressed in HT29 cells for 24 h, and the cells were stimulated with 100 ng/mL TNF-α for another 24 h. Apoptosis signaling was assessed by Western blotting. Fig. 6F and G were repeated three times (2 replicates were used each) on three different experimental days.
3.7. H2S was not responsible for MPST deficiency-induced colitis
The data described above suggested that the downregulation of MPST expression aggravated IEC apoptosis in colitis. MPST is considered a third route of H2S synthesis in addition to cystathionine-β-synthase (CBS)/cystathionine -γ-synthase (CSE) [27]. Following DSS treatment, CSE expression was also markedly decreased in MPST−/− mice, while the expression of another important H2S producer, CBS, showed no significant change, with relatively low expression in the colon (Fig. 7A–B), indicating that MPST deficiency may impair gut H2S synthesis. In this study, NaSH was applied simultaneously with DSS treatment. For this purpose, exogenous NaSH was i.p. injected into mice, and mice that received the same amount of normal saline (NS) served as controls. However, the NaSH-treated MPST ± or MPST −/− mice displayed the same histological performance as the NS-treated controls (Fig. 7C and D). That is, H2S supplementation could not rescue the aggravated intestinal inflammation caused by MPST deficiency in vivo.
Fig. 7.

H2S was not responsible for MPST deficiency-induced colitis. (A) CSE and CBS protein expression and densitometry analysis among MPST+/+, MPST ± and MPST−/− mice treated with 3.5% DSS. (B) mRNA level of CSE in the above mouse colons after treatment with 3.5% DSS. (C and D) Histological changes and scores in colon tissue samples from DSS-treated mice after the addition of 4 mg/kg NaHS. (E) The protein levels of active caspase 3 and caspase 7 in MPST knockdown HT29 cells treated with 1 mM NaHS were probed by Western blotting. (F) mRNA expression of inflammatory cytokines and chemokines after MPST knockdown and supplementation with NaHS. (G–H) mRNA expression of inflammatory cytokines (H) and active caspase 3 (G) after MPST knockdown and supplementation with GYY4137 (400 μM) was measured.
Consistently, the activated expression of cleaved caspase 3 by MPST deficiency was observed in the siMPST group, whereas these adverse effects were not blunted with the H2S supplementation by NaHS (Fig. 7E) or GYY4137 (Fig. 7G). The toxicity of NaHS was determined by CCK8, and the results indicated that 1 mM NaHS did not cause cell death (Fig. S6). Moreover, aggravated expression of proinflammatory genes induced by MPST knockdown could not be rescued through NaHS or GYY4137 supplementation (Fig. 7F and H). Ultimately, endogenous H2S may not be responsible for MPST deficiency-induced aggravated colitis. The decrease in H2S may be a consequence but not a cause of MPST deficiency-induced colitis.
3.8. MPST participated in IBD through an AKT-dependent mechanism
We analyzed the RNA-seq results and found that the PI3K/AKT pathway is closely associated with apoptosis mediated by MPST. Upregulation of AKT expression is critical for the suppression and maintenance of cellular apoptosis [28,29]. We first analyzed MPST and AKT expression in public gene datasets of CD and UC samples. The results indicate that MPST positively correlated with AKT expression levels in human samples (Fig. 8A). Next, the phosphorylation of AKT (Ser 473) was decreased in MPST+/+ organoids treated with TNF-α, and the decrease in p-AKT (Ser 473) was substantially exacerbated in MPST-partly deficient organoids (Fig. 8B). In addition, the levels of cleaved, activated forms of caspase 3 and caspase 7 were significantly higher in MPST-deficient organoids than in MPST+/+ organoids after exposure to TNF-α (Fig. 8B). Moreover, knocking down MPST significantly decreased p-AKT (Ser 473) expression in TNF-α-treated HT29 cells (Fig. 8C). To further test the hypothesis that AKT plays an essential role in mediating colitis in the MPST deficiency group, a rescue experiment was conducted using an AKT overexpression plasmid. Overexpression of AKT via plasmid transfection decreased the cleaved caspase 3 protein level and increased the BCL-XL protein level (Fig. 8D). We also used a specific AKT inhibitor (MK-2208) in the siMPST group and observed a more pronounced apoptosis phenotype than MPST knockdown (Fig. 8E). Flow cytometry analysis showed that the apoptotic cell ratio was significantly decreased after overexpression of AKT (Fig. 8F). As shown in Fig. 8G, overexpression of AKT significantly reduced the levels of proinflammatory cytokines (TNF-α) and chemokines (IL-8 and CCL4) induced by MPST deficiency.
