

Abstract
Pulmonary alveolar microlithiasis is a rare diffuse lung disease characterised by deposition of calcium phosphate within the alveolar airspaces. The disease is usually discovered from birth up to 40 yrs of age and is often diagnosed incidentally during radiography of the chest for other reasons. Many patients are asymptomatic and the majority of patients either have normal or restrictive pulmonary function. The clinical course of the disease varies. While it remains static in some patients, it progresses into pulmonary fibrosis, respiratory failure and cor pulmonale in others. With the exception of lung transplantation, there is no known effective treatment for the disease. Although the aetiology remains unclear, mutations of the solute carrier family 34 (sodium phosphate), member 2 gene (the SLC34A2 gene), which encodes a sodium/phosphate co-transporter, are considered to be the cause of the disease.
We present two cases of pulmonary alveolar microlithiasis with different mutations in the SLC34A2 gene that have not been previously described, and a review of the literature.
Keywords: Alveolar microlithiasis, genetic polymorphisms, type IIb sodium-dependent phosphate co-transporter
Pulmonary alveolar microlithiasis (PAM) is a rare diffuse lung disease. It was first named in 1933 by Puhr [1], and is characterised by deposition of spherical calcium phosphate concretions (microliths) in the alveoli [2–4]. Although the aetiology remains unclear, PAM is thought to be an autosomal recessive disease caused by mutations of the solute carrier family 34 (sodium phosphate), member 2 gene (the SLC34A2 gene), which encodes a sodium phosphate co-transporter [[5].
Herein, we present two cases of PAM with different mutations in the SLC34A2 gene that have not been previously described and, in addition, we review the literature.
CASE ONE
A 49-yr old male from Denmark was referred to the Dept of Respiratory Medicine, Aarhus University Hospital (Aarhus, Denmark) for diagnostic investigation in April 2005. The patient was adopted, and therefore the family history was unknown. He was complaining of fatigue and progressively worsening dyspnoea over the last 4 yrs. He had no cough or sputum. He was a heavy smoker (30 pack-yrs).
Upon first admission, the ECG showed signs of serious ischaemia and he was transferred to the Dept of Cardiology (Aarhus University Hospital), where he underwent coronary stenting and was started on clopidogrel. The physical examination revealed basal fibrotic rales bilaterally and hourglass nails. Pulmonary function tests showed a restrictive pattern (table 1). High-resolution computed tomography (HRCT) and conventional radiographs revealed typical radiological findings for PAM (figs 1 and 2).
Table 1. Patient characteristics.
| Case one | Case two | |
| Sex | Male | Male |
| Age at diagnosis yrs | 50 | 16 |
| Comorbidity | Ischaemic heart disease | Nephrolithiasis Significant haemodynamic aortic valve stenosis |
| HRCT | Bilateral lobular, cavitating, nodular infiltrates, ground-glass attenuation Intra- and interlobular septal thickening and consolidation of basal areas Enlarged lymph nodes in the mediastinum Paraseptally emphysema Punctuate calcifications in the lung parenchyma |
Bilateral increased attenuation involving alveoli, intra- and interlobular septa, fissures and pleura Signs of fibrosis and calcifications A kidney stone |
| Peripheral O2 without supplemental O2 PFT | Normal saturation (98%) | Normal saturation (98%) |
| FEV1 L (% pred) | 2.97 (72.5) | 3.55 (74.6) |
| FVC L | 3.61 (71.8) | 4.14 (73.3) |
| TLC L | 5.22 (70.8) | 5.03 (74.1) |
| Diffusing coefficient mmol·min−1·kPa−1·L−1 (% pred) | 1.05 (71.7) | 4.39 (93.4) |
| Diffusing capacity mmol·min−1·kPa−1 (% pred) | 5.36 (49.4) | 26.11 (81.8) |
| 6-min walk without oxygen | ||
| Distance walked m | 600 | 480 |
| Lowest O2 saturation | No desaturation | No desaturation |
| BAL fluid examination | Alveolar macrophages | Many giant cells |
| Histopathology | Numerous intra-alveolar rounded calcificated bodies with concentrically lamels in the periphery | Numerous concentrically calcified concretions Incrustations and calcifications close to the walls of the alveoli |
| Gene mutation in exon 12 of SLC34A2 gene# | Homozygous c.1393-1404delACC | Homozygous c.1402-1404delACC |
| Treatment | Prednisolone/didronate/no treatment | Didronate/no treatment |
HRCT: high-resolution computed tomography; PFT: pulmonary function test; FEV1: forced expiratory volume in 1 s; % pred: % predicted; FVC: forced vital capacity; TLC: total lung capacity; BAL: bronchoalveolar lavage. #: DNA sequencing analysis of whole coding regions of the SLC34A2 (OMIM *604217) gene T chromosome 4P15.31-P15.2 region (reference sequence: NM_006424.2).
Figure 1.
