

Abstract
Despite major advances in the understanding of the pathogenesis of idiopathic pulmonary fibrosis (IPF), diagnosis and management of the condition continue to pose significant challenges. Clinical management of IPF remains unsatisfactory due to limited availability of effective drug therapies, a lack of accurate indicators of disease progression, and an absence of simple short-term measures of therapeutic response. The identification of more accurate predictors of prognosis and survival in IPF would facilitate counseling of patients and their families, aid communication among clinicians, and would guide optimal timing of referral for transplantation. Improvements in molecular techniques have led to the identification of new disease pathways and a more targeted approach to the development of novel anti-fibrotic agents. However, despite an increased interest in biomarkers of IPF disease progression there are a lack of measures that can be used in early phase clinical trials. Careful longitudinal phenotyping of individuals with IPF together with the application of novel omics-based technology should provide important insights into disease pathogenesis and should address some of the major issues holding back drug development in IPF. The PROFILE (Prospective Observation of Fibrosis in the Lung Clinical Endpoints) study is a currently enrolling, prospective cohort study designed to tackle these issues.
Keywords: Biomarkers, clinical management, idiopathic pulmonary fibrosis, pharmacological treatment, PROFILE study, prognosis
Idiopathic pulmonary fibrosis (IPF) is a progressive, chronic, debilitating disease of unknown aetiology and a median survival from diagnosis of only 2.8–4.2 years [1]. Epidemiology studies suggest that the incidence of IPF has been increasing steadily over the last two to three decades [2, 3]. The reasons for this are unknown but potential risk factors for the development of IPF include cigarette smoking, wood, mineral and metal dust exposure, past viral infection, and chronic gastro-oesophageal reflux with micro-aspiration [4]. Although the pathogenesis of the condition is incompletely understood, current paradigms highlight the role of an aberrant wound healing response following repetitive alveolar injury in genetically susceptible individuals [1]. Whilst understanding of the clinical and molecular characteristics of IPF have increased exponentially since the publication of the first American Thoracic Society (ATS)/European Respiratory Society (ERS) consensus guidelines in 2002 [5], there remain a number of significant barriers to the development of novel therapies for this devastating condition.
DIAGNOSING IPF: THE LACK OF A GOLD STANDARD
The reclassification of the idiopathic interstitial pneumonias (IIPs) in 2002 represented a watershed in the clinical understanding of IPF [5]. For the first time, a diagnosis of IPF was confined to individuals with the histological lesion of usual interstitial pneumonia (UIP). Better prognosis histological lesions, including nonspecific interstitial pneumonia (NSIP) and desquamative interstitial pneumonitis, were no longer considered part of the spectrum of IPF. In the decade since the publication of the ATS/ERS consensus guidelines, the IPF clinical community has generated considerable research that has led to better characterisation of the IPF disease phenotype. This has enabled a clearer understanding of the role of high-resolution computed tomography (HRCT), surgical lung biopsy and multi-disciplinary team assessment in the diagnosis of IPF [6].
Despite diagnostic improvements a number of challenges remain when it comes to diagnosing IPF. First, only about two-thirds of patients with IPF have a “classical” HRCT appearance. Patients with atypical HRCT features require a surgical lung biopsy. Secondly, there is considerable overlap in the histological appearances of the different IIPs. UIP shares a number of features with NSIP. In some cases of UIP/IPF there are areas of organising pneumonia or even NSIP on biopsy. In cases of acute exacerbation of IPF the finding of diffuse alveolar damage is identical to that seen in acute interstitial pneumonia and adult respiratory distress syndrome (albeit occurring on a background of established fibrosis). As a consequence of this overlap in histological appearances, there is considerable inter-observer variation in the reporting of IIP biopsies, with only modest agreement between expert interstitial lung disease (ILD) pathologists [7]. Finally, UIP is seen not only in IPF but also in other conditions, particularly connective tissue disease-associated ILD and hypersensitivity pneumonitis. A number of groups have shown that a small proportion of individuals who, at first presentation, appear to have IPF, subsequently manifest features of systemic autoimmune disease (sometimes many years after the onset of ILD) [8, 9]. For these reasons, and because of the potential morbidity and mortality associated with surgical lung biopsy, there is a great need for better, minimally invasive, diagnostic tests to discriminate IPF from other ILDs.
