

Abstract
Chimeric antigen receptor (CAR) T cell therapy, in which a patient’s own T lymphocytes are engineered to recognize and kill cancer cells, has achieved remarkable success in some hematological malignancies in preclinical and clinical trials, resulting in six FDA-approved CAR-T products currently available in the market. Once equipped with a CAR construct, T cells act as living drugs and recognize and eliminate the target tumor cells in an MHC-independent manner. In this review, we first described all structural modular of CAR in detail, focusing on more recent findings. We then pointed out behind-the-scene elements contributing to CAR expression and reviewed how CAR expression can be drastically affected by the elements embedded in the viral vector backbone.
Keywords: Chimeric antigen receptor, Cancer immunotherapy, Single-chain variable fragment, Nanobody, Lentiviral vectors, Signal peptide
Introduction
Chimeric antigen receptor (CAR) T cell therapy, in which a patient’s own T lymphocytes are engineered to recognize and kill cancer cells, has achieved remarkable success in some hematological malignancies in preclinical and clinical trials, resulting in six FDA-approved CAR-T products currently available in the market [1–6] (Table 1) (Fig. 1). CARs are synthetic immune receptors that connect a single-chain variable fragment (scFv), derived from a monoclonal antibody, to T cell signaling domains to eradicate tumor cells independent of the major histocompatibility complex (MHC). Despite the impressive rate of complete remission (CR) in patients with certain B-cell malignancies [7–9], there are still some concerns about treatment failure associated with the low efficacy of CAR-T cells [10–13]. In the current review, we will discuss how the molecular components of CAR construct and elements of lentiviral vector backbone plasmid transferring CAR expression cassette can contribute to CAR-T cell therapy success or failure.
Table 1.
FDA-approved CAR-T products
| Generic Name | Tisagenlecleucel | Axicabtagene ciloleucel | Brexucabtagene autoleucel | Lisocabtagene maraleucel | Idecabtagene vicleucel | Ciltacabtagene autoleucel |
|---|---|---|---|---|---|---|
| Trade name | KYMRIAH™ | YESCARTA™ | TECARTUS™ | BREYANZI® | ABECMA® | CARVYKTI |
| Company | Novartis | Gilead/Kite | Gilead/Kite | Bristol-Myers Squibb | Bristol-Myers Squibb | Janssen-Cilag International NV |
| Approval date | Aug-2017 | Oct-2017 | Jul-2020 | Feb-2021 | Mar-2021 | Feb-2022 |
| Target patients |
R/R B-ALL R/R DLBCL |
R/R DLBCL R/R NHL |
R/R MCL | R/R LBCL | R/R MM | R/R MM |
| Targeted antigen | CD19 | CD19 | CD19 | CD19 | BCMA | BCMA |
R/R Refractory/Relapsed, ALL Acute Lymphoblastic Leukemia, DLBCL Diffuse large B cell lymphoma, NHL Non-Hodgkin lymphoma, MCL Mantle cell lymphoma, LBCL Large B cell lymphoma, MM Multiple myeloma, BCMA B cell maturation antigen
Fig. 1.

A schematic picture of the structural elements of six FDA-approved CAR-T products. Tisagenlecleucel (a), Axicabtagene ciloleucel (b), Brexucabtagene autoleucel (c), Lisocabtagene maraleucel (d), Idecabtagene vicleucel (e), Ciltacabtagene autoleucel (f)
Structural elements contribute to CAR’s potency
Once equipped with a CAR, T cells, known as CAR-T cells, act as living drugs and recognize and eliminate the target tumor cells. The conventional CAR structure consists of three modular components: the ectodomain, the transmembrane domain, and the endodomain, each of which has specific components and functions and thus the potential to be optimized.
Ectodomain
The ectodomain is the domain of a membrane protein that is outside the cytoplasm and exposed to the extracellular space. The ectodomain in the case of CAR consists of the antigen recognition region and hinge domain.
Antigen recognition domain
The predominant type of antigen-recognition domain in CARs is variable fragments of a conventional monoclonal antibody, commonly an IgG type, connected by a short peptide linker or disulfide bond and then called single-chain variable fragment or scFv [14]. Four crucial features of scFv that may affect CAR-T cell therapy clinical outcomes are affinity, immunogenicity, specificity, and structure. CAR binding affinity, along with its expression levels, determines the antigen-binding characteristics of the CAR and the efficacy of target cell recognition [15]. The CAR affinity must be sufficiently high to recognize the target antigen [16] but not too high to trigger on-target off-tumor toxicities [17]. It can be fine-tuned according to the target antigen density on tumor cells [18, 19]. Constructing CARs with the appropriate affinity to discriminate between malignant and normal cells without rendering any toxicity is crucial. Several studies have demonstrated that CAR with reduced affinity can efficiently distinguish tumors from normal tissues that express the same antigen at lower levels while maintaining potent antitumor activity and prolonged persistence [18–22].
Most CARs developed and tested in clinical studies have utilized scFvs, usually derived from murine monoclonal antibodies [23, 24]. Both humoral and cellular responses triggered by the murine-derived scFvs included in the CAR structure may lead to quick clearance of the CAR-T cells from circulation and thus increase the risk of relapse [25, 26]. Therefore, it seems that developing humanized or fully human scFvs, which are likely to be less immunogenic, would avert the anti-CAR responses and, consequently, circumvent treatment failure [27]. However, these scFvs may still have non-self-sequences since these variable fragments are usually generated through multiple recombination events and somatic hypermutation or fused at junctions that do not typically exist [25]. Lymphodepleting chemotherapy treatment before CAR-T cell infusion has a beneficial effect on reducing anti-CAR-T immune responses [28, 29].
An ideal target antigen must be expressed with high specificity and coverage in tumor cells. Most antigens recognized by CARs are not tumor-specific (TSA), restricted to the tumor cells, but are tumor-associated (TAA), expressed on the surface of normal tissues as well, albeit at a low level. Targeting a TAA, in most cases, leads to unwanted on-target off-tumor toxicities, such as B cell aplasia, resulting from a direct attack on healthy tissues having a shared expression of the targeted antigen [30–32]. Therefore, finding a target with high specificity to tumor cells requires researchers to implement a comprehensive assessment. Tumor antigen heterogeneity, observed predominantly in solid tumors such as malignant mesothelioma (MM), glioblastoma multiforme (GBM), and so forth, is also one of the main impediments restricting the efficacy of monovalent immunotherapeutic strategies directed against only one particular antigen [33]. Therefore, efforts to develop CAR-T cell immunotherapy must confront this high diversity of potential target antigen expression; otherwise, treatment failure or tumor recurrence may occur [33]. Designing CARs with two scFvs in which two corresponding scFvs target two different antigens, such as tandem CARs (TanCAR), dual CARs, loop CARs, AND-gate CARs (synNotch-CAR), and inhibitory CARs (iCARs), is a common strategy to improve the specificity of CARs [30, 34]. In the TanCAR concept, also referred to as OR-gate CAR, two different scFvs are connected outside the cell (in series), usually by a glycine-serine linker [35, 36]. The TanCAR can be activated when any one of the scFvs binds to a target antigen. When two scFvs simultaneously bind to their respective target antigens, the TanCAR will be activated and produce synergistic effects, which results in further activation of CAR-T cells and boosting their tumor-killing ability [35–37]. Dual CAR-T cells refer to the expression of two CARs in the same T cell, with each CAR having its own signaling function and distinct extracellular antigen recognition domains [38–40]. Wang et al. showed that dual CAR-T cells targeting IL-23 and PSMA secreted more cytokines in vitro and functioned significantly better in mouse models of prostate cancer compared to TanCAR-T cells expressing the same scFvs in a single CAR [37]. Like TanCAR-T cells, loop CAR-T cells consist of two scFvs in a single CAR molecule. In TanCARs, the VL-VH of one scFv is directly linked to the VL-VH of the other scFv, whereas the loop structure is formed with the VL-VH of one scFv separated by the VL-VH of the other scFv [41]. It has been shown that loop CAR-T cells are more effective than the TanCAR-T cells in eradicating tumor cells and prolonging survival in xenograft models [42]. Loop CAR-T cells targeting CD19 and CD22 showed promising results in phase II clinical trial (NCT03196830) of patients with relapsed/refractory (R/R) non-Hodgkin lymphoma (NHL) [43] and in phase I clinical trial (NCT03233854) of adults with R/R acute lymphoblastic leukemia (ALL) and large B-cell lymphoma (LBCL) [44]. A schematic picture of TanCAR, dual CAR, and loop CAR has been illustrated in Fig. 2.
Fig. 2.

A schematic picture of TanCAR (a), DualCAR (b), and Loop CAR (c). In TanCAR, two different scFvs are connected outside the cell (in series), usually by a glycine-serine linker. TanCAR can be activated when any one of the scFvs binds to a target antigen. Dual CAR-T cells refer to the expression of two CARs in the same T cell, with each CAR having its own signaling function and distinct extracellular antigen recognition domains. Like TanCAR-T cells, loop CAR-T cells consist of two scFvs in a single CAR molecule. In TanCARs, the VL-VH of one scFv is directly linked to the VL-VH of the other scFv, whereas the loop structure is formed with the VL-VH of one scFv separated by the VL-VH of the other scFv
In contrast to OR-gate CARs, AND-gate CARs, with synthetic Notch (synNotch) receptors [45] as the core element, requires T cells to sense two antigens to activate (Fig. 3a). When the synNotch receptor recognizes a target antigen by its extracellular recognition domain, the transcriptional activator domain of the receptor is released, which can then enter the nucleus and drive the expression of a CAR for a second antigen. These combinatorially gated T cells has shown a remarkable degree of therapeutic discrimination both in vitro and in vivo [46]. Like dual CAR-T cells, iCAR-T cells express two CARs on the same T cells, including a typical tumor-antigen-specific CAR and an iCAR [47]. The iCAR consists of an scFv specific to the antigens expressed exclusively on normal tissue and an inhibitory signaling domain of immunoinhibitory receptors (programmed cell death protein-1 (PD-1) and cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4)) to restrict T cell activity despite concurrent engagement of activating receptors, allowing T cells to distinguish target cells from the off-target cells [47, 48] (Fig. 3b).
Fig. 3.

A schematic picture of synNotch-CAR (a) and iCAR (b). SynNotch-CARs require T cells to sense two antigens to activate. SynNotch receptors are engineered to sense a target antigen on the surface of tumor cells and induce the expression of a CAR specific to a second tumor antigen. iCAR-T cells express two CARs on the same T cells, including a typical tumor-antigen-specific CAR and an iCAR. Recognizing a target antigen on the surface of a normal cell by iCAR leads to the inhibition of the second CAR. iCAR, Inhibitory CAR; TAA, Tumor-Associated Antigen; A and B, Target Antigens
ScFvs have a high propensity for self-aggregation, resulting from their structure, which leads to ligand-independent constitutive signaling, known as tonic signaling [49]. This tonic activation can induce early exhaustion of CAR-T cells and, consequently, limit its anti-tumor efficacy [50]. Amino acid point mutations or substitutions can partially correct the tonic signaling of CARs caused by its scFv [50, 51]. Although scFvs are currently the most often used antigen-recognition domain in CARs, some associated drawbacks, such as immunogenicity or their tendency for aggregation, may pose potential risks and challenges in their applications. Therefore, alternative antigen-binding domains would be beneficial [52]. The nanobody, also known as the VHH domain, is the variable domain of the heavy-chain-only antibodies (HcAb) naturally found in sera of camelids [53]. The VHH domain is the smallest fragment with antigen-binding capability, comparable to conventional antibodies in affinity and specificity [54]. Properties such as small size, high solubility and stability, low immunogenicity, high tissue penetration, and no need for the additional folding and assembly steps or linker optimization due to the lack of variable light chain make nanobodies a promising alternative to scFvs in CARs [54]. Using the VHHs as antigen recognition domains for CAR-T cells appears to be more favorable than scFv, particularly for solid tumors, as they can access epitopes hard or impossible to reach by scFvs [55–57]. The first report about the successful use of nanobodies in the CAR constructs emerged from our lab, where CAR-modified T cells used an anti-MUC1 VHH as the target-binding domain [55]. Anti-MUC1 CAR-T cells showed increased proliferation and IL2 secretion upon the stimulation and could effectively kill MUC1-positive tumor cell lines [55]. For creating more complicated CARs, nanobodies are more favorable than scFvs, due to their compact size and lack of VL chain. Furthermore, the potential cross-pairing of VH and VL, commonly observed among two independent scFv molecules but not among nanobodies, may result in the affinity loss of these scFv-based CARs [58]. Interestingly, the sixth CAR-T product, recently approved by the US FDA for medical use in patients with R/R multiple myeloma (MM), utilizes two VHHs targeting two different B-cell maturation antigen (BCMA) epitopes [59] (Fig. 1). Search for finding antigen binding domains other than scFv has led to new ligand-based CARs. Recently, Wang et al. used the intrinsic binding properties of a natural toxin to develop targeted CAR-T cells. They incorporated chlorotoxin (CLTX), a 36-amino acid peptide isolated from the deathstalker scorpion venom previously used to deliver radiation therapy and imaging reagents to tumor sites of patients with GBM, into a CAR construct to redirect cytotoxic T cells against GBM cells. They found the antitumor effects of CLTX-CAR-T cells robust and specific while making negligible off-tumor toxicity [33]. Zetakine-CARs employ a membrane-tethered cytokine ligand as the antigen recognition domain [60, 61]. Brown et al. developed a zetakine-CAR targeting IL-13 receptor α2 (IL-13Rα2) by fusing a membrane-tethered IL13 ligand mutated at a single site (E13Y) domain to intracellular signaling domains to use for the treatment of recurrent GBM [62]. Moeller et al. also developed an IL3- specific zetakine-CAR targeting the alpha-chain of the IL3 cytokine receptor (CD123), a promising target for acute myeloid leukemia (AML) as it is overexpressed in both leukemic stem cells (LSCs) and blasts [61]. Other ligand-based CARs have been developed and tested in preclinical and clinical studies across a range of malignancies, including those incorporating FMS-like tyrosine kinase 3 ligand (FLT3L) to target FLT3-positive AML [63, 64], Natural Killer Group 2D (NKG2D) receptor to target NKG2D ligands on the surface of tumor cells [65], a proliferation-inducing ligand (APRIL), also known as TNFSF13, to target BCMA and transmembrane activator and calcium-modulator and cyclophilin ligand (TACI), two receptors implicated in the pathogenesis of multiple myeloma [66], and granulocyte-macrophage colony-stimulating factor (GM-CSF) to target the GM-CSF receptor (CD116) involved in the pathogenesis of juvenile myelomonocytic leukemia (JMML) [67].
Linker
The antigen-binding domain of CARs is composed of two distinct critical components; the scFv and the linker, whose rational design is often neglected while designing a CAR construct. The content and length of linkers strongly influence the structural and functional properties of CARs. Although linkers are highly divergent in their sequences, those commonly used in CARs contain repeats of glycine (Gly) and serine (Ser) residues to provide the flexibility necessary for antigen-binding sites to change conformation [68] and maintain good stability in aqueous solutions [69]. The combination of Gly and Ser residues also prevents the formation of secondary structures and reduces the likelihood of the linker interfering with the folding and function of the scFv [70]. The length of the linker between V-domains affects various structural characteristics of scFv, such as size, flexibility, and valency. A linker greater than 12 residues helps V-domains fold in the natural orientation and form a monovalent antigen-binding site [71]. In contrast, shorter linkers restrict V-domain flexibility, prevent intramolecular variable chain pairing, and instead favor intermolecular oligomerization and formation of scFv multimers [72–74]. A recent study, through detailed interrogation, demonstrated how linker length drastically affects the clinical outcomes of CAR-T cell therapy. Singh et al. set out a study at the University of Pennsylvania (Penn)/ Children’s Hospital of Philadelphia (CHOP) to evaluate CD22 as a target for CAR-T cell therapy. After analyzing the results of two pilot clinical trials (NCT02650414 and NCT02588456) using anti-CD22 CAR-T in six pediatric and three adult patients with R/R ALL, they found that response rates were unexpectedly lower compared with results from a similar trial at the National Cancer Institute (NCI) [11, 75]. Further investigation revealed similar patient characteristics, manufacturing processes, and CAR constructs in these clinical trials; the only difference was the length of the scFv linker; the anti-CD22 CAR evaluated by the NCI joined the V-domains using a five-amino acid linker ((Gly4Ser), CAR22-short), while the trials at the Penn / CHOP connected the V-chains using a 20-aa linker ((Gly4Ser)4, CAR22-long) [76]. They found that the CAR22-long was monomeric, distributed uniformly on the cell surface, while the CAR22-short was homodimerized and clustered on the cell surface. The CAR22-short demonstrated greater F-actin polarization and perforin accumulation at the immune synapse (IS), more durable immune synapse formation, and enhanced downstream receptor activation. The CAR22-short also secreted more IFN-γ and drove enhanced cytotoxicity against target tumor cells than its long counterpart. Antigen-independent signaling associated with linker-driven clustering causes CAR22-short T cells to be ‘primed’ and rapidly initiate intracellular signaling and activate immune response programs upon target engagement. Interestingly, the tonic signaling associated with the shortened linker was beneficial for CD22 CARs bearing the 4-1BB costimulatory domain, while replacing 4-1BB with CD28 led to T cell exhaustion and dysfunction. Furthermore, they found that the association between linker length and clustering is not a universal phenomenon. In CARs targeting CD19, shortening the scFv linker did not result in cell surface clustering and tonic signaling [76].