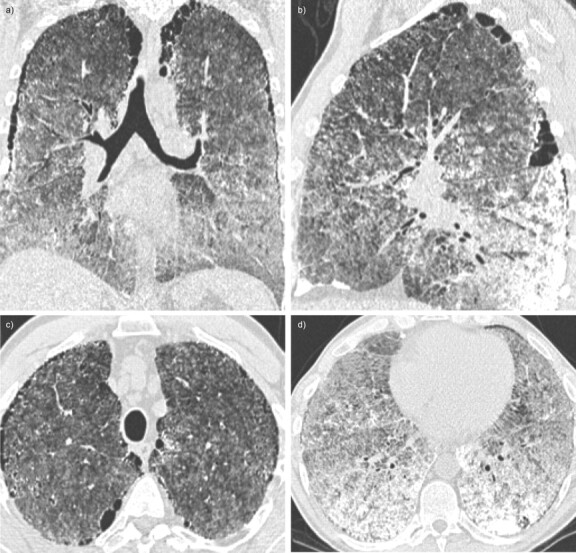
Case 1. High-resolution computed tomography imaging of the lungs showing intra-alveolar calcifications, mainly subpleural, in posterior and basal parts of the lungs. Paraseptal emphysema is visible. a) Coronal view, b) sagittal view and c and d) axial view of the chest.
Figure 2.

Case 1. Chest radiograph of the lungs showing bilateral sand-like calcific micronodules <1 mm (sandstorm lung), mainly involving the lower zones (as a result of larger volumina rather than selective predilection).
Due to clopidogrel treatment, invasive tests were delayed and the patient, therefore, started prednisolone 50 mg daily. The treatment was tapered after 4 months due to lack of efficacy.
Histopathological examination of a lung biopsy showed numerous intra-alveolar rounded calcificated bodies with concentric lamels in the periphery.
In October 2007 the patient started didronate treatment, but it was ceased after 6 months due to lack of effect on symptoms and pulmonary function. He has now been stable for 3.5 yrs without treatment.
In March 2008, a DNA sequencing analysis of whole coding regions of SLC34A2 was performed at Acibadem Genetic Diagnosis Center, Istanbul, Turkey. It showed that the patient had a genotype with a homozygous c.1393-1404delACC mutation in exon 12 of the SLC34A2 gene. The mutation, which results in a deletion of one of the four sequential threonine codons, may have caused the disease by interfering with the structure and function of the protein. Since the mutation has not been reported previously, its association with PAM is unknown. However, given the structure of the mutation, it was considered very likely that this mutation plays a role in the disease of the patient. Family testing could not be performed due to adoption.
CASE TWO
In March 2003, a 16-yr-old Danish male was referred for evaluation of diffuse bilateral infiltrates detected on HRCT. The father of the patient was unknown and, therefore, a complete family history could not be obtained. He presented with fever, cough without sputum, chest pain and fatigue, and had a history of shortness of breath on exertion for as long as he could remember. In childhood, he suffered from several lower respiratory tract infections and asthma. He was a smoker. There was no known family history of respiratory diseases.
Apart from a systolic murmur, the clinical examination was normal. Echocardiography showed bicuspid aortic valve with discrete degenerative changes.
Pulmonary function tests showed a mild restrictive pattern (table 1). Typical radiological findings for PAM were seen on HRCT imaging (fig. 3). In addition, the computed tomography scan revealed a kidney stone.
Figure 3.

Case 2. High-resolution computed tomography of the lungs showing bilateral calcifications with increased attenuation involving alveoli, intra- and interlobular septa, fissures and pleura. Signs of fibrosis are visible. Hounsfield unit measurements were very high, representing areas with calcifications.
Bronchoalveolar lavage (BAL) fluid examination showed no microliths. Histopathological examination of a transbronchial lung biopsy revealed numerous concentric calcified concretions. The histopathology and the typical radiological picture on HRCT confirmed the final diagnosis of PAM.
In June 2005 the patient started didronate treatment which was discontinued 2 yrs later by the patient himself due to a lack of effect on symptoms.
In February 2009, a DNA sequencing analysis of whole coding regions of SLC34A2 was performed at Acibadem Genetic Diagnosis Center and showed a genotype with a homozygous c.1402-1404delACC mutation in exon 12 of the SLC34A2 gene. As for case one, the mutation results in a deletion of one of the four sequential threonine codons, and may have caused the disease by interfering with the structure and function of the protein. This mutation has also never been reported, and its association with PAM is also unknown. However, as for case one, given the structure of the mutation it was considered very likely that this mutation plays a role in the disease of the patient. Family genetic counselling was recommended.
REVIEW
Data source and study selection
A PubMed search with the terms “pulmonary alveolar microlithiasis” was performed. Articles were restricted to those published in the English language with the exception of the study by Puhr [[1]. Additional articles were identified from the reference lists of the identified papers.
Epidemiology
PAM is thought to be an autosomal recessive disease with <600 patients reported worldwide [[6–11]. The disease predominates in a few countries, particularly Turkey, Italy and the USA [[11, 12]. Sporadic and familial cases are reported. Familial cases account for ∼30–50% of the reported cases [[7, 11, 12]. In most reports, no sex predilection was seen [[11]. However, both female [[8] and male [[7, 12] predominance have been described. An excess of males are reported in sporadic cases [[10]. In familial cases, both sexes are affected with equal frequency [[7, 10].
