

Abstract

Nonribosomal peptide synthetases (NRPSs) are a vast source of valuable natural products, and re-engineering them is an attractive path toward structurally diversified active compounds. NRPS engineering often requires heterologous expression, which is hindered by the enormous size of NRPS proteins. Protein splitting and docking domain insertion have been proposed as a strategy to overcome this limitation. Here, we have applied the splitting strategy to the gramicidin S NRPS: Despite better production of the split proteins, gramicidin S production almost ceased. However, the addition of type II thioesterase GrsT boosted production. GrsT is an enzyme encoded in the gramicidin S biosynthetic gene cluster that we have produced and characterized for this purpose. We attribute the activity enhancement to the removal of a stalled intermediate from the split NRPS that is formed due to misinitiation. These results highlight type II thioesterases as useful tools for NRPS engineering.
Nonribosomal peptide synthetases (NRPSs) are multienzymes responsible for the biosynthesis of a broad range of pharmaceutically valuable natural products.1 However, exploitation of nonribosomal peptides as drugs is plagued by several problems. Toxic side effects sometimes limit the applicability (e.g., polymyxins).2 Resistance development diminishes the therapeutic value of long trusted compounds (e.g., penicillin),3 or low biosynthetic yields prevent cost-efficient production.4 Hence, biosynthetic engineering of nonribosomal peptides is a crucial technology to introduce structural variation, optimize bioactivities, increase yields, and thus maintain and expand their usefulness as drugs.
A highly organized modular architecture has made NRPSs a popular target for re-engineering. An evolutionary history of diversity generation by gene recombination supports this approach.5−7 NRPSs operate in a linear assembly line manner, where each module activates, modifies, and incorporates a single amino acid into the growing peptide chain.8 A minimal NRPS module consists of three core domains. The adenylation (A)-domain selects and activates a specific substrate. The thiolation (T)-domain acts as an inter- and intramodular substrate translocator, and the condensation (C)-domain forms the peptide bond.8 NRPS re-engineering through module recombination and specificity engineering has been explored by many groups with the intention to obtain novel bioactive compounds.5,9−12 However, the highly dynamic structure of NRPSs and the complex protein–protein interactions pose challenges for engineering.5 Another obstacle is the enormous size of NRPS proteins which hinders heterologous expression. In recent studies, protein expression could be increased by introducing split sites and docking domains between modules of multimodular proteins.13
As a model system to develop better NRPS engineering strategies, we have harnessed the NRPS responsible for the biosynthesis of gramicidin S (GS).14−17 GS is a membrane-active, broad-spectrum antimicrobial peptide produced by the bacterium Aneurinibacillus migulanus. The GS biosynthetic gene cluster comprises three genes, grsT, grsA, and grsB, arranged in one transcriptional unit (Figure 1A).18 The corresponding NRPS proteins GrsA and GrsB (Figure 1B) are directly responsible for the biosynthesis of GS.19 In several engineering campaigns, the specificity of GrsA has been switched through A-domain mutagenesis14,15 and “subdomain swapping”.17,20 Recently, GrsB has been converted into a DNA-templated NRPS in which the interaction of the NRPS modules is under control of a DNA strand.16
Figure 1.

(A) The GS biosynthetic gene cluster (B) encodes the NRPS proteins GrsA and GrsB as well as GrsT, a putative TEII.18 The Phe-ATE module GrsA initiates GS formation by activation of l-Phe (F), followed by epimerization in the E-domain to d-Phe (f). The successive activation and incorporation of l-Pro by GrsB1, l-Val by GrsB2, l-Orn by GrsB3, and l-Leu by GrsB4 into the nascent pentapeptide is followed by dimerization and cyclization in the type I thioesterase (TEI-) domain. (C) TEIIs free T-domains from stalled substrates.
Although the GS NRPS has been heavily investigated and engineered, the role of the third protein encoded by the GS biosynthetic gene cluster, GrsT, has never been clarified.18 The presence of a conserved motif (GHSXG) suggests type II thioesterase (TEII) activity.21 TEIIs perform housekeeping functions and maintain operation of the NRPS assembly line through various actions (Figure 1C).22,23 The need for TEIIs arises, for instance, from the sloppy action of phosphopantetheine transferases (PPTases) mispriming T-domains with already acylated coenzyme A (CoA). Several studies show a significant decrease in product yield after deletion of TEII genes.22,24−28 While TEIIs are widespread in NRPS pathways, their impact on engineered systems has not been systematically investigated.
The question arises whether TEIIs enhance the activity of engineered NRPSs like in native systems, or even hinder the incorporation of non-natural substrates, which might fall victim to unspecific proofreading. In this study, we address this question by investigating the impact of GrsT on engineered NRPSs derived from the GS NRPS. After introducing a split site in GrsB, protein quality of the heterologously produced protein increased, but GS synthesis suffered. In this split system, GrsT boosts GS synthesis. Our findings show that TEIIs are valuable tools for NRPS engineering.