Fig. 8.

MPST participated in IBD through an AKT-dependent mechanism. (A) Correlative analysis expression of MPST with AKT in the dataset is shown (using dataset GSE59071; control, n = 11; CD, n = 8; UC, n = 97). (B) The protein levels of p-AKT, AKT, CL-caspase7, CL-caspase3 and MPST were measured in MPST-deficient and MPST+/+ organoids by WB. (C) The protein levels of p-AKT, AKT and MPST were measured by WB after MPST knockdown in HT29 cells. (D) The protein levels of p-AKT, AKT, cleaved caspase3, BCL-XL and p-p38 (Thr180/Tyr182) were measured after AKT overexpression in MPST-deficient HT29 cells. (E) MPST-deficient HT29 cells were treated with or without MK2208 (1 μM), and the protein levels of p-AKT, AKT, cleaved caspase3 and MPST were measured by WB. (F) AKT was overexpressed in MPST-deficient HT29 cells, and then the cells were stimulated with 100 ng/mL TNF-α for 24 h. Apoptotic cells were analyzed by PI and Annexin V double staining. (G) mRNA expression of proinflammatory cytokines and chemokines (TNF-α, IL-8 and CCL4) after AKT overexpression was detected in MPST-deficient HT29 cells (n = 3–4/per group). Fig. 8B–E was repeated three times (2 replicates were used each) on three different experimental days.
Next, we investigated how MPST regulates the function of AKT. We transiently transfected HEK293T cells with Flag-MPST and His-AKT, and co-immunoprecipitation experiments found that MPST could directly bind with AKT (Fig. 9A), and then sustain the phosphorylation of AKT (Fig. 8C). Further investigation of the binding site of the two proteins would be of great value to future functional studies, so we explored the domain required for the MPST-AKT interaction. A series of AKT deletion mutants were structured according to their functions, and domain-mapping experiments were performed. Co-IP experiments showed that amino acids (aa) 409–480, a carboxyl-terminal regulatory domain of AKT, were required for the MPST-AKT interaction (Fig. 9B). We then transiently transfected HEK293T cells with Flag-MPST or Flag-mut-MPST (C248A) and His-AKT, and conducted co-immunoprecipitation experiments, which showed that only wild type MPST, not mutated MPST (C248A), could bind to AKT (Fig. 9C). Additionally, in MPST-knockdown HT29 cells, the decreased PP2A level induced by MPST overexpression could be disrupted by the overexpression of mut-MPST, indicating that the disruption of the AKT-MPST association could induce PP2A protein accumulation (Fig. S7). We also found that purified recombinant MPST could bind with purified recombinant AKT in vitro (Fig. 9D). To test how MPST affects the phosphorylation of AKT, the protein levels of phosphorylation-related proteins (PI3K and PTEN) and the dephosphorylation enzyme (PP2A) were measured in MPST-deficient HT-29 cells. We found that PP2A was significantly increased in the MPST-knockdown group compared with the control group, indicating that MPST might affect the dephosphorylation of p-AKT (Ser 473) (Fig. 9E). LB100, a PP2A inhibitor, was used to inhibit PP2A activity in vitro. Consistently, lower AKT phosphorylation levels were observed in MPST-silenced HT-29 cells, whereas these adverse effects could be rescued by LB100 (Fig. 9F). Then, an immunofluorescence staining assay showed that MPST and AKT colocalized in the cytoplasm in HT29 cells (Fig. 9 G).
Fig. 9.

Interaction with AKT is required for MPST function in IBD in vitro. (A) HEK293T cells infected with a Flag-tagged MPST overexpression plasmid and an HA-tagged AKT overexpression plasmid (or the empty vector) for 48 h were collected, and total Flag-tagged MPST was immunoprecipitated using anti-Flag beads. (B) Interaction domains of AKT and MPST were explored using full-length and truncated AKT based on co-immunoprecipitation assays in HEK293T cells. (C) HEK293T cells infected with a Flag-tagged MPST (or Flag-tagged mut MPST) overexpression plasmid and an HA-tagged AKT overexpression plasmid (or the empty vector) for 48 h were collected, and total Flag was immunoprecipitated using anti-Flag beads. (D) Co-IP analyses showing the interaction between recombinant His-MPST and recombinant GST-AKT. (E) The protein levels of phosphorylation-related proteins (PI3K and PTEN) and the dephosphorylation enzyme (PP2A) were measured in MPST-deficient HT-29 cells by WB. (F) MPST-deficient HT29 cells were stimulated with 100 ng/mL TNF-α with or without LB100 (1 μM) for 24 h, and the protein level of p-AKT was measured by WB. (G) Representative immunofluorescent images of DAPI, MPST, AKT and the merge of all staining in HT29 cells.