Hinge domain
A domain usually overlooked while evaluating CAR functionality is the hinge domain (HD). The HD in CARs serves as a spacer that holds scFvs beyond the plasma membrane and gives them the flexibility necessary to access antigen epitopes on the surface of target cells [77, 78]. The origin, length, flexibility, and composition of the HD influence CAR antitumor activity and the occurrence of side effects as well [79–81]. The HDs typically used in CARs are Ig-based, originating from the constant region of human immunoglobulin molecules, or non-Ig based derived from the components naturally expressed on T cells. Ig-based spacers commonly use the hinge-CH2-CH3 regions of IgG molecules, mostly IgG1 and IgG4 [82–85]. Accumulated research studies have shown that effective antigen recognition by CAR relies on the length of HD and the target epitope distance from the cell membrane [78, 86, 87]. A study by Qin et al. showed that hinge-containing anti-CD19 CAR-T cells had a tumor-eradication capacity similar to their hinge-free counterparts. These results suggested that for those antigen epitopes like CD19, which are membrane-distal and expressed on the cell surface with high density, embedding a hinge domain in CAR construct does not improve their killing activities [78]. However, they found that hinge incorporation improved the expansion and migratory capacity of anti-CD19 CAR-T cells. They also achieved similar results with anti-mesothelin CAR-T cells [78]. To access antigen epitopes residing closer to the membrane of target cells or those embedded within heavily glycosylated structures, a hinge domain with proper length is needed to decrease the distance or amend the steric inhibitory effects between the scFv and its epitope [78, 86]. Hudecek et al. designed three CAR constructs bearing either a full-length or truncated IgG4-Fc as a spacer to study the effect of HD length on CAR antitumor activity [87]. All CAR constructs recognize a membrane-distal epitope of the receptor-tyrosine kinase-like orphan receptor 1 (ROR1). They found that HD length did not affect the CAR expression levels; however, CARs with intermediate and short spacer showed superior T-cell cytokine secretion and proliferation after target-antigen recognition [87]. Therefore, the HD length in CARs needs to be carefully tailored regarding the target epitope distance from the cell membrane to achieve an improved antitumor efficacy.
Several clinical studies have reported a lack of persistence in CAR-T cells bearing IgG-derived spacers [82, 88]. Studies have shown that the IgG-derived spacers have ligand-binding capacity. Several amino acid sequences within the CH2 domain can bind to Fcγ receptors (FcγRs) on the innate immune cells, including monocytes/ macrophages, dendritic cells (DC), neutrophils, and natural killer (NK) cells [89]. The Fc: FcγR binding may result in unwanted innate immune response, including antibody-dependent cell-mediated cytotoxicity (ADCC) and phagocytosis, which may, in turn, result in depletion of CAR-T cells having Ig-based spacers in their CAR constructs [90, 91]. The Fc: FcγR interaction may also lead to ligand-independent tonic signaling and subsequently activation-induced T-cell death (AICD) [90, 92]. Therefore, modification of the IgGs-derived spacers, such as replacing the IgG1-CH2 framework with the corresponding IgG2 amino acids [26, 29], which has a lower binding capacity to both human [32] and murine [31] FcγRs, or complete deletion of CH2 region may solve the problems [22]. Jonnalagadda et al. generated several anti-CD19 CARs with different IgG4-derived spacers; one with a nonmutated CH2 domain, one with complete deletion of the CH2 region, and the others with single- or double-point mutations in the CH2 region. They found that CAR with nonmutated CH2 failed to engraft and persist in xenograft models, probably due to Fc binding to FcγR. However, the engraftment and persistence were partially restored by blocking this interaction using intravenous immunoglobulin (IVIG) administration [91]. Moreover, they observed that double point mutations or complete deletion of the CH2 region resulted in an improved persistence and antitumor efficacy in xenograft models compared with CARs containing a nonmutated or single-mutated CH2 [91]. Thus, introducing mutations, truncations, or complete deletion in the IgG-derived spacers is inevitable to diminish the adverse consequences of Fc: FcγR binding and improve CAR persistence and efficacy in vivo [91, 93]. However, CAR-T cells with no in vivo therapeutic efficacy or persistence were reported even after similar modifications in their IgG1-derived spacers [94]. Breyanzi, the FDA-approved CAR product of Juno Therapeutics/Bristol Meyers Squibbs, of note, harbors a 12-amino acid IgG4-derived spacer without the CH2-CH3 sequence (IgG4 hinge only) [93, 95].
To minimize the possibility of potential immunological interactions elicited by the Ig-based spacers and attain the safety needed for clinical use, spacers derived from components naturally expressed on T cells, such as CD8 and CD28, can be incorporated into CAR structure [96]. Alabanza et al. investigated the effect of the hinge and transmembrane (TM) domains derived from human CD28 or CD8α on the biology of fully human or murine-derived CD28-based CARs targeting CD19. They found that regardless of scFv origin, both CARs with CD8α or CD28 HD/TMD showed similar expression levels on the T cell surface. Compared with CARs containing HD/TMD of CD8α, those with CD28 HD/TMD produced significantly higher inflammatory cytokines and underwent more AICD [79]. T cell exhaustion markers such as PD-1 and lymphocyte activation gene-3 (LAG-3) were also higher in CAR-T cells containing CD28 HD/TMD. Based on crystal structures, they realized that enhanced inflammatory cytokine production and AICD observed in CAR-T cells with CD28 HD/TMD resulted from an increased tendency of these CARs to dimerize compared with CARs containing CD8α HD/TMD. They also found that HD/TMD does not affect T cell memory phenotype [79].
In patients with a high tumor burden, CAR-T cell therapy may result in adverse reactions and side effects, such as cytokine release syndrome (CRS), associated with the over-activation of CAR-T cells [97–99]. Considering that the spacer appears to be involved in T cell activation and cytokine production, its modification may provide the safety and efficacy needed for patients with a high tumor burden. To decrease the over-activation of CAR-T cells, Zhang et al. removed two consecutive Gly residues in the CD8-derived spacer of a second-generation anti-CD19 4-1BB-based CAR to reduce spacer flexibility. They found that this modification resulted in better tumor control and lower release of inflammatory cytokines in vivo. Also, they observed a downward trend in tumor load and prolonged survival in xenograft models treated with CAR-T cells bearing a less flexible spacer [100]. In an endeavor to develop long spacer domains with a favorable functionality profile for membrane-proximal targets, Schafer et al. introduced a novel class of spacer derived from the Sialic acid-binding immunoglobulin-type lectins (Siglecs). A long spacer derived from Siglec showed potential cytotoxicity, and its performance was similar to the CD8α spacer in a CAR targeting CD20 in vitro and in vivo while maintaining a favorable cell phenotype profile and cytokine release pattern [94].
The spacer in the CAR construct can also be used for identification, purification, and in vivo tracking of CAR-positive subsets of T cells after engineering [96, 101]. Casucci et al. demonstrated that the incorporation of the nerve growth factor receptor (NGFR) as a spacer into the CAR backbone enables the enrichment of CAR-T cells before infusion into patients and facilitates the in vivo tracking, phenotypic characterization, and isolation of CAR-T cells for ex vivo analysis [101]. They also showed that NGFR, when incorporated into the CAR molecule, cannot trigger signaling upon in vivo growth factor encounter [101]. Bister et al. also inserted a CD34-derived spacer into the CAR backbone to facilitate the detection and enrichment of CAR-T cells before infusion. This spacer was functionally similar to the CD8 spacer in in-vitro and in-vivo experiments [96].
Consistent with studies concerning scFv-based CARs, spacers can affect the expression level and functional activity of nanobody-based CARs [102]. In our previous study, we incorporated three different spacers, including CH3–CH2-hinge and CH3–CH2-hinge-hinge regions derived from human IgG3 and the hinge region of the FcγRII, into anti-MUC1 CARs and observed a greater expression of CARs containing those spacers derived from human IgG3. We also observed that CARs having two repeats of hinge sequence in their spacers (CH3–CH2-hinge-hinge) showed more flexibility which may induce homodimerization and increase the avidity of CAR for the target antigen [102, 103].
Transmembrane domain
The transmembrane domain (TMD), like the hinge domain (HD), is a component in the CAR structure that connects the antigen recognition moiety to the intracellular signaling domain. It is mainly derived from type-I single-spanning proteins, such as CD3ζ, CD4, CD8α, or CD28. The TMD is primarily considered a structural block in the CAR that anchors the receptor in the cell membrane. However, the functional importance of TMD in CAR expression level and stability has been well-established [104, 105]. Fujiwara et al. studied the effect of HD and TMD, derived from various molecules, such as CD4, CD8α, or CD28, on the expression level and antigen-specific cytotoxic activity of CAR [104]. They found that the CAR expression level was enhanced much more in HD/TMD-modified than in HD-modified CARs, suggesting that CAR expression level and stability on the T cells were highly affected by TMD rather than HD [104]. A high surface expression was also reported by Zhang et al. following the incorporation of either CD8α or CD28 TMD into the CAR construct [106]. The TMD of CARs mediates CAR dimerization and interaction with endogenous proteins, forming dimers or trimers [79, 80, 105]. Annenkov et al. showed that CARs containing the transmembrane region of FcεRIγ mediate T cell activation by heterodimerizing with CD3ζ [107]. Bridgeman et al. demonstrated that embedding the transmembrane region of CD3ζ in the CAR structure facilitates signal transmitting and T cell activation via mediating the homodimerization of chimeric receptors or their interaction with the endogenous TCR [105, 108]. Alabanza et al. also showed that CD28-derived HD/TMD has a greater tendency to drive homodimer formation compared to CD8α HD/TMD. This homodimerization can cause an increased tonic signal and AICD in T cells expressing CD28-HD/TMD CARs [79]. Muller et al. found that CD28-TMD can also interact with the endogenous CD28 receptor and form CD28-CAR heterodimers [80]. The CD28-CAR dimers may cause higher on-target off-tumor toxicities by enhancing CAR sensitivity to ectopically expressed low-density antigens, such as the CD19 on brain mural cells [80, 109]. Nevertheless, Majzner et al. demonstrated that the CD28 HD/TMD provides a more stable and efficient immune synapse and decreases the antigen-density threshold for T-cell activation in CD19-specific CARs compared to their CD8 counterparts [110].
The CAR hinge and transmembrane regions can also influence CAR-T cell cytokine production [81]. To find how HD/TMD affects the expansion, cytokine production, and memory generation of CAR-T cells, Ying et al. incorporated CD8α-derived HD/TMD with different lengths into CAR. They found that a CAR harboring an 86-amino-acid HD/TMD produces potent antitumor responses without enhancing serum cytokine concentrations responsible for CRS and neurological toxicity. Thus, their results suggest that modification of CAR hinge and transmembrane regions can modulate cytokine secretion and help ameliorate CAR-T cell-associated toxicities [81]. Guedan et al. also showed that a third-generation CAR composed of ICOS and 4-1BB intracellular domains (ICDs) displayed superior antitumor activity and increased persistence in vivo only when the ICOS ICD was directly fused to an ICOS transmembrane domain [111]. The TMD of ICOS has a constitutive, albeit weak, association with the tyrosine kinase Lck. This association facilitates p85 recruitment to ICOS and subsequent PI3K activation. When incorporated into the CAR, ICOS TMD augments the proximal signaling output by providing an extra pool of Lck [112].
The TMD in the CAR structure can be designed according to the transmembrane-mediated interaction and functionality desired. Schmith et al. used the TMD of 4-1BB to form trimeric CARs, thus enhancing the antigen-binding capacity of the CAR and reducing antigen escape [113]. Wang et al. also generated a novel chimeric receptor in which the transmembrane and cytoplasmic domains of KIR2DS2, a stimulatory killer immunoglobulin-like receptor (KIR), expressed naturally in CD4 and CD8 T and NK cells, fused to an scFv. They then introduced this KIR-based CAR into human T cells expressing DAP12, an immunoreceptor tyrosine-based activation motif (ITAM)-containing adaptor. They found that T cells expressing KIR-CAR fail to trigger cytotoxic activity without KIR/DAP12 association. They also found that the KIR-based CAR exhibited more potent antitumor activity in vivo compared with 2nd-generation CD3ζ-based CARs due to enhanced stability of the KIR/DAP12 complex within the plasma membrane following antigen engagement [114].
Endodomain
Costimulatory domain
T cells require at least two distinct signals for full activation [115]. The first is delivered into T cells when TCR binds to its cognate antigenic peptides bound to MHC molecules on an antigen-presenting cell (APC). The second signal is provided when the costimulatory receptor on the T cell binds to its cognate ligand on the APC [115]. Antigenic stimulation without costimulation results in T cell anergy and unresponsiveness [116, 117]. CARs in which the antigen recognition domain is linked to the Fc receptor gamma chain (FcγR) or TCR zeta chain (CD3ζ) alone are known as “first-generation” CARs [118, 119]. Although the 1st-generation CARs exhibited cytotoxicity against target cells in-vitro and in-vivo, limited antitumor efficacy and poor persistence were observed in early-phase clinical trials [82, 120–122]. The insertion of a costimulatory unit into 1st-generation CARs, introduced as the second-generation CARs, enhanced T cell proliferation and in vivo persistence [122]. Third-generation CARs were also developed by fusing two costimulatory domains in a series. Most costimulatory molecules used in CARs belong to the Ig superfamily, such as CD28 and ICOS, or the TNF receptor superfamily (TNFRSF), such as 4-1BB, OX40, and CD27 [123]. A schematic picture of above-mentioned costimulatory molecules and their ligands has been illustrated in Fig. 4a.
Fig. 4.

A schematic picture of costimulatory molecules commonly used in CAR construct and their ligands on antigen presenting cells (a), Three CAR configurations (19BBz-CD80, 1928z-41BBL, and 19z1-CD80-41BBL) in Zhao’s study that are coupled with complementary costimulatory ligands (b). The 19BBz-CD80 and 1928z-41BBL are two 2nd-generation CARs coupled with CD80 and 41BBL, respectively. The 19z1-CD80-41BBL is a 1st-generation CAR that combined with both CD80 and 41BBL
Perhaps the best-characterized T cell costimulatory molecule is CD28, constitutively expressed on the surface of naïve T cells and some subsets of memory T cells [124]. It was first described in the 1980s as a co-receptor that enhanced TCR-induced proliferation and stimulated the differentiation of naive CD4 T cells [125, 126]. CD28, encoded by the CD28 gene located on human chromosome 2 (2q33), is a 44 kDa type I integral membrane glycoprotein that usually forms homodimers via disulfide bonds between cysteine residues positioned on the juxta transmembrane domain [125–127]. A single Ig-V-like domain on the extracellular portion of CD28 provides the structural specificity for interactions with its ligands, CD80 (B7–1) and CD86 (B7–2), expressed on APC upon activation [128]. CD28 contributes to proliferation [125, 129], IL-2 production [130, 131], survival [132–134], and metabolic activity [135] of naïve T cells by regulating the expression and activity of nuclear factor-κB (NF-κB), nuclear factor of activated T cells (NFAT), and activator protein 1 (AP-1) [136–138]. It is also involved in the cytoskeletal rearrangement, actin polymerization, and membrane rafts recruitment into the immune synapse, which maintains and boosts TCR-induced signaling [139–141].
The inducible T cell co-stimulator (ICOS) (CD278), the third member of the CD28/cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) family, is expressed on activated T cells [123, 142]. ICOS interacts with another B7-related molecule, ICOSL, also known as B7H, B7RP-1, and GL-50, expressed by APCs. Although ICOS and CD28 are similar in structure and downstream pathways, they are not identical [123, 143]. The CD28/B7 engagement is required for the primary immune responses, whereas ICOS/B7RP-1 is essential for secondary immune responses [144, 145]. ICOS is crucial for the development and maintenance of human T helper 17 (Th17) cells [146] and also directs immunity towards humoral or inflammatory responses [144]. Although their function, both, is costimulatory for T-cell proliferation and cytokine secretion, the effects of ICOS on the costimulation of T-cells appear less potent than those exerted by CD28, probably because ICOS cross-linking does not induce IL-2 production [147].