Production of Type II Thioesterase GrsT
To test the influence of TEII activity on engineered versions of the GS NRPS, we wanted to use the cognate enzyme GrsT in established in vitro assays. Therefore, GrsT was heterologously produced in E. coli and the TEII function of the purified enzyme verified. The grsT gene uses the start codon GUG, the most abundant non-AUG start codon in prokaryotes, instead of canonical AUG.29 GUG start codons are also common in E. coli and are translated as Met,30 so we tested expression with AUG or GUG as a start codon. For protein purification, a C-terminal His6-tag was added. Only the construct with the AUG start codon yielded a visible band on an SDS-PAGE gel, but purity remained low (Figure S1A). Next, GrsT was N-terminally fused to maltose binding protein (MBP), glutathione S-transferase (GST), or a small ubiquitin-like modifier (SUMO) to improve solubility (Figure S1B and C). Only the MBP-tag provided acceptable yield (17 mg L–1) and purity.
GrsT Hydrolyzes Coenzyme A Esters
With MBP-GrsT successfully purified, we measured its hydrolytic activity on acetyl-CoA, which is a standard substrate for TEIIs and resembles the expected cognate substrate, the acylated T-domain. As a negative control, the catalytic Ser95 residue identified by homology modeling (Figure S2) was mutated to Ala to verify its catalytic role (MBP-GrsT-S95A, Figure S3). Hydrolysis of acetyl-CoA was detected via CoA formation measured and quantified by UPLC-MS/MS. A catalytic efficiency of 28 ± 3 M–1 s–1 was determined for MBP-GrsT (Table 1). This value is in a similar range as those previously reported for RifR (11 ± 0.2 M–1 s–1)22 and SrfAD (43 M–1 s–1)31 for the same substrate. To test the influence of the MBP-tag on catalysis, the hydrolysis experiment was repeated after proteolytic cleavage of the MBP-tag (Figure S4), but GrsT without MBP-tag could not be purified to homogeneity. The impure protein showed an apparent catalytic efficiency of 17 ± 3 M–1 s–1, similar to MBP-GrsT, indicating that the stabilizing MBP-tag does not strongly interfere with activity. As the MBP-tag increases yield, purity, and catalytic efficiency, the fusion protein MBP-GrsT was used for all further enzymatic assays. Although the requirement for MBP-tagging possibly indicates poor stability of GrsT, MBP-GrsT shows no decrease of acetyl-CoA hydrolysis between 20 and 30 °C (Figure S5). Catalytic efficiency of MBP-GrsT slightly increases with the longer acyl chains of propionyl-CoA (58 ± 3 M–1 s–1) and butyryl-CoA (120 ± 30 M–1 s–1), which has been similarly observed with RifR22 and points toward a more spacious hydrophobic binding pocket. In conclusion, MBP-GrsT shows catalytic behavior typical of a TEII.
Table 1. Catalytic Efficiency of Acyl-CoA Hydrolysisa.
GrsT Influences Stereoselectivity
First, we investigated the impact of GrsT on an engineered minimal NRPS with inactivated epimerization (E)-domain (Figure 2A). GS contains a d-Phe residue in the first position. The E-domain of GrsA is responsible for the epimerization of l-Phe to d-Phe. Since the equilibrium constant of epimerization is close to one, both l- and d-Phe are present on the T-domain of GrsA, but the donor site of GrsB1’s C-domain is stereoselective toward d-Phe.32 This stereoselectivity hinders peptide formation when the E-domain of GrsA is inactivated, and l-Phe accumulates on the T-domain. The GrsA/GrsB1 modules excised from the GS NRPS generate the cyclic dipeptide d-Phe-l-Pro-diketopiperazine (fP-DKP) through Pro-promoted self-cyclization, which is convenient for kinetic analysis.33,34 We inactivated the E-domain of GrsA by mutating His753 to Ala35 to test the effect of GrsT on the stalled NRPS (Figures 2A and S3). It has been previously shown that TEIIs SrfAD and TycF hydrolyze l-Phe from GrsA-H753A, restoring the availability of the Ppant arm to react with d-Phe added later in the experiment. This enhanced the fP-DKP formation compared to control samples with no TEII.34 To probe the effect of MBP-GrsT, an excess of GrsB1 was used to make the first module rate limiting (Figure S7). GrsA-H753A was loaded with a mixture of d- and l-Phe. The concentrations of DKPs were measured in the presence and absence of MBP-GrsT in 5 min intervals using UPLC-MS/MS (Figures 2B and S8). MBP-GrsT reduced the formation rate of l/l configured FP-DKP 2-fold, which is consistent with our hypothesis that stalled l-Phe is removed by TEII activity. Accordingly, hydrolytic activity of MBP-GrsT was detectable with l-Phe-CoA at a level similar to the acyl-CoA’s (kcat/KM = 25 ± 6 M–1 s–1; Table 1).
Figure 2.