Aetiology
Although the aetiology of PAM remains unclear, mutations of the SLC34A2 gene, which encodes a type IIb sodium-dependent phosphate co-transporter (NaPi-IIb), are considered to be the cause of the disease [[5]. SLC34A2 is primarily expressed in the lung and here only in alveolar type II cells [[5, 13]. These cells are responsible for production of surfactant. Loss of function of the gene due to mutations may lead to a decreased cell uptake of phosphate, which in turn may lead to formation of intra-alveolar microliths as a result of phosphate-chelating calcium in the extracellular fluid [[5, 14].
NaPi-IIb is a member of the solute carrier family SLC34, the sodium-phosphate co-transporter family. The family consists of three members; NaPi-lla (SLC34A1), NaPi-llb (SLC34A2) and NaPi-llc (SLC34A3), all representing type ll sodium/phosphate co-transporters. Type II Na/Pi-co-transporters are expressed in several tissues and play a major role in the homeostasis of inorganic phosphate [15].
Genetic characteristics
There are four earlier reports describing mutations in patients with PAM. Corut et al. [5] examined a large consanguineous family with six affected individuals, and six other unrelated affected patients. A total of six different homozygous mutations in the SLC34A2 gene were reported; two frame-shifts, one chain termination, one amino acid substitution, one deletion and, in the family, a deletion spanning the minimal promoter and exon-1 was detected [5]. Huqun et al. [13] identified two different homozygous mutations in the SLC34A2 gene in six patients, which were unrelated except for two siblings. Inbreeding was certain in three of the patients. A frame-shift mutation was identified in two unrelated patients, and a mutation causing splicing failure, leading to a premature termination of the protein, in the remaining four patients [13]. Ishihara et al. [16] found a homozygous deletion in SLC34A2 gene in one case. Dogan et al. [17] identified a frame-shift mutation in the SLC34A2 gene in a 10-member family. Three siblings were homozygous, and both parents (consanguineous marriage) and one sibling were heterozygous for the mutation. All siblings with the homozygous mutation were diagnosed with PAM [17].
Olauson et al. [18] reported one case with PAM where no disease-causing mutations or single nucleotide polymorphism were found in the SLC34A2 gene. The authors suggested that PAM might be a polygenic disorder arising from mutations other than those in SLC34A2 [18].
The severity of the disease may depend on the type of mutation. Thus, it was found that the mutation in exon 12 causes a truncation of the protein and a possible decreased activity of the protein [5], while mutations in exons 7 and 8 cause total loss of function of the protein [13]. PAM was found to have full penetrance since none of the unaffected members in the large family studied were homozygous for the disease haplotype [5]. Assuming full penetrance, an increased turnover of the alveolar epithelial cells, e.g. due to inflammation or smoking, may influence the onset of the symptoms in patients with a decreased activity of the SLC34A2, and thereby accelerate the development of symptoms. In support of this, smokers had a more severe clinical phenotype than nonsmokers, as observed by Corut et al. [5]. In the two cases presented here, both had a deletion in exon 12, and one may speculate that the early onset of the disease in case two may be due to smoking. However, these associations remain to be investigated in a larger number of patients with PAM.
Clinical features
The disease is usually discovered from birth up to 40 yrs of age, and is mostly diagnosed incidentally during pulmonary radiography of the chest for other reasons [7, 8, 12, 19]. Many patients have no clinical symptoms, and most of the cases either have normal pulmonary function or a mild restrictive pattern [11, 19].
In symptomatic patients, typical complaints are dyspnoea, nonproductive cough, chest pain and asthenia [11]. Although the clinical course varies, the disease is usually slowly progressive. In some patients, the disease remains static, while it in others worsens over time, progressing into pulmonary fibrosis, respiratory failure and cor pulmonale [12, 19, 20].
Elevated serum concentrations of surfactant protein (SP)-A and SP-D have been seen in PAM, with a tendency of increasing levels as the disease progress. Therefore, SP-A and SP-D have been suggested to be useful serum markers in monitoring the disease activity and progression [21].
Although the SLC34A2 gene is involved in phosphate homeostasis in several organs, including the lung [14, 15], blood levels of calcium and phosphate are usually normal in PAM [2, 19, 22, 23]. However, one case has been reported with slightly low normal serum phosphate and low 24-h urinary excretion of phosphate and calcium [18], and another with serum calcium below normal [24]. In addition, a few cases with mild hypercalcaemia [25–27], and one case with hyperphosphataemia have been reported [25]. Interestingly, Olauson et al. [18] found a reduction in the phosphaturic hormone fibroblast growth factor 23 in a case with PAM, suggesting the possibility of a compensatory mechanism for maintaining normal serum phosphate through increased renal reabsorption of phosphate.