Collectively, these results show that AKT plays an important role in MPST-regulated colitis.
4. Discussion
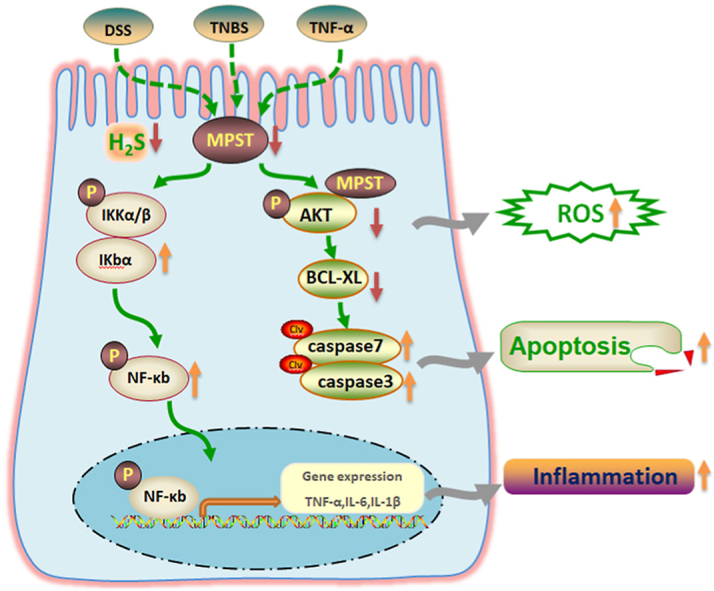
In this study, we propose that gut epithelial MPST functions as an essential checkpoint for intestinal dysfunction and inflammation by regulating the AKT signaling pathway. Compelling evidence from this study demonstrates that epithelial MPST deficiency is associated with aggravated intestinal inflammation and ROS. The deletion of MPST in the colon also contributed to increased apoptosis in intestine of the DSS-treated mice, which may be a consequence of acute damage [30]. Furthermore, our results suggest that the epithelial MPST/AKT/apoptosis axis is an essential circuit in the pathogenesis of IBD (Graphical Abstract).
In our study, MPST whole-body-knockout mice, but not IEC-specific or marrow-specific knockout mice, were used. Whole-body MPST deficiency aggravated DSS-induced experimental colitis (Fig. 2), while MPST deficiency in hematopoietic cells not aggravated that (Fig.S4), indicating that MPST in IECs may play a more important role in IBD. After ruling out the hematopoietic contribution in the aggravated colitis caused by MPST deficiency, we focused on MPST in IECs, which seemed to play an important regulatory role in IBD pathogenesis.
Our colonic IHC staining revealed a broad spectrum of MPST-expressing cell types, including epithelial cells and intestinal immune cells (Fig. 1). This led us to identify which of them plays a relatively more important role in IBD pathogenesis. For epithelial cells, we are interested in the role of MPST in regulating general barrier function, immunological function in host enteric defense and cell lineage-specific functions that contribute to the development of intestinal inflammation. In this study, we focused on the role of MPST in the intestinal epithelium. We also found that MPST in hematopoietic cells participated in IBD (Supplement Figure 4). Further work is needed to investigate the mechanism of MPST in immune cells. We wondered which types of regulatory immune cells were regulated by MPST and how they functioned in IBD as well as their regulatory mechanisms. Myeloid cells and lymphoid cells, originating from multipotent hematopoietic stem cells located in the red bone marrow, can differentiate into several types of cells present in the GI tract, such as mast cells and T cells [31]. Monocytes can develop into macrophages that are classically divided into two main subgroups, proinflammatory macrophages (M1) and anti-inflammatory and/or wound healing macrophages (M2), which may play different roles in IBD [32]. Whether MPST regulates myeloid cell and lymphoid cell differentiation, macrophage differentiation, and the GI M1/M2 ratio requires further investigation. Apart from macrophages, dendritic cells, monocytes and granulocytes are also important players in the innate immune system. Whether MPST influences the functions of these cells also needs further exploration.