(A) Mechanism of DKP formation by GrsA-H753A/GrsB1 with inactivated E-domain. (B) The rate of ll-DKP formation in the presence or absence of MBP-GrsT. (C) sdVGrsA-STAP forms DKPs from a 5:1 mixture of Val and Phe. (D) Effect of MBP-GrsT and MBP-GrsT-S95A on DKP formation by sdVGrsA-STAP. (B and D) Each data point represents the mean of two biological replicates with the standard deviation as error bars.
GrsT Influences Side-Chain Selectivity
Next, we investigated the impact of GrsT on a DKP synthetase where an alternative side chain becomes incorporated. We have previously created the chimeric initiation module sdVGrsA-STAP17,20 from GrsA through “subdomain-swapping”17,36,37 with another module and directed evolution (Figure 2C). sdVGrsA-STAP has a slight preference for l-Val over the cognate GrsA substrate l-Phe.20 Similar to the previously discussed E-domain knockout, A-domain engineering in sdVGrsA-STAP causes a noncognate substrate, in this case Val, to accumulate on the T-domain because there is a selective downstream C-domain refusing the “engineered” substrate.20 It was expected that stalling on the T-domain would expose Val to TEII activity. Hence, we quantified the impact of MBP-GrsT on the sdVGrsA-STAP/GrsB1 dimodule with competing l-Val and l-Phe as substrates (Figure 2C). We found that MBP-GrsT inverts the product preference back to wild-type by reducing d-Val-l-Pro-DKP (vP-DKP) and increasing fP-DKP formation (Figure 2D), thereby counteracting the effect of A-domain engineering.
NRPS Splitting Improves Protein Purity
Subsequently, we tested the impact of GrsT on in vitro biosynthesis of GS with either wild-type GrsB or an engineered split variant of GrsB. When reconstituting GS synthesis in vitro, the large size of the GrsB protein (510 kDa) renders expression and purification difficult. Split NRPS proteins have previously attracted considerable interest and enhanced the yields of heterologously produced constructs.13,16 Therefore, we split GrsB into two smaller subunits, GrsB12 and GrsB34, and recombined the two fragments using docking domains (DDs) from Xenorhabdus innexi DSM 16336 (Figure 3A). The C-terminal docking domain (Dc) from InxA and the N-terminal docking domain (Dn) from InxB were introduced at the C-terminus of GrsB12 and N-terminus of GrsB34, respectively. This resulted in two smaller proteins, GrsB12-Dc (243 kDa) and Dn-GrsB34 (279 kDa), with the docking domains facilitating the interaction between them. As intended, yield and purity of the proteins were higher compared to intact GrsB (Figure 3B).
Figure 3.
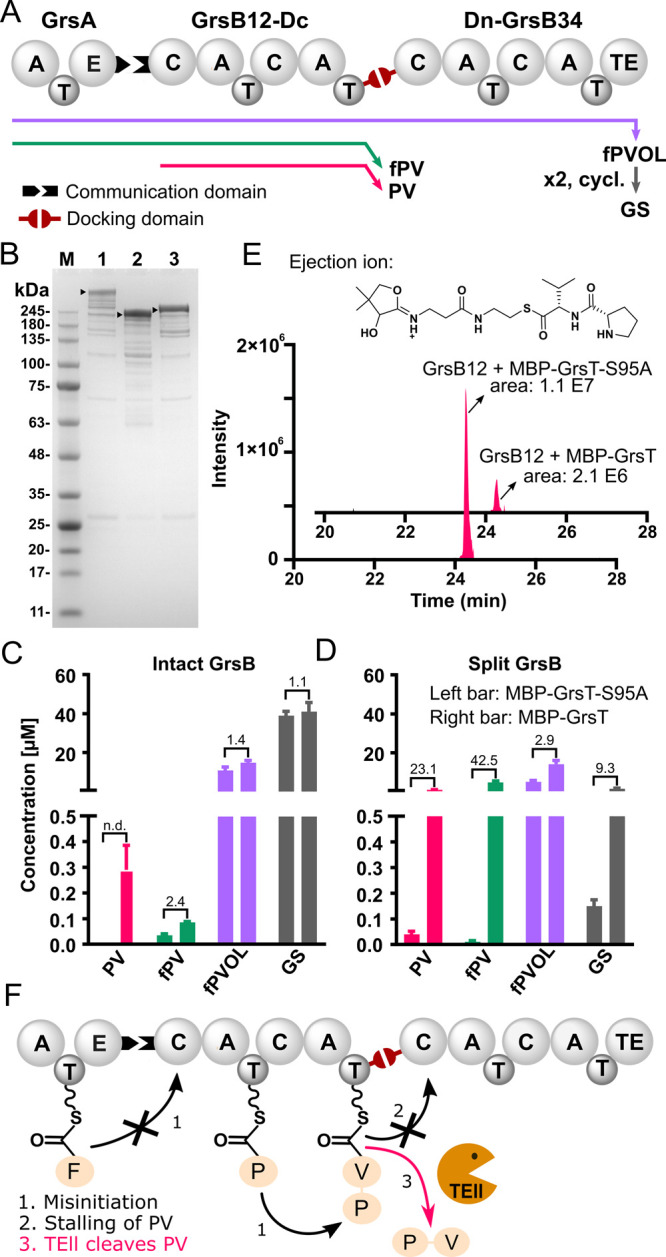
In vitro biosynthesis of GS using split GrsB. (A) GS synthetase GrsB is split into GrsB12 and GrsB34, which communicate through docking domains Dc and Dn. GrsA and GrsB12 interact via communication (COM) domains. (B) SDS-PAGE analysis of intact GrsB (1), GrsB12-Dc (2), and Dn-GrsB34 (3). On each lane, 2 μg of protein has been loaded. M: BlueEye Prestained Protein Marker. The bands corresponding to the desired protein are marked with black arrows. Peptide formation with (C) intact GrsB and (D) split GrsB quantified by triple quad UPLC-MS. Error bars represent the standard deviation from the mean of two biological replicates. The numbers above bars indicate the fold increase of peptide production with MBP-GrsT compared to the inactivated enzyme (n.d.: no defined). For yields of additional peptides fP-DKP and PVOL, see Figure S11. (E) UPLC-HRMS chromatogram of the PV loaded peptide (m/z [M+2H]2+ calc., 1155.5712; found, 1155.5697) obtained after tryptic digest of GrsB12 and structure of the ejected fragment used to verify the peak identity.38 (F) Misinitiation on GrsB1 causes stalling of PV on GrsB2, and GrsT clears this roadblock.