PREDICTING DISEASE PROGRESSION
The clinical course of individual patients with IPF is highly variable and unpredictable. Insights gained from clinical trials have demonstrated that IPF patients may, as shown in figure 1, exhibit any of several distinct patterns of disease progression [10, 11]. In most patients, IPF progresses slowly with inexorable decline in lung function occurring only over a period of years. However, 10–15% of individuals have a much more rapid disease course progressing from first symptoms to death from respiratory failure over a period of months. The third pattern of disease progression is that of periods of relatively slow decline punctuated by episodes of rapid acute deterioration (acute exacerbations). These episodes often prove to be fatal but, if survived, invariably lead to a permanent step down in pulmonary function [12].
Figure 1.

Schematic representation of the potential clinical courses of idiopathic pulmonary fibrosis. As disease progresses there is a subclinical period in which only radiographic findings of disease may be present, followed by a symptomatic period consisting of both pre- and post-diagnosis clinical phases. The rate of decline and progression to death may be rapid (A), slow (C and D) or mixed (B), with periods of relative stability interposed with periods of acute decline (#). Reproduced from [11] with permission.
Whilst it is possible to recognise these dramatically different patterns of disease behaviour retrospectively, it is not possible, with currently available clinical tools, to identify at diagnosis how any given individual's disease will progress in the future. Prospective identification of disease behaviour would enable better tailoring of therapeutic regimens and would permit the development of strategies to prevent or ameliorate acute exacerbations.
IDENTIFYING KEY DISEASE MECHANISMS
As already alluded to, IPF is a complex, multi-pathway disease and the pathologic processes underlying both the initiation and progression of the condition are not well understood [1]. It is clear that, at a molecular level, there is activation and persistence of many, if not all, of the key pathways involved in the normal wound healing response. Furthermore, there is involvement of a diverse range of cell types including epithelial cells, fibroblasts, endothelial cells, and circulating and tissue resident inflammatory cells. As such the process of fibrogenesis involves the complex interplay of a vast array of chemical messengers and signalling pathways, many of which display considerable pleiotropism and redundancy.
The enormous complexity of the molecular processes driving the development of fibrosis, and the fact that it is usually only possible to study established disease, makes it is very hard to be certain which of the activated pathways seen in IPF represent primary drivers of the condition and which are simply secondary or bystander phenomenon. Because of this there is a bewildering array of potential therapeutic targets in IPF but a lack of pre-clinical tools to enable selection of those that are most likely to lead to the development of effective therapies [13]. Only by developing better pre-clinical assays will it be possible to intelligently select drugs and targets that should be developed through to therapeutic trials.
CLINICAL TRIAL DESIGN
Clinical trials in IPF remain in their infancy, with the first true multicentre, randomised controlled trial only being published as recently as 2004 [14]. Since then the majority of studies undertaken in IPF have been opportunistic; that is they have been carried out with compounds generally developed for other indications rather than designed specifically as anti-fibrotic agents (pirfenidone can be considered the one exception to this phenomenon) [15]. In most cases these drugs have been tested directly in phase IIB or even phase III trials [16, 17]. Whilst there has been a clear evolution in late-phase clinical trial design in IPF, with better understanding of the relative value of the different end-points measured (notwithstanding the current vigorous debate about the preferable use of forced vital capacity over mortality as an end-point in registration trials) [18–21], the same cannot be said for proof-of-concept or proof-of-mechanism studies. Appropriate, validated, short-term read-outs that provide confidence that a novel therapy has engaged both its target and is blocking key fibrotic pathways are lacking. Until such markers are available it will be difficult to design short, early phase and dose-validation studies for IPF. Given the large number of potential targets in IPF there is a major need for proof-of-concept studies as a means of narrowing down candidate target and molecule selection and to expedite the passage of the most promising drugs from early clinical development through to registration studies and then the clinic.
DISEASE STRATIFICATION
The heterogeneity seen in IPF phenotypes and disease progression, and the complex pathogenetic mechanisms at play, make it unlikely that a single therapy will be effective for all patients with the condition. In the oncology field, disease stratification and the tailoring of therapies to individual patients has seen remarkable improvements in outcome for many cancers [22]. In asthma it has recently been demonstrated that serum periostin levels predict a group of patients more likely to respond to anti-interleukin-13 therapy [23]. It is to be anticipated that a similar approach to disease stratification in IPF could be used to identify individuals most likely to benefit from specific therapeutic approaches. Whilst the treatment options for IPF remain limited there is little need for stratification, but as new (and expensive) treatments emerge, personalisation of therapeutic regimens will become increasingly important; so as to maximise benefit for individuals whilst at the same time minimise harm from adverse effects.