Radiological characteristics
The characteristic chest radiograph reveals a picture of fine sand-like micronodulation of calcific densities bilaterally, described as a sandstorm, involving mainly the middle and lower zones [2, 7, 8, 12]. Calcified micronodules, ground-glass opacities, interlobular septal thickening, pleural and subpleural calcification and cysts are typical radiological findings seen in HRCT [17, 28–35].
Pathological characteristics
The radiographic appearance is pathognomonic. However, fibreoptic broncoscopy with BAL showing microliths or histological examination of a lung biopsy (fig. 4) is sometimes required to confirm the diagnosis [7, 8, 12]. Macroscopic examination at autopsy shows heavy and large lungs, and the cut surface is hard due to numerous microliths. Typical findings within the alveolar spaces are numerous calcified bodies, concentrically laminated with an onion skin-like appearance. The microliths usually range from 50 to 1,000 μm in diameter [22, 25, 35–39], although microliths up to 5,000 μm in size have been reported [40]. The microliths are mainly composed of calcium and phosphorus and, to a lesser degree, of varying amounts of iron, magnesium, potassium and copper. Variable degrees of fibrosis are shown in the lung interstitium [22, 25, 36–38, 40].
Figure 4.

Case 1. Lung section from a patient with pulmonary alveolar microlithiasis showing calcified microliths localised in the intra-alveolar spaces. Haematoxylin and eosin staining. Original magnification: a) ×40 and b) ×200.
Extrapulmonary manifestations and comorbidity
As SLC34A2 is expressed in other tissues, mutations in the gene might be involved in extra-pulmonary calcifications. NaPi-IIb has been detected in various tissues such as kidney, prostate, pancreas, small intestine, uterus, ovary, testis, mammary glands, thyroid gland, salivary glands, trachea, liver and placenta [14, 41]. Recently, NaPi-IIb has been identified in a human aortic valve [42].
Extrapulmonary calcifications have been reported in different cases of PAM, such as medullary nephrocalcinosis and/or nephrolithiasis [6, 18, 25, 26, 43], calcification of the lumbar sympathetic chain and possible testicular involvement [23], punctate calcifications in the seminal vesicles [44], and epididymal and periurethral calcifications causing obstructive azospermia [45].
Cardiac comorbidity has only been described in a very few cases of PAM. Four cases with mitral stenosis have been reported, of whom one also had aortic valve stenosis [38, 46, 47]. Furthermore, one case with mitral and aortic valve calcification has been described in a renal transplant recipient [48]. In addition, a case with concomitant valve calcification, cholelithiasis and idiopathic medullary nephrocalcinosis has been described [49]. Two cases with pericardial calcification have also been reported [50, 51].
Reported comorbidities in PAM include milk-alkali syndrome [25], diaphyseal aclasia [52], autosomal recessive Waardenburg-anophthalmia syndrome [53], hypertrophic pulmonary osteoarthropathy [54], pectus excavatum [55] and lymphocytic interstitial pneumonitis [56]. Furthermore, a case with PAM after varicella zoster virus infection, in a patient with antiphospholipid syndrome and discoid lupus erythematosus, has been reported [57].
Extrapulmonary calcifications are not commonly described in earlier reports. This may be due to under-reporting. Another possible explanation could be variability in the penetrance of the mutation. Whether extrapulmonary calcifications are caused by mutations in the SLC34A2 gene remains unclear, further investigations are needed to elucidate the possible connection.
Treatment
There is no known effective treatment for PAM, with the exception of lung transplantation [3, 31, 35, 58–61]. To our knowledge, recurrence of the disease after lung transplantation has not been reported.
Observational studies have found therapeutic BAL to be ineffective [62–64], except for symptomatic improvement in one case, without improvement in conventional pulmonary radiography and HRCT [36].
Corticosteroids are generally considered to be ineffective. However, only a few authors have reported on the effect of corticosteroids. Badger et al. [26] found steroids ineffective after 48 days of treatment. Freiberg et al. [65] reported on one case, in which no additional clinical improvement was seen after 2 weeks of treatment with prednisolone (40 mg daily). In one case, treatment with prednisolone (20 mg daily) for 6 months showed improvement on wheezing and chest tightness, but symptoms recurred when the treatment was discontinued. The patient had normal lung function and the authors discussed the possibility that the patient was suffering from asthma in addition to PAM, and that the symptoms might be caused by an inflammatory reaction due to the microliths or to some other respiratory irritant [36].
Treatment with corticosteroid and hydroxychloroquine showed clinical improvement in an 8-month-old infant [66], and improvement of pulmonary function and exercise tolerance was seen with corticosteroid therapy only in another child with PAM and lymphocytic interstitial pneumonitis [56]. A patient with PAM, after a varicella zoster virus infection, and concomitant antiphospholipid syndrome and discoid lupus erythematosus was treated with inhaled steroids (200 μg budesonide per day) and showed symptomatic improvement [57]. One of our cases (case one) was treated with high doses of prednisolone, before the diagnosis was proven, without any benefits.