The expression of 4-1BB (TNFRSF9, CD137, ILA), first discovered in the late 80 as an inducible costimulatory molecule on activated T cells, is mainly activation-induced and not restricted to T cells and detected on various types of non-T cells as well [148–150]. 4-1BB interaction with its ligand, 4-1BBL, can costimulate T cells by activating the NF-κB, c-Jun, and p38 downstream pathways independently of CD28 signals. In contrast to CD28, 4-1BB enhances T cell effector responses by stimulating proliferation, cytokine production, and cytolytic activity and inhibiting AICD of effector T cells, not naïve T cells [151, 152]. Furthermore, numerous studies have suggested that 4-1BB predominantly promotes CD8 T cell-mediated response [153, 154], and some also described a dual immunoregulatory function for 4-1BB. While it supports both CD4 and CD8 T cell-mediated responses in vitro, it preferentially augments clonal expansion and survival of CD8 T cells in vivo, suppressing CD4 + T-cell function [155, 156].
The expression of OX40 (TNFRSF4, CD134), first described as a T cell activation marker, can be induced on activated CD4 and CD8 T cells as well as on several other lymphoid and non-lymphoid cells. A slight expression can be noticed within the first hours of T cell activation in vitro and in vivo, peaking anywhere from 1 to 5 days after initial stimulation. Although TCR signals are sufficient for inducing OX40, the interaction of CD28 with its ligands can increase and maintain OX40 expression; T cell and APC-derived cytokines such as IL-1, IL-2, and TNF may also modulate its amount and length [157]. Its ligand, OX40L (TNFSF4), is not constitutively expressed but can be induced on APCs upon activation [157]. Known as the late costimulatory molecules of the TNFR family, 4-1BB and OX40 prolong T-cell persistence and promote the generation and survival of effector and memory T cells [158–160]. Hombach et al. demonstrated that in contrast with CD28-based CARs, OX40-costimulated CARs do not secrete IL-2 and IL-10. They also found that in a CD28-OX40 dual costimulatory CAR, OX40 represses IL-10 secretion induced by CD28 without affecting IL-2 and IFNγ production. This favorable feature of OX40 can be employed, particularly for treating solid tumors in which IL-10, an immunosuppressive cytokine, is secreted into the TME by tumor and stromal cells [161]. Concerning nanobody-based CAR-T cells, we observed that anti-HER2 nanobody-based CAR-T cells containing a combination of CD28-OX40 showed increased expansion level and cytotoxicity in vitro compared to CAR-T cells lacking OX40 [56]. Recently, Zhang et al. showed that OX40 signaling enhanced CAR-T cell survival through up-regulation of anti-apoptotic Bcl-2-like molecules and improved proliferation through increased activation of the NF-κB, MAPK, and PI3K-AKT pathways [162].
The CD27 (TNFRSF7) is constitutively expressed on T cells, NKT cells, NK cells, and other immune cells. Its expression on T cells is strongly upregulated following activation [158]. CD27 engagement by its ligand, CD70, supports CD28-mediated costimulation and the survival of proliferating T cells [163]. CD27/CD70 interaction can also augment the expansion and survival of effector cells and enhance the development of memory CD8 T cells. Likewise, it can promote proliferation, polarization, and cytokine production by CD4 T cells [164]. It has been reported that CD27-costimulated CARs exhibited an antitumor activity similar to 4-1BB or CD28-based CAR-T cells while providing a persistent comparable to that of 4-1BB-based CAR-T cells. The combination of CD27 and CD28 costimulatory domains in a 3rd-generation CAR produced encouraging clinical results in neuroblastoma, AML, and lymphoma patients [165].
Most clinical trials have used CD28 or 4-1BB-costimulated CARs to date [166]. Although T cells expressing CARs with either a 4-1BB or CD28 costimulatory domain have demonstrated similar antitumor activity, particularly against lymphomas [7, 10, 167, 168], T cells expressing CD28-costimulated CARs display higher cytokine production but lower persistence [111, 169–171]. The persistence of CAR costimulated by CD28 was identical to that achieved with CD3ζ alone, indicating that CD28 does not support human T-cell survival in vivo [172, 173]. The persistence of CD28-costimulated CAR-T cells can be improved by replacing CD28 with 4-1BB [174, 175] or CD27 [176] or by adding 4-1BB alone [175] or a combination of 4-1BB and CD27 [177]. CARs harboring the 4-1BB costimulatory domain mediate long-term survival of T cells in the circulation by maintaining central memory phenotype and relying on oxidative metabolism, whereas CD28-costimulated CARs promote effector memory differentiation and rely on aerobic glycolysis [50, 169, 178]. In efforts to find an ideal combination of CD28 and 4-1BB that would preserve the superior tumoricidal capacity of CD28-based CARs with the sustenance afforded by the 4-1BB-based CARs, Zhao et al. developed seven different structural configurations of CARs, three of which (1928z-41BBL, 19BBz-CD80, and 19z1-CD80-41BBL) coupled with costimulation ligands [179] (Fig. 4b). They found that the 1928z-41BBL and 19BBz-CD80 configurations showed more favorable properties regarding in vivo tumoricidal cytotoxicity, proliferation, persistence, and IRF7/IFNβ pathway induction. The 1928z-41BBL CAR, however, consistently outperformed the 19BBz-CD80 and emerged as the most potent configuration. They also observed that CD28 downregulation, occurring following activation, averts the activity of its constitutively expressed ligand (CD80) provided by the 19BBz-CD80 configuration [179]. Furthermore, the 19z1-CD80-41BBL configuration, in which CAR coupled with both CD80 and 41BBL simultaneously, was the least effective, expanding steadily but exerting inferior tumor control [179]. The lack of durable antitumor responses in CD28-costimulated CAR-T cells may also be due to tonic signaling and T cell exhaustion mediated by the CD28 costimulatory domain. Recently, Guedan et al. reduced T cell exhaustion, driven by CD28, and thus enhanced in vivo persistence of the CD28-based CARs targeting mesothelin by disrupting the interaction between the CD28 signaling domain and the SH2-domain of Grb2 via a single amino acid alteration [180]. CARs containing ICOS also showed better persistence when compared with CD28-based CARs. It is likely because ICOS activates PI3K signaling and, consequently, Akt signaling more potently than CD28. As shown by Guedan et al., expressing ICOS-based CAR in CD4 T cells not only improves the persistence of these cells but also enhances the in vivo persistence of CD8 T cells expressing either 4-1BB– or CD28-based CAR [111]. Despite accumulated data indicating poor persistence of CD28-based CARs, prolonged survival of anti-CD19 CARs containing CD28 costimulatory domains has been reported in more recent trials, suggesting that, aside from CD28, other elements incorporated into the CAR structure may also affect the CAR-T cells in vivo persistence [7, 79, 81, 111, 181, 182].
Different costimulatory domains can induce various downstream signaling pathways. Selecting a costimulatory unit or a combination that would heighten antitumor activity and maintain the long-term persistence of CAR-T cells is crucial. Besides costimulatory molecules discussed in this review, alternative costimulatory molecules, such as CD40 (TNFRSF5) [183], Herpes Virus Entry Mediator (HVEM) (TNFRSF14, CD270) [184], Glucocorticoid-Induced TNFR-Related protein (GITR) (TNFRSF18, CD357) [185], Myeloid Differentiation Primary Response 88 (MYD88)/CD40 [186], Toll-Like Receptor 2 (TLR2) [187], and Dectin-1, a C-type Lectin Receptor [188], have been explored to determine whether the incorporation of these molecules into CAR structure could improve clinical outcomes in hematological malignancies or heighten potential applications of CAR-T cell therapy in patients with solid tumors.
Activation domain
In the CAR construct, the activation motif is the critical component to trigger T cell activation signaling, including the initiation of cytotoxicity. The CD3ζ is the most common activation molecule used in CAR-T cells [189]; however, early studies also utilized the FcγR as the primary activating domain in CARs [118, 190]. All FDA-approved CARs have employed CD3ζ as the cytoplasmic activation domain (Fig. 1). The CD3ζ (CD247) is a component of the TCR complex comprising three tyrosine-rich sequences known as ITAMs, named ITAM1, ITAM2, and ITAM3, from the membrane-proximal to the membrane-distal direction. The ITAMs presented on the cytoplasmic region of CD3ζ are the phosphorylation sites recruiting the tyrosine-protein kinase ZAP70, which, in turn, triggers downstream signaling cascades. The two distal ITAMs (ITAM2 and ITAM3) display a lower binding affinity for ZAP-70 compared to ITAM1. In T cells, the quantity and diversity of ITAMs affect optimal signaling; however, Feucht et al. demonstrated that a single functional ITAM is sufficient for potent antitumor efficacy. CAR containing a single ITAM (either ITAM1, 2, or 3) outperformed the triple- and double-ITAM-containing CARs in vivo and limited T cell differentiation, resulting in an increased fraction of central memory CAR-T cells and increased persistence as well [191]. Fisher et al. designed a novel CAR construct in which an scFv targeting GD2 was linked to DAP10 as the sole signaling domain and transduced into γ/δ T cells. Due to the absence of the CD3ζ, these CAR-T cells depended on their native γ/δ TCR for activation. They could efficiently eradicate neuroblastoma cells; however, those target cells lacking γ/δ TCR ligands could escape [192]. A summary of all structural elements of CAR reviewed here has been shown in Fig. 5.
Fig. 5.
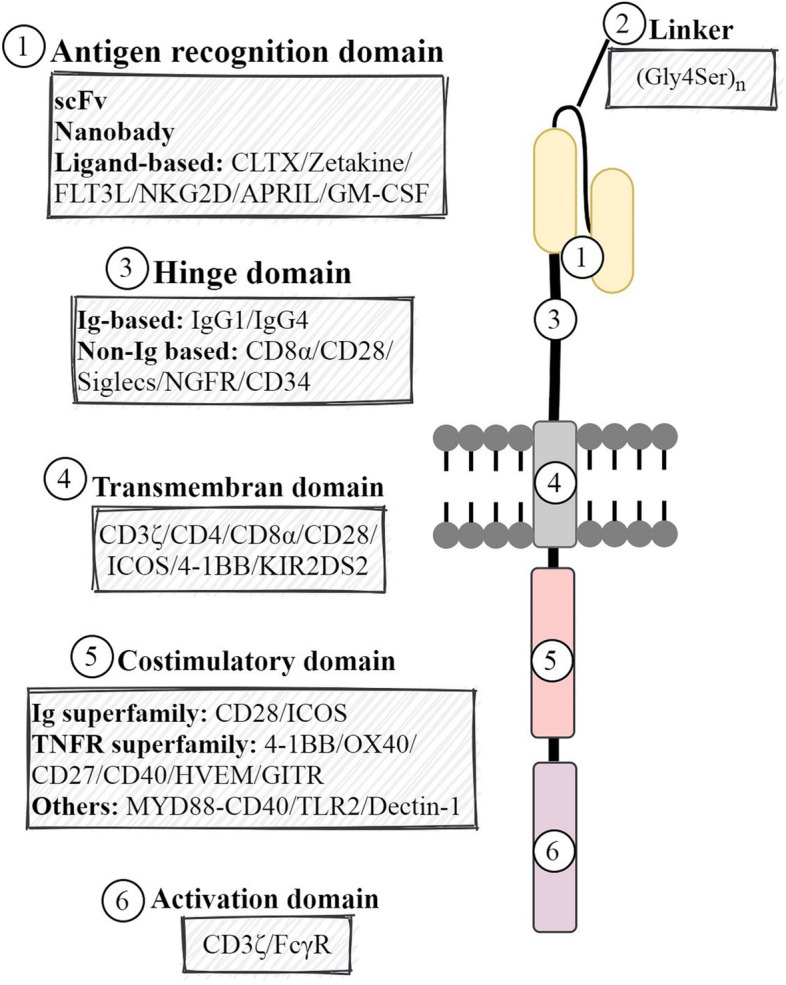
A schematic picture of all structural elements of CAR reviewed here
Vector backbone
Retroviral vectors are attractive tools for gene therapy due to the ability to integrate into the genome of target cells, a requisite for long-term expression, and maintaining a large cloning capacity, which is ideal for most clinical situations. In addition to the above-stated features common to all retroviral vectors, vectors derived from lentiviruses offer a unique advantage over their oncoretroviral counterparts: they can translocate across an intact nuclear membrane to transduce nondividing cells, the main potential targets of most gene therapies [193]. Lentiviral (LV) vectors used for gene therapy are predominantly derived from Human Immunodeficiency Virus type 1 (HIV-1). Two components are needed to make a virus-based gene delivery system: first, the packaging elements, encompassing the structural proteins and the enzymes required to generate an infectious particle, and second, the vector itself, that is, the genetic material transferred to the target cell [194]. The viral coding sequences are usually removed from the viral backbone and replaced by the gene of interest, such as the CAR. In this way, the transgene sequence is flanked by two long terminal repeats (LTRs), which are essential for the transgene integration into the host genome. The LTRs are identical in nucleotide sequence and organization and consist of U3-R-U5 regions in which the U3 is necessary for retroviral RNA transcription due to having endogenous enhancer/promoter sequences. Therefore, in wild-type retroviral-based vectors, transgene expression can be driven by the transcriptional sequences within the LTRs. However, to meet the safety requirements for clinical applications and minimize the risk of arising replication-competent recombinants (RCRs), self-inactivating (SIN) vectors are developed by removing a majority of the U3 region of the 3′ LTR. This deletion is transferred to the 5′ LTR of the proviral DNA during reverse transcription. Thus, SIN LV vectors retain all properties of their parent while lacking the ability to produce full-length vector RNA in transduced cells [193]. SIN LV vectors are the gold standard for CAR-T cell production due to the highest transduction/transfection rate. However, the efficiency of gene delivery by these vectors relies on some elements that should be present in the vector backbone or expression cassette (Fig. 6).
Fig. 6.

A schematic picture of a SIN LV vector backbone indicating where modular regulatory elements should be placed. LTR: Long Terminal Repeat, RRE: Rev-Response Element, cPPT: Central Polypurine Tract, IP: Internal Promoter, WPRE: Woodchuck Hepatitis Virus (WHV) Posttranscriptional Regulatory Element, USE: Upstream Sequence Element, PA: Polyadenylation Signal
Promoter
A gene transfer approach must be effectively directed to the specific tissues/cells where is desired, and the resulting transgene expression should be at a level required for a specific application [195]. Regarding CAR-T cell therapy, the CAR density on the T cell surface must be high enough to recognize the target antigen but not too high to trigger antigen-independent tonic signaling or drive off-tumor on-target cytotoxicity as a result of inappropriate recognition of target antigen on cells rather than tumor cells. Lentiviral vectors can transfer CAR construct into activated CD4 and CD8 human T cells with high efficiency, but its expression level depends on the promoter that drives its transcription. The U3 region of the 5′ LTR possesses endogenous enhancer/promoter sequences; however, it is removed in SIN LV vectors to enhance safety. Accordingly, for transgene expression in SIN vectors, internal promoters are employed. Selecting an appropriate promoter is a fundamental step toward a successful CAR-T cell therapy [196]. Promoter is a cis-acting element within the vector backbone that can dictate the level and duration of transgene expression and restrict expression to the specific tissues/cells, two critical goals desired for clinical applications. Various viral promoters (immediate-early cytomegalovirus [CMV], murine stem cell virus [MSCV] or spleen focus forming virus [SFFV] promoter), and cellular promoters (human elongation factor 1α-subunit [EF1α], human phosphoglycerate kinase1 [PGK], human ubiquitin C [UbiC] or the chicken β-actin and its derivative CAG) have been evaluated for transgene expression in lentiviral vectors. CMV and EF1α are the most commonly used promoters for CAR expression in T cells.
During productive infection of Human CMV (HCMV), viral genes are expressed, from immediate-early (IE) to early (E) and late (L) genes, respectively, in a temporal cascade [197]. The expression of IE genes is driven by a potent enhancer-containing promoter. This Major IE Enhancer and Promoter (MIEP) is active in various cell types and is the most commonly used promoter in mammalian expression plasmids [197]. Successful gene therapy with CAR-expressing T cells relies on the ability of T cells to maintain adequate receptor expression for a long time. The EF1a promoter is a strong promoter frequently used for CAR expression, as it often drives a strong and stable expression regardless of tissue specificity or the T cell activation state [169, 198]. Milone et al. evaluated several promoters, including EF1α, CMV, PGK, and UbiC, to identify the one that can drive the highest stable expression of a transgene in primary CD4 and CD8 T cells [169]. They found that although the CMV promoter caused a high expression level of the green fluorescent protein (GFP) early after transduction, expression dropped to < 25% of the initial expression level after 10 days of culture. In contrast, the EF1α promoter induced the highest level of GFP expression and optimally maintained it in both CD4 and CD8 T cells [169]. The sustained proliferation of CAR-T cells depends on CAR structure and high expression, the latter of which is required but not sufficient. When CARs having the CD28 transmembrane and cytosolic domain is expressed under the control of the EF1α promoter, they display a constitutive growth phenotype. Despite the constitutive growth phenotype, these CARs showed inferior antitumor effects and engraftment in vivo [199]. That is probably the explanation for why the two FDA-approved CD28-based CAR products, including Yescarta (axicabtagene ciloleucel, Kite Pharma Inc.) and Tecartus (brexucabtagene autoleucel, Kite Pharma Inc.) use the MSCV promoter instead of EF1α for CAR expression. The retroviral vectors incorporating MSCV LTR have been widely used in pre-clinical and clinical studies to drive high-level and long-term maintenance of CAR expression [200, 201].