NRPS Splitting Harms Activity
In vitro production of GS was performed by coincubation of GrsA, GrsB12-Dc, and Dn-GrsB34 and all necessary substrates. Although splitting GrsB improved protein production, GS production plummeted (Figure 3C and D). To understand the detrimental effect of NRPS splitting, we recorded comparative UPLC-HRMS data on the product mixtures obtained with split and intact GrsB (Figure S9). Due to protein splitting, a strong increase was observed for the mass feature m/z 362.2076 assigned as the shunt product fPV. The assignment of fPV was corroborated by the observation of several characteristic fragment ions (Figure S9C). Strongly enhanced production of the fPV peptide seems to indicate poor processivity at the split site between GrsB12-Dc and Dn-GrsB34 and premature hydrolysis from the T-domain of GrsB2.
GrsT Boosts Activity of Split NRPS
Accumulation of the shunt product fPV indicated stalling of intermediates on the split GS assembly line and, possibly, the need for TEII maintenance. When GS synthesis was tested in the presence of MBP-GrsT, a concentration of 2.5 μM provided the highest amount of GS (Figure S10). Gratifyingly, MBP-GrsT increased GS biosynthesis 9.3-fold compared to the inactivated counterpart (Figure 3D). At the same time, MBP-GrsT enhanced production of the pentapeptide shunt product fPVOL 2.9-fold back to the level also found with intact GrsB (14 μM, Figures 3C and D). It is noteworthy that GS biosynthesis requires that two fPVOL pentapeptides meet at the TEl domain of GrsB4 (Figure 1B). Splitting slows the synthesis down, which in turn reduces the likelihood of two fPVOL pentapeptides arriving at the last module without hydrolyzing prematurely. Although the net effect on GS formation is positive, unspecific MBP-GrsT also causes losses of the on-pathway intermediates fPV and fPVOL.
We went on to investigate whether GrsT has a beneficial effect on GS production also in vivo, although these experiments were complicated by the lack of control over protein concentrations in the heterologous host E. coli and a high biological error. We compared four E. coli strains transformed with plasmids encoding intact or split GrsB and active or inactive GrsT (grsTAB, grsT[S95A]AB, grsTAB12-B34, and grsT[S95A]AB12-B34). Active GrsT enhanced the average GS production 1.5- and 3-fold, with intact and split GrsB, respectively (Figure S12). However, the increases were not statistically significant with three biological replicates. Possibly, alternative thioesterases or poor expression attenuate the effect of GrsT in vivo.
Misinitiation Product Blocks Split NRPS
To rationalize the beneficial effect of the TEII on in vitro reactions, we compared the metabolite profiles with and without active MBP-GrsT by UPLC-HRMS (Figure S9B). Production of the tripeptide shunt product fPV was strongly enhanced in the sample containing MBP-GrsT, but this cannot explain improved GS formation, because fPV is a hydrolyzed on-pathway intermediate. Furthermore, we observed an increase in a mass feature that we misassigned at first as the dipeptide PV, but an authentic standard revealed that this was a product of in-source fragmentation. Serendipitously, analysis by triple quad UPLC-MS detected a peak for the actual PV peptide with a different retention time that was 23-fold increased by MBP-GrsT (Figure 3D). Misinitiation of synthesis on GrsB1 would yield PV on module GrsB2 (Figure 3A), and PV is probably accepted at a much lower rate by GrsB3 than the cognate peptide, fPV, explaining the requirement for TEII activity. The presence of stalled PV on GrsB2 was confirmed by UPLC-HRMS detection of a tryptic fragment encompassing the pantetheine attachment site of the T-domain. PV peptide was detected on freshly purified GrsB2 and the peak area was reduced 5-fold through incubation with 2.5 μM MBP-GrsT for 2 h at 37 °C (Figures 3E and S13). Blockage of freshly purified GrsB12 by PV may explain a lag phase in the kinetics of GS formation alleviated by MBP-GrsT (Figure S14). Removal of stalled PV by MBP-GrsT (Figure 3F) results in 0.9 μM of the free dipeptide and is therefore a plausible explanation for the beneficial effect on GS synthesis.