A recent report describes beneficial effects of long-term treatment with disodium etidronate in two cases with PAM, diagnosed in childhood (9 and 11 yrs of treatment, respectively) [67]. Treatment with disodium etidronate showed improvement in the general condition and radiological stabilisation in a child with PAM [68]. Other studies have found no significant clinical or radiological improvement with the treatment [69, 70]. Didronate did not have an effect in either of our patients. The lack of efficacy in our patients might be due to an insufficient duration of treatment, the maximum duration of treatment being 2 yrs in case two. Another possible explanation could be that the chelating agents only inhibit the development of microliths, rather than dissolve already formed elements.
In severe PAM with respiratory failure and hypoxia, palliative treatment with oxygen improves subjective daytime function and oxygenation [65].
Long-term course
Long-term survival is still uncertain. There are only a few reports with long-term follow-up data. Furthermore, due to the variability of the clinical course of the disease, the time of the diagnosis may not reflect the onset of the disease.
Some authors have reported survival of >20 yrs [3, 6, 10, 18, 39, 52, 58, 62, 64, 67, 71], and only a few cases with a follow-up period of >40 yrs after the diagnosis have been reported [10, 12, 22]. However, most cases only have up to 10 yrs of follow-up time after the initial diagnosis [3, 6, 9, 23, 25, 31, 36, 38, 47, 48, 54, 56, 58, 59, 61, 66, 68–70, 72, 73]. Tachibana et al. [74] presented data on the long-term course of 53 Japanese patients with PAM. 28 of these patients were diagnosed during childhood. During a follow-up period of 10–49 yrs, 18 out of 53 patients died of respiratory insufficiency due to PAM [74].
CONCLUSION
PAM is a rare parenchymal lung disease often diagnosed incidentally during pulmonary radiography for other reasons. It has only been a few years since its genetic background was discovered, showing that sporadic or familial mutations of the SLC34A2 gene that encodes a type IIb sodium/phosphate co-transporter can cause PAM. In both our patients a new mutation of the SLC34A2 gene has been proven, and in both patients it was considered very likely that the mutations played a role in the disease. Unfortunately, a full genetic family investigation was not possible, due to adoption in case one and the lack of contact to the father of case two. Genetic counselling of families of individuals affected with PAM is important, since better knowledge and understanding of the aetiology and genotype–phenotype correlation may help to develop specific treatment in the future. There are no known prognostic indices and, therefore, identification of specific genotypes combined with long-term follow-up might add to our knowledge, helping us to predict who might progress to end-stage lung disease.
Acknowledgments
We thank H. Hager (Dept of Pathology, Aarhus University Hospital, Aarhus, Denmark) for kindly providing the histological images.
Footnotes
Statement of Interest
None declared.
Provenance
Submitted article, peer reviewed.
REFERENCES
- 1.Puhr L. Mikrolithiasis alveolaris pulmonum. [Pulmonary alveolar microlithiasis]. Virchows Arch (Pathol Anat) 1933; 290: 156. [Google Scholar]
- 2.Chan ED, Morales DV, Welsh CH, et al. Calcium deposition with or without bone formation in the lung. Am J Respir Crit Care Med 2002; 165: 1654–1669. [DOI] [PubMed] [Google Scholar]
- 3.Edelman JD, Bavaria J, Kaiser LR, et al. Bilateral sequential lung transplantation for pulmonary alveolar microlithiasis. Chest 1997; 112: 1140–1144. [DOI] [PubMed] [Google Scholar]
- 4.Ozcelik U, Gulsun M, Gocmen A, et al. Treatment and follow-up of pulmonary alveolar microlithiasis with disodium editronate: radiological demonstration. Pediatr Radiol 2002; 32: 380–383. [DOI] [PubMed] [Google Scholar]
- 5.Corut A, Senyigit A, Ugur SA, et al. Mutations in SLC34A2 cause pulmonary alveolar microlithiasis and are possibly associated with testicular microlithiasis. Am J Hum Genet 2006; 79: 650–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sosman MC, Dodd GD, Jones WD, et al. The familial occurrence of pulmonary alveolar microlithiasis. Am J Roentgenol Radium Ther Nucl Med 1957; 77: 947–1012. [PubMed] [Google Scholar]