Since heterogeneous expression of the CAR makes it challenging to ensure consistent behavior among individual CAR-T cells as their avidity toward the antigen can vary, targeted insertion of CAR into the first exon of the constant chain of the TCRα gene (TRAC) allows for a homogenous and consistent TCR-like expression, thus dodging issues related to the variegated transgene expression [202]. Eyquem et al. used the CRISPR/Cas9 paired with an adeno-associated virus (AAV) vector repair matrix to insert a CD19-specific CAR construct into the TRAC locus. CAR-T cells generated by CRISPR knock-in strategy outperformed the CAR-T cells generated via retroviral infection, both in vitro and in vivo [203]. These studies also demonstrated that targeting the CAR into the TRAC locus results in receptor internalization and re-expression cycle that is much more closely matched to the normal receptor, ultimately leading to a more sustained and effective antitumor response. Furthermore, this elegant CAR-knock-in and TCR-knockout strategy not only averts tonic CAR signaling by precisely regulating the expression of the CAR but also minimizes the risk of graft versus host disease (GVHD) by diminishing the expression of αβ TCRs on the T cell surface [203].
The expression of multiple genes requires self-cleaving 2A-like peptides of the Foot and Mouth Disease Virus (FMDV) [204], an internal ribosome entry site (IRES) [205], or the use of several promoters. The two latter strategies are the most widely used. It has been shown that the expression levels of the genes upstream and downstream of the IRES element vary, with the downstream gene typically expressed at lower levels [206]. Using two separate, divergent promoters may significantly increase the size of expression cassettes, and also probable different tissue specificity and mutual interference between them may prevent efficient co-expression in the same target cell [207, 208]. However, an alternative approach is to employ a single, compact bidirectional promoter [209]. He et al. could successfully use a bidirectional promoter to express dual CAR cassettes in the Sleeping Beauty system; however, they found that the investigated bidirectional promoters are sub-optimal for lentiviral production of long RNA encoding long dual-CAR constructs [210]. EF1α can be the best choice for driving lentiviral-based vectors containing a long RNA in CAR-T cells. Hosseini Rad et al. evaluated four strong well-characterized promoters, including EF1α, CMV, hPGK, and RPBSA, for optimal expression of a long and complicated RNA encoding multiple gene products in CAR-T cells and found that EF1α is the best choice for driving short as well as long RNA in CAR-T cells [196]. They also discovered that the EF1α promoter exhibited the best transduction efficiency, killing ability, and cytokine production. Furthermore, the authors observed a reduction in CAR expression driven by the hPGK and RPBSA promoters, which retained acceptable killing ability but reduced cytokine production [196].
Signal peptide
Recognition of target antigen typically requires CAR expression on the T cell surface. CAR protein is a type I membrane protein that must be trafficked through the secretory pathway to the plasma membrane, where it can be anchored to and exert its function. Accordingly, the CAR coding sequence starts with a signal peptide (SP). The SP, also known as the leading peptide, was first described in 1975 as a short transient peptide, predominantly found at the N-terminal of secretory and type I membrane proteins that direct the nascent polypeptide chain to the endoplasmic reticulum (ER) membrane [211, 212]. The SPs typically contain 25-30aa; however, longer SPs (up to 140aa) are also seen in eukaryotes; they are predominantly organelle-targeting, which remain stable even after protein maturation, and usually add extra functions to the protein targeting [213]. The SPs act like address tags and mediate the translocation of secretory proteins across intracellular membranes and final localization. They are more precisely required for protein translocation across the first membrane on the secretory pathway and thus universally control the entry of all proteins to the secretory pathway [211]. Although SPs may vary in length and sequence, they are found in both prokaryotes and eukaryotes and share a conserved tripartite structure, commonly characterized by a positively charged N-terminal region, a 9-to 12-residue-long hydrophobic stretch in the center that forms an α-helical conformation, and a polar C-terminal region with the cleavage site for signal peptidase [212]. As a nascent protein emerges from the ribosome, the signal peptide is recognized by the signal recognition particle (SRP), a cytoplasmic ribonucleoprotein [214]. SRP pauses elongation of the nascent polypeptide chain until the SRP–SP–ribosome complex interacts with an SRP receptor on the ER membrane. Upon interaction, the nascent chain is inserted into the ER translocon, and polypeptide chain elongation resumes. The SP is then cleaved off by a signal peptidase residing in the ER while the growing protein passes through the ER membrane [215, 216]. The SPs usually incorporated into the CARs are derived from human CD8α [217], IL-2 [218], GM-CSF receptor (GM-CSF) α chain [219], or murine Ig-kappa (IgK) [220]. Wang et al. chose the leader sequence of GM-CSFRα at the beginning of a CD19-specific CAR and a truncated form of human epidermal growth factor receptor (huEGFRt) coding sequences, based on its capacity to sort type I transmembrane proteins to the plasma membrane in T cells [221]. Recently, Ping et al. designed CAR-T cells that could secret α-PD-1 scFv in solid tumors. They compared six signal peptides frequently used in engineering secretory proteins, including Secrecon, murine IgK V-III region (IgKVIII), human IgKVIII, CD33, human tissue plasminogen activator (TPA), a consensus 16aa signal peptide, and native secreted alkaline phosphatase (SEAP) [222], to find the one that can enhance extracellular accumulations of anti-PD-1 scFv. They observed that the one derived from human IgK VIII was the best choice as the secreting capacity of anti-PD-1 scFv was significantly enhanced by this leading peptide [223].
RRE
Retroviruses such as lentiviruses employ various mechanisms to regulate the expression of alternatively spliced viral mRNAs. The presence of suboptimal splice sites allows for differential expression of several mRNAs from a single pre-RNA. Lentiviruses, including HIV-1, utilize Rev/Rex proteins that act in trans to regulate the nuclear export of unspliced or singly-spliced mRNAs required for the expression of structural and enzymatic proteins and progeny viral RNA genomes as well [224]. After integration into the host genome, during the early phase of HIV-1 life, the viral DNA called the provirus now on, is acted upon by cellular transcription factors to express viral genes. The early population of the transcripts is a fully-spliced mRNA (2 kb) exported to the cytoplasm to be translated into viral regulatory proteins, including Tat, Nef, and Rev. The Rev is a nucleocytoplasmic shuttle protein necessary for virus replication. Rev, later in the viral life cycle, is imported to the nucleus to mediate the export of the Rev Response Element (RRE)-containing unspliced (9 kb) or partially spliced (4 kb) mRNAs to the cytoplasm, where they are translated into viral proteins, such as Gag, Gag-Pol, Env, and accessory proteins, or packaged as the viral genome into newly budding virions [225]. The interaction between Rev and RRE is necessary for exporting such intron-containing mRNAs, which are usually kept in the nucleus to be spliced or degraded. The RRE is a cis-acting element located at the junction between the SU (gp120) and TM (gp41) domains of the ENV gene on viral genomic RNA [226], with a well-conserved sequence (351 nucleotides) and highly branched structure (an approximately equimolar mixture of 4 and 5 stem-loop conformations) [227] that provides an architectural scaffold with a high affinity for Rev binding. DiMattia et al., using X-ray crystallography and cryo-EM, described an “A”-shaped architecture for RRE, which allows 8–12 Rev-subunits to bind to and mediate nuclear export [228]. Both Rev oligomerization and its interaction with RRE, initiated by binding the Rev arginine-rich motif (ARM) to a high-affinity Rev-binding site known as stem-loop IIB on the RRE, are critical for viral RNAs export and, consequently, virus replication [229]. After assembly of the Rev-RRE ribonucleoprotein (RNP) complex, the nuclear export sequences (NESs), displayed on the Rev, interact with the host Crm1/RanGTP nuclear-export machinery and facilitate the nuclear export of viral intron-containing mRNAs [225, 230, 231].
cPPT
To be inserted into the host genome, the single-stranded RNA genome needs to be converted into double-stranded DNA before nuclear import. While synthesis of the first (minus) strand of DNA is initiated by a cellular tRNA molecule already packaged into the retroviral particles, synthesis of the plus-strand DNA is primed by a short purine-rich remnant of the viral RNA known as the polypurine tract (PPT), selectively preserved when the RT digest the viral RNA from the nascent RNA/DNA hybrid [232]. Besides this copy of PPT, shared by all retroviruses, HIV-1 carries a second copy of the PPT, known as cPPT, placed near the center of the genome in the integrase open reading frame [233]. Thus, synthesis of the plus-strand DNA in HIV-1 is primed by both PPT and cPPT, resulting in two discrete plus-strand segments, each covering half of the viral genome [234]. Synthesis of upstream plus-strand DNA initiated at the PPT continues for approximately 99 nucleotides downstream of cPPT, expelling the centrally initiated downstream plus-strand DNA, and is then stopped at the central termination sequence (CTS), creating a triple-stranded DNA structure at the cPPT. The 99-nucleotide-long overlapping DNA, also known as the central DNA flap, is vital for the pre-integration complex (PIC) formation and thus nuclear import [235]. Defects in the DNA flap formation result in the trapping of the PIC at the cytoplasmic side of the nuclear pore, prohibiting nuclear entry of the HIV-1 genome [236]. It has been shown that the insertion of a 118-bp sequence of the HIV-1 containing the cPPT/CTS element into lentiviral vectors enhances transduction efficiency, up to 85% in T cells pre-activated with IL-2 and PHA, by facilitating the nuclear import of transgene through a central DNA flap [237, 238]. Also, studies demonstrated that the presence of the cPPT/CST in lentiviral vectors leads to a 10-fold increase in the amount of integrated DNA and a 5- to 10-fold increase in the transduction efficiency, differing based on cell type [239]. Studies also revealed that initiating plus-strand synthesis at two separate sites in HIV-1 derived vectors would decrease the time during which minus-strand viral DNA remains single-stranded, thus averting its exposure to cellular enzymes, particularly cytidine deaminases of the APOBEC family known as natural defense barriers against HIV infection, that act on newly synthesized single-stranded viral DNA [232].
Poly(a) signal and USE
A critical step in the mRNA 3′-end processing is polyadenylation, the consequences of which may impact mRNA stability and translation efficiency. Like eukaryotes, retroviruses need two cis-acting elements (together known as the core polyA site) for transcription termination and polyadenylation, including an almost invariant AAUAAA sequence, placed 10–30 nucleotides upstream of the cleavage site, and a more variant GU/U-rich sequence positioned immediately downstream [240]. In some retroviruses, the AAUAAA sequence is placed in the U3 region, while in all lentiviruses, including HIV-1, it is present in the R region [241].
HIV-1, in common with all retroviruses, has two LTRs (U3-R-U5), which are identical in nucleotide sequence and organization. Transcription starts at the junction of U3-R, and polyadenylation occurs at the junction of R-U5. Therefore, it is expected that the transcript initiated at + 1 nucleotide in the 5′ LTR would be polyadenylated at the end of the R sequence in the 5′ LTR itself, resulting in a truncated viral RNA [240]. To prevent premature termination and polyadenylation, the promoter-proximal polyA site in the 5′ LTR must be suppressed, while the promoter-distal polyA site in the 3′ LTR has to be selectively and efficiently used to polyadenylate all resulting viral RNAs [241]. Therefore, lentiviruses have evolved with additional sequences to support and ensure efficient transcriptional termination and prevent the possibility of read-through into cellular genes. In SIN vectors, most of the U3 region is deleted from LTRs to enhance safety. However, in the case of HIV, this deletion results in leaky polyadenylation suggesting that U3 contains termination enhancer motifs or USEs. These USEs are U-rich sequences placed upstream of the AAUAAA sequence serving a critical role in selecting the 3′ polyA site in preference to an identical polyA site at the 5′ end of mRNA [242, 243]. Efficient 3′-end processing of HIV-1 has been shown to depend on a stem-loop structure that places the USE and the core polyA site close together [244].
Schambach et al. incorporated seven different USEs derived from various viral or cellular genes into SIN LV vectors to find the best one for improving viral titer and gene expression. They observed that the USE derived from simian virus 40 (SV40) late mRNA, especially when duplicated (2xSV), provided the best results, improving viral titer (up to threefold) and gene expression (by 45–100%) [244]. Interestingly, the relatively small 2xSV USE (100 bp) was nearly as potent as the WPRE (600 bp) in enhancing viral titer and transgene expression. However, the level of the effects depended on other elements, such as promoter and target cell type. They also found that the 2xSV USE was superior to the WPRE in suppressing transcriptional read-through, thus improving vector efficiency and biosafety [244]. Hager et al. investigated whether the inclusion of a strong polyA signal as part of an internal transcription unit has the potential to overcome problems regarding insufficient termination. They found that an internal polyA signal increases transgene expression to levels comparable to that typically observed in the presence of the WPRE but decreases viral titer in a promoter-dependent manner [245]. Studies have shown that an expression cassette containing a combination of WPRE (a shortened version with 247 bp), an SV40 late USE, and a full-length SV40 late polyA signal drives the highest levels of gene expression compared to vectors containing each element alone [246] (Fig. 6).
WPRE
The WPRE, a post-transcriptional regulatory element (PRE) of the woodchuck hepatitis virus (WHV), is primarily known as an RNA export element that facilitates the accumulation of surface antigen transcripts in the cytoplasm from the intronless hepadnavirus genome [247]. WPRE can substantially enhance viral titers and transgene expression from various RNA and DNA viral vectors [248–250] when placed in the sense orientation in the 3′-untranslated region (3′-UTR), upstream of the polyadenylation signal [251]. Xu et al. observed that the insertion of WPRE in viral vectors could increase the expression of the reporter gene up to 7-fold in vitro and up to 50-fold in vivo [250]. The positive impact of the WPRE on gene expression is not due to an enhanced rate of transcription, viral mRNA half-life, or nuclear export; it instead acts post-transcriptionally and increases the efficiency of mRNA 3′-end processing [248, 251]. Studies demonstrated that oncoretroviruses, but not lentiviruses, usually display a high transcriptional readthrough activity in the 3′ LTR [252]. This reduced 3′ termination efficiency may cause RNA instability during transgene expression and also raise safety issues regarding the increased risk of activating or capturing downstream cellular oncogenes [252]. SIN LV vectors, which lack the U3 region, also suffer from a leaky transcription termination and exhibit high transcriptional readthrough activity, suggesting that additional termination signals must be present within the U3 sequence [253]. Further studies showed that besides USE, two additional elements in U3, including the transcriptional control region and the nuclear factor of activated T cells/upstream stimulatory factor (NFAT/USF) binding region, contribute significantly to lentiviral LTR transcriptional termination (Fig. 7). Restoration of the transcriptional control region alone reduces readthrough by 70–80%, while insertion of the NFAT/USF binding region reduces RNA readthrough to a level even lower than that of the wild type LTR [254]. However, instead of restoring the U3, which causes safety issues, genetic elements, such as WPRE, can be inserted into SIN LV vectors to reduce readthrough without causing any adverse effects.
Fig. 7.

A schematic picture of the LTR and also termination signals present within the U3 region. Studies showed that besides USE, two additional elements in U3, the transcriptional control region and the nuclear factor of activated T cells/upstream stimulatory factor (NFAT/USF) binding region, contribute significantly to lentiviral LTR transcriptional termination
Like the WPRE, PRE found in the human hepatitis B virus (HBV) (HPRE) can increase the level of gene expression; however, studies showed that WPRE displays two to three times more potent activity [247]. WPRE (600 bps) and HPRE (553 bp) have two homologous subelements, called α and β, while WPRE contains an additional subelement named gamma [247]. Both WPRE and HPRE contain AU-rich motifs in the β-loop that resemble the USE core sequence. WPRE also possesses a second USE core sequence-like motif in the γ-loop, which may explain its potency of transcript termination and increased total viral mRNA over HPRE [251]. However, Choi et al. discovered that the insertion of a shortened WPRE sequence (247 bp), containing approximately 41.2% of the original size, into adeno-associated virus (AAV) vectors causes a level of transgene expression comparable to that of wild-type WPRE while providing more space for larger transgenes [246].
