

Abstract

The controlled binding of the catalytically dead CRISPR nuclease (dCas) to DNA can be used to create complex, programmable transcriptional genetic circuits, a fundamental goal of synthetic biology. This approach, called CRISPR interference (CRISPRi), is advantageous over existing methods because the programmable nature of CRISPR proteins in principle enables the simultaneous regulation of many different targets without crosstalk. However, the performance of dCas-based genetic circuits is limited by both the sensitivity to leaky repression within CRISPRi logic gates and retroactive effects due to a shared pool of dCas proteins. By utilizing antisense RNAs (asRNAs) to sequester gRNA transcripts as well as CRISPRi feedback to self-regulate asRNA production, we demonstrate a mechanism that suppresses unwanted repression by CRISPRi and improves logical gene circuit function in Escherichia coli. This improvement is particularly pronounced during stationary expression when CRISPRi circuits do not achieve the expected regulatory dynamics. Furthermore, the use of dual CRISPRi/asRNA inverters restores the logical performance of layered circuits such as a double inverter. By studying circuit induction at the single-cell level in microfluidic channels, we provide insight into the dynamics of antisense sequestration of gRNA and regulatory feedback on dCas-based repression and derepression. These results demonstrate how CRISPRi inverters can be improved for use in more complex genetic circuitry without sacrificing the programmability and orthogonality of dCas proteins.
Keywords: CRISPRi, inverter, circuit, single-cell, sequestration, antisense
Introduction
A primary goal of biological engineering is the implementation of entirely new transcriptional regulatory interactions and gene networks inside a cell. Controlling such networks allows us to endow microorganisms with original engineered genetic components, such as oscillators,1 memory elements,2 and complex logic functions.3 Despite advances in the standardization of various genetic elements in bacteria, including molecular sensors4 and terminators,5 we still lack standard universal transcriptional processing components that can be reprogrammed to interact with arbitrary regulatory components or reused within large-scale synthetic gene networks.
It has been shown that synthetic transcription factors based on CRISPR interference (CRISPRi)6−10 can be used to reprogram cellular function. A catalytically dead CRISPR protein, designated dCas, can be utilized in bacteria as a programmable transcriptional repressor by obstructing transcription at the dCas binding site (Figure 1A). Using CRISPR as a regulatory element is advantageous because repression can be targeted to any arbitrary DNA sequence as long as a protospacer adjacent motif (PAM) site is present. Furthermore, dCas proteins can be combined with other components to create CRISPR activators (CRISPRa)11,12 in addition to repressors. Due to their practically infinite potential for programmability and orthogonality (simultaneous expression without crosstalk between the gRNA/dCas complex and unmatched targets), gene circuits using CRISPRi stand to drastically expand the capabilities of synthetic gene networks.
Figure 1.
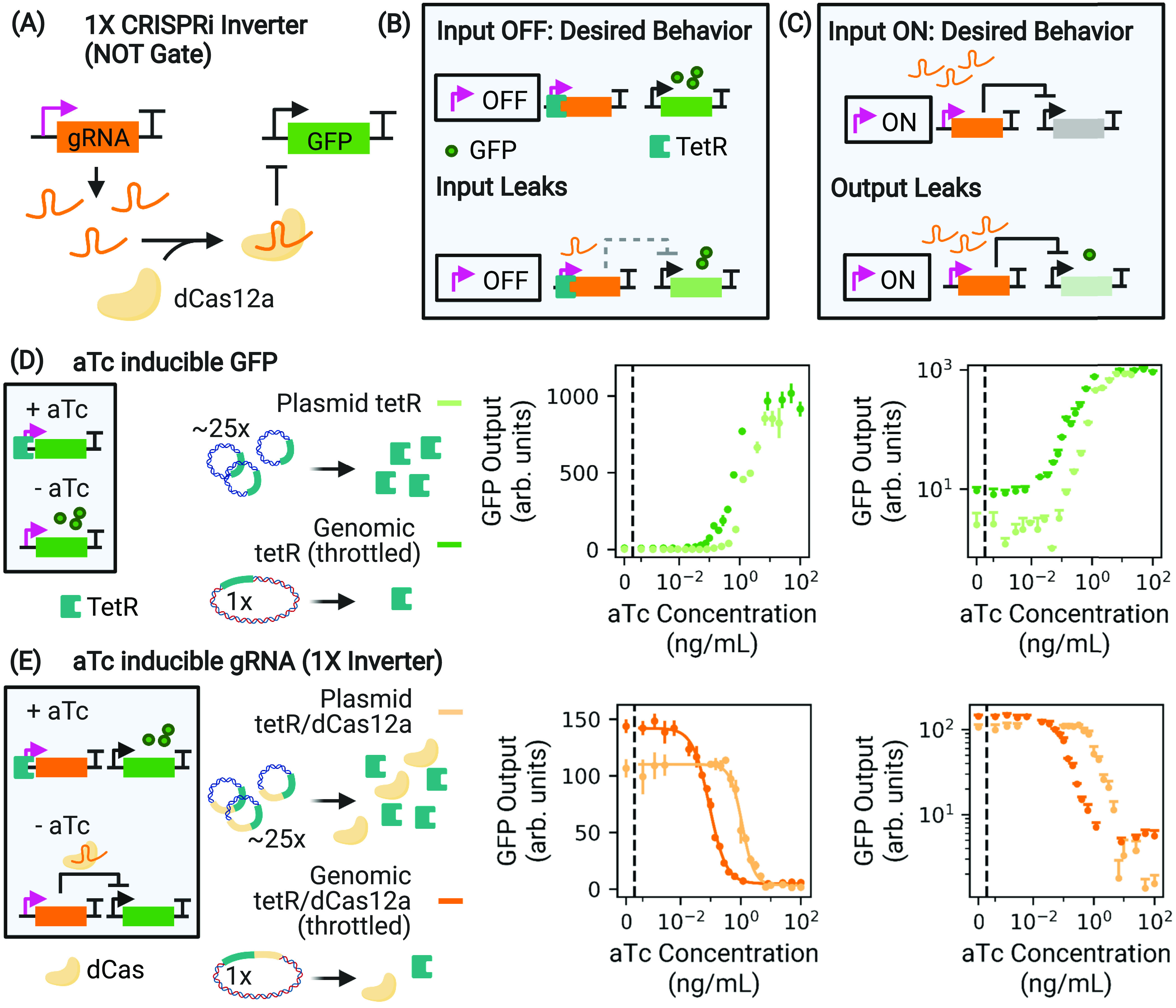
Leaks in dCas-based transcriptional circuits. (A) A CRISPRi-based NOT gate drives the production of a gRNA that programs dCas to bind to and repress expression from the target promoter, here inhibiting GFP production. (B) If the input module is an inducible sensor, any basal expression allows the unwanted production of a few gRNA that can efficiently repress the output (input leak). (C) Downstream applications can be hindered by incomplete repression by dCas (output leak). (D) We throttle tetR availability by expressing it in the genome (dark green), which causes leaky pTet expression at low aTc concentration compared to 20- to 30-fold plasmid (p15A origin, light green) expression. When used as an input promoter in an inverter, such a leaky pTet causes input leak. (E) We throttle both tetR and dCas availability, now for the 1× inverter. Throttling dCas decreases the sensitivity to leaked gRNA at low aTc concentration, increasing the overall dynamic range (dark orange) with respect to high copy plasmid expression of dCas (light orange). However, this decreases the absolute off level of GFP expression, as is evident in log space. Throttling the availability of TetR and dCas increases the leak of transcripts that they repress (gRNA and GFP mRNA, respectively), facilitating study of how these impacts can be mitigated. The curves depicted in (D) and (E) were taken during exponential growth. In linear space, the displayed error bars are ±1 standard deviation from threefold biological replicates.