A PATHWAY TO BETTER UNDERSTANDING
Having identified the key barriers to the development of effective treatments for IPF what can be done to rectify these problems? For most diseases, development of the condition and the subsequent clinical phenotype are influenced by the complex interaction of host and environment factors. At the simplest level, an individual's genetic make-up determines how their genes will be transcribed into RNA and then translated into proteins. This in turn influences disease susceptibility and progression. However, this uni-dimensional view of the genotype–phenotype relationship fails to account for the effect of environmental exposures and host responses in influencing the development of disease. It is now recognised that the intricate interplay of a number of important processes can influence and change an individual's phenotype overtime. Processes such as epigenetic and microRNA regulation of DNA and RNA can lead to alterations in gene transcription that then change an individual's likelihood of developing disease. In turn the microbiome (the totality of the bacteria inhabiting the human body) increasingly appears to have important effects in influencing both health and disease. All these elements can, in themselves, be affected by environmental factors, e.g. pollution or cigarette smoke. Figure 2 illustrates the combination of host and environment factors that interact to determine an individual's clinical phenotype.
Figure 2.

A representation of the key factors controlling host–environment interactions and influencing individual phenotypes and, therefore, susceptibility to disease. Transcription of genes to RNA with translation to proteins is controlled by epigenetic modification and is subject to regulation by microRNAs. The role played by proteins (as receptors, growth factors and second messengers) is in turn influenced by a variety of post-translational modifications including glycosylation, citrullination and phosphorylation, all of which may be altered by disease (e.g. diabetes) or the environment (e.g. the host microbiome, pollution, cigarette smoke, etc.).
Modern biological techniques combined with computational biology methods are enabling a much clearer understanding of host–environment interactions and their relationship to disease. The application of omics-based technology (defined as the study of all constituents of a specific area collectively, e.g. genomics is the study of all of the genes in an organism) to carefully phenotyped, prospectively identified and longitudinally followed disease cohorts affords the promise of deeper understanding of the factors that contribute to disease susceptibility and progression. This amalgamation of clinical information and omic data, as shown in figure 3, provides identification of a specific “fingerprint” of a disease. Such a fingerprint should better enable the identification of key pathogenic mechanisms, and provide a guide to potential therapeutic targets. It is also hoped that disease fingerprinting will permit the discovery of appropriate disease biomarkers that might, in turn, enable better measurement of future disease behaviour and short-term measurement of response to therapy.
Figure 3.
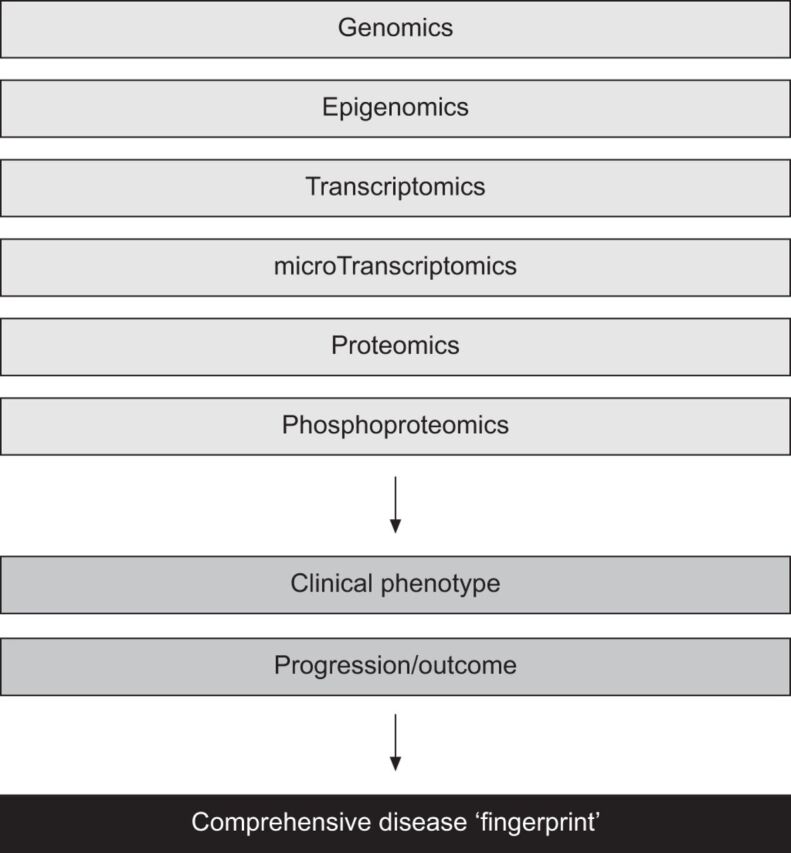
Developing a disease fingerprint. The combination of modern omic-based techniques together with careful longitudinal phenotyping of disease cohorts permits the development of a specific disease signature or fingerprint. Adopting such an approach should provide novel disease insights that inform understanding of disease biology and identification of therapeutic targets. Disease fingerprinting will also enable the development of diagnostic tools and should facilitate biomarker discovery, which in turn should improve early phase clinical trial design.