Conclusion and future perspectives
The optimal design of a CAR construct requires a comprehensive understanding of the features of each component alone and in combination with others within the CAR. As CAR constructs become more complex and more elements come into play, a deep understanding of the impact of distinct domains would likely improve the rational design of CAR-T cells to fit the specific needs of individual patients. However, even an optimized CAR may not overcome all the hindrances presented by the complex nature of tumors. Furthermore, viral vectors, particularly lentiviral vectors, may be highly efficient in CAR-T cell production, but several critical features discourage their use in the clinic, supporting nonviral approaches [255]. The possibility of insertional mutagenesis, caused by the random integration of viral DNA into the host genome, the limited cargo size capacity, and the high manufacturing complexity and cost are some drawbacks associated with viral vectors, prompting researchers to look for other alternatives. Transposable elements (transposons) are the most common alternatives to viral vectors with a vast potential for diverse applications in genetic engineering, including CAR-T cell therapies, since they are easier and less expensive to manufacture due to their plasmid-based nature and offer a larger cargo size capacity to deliver multiple transgenes. Various transposon-based systems, including the Sleeping Beauty (SB) [256], the piggyBac (PB) [257], and Tol2 transposon [258], have been reported for CAR-T cell production as these systems provide safe and reliable DNA transfer into T-cells. Like viral-based CAR-T cells, transposon-mediated CAR-T cell clinical applications mainly focus on blood malignancies and target the CD19 antigen [259–262]. Kebriaei et al. (2016) reported the first human application of the SB system for 26 patients with advanced NHL or ALL. All patients received a single dose of patient- or donor-derived CD19-specific CAR-T cells generated with SB in the phase I adjuvant setting following autologous (NCT00968760) or allogeneic (NCT01497184) hematopoietic stem cell transplantation (HSCT). They utilized high-throughput sequencing to analyze the CAR integration patterns in T cells, genetically modified by SB transposon/transposase plasmids, and observed that integrations were widely distributed throughout the genome with no bias [259]. For patients who received CAR-T cells after autologous HSCT (n = 7), the 30-month progression-free survival (PFS) and overall survival (OS) rates were around 83 and 100%, respectively. For those who received CAR-T cells after allogeneic HSCT (n = 19), the respective 12-month rates were 53 and 63% [259]. Recently, Li et al. used piggyBac-produced CD19-specific CAR-T cell therapy for a male patient of triple-hit R/R DLBCL with TP53 mutation (ChiCTR1800018111). Triple-hit lymphoma (THL), a relatively rare subset of DLBCL identified in approximately 1% of DLBCL cases, carries concurrent MYC, BCL2, and BCL6 rearrangements. Despite grade 2 cytokine release syndrome, the patient achieved CR two-month after CAR infusion and has still kept sustained CR for over 24 months [262]. Recent studies on transposon-mediated CAR-T cell therapy have mainly focused on two areas: 1) developing novel transposon systems to deliver multiple transgenes, such as the hyperactive Tc Buster, which has been originally isolated from the red flour beetle and shown comparable transposition efficiency to the SB and PB [263], 2) designing novel platforms to generate universal allogeneic CAR-T cells, such as the CRISPR-Cas9 ribonucleoparticles (RNP)-minicircles (mc) SB transposon platform optimized by Tipanee et al. to express CD19-specific CAR while inactivating allogeneic TCRs [264]. Although viral and nonviral techniques have their benefits and drawbacks, the question that needs to be answered is whether nonviral methods will hold the potential to meet these challenges and take CAR technology beyond the hematological malignancies.
Acknowledgments
This work was partly supported by the Council for Development of Stem Cell Sciences and Technologies [Grant No. 11/56623], and also by faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran.
Abbreviations
- CAR
Chimeric Antigen Receptor
- FDA
Food and Drug Administration
- MHC
Major Histocompatibility Complex
- scFv
Single-Chain Variable Fragment
- CR
Complete Remission
- TSA
Tumor-Specific Antigen
- TAA
Tumor-Associated Antigen
- MM
Malignant Mesothelioma
- GBM
Glioblastoma Multiforme
- TanCAR
Tandem CAR
- iCAR
Inhibitory CAR
- PSMA
Prostate-Specific Membrane Antigen
- R/R
Relapsed/Refractory
- NHL
Non-Hodgkin Lymphoma
- ALL
Acute Lymphoblastic Leukemia
- LBCL
Large B-cell Lymphoma
- PD-1
Programmed Cell Death Protein-1
- CTLA-4
Cytotoxic T-Lymphocyte-Associated Antigen 4
- HcAb
Heavy-chain-only Antibodies
- MUC1
Mucin 1
- CLTX
Chlorotoxin
- AML
Acute Myeloid Leukemia
- LSCs
Leukemic Stem Cells
- FLT3L
FMS-like Tyrosine Kinase 3 Ligand
- NKG2D
Natural Killer Group 2D
- APRIL
A Proliferation-Inducing Ligand
- TNFSF13
Tumor Necrosis Factor Ligand Superfamily Member 13
- BCMA
B-cell Maturation Antigen
- TACI
Transmembrane Activator and Calcium-Modulator and Cyclophilin Ligand
- GM-CSF
Granulocyte-Macrophage Colony-Stimulating Factor
- JMML
Juvenile Myelomonocytic Leukemia
- Gly
Glycine
- Ser
Serine
- V-domain
Variable domain
- Penn
University of Pennsylvania
- CHOP
Children’s Hospital of Philadelphia
- NCI
National Cancer Institute
- IS
Immune Synapse
- HD
Hinge Domain
- ROR1
Receptor-Tyrosine Kinase-Like Orphan Receptor 1
- FcγRs
Fc-gamma Receptors
- DC
Dendritic Cells
- NK cells
Natural Killer Cells
- ADCC
Antibody-Dependent Cell-Mediated Cytotoxicity
- AICD
Activation-Induced T-Cell Death
- IVIG
Intravenous Immunoglobulin
- TM
Transmembrane
- TMD
Transmembrane Domain
- LAG-3
Lymphocyte Activation Gene-3
- CRS
Cytokine Release Syndrome
- Siglecs
Sialic Acid-Binding Immunoglobulin-Type Lectins
- NGFR
Nerve Growth Factor Receptor
- ICD
Intracellular Domain
- KIR
Killer Immunoglobulin-Like Receptor
- ITAM
Immunoreceptor Tyrosine-Based Activation Motif
- TCR
T-cell Receptor
- APC
Antigen-Presenting Cell
- CD3ζ
TCR Zeta Chain
- TNF
Tumor Necrosis Factor
- TNFRSF
TNF Receptor Superfamily
- NF-κB
Nuclear Factor-κB
- NFAT
Nuclear Factor of Activated T Cells
- AP-1
Activator Protein 1
- ICOS
Inducible T-Cell Co-Stimulator
- Th17
T Helper 17
- TME
Tumor Microenvironment
- NKT cells
Natural Killer T Cells
- HVEM
Herpes Virus Entry Mediator
- GITR
Glucocorticoid-Induced TNFR-Related Protein
- MYD88
Myeloid Differentiation Primary Response 88
- TLR2
Toll-Like Receptor 2
- LV
Lentiviral
- HIV-1
Human Immunodeficiency Virus Type 1
- LTRs
Long Terminal Repeats
- RCRs
Replication-Competent Recombinants
- SIN vectors
Self-Inactivating Vectors
- CMV
Cytomegalovirus
- MSCV
Murine Stem Cell Virus
- SFFV
Spleen Focus Forming Virus
- EF-1α
Elongation Factor 1α-Subunit
- PGK
Phosphoglycerate Kinase
- UbiC
Ubiquitin C
- IE
Immediate-Early
- E
Early
- L
Late
- MIEP
Major IE Enhancer and Promoter
- GFP
Green Fluorescent Protein
- TRAC
TCRα Subunit Constant Gene
- AAV
Adeno-Associated Virus
- GVHD
Graft Versus Host Disease
- FMDV
Foot and Mouth Disease Virus
- IRES
Internal Ribosome Entry Site
- SP
Signal Peptide
- ER
Endoplasmic Reticulum
- SRP
Signal Recognition Particle
- IgK
Ig-Kappa
- huEGFRt
Human Epidermal Growth Factor Receptor
- TPA
Human Tissue Plasminogen Activator
- SEAP
Secreted Alkaline Phosphatase
- RRE
Rev Response Element
- Cryo-EM
Cryogenic Electron Microscopy
- ARM
Arginine-Rich Motif
- RNP
Ribonucleoprotein
- NESs
Nuclear Export Sequences
- RT
Reverse Transcriptase
- PPT
Polypurine Tract
- cPPT
Central Polypurine Tract
- CTS
Central Termination Sequence
- PIC
Pre-Integration Complex
- APOBEC family
Apolipoprotein B mRNA Editing Catalytic Polypeptide-like family
- USEs
Upstream Enhancer Elements
- SV40
Simian Virus 40
- WPRE
Post-Transcriptional Regulatory Element (PRE) of the Woodchuck Hepatitis Virus (WHV)
- UTR
Untranslated Region
- NFAT/USF
Nuclear Factor of Activated T Cells/Upstream Stimulatory Factor
- HBV
Human Hepatitis B Virus
- HPRE
Post-Transcriptional Regulatory Element of Human Hepatitis B Virus
- SB
Sleeping Beauty
- PB
PiggyBac
- HSCT
Hematopoietic Stem Cell Transplantation
- PFS
Progression-Free Survival
- OS
Overall Survival
- DLBCL
Diffuse Large B-cell Lymphoma
- THL
Triple-hit Lymphoma
- RNP
Ribonucleoparticles
- mc
Minicircles
Authors’ contributions
All authors contributed to the conception and the main idea of the work. MM and FR wrote the original draft. MM collected the data and drew the figs. FR edited the final draft for intellectual content. All authors read and approved the final manuscript.
Funding
None.
Availability of data and materials
Not applicable.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Marzieh Mazinani, Email: marzieh.mazinani@yahoo.com.
Fatemeh Rahbarizadeh, Email: rahbarif@modares.ac.ir.
References
- 1.US Food & Drug Administration . FDA approves tisagenlecleucel for adults with relapsed or refractory large B cell lymphoma. 2018. [Google Scholar]
- 2.US Food & Drug Administration . FDA approves axicabtagene ciloleucel for large B-cell lymphoma. 2017. [Google Scholar]
- 3.US Food & Drug Administration . FDA approves brexucabtagene autoleucel for relapsed or refractory mantle cell lymphoma. 2020. [Google Scholar]
- 4.US Food & Drug Administration. FDA approves lisocabtagene maraleucel for second-line treatment of large B-cell lymphoma. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lisocabtagene-maraleucel-second-line-treatment-large-b-cell-lymphoma. 2022.
- 5.US Food & Drug Administration . FDA approves idecabtagene vicleucel for multiple myeloma. 2021. [Google Scholar]
- 6.US Food & Drug Administration . FDA approves ciltacabtagene autoleucel for relapsed or refractory multiple myeloma. 2022. [Google Scholar]
- 7.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med. 2018;378(5):439–448. doi: 10.1056/NEJMoa1709866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric antigen receptor T cells in refractory B-cell lymphomas. N Engl J Med. 2017;377(26):2545–2554. doi: 10.1056/NEJMoa1708566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018;378(5):449–459. doi: 10.1056/NEJMoa1709919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med. 2018;24(1):20–28. doi: 10.1038/nm.4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127(26):3321–3330. doi: 10.1182/blood-2016-04-703751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. doi: 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glockshuber R, Malia M, Pfitzinger I, Plückthun A. A comparison of strategies to stabilize immunoglobulin Fv-fragments. Biochemistry. 1990;29(6):1362–1367. doi: 10.1021/bi00458a002. [DOI] [PubMed] [Google Scholar]
- 15.van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14(7):499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramakrishna S, Highfill SL, Walsh Z, Nguyen SM, Lei H, Shern JF, et al. Modulation of target antigen density improves CAR T-cell functionality and persistence. Clin Cancer Res. 2019;25(17):5329–5341. doi: 10.1158/1078-0432.CCR-18-3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M, et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3 ζ chimeric antigen receptor-modified effector CD8+ T cells. J Immunol. 2015;194(3):911–920. doi: 10.4049/jimmunol.1402346. [DOI] [PubMed] [Google Scholar]
- 18.Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning sensitivity of CAR to EGFR density limits recognition of Normal tissue while maintaining potent antitumor activity. Cancer Res. 2015;75(17):3505–3518. doi: 10.1158/0008-5472.CAN-15-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 2015;75(17):3596–3607. doi: 10.1158/0008-5472.CAN-15-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Olson ML, Mause ERV, Radhakrishnan SV, Brody JD, Rapoport AP, Welm AL, et al. Low-affinity CAR T cells exhibit reduced trogocytosis, preventing rapid antigen loss, and increasing CAR T cell expansion. Leukemia. 2022;36(7):1943-6. 10.1038/s41375-022-01585-2. [DOI] [PMC free article] [PubMed]
- 21.Ghorashian S, Kramer AM, Onuoha S, Wright G, Bartram J, Richardson R, et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat Med. 2019;25(9):1408–1414. doi: 10.1038/s41591-019-0549-5. [DOI] [PubMed] [Google Scholar]
- 22.Drent E, Themeli M, Poels R, de Jong-Korlaar R, Yuan H, de Bruijn J, et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol Ther. 2017;25(8):1946–1958. doi: 10.1016/j.ymthe.2017.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han L, Zhang J-S, Zhou J, Zhou K-S, Xu B-L, Li L-L, et al. Single VHH-directed BCMA CAR-T cells cause remission of relapsed/refractory multiple myeloma. Leukemia. 2021;35(10):3002–3006. doi: 10.1038/s41375-021-01269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagner DL, Fritsche E, Pulsipher MA, Ahmed N, Hamieh M, Hegde M, et al. Immunogenicity of CAR T cells in cancer therapy. Nat Rev Clin Oncol. 2021;18(6):379–393. doi: 10.1038/s41571-021-00476-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, Chen L-J, Yang S-S, Sun Y, Wu W, Liu Y-F, et al. Exploratory trial of a biepitopic CAR T-targeting B cell maturation antigen in relapsed/refractory multiple myeloma. Proc Natl Acad Sci. 2019;116(19):9543–9551. doi: 10.1073/pnas.1819745116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao J, Wang G, Cheng H, Wei C, Qi K, Sang W, et al. Potent anti-leukemia activities of humanized CD19-targeted chimeric antigen receptor T (CAR-T) cells in patients with relapsed/refractory acute lymphoblastic leukemia. Am J Hematol. 2018;93(7):851–858. doi: 10.1002/ajh.25108. [DOI] [PubMed] [Google Scholar]
- 28.Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8(355):355ra116. doi: 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gorovits B, Koren E. Immunogenicity of chimeric antigen receptor T-cell therapeutics. BioDrugs. 2019;33(3):275–284. doi: 10.1007/s40259-019-00354-5. [DOI] [PubMed] [Google Scholar]
- 30.Han X, Wang Y, Wei J, Han W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J Hematol Oncol. 2019;12(1):128. doi: 10.1186/s13045-019-0813-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei J, Han X, Bo J, Han W. Target selection for CAR-T therapy. J Hematol Oncol. 2019;12(1):62. doi: 10.1186/s13045-019-0758-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang D, Starr R, Chang WC, Aguilar B, Alizadeh D, Wright SL, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med. 2020;12(533):eaaw2672. 10.1126/scitranslmed.aaw2672. [DOI] [PMC free article] [PubMed]
- 34.Miao L, Zhang J, Huang B, Zhang Z, Wang S, Tang F, et al. Special chimeric antigen receptor (CAR) modifications of T cells: a review. Front Oncol. 2022;12:832765. doi: 10.3389/fonc.2022.832765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider D, Xiong Y, Wu D, Nӧlle V, Schmitz S, Haso W, et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer. 2017;5:42. doi: 10.1186/s40425-017-0246-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol Ther Nucleic Acids. 2013;2(7):e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang D, Shao Y, Zhang X, Lu G, Liu B. IL-23 and PSMA-targeted duo-CAR T cells in prostate Cancer eradication in a preclinical model. J Transl Med. 2020;18(1):23. doi: 10.1186/s12967-019-02206-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016;126(10):3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira ACP, et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol. 2012;32(5):1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 41.Xie B, Li Z, Zhou J, Wang W. Current Status and Perspectives of Dual-Targeting Chimeric Antigen Receptor T-Cell Therapy for the Treatment of Hematological Malignancies. Cancers (Basel). 2022;14(13):3230. 10.3390/cancers14133230. [DOI] [PMC free article] [PubMed]
- 42.Qin H, Ramakrishna S, Nguyen S, Fountaine TJ, Ponduri A, Stetler-Stevenson M, et al. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol Ther Oncolytics. 2018;11:127–137. doi: 10.1016/j.omto.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y, Li J, Lou X, Chen X, Yu Z, Kang L, et al. A prospective investigation of bispecific CD19/22 CAR T cell therapy in patients with relapsed or refractory B cell non-Hodgkin lymphoma. Front Oncol. 2021;11:664421. doi: 10.3389/fonc.2021.664421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spiegel JY, Patel S, Muffly L, Hossain NM, Oak J, Baird JH, et al. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: a phase 1 trial. Nat Med. 2021;27(8):1419–1431. doi: 10.1038/s41591-021-01436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell. 2016;164(4):780–791. doi: 10.1016/j.cell.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roybal Kole T, Rupp Levi J, Morsut L, Walker Whitney J, McNally Krista A, Park Jason S, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164(4):770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med. 2013;5(215):215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu S, Yi M, Qin S, Wu K. Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol Cancer. 2019;18(1):125. doi: 10.1186/s12943-019-1057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calderon H, Mamonkin M, Guedan S. Analysis of CAR-mediated tonic signaling. Methods Mol Biol. 2020;2086:223–236. doi: 10.1007/978-1-0716-0146-4_17. [DOI] [PubMed] [Google Scholar]
- 50.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Landoni E, Fucá G, Wang J, Chirasani VR, Yao Z, Dukhovlinova E, et al. Modifications to the framework regions eliminate chimeric antigen receptor tonic signaling. Cancer Immunol Res. 2021;9(4):441–453. doi: 10.1158/2326-6066.CIR-20-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asaadi Y, Jouneghani FF, Janani S, Rahbarizadeh F. A comprehensive comparison between camelid nanobodies and single chain variable fragments. Biomark Res. 2021;9(1):87. doi: 10.1186/s40364-021-00332-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rahbarizadeh F, Rasaee MJ, Forouzandeh Moghadam M, Allameh AA, Sadroddiny E. Production of novel recombinant single-domain antibodies against tandem repeat region of MUC1 mucin. Hybrid Hybridomics. 2004;23(3):151–159. doi: 10.1089/1536859041224334. [DOI] [PubMed] [Google Scholar]