However, despite successes in creating dCas-based endpoint3,13,14 and dynamic15−17 circuits, challenges remain when circuits are scaled up from just a few CRISPRi elements.18 In fact, CRISPR’s reprogrammability is also the source of its greatest weaknesses: because a shared pool of dCas proteins is drawn on simultaneously from all active elements (or nodes) in a circuit, it is possible for downstream nodes to interfere with the regulatory activity of those further upstream, an effect called retroactivity.19−21 Even if we neglect the effects of retroactivity entirely, CRISPRi circuits are extremely sensitive to transcription leaks22 because they lack the nonlinear cooperative response that is necessary to mitigate the impact of leaky repression.18 We quantify “input leak” as the amount of transcripts expressed when the input promoter driving gRNA production is in the off state (e.g., when tetR binding should block all expression from an inducible pTet promoter, reducing GFP production in Figure 1B). These leaked gRNAs are processed by dCas and may bind to their target, reducing output gene expression from the expected maximum. Alternatively, “output leak” is the instance where the node-processing module (dCas12a + gRNA) ineffectively represses the output, increasing output expression above its expected minimum even at full gRNA induction (Figure 1C).
In this work, we use 1× CRISPRi inverters derived from Francisella novicida Cas12a8,9,23 to demonstrate that CRISPRi in combination with antisense sequestration reduces the impact of retroactivity and the sensitivity to transcription leaks. The benefits are particularly pronounced during postexponential growth and stationary expression, when accumulation of dCas with leaked gRNA transcripts in a simple inverter cripples circuit performance. We further show that CRISPR’s unique mechanism of action can be used to regulate its own antisense regulation, yielding a regulatory feedback mechanism that further increases the dynamic range. Extending this to two inverters connected in series (i.e., a 2× inverter), we also show that antisense sequestration drastically improves the dynamic range of a layered genetic circuit. Finally, we use microfluidics to study the behavior of these inverters at the single-cell level, enabling us to observe how sequestration affects the long equilibration time of CRISPRi circuits. Our results show that antisense sequestration can be used to reduce the impact of leakiness in dCas-based nodes without sacrificing the programmability or orthogonality of CRISPRi synthetic gene circuits.
Results
Creating a High-Leak Model 1× CRISPRi Inverter
A single dCas CRISPRi inverter functions by driving the production of a gRNA that programs dCas to bind to and suppress expression from a targeted output promoter.9 In our work, the inverter drives GFP production (Figure 1A). Since the nuclease-dead Cas12a (dCas12a) is expressed constitutively, the output is manipulated via the induction of gRNA transcription using an anhydrotetracycline (aTc)-inducible pTet promoter. Thus, in this model system, cells turn from green to white when aTc is added and GFP production ceases.
To better understand ways to mitigate leak sensitivity, we designed a single CRISPRi inverter that purposely suffers from increased leakiness. We achieved this by throttling it twice. First, we reduced the availability of TetR, which causes the pTet promoter to “leak”, raising the basal expression level and producing gRNA transcripts even when the aTc concentration is low. We compared the performance of pTet-driven GFP expression against a system where TetR is expressed by the same promoter but at 20- to 30-fold higher copy number using a plasmid (p15A origin; Figure 1D).24 This results in significantly weakened repression at low aTc levels due to limited TetR availability, which prevents total suppression of GFP production (Figure 1D). Next, we reduced the availability of dCas12a, which increases GFP mRNA production at all aTc levels (Figure 1E) by moving dCas12a from the medium-copy plasmid to the genome. This throttles the output such that there is imperfect repression at high levels of aTc (Figure 1E). Thus, the performance of the 1× inverter is limited by both the availability of TetR at low aTc induction and the availability of dCas12a at high aTc concentration.
Interestingly, because the 1× performance is limited by unwanted repression by dCas12a in the zero-aTc state, limiting the availability of dCas12a increases the absolute dynamic range of the 1× inverter. This is similar to the effect observed in ref (21), where the presence of a competitor CRISPRi module increases the dynamic range of a basic inverter by utilizing available dCas12a space when gRNA production is low. Because of this, all of the circuits in this work were throttled with low genomic dCas12a and tetR availability unless otherwise noted. GFP mRNA- and gRNA-producing nodes were expressed from a plasmid with low, stringently controlled copy number (pSC101).
Sequestration of gRNA Reduces Circuit Sensitivity to gRNA Leaks during Stationary Expression
To decrease the impact of leaked gRNA transcripts on the performance of the 1× inverter, we used a format inspired by ref (25) and antisense RNA (asRNA) design rules from ref (26) to create a hybrid CRISPRi/asRNA system that pairs each CRISPRi node with an asRNA node specifically designed to orthogonally sequester the corresponding gRNA (Figures 2A and S1C). The gRNA is designed to target the −10 site of the target promoter, which we know effectively represses transcription based on our previous work.9 The asRNA includes a tag that recruits Hfq, a protein which facilitates RNA–RNA interactions.27 While previous authors derepressed dCas9 by binding to a linker located between the sgRNA hairpin and the terminator,25 we sequester dCas12a-based gRNAs by binding to a longer sequence that occludes the 20 bp spacer, a 40 bp unique tag, and a portion of the CRISPR repeat sequence. A comparison of occluding different lengths of the gRNA is included in Figure 2B. The efficacy of sequestration depends on disruption of the repeat hairpin, but this comes at the cost of orthogonal sequestration of unique gRNAs if too many nucleotides of the shared repeat sequence are occluded. Ultimately, we chose to occlude only nine base pairs of the repeat sequence (Partial Repeat Occlusion in Figure 2B) in order to minimize the likelihood of nonorthogonal interactions between asRNAs and noncognate gRNA. We also designed and tested a system to sequester mRNA output in parallel to gRNA sequestration, which is discussed further in the Supplementary Text.