- 7.Ucan ES, Keyf AI, Aydilek R, et al. Pulmonary alveolar microlithiasis: review of Turkish reports. Thorax 1993; 48: 171–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mariotta S, Guidi L, Papale M, et al. Pulmonary alveolar microlithiasis: review of Italian reports. Eur J Epidemiol 1997; 13: 587–590. [DOI] [PubMed] [Google Scholar]
- 9.Senyigit A, Yaramis A, Gurkan F, et al. Pulmonary alveolar microlithiasis: a rare familial inheritance with report of six cases in a family. Contribution of six new cases to the number of case reports in Turkey. Respiration 2001; 68: 204–209. [DOI] [PubMed] [Google Scholar]
- 10.Castellana G, Gentile M, Castellana R, et al. Pulmonary alveolar microlithiasis: clinical features, evolution of the phenotype, and review of the literature. Am J Med Genet 2002; 111: 220–224. [DOI] [PubMed] [Google Scholar]
- 11.Mariotta S, Ricci A, Papale M, et al. Pulmonary alveolar microlithiasis: report on 576 cases published in the literature. Sarcoidosis Vasc Diffuse Lung Dis 2004; 21: 173–181. [PubMed] [Google Scholar]
- 12.Castellana G, Lamorgese V. Pulmonary alveolar microlithiasis. World cases and review of the literature. Respiration 2003; 70: 549–555. [DOI] [PubMed] [Google Scholar]
- 13.Huqun, Izumi S, Miyazawa H, et al. Mutations in the SLC34A2 gene are associated with pulmonary alveolar microlithiasis. Am J Respir Crit Care Med 2007; 175: 263–268. [DOI] [PubMed] [Google Scholar]
- 14.Feild JA, Zhang L, Brun KA, et al. Cloning and functional characterization of a sodium-dependent phosphate transporter expressed in human lung and small intestine. Biochem Biophys Res Commun 1999; 258: 578–582. [DOI] [PubMed] [Google Scholar]
- 15.Murer H, Forster I, Biber J. The sodium phosphate cotransporter family SLC34. Pflugers Arch 2004; 447: 763–767. [DOI] [PubMed] [Google Scholar]
- 16.Ishihara Y, Hagiwara K, Zen K, et al. A case of pulmonary alveolar microlithiasis with an intragenetic deletion in SLC34A2 detected by a genome-wide SNP study. Thorax 2009; 64: 365–367. [DOI] [PubMed] [Google Scholar]
- 17.Dogan OT, Ozsahin SL, Gul E, et al. A frame-shift mutation in the SLC34A2 gene in three patients with pulmonary alveolar microlithiasis in an inbred family. Intern Med 2010; 49: 45–49. [DOI] [PubMed] [Google Scholar]
- 18.Olauson H, Brandenburg V, Larsson TE. Mutation analysis and serum FGF23 level in a patient with pulmonary alveolar microlithiasis. Endocrine 2010; 37: 244–248. [DOI] [PubMed] [Google Scholar]
- 19.Prakash UB. Pulmonary alveolar microlithiasis. Semin Respir Crit Care Med 2002; 23: 103–113. [DOI] [PubMed] [Google Scholar]
- 20.Terada T. Pulmonary alveolar microlithiasis with cor pulmonale: an autopsy case demonstrating a marked decrease in pulmonary vascular beds. Respir Med 2009; 103: 1768–1771. [DOI] [PubMed] [Google Scholar]
- 21.Takahashi H, Chiba H, Shiratori M, et al. Elevated serum surfactant protein A and D in pulmonary alveolar microlithiasis. Respirology 2006; 11: 330–333. [DOI] [PubMed] [Google Scholar]
- 22.Moran CA, Hochholzer L, Hasleton PS, et al. Pulmonary alveolar microlithiasis. A clinicopathologic and chemical analysis of seven cases. Arch Pathol Lab Med 1997; 121: 607–611. [PubMed] [Google Scholar]
- 23.Coetzee T. Pulmonary alveolar microlithiasis with involvement of the sympathetic nervous system and gonads. Thorax 1970; 25: 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malhotra B, Sabharwal R, Singh M, et al. Pulmonary alveolar microlithiasis with calcified pleural plaques. Lung India 2010; 27: 250–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Portnoy LM, Amadeo B, Hennigar GR. Pulmonary alveolar microlithiasis. an unusual case (associated with milk-alkali syndrome). Am J Clin Pathol 1964; 41: 194–201. [DOI] [PubMed] [Google Scholar]
- 26.Badger TL, Gottlieb L, Gaensler EA. Pulmonary alveolar microlithiasis, or calcinosis of the lungs. N Engl J Med 1955; 253: 709–715. [DOI] [PubMed] [Google Scholar]
- 27.Pant K, Chawla R, Chhabra SK, et al. Pulmonary alveolar microlithiasis – a case report and review of literature. Indian J Chest Dis Allied Sci 1988; 30: 141–147. [PubMed] [Google Scholar]
- 28.Deniz O, Ors F, Tozkoparan E, et al. High resolution computed tomographic features of pulmonary alveolar microlithiasis. Eur J Radiol 2005; 55: 452–460. [DOI] [PubMed] [Google Scholar]