ADDRESSING THE PROBLEMS IN IPF: THE PROFILE STUDY
A number of omics-based studies have been undertaken in IPF [24–27]. However, in almost all cases these have relied on single point of diagnosis samples gathered opportunistically in relatively small, single centre populations. Furthermore, none of these studies has integrated the results of genetic, epigenetic and transcriptomic analysis with longitudinal phenotype to provide a true IPF disease fingerprint. To address this deficit the PROFILE (Prospective Observation of Fibrosis in the Lung Clinical Endpoints) study (www.clinicaltrials.gov NCT01110694 and NCT01134822), a UK-based, multicentre, prospective cohort study of individuals newly diagnosed with IPF, was launched in 2010. The study is recruiting individuals from both tertiary specialist ILD units and from local secondary care hospitals. Enrolled subjects are undergoing routine clinical assessment and blood sample collection at seven pre-specified time-points from diagnosis through to 3 years.
The aims of the PROFILE study are: 1) to prospectively validate a panel of previously published biomarkers in patients with well-characterised IPF; 2) to undertake open platform omic-based analysis of biological samples with a view to applying computational biology methodology to deliver a comprehensive disease fingerprint; and 3) to prospectively evaluate longitudinal disease behaviour in patients with IPF with a view to developing composite biological and clinical end-points for subsequent use in intervention studies. Overall it is hoped that the study cohort will inform understanding of IPF pathogenesis, permit development of better clinical tools to aid diagnosis and prognostication, enable better early phase clinical trial design and drive the development of novel therapeutics. As of January 2013 the study has enrolled over 300 participants.
CONCLUSION
The increasing incidence and devastating prognosis of IPF makes the search for new and effective therapies a priority for the respiratory research community. Better understanding of IPF, through careful longitudinal disease phenotyping and the application of omics-based technologies to large cohorts of patients, affords the opportunity to overcome the barriers hindering the development of new treatments for IPF. The ongoing PROFILE study will hopefully provide insight into IPF disease pathogenesis whilst also enabling biomarker discovery. It is hoped that as the results from the PROFILE study become available they will provide impetus to the translational development of novel and effective treatments for IPF.
Supplementary Material
Acknowledgments
This talk was presented at the 17th International Colloquium on Lung and Airway Fibrosis (ICLAF), Modena, Italy. It can be viewed online at: http://bit.ly/ZubFdu
I would like to thank all the individuals who have participated in the development of the PROFILE study, especially: Gisli Jenkins the principal investigator of PROFILE Central England (University of Nottingham, Nottingham, UK), and Pauline Lukey, Juliet Simpson and Richard Marshall (GSK R&D, Stevenage, UK).
Footnotes
Provenance
Publication of this peer-reviewed article was supported by InterMune Inc., USA (principal sponsor, European Respiratory Review issue 128).
Support Statement
The PROFILE study was funded through an unrestricted academic industry grant (COL29296) from the Fibrosis DPU (GlaxoSmithKline, Stevenage, UK) as part of the CRAFT consortium.