Figure 2.
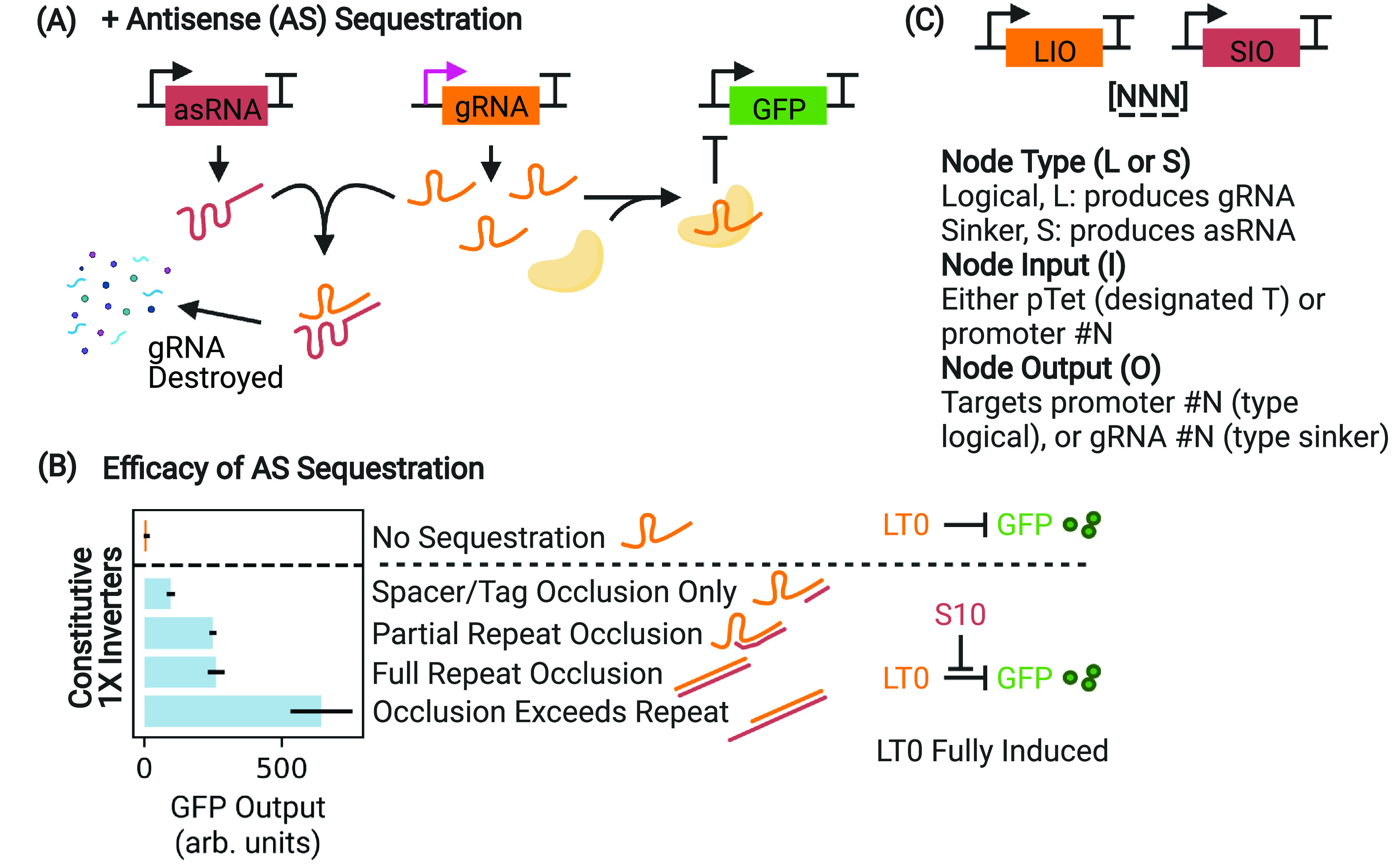
Efficacy of gRNA sequestration measured via interference with a 1× inverter. (A) By soaking up and destroying leaked gRNA transcripts using a matching asRNA sequence, the upstream circuit leak that limits the circuit dynamic range can be nullified. (B) Full expression of a 1× inverter (orange, corresponding to high aTc concentration in Figure 1D) produces cells that are white, as GFP expression is suppressed by dCas binding. Occluding portions of the gRNA (occluding only the spacer/tag, partial or full occlusion of the repeat, and occlusion that exceeds the repeat sequence, light blue) results in a demonstrable difference in sequestration efficacy as a function of interference with the function of CRISPRi, which increases GFP output. Occlusion of the complete gRNA sequence, exceeding the full length of the repeat, results in the most effective sequestration. Ultimately, partial repeat occlusion is used in all subsequent experiments in order to minimize potential nonorthogonality with asRNAs intended to target different gRNAs. Differences in GFP output are measured during exponential growth. For clarity, the HFQ recruitment tag on the asRNA is not depicted. (C) Nodes are notated with a three-character system designating the node type (logic or sinker), the promoter number, and the output number (either CRISPRi target or asRNA tag). This is useful for specifying the node order as circuits get larger and more complex. In this work, GFP is always driven by promoter 0. A pTet-driven node input is designated “T”.
For both gRNA- and asRNA-producing nodes, we use sets of three-character codes in order to specify nodes and their ordering (Figure 2C). This is helpful to notate circuit design and node order as multiple levels of feedback are added. The first character indicates whether the node produces a gRNA (L for logic node) or an asRNA (S for sinker node). The second character indicates the identity of the promoter driving the node, and the third character indicates the identity of its output. Promoters, gRNA, and asRNA pairings are indicated with a number, except for T, which refers to the inducible pTet promoter. For example, node L10 is driven by promoter 1 and produces a gRNA that targets promoter 0. Node S00 is driven by promoter 0 and produces an asRNA that targets gRNA sequence 0. “Dud” nodes are nontargeted stand-ins that constitutively express a gRNA or asRNA sequence as appropriate but do not target anything in the system. This means that in any given system of inverters being compared, the number of gRNA and asRNA nodes as well as their compositional context and number of competitor gRNA/asRNAs is preserved. Promoter 0 is always used to drive GFP production in these experiments.
CRISPRi inverters are extremely sensitive to leaked gRNAs, as the presence of just a few may allow them to bind to otherwise unoccupied dCas12a and persistently repress their targets until diluted away. As a result, the performance of a 1× inverter plummets with respect to a control (constitutive expression of GFP with controlled compositional context; Figure 3A), especially during stationary expression, resulting in inverter fold change that falls off with growth. During stationary expression, the 1× inverter output covers only 45.3% of the expected range (orange, with respect to the dotted green control; Figure 3A,B). During exponential growth, this effect is less extreme, as a basic 1× inverter (orange, Figure S2) covers 75.0% of the expected maximal dynamic range. In both instances, poor performance is driven by low maximal GFP expression under the low-induction conditions. As expected, the basic inverter shows very low, although nonzero, leakage of mRNA (and thus GFP) with respect to the background (3.3% of the maximum GFP expression during exponential growth, 3.7% during stationary expression; Figures 3B and S2) due to the high effectiveness of dCas binding.
Figure 3.
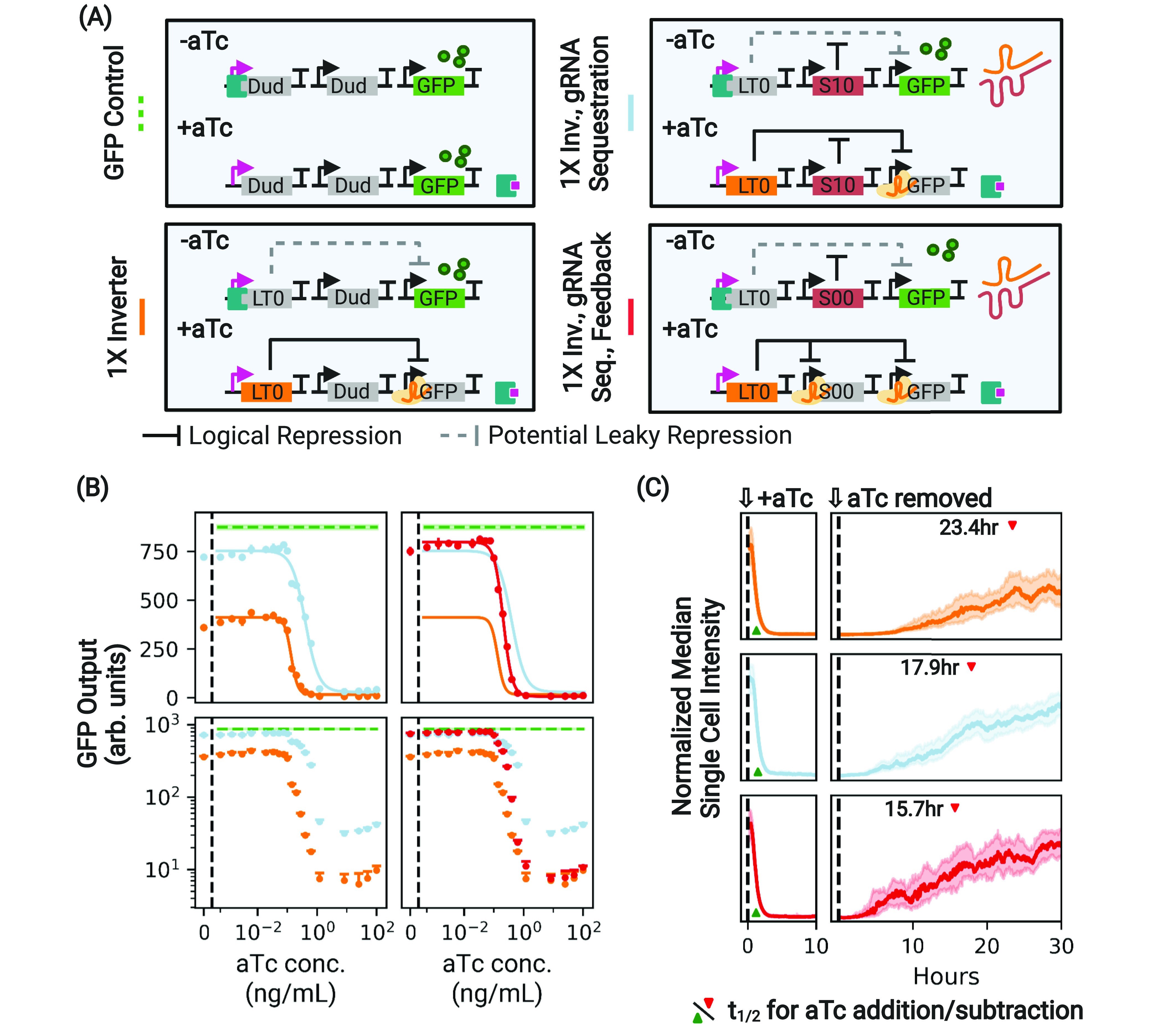
Antisense sequestration of gRNA increases the dynamic range of a 1× inverter. (A) A control and variants of the 1× inverter with gRNA sequestration designed to have the same compositional context. The additional “Dud” node upstream of the first node in each depicted circuit that constitutively expresses nontargeted asRNA has been omitted for simplicity. (B) During stationary expression, the absolute dynamic range of the basic 1× inverter (orange) is greatly limited by circuit leak, which reduces GFP output with respect to the expected maximum (dotted green) when the aTc concentration is low. Antisense sequestration of the gRNA via S10 (light blue) acts to suppress CRISPRi-based repression, expanding the dynamic range of the circuit. However, this comes at the cost of suboptimally higher expression at high induction, as is evident in log space. The addition of the feedback mechanism (red) suppresses production of the asRNA when gRNA production is high, maintaining a high dynamic range while nullifying the unwanted impacts of sequestration at high aTc concentrations. In linear space, the displayed error bars are ±1 standard deviation from threefold biological replicates. Performance is shown relative to the performance of a GFP control with the same compositional context arrangement of nodes (dashed green line) and the basic 1× inverter (orange). For these and all subsequent experiments, dCas12a and tetR are expressed constitutively in the genome. (C) The same constructs, this time under the addition and subtraction of aTc in a microfluidic chamber. The presence of antisense sequestration (light blue) speeds circuit response under aTc removal (derepression by the dCas protein; t1/2 indicated with a red caret) at the cost of some speed in repression (Table 1). Use of the dCas regulatory feedback restores the speed of repression while maintaining improved speed of derepression. Traces show median intensities of single cells across all microfluidic channels. Shaded regions indicate ±1 quartile. t1/2 was calculated using a spline fit to the microfluidic data (Figures S8 and S9).