- 29.Sumikawa H, Johkoh T, Tomiyama N, et al. Pulmonary alveolar microlithiasis: CT and pathologic findings in 10 patients. Monaldi Arch Chest Dis 2005; 63: 59–64. [DOI] [PubMed] [Google Scholar]
- 30.de Laurentiis G, Vitiello L, Racioppi L, et al. CD8+ T-cell alveolitis in familial pulmonary alveolar microlithiasis. Eur Respir J 2007; 30: 165–171. [DOI] [PubMed] [Google Scholar]
- 31.Samano MN, Waisberg DR, Canzian M, et al. Lung transplantation for pulmonary alveolar microlithiasis: a case report. Clinics (Sao Paulo) 2010; 65: 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gasparetto EL, Tazoniero P, Escuissato DL, et al. Pulmonary alveolar microlithiasis presenting with crazy-paving pattern on high resolution CT. Br J Radiol 2004; 77: 974–976. [DOI] [PubMed] [Google Scholar]
- 33.Saleem A, Chaudhary A, Iqbal ZH. Pulmonary alveolar microlithiasis. J Coll Physicians Surg Pak 2009; 19: 391–392. [PubMed] [Google Scholar]
- 34.Marchiori E, Goncalves CM, Escuissato DL, et al. Pulmonary alveolar microlithiasis: high-resolution computed tomography findings in 10 patients. J Bras Pneumol 2007; 33: 552–557. [DOI] [PubMed] [Google Scholar]
- 35.Siddiqui NA, Fuhrman CR. Best cases from the AFIP: pulmonary alveolar microlithiasis. Radiographics 2011; 31: 585–590. [DOI] [PubMed] [Google Scholar]
- 36.Pracyk JB, Simonson SG, Young SL, et al. Composition of lung lavage in pulmonary alveolar microlithiasis. Respiration 1996; 63: 254–260. [DOI] [PubMed] [Google Scholar]
- 37.Saputo V, Zocchi M, Mancosu M, et al. Pulmonary alveolar microlithiasis. A case report with a discussion of differential diagnosis. Helv Paediatr Acta 1979; 34: 245–255. [PubMed] [Google Scholar]
- 38.Sharp ME, Danino EA. An unusual form of pulmonary calcification: microlithiasis alveolaris pulmonum. J Pathol Bacteriol 1953; 65: 389–399. [DOI] [PubMed] [Google Scholar]
- 39.Kawakami M, Sato S, Takishima T. Electron microscopic studies on pulmonary alveolar microlithiasis. Tohoku J Exp Med 1978; 126: 343–361. [DOI] [PubMed] [Google Scholar]
- 40.Barnard NJ, Crocker PR, Blainey AD, et al. Pulmonary alveolar microlithiasis. A new analytical approach. Histopathology 1987; 11: 639–645. [DOI] [PubMed] [Google Scholar]
- 41.Xu H, Bai L, Collins JF, et al. Molecular cloning, functional characterization, tissue distribution, and chromosomal localization of a human, small intestinal sodium-phosphate (Na+-Pi) transporter (SLC34A2). Genomics 1999; 62: 281–284. [DOI] [PubMed] [Google Scholar]
- 42.Jönsson ÅLM, Hilberg O, Bendstrup EM, et al. SLC34A2 gene mutation may explain co-morbidity of pulmonary alveolar microlithiasis and aortic valve sclerosis. Am J Respir Crit Care Med 2012; 185: 464. [DOI] [PubMed] [Google Scholar]
- 43.Pant K, Shah A, Mathur RK, et al. Pulmonary alveolar microlithiasis with pleural calcification and nephrolithiasis. Chest 1990; 98: 245–246. [DOI] [PubMed] [Google Scholar]
- 44.Arslan A, Yalin T, Akan H, et al. Pulmonary alveolar microlithiasis associated with calcifications in the seminal vesicles. J Belge Radiol 1996; 79: 118–119. [PubMed] [Google Scholar]
- 45.Kanat F, Teke T, Imecik O. Pulmonary alveolar microlithiasis with epididymal and periurethral calcifications causing obstructive azospermia. Int J Tuberc Lung Dis 2004; 8: 1275. [PubMed] [Google Scholar]
- 46.Yesner R, Skorneck AB. Pulmonary alveolar microlithiasis in mitral stenosis. Am J Pathol 1956; 32: 611. [Google Scholar]
- 47.Lawson HM. Disseminated ossification of the lungs in association with mitral stenosis. Br Med J 1949; 1: 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Richardson J, Slovis B, Miller G, et al. Development of pulmonary alveolar microlithiasis in a renal transplant recipient. Transplantation 1995; 59: 1056–1057. [PubMed] [Google Scholar]
- 49.Giallauria F, Giallauria G. Pulmonary alveolar microlithiasis. Thorax 2011; 66: 840. [DOI] [PubMed] [Google Scholar]
- 50.Verma BN. Pulmonary alveolar microlithiasis in a child of thirteen years. Br J Dis Chest 1963; 57: 213–215. [DOI] [PubMed] [Google Scholar]
- 51.Kiatboonsri S, Charoenpan P, Vathesatogkit P, et al. Pulmonary alveolar microlithiasis: report of five cases and literature review. J Med Assoc Thai 1985; 68: 672–677. [PubMed] [Google Scholar]