Misinitiation on GrsB1 should be equally likely with split or intact GrsB, so why does intact GrsB not benefit from a TEII? Stalling of PV may be exacerbated by the artificial, noncovalent module connection in split GrsB. To clarify the fate of the erroneous intermediate PV in intact GrsB, we looked for the potential elongation product PVOL (Figure S11). Similar quantities of PVOL were measured in the presence or absence of MBP-GrsT (0.23 and 0.28 μM, respectively). Apparently, intact GrsB also performs misinitiation but does not require TEII proof-reading because it directly converts PV into PVOL, which is then cleaved by GrsB4-TEI.
The popularity of NRPS engineering5,9−12 urges the question of how proof-reading TEIIs will co-operate with modified enzymes. Since TEIIs have evolved to free T-domains from stalled, noncognate substrates, it would be expected that TEIIs also hinder attempts to augment conversion of alternative substrates. To test this assumption, we have investigated the impact of TEII activity on variants of the GS NRPS, where (I) the epimerization domain (E-domain) in GrsA has been inactivated or (II) GrsA specificity has been changed from Phe to Val. To test the engineered derivatives of the GS NRPS together with a TEII, we have newly produced and characterized GrsT. As expected, GrsT has a broad substrate spectrum and therefore behaves as a typical TEII (Table 1). The proof-reading activity apparently results from the longer dwell time of noncognate, slowly converted substrates—the longer an intermediate resides on the T-domain, the higher the likelihood of a TEII encounter. It makes no difference if a substrate is poorly converted because it was loaded accidentally or due to semisuccessful engineering. Accordingly, production of FP-DKP by the E-domain knockout GrsA-H753A/GrsB1 is halved because GrsT decimates the slowly converted l-Phe-intermediate.
By continuously sweeping lingering acyl residues from T-domains, TEIIs can not only maintain activity but also shift the product spectrum of an NRPS. One case in glycopeptide antibiotic biosynthesis has been described, where substrate specificity is controlled by a C-domain behind a multispecific A-domain, leaving undesired substrates on the T-domain for TEII cleavage.39 A-domain engineering creates a similar situation. We have previously observed that an engineered, bispecific A-domain in sdVGrsA-STAP loads Phe and Val. Without an active TEII present, the Val substrate disfavored by the C-domain then accumulates on the T-domain until the C-domain selectivity is overridden.20 Here, we demonstrate that a TEII continuously deacylating the T-domain disrupts this mechanism by preventing accumulation of Val. Therefore, the TEII shifts the product specificity toward the wild-type substrate Phe, which reflects the innate C-domain preference and thus counteracts the Val preference brought about by A-domain engineering. Exclusive control of A-domains over product specificity would be desirable for NRPS engineering, but TEII activity transfers control over the product specificity to the C-domain (Figure 2D). In conclusion, when structural changes are desired that challenge the C-domain specificity, A-domain engineering will be more successful in the absence of TEII domains. Consequently, inactivation of TEII genes should be considered in the context of A-domain engineering.
Modifying stereoconfiguration or side-chains of a residue has been hindered by TEII activity (Figure 2). In contrast, split GrsB showcases a favorable effect of TEII activity on an engineered NRPS. Splitting of GrsB into two fragments expectedly enhanced protein quality but unfortunately reduced the efficiency of GS formation as well (Figure 3). Presumably, this reduction is caused by poor communication between the two enzymes at the artificial, noncovalent module interface. This poor communication results in large amounts of the shunt product fPV being formed, which reduces the flow of intermediates toward the desired decapeptide. Worse still, the dipeptide PV, a product of misinitiation, accumulates on GrsB12 and blocks synthesis unless it is removed by GrsT.
Splitting is an enticing strategy to make the size of gigantic NRPS proteins more manageable for heterologous expression. This strategy is not generally put into question by the activity losses observed with split GrsB because alternative split sites or docking domains have not been thoroughly explored. These results do indicate, however, that TEII domains can bolster activity when engineering has uncoupled the assembly line. This success is clearly related to the fact that splitting does not modify the product sequence. Other scenarios are conceivable where TEIIs bolster the activity of recombined NRPSs making new sequences, but this will depend on the complicated interplay of A- and C-domains and the resulting residence times of unnatural intermediates on T-domains. The successful combination of a TEII with an artificial split NRPS perhaps suggests that TEIIs also play a role in facilitating the making and breaking of connections between NRPS modules in natural evolution.