We sought to determine whether antisense sequestration (light blue, Figure 3A,B) had a substantial effect on circuit performance. We found that antisense sequestration significantly improves the performance with respect to the basic 1× inverter, especially during stationary expression, where the dynamic range of the circuit increases from 45.3% to 82.8% of the expected range. During exponential growth, however, the overall dynamic range is relatively unaffected (Figure S2). Thus, the effect of asRNA during stationary expression is to translate the entire induction curve toward higher levels of GFP production, consistent with the expectation that gRNAs are sequestered at all levels of aTc induction. This effectively reduces the inverter fold change below that of the original inverter for exponential through most measured stationary growth.
Utilization of Positive Feedback Reduces mRNA Leak at High Levels of Induction
While the absolute dynamic range of a 1× inverter is vastly improved by the use of antisense sequestration, this comes at some significant cost (first column of Figures 3B and S2): at high levels of induction, the presence of antisense sequestration also increases the leakage of output mRNA, thereby reducing CRISPRi effectiveness. Because antisense RNA expression is unregulated, sequestration of the gRNA inhibits gRNA function at all levels of gRNA expression, even when maximal gRNA expression is desirable. It is important that we mitigate this leak, especially for the instance where the node output is the circuit output (i.e., GFP rather than another processing node). CRISPRi’s inherently programmable mechanism of action and compact regulatory footprint gives us a way to introduce feedback into the system such that asRNA is produced only when it is desired. Specifically, we implement positive feedback by having the 1× inverter regulate the production of its own antisense sequestration RNA (Figure 3A, gRNA sequestration + feedback). This system creates a feedback mechanism that reinforces sequestration when it is desirable and suppresses it when it is not. This is a positive feedback mechanism because it is self-reinforcing: when gRNA levels are high, matching asRNA levels are forced lower, increasing the gRNA concentration; when asRNA levels are high, gRNA levels are suppressed, leading to reduced repression of asRNA transcription and thus higher levels of asRNA.
Figure 3 shows how the use of regulatory feedback entirely removes the penalty at high levels of aTc induction introduced by antisense sequestration during stationary expression (red curve). Leak of mRNA output at high levels of aTc is reduced to a level comparable to that of the original 1× inverter. Furthermore, the absolute range of the circuit (90.5% of the expected maximal range) is retained, more than doubling the range of the original inverter. Essentially, the regulatory feedback module allows us to use antisense sequestration to control gRNAs leaked by ineffectual repression by TetR without increasing the number of leaked mRNAs at high aTc induction.
We also attempted to minimize output leak by sequestering mRNA (Figures S3 and S4). This was less successful because of greatly slowed circuit dynamics and increased noise when used in conjunction with gRNA sequestration, as discussed in the Supplementary Text.
Sequestration with Regulatory Feedback Speeds Derepression at No Cost to Repression Speed
Although the population-wide induction dynamics measured using microplate fluorescence allows us to study equilibrium expression in high density culture, it gives us a limited ability to measure alterations of the induction dynamics due to antisense sequestration. Thus, to more precisely understand how asRNA sequestration interacts with our 1× inverter variants, we used a “mother machine” device (Materials and Methods, Figures S6 and S7, and Movie S1) in order to track the expression level of individual cells in response to induction and repression of the 1× inverter.
We first considered the impact of gRNA sequestration on the repression dynamics when cells are exposed to aTc and on the derepression time scale when aTc is removed from the system. In agreement with previous authors,25 we find that the use of antisense sequestration speeds up derepression, reducing t1/2 by 24% from 23 to 17 h (Figure 3C, noting the position of the red caret, and Movie S1). However, the use of gRNA sequestration increases the response time following the addition of inducer by 15%, suggesting that antisense sequestration may interfere with the repression dynamics even at high gRNA levels.
We next investigated whether regulatory feedback further improves the induction/repression dynamics. Figure 3C shows that gRNA sequestration further reduces the derepression time when aTc is removed, decreasing it by 33% with respect to the original inverter. In addition, the use of the positive feedback mechanism completely restores the induction response time and appears to nullify the increase in response time associated with gRNA sequestration. Thus, our single-cell results show that a circuit that contains both antisense sequestration and regulatory feedback displays the highest dynamic range and responds up to 33% faster than a basic 1× inverter.
Use of Antisense Sequestration Partially Restores the Logical Behavior of a Double Inverter
To evaluate whether antisense sequestration also improves the dynamic range and response time of multilayered circuits (i.e., circuits where the output of one logic node is used as the input of another logic node), we constructed a double inverter using the same basic approach to node arrangement as before (i.e., alternating logic and sinker nodes) (Figure 4A). Other authors have previously observed signal loss in multiple inverters,13,18 and such a result is expected due to dCas leak sensitivity.22 The double inverter, while in principle a simple circuit, is an excellent testbed for measuring how our antisense sequestration system alters circuit performance when nodes are used in series.
Figure 4.