IKK/NF-κB signaling is recognized as a critical determinant of intestinal homeostasis and inflammation, which also controls immune responses and cell survival by regulating the expression of proinflammatory and prosurvival genes [33]. In addition, TNF is a key pathogenic factor in intestinal inflammation, and TNF-neutralizing antibodies are highly effective in the treatment of IBD, including CD and UC. Consistent with these findings, the mRNA levels of the chemokines TNF-α, IL-1β and CCL-3 were elevated in the intestines of DSS-treated MPST +/- and MPST−/− mice. Furthermore, the IKK/NF-κB pathway showed stronger activation in the DSS-treated MPST +/- and MPST−/− mice than in the WT mice. Therefore, MPST seems to be a major contributor to the protection of the intestine from inflammation.
In addition to the CBS/CSE system, MPST is also an important H2S-generating enzyme regulating endogenous H2S production. Recent studies have shown that H2S functions as a signaling molecule in the pathogenesis of inflammatory and neoplastic colonic diseases [24,34]. Several lines of evidence suggest that excessive bacterial production of H2S participates in the progression of colitis [35,36]. In this study, we assessed whether MPST regulates intestinal dysfunction in the pathogenesis of IBD. Consistent with previous studies, following DSS treatment, colonic CSE was significantly decreased, indicating that H2S may be involved in the pathogenesis of colitis. Lack of MPST indeed impaired the endogenous synthesis of colonic H2S. Physiological H2S might act as an anti-inflammatory molecule in colitis [[37], [38], [39]]. However, whether decreasing H2S is the cause or consequence of MPST-related colitis needs to be explored. Thus, we replenished H2S via i.p. NaSH, which is a commonly used H2S donor. H2S supplementation did not rescue the inducible aggravation of colitis caused by MPST deficiency. Among the TNF-α-induced colitis models in vitro, NaHS addition did not ameliorate intestinal inflammation; there are several potential mechanisms to explain this phenomenon. NaHS releases H2S suddenly and within a short interval of time, and the effect of H2S on tight junction integrity is still unknown [37]. In addition, DSS contains a nonabsorbable carbohydrate-bound sulfate that can be metabolized to H2S by sulfate-reducing bacteria [40]. Previous studies suggested that MPST inhibits H2S production via the downregulation of CSE expression in the liver [16]. We believe that the downregulated H2S may be a consequence of MPST deficiency, which may not participate in MPST-related colitis. Thus, we assumed that decreased endogenous H2S may not be responsible for MPST deficiency-induced colitis.
To gain insight into the molecular mechanisms driving the aggravated colonic damage induced by MPST knockout, we performed RNA-seq analysis of HT29 cells. As shown in the heatmap, apoptosis signaling of a specific set of genes was strong, indicating that intestinal apoptotic activity may account for the aggravated colitis caused by MPST deficiency. Excessive apoptosis of IECs destroys the intestinal epithelial barrier and leads to the development of colitis [41]. It has become increasingly evident that apoptosis is closely related to the intestinal barrier and is involved in the pathogenesis of colitis. In this study, we found that MPST played an important role in IEC apoptosis. When MPST expression was knocked down in HT-29 cells, both western blot and flow cytometry assays showed that cell apoptosis was significantly more prevalent. Moreover, MPST overexpression was associated with elevated expression of BCL-XL and decreased expression of cleaved caspase 3. In contrast, MPST knockdown induced increased levels caspase 3/7 cleavage activity. In addition, increased MPST activity alleviated intestinal apoptosis in a TNF-α-induced cell model. Consistent with our results, a previous study also showed that apoptosis was efficiently induced through the addition of S-Allyl-L-Cysteine, while there was a significant decrease in MPST activity in the MCF-7-cell line [15]. Thus, MPST might play an important role in regulating intestinal apoptosis activity and might be a potential therapeutic target for IBD.