- 54.Arbabi-Ghahroudi M. Camelid single-domain antibodies: historical perspective and future outlook. Front Immunol. 2017;8:1589. doi: 10.3389/fimmu.2017.01589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bakhtiari SH, Rahbarizadeh F, Hasannia S, Ahmadvand D, Iri-Sofla FJ, Rasaee MJ. Anti-MUC1 nanobody can redirect T-body cytotoxic effector function. Hybridoma (Larchmt) 2009;28(2):85–92. doi: 10.1089/hyb.2008.0079. [DOI] [PubMed] [Google Scholar]
- 56.Jamnani FR, Rahbarizadeh F, Shokrgozar MA, Mahboudi F, Ahmadvand D, Sharifzadeh Z, et al. T cells expressing VHH-directed oligoclonal chimeric HER2 antigen receptors: towards tumor-directed oligoclonal T cell therapy. Biochim Biophys Acta. 2014;1840(1):378–386. doi: 10.1016/j.bbagen.2013.09.029. [DOI] [PubMed] [Google Scholar]
- 57.Hassani M, Hajari Taheri F, Sharifzadeh Z, Arashkia A, Hadjati J, van Weerden WM, et al. Construction of a chimeric antigen receptor bearing a nanobody against prostate a specific membrane antigen in prostate cancer. J Cell Biochem. 2019;120(6):10787–10795. doi: 10.1002/jcb.28370. [DOI] [PubMed] [Google Scholar]
- 58.Ref. 58: Bao C, Gao Q, Li LL, Han L, Zhang B, Ding Y, et al. The Application of Nanobody in CAR-T Therapy. Biomolecules. 2021;11(2):238. 10.3390/biom11020238. [DOI] [PMC free article] [PubMed]
- 59.Zhao WH, Liu J, Wang BY, Chen YX, Cao XM, Yang Y, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol. 2018;11(1):141. doi: 10.1186/s13045-018-0681-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–9166. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 61.Moeller R, Scherer J, Kassim S. 871 Construction and evaluation of interleukin 3 (IL3)-zetakine redirected cytolytic T Cells for the treatment of CD123 expressing acute myeloid leukemia. Journal for ImmunoTherapy of Cancer. 2021;9(Suppl 2):A912-A. doi: 10.1136/jitc-2021-SITC2021.871. [DOI] [Google Scholar]
- 62.Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–4072. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang Y, Xu Y, Li S, Liu J, Xing Y, Xing H, et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J Hematol Oncol. 2018;11(1):60. doi: 10.1186/s13045-018-0603-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maiorova V, Mollaev MD, Vikhreva P, Kulakovskaya E, Pershin D, Chudakov DM, et al. Natural Flt3Lg-Based Chimeric Antigen Receptor (Flt3-CAR) T Cells Successfully Target Flt3 on AML Cell Lines. Vaccines (Basel). 2021;9(11):1238. 10.3390/vaccines9111238. [DOI] [PMC free article] [PubMed]
- 65.Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. 2019;7(1):100–112. doi: 10.1158/2326-6066.CIR-18-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee L, Draper B, Chaplin N, Philip B, Chin M, Galas-Filipowicz D, et al. An APRIL-based chimeric antigen receptor for dual targeting of BCMA and TACI in multiple myeloma. Blood. 2018;131(7):746–758. doi: 10.1182/blood-2017-05-781351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakazawa Y, Matsuda K, Kurata T, Sueki A, Tanaka M, Sakashita K, et al. Anti-proliferative effects of T cells expressing a ligand-based chimeric antigen receptor against CD116 on CD34(+) cells of juvenile myelomonocytic leukemia. J Hematol Oncol. 2016;9:27. doi: 10.1186/s13045-016-0256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan BX, Sun YQ. Glycine residues provide flexibility for enzyme active sites. J Biol Chem. 1997;272(6):3190–3194. doi: 10.1074/jbc.272.6.3190. [DOI] [PubMed] [Google Scholar]
- 69.Chen X, Zaro JL, Shen WC. Fusion protein linkers: property, design and functionality. Adv Drug Deliv Rev. 2013;65(10):1357–1369. doi: 10.1016/j.addr.2012.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.van Rosmalen M, Krom M, Merkx M. Tuning the flexibility of Glycine-serine linkers to allow rational Design of Multidomain Proteins. Biochemistry. 2017;56(50):6565–6574. doi: 10.1021/acs.biochem.7b00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hudson PJ, Kortt AA. High avidity scFv multimers; diabodies and triabodies. J Immunol Methods. 1999;231(1–2):177–189. doi: 10.1016/S0022-1759(99)00157-X. [DOI] [PubMed] [Google Scholar]
- 72.Whitlow M, Filpula D, Rollence ML, Feng SL, Wood JF. Multivalent Fvs: characterization of single-chain Fv oligomers and preparation of a bispecific Fv. Protein Eng. 1994;7(8):1017–1026. doi: 10.1093/protein/7.8.1017. [DOI] [PubMed] [Google Scholar]
- 73.Dolezal O, Pearce LA, Lawrence LJ, McCoy AJ, Hudson PJ, Kortt AA. ScFv multimers of the anti-neuraminidase antibody NC10: shortening of the linker in single-chain Fv fragment assembled in V(L) to V(H) orientation drives the formation of dimers, trimers, tetramers and higher molecular mass multimers. Protein Eng. 2000;13(8):565–574. doi: 10.1093/protein/13.8.565. [DOI] [PubMed] [Google Scholar]
- 74.Schirrmann T, Menzel C, Hust M, Prilop J, Jostock T, Dübel S. Oligomeric forms of single chain immunoglobulin (scIgG) MAbs. 2010;2(1):73–76. doi: 10.4161/mabs.2.1.10784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-cell selection affects chimeric antigen receptor (CAR) T-cell potency and toxicity: updated results from a phase I anti-CD22 CAR T-cell trial. J Clin Oncol. 2020;38(17):1938–1950. doi: 10.1200/JCO.19.03279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Singh N, Frey NV, Engels B, Barrett DM, Shestova O, Ravikumar P, et al. Antigen-independent activation enhances the efficacy of 4-1BB-costimulated CD22 CAR T cells. Nat Med. 2021;27(5):842–850. doi: 10.1038/s41591-021-01326-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moritz D, Groner B. A spacer region between the single chain antibody- and the CD3 zeta-chain domain of chimeric T cell receptor components is required for efficient ligand binding and signaling activity. Gene Ther. 1995;2(8):539–546. [PubMed] [Google Scholar]
- 78.Qin L, Lai Y, Zhao R, Wei X, Weng J, Lai P, et al. Incorporation of a hinge domain improves the expansion of chimeric antigen receptor T cells. J Hematol Oncol. 2017;10(1):68. doi: 10.1186/s13045-017-0437-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther. 2017;25(11):2452–2465. doi: 10.1016/j.ymthe.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Muller YD, Nguyen DP, Ferreira LMR, Ho P, Raffin C, Valencia RVB, et al. The CD28-transmembrane domain mediates chimeric antigen receptor Heterodimerization with CD28. Front Immunol. 2021;12:639818. doi: 10.3389/fimmu.2021.639818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ying Z, Huang XF, Xiang X, Liu Y, Kang X, Song Y, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med. 2019;25(6):947–953. doi: 10.1038/s41591-019-0421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant. 2010;16(9):1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119(17):3940–3950. doi: 10.1182/blood-2011-10-387969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O'Neill A, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J Immunother. 2005;28(3):203–211. doi: 10.1097/01.cji.0000161397.96582.59. [DOI] [PubMed] [Google Scholar]
- 87.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19(12):3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gargett T, Yu W, Dotti G, Yvon ES, Christo SN, Hayball JD, et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol Ther. 2016;24(6):1135–1149. doi: 10.1038/mt.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hombach A, Hombach AA, Abken H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc ‘spacer’ domain in the extracellular moiety of chimeric antigen receptors avoids ‘off-target’ activation and unintended initiation of an innate immune response. Gene Ther. 2010;17(10):1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 90.Strohl WR. Optimization of fc-mediated effector functions of monoclonal antibodies. Curr Opin Biotechnol. 2009;20(6):685–691. doi: 10.1016/j.copbio.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 91.Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23(4):757–768. doi: 10.1038/mt.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Watanabe N, Bajgain P, Sukumaran S, Ansari S, Heslop HE, Rooney CM, et al. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology. 2016;5(12):e1253656. doi: 10.1080/2162402X.2016.1253656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C, et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol Res. 2015;3(2):125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schäfer D, Henze J, Pfeifer R, Schleicher A, Brauner J, Mockel-Tenbrinck N, et al. A novel Siglec-4 derived spacer improves the functionality of CAR T cells against membrane-proximal epitopes. Front Immunol. 2020;11:1704. doi: 10.3389/fimmu.2020.01704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Turtle CJ, Hanafi LA, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126(6):2123–2138. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bister A, Ibach T, Haist C, Smorra D, Roellecke K, Wagenmann M, et al. A novel CD34-derived hinge for rapid and efficient detection and enrichment of CAR T cells. Mol Ther Oncolytics. 2021;23:534–546. doi: 10.1016/j.omto.2021.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Aldoss I, Bargou RC, Nagorsen D, Friberg GR, Baeuerle PA, Forman SJ. Redirecting T cells to eradicate B-cell acute lymphoblastic leukemia: bispecific T-cell engagers and chimeric antigen receptors. Leukemia. 2017;31(4):777–787. doi: 10.1038/leu.2016.391. [DOI] [PubMed] [Google Scholar]
- 98.Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singh N, Frey NV, Grupp SA, Maude SL. CAR T cell therapy in acute lymphoblastic leukemia and potential for chronic lymphocytic leukemia. Curr Treat Options Oncol. 2016;17(6):28. doi: 10.1007/s11864-016-0406-4. [DOI] [PubMed] [Google Scholar]
- 100.Zhang A, Sun Y, Du J, Dong Y, Pang H, Ma L, et al. Reducing Hinge Flexibility of CAR-T Cells Prolongs Survival In Vivo With Low Cytokines Release. Front Immunol. 2021;12:724211. 10.3389/fimmu.2021.724211. [DOI] [PMC free article] [PubMed]
- 101.Casucci M, Falcone L, Camisa B, Norelli M, Porcellini S, Stornaiuolo A, et al. Extracellular NGFR spacers allow efficient tracking and enrichment of fully functional CAR-T cells co-expressing a suicide gene. Front Immunol. 2018;9:507. doi: 10.3389/fimmu.2018.00507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Iri-Sofla FJ, Rahbarizadeh F, Ahmadvand D, Rasaee MJ. Nanobody-based chimeric receptor gene integration in Jurkat cells mediated by φC31 integrase. Exp Cell Res. 2011;317(18):2630–2641. doi: 10.1016/j.yexcr.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 103.Sharifzadeh Z, Rahbarizadeh F, Shokrgozar MA, Ahmadvand D, Mahboudi F, Jamnani FR, et al. Genetically engineered T cells bearing chimeric nanoconstructed receptors harboring TAG-72-specific camelid single domain antibodies as targeting agents. Cancer Lett. 2013;334(2):237–244. doi: 10.1016/j.canlet.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 104.Fujiwara K, Tsunei A, Kusabuka H, Ogaki E, Tachibana M, Okada N. Hinge and Transmembrane Domains of Chimeric Antigen Receptor Regulate Receptor Expression and Signaling Threshold. Cells. 2020;9(5):1182. 10.3390/cells9051182. [DOI] [PMC free article] [PubMed]
- 105.Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3zeta transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J Immunol. 2010;184(12):6938–6949. doi: 10.4049/jimmunol.0901766. [DOI] [PubMed] [Google Scholar]
- 106.Zhang T, Wu MR, Sentman CL. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol. 2012;189(5):2290–2299. doi: 10.4049/jimmunol.1103495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Annenkov AE, Moyes SP, Eshhar Z, Mageed RA, Chernajovsky Y. Loss of original antigenic specificity in T cell hybridomas transduced with a chimeric receptor containing single-chain Fv of an anti-collagen antibody and fc epsilonRI-signaling gamma subunit. J Immunol. 1998;161(12):6604–6613. [PubMed] [Google Scholar]
- 108.Bridgeman JS, Ladell K, Sheard VE, Miners K, Hawkins RE, Price DA, et al. CD3ζ-based chimeric antigen receptors mediate T cell activation via cis- and trans-signalling mechanisms: implications for optimization of receptor structure for adoptive cell therapy. Clin Exp Immunol. 2014;175(2):258–267. doi: 10.1111/cei.12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ferreira LMR, Muller YD. CAR T-cell therapy: is CD28-CAR Heterodimerization its Achilles’ heel? Front Immunol. 2021;12:766220. doi: 10.3389/fimmu.2021.766220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discov. 2020;10(5):702–723. doi: 10.1158/2159-8290.CD-19-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Guedan S, Posey AD, Jr., Shaw C, Wing A, Da T, Patel PR, et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight. 2018;3(1):e96976. 10.1172/jci.insight.96976. [DOI] [PMC free article] [PubMed]
- 112.Wan Z, Shao X, Ji X, Dong L, Wei J, Xiong Z, et al. Transmembrane domain-mediated Lck association underlies bystander and costimulatory ICOS signaling. Cell Mol Immunol. 2020;17(2):143–152. doi: 10.1038/s41423-018-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schmidts A, Ormhøj M, Choi BD, Taylor AO, Bouffard AA, Scarfò I, et al. Rational design of a trimeric APRIL-based CAR-binding domain enables efficient targeting of multiple myeloma. Blood Adv. 2019;3(21):3248–3260. doi: 10.1182/bloodadvances.2019000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon E, et al. Generation of potent T-cell immunotherapy for CANCER using DAP12-based, multichain, Chimeric Immunoreceptors. Cancer Immunol Res. 2015;3(7):815–826. doi: 10.1158/2326-6066.CIR-15-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13(4):227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jenkins MK, Ashwell JD, Schwartz RH. Allogeneic non-T spleen cells restore the responsiveness of normal T cell clones stimulated with antigen and chemically modified antigen-presenting cells. J Immunol. 1988;140(10):3324–3330. [PubMed] [Google Scholar]
- 117.Schwartz RH, Mueller DL, Jenkins MK, Quill H. T-cell clonal anergy. Cold Spring Harb Symp Quant Biol. 1989;54(Pt 2):605–610. doi: 10.1101/SQB.1989.054.01.072. [DOI] [PubMed] [Google Scholar]
- 118.Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci U S A. 1993;90(2):720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hwu P, Shafer GE, Treisman J, Schindler DG, Gross G, Cowherd R, et al. Lysis of ovarian cancer cells by human lymphocytes redirected with a chimeric gene composed of an antibody variable region and the fc receptor gamma chain. J Exp Med. 1993;178(1):361–366. doi: 10.1084/jem.178.1.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hwu P, Yang JC, Cowherd R, Treisman J, Shafer GE, Eshhar Z, et al. In vivo antitumor activity of T cells redirected with chimeric antibody/T-cell receptor genes. Cancer Res. 1995;55(15):3369–3373. [PubMed] [Google Scholar]
- 121.Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6106–6115. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lim WA, June CH. The principles of engineering immune cells to treat Cancer. Cell. 2017;168(4):724–740. doi: 10.1016/j.cell.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Watts TH, DeBenedette MA. T cell co-stimulatory molecules other than CD28. Curr Opin Immunol. 1999;11(3):286–293. doi: 10.1016/S0952-7915(99)80046-6. [DOI] [PubMed] [Google Scholar]
- 124.Redmond WL, Ruby CE, Weinberg AD. The role of OX40-mediated co-stimulation in T-cell activation and survival. Crit Rev Immunol. 2009;29(3):187–201. doi: 10.1615/CritRevImmunol.v29.i3.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Gmünder H, Lesslauer W. A 45-kDa human T-cell membrane glycoprotein functions in the regulation of cell proliferative responses. Eur J Biochem. 1984;142(1):153–160. doi: 10.1111/j.1432-1033.1984.tb08263.x. [DOI] [PubMed] [Google Scholar]
- 126.Lesslauer W, Koning F, Ottenhoff T, Giphart M, Goulmy E, van Rood JJ. T90/44 (9.3 antigen). A cell surface molecule with a function in human T cell activation. Eur J Immunol. 1986;16(10):1289–1296. doi: 10.1002/eji.1830161017. [DOI] [PubMed] [Google Scholar]
- 127.Riha P, Rudd CE. CD28 co-signaling in the adaptive immune response. Self Nonself. 2010;1(3):231–240. doi: 10.4161/self.1.3.12968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.van der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med. 1997;185(3):393–403. doi: 10.1084/jem.185.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Harding FA, McArthur JG, Gross JA, Raulet DH, Allison JP. CD28-mediated signalling co-stimulates murine T cells and prevents induction of anergy in T-cell clones. Nature. 1992;356(6370):607–609. doi: 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- 130.Lindstein T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244(4902):339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 131.Fraser JD, Irving BA, Crabtree GR, Weiss A. Regulation of interleukin-2 gene enhancer activity by the T cell accessory molecule CD28. Science. 1991;251(4991):313–316. doi: 10.1126/science.1846244. [DOI] [PubMed] [Google Scholar]
- 132.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, et al. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3(1):87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 133.Noel PJ, Boise LH, Green JM, Thompson CB. CD28 costimulation prevents cell death during primary T cell activation. J Immunol. 1996;157(2):636–642. [PubMed] [Google Scholar]
- 134.Radvanyi LG, Shi Y, Vaziri H, Sharma A, Dhala R, Mills GB, et al. CD28 costimulation inhibits TCR-induced apoptosis during a primary T cell response. J Immunol. 1996;156(5):1788–1798. [PubMed] [Google Scholar]