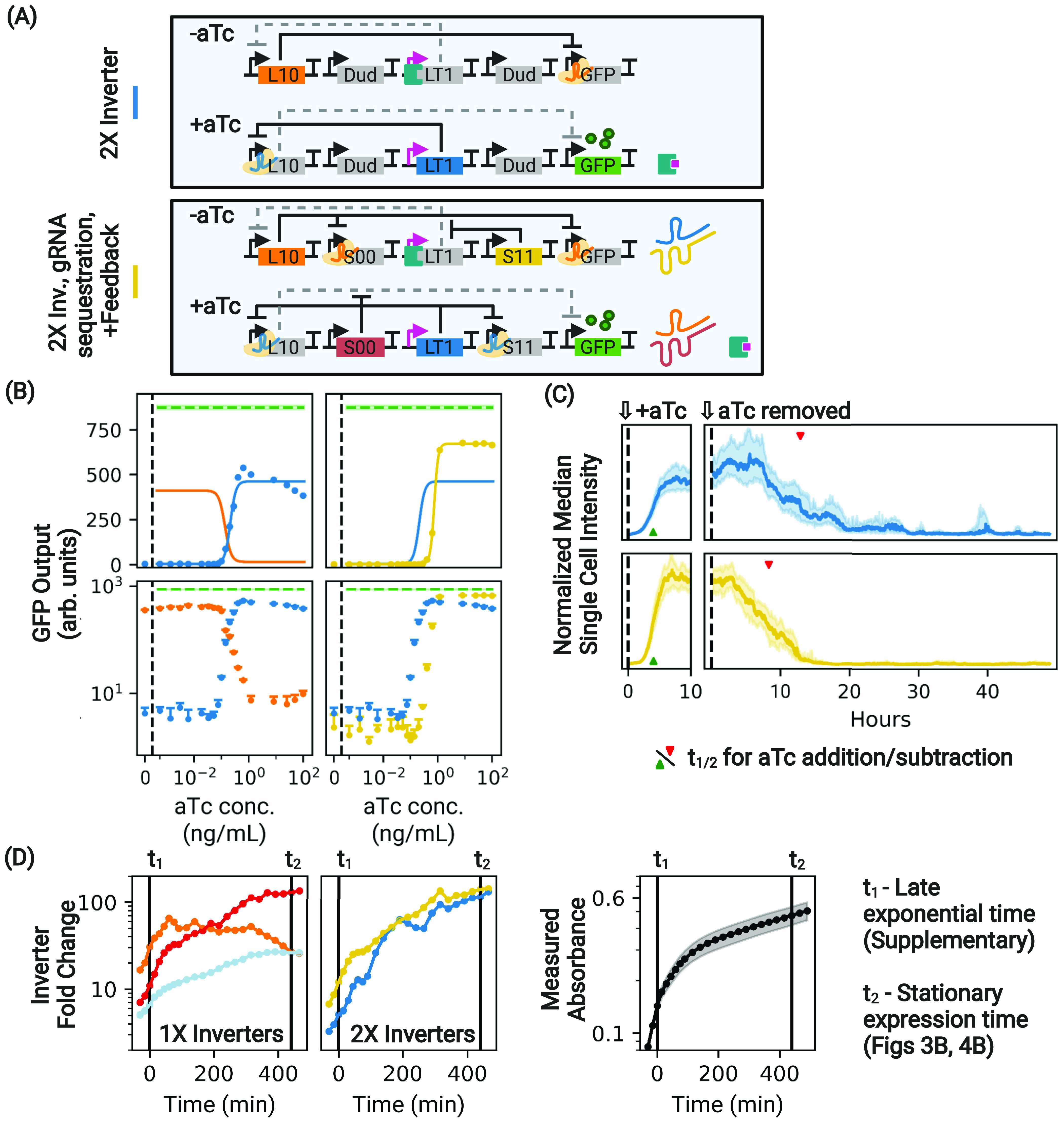
Antisense sequestration of gRNA partially restores the dynamic range of a 2× inverter. (A) Two variants of the 2× inverter controlled to have the same compositional context. For simplicity, the additional asRNA Dud node upstream of the circuit is not depicted. (B) Antisense sequestration of gRNA of the 2× inverter (yellow) partially restores the dynamic range by expanding the range of expression in both the ON and OFF states compared to the basic 2× inverter with no sequestration (blue). In linear space, the displayed error bars are ±1 standard deviation from threefold biological replicates. Performance is compared to the same GFP control (dotted green) used in Figure 3. Due to the extremely slow equilibration time of the 2× inverter, these constructs were run for additional time in order to be allowed to reach equilibrium (see Materials and Methods). (C) The same constructs, this time under the addition and subtraction of aTc in a microfluidic chamber. The presence of antisense sequestration speeds circuit response under aTc addition and removal with respect to the basic 2× inverter. Traces show the median intensities of single cells across all microfluidic channels. Shaded regions indicate ± quartiles. (D) Changes in inverter fold change (calculated as the maximum:minimum ratio of the Hill function fit) as functions of time. The indicated times (t1 and t2) correspond to measurements during the late exponential phase (see Figures S2 and S5) and stationary phase (Figures 3B and 4B), respectively.
We first observe that the basic double inverter performs extremely poorly during exponential growth, covering only 10.8% of the expected maximal range with respect to the control (Figure S2, blue), and vastly underperforms the single inverter. The performance of the double inverter does recover during stationary expression, although the maximal expression of a 2× inverter is comparable to that of the single inverter but still less than the expected maximal expression (Figure 4B, in blue).
Poor performance in a double inverter is expected to be the result of leaked gRNAs from the first node without inducer, which in turns drives leaky expression of the second node when it should normally be turned off. Thus, we next sought to determine whether the addition of antisense sequestration with feedback, our best-performing system for the 1× inverter, could improve a basic 2× inverter (Figure 4). Figure 4B shows that antisense sequestration restores a significant fraction of the dynamic range by expanding the span of expression at both low and high levels of induction. While the absolute dynamic range during exponential growth (42.1%) remains low, its dynamic range is nearly quadrupled compared to the original 2× inverter (Figure S5). The inverter fold change is essentially unchanged between these two 2× inverter variants but increases as cells transition from exponential to stationary expression (Figure 4D).
In addition, the results of single-cell measurements presented in Figure 4C show that the use of sequestration significantly speeds up the response of the 2× inverter circuit under aTc removal, reducing t1/2 by 36%, without significantly affecting its performance under aTc addition (Table 1). Therefore, even though the double inverter is extremely susceptible to slow dCas12a dynamics because a large population of programmed dCas12a needs to be replaced in order to reach equilibrium, our single-cell results show that antisense sequestration and regulatory feedback significantly improve both the dynamic range and response time of the 2× inverter when the inducer is added or removed.
Table 1. Circuit Response to Induction as Observed via Microfluidicsa.
|
t1/2 (min) |
||
|---|---|---|
| circuit | aTc addition | aTc removal |
| 1× inverter | 77 | 1405 |
| 1× inverter + gRNA sequestration | 89 | 1072 |
| 1× inverter + gRNA sequestration, Feedback | 73 | 940 |
| 1× inverter + mRNA sequestration, feedback | 69 | 2903 |
| 1× inverter + gRNA/mRNA sequestration, feedback | 68 | 1220 |
| 2× inverter | 240 | 772 |
| 2× inverter + gRNA sequestration, feedback | 242 | 498 |
The circuit response time t1/2 was observed in response to both aTc addition and aTc removal. t1/2 was calculated using a spline fit to the median induction curve (see Figures S8 and S9).
Discussion
The use of dCas proteins as programmable repressors holds great promise in synthetic biology, given that they are effective, highly engineerable, and orthogonal. However, these nucleases did not evolve for the purpose of transcriptional repression and suffer as an all-purpose transcription factor, particularly due to sensitivity to leak. Inheritance of leak between upstream and downstream nodes drives poor performance in dCas-based systems, causing oscillators to not oscillate and toggle switches to not toggle.22 Hence, a CRISPRi-based system that improves leak tolerance could fix these problems without sacrificing programmability or orthogonality. Furthermore, increasing the effectiveness of dCas-based transcriptional regulators allows us to free up the use of a diverse but limited set of inducible sensors4 for sensing rather than internal logic processing components.
Our system improves on the suboptimal performance of CRISPRi-based circuits by dealing with circuit leak directly by removing leaked transcripts from the system, similar to “sponge sites” present in natural systems that sequester excess transcription factors via DNA sites in the genome28−31 or RNAs.32 Because dCas12a associates functionally irreversibly33 with its target until displaced by DNA replication, just a few leaked gRNA transcripts can cause significant repression. It is advantageous to regulate gRNA directly, as RNA-based sequestration benefits from a separation of time scales since gRNA and asRNA diffusion is a fast process that should equilibrate before the demonstrably slow dCas search mechanic.34 Furthermore, in the context of CRISPRi-based gene circuits, dCas is physiologically expensive, and its maximum concentration is a limiting factor;18 nucleases such as dCas9 are even toxic in some bacteria when highly expressed.35 By contrast, RNA transcripts are physiologically inexpensive to produce and destroy, unlike dCas proteins, a costly resource.