However, the mechanism by which MPST regulates apoptosis is still unknown. The role of PI3K-AKT in IBD is multiplex. They can regulate epithelium inflammation, epithelium apoptosis, cell proliferation and dendritic cells maturation by regulating different downstream targeted genes [42], such as NF-κB [43,44], mTOR [45,46], FOXO1, BAD and caspase. Early studies revealed that PI3K-AKT phosphorylation is a key event in the progression of colitis via the activation of inflammatory signaling by regulating NF-κB or mTOR. In those cases, the p-AKT protein level was increased in the DSS-treated group compared with the control group, subsequently activating its downstream NF-κB pathway. On the other hand, the PI3K/AKT pathway was reported to ameliorate cell apoptosis, which may improve the intestinal barrier integrity in IBD. A previous study demonstrated that the activation of PI3K/AKT by selective 5-HT1A agonist can prevent apoptosis of IECs in IBD [47]. Recent studies also elucidated that increased p-AKT expression can improve colonic mucosal damage in HFD + DSS treated mice [48]. Furthermore, activating the PI3K/AKT pathway by oxytocin can suppress dendritic cells maturation, thus alleviating DSS-induced colitis [49]. Herein, we show that p-AKT (S473) was decreased in MPST-deficient cells, and lower p-AKT contributed to IEC apoptosis by activating the caspase family. The results were consistent with the reported function of PI3K-AKT in regulating cell apoptosis in IBD. We also show that the NF-κB signaling pathway was activated in MPST-deficient cells, further contributing to IEC inflammation. Taken together, activated NF-κB signaling but decreased p-AKT level indicated that the PI3K-AKT signaling may not serve as the upstream of NF-κB in this MPST-deficient scenario. Most importantly, co-IP indicated a marked interaction between AKT and MPST. Combined with previous data, these findings indicated that AKT signaling, not H2S signaling, plays an essential role in MPST-mediated colitis.
Based on our findings, further study is needed to clarify the mechanism leading to MPST downregulation in IBD. Furthermore, we found that MPST might play different roles in different cell types. The oxidative stress response play an important role in stromal cells during inflammation and carcinogenesis [[50], [51], [52]].Research concerning IEC-specific MPST KO mice, bone marrow-specific MPST KO mice, and colonic MPST overexpression awaits further exploration in future studies. In summary, our data demonstrated that a defect in MPST expression might aggravate inflammation and epithelial cell apoptosis through AKT signaling in colitis, suggesting that the modulation of MPST expression might be a potential therapeutic approach for colitis treatment.
Grant support
This study was supported by the National Natural Science Foundation of China (No.82030019 to Chaohui Yu, No.82170533 and No. U1803121 to Zhe Shen, No. 82100541 to Jie Zhang, No. 82000489 to Juan Du, No. 82001710 to Danyi Xu) and the Natural Science Foundation of Zhejiang Province (No. LY20H030005 to Chao Lu and No.LQ22H030014 to Jie Zhang).
Transcript profiling
SAR (PRJNA725951).
Author contributions
J.Z., L.C., and XF.Z. conceived and designed research; CX. T., Ys. C. and Yw. Z. performed experiments; Ml. Y.,C. L. X.S, TTZ and M. L. analyzed data; S. L. and Br. L. interpreted results of experiments; Ch. Y. prepared figures and drafted manuscript; H. W. and Z. S. edited and revised manuscript. All authors approved final version of manuscript.
Declaration of competing interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We thank Professor Chenfu Xu, Juan Du and Nan Lin for help with this study.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102469.
Contributor Information
Chaohui Yu, Email: zyyyych@zju.edu.cn.
Hao Wu, Email: wu.hao@zs-hospital.sh.cn.
Zhe Shen, Email: sz8239@zju.edu.cn.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Baumgart D.C., Carding S.R. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. doi: 10.1016/S0140-6736(07)60750-8. [DOI] [PubMed] [Google Scholar]
- 2.Weissman S., Systrom H.K., Aziz M., et al. Colorectal cancer prevention in inflammatory bowel disease: a systematic analysis of the overall quality of guideline recommendations. Inflamm. Bowel Dis. 2021;28(5):745–754. doi: 10.1093/ibd/izab164. [DOI] [PubMed] [Google Scholar]