- 135.Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16(6):769–777. doi: 10.1016/S1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 136.Granelli-Piperno A, Nolan P. Nuclear transcription factors that bind to elements of the IL-2 promoter. Induction requirements in primary human T cells. J Immunol. 1991;147(8):2734–2739. [PubMed] [Google Scholar]
- 137.Shapiro VS, Truitt KE, Imboden JB, Weiss A. CD28 mediates transcriptional upregulation of the interleukin-2 (IL-2) promoter through a composite element containing the CD28RE and NF-IL-2B AP-1 sites. Mol Cell Biol. 1997;17(7):4051–4058. doi: 10.1128/MCB.17.7.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Miller J, Baker C, Cook K, Graf B, Sanchez-Lockhart M, Sharp K, et al. Two pathways of costimulation through CD28. Immunol Res. 2009;45(2–3):159–172. doi: 10.1007/s12026-009-8097-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Viola A, Schroeder S, Sakakibara Y, Lanzavecchia A. T lymphocyte costimulation mediated by reorganization of membrane microdomains. Science. 1999;283(5402):680–682. doi: 10.1126/science.283.5402.680. [DOI] [PubMed] [Google Scholar]
- 140.Boomer JS, Green JM. An enigmatic tail of CD28 signaling. Cold Spring Harb Perspect Biol. 2010;2(8):a002436. doi: 10.1101/cshperspect.a002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Zumerle S, Molon B, Viola A. Membrane Rafts in T Cell Activation: A Spotlight on CD28 Costimulation. Front Immunol. 2017;8:1467. 10.3389/fimmu.2017.01467. [DOI] [PMC free article] [PubMed]
- 142.Yoshinaga SK, Whoriskey JS, Khare SD, Sarmiento U, Guo J, Horan T, et al. T-cell co-stimulation through B7RP-1 and ICOS. Nature. 1999;402(6763):827–832. doi: 10.1038/45582. [DOI] [PubMed] [Google Scholar]
- 143.Riley JL, Mao M, Kobayashi S, Biery M, Burchard J, Cavet G, et al. Modulation of TCR-induced transcriptional profiles by ligation of CD28, ICOS, and CTLA-4 receptors. Proc Natl Acad Sci U S A. 2002;99(18):11790–11795. doi: 10.1073/pnas.162359999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Coyle AJ, Lehar S, Lloyd C, Tian J, Delaney T, Manning S, et al. The CD28-related molecule ICOS is required for effective T cell-dependent immune responses. Immunity. 2000;13(1):95–105. doi: 10.1016/S1074-7613(00)00011-X. [DOI] [PubMed] [Google Scholar]
- 145.Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH, et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature. 2001;409(6816):97–101. doi: 10.1038/35051100. [DOI] [PubMed] [Google Scholar]
- 146.Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN, et al. The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells. Sci Transl Med. 2010;2(55):55ra78. doi: 10.1126/scitranslmed.3000448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Harada Y, Ohgai D, Watanabe R, Okano K, Koiwai O, Tanabe K, et al. A single amino acid alteration in cytoplasmic domain determines IL-2 promoter activation by ligation of CD28 but not inducible costimulator (ICOS) J Exp Med. 2003;197(2):257–262. doi: 10.1084/jem.20021305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kwon BS, Weissman SM. cDNA sequences of two inducible T-cell genes. Proc Natl Acad Sci U S A. 1989;86(6):1963–1967. doi: 10.1073/pnas.86.6.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Schwarz H, Blanco FJ, von Kempis J, Valbracht J, Lotz M. ILA, a member of the human nerve growth factor/tumor necrosis factor receptor family, regulates T-lymphocyte proliferation and survival. Blood. 1996;87(7):2839–2845. doi: 10.1182/blood.V87.7.2839.bloodjournal8772839. [DOI] [PubMed] [Google Scholar]
- 150.Vinay DS, Kwon BS. 4-1BB signaling beyond T cells. Cell Mol Immunol. 2011;8(4):281–284. doi: 10.1038/cmi.2010.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cannons JL, Lau P, Ghumman B, DeBenedette MA, Yagita H, Okumura K, et al. 4-1BB ligand induces cell division, sustains survival, and enhances effector function of CD4 and CD8 T cells with similar efficacy. J Immunol. 2001;167(3):1313–1324. doi: 10.4049/jimmunol.167.3.1313. [DOI] [PubMed] [Google Scholar]
- 152.Hurtado JC, Kim Y-J, Kwon BS. Signals through 4-1BB are costimulatory to previously activated splenic T cells and inhibit activation-induced cell death. J Immunol. 1997;158(6):2600–2609. [PubMed] [Google Scholar]
- 153.Takahashi C, Mittler RS, Vella AT. Cutting edge: 4-1BB is a bona fide CD8 T cell survival signal. J Immunol. 1999;162(9):5037–5040. [PubMed] [Google Scholar]
- 154.Shuford WW, Klussman K, Tritchler DD, Loo DT, Chalupny J, Siadak AW, et al. 4-1BB costimulatory signals preferentially induce CD8+ T cell proliferation and lead to the amplification in vivo of cytotoxic T cell responses. J Exp Med. 1997;186(1):47–55. doi: 10.1084/jem.186.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Myers L, Takahashi C, Mittler RS, Rossi RJ, Vella AT. Effector CD8 T cells possess suppressor function after 4-1BB and toll-like receptor triggering. Proc Natl Acad Sci U S A. 2003;100(9):5348–5353. doi: 10.1073/pnas.0837611100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Vinay DS, Cha K, Kwon BS. Dual immunoregulatory pathways of 4-1BB signaling. J Mol Med (Berl) 2006;84(9):726–736. doi: 10.1007/s00109-006-0072-2. [DOI] [PubMed] [Google Scholar]
- 157.Croft M, So T, Duan W, Soroosh P. The significance of OX40 and OX40L to T-cell biology and immune disease. Immunol Rev. 2009;229(1):173–191. doi: 10.1111/j.1600-065X.2009.00766.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. 2003;3(8):609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 159.Dawicki W, Bertram EM, Sharpe AH, Watts TH. 4-1BB and OX40 act independently to facilitate robust CD8 and CD4 recall responses. J Immunol. 2004;173(10):5944–5951. doi: 10.4049/jimmunol.173.10.5944. [DOI] [PubMed] [Google Scholar]
- 160.Serghides L, Bukczynski J, Wen T, Wang C, Routy JP, Boulassel MR, et al. Evaluation of OX40 ligand as a costimulator of human antiviral memory CD8 T cell responses: comparison with B7.1 and 4-1BBL. J Immunol. 2005;175(10):6368–6377. doi: 10.4049/jimmunol.175.10.6368. [DOI] [PubMed] [Google Scholar]
- 161.Hombach AA, Heiders J, Foppe M, Chmielewski M, Abken H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+ T cells. OncoImmunology. 2012;1(4):458–466. doi: 10.4161/onci.19855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang H, Li F, Cao J, Wang X, Cheng H, Qi K, et al. A chimeric antigen receptor with antigen-independent OX40 signaling mediates potent antitumor activity. Sci Transl Med. 2021;13(578):eaba7308. 10.1126/scitranslmed.aba7308. [DOI] [PubMed]
- 163.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9(4):271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Bullock TN. Stimulating CD27 to quantitatively and qualitatively shape adaptive immunity to cancer. Curr Opin Immunol. 2017;45:82–88. doi: 10.1016/j.coi.2017.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.He Y, Vlaming M, van Meerten T, Bremer E. The Implementation of TNFRSF Co-Stimulatory Domains in CAR-T Cells for Optimal Functional Activity. Cancers (Basel). 2022;14(2):299. 10.3390/cancers14020299. [DOI] [PMC free article] [PubMed]
- 166.Cappell KM, Kochenderfer JN. A comparison of chimeric antigen receptors containing CD28 versus 4-1BB costimulatory domains. Nat Rev Clin Oncol. 2021;18(11):715–727. doi: 10.1038/s41571-021-00530-z. [DOI] [PubMed] [Google Scholar]
- 167.Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56. doi: 10.1056/NEJMoa1804980. [DOI] [PubMed] [Google Scholar]
- 168.Abramson JS, Palomba ML, Gordon LI, Lunning MA, Wang M, Arnason J, et al. Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study. Lancet. 2020;396(10254):839–852. doi: 10.1016/S0140-6736(20)31366-0. [DOI] [PubMed] [Google Scholar]
- 169.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther. 2009;17(8):1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Priceman SJ, Gerdts EA, Tilakawardane D, Kennewick KT, Murad JP, Park AK, et al. Co-stimulatory signaling determines tumor antigen sensitivity and persistence of CAR T cells targeting PSCA+ metastatic prostate cancer. Oncoimmunology. 2018;7(2):e1380764. doi: 10.1080/2162402X.2017.1380764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Amatya C, Pegues MA, Lam N, Vanasse D, Geldres C, Choi S, et al. Development of CAR T cells expressing a suicide gene plus a chimeric antigen receptor targeting signaling lymphocytic-activation molecule F7. Mol Ther. 2021;29(2):702–717. doi: 10.1016/j.ymthe.2020.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Powell DJ, Jr, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105(1):241–250. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Song D-G, Powell DJ. Pro-survival signaling via CD27 costimulation drives effective CAR T-cell therapy. Oncoimmunology. 2012;1(4):547–549. doi: 10.4161/onci.19458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Locke FL, Neelapu SS, Bartlett NL, Siddiqi T, Chavez JC, Hosing CM, et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol Ther. 2017;25(1):285–295. doi: 10.1016/j.ymthe.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Porter DL, Hwang WT, Frey NV, Lacey SF, Shaw PA, Loren AW, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med. 2015;7(303):303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Song DG, Ye Q, Poussin M, Harms GM, Figini M, Powell DJ., Jr CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood. 2012;119(3):696–706. doi: 10.1182/blood-2011-03-344275. [DOI] [PubMed] [Google Scholar]
- 177.Wutti-In Y, Sujjitjoon J, Sawasdee N, Panya A, Kongkla K, Yuti P, et al. Development of a novel anti-CD19 CAR containing a fully human scFv and three costimulatory domains. Front Oncol. 2021;11:802876. doi: 10.3389/fonc.2021.802876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD, Jr, et al. Distinct signaling of Coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380–390. doi: 10.1016/j.immuni.2016.01.021. [DOI] [PubMed] [Google Scholar]
- 179.Zhao Z, Condomines M, van der Stegen SJC, Perna F, Kloss CC, Gunset G, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T cells. Cancer Cell. 2015;28(4):415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Guedan S, Madar A, Casado-Medrano V, Shaw C, Wing A, Liu F, et al. Single residue in CD28-costimulated CAR-T cells limits long-term persistence and antitumor durability. J Clin Invest. 2020;130(6):3087–3097. doi: 10.1172/JCI133215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2020;382(14):1331–1342. doi: 10.1056/NEJMoa1914347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Brudno JN, Lam N, Vanasse D, Shen YW, Rose JJ, Rossi J, et al. Safety and feasibility of anti-CD19 CAR T cells with fully human binding domains in patients with B-cell lymphoma. Nat Med. 2020;26(2):270–280. doi: 10.1038/s41591-019-0737-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Munroe ME, Bishop GA. A costimulatory function for T cell CD40. J Immunol. 2007;178(2):671–682. doi: 10.4049/jimmunol.178.2.671. [DOI] [PubMed] [Google Scholar]
- 184.Nunoya JI, Masuda M, Ye C, Su L. Chimeric antigen receptor T cell bearing herpes virus entry mediator co-stimulatory signal domain exhibits high functional potency. Mol Ther Oncolytics. 2019;14:27–37. doi: 10.1016/j.omto.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Golubovskaya VM, Berahovich R, Xu Q, Zhou H, Xu S, Guan J, et al. GITR domain inside CAR co-stimulates activity of CAR-T cells against cancer. Front Biosci (Landmark Ed) 2018;23:2245–2254. doi: 10.2741/4703. [DOI] [PubMed] [Google Scholar]
- 186.Collinson-Pautz MR, Chang WC, Lu A, Khalil M, Crisostomo JW, Lin PY, et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia. 2019;33(9):2195–2207. doi: 10.1038/s41375-019-0417-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Lai Y, Weng J, Wei X, Qin L, Lai P, Zhao R, et al. Toll-like receptor 2 costimulation potentiates the antitumor efficacy of CAR T cells. Leukemia. 2018;32(3):801–808. doi: 10.1038/leu.2017.249. [DOI] [PubMed] [Google Scholar]
- 188.Liang X, Huang Y, Li D, Yang X, Jiang L, Zhou W, et al. Distinct functions of CAR-T cells possessing a dectin-1 intracellular signaling domain. Gene Ther. 2021. 10.1038/s41434-021-00257-7. [DOI] [PubMed]
- 189.Sadelain M, Brentjens R, Rivière I. The promise and potential pitfalls of chimeric antigen receptors. Curr Opin Immunol. 2009;21(2):215–223. doi: 10.1016/j.coi.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Hombach A, Muche JM, Gerken M, Gellrich S, Heuser C, Pohl C, et al. T cells engrafted with a recombinant anti-CD30 receptor target autologous CD30(+) cutaneous lymphoma cells. Gene Ther. 2001;8(11):891–895. doi: 10.1038/sj.gt.3301467. [DOI] [PubMed] [Google Scholar]
- 191.Feucht J, Sun J, Eyquem J, Ho YJ, Zhao Z, Leibold J, et al. Calibration of CAR activation potential directs alternative T cell fates and therapeutic potency. Nat Med. 2019;25(1):82–88. doi: 10.1038/s41591-018-0290-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fisher J, Abramowski P, Wisidagamage Don ND, Flutter B, Capsomidis A, Cheung GW, et al. Avoidance of on-target off-tumor activation using a co-stimulation-only chimeric antigen receptor. Mol Ther. 2017;25(5):1234–1247. doi: 10.1016/j.ymthe.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, et al. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72(12):9873–9880. doi: 10.1128/JVI.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Sertkaya H, Ficarelli M, Sweeney NP, Parker H, Vink CA, Swanson CM. HIV-1 sequences in lentiviral vector genomes can be substantially reduced without compromising transduction efficiency. Sci Rep. 2021;11(1):12067. doi: 10.1038/s41598-021-91309-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Powell SK, Rivera-Soto R, Gray SJ. Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov Med. 2015;19(102):49–57. [PMC free article] [PubMed] [Google Scholar]
- 196.Rad SMAH, Poudel A, Tan GMY, McLellan AD. Promoter choice: who should drive the CAR in T cells? Plos One. 2020;15(7):e0232915-e. doi: 10.1371/journal.pone.0232915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Isomura H, Stinski MF. The human cytomegalovirus major immediate-early enhancer determines the efficiency of immediate-early gene transcription and viral replication in permissive cells at low multiplicity of infection. J Virol. 2003;77(6):3602–3614. doi: 10.1128/JVI.77.6.3602-3614.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Fang Y, Zhang Y, Guo C, Chen C, Gao H, Zhou X, et al. Safety and efficacy of an immune cell-specific chimeric promoter in regulating anti-PD-1 antibody expression in CAR T cells. Mol Ther Methods Clin Dev. 2020;19:14–23. doi: 10.1016/j.omtm.2020.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res. 2015;3(4):356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Kochenderfer JN, Feldman SA, Zhao Y, Xu H, Black MA, Morgan RA, et al. Construction and preclinical evaluation of an anti-CD19 chimeric antigen receptor. J Immunother. 2009;32(7):689–702. doi: 10.1097/CJI.0b013e3181ac6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Hughes MS, Yu YY, Dudley ME, Zheng Z, Robbins PF, Li Y, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16(4):457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Brandt LJB, Barnkob MB, Michaels YS, Heiselberg J, Barington T. Emerging approaches for regulation and control of CAR T cells: a Mini review. Front Immunol. 2020;11:326. doi: 10.3389/fimmu.2020.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Klump H, Schiedlmeier B, Vogt B, Ryan M, Ostertag W, Baum C. Retroviral vector-mediated expression of HoxB4 in hematopoietic cells using a novel coexpression strategy. Gene Ther. 2001;8(10):811–817. doi: 10.1038/sj.gt.3301447. [DOI] [PubMed] [Google Scholar]
- 205.Martínez-Salas E. Internal ribosome entry site biology and its use in expression vectors. Curr Opin Biotechnol. 1999;10(5):458–464. doi: 10.1016/S0958-1669(99)00010-5. [DOI] [PubMed] [Google Scholar]
- 206.Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E, Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol Ther. 2000;1(4):376–382. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- 207.Emerman M, Temin HM. Quantitative analysis of gene suppression in integrated retrovirus vectors. Mol Cell Biol. 1986;6(3):792–800. doi: 10.1128/mcb.6.3.792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Curtin JA, Dane AP, Swanson A, Alexander IE, Ginn SL. Bidirectional promoter interference between two widely used internal heterologous promoters in a late-generation lentiviral construct. Gene Ther. 2008;15(5):384–390. doi: 10.1038/sj.gt.3303105. [DOI] [PubMed] [Google Scholar]
- 209.Amendola M, Venneri MA, Biffi A, Vigna E, Naldini L. Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat Biotechnol. 2005;23(1):108–116. doi: 10.1038/nbt1049. [DOI] [PubMed] [Google Scholar]
- 210.He K, Rad SMAH, Poudel A, McLellan AD. Compact Bidirectional Promoters for Dual-Gene Expression in a Sleeping Beauty Transposon. Int J Mol Sci. 2020;21(23):9256. 10.3390/ijms21239256. [DOI] [PMC free article] [PubMed]
- 211.Mølhøj M, Degan FD. Leader sequences are not signal peptides. Nat Biotechnol. 2004;22(12):1502. doi: 10.1038/nbt1204-1502. [DOI] [PubMed] [Google Scholar]
- 212.von Heijne G. Signal sequences. The limits of variation. J Mol Biol. 1985;184(1):99–105. doi: 10.1016/0022-2836(85)90046-4. [DOI] [PubMed] [Google Scholar]
- 213.Owji H, Nezafat N, Negahdaripour M, Hajiebrahimi A, Ghasemi Y. A comprehensive review of signal peptides: structure, roles, and applications. Eur J Cell Biol. 2018;97(6):422–441. doi: 10.1016/j.ejcb.2018.06.003. [DOI] [PubMed] [Google Scholar]
- 214.Walter P, Blobel G. Purification of a membrane-associated protein complex required for protein translocation across the endoplasmic reticulum. Proc Natl Acad Sci. 1980;77(12):7112–7116. doi: 10.1073/pnas.77.12.7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kapp K SS, Lemberg MK, et al. Post-Targeting functions of signal peptides. madame curie bioscience database, Austin: Landes Bioscience; 2000-2013, Available from https://www.ncbi.nlm.nih.gov/books/NBK6322/.