Since the reduction in leaked mRNAs in the 1× inverter is less dramatic during exponential growth (Figure S2), our results support the hypothesis that circuit performance is driven by cellular division time. When cells are rapidly dividing, dCas12a is kicked off its target by the DNA replication machinery at a higher rate, which in turn impedes complete repression of asRNA in the high-aTc state. However, when replication rates are low, the system reaches an equilibrium where asRNA production is kept at a low enough level to totally restore the full repression of the original inverter. This is evident in the evolution of the inverter fold change over the course of the experiment, depicted in Figure 4D, where the 1× inverter with sequestration and feedback dramatically outperforms the basic 1× inverter as the system transitions from expontential to stationary expression.
Dual CRISPRi/antisense RNA (asRNA) elements have been created previously as a means to more rapidly “derepress” CRISPRi nodes,25,36 counteracting excessive memory of initial repression by dCas. Microfluidic experiments allow us to study the performance of cells over long periods of time under constant growth conditions and variable exposure to induction. Our results show that dCas-based regulatory feedback can counteract the slowing of dCas repression by antisense sequestration without cost to beneficial speeding of derepression. Ultimately, we show that a 2× inverter that uses asRNA to limit the impact of transcription leaks responds far more quickly to aTc removal.
Remaining challenges include the sensitivity to the growth phase and the intrinsic complexity of adding an additional layer of feedback to CRISPRi. Recent work on this subject has often only considered expression during exponential growth stages. However, we believe that it is important to consider the performance of dCas circuits under dynamic growth conditions and at high densities, given the importance of host/circuit interactions in the performance of synthetic gene circuits.37,38 During stationary-phase growth with constant protein production,39 more dCas proteins find their cognate target, strengthening repression and thus leak sensitivity. Ultimately, it is important to understand how these circuits perform and can be improved under conditions that are more relevant in industrial applications.40
Our solution improves the performance of dCas circuits without sacrificing the attribute that makes them so desirable: programmability. However, this does come at the cost of increased complexity. Despite this, we believe that merging antisense sequestration with CRISPRi is necessary in order to mitigate potentially crippling issues with CRISPRi and other dCas-based transcription factors. Furthermore, the use of feedback in essential for robustness in engineered circuits.41 Especially with further advancement in the cell-free assembly of large plasmids (e.g., OriCiro42), it will only become easier to create larger systems of dCas synthetic gene networks.
In this work, we have demonstrated that antisense-RNA-based sponge sites can be used to reduce leak sensitivity for CRISPRi-based gene circuits, particularly during slow growth when leaked gRNA transcripts drive unintended repression and reduced dynamic range. While this work explores only the impacts of sequestration on NOT gate CRISPRi repressors, in principle this technique could also be used with CRISPRa.11,12 Additionally, this work could be combined with dCas degradation, which should in principle further reduce leak sensitivity,22 or dCas self-regulation, which could further fortify circuit performance.21 Overall, our system reduces leak sensitivity in CRISPRi systems, which will help to realize their potential to create complex and engineerable genetic systems.
Materials and Methods
Plasmid Assembly
The original CRISPRi and sinker nodes were ordered as gene blocks from IDT and inserted into pUC19 plasmid for ease of modification. Site-directed mutagenesis to modify the sequences was done exclusively using NEB’s Q5 Hot Start and NEB’s KLD (kinase, ligase, and dpn1 digestion) prior to transformation. Verification of correct sequences was done using Sanger sequencing via Cornell’s Genomics Facility. Modification of individual nodes was completed in pUC19 before digestion and insertion into the main experimental plasmid containing the circuit. Assembly of multinode circuits was done using standard molecular biology techniques for Escherichia coli using restriction enzyme digestion with AarI (Thermo Fisher) and BsaI-HFv2 (NEB). Ligation of digested components was done using NEB’s Instant Ligase. Colony PCR using NEB’s Taq polymerase was used to check for successful node insertion.
All cloning was done in NEB Stable in order to minimize possible recombination events due to the use of repetitive sequences. Cells used for cloning were cultured using liquid Terrific Broth (TB) (VWR) prepared using the manufacturer’s instructions. Plasmids were maintained using antibiotics (kanamycin, chloramphenicol, and ampicillin) as appropriate. Lysogeny broth (LB) (VWR) agar plates were used as a solid medium.
FnCas12a was made catalytically dead via D917A and E1006A mutations. All GFP sequences had an orthogonal ssrA tag43 for degradation, although this was not induced in the course of these experiments. Annotated plasmid sequences used in this study can be found in the Supporting Information and are hosted by Benchling.
Node Design
Generally, CRISPRi and antisense sequestration nodes are designed to be standard parts with comparable expression strength. The design of nodes used in this system is illustrated in Figure S1. Every gRNA-producing node (including the pTet logic node), termed a “logic” node, and every asRNA-producing node, termed a “sinker” node, shares the same strong promoter with conserved −35 and −10 promoter sites TTGACA and TAAAAT. The output promoter that drives GFP is designed to be weaker (−35 and −10 sites TTGTCA and TAAAAT), as expression of GFP by the strong promoter causes a fitness penalty. All of the nodes except those driven by pTet have a PAM site TTTG, which is necessary for dCas binding for logical control or feedback.
All of the nodes were inserted in a tandem orientation as indicated in Figure S1. Annotated versions of the logic node (https://benchling.com/s/seq-JItVzOqT6qBnrCk0aLcn), pTet-driven logic node (https://benchling.com/s/seq-gdhKvLmV6Xs6u2lbNLFb), and sinker node (https://benchling.com/s/seq-s6qBkmmvJ38VzyDX1RHB) are hosted via Benchling. Randomized 40 bp tags with fixed GC content were used to produce cognate asRNAs that orthogonally sequester gRNA sequences. Two asRNA sequences exhibited toxic effects when expressed and were not used further in the study (see Sinker Node Archetype in the Supporting Information). The HFQ tag used to facilitate sRNA sequestration was the micF M7.4 tag from ref (26).
Microplate Fluorescence Assays
All measurements of fluorescence were conducted using the GL002 strain unless otherwise noted. GL002 is a variant of the F3 strain with genomically integrated44 expression of tetR, lacI, and dCas12a. The F3 strain (Wakamoto Lab, University of Tokyo) contains knockouts of fliC, fimA, and flu that decrease cell aggregation and adhesion to surfaces.45 Cells were made electrocompetent, electroporated at 1800 V (BTX ECM399), and recovered in NEB Stable medium prior to plating for colony selection.
The experimental procedure was as follows. Electrocompetent GL002 cells were electroporated with the pSC101 plasmid containing a complete circuit and plated as described above. After colonies were visible on the next day, three different colonies were selected to inoculate three different 10 mL cell culture tubes, each with 2 mL of H medium with antibiotic (kanamycin), and grown to saturation overnight for 18–19 h in a shaker held at 37 °C. A 100 μL aliquot of this culture was then used to inoculate a tray containing 2 mL of H medium with antibiotic, which was shaken until homogeneous. Then 1 μL volumes were taken from this tray using a multipipettor and used to inoculate wells of a 96-well plate (VWR, cat. no. 10062-900) with 200 μL volumes of H medium, appropriate antibiotics, and inducer. A sacrificial border of 36 200 μL volumes surrounded the 60 wells used for each experiment on the plate to minimize evaporative losses. Quantitative measurements of fluorescence were made using a Synergy H1 hybrid multimode microplate reader (BioTek) with the temperature held at 37 °C and linear shaking at 10 s intervals. Top and bottom fluorescence measurements and 600 nm absorbance measurements were taken at 3 min intervals, although only the top measurements are reported here.