Our results show how the newly characterized TEII GrsT shifts the product specificity of engineered NRPSs with a knocked-out E-domain or subdomain-swapped A-domain back to wild-type. However, in split GrsB, where the product sequence is unchanged, but module communication disturbed, GrsT boosts production of the desired product through removal of an erroneous intermediate resulting from misinitiation. These activities highlight the ambivalent role of TEIIs and the need to consider them as important contributors to successful NRPS engineering.
Acknowledgments
We acknowledge financial support by the Daimler und Benz Stiftung and the Fonds der Chemischen Industrie. Furthermore, this work has been supported by a Max-Buchner-Fellowship (H.K.) and a Ph.D. fellowship (F.P.) cofinanced by the German Academic Exchange Service (DAAD) and the Jena School for Microbial Communication (JSMC). Financial support by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) by SFB 1127/2 ChemBioSys (239748522) (to F.T., and C.H.), the Leibniz Award (to C.H.), and under Germany ´s Excellence Strategy – EXC 2051 – Project-ID 390713860 is gratefully acknowledged. We would like to thank Y. Li (Leibniz-HKI) for determining high-resolution masses.
Glossary
Abbreviations
- NRPS
nonribosomal peptide synthetase
- A-domain
adenylation domain
- T-domain
thiolation domain
- C-domain
condensation domain
- TEII
type II thioesterase
- GS
gramicidin S
- MBP
maltose binding protein
- CoA
coenzyme A
- DKP
diketopiperazine
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschembio.2c00341.
Complete description of experimental methods for cloning, protein expression, chemical synthesis, and enzymatic assays. Additional data including gel images, enzyme kinetics, volcano plots, peptide quantification (Figures S1–S14), oligo sequences, protein sequences, instrument parameters (Tables S1–S4), and additional references (PDF)
Author Contributions
∥ F.P. and S.D.: These authors contributed equally. F.P. and S.D.: conceptualization, formal analysis, investigation, project administration, visualization, writing—original draft, review, and editing. H.P.: conceptualization, formal analysis, investigation, visualization. F.T.: formal analysis, investigation, visualization. C.H.: conceptualization, writing—review and editing. H.K.: conceptualization, funding acquisition, resources, supervision, writing—review and editing.
The authors declare no competing financial interest.
Supplementary Material
References
- Felnagle E. A.; Jackson E. E.; Chan Y. A.; Podevels A. M.; Berti A. D.; McMahon M. D.; Thomas M. G. Nonribosomal Peptide Synthetases Involved in the Production of Medically Relevant Natural Products. Mol. Pharmaceutics 2008, 5 (2), 191–211. 10.1021/mp700137g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavascki A. P.; Goldani L. Z.; Li J.; Nation R. L. Polymyxin B for the Treatment of Multidrug-Resistant Pathogens: A Critical Review. J. Antimicrob. Chemother. 2007, 60 (6), 1206–1215. 10.1093/jac/dkm357. [DOI] [PubMed] [Google Scholar]
- Hutchings M.; Truman A.; Wilkinson B. Antibiotics: Past, Present and Future. Curr. Opin. Microbiol. 2019, 51, 72–80. 10.1016/j.mib.2019.10.008. [DOI] [PubMed] [Google Scholar]
- Li J.; Jaitzig J.; Hillig F.; Süssmuth R.; Neubauer P. Enhanced Production of the Nonribosomal Peptide Antibiotic Valinomycin in Escherichia coli through Small-Scale High Cell Density Fed-Batch Cultivation. Appl. Microbiol. Biotechnol. 2014, 98 (2), 591–601. 10.1007/s00253-013-5309-8. [DOI] [PubMed] [Google Scholar]
- Brown A. S.; Calcott M. J.; Owen J. G.; Ackerley D. F. Structural, Functional and Evolutionary Perspectives on Effective Re-Engineering of Non-Ribosomal Peptide Synthetase Assembly Lines. Nat. Prod. Rep. 2018, 35 (11), 1210–1228. 10.1039/C8NP00036K. [DOI] [PubMed] [Google Scholar]