- 52.Ritchie DA, O'Connor SA, McGivern D. An unusual presentation of pulmonary alveolar microlithiasis and diaphyseal aclasia. Br J Radiol 1992; 65: 178–181. [DOI] [PubMed] [Google Scholar]
- 53.Schmidt H, Lorcher U, Kitz R, et al. Pulmonary alveolar microlithiasis in children. Pediatr Radiol 1996; 26: 33–36. [DOI] [PubMed] [Google Scholar]
- 54.Emri S, Coplu L, Selcuk ZT, et al. Hypertrophic pulmonary osteoarthropathy in a patient with pulmonary alveolar microlithiasis. Thorax 1991; 46: 145–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hasan S, Cevat O, Sami C, et al. Pulmonary alveolar microlithiasis with pectus excavatum. Case report. Indian J Med Sci 1994; 48: 199–200. [PubMed] [Google Scholar]
- 56.Ratjen FA, Schoenfeld B, Wiesemann HG. Pulmonary alveolar microlithiasis and lymphocytic interstitial pneumonitis in a ten year old girl. Eur Respir J 1992; 5: 1283–1285. [PubMed] [Google Scholar]
- 57.Yilmaz MI, Koc B, Kantarcioglu M, et al. Pulmonary alveolar microlithiasis after Varicella zoster infection in a patient presenting with antiphospholipid syndrome and discoid lupus. Rheumatol Int 2002; 22: 213–215. [DOI] [PubMed] [Google Scholar]
- 58.Shigemura N, Bermudez C, Hattler BG, et al. Lung transplantation for pulmonary alveolar microlithiasis. J Thorac Cardiovasc Surg 2010; 139: e50–e52. [DOI] [PubMed] [Google Scholar]
- 59.Raffa H, El-Dakhakhny M, Al-Ibrahim K, et al. Single lung transplantation for alveolar micro-lithiasis: the first clinical report. Saudi J Kidney Dis Transpl 1996; 7: 189–193. [PubMed] [Google Scholar]
- 60.Bonnette P, Bisson A, el Kadi NB, et al. Bilateral single lung transplantation. Complications and results in 14 patients. Eur J Cardiothorac Surg 1992; 6: 550–554. [DOI] [PubMed] [Google Scholar]
- 61.Jackson KB, Modry DL, Halenar J, et al. Single lung transplantation for pulmonary alveolar microlithiasis. J Heart Lung Transplant 2001; 20: 226. [DOI] [PubMed] [Google Scholar]
- 62.Mascie-Taylor BH, Wardman AG, Madden CA, et al. A case of alveolar microlithiasis: observation over 22 years and recovery of material by lavage. Thorax 1985; 40: 952–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Palombini BC, da Silva Porto N, Wallau CU, et al. Bronchopulmonary lavage in alveolar microlithiasis. Chest 1981; 80: 242–243. [DOI] [PubMed] [Google Scholar]
- 64.Chalmers AG, Wyatt J, Robinson PJ. Computed tomographic and pathological findings in pulmonary alveolar microlithiasis. Br J Radiol 1986; 59: 408–411. [DOI] [PubMed] [Google Scholar]
- 65.Freiberg DB, Young IH, Laks L, et al. Improvement in gas exchange with nasal continuous positive airway pressure in pulmonary alveolar microlithiasis. Am Rev Respir Dis 1992; 145: 1215–1216. [DOI] [PubMed] [Google Scholar]
- 66.Dahabreh M, Najada A. Pulmonary alveolar microlithiasis in an 8-month-old infant. Ann Trop Paediatr 2009; 29: 55–59. [DOI] [PubMed] [Google Scholar]
- 67.Ozcelik U, Yalcin E, Ariyurek M, et al. Long-term results of disodium etidronate treatment in pulmonary alveolar microlithiasis. Pediatr Pulmonol 2010; 45: 514–517. [DOI] [PubMed] [Google Scholar]
- 68.Gocmen A, Toppare MF, Kiper N, et al. Treatment of pulmonary alveolar microlithiasis with a diphosphonate – preliminary results of a case. Respiration 1992; 59: 250–252. [DOI] [PubMed] [Google Scholar]
- 69.Jankovic S, Pavlov N, Ivkosic A, et al. Pulmonary alveolar microlithiasis in childhood: clinical and radiological follow-up. Pediatr Pulmonol 2002; 34: 384–387. [DOI] [PubMed] [Google Scholar]
- 70.Mariotta S, Guidi L, Mattia P, et al. Pulmonary microlithiasis. Report of two cases. Respiration 1997; 64: 165–169. [DOI] [PubMed] [Google Scholar]
- 71.Prakash UB, Barham SS, Rosenow EC, 3rd, et al. Pulmonary alveolar microlithiasis a review including ultrastructural and pulmonary function studies. Mayo Clin Proc 1983; 58: 290–300. [PubMed] [Google Scholar]
- 72.Thurairajasingam S, Dharmasena BD, Kasthuriratna T. Pulmonary alveolar microlithiasis. Australas Radiol 1975; 19: 175–180. [DOI] [PubMed] [Google Scholar]
- 73.Volle E, Kaufmann HJ. Pulmonary alveolar microlithiasis in pediatric patients – review of the world literature and two new observations. Pediatr Radiol 1987; 17: 439–442. [DOI] [PubMed] [Google Scholar]
- 74.Tachibana T, Hagiwara K, Johkoh T. Pulmonary alveolar microlithiasis: review and management. Curr Opin Pulm Med 2009; 15: 486–490. [DOI] [PubMed] [Google Scholar]