In experiments studying the 2× inverter, only 1 μL of overnight culture was used to inoculate the plate because of the anticipated extremely long equilibration time under aTc addition. All other experimental parameters were held constant.
H medium46 was used throughout these experiments because it is sufficiently rich yet minimally autofluorescent, easing microfluidic study. H medium is LB without yeast extract with 10 g/L tryptone (BD) and 8 g/L NaCl (VWR).
In order to account for small variations in inoculation volume, fluorescence curves were aligned using the absorbance measure. Curves were aligned to the time when they crossed an absorbance (corrected for medium absorbance) of 0.04, which corresponds to a standard optical density at 600 nm (OD600) of 0.16. Background fluorescence, calculated via the fluorescence of GL002 cells containing plasmid w37, which contains the pSC101 origin but lacks GFP, was subtracted. Fluorescence readings were smoothed using SciPy’s implementation of the Wiener filter.
Anhydrotetracycline (Alfa Aesar), used for pTet induction, was kept at a stock concentration of 100 ng/μL in a 50% ethanol solution and protected from light.
Induction Analysis
Induction curves were taken at slices in time with respect to the time when the alignment OD600 was reached (see Microplate Fluorescence Assays). The times were 60 and 500 min with respect to the alignment time for exponential and stationary expression, respectively. The induction curves were fit to a Hill function of form
| 1 |
The values of ymin and ymax were used for calculations of the absolute dynamic range and fold change.
Microfluidic Experimental Design
Dynamic induction experiments were performed in a microfluidic device with chambers of two sizes, with lateral dimensions (L × W) of 40.5 μm × 7.1 μm and 35 μm × 7.1 μm. For clarity, only data from the shorter chambers are reported in this study.
As in prior experiments, plasmids containing the circuit of interest were electroporated into the GL002 strain of E. coli and grown on a plate overnight. The following morning, the cells were inoculated and grown in 3 mL of H medium with kanamycin to an OD600 of 0.5, concentrated by centrifugation, and pipetted into the plasma-cleaned microfluidic device. The device was then placed in an imaging setup with the temperature held at 37 °C. A bottle of fresh medium (H medium + 1× kanamycin + bovine serum albumin (100 mg/L) ± aTc) pressurized to 5 psi was used to deliver a constant flow of fresh medium (100 mL/day) to the cells.
Cells were introduced and allowed to populate microfluidic chambers. For all constructs, cells were inoculated in the absence of inducer and allowed to equilibrate. After the inducer was added, the system was allowed to equilibrate again, and then the inducer was taken away so that we could measure the return to the initial state (Figure S7). The full induction was run over the course of at least 80 h in order to allow the system to reach equilibrium initially, after aTc induction, and allow us to measure the response time when aTc was removed. The response time t1/2 is defined as the time taken to reach halfway between the equilibrium minimum and maximum levels of expression in linear space. Expression levels (as observed in the microfluidic device) and the measured response times are included in Table 1.
Single cells were resolved by epifluorescence microscopy of sf-GFP with a 100×, 1.4 NA apochromat Leica objective. We typically observed the circuits in their noninduced state overnight, then added aTc to induce the circuit for 23 h ± 15 min, and finally recorded the recovery after removal of aTc for 40 to 60 h. We monitored 20 to 40 chambers in parallel in a given experiment.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.2c00155.
Movie S1 showing a side-by-side comparison of circuit performance under induction (MP4)
Supplementary text on mRNA sequestration; supplementary figures showing node design, additional results describing the effects of sequestration during exponential growth, microfluidic device design, and additional traces from microfluidic study of inverter induction; and a description of Movie S1 (PDF)
Oligonucleotides used in this study and links to plasmids and node sequences hosted on Benchling (XLSX)
Author Contributions
D.A.S. and G.L. designed the study. Experiments were performed by D.A.S. and L.B.C. D.A.S. wrote the manuscript. All of the authors participated in editing and proofreading the manuscript.
Support for this work was provided via NIH 1R35 GM133759 Maximizing Investigators’ Research Award (MIRA). This work was performed in part at the Cornell NanoScale Facility, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant NNCI-2025233).
The authors declare no competing financial interest.
Supplementary Material
References
- Potvin-Trottier L.; Lord N. D.; Vinnicombe G.; Paulsson J. Synchronous long-term oscillations in a synthetic gene circuit. Nature 2016, 538, 514–517. 10.1038/nature19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner T. S.; Cantor C. R.; Collins J. J. Construction of a genetic toggle switch in Escherichia coli. Nature 2000, 403, 339–342. 10.1038/35002131. [DOI] [PubMed] [Google Scholar]
- Nielsen A. A. K.; Voigt C. A. Multi-input CRISPR/Cas genetic circuits that interface host regulatory networks. Mol. Syst. Biol. 2014, 10, 763. 10.15252/msb.20145735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer A. J.; Segall-Shapiro T. H.; Glassey E.; Zhang J.; Voigt C. A. Escherichia coli “Marionette” strains with 12 highly optimized small-molecule sensors. Nat. Chem. Biol. 2019, 15, 196–204. 10.1038/s41589-018-0168-3. [DOI] [PubMed] [Google Scholar]
- Chen Y.-J.; Liu P.; Nielsen A. A. K.; Brophy J. A. N.; Clancy K.; Peterson T.; Voigt C. A. Characterization of 582 natural and synthetic terminators and quantification of their design constraints. Nat. Methods 2013, 10, 659–664. 10.1038/nmeth.2515. [DOI] [PubMed] [Google Scholar]
- Qi L. S.; Larson M. H.; Gilbert L. A.; Doudna J. A.; Weissman J. S.; Arkin A. P.; Lim W. A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson M. H.; Gilbert L. A.; Wang X.; Lim W. A.; Weissman J. S.; Qi L. S. CRISPR interference (CRISPRi) for sequence-specific control of gene expression. Nat. Protoc. 2013, 8, 2180–2196. 10.1038/nprot.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. K.; Kim H.; Ahn W.-C.; Park K.-H.; Woo E.-J.; Lee D.-H.; Lee S.-G. Efficient Transcriptional Gene Repression by Type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synth. Biol. 2017, 6, 1273–1282. 10.1021/acssynbio.6b00368. [DOI] [PubMed] [Google Scholar]
- Specht D. A.; Xu Y.; Lambert G. Massively parallel CRISPRi assays reveal concealed thermodynamic determinants of dCas12a binding. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 11274–11282. 10.1073/pnas.1918685117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jusiak B.; Cleto S.; Perez-Piñera P.; Lu T. K. Engineering Synthetic Gene Circuits in Living Cells with CRISPR Technology. Trends Biotechnol. 2016, 34, 535–547. 10.1016/j.tibtech.2015.12.014. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Wan X.; Wang B. Engineered CRISPRa enables programmable eukaryote-like gene activation in bacteria. Nat. Commun. 2019, 10, 3693. 10.1038/s41467-019-11479-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana J.; Dong C.; Kiattisewee C.; Chavali V. P.; Tickman B. I.; Carothers J. M.; Zalatan J. G. Effective CRISPRa-mediated control of gene expression in bacteria must overcome strict target site requirements. Nat. Commun. 2020, 11, 1618. 10.1038/s41467-020-15454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didovyk A.; Borek B.; Hasty J.; Tsimring L. Orthogonal Modular Gene Repression in Escherichia coli Using Engineered CRISPR/Cas9. ACS Synth. Biol. 2016, 5, 81–88. 