- Baunach M.; Chowdhury S.; Stallforth P.; Dittmann E. The Landscape of Recombination Events That Create Nonribosomal Peptide Diversity. Mol. Biol. Evol. 2021, 38 (5), 2116–2130. 10.1093/molbev/msab015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götze S.; Arp J.; Lackner G.; Zhang S.; Kries H.; Klapper M.; García-Altares M.; Willing K.; Günther M.; Stallforth P. Structure Elucidation of the Syringafactin Lipopeptides Provides Insight in the Evolution of Nonribosomal Peptide Synthetases. Chem. Sci. 2019, 10 (48), 10979–10990. 10.1039/C9SC03633D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Süssmuth R. D.; Mainz A. Nonribosomal Peptide Synthesis-Principles and Prospects. Angew. Chem., Int. Ed. 2017, 56 (14), 3770–3821. 10.1002/anie.201609079. [DOI] [PubMed] [Google Scholar]
- Baltz R. H. Synthetic Biology, Genome Mining, and Combinatorial Biosynthesis of NRPS-Derived Antibiotics: A Perspective. J. Ind. Microbiol. Biotechnol. 2018, 45 (7), 635–649. 10.1007/s10295-017-1999-8. [DOI] [PubMed] [Google Scholar]
- Alanjary M.; Cano-Prieto C.; Gross H.; Medema M. H. Computer-Aided Re-Engineering of Nonribosomal Peptide and Polyketide Biosynthetic Assembly Lines. Nat. Prod. Rep. 2019, 36 (9), 1249–1261. 10.1039/C9NP00021F. [DOI] [PubMed] [Google Scholar]
- Bozhüyük K. A. J.; Linck A.; Tietze A.; Kranz J.; Wesche F.; Nowak S.; Fleischhacker F.; Shi Y. N.; Grün P.; Bode H. B. Modification and de Novo Design of Non-Ribosomal Peptide Synthetases Using Specific Assembly Points within Condensation Domains. Nat. Chem. 2019, 11 (7), 653–661. 10.1038/s41557-019-0276-z. [DOI] [PubMed] [Google Scholar]
- Beck C.; Garzón J. F. G.; Weber T. Recent Advances in Re-Engineering Modular PKS and NRPS Assembly Lines. Biotechnol. Bioprocess Eng. 2020, 25 (6), 886–894. 10.1007/s12257-020-0265-5. [DOI] [Google Scholar]
- Kegler C.; Bode H. B. Artificial Splitting of a Non-Ribosomal Peptide Synthetase by Inserting Natural Docking Domains. Angew. Chem. Int. Ed. 2020, 59 (32), 13463–13467. 10.1002/anie.201915989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kries H.; Wachtel R.; Pabst A.; Wanner B.; Niquille D.; Hilvert D. Reprogramming Nonribosomal Peptide Synthetases for “Clickable” Amino Acids. Angew. Chem. Int. Ed 2014, 53 (38), 10105–10108. 10.1002/anie.201405281. [DOI] [PubMed] [Google Scholar]
- Niquille D. L.; Hansen D. A.; Mori T.; Fercher D.; Kries H.; Hilvert D. Nonribosomal Biosynthesis of Backbone-Modified Peptides. Nat. Chem. 2018, 10 (3), 282–287. 10.1038/nchem.2891. [DOI] [PubMed] [Google Scholar]
- Huang H. M.; Stephan P.; Kries H. Engineering DNA-Templated Nonribosomal Peptide Synthesis. Cell Chem. Biol. 2021, 28 (2), 221–227. 10.1016/j.chembiol.2020.11.004. [DOI] [PubMed] [Google Scholar]
- Kries H.; Niquille D. L.; Hilvert D. A Subdomain Swap Strategy for Reengineering Nonribosomal Peptides. Chem. Biol. 2015, 22 (5), 640–648. 10.1016/j.chembiol.2015.04.015. [DOI] [PubMed] [Google Scholar]
- Krätzschmar J.; Krause M.; Marahiel M. A. Gramicidin S Biosynthesis Operon Containing the Structural Genes grsA and grsB Has an Open Reading Frame Encoding a Protein Homologous to Fatty Acid Thioesterases. J. Bacteriol. 1989, 171 (10), 5422–5429. 10.1128/jb.171.10.5422-5429.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipmann F. Nonribosomal Polypeptide Synthesis on Polyenzyme Templates. Acc. Chem. Res. 1973, 6 (11), 361–367. 10.1021/ar50071a001. [DOI] [Google Scholar]
- Stanišić A.; Hüsken A.; Stephan P.; Niquille D. L.; Reinstein J.; Kries H. Engineered Nonribosomal Peptide Synthetase Shows Opposite Amino Acid Loading and Condensation Specificity. ACS Catal. 2021, 11 (14), 8692–8700. 10.1021/acscatal.1c01270. [DOI] [Google Scholar]
- Turgay K.; Krause M.; Marahiel M. A. Four Homologous Domains in the Primary Structure of GrsB Are Related to Domains in a Superfamily of Adenylate-Forming Enzymes. Mol. Microbiol. 1992, 6 (18), 529–546. 10.1111/j.1365-2958.1992.tb01498.x. [DOI] [PubMed] [Google Scholar]
- Claxton H. B.; Akey D. L.; Silver M. K.; Admiraal S. J.; Smith J. L. Structure and Functional Analysis of RifR, the Type II Thioesterase from the Rifamycin Biosynthetic Pathway. J. Biol. Chem. 2009, 284 (8), 5021–5029. 10.1074/jbc.M808604200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Brittain W. D. G.; Zhang Q.; Lu Z.; Tong M. H.; Wu K.; Kyeremeh K.; Jenner M.; Yu Y.; Cobb S. L.; et al. Aminoacyl Chain Translocation Catalysed by a Type II Thioesterase Domain in an Unusual Non-Ribosomal Peptide Synthetase. Nat. Commun. 2022, 13 (1), 1–14. 10.1038/s41467-021-27512-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linne U.; Schwarzer D.; Schroeder G. N.; Marahiel M. A. Mutational Analysis of a Type II Thioesterase Associated with Nonribosomal Peptide Synthesis. Eur. J. Biochem. 2004, 271 (8), 1536–1545. 10.1111/j.1432-1033.2004.04063.x. [DOI] [PubMed] [Google Scholar]