- 3.Clarke W.T., Feuerstein J.D. Updates in colorectal cancer screening in inflammatory bowel disease. Curr. Opin. Gastroenterol. 2018;34:208–216. doi: 10.1097/MOG.0000000000000448. [DOI] [PubMed] [Google Scholar]
- 4.Magro F., Gionchetti P., Eliakim R., et al. Third european evidence-based consensus on diagnosis and management of ulcerative colitis. Part 1: definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J Crohns Colitis. 2017;11:649–670. doi: 10.1093/ecco-jcc/jjx008. [DOI] [PubMed] [Google Scholar]
- 5.Papamichael K., Cheifetz A.S., Melmed G.Y., et al. Appropriate therapeutic drug monitoring of biologic agents for patients with inflammatory bowel diseases. Clin. Gastroenterol. Hepatol. : the official clinical practice journal of the American Gastroenterological Association. 2019;17:1655–16568 e3. doi: 10.1016/j.cgh.2019.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Papamichael K., Afif W., Drobne D., et al. Therapeutic drug monitoring of biologics in inflammatory bowel disease: unmet needs and future perspectives. The lancet Gastroenterology & hepatology. 2022;7:171–185. doi: 10.1016/S2468-1253(21)00223-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vereecke L., Beyaert R., van Loo G. Enterocyte death and intestinal barrier maintenance in homeostasis and disease. Trends Mol. Med. 2011;17:584–593. doi: 10.1016/j.molmed.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 8.Zhang J., Xu M., Zhou W., et al. Deficiency in the anti-apoptotic protein dj-1 promotes intestinal epithelial cell apoptosis and aggravates inflammatory bowel disease via p53. J. Biol. Chem. 2020;295:4237–4251. doi: 10.1074/jbc.RA119.010143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuo W.T., Shen L., Zuo L., et al. Inflammation-induced occludin downregulation limits epithelial apoptosis by suppressing caspase-3 expression. Gastroenterology. 2019;157:1323–1337. doi: 10.1053/j.gastro.2019.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh A.K., Hertzberger R.Y., Knaus U.G. Hydrogen peroxide production by lactobacilli promotes epithelial restitution during colitis. Redox Biol. 2018;16:11–20. doi: 10.1016/j.redox.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flannigan K.L., Agbor T.A., Blackler R.W., et al. Impaired hydrogen sulfide synthesis and il-10 signaling underlie hyperhomocysteinemia-associated exacerbation of colitis. Proc. Natl. Acad. Sci. U. S. A. 2014;111:13559–13564. doi: 10.1073/pnas.1413390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y., Liao R., Qiang Z., et al. Exogenous h2s protects colon cells in ulcerative colitis by inhibiting nlrp3 and activating autophagy. DNA Cell Biol. 2021;40:748–756. doi: 10.1089/dna.2020.6380. [DOI] [PubMed] [Google Scholar]
- 13.Flannigan K.L., Ferraz J.G., Wang R., Wallace J.L. Enhanced synthesis and diminished degradation of hydrogen sulfide in experimental colitis: a site-specific, pro-resolution mechanism. PLoS One. 2013;8 doi: 10.1371/journal.pone.0071962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pedre B., Dick T.P. 3-mercaptopyruvate sulfurtransferase: an enzyme at the crossroads of sulfane sulfur trafficking. Biol. Chem. 2020 doi: 10.1515/hsz-2020-0249. [DOI] [PubMed] [Google Scholar]
- 15.Bronowicka-Adamska P., Bentke A., Lasota M., Wrobel M. Effect of s-allyl -l-cysteine on mcf-7 cell line 3-mercaptopyruvate sulfurtransferase/sulfane sulfur system, viability and apoptosis. Int. J. Mol. Sci. 2020:21. doi: 10.3390/ijms21031090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M., Xu C., Shi J., et al. Fatty acids promote fatty liver disease via the dysregulation of 3-mercaptopyruvate sulfurtransferase/hydrogen sulfide pathway. Gut. 2018;67:2169–2180. doi: 10.1136/gutjnl-2017-313778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wirtz S., Popp V., Kindermann M., et al. Chemically induced mouse models of acute and chronic intestinal inflammation. Nat. Protoc. 2017;12:1295–1309. doi: 10.1038/nprot.2017.044. [DOI] [PubMed] [Google Scholar]