- 216.Walter P, Gilmore R, Blobel G. Protein translocation across the endoplasmic reticulum. Cell. 1984;38(1):5–8. doi: 10.1016/0092-8674(84)90520-8. [DOI] [PubMed] [Google Scholar]
- 217.Zhang E, Gu J, Xue J, Lin C, Liu C, Li M, et al. Accurate control of dual-receptor-engineered T cell activity through a bifunctional anti-angiogenic peptide. J Hematol Oncol. 2018;11(1):44. doi: 10.1186/s13045-018-0591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Moot R, Raikar SS, Fleischer L, Querrey M, Tylawsky DE, Nakahara H, et al. Genetic engineering of chimeric antigen receptors using lamprey derived variable lymphocyte receptors. Mol Ther Oncolytics. 2016;3:16026. doi: 10.1038/mto.2016.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101(4):1637–1644. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 220.Feldmann A, Hoffmann A, Bergmann R, Koristka S, Berndt N, Arndt C, et al. Versatile chimeric antigen receptor platform for controllable and combinatorial T cell therapy. OncoImmunology. 2020;9(1):1785608. doi: 10.1080/2162402X.2020.1785608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wang X, Chang WC, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Güler-Gane G, Kidd S, Sridharan S, Vaughan TJ, Wilkinson TC, Tigue NJ. Overcoming the refractory expression of secreted recombinant proteins in mammalian cells through modification of the signal peptide and adjacent amino acids. Plos One. 2016;11(5):e0155340. doi: 10.1371/journal.pone.0155340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ping Y, Li F, Nan S, Zhang D, Shi X, Shan J, et al. Augmenting the effectiveness of CAR-T cells by enhanced self-delivery of PD-1-neutralizing scFv. Front Cell Dev Biol. 2020;8:803. doi: 10.3389/fcell.2020.00803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Lee J-H, Culver G, Carpenter S, Dobbs D. Analysis of the EIAV rev-responsive element (RRE) reveals a conserved RNA motif required for high affinity rev binding in both HIV-1 and EIAV. Plos One. 2008;3(6):e2272. doi: 10.1371/journal.pone.0002272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Fernandes J, Jayaraman B, Frankel A. The HIV-1 rev response element: an RNA scaffold that directs the cooperative assembly of a homo-oligomeric ribonucleoprotein complex. RNA Biol. 2012;9(1):6–11. doi: 10.4161/rna.9.1.18178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zapp ML, Green MR. Sequence-specific RNA binding by the HIV-1 rev protein. Nature. 1989;342(6250):714–716. doi: 10.1038/342714a0. [DOI] [PubMed] [Google Scholar]
- 227.Sherpa C, Rausch JW, Le Grice SF, Hammarskjold ML, Rekosh D. The HIV-1 rev response element (RRE) adopts alternative conformations that promote different rates of virus replication. Nucleic Acids Res. 2015;43(9):4676–4686. doi: 10.1093/nar/gkv313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.DiMattia MA, Watts NR, Cheng N, Huang R, Heymann JB, Grimes JM, et al. The structure of HIV-1 rev filaments suggests a bilateral model for rev-RRE assembly. Structure. 2016;24(7):1068–1080. doi: 10.1016/j.str.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Hoffmann D, Schwarck D, Banning C, Brenner M, Mariyanna L, Krepstakies M, et al. Formation of trans-activation competent HIV-1 rev:RRE complexes requires the recruitment of multiple protein activation domains. Plos One. 2012;7(6):e38305. doi: 10.1371/journal.pone.0038305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Booth DS, Cheng Y, Frankel AD. The export receptor Crm1 forms a dimer to promote nuclear export of HIV RNA. Elife. 2014;3:e04121-e. doi: 10.7554/eLife.04121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Daugherty MD, Booth DS, Jayaraman B, Cheng Y, Frankel AD. HIV rev response element (RRE) directs assembly of the rev homooligomer into discrete asymmetric complexes. Proc Natl Acad Sci U S A. 2010;107(28):12481–12486. doi: 10.1073/pnas.1007022107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Wurtzer S, Goubard A, Mammano F, Saragosti S, Lecossier D, Hance AJ, et al. Functional central polypurine tract provides downstream protection of the human immunodeficiency virus type 1 genome from editing by APOBEC3G and APOBEC3B. J Virol. 2006;80(7):3679–3683. doi: 10.1128/JVI.80.7.3679-3683.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Charneau P, Clavel F. A single-stranded gap in human immunodeficiency virus unintegrated linear DNA defined by a central copy of the polypurine tract. J Virol. 1991;65(5):2415–2421. doi: 10.1128/jvi.65.5.2415-2421.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Charneau P, Alizon M, Clavel F. A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J Virol. 1992;66(5):2814–2820. doi: 10.1128/jvi.66.5.2814-2820.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Arhel N, Munier S, Souque P, Mollier K, Charneau P. Nuclear import defect of human immunodeficiency virus type 1 DNA flap mutants is not dependent on the viral strain or target cell type. J Virol. 2006;80(20):10262–10269. doi: 10.1128/JVI.00974-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Arhel NJ, Souquere-Besse S, Munier S, Souque P, Guadagnini S, Rutherford S, et al. HIV-1 DNA flap formation promotes uncoating of the pre-integration complex at the nuclear pore. EMBO J. 2007;26(12):3025–3037. doi: 10.1038/sj.emboj.7601740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Manganini M, Serafini M, Bambacioni F, Casati C, Erba E, Follenzi A, et al. A human immunodeficiency virus type 1 pol gene-derived sequence (cPPT/CTS) increases the efficiency of transduction of human nondividing monocytes and T lymphocytes by lentiviral vectors. Hum Gene Ther. 2002;13(15):1793–1807. doi: 10.1089/104303402760372909. [DOI] [PubMed] [Google Scholar]
- 238.Johnson NM, Alvarado AF, Moffatt TN, Edavettal JM, Swaminathan TA, Braun SE. HIV-based lentiviral vectors: origin and sequence differences. Mol Ther Methods Clin Dev. 2021;21:451–465. doi: 10.1016/j.omtm.2021.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Van Maele B, De Rijck J, De Clercq E, Debyser Z. Impact of the central polypurine tract on the kinetics of human immunodeficiency virus type 1 vector transduction. J Virol. 2003;77(8):4685–4694. doi: 10.1128/JVI.77.8.4685-4694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Guntaka RV. Transcription termination and polyadenylation in retroviruses. Microbiol Rev. 1993;57(3):511–521. doi: 10.1128/mr.57.3.511-521.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Furger A, Monks J, Proudfoot NJ. The retroviruses human immunodeficiency virus type 1 and Moloney murine leukemia virus adopt radically different strategies to regulate promoter-proximal polyadenylation. J Virol. 2001;75(23):11735–11746. doi: 10.1128/JVI.75.23.11735-11746.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Brown PH, Tiley LS, Cullen BR. Efficient polyadenylation within the human immunodeficiency virus type 1 long terminal repeat requires flanking U3-specific sequences. J Virol. 1991;65(6):3340–3343. doi: 10.1128/jvi.65.6.3340-3343.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Gilmartin GM, Fleming ES, Oetjen J, Graveley BR. CPSF recognition of an HIV-1 mRNA 3′-processing enhancer: multiple sequence contacts involved in poly(a) site definition. Genes Dev. 1995;9(1):72–83. doi: 10.1101/gad.9.1.72. [DOI] [PubMed] [Google Scholar]
- 244.Schambach A, Galla M, Maetzig T, Loew R, Baum C. Improving transcriptional termination of self-inactivating gamma-retroviral and lentiviral vectors. Mol Ther. 2007;15(6):1167–1173. doi: 10.1038/sj.mt.6300152. [DOI] [PubMed] [Google Scholar]
- 245.Hager S, Frame FM, Collins AT, Burns JE, Maitland NJ. An internal polyadenylation signal substantially increases expression levels of lentivirus-delivered transgenes but has the potential to reduce viral titer in a promoter-dependent manner. Hum Gene Ther. 2008;19(8):840–850. doi: 10.1089/hum.2007.165. [DOI] [PubMed] [Google Scholar]
- 246.Choi J-H, Yu N-K, Baek G-C, Bakes J, Seo D, Nam HJ, et al. Optimization of AAV expression cassettes to improve packaging capacity and transgene expression in neurons. Mol Brain. 2014;7(1):17. doi: 10.1186/1756-6606-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Donello JE, Loeb JE, Hope TJ. Woodchuck hepatitis virus contains a tripartite posttranscriptional regulatory element. J Virol. 1998;72(6):5085–5092. doi: 10.1128/JVI.72.6.5085-5092.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Zufferey R, Donello JE, Trono D, Hope TJ. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J Virol. 1999;73(4):2886–2892. doi: 10.1128/JVI.73.4.2886-2892.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Loeb JE, Cordier WS, Harris ME, Weitzman MD, Hope TJ. Enhanced expression of transgenes from adeno-associated virus vectors with the woodchuck hepatitis virus posttranscriptional regulatory element: implications for gene therapy. Hum Gene Ther. 1999;10(14):2295–2305. doi: 10.1089/10430349950016942. [DOI] [PubMed] [Google Scholar]
- 250.Xu ZL, Mizuguchi H, Mayumi T, Hayakawa T. Woodchuck hepatitis virus post-transcriptional regulation element enhances transgene expression from adenovirus vectors. Biochim Biophys Acta. 2003;1621(3):266–271. doi: 10.1016/S0304-4165(03)00078-3. [DOI] [PubMed] [Google Scholar]
- 251.Higashimoto T, Urbinati F, Perumbeti A, Jiang G, Zarzuela A, Chang LJ, et al. The woodchuck hepatitis virus post-transcriptional regulatory element reduces readthrough transcription from retroviral vectors. Gene Ther. 2007;14(17):1298–1304. doi: 10.1038/sj.gt.3302979. [DOI] [PubMed] [Google Scholar]
- 252.Zaiss AK, Son S, Chang LJ. RNA 3′ readthrough of oncoretrovirus and lentivirus: implications for vector safety and efficacy. J Virol. 2002;76(14):7209–7219. doi: 10.1128/JVI.76.14.7209-7219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Iwakuma T, Cui Y, Chang LJ. Self-inactivating lentiviral vectors with U3 and U5 modifications. Virology. 1999;261(1):120–132. doi: 10.1006/viro.1999.9850. [DOI] [PubMed] [Google Scholar]
- 254.Yang Q, Lucas A, Son S, Chang L-J. Overlapping enhancer/promoter and transcriptional termination signals in the lentiviral long terminal repeat. Retrovirology. 2007;4(1):4. doi: 10.1186/1742-4690-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Lukjanov V, Koutná I, Šimara P. CAR T-cell production using nonviral approaches. J Immunol Res. 2021;2021:6644685. doi: 10.1155/2021/6644685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Singh H, Moyes JS, Huls MH, Cooper LJ. Manufacture of T cells using the sleeping beauty system to enforce expression of a CD19-specific chimeric antigen receptor. Cancer Gene Ther. 2015;22(2):95–100. doi: 10.1038/cgt.2014.69. [DOI] [PubMed] [Google Scholar]
- 257.Cary LC, Goebel M, Corsaro BG, Wang HG, Rosen E, Fraser MJ. Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology. 1989;172(1):156–169. doi: 10.1016/0042-6822(89)90117-7. [DOI] [PubMed] [Google Scholar]
- 258.Tsukahara T, Iwase N, Kawakami K, Iwasaki M, Yamamoto C, Ohmine K, et al. The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene Ther. 2015;22(2):209–215. doi: 10.1038/gt.2014.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using sleeping beauty to generate CD19-specific CAR T cells. J Clin Invest. 2016;126(9):3363–3376. doi: 10.1172/JCI86721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Kebriaei P, Huls H, Neel SL, Olivares S, Orozco AF, Su S, et al. Shortening the time to manufacture CAR+ T cells with sleeping beauty system supports T-cell engraftment and anti-tumor effects in patients with refractory CD19+ tumors. Blood. 2017;130:2060. doi: 10.1182/blood.V130.Suppl_1.886.886. [DOI] [Google Scholar]
- 261.Magnani CF, Gaipa G, Belotti D, Matera G, Tettamanti S, Cabiati B, et al. Donor-derived CD19 CAR cytokine induced killer (CIK) cells engineered with sleeping beauty transposon for relapsed B-cell acute lymphoblastic leukemia (B-ALL) Blood. 2019;134:200. doi: 10.1182/blood-2019-125894. [DOI] [Google Scholar]
- 262.Li C, Sun Y, Wang J, Tang L, Jiang H, Guo T, et al. PiggyBac-generated CAR19-T cells plus Lenalidomide cause durable complete remission of triple-hit refractory/relapsed DLBCL: a case report. Front Immunol. 2021;12:599493. doi: 10.3389/fimmu.2021.599493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Pomeroy EJ, Lahr WS, Chang JW, Krueger JB, Wick BJ, Slipek NJ, et al.. Non-Viral Engineering of CAR-NK and CAR-T Cells Using the Tc Buster Transposon System™. bioRxiv. 2021. 10.1101/2021.08.02.454772.
- 264.Tipanee J, Samara-Kuko E, Gevaert T, Chuah MK, VandenDriessche T. Universal allogeneic CAR T cells engineered with Sleeping Beauty transposons and CRISPR-CAS9 for cancer immunotherapy. Mol Ther. 2022:S1525-0016(22)00366-5. 10.1016/j.ymthe.2022.06.006. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.