- Ohlemacher S. I.; Xu Y.; Kober D. L.; Malik M.; Nix J. C.; Brett T. J.; Henderson J. P. YbtT Is a Low-Specificity Type II Thioesterase That Maintains Production of the Metallophore Yersiniabactin in Pathogenic Enterobacteria. J. Biol. Chem. 2018, 293 (51), 19572–19585. 10.1074/jbc.RA118.005752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotowska M.; Pawlik K. Roles of Type II Thioesterases and Their Application for Secondary Metabolite Yield Improvement. Appl. Microbiol. Biotechnol. 2014, 98 (18), 7735–7746. 10.1007/s00253-014-5952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang L.; Huang S.; Liu W. Q.; Karim A. S.; Jewett M. C.; Li J. Total in Vitro Biosynthesis of the Nonribosomal Macrolactone Peptide Valinomycin. Metab. Eng. 2020, 60, 37–44. 10.1016/j.ymben.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Jaitzig J.; Theuer L.; Legala O. E.; Süssmuth R. D.; Neubauer P. Type II Thioesterase Improves Heterologous Biosynthesis of Valinomycin in Escherichia Coli. J. Biotechnol. 2015, 193, 16–22. 10.1016/j.jbiotec.2014.10.037. [DOI] [PubMed] [Google Scholar]
- Watanabe K.; Suzuki T. Genetic Code and Its Variants. Encycl. Life Sci. 2001, 1–9. 10.1038/npg.els.0000810. [DOI] [Google Scholar]
- Blattner F. R.; Plunkett G.; Bloch C. A.; Perna N. T.; Burland V.; Riley M.; Collado-Vides J.; Glasner J. D.; Rode C. K.; Mayhew G. F.; et al. The Complete Genome Sequence of Escherichia coli K-12. Science 1997, 277 (5331), 1453–1462. 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Schwarzer D.; Mootz H. D.; Linne U.; Marahiel M. A. Regeneration of Misprimed Nonribosomal Peptide Synthetases by Type II Thioesterases. Proc. Natl. Acad. Sci. U.S.A. 2002, 99 (22), 14083–14088. 10.1073/pnas.212382199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L.; Walsh C. T. Kinetic Analysis of Three Activated Phenylalanyl Intermediates Generated by the Initiation Module PheATE of Gramicidin S Synthetase. Biochemistry 2001, 40 (18), 5329–5337. 10.1021/bi015518+. [DOI] [PubMed] [Google Scholar]
- Bergendahl V.; Linne U.; Marahiel M. A. Mutational Analysis of the C-Domain in Nonribosomal Peptide Synthesis. Eur. J. Biochem. 2002, 269 (2), 620–629. 10.1046/j.0014-2956.2001.02691.x. [DOI] [PubMed] [Google Scholar]
- Yeh E.; Kohli R. M.; Bruner S. D.; Walsh C. T. Type II Thioesterase Restores Activity of a NRPS Module Stalled with an Aminoacyl-S-Enzyme That Cannot Be Elongated. ChemBioChem. 2004, 5 (9), 1290–1293. 10.1002/cbic.200400077. [DOI] [PubMed] [Google Scholar]
- Stachelhaus T.; Walsh C. T. Mutational Analysis of the Epimerization Domain in the Initiation Module PheATE of Gramicidin S Synthetase. Biochemistry 2000, 39 (19), 5775–5787. 10.1021/bi9929002. [DOI] [PubMed] [Google Scholar]
- Crüsemann M.; Kohlhaas C.; Piel J. Evolution-Guided Engineering of Nonribosomal Peptide Synthetase Adenylation Domains. Chem. Sci. 2013, 4 (3), 1041–1045. 10.1039/C2SC21722H. [DOI] [Google Scholar]
- Thong W. L.; Zhang Y.; Zhuo Y.; Robins K. J.; Fyans J. K.; Herbert A. J.; Law B. J. C.; Micklefield J. Gene Editing Enables Rapid Engineering of Complex Antibiotic Assembly Lines. Nat. Commun. 2021, 12 (1), 6872. 10.1038/s41467-021-27139-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrestein P. C.; Bumpus S. B.; Calderone C. T.; Garneau-Tsodikova S.; Aron Z. D.; Straight P. D.; Kolter R.; Walsh C. T.; Kelleher N. L. Facile Detection of Acyl and Peptidyl Intermediates on Thiotemplate Carrier Domains via Phosphopantetheinyl Elimination Reactions during Tandem Mass Spectrometry. Biochemistry 2006, 45 (42), 12756–12766. 10.1021/bi061169d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaniusaite M.; Tailhades J.; Marschall E. A.; Goode R. J. A.; Schittenhelm R. B.; Cryle M. J. A Proof-Reading Mechanism for Non-Proteinogenic Amino Acid Incorporation into Glycopeptide Antibiotics. Chem. Sci. 2019, 68 (20), 42–61. 10.1039/C9SC03678D. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.