- 18.Gracz A.D., Puthoff B.J., Magness S.T. Identification, isolation, and culture of intestinal epithelial stem cells from murine intestine. Methods Mol. Biol. 2012;879:89–107. doi: 10.1007/978-1-61779-815-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karki R., Man S.M., Malireddi R.K.S., et al. Nlrc3 is an inhibitory sensor of pi3k-mtor pathways in cancer. Nature. 2016;540:583–587. doi: 10.1038/nature20597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang L.Y., He Q., Liang S.J., et al. Clc-3 chloride channel/antiporter defect contributes to inflammatory bowel disease in humans and mice. Gut. 2014;63:1587–1595. doi: 10.1136/gutjnl-2013-305168. [DOI] [PubMed] [Google Scholar]
- 21.Zaki M.H., Boyd K.L., Vogel P., et al. The nlrp3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin W., Ma C., Su F., et al. Raf kinase inhibitor protein mediates intestinal epithelial cell apoptosis and promotes ibds in humans and mice. Gut. 2017;66:597–610. doi: 10.1136/gutjnl-2015-310096. [DOI] [PubMed] [Google Scholar]
- 23.Liu Y., Peng J., Sun T., et al. Epithelial ezh2 serves as an epigenetic determinant in experimental colitis by inhibiting tnfalpha-mediated inflammation and apoptosis. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E3796–E3805. doi: 10.1073/pnas.1700909114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo F.F., Yu T.C., Hong J., Fang J.Y. Emerging roles of hydrogen sulfide in inflammatory and neoplastic colonic diseases. Front. Physiol. 2016;7:156. doi: 10.3389/fphys.2016.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kushkevych I., Castro Sangrador J., Dordević D., et al. Evaluation of physiological parameters of intestinal sulfate-reducing bacteria isolated from patients suffering from ibd and healthy people. J. Clin. Med. 2020;9 doi: 10.3390/jcm9061920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butin-Israeli V., Bui T.M., Wiesolek H.L., et al. Neutrophil-induced genomic instability impedes resolution of inflammation and wound healing. J. Clin. Invest. 2019;129:712–726. doi: 10.1172/JCI122085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jimenez M., Gil V., Martinez-Cutillas M., Mañé N., Gallego D. Hydrogen sulphide as a signalling molecule regulating physiopathological processes in gastrointestinal motility. Br. J. Pharmacol. 2017;174:2805–2817. doi: 10.1111/bph.13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shariati M., Meric-Bernstam F. Targeting akt for cancer therapy. Expet Opin. Invest. Drugs. 2019;28:977–988. doi: 10.1080/13543784.2019.1676726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wright K., Kolios G., Westwick J., Ward S.G. Cytokine-induced apoptosis in epithelial ht-29 cells is independent of nitric oxide formation. Evidence for an interleukin-13-driven phosphatidylinositol 3-kinase-dependent survival mechanism. J. Biol. Chem. 1999;274:17193–17201. doi: 10.1074/jbc.274.24.17193. [DOI] [PubMed] [Google Scholar]
- 30.Araki Y., Mukaisyo K., Sugihara H., Fujiyama Y., Hattori T. Increased apoptosis and decreased proliferation of colonic epithelium in dextran sulfate sodium-induced colitis in mice. Oncol. Rep. 2010;24:869–874. doi: 10.3892/or.2010.869. [DOI] [PubMed] [Google Scholar]
- 31.Weiskopf K., Schnorr P.J., Pang W.W., et al. Myeloid cell origins, differentiation, and clinical implications. Microbiol. Spectr. 2016;4 doi: 10.1128/microbiolspec.MCHD-0031-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Binienda A., Ziolkowska S., Hauge I.H., Salaga M. The role of immune and epithelial stem cells in inflammatory bowel disease therapy. Curr. Drug Targets. 2020;21:1405–1416. doi: 10.2174/1389450121666200504074922. [DOI] [PubMed] [Google Scholar]
- 33.Garvey M.I., Bradley C.W., Holden E. Blossoming vancomycin-resistant enterococci infections. J. Hosp. Infect. 2017;97:421–423. doi: 10.1016/j.jhin.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 34.Figliuolo V.R., Coutinho-Silva R., Coutinho C. Contribution of sulfate-reducing bacteria to homeostasis disruption during intestinal inflammation. Life Sci. 2018;215:145–151. doi: 10.1016/j.lfs.2018.11.009. [DOI] [PubMed] [Google Scholar]
- 35.Furne J.K., Suarez F.L., Ewing S.L., Springfield J., Levitt M.D. Binding of hydrogen sulfide by bismuth does not prevent dextran sulfate-induced colitis in rats. Dig. Dis. Sci. 2000;45:1439–1443. doi: 10.1023/a:1005580709390. [DOI] [PubMed] [Google Scholar]