10.1021/acssynbio.5b00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cress B. F.; Jones J. A.; Kim D. C.; Leitz Q. D.; Englaender J. A.; Collins S. M.; Linhardt R. J.; Koffas M. Rapid generation of CRISPR/dCas9-regulated, orthogonally repressible hybrid T7-lac promoters for modular, tuneable control of metabolic pathway fluxes in Escherichia coli. Nucleic Acids Res. 2016, 44, 4472–4485. 10.1093/nar/gkw231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Moreno J.; Tasiudi E.; Stelling J.; Schaerli Y. Multistable and dynamic CRISPRi-based synthetic circuits. Nat. Commun. 2020, 11, 2746. 10.1038/s41467-020-16574-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo J.; Yuan R.; Sánchez C.; Paulsson J.; Silver P. A. Toward a translationally independent RNA-based synthetic oscillator using deactivated CRISPR-Cas. Nucleic Acids Res. 2020, 48, 8165–8177. 10.1093/nar/gkaa557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henningsen J.; Schwarz-Schilling M.; Leibl A.; Gutierrez J.; Sagredo S.; Simmel F. C. Single Cell Characterization of a Synthetic Bacterial Clock with a Hybrid Feedback Loop Containing dCas9-sgRNA. ACS Synth. Biol. 2020, 9, 3377–3387. 10.1021/acssynbio.0c00438. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Voigt C. A. Engineered dCas9 with reduced toxicity in bacteria: implications for genetic circuit design. Nucleic Acids Res. 2018, 46, 11115–11125. 10.1093/nar/gky884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayanthi S.; Nilgiriwala K. S.; Del Vecchio D. Retroactivity Controls the Temporal Dynamics of Gene Transcription. ACS Synth. Biol. 2013, 2, 431–441. 10.1021/sb300098w. [DOI] [PubMed] [Google Scholar]
- Brophy J. A. N.; Voigt C. A. Principles of genetic circuit design. Nat. Methods 2014, 11, 508–520. 10.1038/nmeth.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-H.; Bellato M.; Qian Y.; Cárdenas P.; Pasotti L.; Magni P.; Del Vecchio D. dCas9 regulator to neutralize competition in CRISPRi circuits. Nat. Commun. 2021, 12, 1692. 10.1038/s41467-021-21772-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clamons S. E.; Murray R. M. Modeling Dynamic Transcriptional Circuits with CRISPRi. bioRxiv 2017, 10.1101/225318. [DOI] [Google Scholar]
- Zetsche B.; Gootenberg J. S.; Abudayyeh O. O.; Slaymaker I. M.; Makarova K. S.; Essletzbichler P.; Volz S. E.; Joung J.; van der Oost J.; Regev A.; Koonin E. V.; Zhang F. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz R.; Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997, 25, 1203–1210. 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. J.; Hoynes-O’Connor A.; Leong M. C.; Moon T. S. Programmable control of bacterial gene expression with the combined CRISPR and antisense RNA system. Nucleic Acids Res. 2016, 44, 2462–2473. 10.1093/nar/gkw056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoynes-O’Connor A.; Moon T. S. Development of Design Rules for Reliable Antisense RNA Behavior in E. coli. ACS Synth. Biol. 2016, 5, 1441–1454. 10.1021/acssynbio.6b00036. [DOI] [PubMed] [Google Scholar]
- Møller T.; Franch T.; Højrup P.; Keene D. R.; Bächinger H. P.; Brennan R. G.; Valentin-Hansen P. Hfq: A Bacterial Sm-like Protein that Mediates RNA-RNA Interaction. Mol. Cell 2002, 9, 23–30. 10.1016/S1097-2765(01)00436-1. [DOI] [PubMed] [Google Scholar]
- Lee T.-H.; Maheshri N. A regulatory role for repeated decoy transcription factor binding sites in target gene expression. Mol. Syst. Biol. 2012, 8, 576. 10.1038/msb.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster R. C.; Weinert F. M.; Garcia H. G.; Song D.; Rydenfelt M.; Phillips R. The Transcription Factor Titration Effect Dictates Level of Gene Expression. Cell 2014, 156, 1312–1323. 10.1016/j.cell.2014.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemme C. A.; Esadze A.; Iwahara J. Influence of Quasi-Specific Sites on Kinetics of Target DNA Search by a Sequence-Specific DNA-Binding Protein. Biochemistry 2015, 54, 6684–6691. 10.1021/acs.biochem.5b00967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemme C. A.; Nguyen D.; Chattopadhyay A.; Iwahara J. Regulation of transcription factors via natural decoys in genomic DNA. Transcription 2016, 7, 115–120. 10.1080/21541264.2016.1188873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriss G. R.; Cooper T. A. Protein sequestration as a normal function of long noncoding RNAs and a pathogenic mechanism of RNAs containing nucleotide repeat expansions. Hum. Genet. 2017, 136, 1247–1263. 10.1007/s00439-017-1807-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strohkendl I.; Saifuddin F. A.; Rybarski J. R.; Finkelstein I. J.; Russell R. Kinetic Basis for DNA Target Specificity of CRISPR-Cas12a. Mol. Cell 2018, 71, 816–824. 10.1016/j.molcel.2018.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. L.; Leroy P.; Unoson C.; Fange D.; Ćurić V.; Lawson M. J.; Elf J. Kinetics of dCas9 target search in Escherichia coli. Science 2017, 357, 1420–1424. 10.1126/science.aah7084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S.; Choe D.; Lee E.; Kim S. C.; Palsson B.; Cho B.-K. High-Level dCas9 Expression Induces Abnormal Cell Morphology in Escherichia coli. ACS Synth. Biol. 2018, 7, 1085–1094. 10.1021/acssynbio.7b00462. [DOI] [PubMed] [Google Scholar]
- Mückl A.; Schwarz-Schilling M.; Fischer K.; Simmel F. C. Filamentation and restoration of normal growth in Escherichia coli using a combined CRISPRi sgRNA/antisense RNA approach. PLoS One 2018, 13, e0198058 10.1371/journal.pone.0198058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolados E.-M.; Weisse A. Y.; Ceroni F.; Oyarzún D. A. Growth Defects and Loss-of-Function in Synthetic Gene Circuits. ACS Synth. Biol. 2019, 8, 1231–1240. 10.1021/acssynbio.8b00531. [DOI] [PubMed] [Google Scholar]
- Tan C.; Marguet P.; You L. Emergent bistability by a growth-modulating positive feedback circuit. Nat. Chem. Biol. 2009, 5, 842–848. 10.1038/nchembio.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gefen O.; Fridman O.; Ronin I.; Balaban N. Q. Direct observation of single stationary-phase bacteria reveals a surprisingly long period of constant protein production activity. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 556–561. 10.1073/pnas.1314114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser F.; Broers N. J.; Hartmans S.; Tamsir A.; Kerkman R.; Roubos J. A.; Bovenberg R.; Voigt C. A. Genetic Circuit Performance under Conditions Relevant for Industrial Bioreactors. ACS Synth. Biol. 2012, 1, 555–564. 10.1021/sb3000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall-Shapiro T. H.; Sontag E. D.; Voigt C. A. Engineered promoters enable constant gene expression at any copy number in bacteria. Nat. Biotechnol. 2018, 36, 352–358. 10.1038/nbt.4111. [DOI] [PubMed] [Google Scholar]
- Hasebe T.; Narita K.; Hidaka S.; Su’etsugu M. Efficient Arrangement of the Replication Fork Trap for In Vitro Propagation of Monomeric Circular DNA in the Chromosome-Replication Cycle Reaction. Life 2018, 8, 43 10.3390/life8040043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron D. E.; Collins J. J. Tunable protein degradation in bacteria. Nat. Biotechnol. 2014, 32, 1276–1281. 10.1038/nbt.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre F.; Cui L.; Priest D. G.; Endy D.; Dodd I. B.; Shearwin K. E. One-Step Cloning and Chromosomal Integration of DNA. ACS Synth. Biol. 2013, 2, 537–541. 10.1021/sb400021j. [DOI] [PubMed] [Google Scholar]
- Lambert G.; Kussell E. Memory and Fitness Optimization of Bacteria under Fluctuating Environments. PLOS Genet. 2014, 10, e1004556 10.1371/journal.pgen.1004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbing K.; Brent R. Recipes and tools for culture of Escherichia coli. Curr. Protoc. Mol. Biol. 2019, 125, e83 10.1002/cpmb.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.