Statement of Interest
Conflict of interest information can be found alongside the online version of this article at err.ersjournals.com
REFERENCES
- 1.Maher TM, Wells AU, Laurent GJ. Idiopathic pulmonary fibrosis: multiple causes and multiple mechanisms? Eur Respir J 2007; 30: 835–839. [DOI] [PubMed] [Google Scholar]
- 2.Navaratnam V, Fleming KM, West J, et al. The rising incidence of idiopathic pulmonary fibrosis in the UK. Thorax 2011; 66: 462–467. [DOI] [PubMed] [Google Scholar]
- 3.Raghu G, Weycker D, Edelsberg J, et al. Incidence and prevalence of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2006; 174: 810–816. [DOI] [PubMed] [Google Scholar]
- 4.Demedts M, Wells AU, Anto JM, et al. Interstitial lung diseases: an epidemiological overview. Eur Respir J 2001; 18: Suppl. 32, 2s–16s. [PubMed] [Google Scholar]
- 5.American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society. International multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002; 165: 277–304. [DOI] [PubMed] [Google Scholar]
- 6.Flaherty KR, King TE, Jr, Raghu G, et al. Idiopathic interstitial pneumonia: what is the effect of a multidisciplinary approach to diagnosis? Am J Respir Crit Care Med 2004; 170: 904–910. [DOI] [PubMed] [Google Scholar]
- 7.Nicholson AG, Addis BJ, Bharucha H, et al. Inter-observer variation between pathologists in diffuse parenchymal lung disease. Thorax 2004; 59: 500–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kinder BW, Collard HR, Koth L, et al. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med 2007; 176: 691–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corte TJ, Copley SJ, Desai SR, et al. Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur Respir J 2012; 39: 661–668. [DOI] [PubMed] [Google Scholar]
- 10.Martinez FJ, Safrin S, Weycker D, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med 2005; 142: 963–967. [DOI] [PubMed] [Google Scholar]
- 11.Ley B, Collard HR, King TE, Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 183: 431–440. [DOI] [PubMed] [Google Scholar]
- 12.Collard HR, Moore BB, Flaherty KR, et al. Acute exacerbations of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2007; 176: 636–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maher TM. Idiopathic pulmonary fibrosis: pathobiology of novel approaches to treatment. Clin Chest Med 2012; 33: 69–83. [DOI] [PubMed] [Google Scholar]
- 14.Raghu G, Brown KK, Bradford WZ, et al. A placebo-controlled trial of interferonγ-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med 2004; 350: 125–133. [DOI] [PubMed] [Google Scholar]
- 15.Maher TM. Pirfenidone in idiopathic pulmonary fibrosis. Drugs Today (Barc) 2010; 46: 473–482. [DOI] [PubMed] [Google Scholar]
- 16.King TE, Jr, Brown KK, Raghu G, et al. BUILD-3: a randomized, controlled trial of bosentan in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2011; 184: 92–99. [DOI] [PubMed] [Google Scholar]
- 17.Demedts M, Behr J, Buhl R, et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N Engl J Med 2005; 353: 2229–2242. [DOI] [PubMed] [Google Scholar]
- 18.Raghu G, Collard HR, Anstrom KJ, et al. Idiopathic pulmonary fibrosis: clinically meaningful primary endpoints in phase 3 clinical trials. Am J Respir Crit Care Med 2012; 185: 1044–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vancheri C, Du Bois RM. A progression-free end-point for idiopathic pulmonary fibrosis trials: lessons from cancer. Eur Respir J 2013; 41: 262–269. [DOI] [PubMed] [Google Scholar]
- 20.Du Bois RM, Nathan SD, Richeldi L, et al. Idiopathic pulmonary fibrosis: lung function is a clinically meaningful endpoint for phase III trials. Am J Respir Crit Care Med 2012; 186: 712–715. [DOI] [PubMed] [Google Scholar]
- 21.Wells AU, Behr J, Costabel U, et al. Hot of the breath: mortality as a primary end-point in IPF treatment trials: the best is the enemy of the good. Thorax 2012; 67: 938–940. [DOI] [PubMed] [Google Scholar]
- 22.Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366: 1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corren J, Mansfield LE, Pertseva T, et al. Efficacy and safety of fluticasone/formoterol combination therapy in patients with moderate-to-severe asthma. Resp Med 2013; 107: 180–195. [DOI] [PubMed] [Google Scholar]
- 24.Rosas IO, Richards TJ, Konishi K, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med 2008; 5: e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Selman M, Carrillo G, Estrada A, et al. Accelerated variant of idiopathic pulmonary fibrosis: clinical behavior and gene expression pattern. PLoS One 2007; 2: e482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Selman M, Pardo A, Barrera L, et al. Gene expression profiles distinguish idiopathic pulmonary fibrosis from hypersensitivity pneumonitis. Am J Respir Crit Care Med 2006; 173: 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang IV, Luna LG, Cotter J, et al. The peripheral blood transcriptome identifies the presence and extent of disease in idiopathic pulmonary fibrosis. PLoS One 2012; 7: e37708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.