- 36.Singh S.B., Lin H.C. Hydrogen sulfide in physiology and diseases of the digestive tract. Microorganisms. 2015;3:866–889. doi: 10.3390/microorganisms3040866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao H., Yan R., Zhou X., Ji F., Zhang B. Hydrogen sulfide improves colonic barrier integrity in dss-induced inflammation in caco-2 cells and mice. Int. Immunopharm. 2016;39:121–127. doi: 10.1016/j.intimp.2016.07.020. [DOI] [PubMed] [Google Scholar]
- 38.Hirata I., Naito Y., Takagi T., et al. Endogenous hydrogen sulfide is an anti-inflammatory molecule in dextran sodium sulfate-induced colitis in mice. Dig. Dis. Sci. 2011;56:1379–1386. doi: 10.1007/s10620-010-1461-5. [DOI] [PubMed] [Google Scholar]
- 39.Chen S.W., Zhu J., Zuo S., et al. Protective effect of hydrogen sulfide on tnf-α and ifn-γ-induced injury of intestinal epithelial barrier function in caco-2 monolayers. Inflamm. Res. : official journal of the European Histamine Research Society [et al. 2015;64:789–797. doi: 10.1007/s00011-015-0862-5. [DOI] [PubMed] [Google Scholar]
- 40.Roediger W.E., Moore J., Babidge W. Colonic sulfide in pathogenesis and treatment of ulcerative colitis. Dig. Dis. Sci. 1997;42:1571–1579. doi: 10.1023/a:1018851723920. [DOI] [PubMed] [Google Scholar]
- 41.Gunther C., Neumann H., Neurath M.F., Becker C. Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut. 2013;62:1062–1071. doi: 10.1136/gutjnl-2011-301364. [DOI] [PubMed] [Google Scholar]
- 42.Liu C., Zeng Y., Wen Y., Huang X., Liu Y. Natural products modulate cell apoptosis: a promising way for the treatment of ulcerative colitis. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.806148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ni L., Lu Q., Tang M., et al. Periplaneta americana extract ameliorates dextran sulfate sodium-induced ulcerative colitis via immunoregulatory and pi3k/akt/nf-kappab signaling pathways. Inflammopharmacology. 2022;30:907–918. doi: 10.1007/s10787-022-00955-7. [DOI] [PubMed] [Google Scholar]
- 44.Zaghloul M.S., Elshal M., Abdelmageed M.E. Preventive empagliflozin activity on acute acetic acid-induced ulcerative colitis in rats via modulation of sirt-1/pi3k/akt pathway and improving colon barrier. Environ. Toxicol. Pharmacol. 2022;91 doi: 10.1016/j.etap.2022.103833. [DOI] [PubMed] [Google Scholar]
- 45.Makled M.N., Serrya M.S., El-Sheakh A.R. Fingolimod ameliorates acetic acid-induced ulcerative colitis: an insight into its modulatory impact on pro/anti-inflammatory cytokines and akt/mtor signalling. Basic Clin. Pharmacol. Toxicol. 2022;130:569–580. doi: 10.1111/bcpt.13720. [DOI] [PubMed] [Google Scholar]
- 46.Dong L., Du H., Zhang M., et al. Anti-inflammatory effect of rhein on ulcerative colitis via inhibiting pi3k/akt/mtor signaling pathway and regulating gut microbiota. Phytother Res. 2022;36:2081–2094. doi: 10.1002/ptr.7429. [DOI] [PubMed] [Google Scholar]
- 47.Walldorf J., Porzner M., Neumann M., et al. The selective 5-ht1a agonist sr57746a protects intestinal epithelial cells and enteric glia cells and promotes mucosal recovery in experimental colitis. Inflamm. Bowel Dis. 2022;28:423–433. doi: 10.1093/ibd/izab191. [DOI] [PubMed] [Google Scholar]
- 48.Lee Y.H., Kim H., Nam S., et al. Protective effects of high-fat diet against murine colitis in association with leptin signaling and gut microbiome. Life. 2022;12 doi: 10.3390/life12070972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dou D., Liang J., Zhai X., et al. Oxytocin signalling in dendritic cells regulates immune tolerance in the intestine and alleviates dss-induced colitis. Clin. Sci. (Lond.) 2021;135:597–611. doi: 10.1042/CS20201438. [DOI] [PubMed] [Google Scholar]
- 50.Perl A., Hanczko R., Telarico T., Oaks Z., Landas S. Oxidative stress, inflammation and carcinogenesis are controlled through the pentose phosphate pathway by transaldolase. Trends Mol. Med. 2011;17:395–403. doi: 10.1016/j.molmed.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Carini F., Mazzola M., Rappa F., et al. Colorectal carcinogenesis: role of oxidative stress and antioxidants. Anticancer Res. 2017;37:4759–4766. doi: 10.21873/anticanres.11882. [DOI] [PubMed] [Google Scholar]
- 52.Mariani F., Sena P., Roncucci L. Inflammatory pathways in the early steps of colorectal cancer development. World J. Gastroenterol. 2014;20:9716–9731. doi: 10.3748/wjg.v20.i29.9716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.